
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 02 April 2015
Sec. Cancer Molecular Targets and Therapeutics
Volume 5 - 2015 | https://doi.org/10.3389/fonc.2015.00069
 Rachana Trivedi
Rachana Trivedi Durga Prasad Mishra*
Durga Prasad Mishra*Resistance to chemotherapeutic drugs is the major hindrance in the successful cancer therapy. The tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) is a member of the tumor necrosis factor (TNF) family of ligands, which initiates apoptosis in cancer cells through interaction with the death receptors DR4 and DR5. TRAIL is perceived as an attractive chemotherapeutic agent as it specifically targets cancer cells while sparing the normal cells. However, TRAIL therapy has a major limitation as a large number of the cancer develop resistance toward TRAIL and escape from the destruction by the immune system. Therefore, elucidation of the molecular targets and signaling pathways responsible for TRAIL resistance is imperative for devising effective therapeutic strategies for TRAIL resistant cancers. Although, various molecular targets leading to TRAIL resistance are well-studied, recent studies have implicated that the contribution of some key cellular processes toward TRAIL resistance need to be fully elucidated. These processes primarily include aberrant protein synthesis, protein misfolding, ubiquitin regulated death receptor expression, metabolic pathways, epigenetic deregulation, and metastasis. Novel synthetic/natural compounds that could inhibit these defective cellular processes may restore the TRAIL sensitivity and combination therapies with such compounds may resensitize TRAIL resistant cancer cells toward TRAIL-induced apoptosis. In this review, we have summarized the key cellular processes associated with TRAIL resistance and their status as therapeutic targets for novel TRAIL-sensitizing agents.
Pre-existing or acquired resistance to chemotherapy is a major obstacle in effective cancer therapy, as it often leads to the therapy failure and the disease relapse (1). Therefore, there is an ever increasing need for development of safe drugs and novel therapeutic strategies for targeting these chemo-resistant cancer cells. It is imperative that these chemotherapeutic agents or strategies should selectively target the cancer cells in an irreversible manner without harming the normal cells (2). However, poor tolerance and chemotherapy associated side effects are still major hurdles in therapeutic targeting of chemo-resistant cancer cells. Chemotherapeutic drugs can target cancer cells through inhibition of cellular proliferation and survival or induction of cell cycle arrest and apoptosis. Among these processes, apoptosis is an evolutionarily conserved and the most widely studied cellular response, essential for maintenance of tissue homeostasis and removal of unwanted cells (3). Apoptosis is triggered by either the intrinsic or extrinsic stimuli. The intrinsic pathway of apoptosis includes cellular damage brought about by stress, ultraviolet (UV) or ionizing radiation, or oncogene activation. On the other hand, the extrinsic pathway of apoptosis is triggered by the binding of extracellular ligands to specific transmembrane receptors resulting in caspase activation and cell death (2, 4). The tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) belongs to the group of chemotherapeutic agents selectively targeting a wide variety of cancer cells without affecting the normal cells (5–8). The therapeutic potential of TRAIL is attributed to its receptor expression in a variety of tissues like lymphocytes, spleen, thymus, ovary, prostate, colon, intestine, and placenta compared to the restricted and transient expression of other ligands of the TNF family (8). Therefore, TRAIL is considered as a promising and effective anticancer agent under clinical investigation (9, 10). The therapeutic usage of other members of the TNF super family like Fas/FasL and TNFα/TNFR1 are limited due to their severe side effects including lethal septic shock like responses (11). The Fas/FasL and TNFα/TNFR1 are known to activate the oncogenic NF-κB pathway while TRAIL shows weak effects on NF-κB activation, and is therefore considered safe as a therapeutic agent. It also plays an important role in the natural killer cell mediated immunosurveillance against the rapidly growing and metastatic cancer cells (8). Pre-clinical studies have shown that administration of the soluble form of the recombinant TRAIL in mice and non-human primates suppressed the proliferation of TRAIL sensitive human tumor xenografts, with no apparent systemic toxicity underscoring the potential utility of rhTRAIL in vivo (6, 7). However, the major limitation of the TRAIL therapy is development of TRAIL resistance through a variety of mechanisms in cancer cells. Therefore, to enhance the TRAIL mediated apoptotic effect, the combination of TRAIL along with novel TRAIL sensitizing agents possibly represents the best clinical option (Table 1).
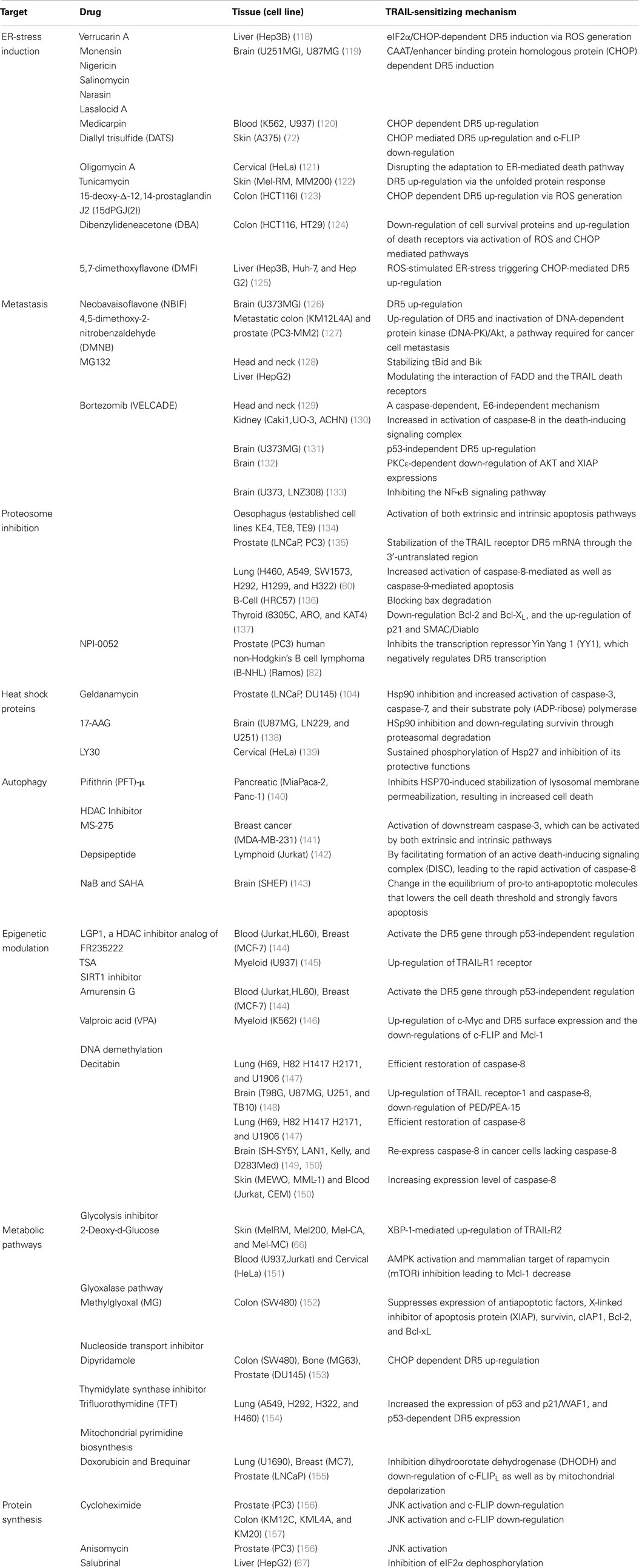
Table 1. Small molecule with TRAIL sensitization ability.
TRAIL is a member of the TNF-related proteins having structural and functional similarity with CD95L. TRAIL is a 20 kDa protein encoded by a gene with five exons and three introns located on the chromosome 3 (12–14). TRAIL is mainly expressed on the cells of the immune system and plays critical roles in T-cell homeostasis and NK or T-cell mediated killing of virally and oncogenically transformed cells (15, 16). TRAIL is a type II transmembrane protein with an extracellular domain which can be cleaved to form its biologically active soluble form (17). Initially TRAIL was identified and cloned based on the sequence homology of its extracellular domain with CD95L (28% homology) and TNFα (23% homology) (17). However, its extracellular carboxy terminal portion is proteolytically cleaved from the cell surface in a vesicle associated or soluble form (17, 18). Previous studies have also shown that TRAIL interacts with two agonistic receptors i.e., (1) TRAIL-R1 (DR4) and (2) TRAIL-R2 (DR5/TRICK2/KILLER) (19–21), and three antagonistic receptors i.e., (1) TRAIL-R3 (DcR1/TRID/LIT), (2) TRAIL-R4 (DcR2/TRUNND), and a soluble receptor i.e., osteoprotegerin (OPG) (22, 23). OPG was identified initially as a receptor for the receptor activator of nuclear factor kappa-B ligand (RANKL) (24) (Figure 1). TRAIL-R1 and TRAIL-R2 are type I transmembrane proteins sharing a sequence homology of 58% (25) with a cytoplasmic or death domain which recruits apoptosis signaling molecules for the induction of cell death (26). The TRAIL-R1 and TRAIL-R2 expression is regulated by p53 and the TRAIL-R2 gene promoter has a p53 responsive element (27). The TRAIL receptors TRAIL-R1 and TRAIL-R2 not only trigger apoptosis in TRAIL-sensitive cells but also activate survival pathways in tumor cells that resist the induction of cell death upon exposure to TRAIL (28). Post-translational modifications such as glycosylation and palmitoylation of DR4 and DR5 death receptors are also important regulators of TRAIL induced signaling (29). There exists a correlation between the expression of glycosylation initiating enzyme polypeptide N-acetylgalactosaminyltransferase 14 (GALNT14) and sensitization toward TRAIL mediated apoptosis in different cancers like pancreatic carcinoma, lung cancer, and malignant melanoma (30). O-glycosylation enhanced ligand-induced clustering of DR4 and DR5, which mediated recruitment and activation of apoptosis-initiating protease caspase-8 (31). TRAIL binds to its receptor as a homotrimer form, which is biologically much more active than the trimeric form. TRAIL-R3 and TRAIL-R4 lack the functional death domain (DD), and therefore are unable to transmit the apoptotic signals induced by binding to TRAIL ligands (23, 32). Therefore, TRAIL-R3 and TRAIL-R4 are believed to be competitive inhibitors regulating TRAIL-induced apoptosis (20). OPG is the only soluble receptor of TRAIL with lower binding affinity as compared to the other death receptors (24).
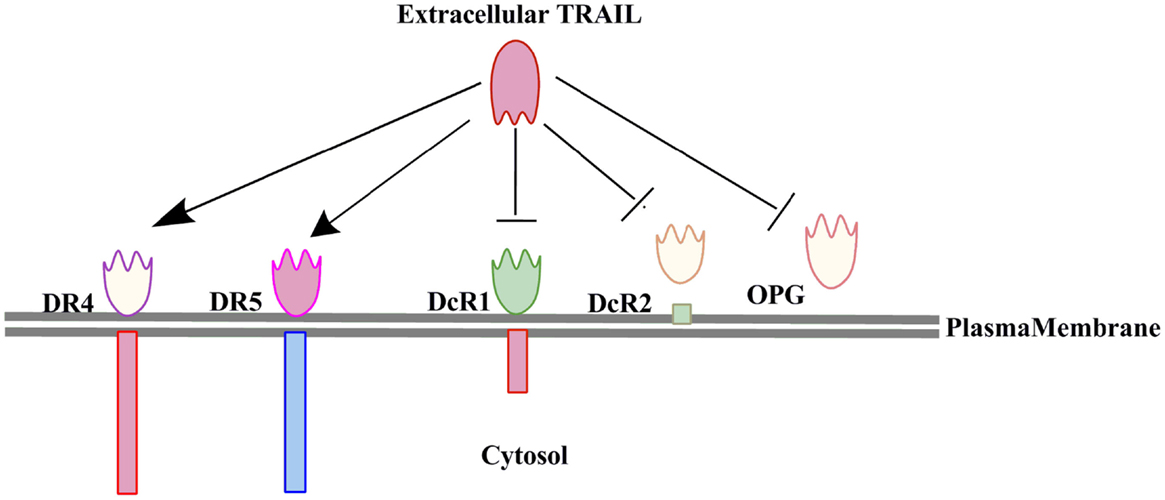
Figure 1. Interaction of TRAIL with its five receptors (two agonostic receptors: DR4, DR5; and three antagonistic receptors: DcR1, DcR2, and OPG). Only the two agonistic receptors DR4 and DR5 can transduce the TRAIL induced cell death signaling.
Depending upon the stimuli, either the extrinsic or the intrinsic pathways of apoptosis are activated (33) in a cancer cell. The cross talk between these two pathways is mediated through the truncation of the pro apoptotic protein Bid (34). The extrinsic pathway is mediated through the binding of TRAIL to its two death receptors DR4 (TRAIL R1) and DR5 (TRAIL R2). Binding of TRAIL to its receptors leads to the trimerization of receptors and formation of the death inducing signaling complex (DISC) (25). An adaptor protein FADD translocates to the DISC, and interacts with the DD, and facilitates the recruitment of procaspase-8/10 through interaction of their respective death effector domains (DED). Self activation of these initiator caspases (35) by DISC is required for the execution of apoptosis via the extrinsic pathway. In some cell types, type I activation of caspase-8 is sufficient for the subsequent activation of the effector caspase-3 and execution of apoptosis (2). But in other cell types, type II involvement of the mitochondrial pathway (intrinsic pathway) is required (4). In case of the intrinsic pathway, activation of caspase-8 leads to cleavage of Bcl-2 inhibitory BH3-domain interacting protein (Bid) (36). Subsequently, the truncated Bid interacts with Bax and Bak and induces their oligomerization in the mitochondrial membrane, which leads to the loss of the mitochondria membrane potential and ultimately release of cytochrome c (37) and Smac/Diablo (38) (Figure 2). At the DISC, activation of caspase-8 and caspase-10 can be inhibited by cellular FLICE-like inhibitory protein (c-FLIP) (39). Type II cells also require the inactivation of intracellular apoptosis inhibitors, such as X-linked inhibitor of apoptosis protein (XIAP), which directly inhibits the effector caspase activity (40). The paradigm-changing model for DISC assembly and structure indicated that FADD is substoichiometric and procaspase-8 is recruited, not only through an interaction with FADD but also by interacting with itself. The DED chain assembly model also presents the intriguing possibility that only a small amount of DISC is required for activation of large amounts of caspase-8 (41). Like caspase-8 and caspase-10, c-FLIP also has two DEDs, and has 13 discrete splice variants, and three of which are expressed as proteins: the 26 KDa short form (c-FLIPS), the 24 KDa form of c-FLIP (c-FLIPR), and the 55 KDa long form (c-FLIPL) (42, 43). The C-terminus of c-FLIPS is smaller than that of c-FLIPL and very much similar to the caspase-8 and caspase-10 structure, but this region of c-FLIPL does not contain a functional caspase domain, which is due to substitution of several amino acids, mainly the crucial cysteine residue in the catalytic domain which is necessary for the catalytic activity of caspases (43, 44). In humans, single nucleotide polymorphism defines the production of c-FLIPS or c-FLIPL in a three′ splice site of the c-FLIP gene. An intact splice site directs production of c-FLIPS, but the splice-dead variant results in production of c-FLIPR. Both c-FLIPL and c-FLIPS isoforms are short-lived proteins and are largely degraded by the ubiquitin–proteasome degradation system. Levels of c-FLIPL and c-FLIPS are regulated by JNK activation via the E3 ubiquitin ligase Itch and also through phosphorylation. The protein kinase C (PKC) phosphorylation at the serine 193 (S193) residue of c-FLIPS inhibits its polyubiquitination, stabilizes c-FLIPS levels, and increases cell survival (45, 46). c-FLIP isoforms are reported to be overexpressed in pancreatic cancer, where as very low or no expression is found in normal pancreatic ducts (47). c-FLIP protein enhances the anti-apoptotic activity of Akt by modulating GSK3β activity and thus induces resistance to TRAIL (48). High-grade prostatic intraepithelial neoplasia (HGPIN) and prostate cancer are found to express high level of c-FLIP as compared to normal prostate epithelium (47). The naturally occurring differences in the levels or states of proteins regulating receptor-mediated apoptosis are the primary causes of cell-to-cell variability in the timing and probability of death (49).
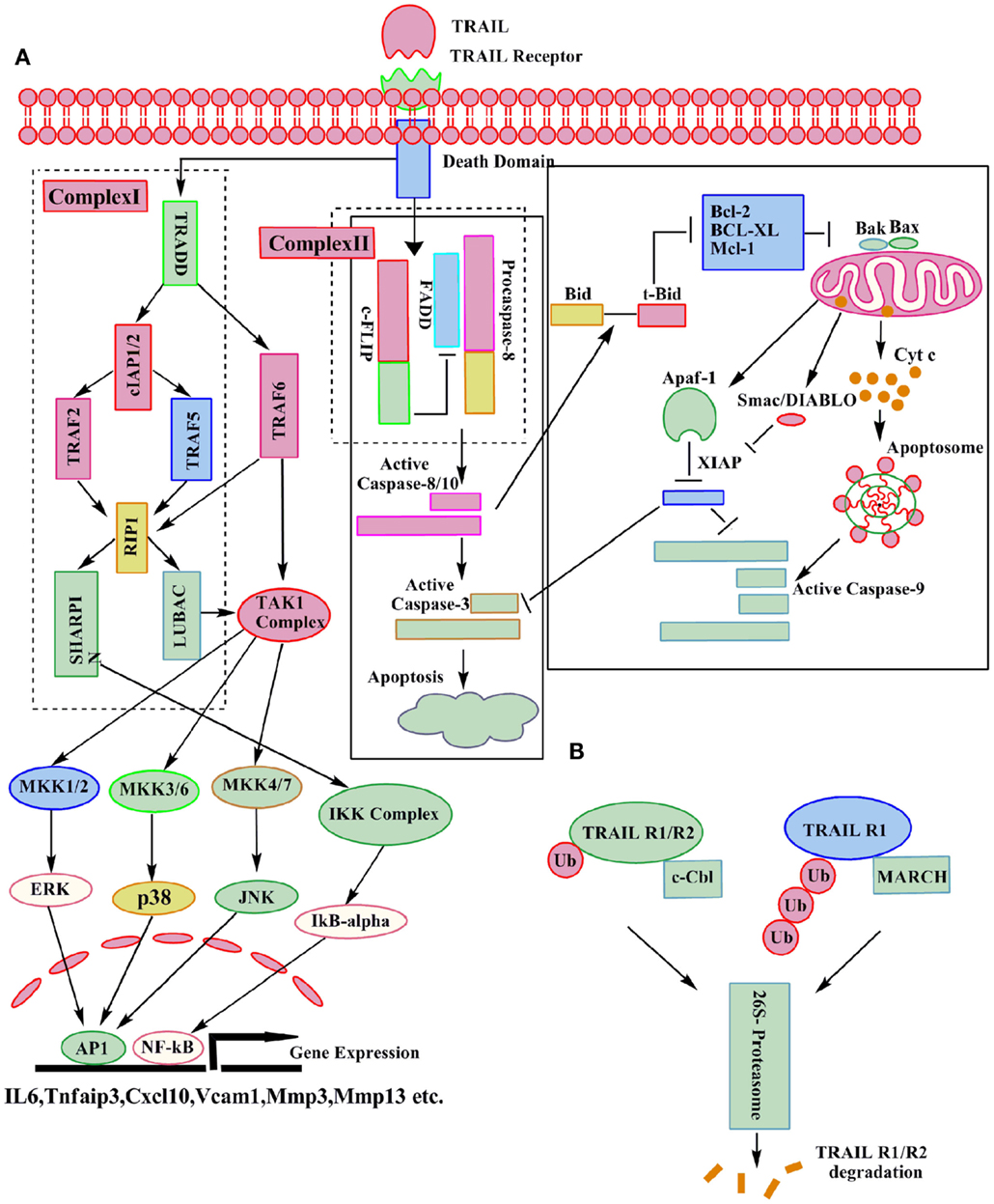
Figure 2. Molecular details of canonical and non-canonical TRAIL signaling. Following TRAIL binding to its death receptors, the DISC can be formed which results in caspase-3 activation and apoptosis. A secondary complex can also be formed after TRAIL receptor activation, leading to the activation of various kinases and the induction of direct or indirect non-apoptotic responses as indicated (A). The ubiquitin–proteasome system can assist in the degradation of TRAIL-Rs (B).
Many disease conditions are attributed to failure in synthesis of a specific active protein (50). Such conditions generally involve a mutation of the gene encoding the protein, leading to an altered protein level or activity (51). Protein translational control is an important strategy in regulation of eukaryotic gene expression. Interestingly, dysregulated translation has now been linked to multiple human cancers (52). A major target of translational control is the eukaryotic translation initiation factor 4E (eIF4E), which interacts with the 7-methylguanosine cap structure located at the 5′ untranslated regions of cellular messenger RNA (mRNA) and transfers these mRNA to the eIF4F translation initiation complex, an assembly of the cap-binding protein eIF4E, the RNA helicase eIF4A, and the scaffolding protein eIF4G (53, 54). Availability of eIF4E is the determining factor for the assembly of eIF4F. As eIF4E is scarce among the initiation factors involved in the eIF4F complex, eIF4E is the rate-limiting factor for cap-dependent translation initiation (54). Several human cancers exhibit inadequate eIF4F activation. Inhibitor of eIF4E/eIF4G interaction can act as a TRAIL sensitizer by down-regulating the levels of cyclin D1 and hypoxia-inducing factor-1α (HIF-1α), and both of which follow the cap-dependent translation regulation mechanism (55). Inhibitors of the eIF4E/eIF4G increase TRAIL-induced apoptosis through the up-regulation of DR5 and inhibition of c-FLIP, independent of inhibition of cap-dependent protein translation (56). JNK signaling induces apoptosis by inducing secretion of death ligands to promote release of cytochrome c from mitochondria to cytosol or by posttranslational modification phosphorylation of downstream pro-apoptotic proteins (57). It has been also reported that JNK activation up-regulates DR5 expression, which leads to apoptosis in cancer cells through caspase-8 activation (58). JNK has also been reported to up-regulate the expression of CHOP via an AP-1 binding site in promoter region in HeLa cells. This JNK dependent CHOP expression leads to DR5 up-regulation and induces TRAIL mediated apoptosis (58).
Endoplasmic Reticulum (ER) is a key intracellular organelle involved in the regulation of protein synthesis, proper folding of newly synthesized proteins and regulation of the intracellular calcium levels (59, 60). The malfunctioning of the aforementioned processes leads to the cellular stress response known as ER stress. ER stress induces signaling pathway which is known as the “unfolding protein response” (UPR). The prolonged and severe ER stress leads to apoptosis (61, 62). ER stress induces activation of the intrinsic apoptotic pathway (63) through the DR5 death receptors (64). DR5 up-regulation by ER stress inducing agents has been suggested to play a crucial role in the sensitization of TRAIL resistant cells (65, 66). Activation of TRAIL receptors induces translocation of pro-apoptotic Par-4/GRP78 complex to the cell surface of cancer cells. Combination of salubrinal and TRAIL leads to dephosphorylation of eIF2-α increased expression of Bim, a CHOP-regulated proapoptotic protein in hepatoma cells for TRAIL sensitization (67). Par-4 is spontaneously secreted by normal and cancer cells in culture and also by Par-4 transgenic mice that are resistant to spontaneous tumors. Par-4 induces apoptosis by binding to glucose regulated protein-78 (GRP78) which results in ER-stress and activation of the FADD/caspase-8/caspase-3 pathway (68). Thus Par-4 activates extrinsic pathway involving cell surface GRP78 receptor for apoptosis induction (69). An increasing number of reports have also demonstrated that inducers of ER stress also sensitize glioblastoma, colon cancer, breast cancer, melanoma, and hepatoma cells to TRAIL induced apoptosis (70–72). ER stress inducers provide a promising option for sensitizing TRAIL resistant cells as ER stress alters the cellular levels of different apoptosis-related proteins responsible for TRAIL resistance, including a decrease in the levels of FLIP and Mcl-1 and the up-regulation of DR5 (70–72).
Protein stabilization is a key regulatory mechanism required for the control of cellular development, cell growth, and regulation of cell cycle, and mediation of apoptosis (73). The selective degradation or stabilization of intracellular proteins through the ubiquitin-dependent pathway is vital for adjusting the regulation of these cellular processes (73). Ubiquitination targets many key regulatory proteins for degradation of these proteins through the 26S proteasome. The NF-κB pathway proteins, p53, and the inhibitors of apoptosis (IAPs) proteins are well-known target of the proteasome (74). Due to these reasons, the proteasome acts as an attractive target for cancer therapeutics. Proteasome inhibitors are a novel class of compounds with promising anticancer effects. Proteasome inhibitors are more selective for cancer cells opposite to normal cells with the unknown reasons (75). Proteasome inhibitors also show additive effects in chemosensitization and radio sensitization of tumor cell lines (76). Proteasome inhibitor PS-341 sensitizes HCT-116 and HC4 cell lines to TRAIL through DR5 up-regulation and activation of extrinsic and intrinsic apoptotic pathway (77). MG132 also sensitizes HCT-116 cells to TRAIL by DR5 up-regulation (78). Bortezomib sensitizes acute myeloid leukemic cells to TRAIL by down-regulation of antiapoptotic proteins Bcl-xL and Mcl-1 and up-regulation of death receptors DR4, DR5, and proapoptotic protein p21, activation of executioner caspases, and a loss of the mitochondrial membrane potential (79). Bortezomib also sensitizes non-small lung carcinoma cells to TRAIL mediated apoptosis through DR5 up-regulation involving both the extrinsic and intrinsic apoptotic pathways (80). b-AP15, a novel inhibitor of proteasome deubiquitinating activity, sensitizes tumor cells to TRAIL mediated apoptosis through DR5 up-regulation and c-FLIP down-regulation (81). Proteasome inhibitor, NPI-0052, sensitizes tumor cells to TRAIL induced apoptosis by inhibiting the transcription repressor Yin Yang 1 (YY1), which regulates TRAIL resistance through the negative regulation of DR5. NPI-0052 up-regulates the DR5 promoter activity along with increase in both surface and total DR5 protein expression (82). The degradation of specific cell proteins is involved in determining cell proliferation or cell death. Inhibition of the ubiquitin–proteasome system by proteasome inhibitors blocks the process of programed cell death in thymocytes and neurons, but induces apoptosis in various human cancer cell lines (83). The ubiquitin–proteasome pathway has been also reported to control TRAIL apoptosis signaling by affecting levels of death domain adaptor molecules including Fas-associated death domain (FADD) and Fas-like inhibitor protein (FLIP) (73). Ubiquitination is considered to be a crucial regulator of DISC activity through recruitment of E3 ligase Cullin3 to the DISC. This recruitment leads to poly-ubiquitination of caspase-8 which results in DISC recruitment of the ubiquitin-binding protein p62, leading to the stabilization of the activated caspase-8, there by facilitating DISC activation (84). Inhibitors of apoptosis proteins (IAPs) are a family of proteins defined by the baculovirus repeat (BIR) domains and inhibit caspase activation; the majority of the caspase-inhibiting IAPs possess a carboxyl-terminal RING zinc-finger motif and exhibit E3 ligase activity (85). The overexpression of cIAP1 results in its autoubiquitination and degradation (86). cIAP2 can encourage monoubiquitination of caspase-3 and -7, and that XIAP catalyzes the ubiquitination and degradation of caspase-3 (87). This is supported by the study that IAPs catalyzed their own ubiquitination in vitro, and this activity requires the RING domain (87). The proteasome inhibitor PS-341 enhances TRAIL killing by increasing the level of DR5 and DR4 receptors, thus increasing caspase-8 activation (77). Proteasome inhibitor MG132 and MG115 sensitizes hepatocellular carcinoma cells to TRAIL by suppressing caspase inhibitors and the AKT signaling pathway (83). The proteasome inhibitors MG132 or Bortezomib sensitize human malignant pleural mesothelioma cells to TRAIL induced apoptosis through Mcl-1 and Akt protein cleavages (88). Bortezomib-mediated proteasome inhibition also sensitizes TRAIL resistant HPV-positive HNSCC cells to TRAIL-induced cell death through both the extrinsic and intrinsic pathways of apoptosis (89). Death-associated protein kinase (DAPK2) is a modulator of TRAIL signaling and inhibition of the expression of DAPK2 results in phosphorylation of NF-κB and transcriptional activity, which leads to induction of NF-κB target genes including DR4 and DR5 (90). Collectively, these findings indicate that the combination of proteasome inhibitors and TRAIL could be a promising strategy for TRAIL sensitization.
Heat shock proteins (Hsp) are a highly conserved group of intracellular proteins classified by molecular weight into groups of Hsp110, Hsp90, Hsp70, Hsp60, small molecular Hsps (<27 kDa), and ubiquitin (91, 92). Hsps are highly abundant cytosolic proteins and function as molecular chaperones. Hsp function is best explained under cellular stress condition like heat, hypoxia under which levels of Hsps are significantly amplified (93). Under these stress conditions, Hsps encourage cell proliferation by inhibiting protein aggregation and enhancing the proper folding of damaged proteins (94, 95). Hsps also play a crucial role in normal cells, especially Hsp70, and to some amount Hsp90 are essentially implicated in protein folding functions (93–95). Hsp70 does so by binding to newly synthesized peptides thereby inhibiting premature protein misfolding, whereas Hsp90 binds to proteins with unstable tertiary structures and hamper protein degradation. Hsp60 and Hsp27 both function in protein folding by making a complex that make use of ATP to form intramolecular interactions required for client protein folding (96, 97). Hsp70 and Hsp90 are also implicated in the DNA-binding activity and stability of mutant p53, thereby resulting in cellular transformation (98). These findings indicate that the usual protein folding functions of Hsps, and in particular Hsp70 and Hsp90, are subverted by tumors to stabilize proteins important for the establishment and preservation of the transformed phenotype. Heat shock proteins such as Hsp70 and Hsp90 interact with Apaf-1, while Hsp27 sequesters cytochrome c from the cytoplasm, thereby preventing formation of apoptosome (99, 100). The heat shock proteins i.e., Hsp60 and Hsp10 promote procaspases three maturation (101), while Hsp90α has been reported to recruit FLIPs to DISC leading to TRAIL resistance. Inhibition of Hsp90 function affects multiple oncogenic substrates simultaneously and has been reported to have a TRAIL sensitizing effect (93). Combination of the Hsp90 inhibitor 17-AAG with “death receptor” targeting agents can synergistically improve their anti-tumor activities and decrease the TRAIL resistance in glioma cells (102). In TRAIL/TNF-resistant prostate cancer cells, pre- or co-treatment to17-AAG with TRAIL/TNF is known to induce high levels of apoptosis (103) through inhibition of the NF-κB or Akt cell survival pathways (104, 105). Synergistic effects between 17-AAG and anti-TRAIL monoclonal antibodies have also been observed (106). Collectively, these studies underscore the critical role of the Hsps in regulation of TRAIL resistance.
Resistance to chemotherapeutic drugs is a universal clinical concern in cancer therapy. Intrinsic or acquired drug resistance can be due to a wide variety of mechanisms including tumor cell heterogeneity, drug efflux and metabolism, tumor microenvironment, or stress-induced genetic or epigenetic alterations as a cellular response to drug exposure (107). Among these mechanisms, the response or adaptation of cancer cell itself to anticancer drug-induced tumor microenvironment stresses is a fundamental cause for chemotherapy resistance. Autophagy is a lysosomal degradation process typically activated in response to adverse microenvironmental stresses (107). Autophagy itself fulfils a dual role, with both tumor-promoting and tumor-suppressing effects. Tumor cells activate autophagy in response to cellular stress and/or increased metabolic load related to enhanced cellular proliferation (108). As a response to anticancer treatments, whether autophagy activation leads to cell survival or cell death remains still unclear. Previous studies have suggested that the induction of autophagy could be a useful therapeutic approach to overcome drug resistance of cancers to some therapeutic agents, particularly those which typically induce an apoptotic response (107, 109). Cytoprotective autophagy circumvents TRAIL sensitivity, and inhibiting autophagy in cancer cells, sensitizes cancer cells to TRAIL (110). However, apoptosis-defective tumor cells can survive TRAIL-mediated stress by eliciting a protective autophagic process coupled with enhanced F-actin polymerization (88). Although the autophagy regulates key processes associated with TRAIL resistance, still more studies are needed to elucidate the molecular mechanisms of autophagy mediated TRAIL resistance and to provide basis for therapeutic approaches that can target autophagy mediated TRAIL resistance.
Epigenetic changes may contribute to both cell survival and chemotherapy resistance in cancer cells. Abnormal DNA methylation at CpG islands and other associated epigenetic deregulations are observed during the acquisition of drug resistance (111). Recent studies suggest that epigenetic deregulation of gene expression by DNA methylation and aberrant histone deacetylation plays a crucial role in tumor development (112, 113). The role of epigenetic mechanisms in the silencing of the death receptor mediated pathway has been demonstrated in cases of medulloblastoma, as the inhibition of DNA methylation restored apoptosis suggesting the crucial role of DNA methylation in caspase-8 inactivation, a critical process in TRAIL resistance (114). Loss of caspase-8 gene expression critical to the process of TRAIL resistance by aberrant DNA methylation has also been supported by a number of other studies (114, 115). Overexpression of FAS/CD95 receptor and its cognate FAS ligand (FASL) are known to develop resistant in brain tumors toward etoposide treatment (116). The expression of DR4 and DR5 is also deregulated in human cancer cells by such mechanisms and it can be reversed by agents that target the DNA methylation or histone deacetylation (Figure 3). Modulation of chromatin by histone acetyltransferases (HATs) and histone deacetylases (HDACs) represents one important regulatory mechanism involved in gene transcription. Importantly, the HDAC inhibitor, MS-275 is effective in inhibiting the proliferation of cancer cells (Daoy and D283) in vitro through MS-275-induced increase in acetylation of histones H3 and H4 in the DR4 promoter and reactivation of DR4 expression in cancer cells. A significant potentiation of apoptosis observed in the presence of both MS-275 and recombinant TRAIL suggests that the up-regulated DR4 receptors are cell-surface associated and functional (111). At the receptor level, somatic mutations in the TRAIL receptors, down-regulation of DR4 and DR5, and over expression of the decoy receptors DcR1 and DcR2 can all confer selective resistance to TRAIL therapy (117). In summary, the reversal of aberrant gene repression with the use of a combination of epigenetic modulators and TRAIL could thus enhance the therapeutic benefit in a wide range of malignancies.
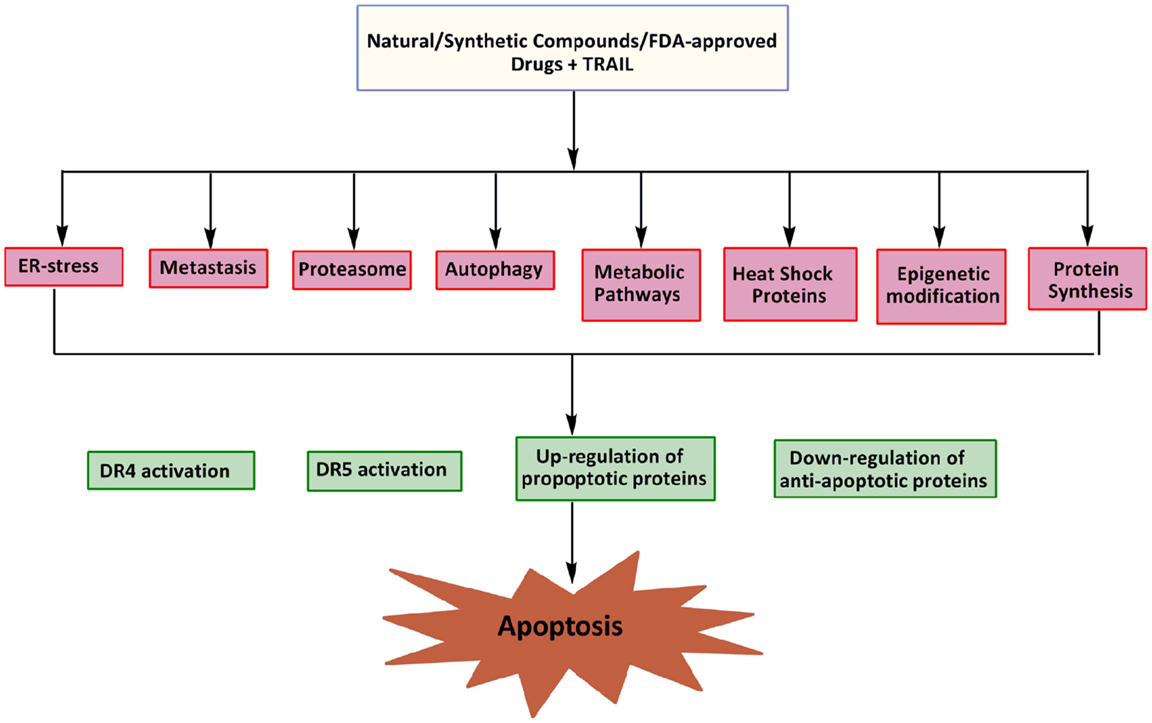
Figure 3. Potential targets for sensitization of cancer cells to TRAIL induced apoptosis.
Metabolic processes and regulation in cancer cells differ significantly from the normal cells (158). Therefore, therapeutic targeting of metabolic pathways is a promising approach for enhancing TRAIL sensitivity in cancer cells. Cancer cells mostly rely on aerobic glycolysis, fatty acid synthesis, and glutaminolysis for their growth and proliferation (159). And this fact suggests that targeting cancer cell metabolism could provide a selective approach for targeting cancer cells without harming normal cells. Aerobic glycolysis or the Warburg effect links the high rate of anaerobic glycolysis to cancer (160). Mitochondrial respiration injury and hypoxia are often associated with resistance to chemotherapeutic drug-induced apoptosis. (161). One probable association between metabolic change and resistance to apoptosis is the association of HKs with the voltage-dependent channel protein (VDAC) under glycolytic metabolism. Tp53-induced glycolysis and apoptosis regulator (TIGAR), a target of p53, reduces the level of Fructose1, 6-biphosphatase, and ultimately inhibits glycolysis. TIGAR, a target of p53, inhibits glycolysis by reducing the level of FBP. Glucose is then diverted into the pentose phosphate pathway (PPP) to make NADH and nucleotides, instigating an increase in glutathione. As such, TIGAR decreases the sensitivity of cells to p53 and other apoptotic signals linked with ROS (162). Similarly, an over-expression of PFK redirects glucose from glycolysis to the PPP and increases the resistance to oxidative stress (163). This aberrant high rate of glycolysis generates microenvironmental acidosis which requires evolution to phenotypes resistant to acid-induced cellular toxicity. Following, cell populations with up-regulated glycolysis and acid resistance have a potent growth advantage, which stimulates unconstrained proliferation and cell invasion (164). Efforts have been made to adjust the metabolic reprograming of cancer cells by treating with glycolysis inhibiting compounds. 2-Deoxyglucose (2-DG) is best known as an inhibitor of glucose metabolism. Inside a cell, it is converted to phosphorylated 2-DG (2-DG-P) by hexokinase, the first and the rate-limiting enzyme in glycolysis (165). Glycolysis inhibitor, 2-DG, accumulates in cells and inhibits Hexokinase (HK). At high concentration, 2-DG leads to depletion of ATP level and results in apoptosis (166). 2-DG has been reported to sensitize tumor cells to death receptor induced apoptosis linking glucose metabolism to Mcl-1 down expression (151). The therapeutic potential of 2-DG has prompted sufficient interest in the United States, and there is an ongoing phase one clinical trial for this compound (ClinicalTrials.gov identifier: NCT00247403) (165). Dichloroacetate (DCA), another glycolysis inhibitor, prevents pyruvate dehyrogenase kinase (PDK) by increasing mitochondrial metabolism through forcing pyruvate in to mitochondria (167). DCA also reduces tumor growth in vitro and in vivo without affecting normal tissue (168). Altered expression of PKM2 is associated with drug resistance in different tumor. This shows that PKM2 is a potential target for adjuvant cancer therapy (169). Silencing of PKM2 intensify the efficacy of docetaxel because of enhanced inhibition of proliferation and apoptosis-inducing activity both in vitro and in vivo (170). It has been also reported that glucose deprivation intensifies TRAIL-induced apoptosis by decreasing the expression of cFLIP through the ceramide-AKT-FLIP pathway (171). Tumor cells tend to have a large pool of glutamate, and this pool is maintained by their ability to convert glutamine into glutamate through glutamine synthase (GLS), a mitochondrial enzyme highly active in tumors. Like glycolysis, this abnormal glutamine metabolism cancer cells makes these cells addicted to glutamine and this leads to increased synthesis of by-products essential for fast cell growth and proliferation (172, 173). Similarly, the salvage pathway of nucleotide synthesis is one of the attractive targets for cancer therapy. Dipyridamole is a known nucleoside transport inhibitor that sensitizes cancer cells to TRAIL induced apoptosis. Thymidylate synthase (TS) is an E2F-1 regulating enzyme, crucial for DNA synthesis and repair. Many cancer cells show elevated expression of this enzyme and have been associated with poor prognosis in various solid cancers including non-small cell lung cancer (174). The novel thymidylate synthase inhibitor trifluorothymidine (TFT) has been reported to enhance TRAIL-induced apoptosis in NSCLC cells by sensitizing the apoptotic machinery at different levels in the TRAIL pathway (154). The mitochondrion is the main power station of the cell that generates most of the cell’s supply of ATP by glycolysis and oxidative phosphorylation. In addition, mitochondria are also involved in a range of intracellular processes, such as cell growth and division, differentiation, apoptosis, and intracellular signaling. Mitochondria participate in de novo biosynthesis of pyrimidines, which is catalyzed by dihydroorotate dehydrogenase (DHODH), an FMN flavoprotein in the inner mitochondrial membrane, which transfers electrons from dihydroorotate to ubiquinone of the ETC for further oxidation (175). Doxorubicin is reported to sensitize cancer cells to TRAIL mediated apoptosis by targeting dihydroorotate dehydrogenase (DHODH) (155). Therefore, the novel inhibitors of metabolic pathways may be promising agents for TRAIL sensitization.
The process of cancer metastasis involves tumor cell invasion at the primary tumor, intravasation, arrest, and extravasation of the circulatory system, followed by angiogenesis and progressive outgrowth at a distant site (176, 177). Metastatic potential is measured by the number and size of large lesions on imaging and by indices of patient survival. Epithelial–mesenchymal transition (EMT) plays an essential role in promoting metastasis in epithelium-derived carcinoma and considered to be the key process driving tumor cell invasiveness and metastasis (178, 179). Recent studies have established the dynamic association of EMT and its reverse program, mesenchymal–epithelial transition (MET), in the metastatic process (179). EMT is characterized by the down-regulation of epithelial proteins, such as E-cadherin, γ-catenin/plakoglobin, α-catenin, and β-catenin (180) and with a stimulation of mesenchymal proteins, including α-smooth muscle actin, fibronectin, N-cadherin, or vimentin (181). This is mediated by transcription factors like Twist, E12/E47, and members of the Snail, and ZEB protein families (182, 183). ZEB1 suppresses E-cadherin expression by recruiting HDAC. EMT transition has been a novel target for TRAIL sensitization, and HDAC inhibitor MS-275 inhibits angiogenesis, reverses EMT, attenuates metastasis, and sensitizes TRAIL-resistant breast cancer MDA-MB-468 xenografts in vivo (178). A recent study has demonstrated that the increased migration and invasion is a crucial factor in regulation of TRAIL resistance in cancer cells (184). A recent study suggests that synergistic co-targeting of oncogenic and death receptor pathways can not only overcome melanoma resistance to different anti-tumor agents in vitro, but can also promote pro-apoptotic effects and inhibition of tumor angiogenesis in vivo (185). These data collectively support that metastatic potential of cancer cells can be a possible target for TRAIL sensitization.
Although TRAIL has high specificity and therapeutic efficacy against cancer cells, the mechanisms involved in TRAIL resistance are not well elucidated. Therefore, recent research efforts have focused on devising strategies to overcome TRAIL resistance in cancer cells in the clinical setting. A prolonged exposure at high concentrations of TRAIL might be required to overcome resistance (186, 187). However, the short plasma half life of TRAIL (7) due to rapid elimination through metabolism (6), achieving prolonged exposure at high concentrations is difficult. Recombinant TRAIL developed by Genentech (San Francisco, CA, USA) and Amgen (Thousand Oaks, CA, USA) is a receptor agonist that directly activates the functional death receptor TRAIL-R1 and TRAIL-R2, and used as a targeted therapy for both hematological malignancies and solid tumors. Pre-clinical studies have been performed using recombinant rhTRAIL and have provided evidence for the use of exogenous TRAIL for suppressing tumor growth both in vitro and in vivo (188, 189). A recent study indicated that the use of non-tagged version of rhTRAIL induces apoptosis in malignant cells but not in normal cells (190). It is also proven that non-tagged native rhTRAIL can reduce tumor growth without damaging human hepatocytes in the chimeric mouse model (191). Monoclonal antibodies targeting DR4 and DR5 have been proven to be clinically effective for cancer treatment because they can selectively bind to specific antigens and have longer half life as compared to rhTRAIL ligands (188). In 2008, a study reported the humanized DR5 agonistic MAb, CS-1008 generated from mouse DR5 MAb TRA-8 through a complementarity-determining region grafting (192) (Table 2). HGS-ETR1 (Mapatumumab; Human Genome Sciences, Rockville, MD, USA) is a fully human agonistic monoclonal antibody that binds TRAIL-R1, and it is in phase-II clinical trial as a single agent in patients with non-small cell lung cancer and colorectal cancer (193). Monoclonal antibodies target distinct receptor expression profile in malignant cells, whereas soluble TRAIL interacts with TRAIL-R1 and TRAIL-R2 as well as the decoy receptors. Therefore, soluble TRAIL may either have a wider therapeutic spectrum or a narrower and more unpredictable therapeutic window compared to that of the highly specific antibodies (194). The efficacy of selective drugs involved in TRAIL sensitization (Table 3) and survival profile of selective genes involved in TRAIL sensitization (Table 4) have been also identified using DRUGSURV and PPISURV respectively.
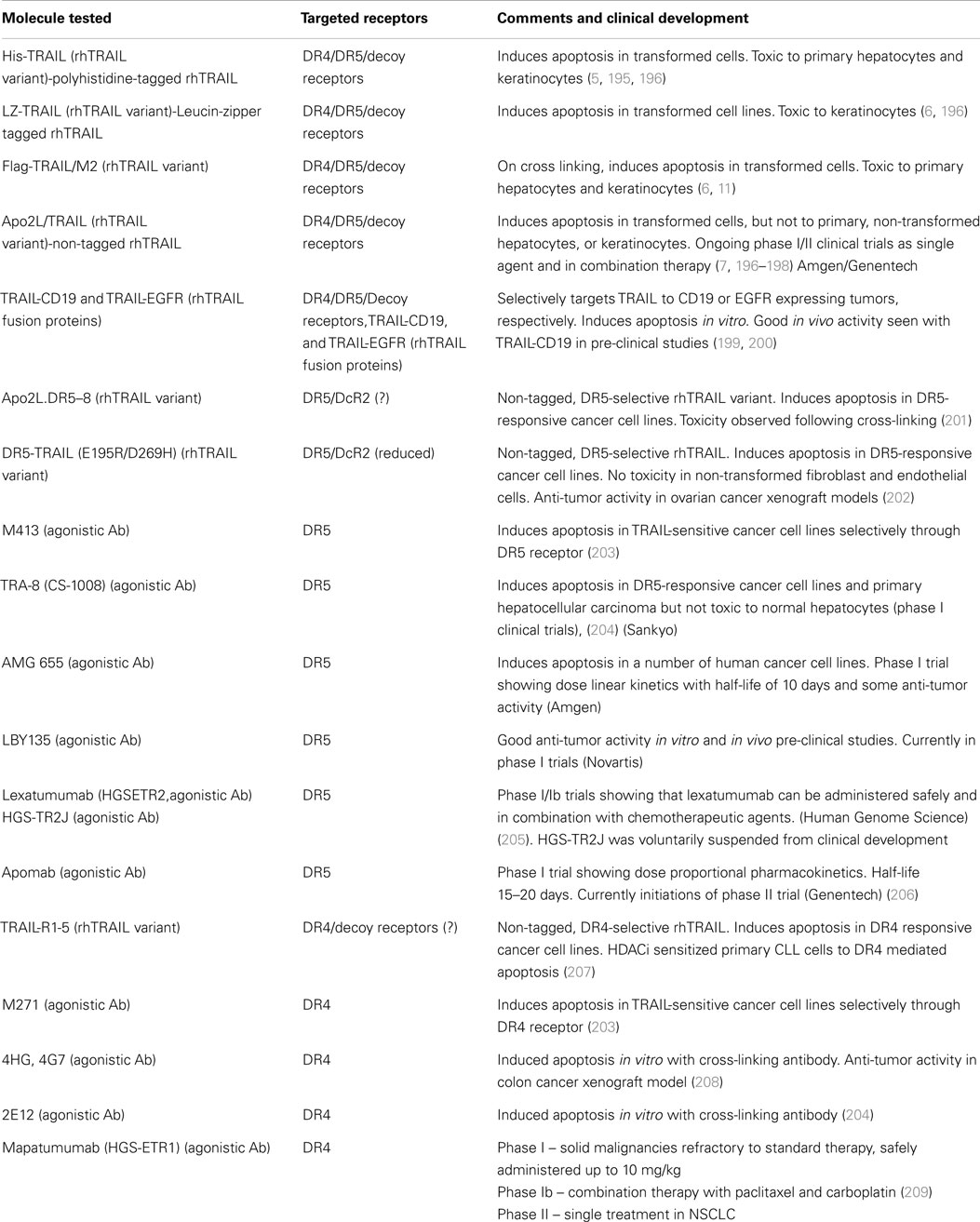
Table 2. Summary of current recombinant human TRAIL variants, agonistic DR4, DR5-specific antibodies, their pre-clinical development and current clinical status.
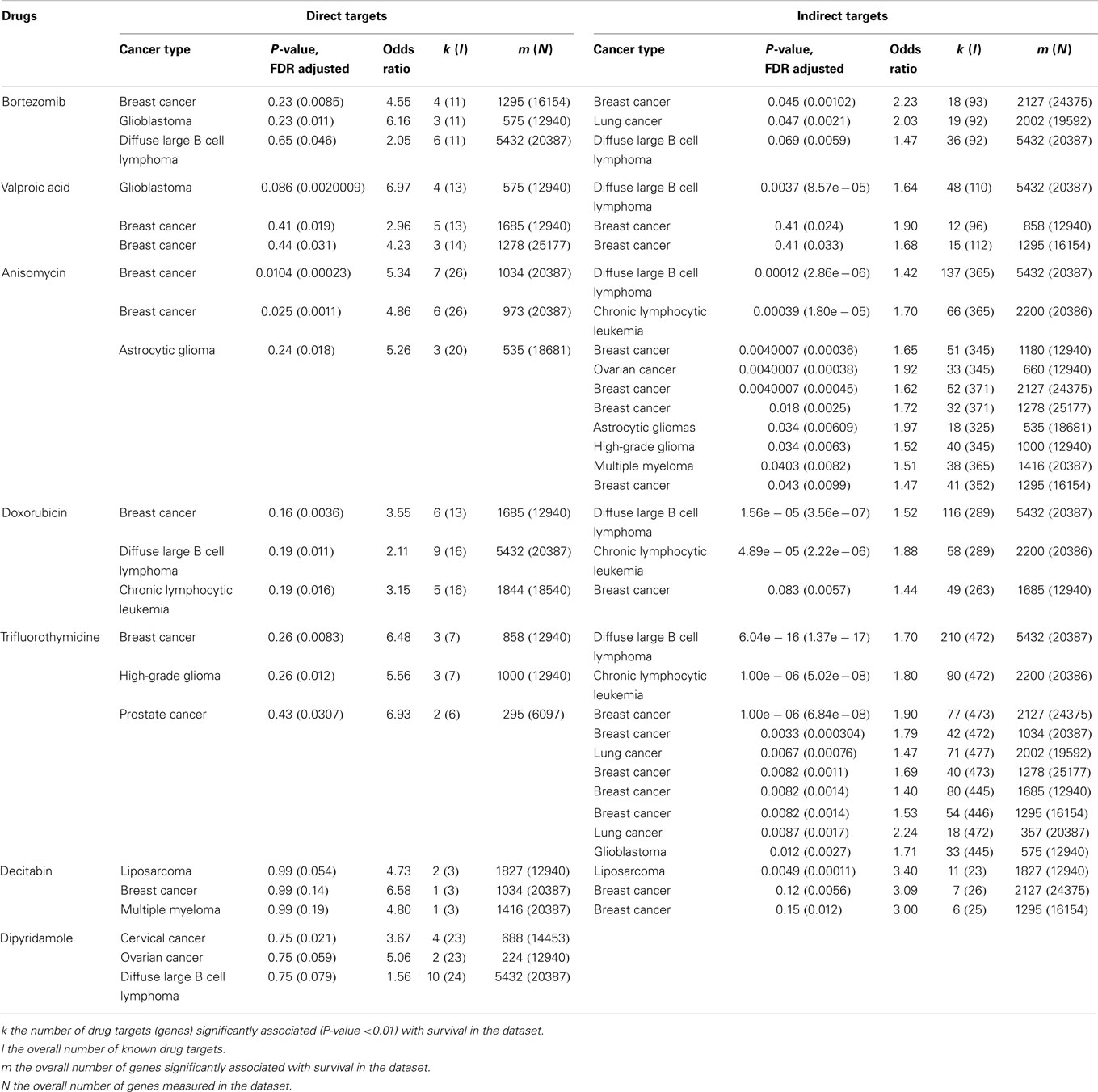
Table 3. The efficacy of selective drugs involved in TRAIL sensitization (212).
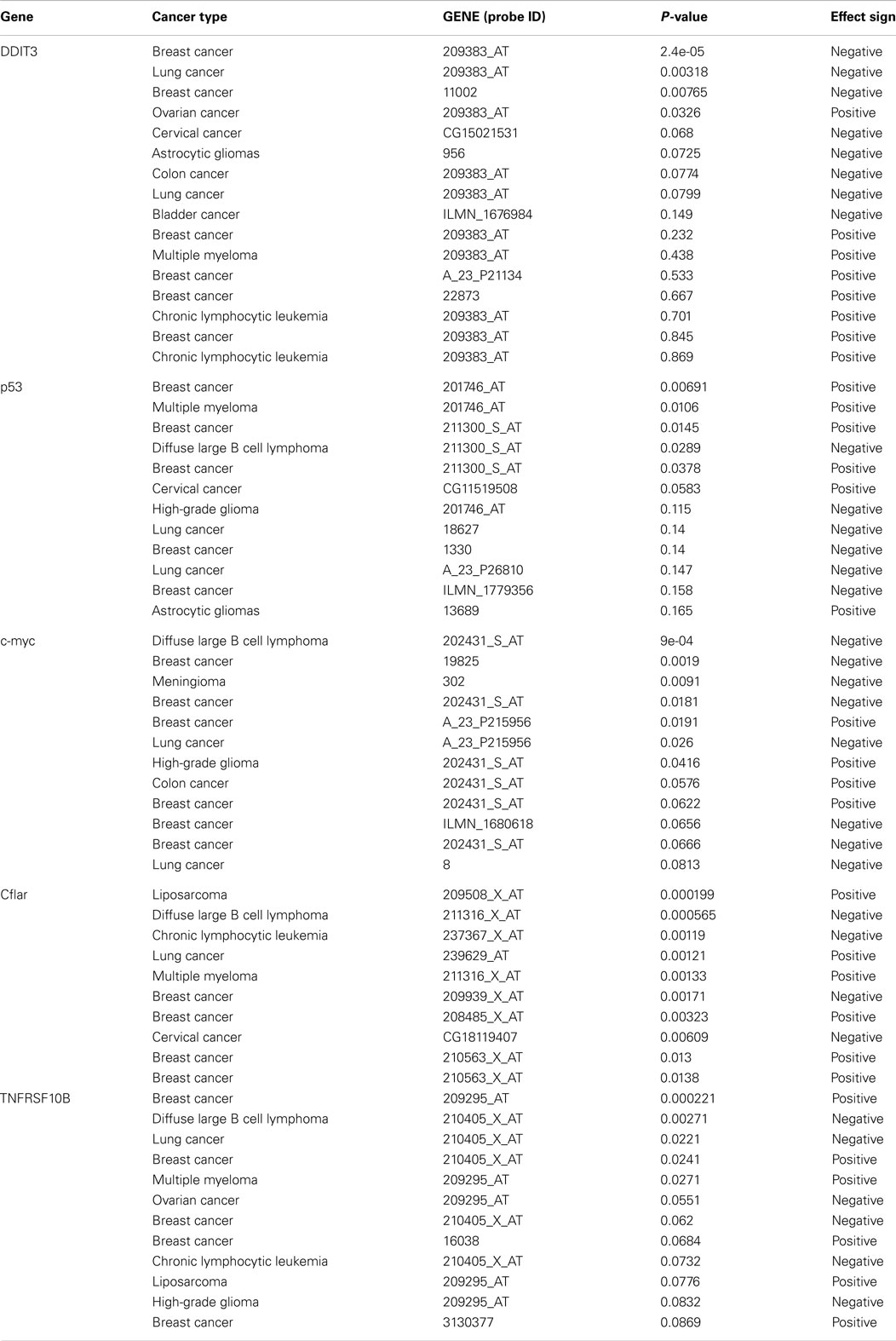
Table 4. Survival profile of selective genes involved in TRAIL sensitization (212).
Administration of soluble TRAIL in animal models of cancer has shown significant antitumor effect without any systemic toxicity. In in vivo studies, a trimerized (6) or a non-tagged (7, 191) form of TRAIL has shown a good toxicity profile, and organ toxicity might be expected at high doses of soluble TRAIL. In TRAIL related cancer therapeutics, recombinant soluble form of TRAIL, rhTRAIL (Dulanermin), the TRAIL R1-targeting agonistic monoclonal antibody mapatumumab; and the TRAIL R2-targeting agonistic monoclonal antibodies conatumumab, tigatuzumab, HGS-ETR-2 (lexatumumab), and DAB4 (PRO95780) against different types of cancer including non-Hodgekin lymphoma and colorectal cancer have been advanced to clinical development as chemotherapeutic agents (210). Mostly non-small cell lung carcinoma and other solid cancers are treated with a combination of rhTRAIL (Dulanermin) and paclitaxel, carboplatin and bevacizumab; mapatumumab and paclitaxel or carboplatin, as well as mapatumumab combined with gemcitabine or cisplatin (211). These combinations are evolving as very effective treatment against TRAIL resistant cancer cells.
In the last decade, search for novel cancer therapeutics has focused on the goal of developing specific, targeted, and less toxic agents for treatment of cancers. In this context, TRAIL as a promising chemotherapeutic agent has attracted much attention, and is currently being evaluated in the phase II clinical trials. However, the dogged pursuit of validating TRAIL as a specific anti-cancer agent has further highlighted its limitations in the clinical setting. The precise mechanisms involved in the escape from TRAIL-induced cytotoxicity and development of TRAIL resistance in some cancer cells is still not well understood. Whether combination of TRAIL receptor agonists with natural or synthetic TRAIL sensitizers will restore cancer cell sensitivity toward TRAIL is still an open question (213). It is still not well-understood whether the cellular processes alone or in combination can induce TRAIL resistance. It is also not known whether different types of tumor undergo TRAIL resistance through similar or specific mechanisms? The question whether and how TRAIL resistance could be measured to monitor therapy response in patients needs further attention. Pre-clinical studies till date suggest that combination therapy with TRAIL and chemotherapeutic drugs, natural compounds, or radiation is undoubtedly a logical way forwards in devising rationalized therapeutic regimens for TRAIL resistant cancers. However, effective therapeutic targeting of TRAIL resistance will essentially need to focus on (1) development of strategies for increasing the half life of TRAIL, (2) identification of suitable biomarkers through pre-selection of patients responsive to rhTRAIL/agonist antibody therapy, (3) development of novel synergistic combinations with TRAIL and inhibitors of cell stress response proteins, and (4) screening and identification of novel TRAIL sensitizers from FDA approved drug libraries. In the future, combination therapies with TRAIL would necessitate targeting the signaling pathways associated with the self-seeding properties of each cancer along with their varying pre-metastatic niches. However, given the complexity of the TRAIL system, further studies in primary tumor cells of diverse origin along with validation studies through syngenic and xenograft mice models and clinical trials would be required to develop personalized medicine on the basis of the TRAIL/TRAIL receptor pathway.
RT: collected and reviewed the literature and wrote the manuscript. DM: corrected and revised the manuscript. Both authors read and approved the final manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank all the members of the DP Mishra laboratory for helpful discussions. This work was supported by the Fast Track Grant from the Department of Science and Technology (GAP-00 56) and CSIR Network Project (BSC0103) to DM. RT acknowledges the support by the senior research fellowship from the Council of Scientific and Industrial Research, New Delhi. The CDRI communication No of this manuscript is 8943.
Rachana Trivedi has received a Master of Technology (M.Tech) degree in Biotechnology from the West Bengal University of Technology, India, in 2008. In 2009, she received a CSIR-Junior Research Fellowship, funded by the Council for Scientific and Innovative Research, Government of India, and is currently pursuing a Ph.D program in Cancer Biology at the Endocrinology Division of the CSIR-Central Drug Research Institute, Lucknow, under the supervision of Dr. Durga Prasad Mishra.
Durga Prasad Mishra, Principal Scientist, is the Head of the Cell Death Research Laboratory (CDRL) at the Endocrinology Division of the CSIR-Central Drug Research Institute, Lucknow, India. He has received his master’s degree in Physiology and Ph.D in Molecular Endocrinology. The prime focus of his laboratory is to study the molecular mechanisms of cell death in cancer, neuronal, and male germ cells. CDRL is actively screening for TRAIL sensitizers using commercially available FDA approved drug libraries and natural compound derived libraries synthesized at the CSIR-Central Drug Research Institute.
c-FLIP, cellular FLICE-like inhibitory protein; DcR, decoy receptor; DD, death domain; DED, death effector domain; DISC, death inducing signaling complex; DR, death receptor; EMT, epithelial mesenchymal transition; HDAC, histone deacetylase; HSPs, heat shock proteins; JNK, c-Jun-N-terminal kinase; NF-κB, nuclear factor kappa B; OPG, osteoprotegerin; RANKL, receptor activator of nuclear factor kappa-B ligand; TNF, tumor necrosis factor; TRAIL, tumor necrosis factor-related apoptosis-inducing ligand; XIAP, X-linked inhibitor of apoptosis protein; UPR, unfolded protein response; UV, ultraviolet.
1. Nagane M, Huang HJ, Cavenee WK. The potential of TRAIL for cancer chemotherapy. Apoptosis (2001) 6(3):191–7. doi:10.1023/A:1011336726649
2. Hall MA, Cleveland JL. Clearing the TRAIL for cancer therapy. Cancer Cell (2007) 12(1):4–6. doi:10.1016/j.ccr.2007.06.011
3. Ashkenazi A, Dixit VM. Death receptors: signaling and modulation. Science (1998) 281(5381):1305–8. doi:10.1126/science.281.5381.1305
4. Takeda K, Stagg J, Yagita H, Okumura K, Smyth MJ. Targeting death-inducing receptors in cancer therapy. Oncogene (2007) 26(25):3745–57. doi:10.1038/sj.onc.1210374
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
5. Pitti RM, Marsters SA, Ruppert S, Donahue CJ, Moore A, Ashkenazi A. Induction of apoptosis by Apo-2 ligand, a new member of the tumor necrosis factor cytokine family. J Biol Chem (1996) 271(22):12687–90. doi:10.1074/jbc.271.22.12687
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
6. Walczak H, Miller RE, Ariail K, Gliniak B, Griffith TS, Kubin M, et al. Tumoricidal activity of tumor necrosis factor-related apoptosis-inducing ligand in vivo. Nat Med (1999) 5(2):157–63. doi:10.1038/5517
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
7. Ashkenazi A, Pai RC, Fong S, Leung S, Lawrence DA, Marsters SA, et al. Safety and antitumor activity of recombinant soluble Apo2 ligand. J Clin Invest (1999) 104(2):155–62.
8. Wiley SR, Schooley K, Smolak PJ, Din WS, Huang CP, Nicholl JK, et al. Identification and characterization of a new member of the TNF family that induces apoptosis. Immunity (1995) 3(6):673–82. doi:10.1016/1074-7613(95)90057-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
9. Fulda S. Tumor-necrosis-factor-related apoptosis-inducing ligand (TRAIL). Adv Exp Med Biol (2014) 818:167–80. doi:10.1007/978-1-4471-6458-6_8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
10. Merino D, Lalaoui N, Morizot A, Solary E, Micheau O. TRAIL in cancer therapy: present and future challenges. Expert Opin Ther Targets (2007) 11(10):1299–314. doi:10.1517/14728222.11.10.1299
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
11. Schneider P, Holler N, Bodmer JL, Hahne M, Frei K, Fontana A, et al. Conversion of membrane-bound Fas(CD95) ligand to its soluble form is associated with downregulation of its proapoptotic activity and loss of liver toxicity. J Exp Med (1998) 187(8):1205–13. doi:10.1084/jem.187.8.1205
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
12. Snell V, Clodi K, Zhao S, Goodwin R, Thomas EK, Morris SW, et al. Activity of TNF-related apoptosis-inducing ligand (TRAIL) in haematological malignancies. Br J Haematol (1997) 99(3):618–24. doi:10.1046/j.1365-2141.1997.4393250.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
13. Herr I, Wilhelm D, Bohler T, Angel P, Debatin KM. JNK/SAPK activity is not sufficient for anticancer therapy-induced apoptosis involving CD95-L, TRAIL and TNF-alpha. Int J Cancer (1999) 80(3):417–24. doi:10.1002/(SICI)1097-0215(19990129)80:3%3C417::AID-IJC14%3E3.0.CO;2-B
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
14. Jeremias I, Herr I, Boehler T, Debatin KM. TRAIL/Apo-2-ligand-induced apoptosis in human T cells. Eur J Immunol (1998) 28(1):143–52. doi:10.1002/(SICI)1521-4141(199801)28:01%3C143::AID-IMMU143%3E3.0.CO;2-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
15. Hayakawa Y, Screpanti V, Yagita H, Grandien A, Ljunggren HG, Smyth MJ, et al. NK cell TRAIL eliminates immature dendritic cells in vivo and limits dendritic cell vaccination efficacy. J Immunol (2004) 172(1):123–9. doi:10.4049/jimmunol.172.1.123
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
16. Janssen EM, Droin NM, Lemmens EE, Pinkoski MJ, Bensinger SJ, Ehst BD, et al. CD4+ T-cell help controls CD8+ T-cell memory via TRAIL-mediated activation-induced cell death. Nature (2005) 434(7029):88–93. doi:10.1038/nature03337
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
17. Mariani SM, Krammer PH. Surface expression of TRAIL/Apo-2 ligand in activated mouse T and B cells. Eur J Immunol (1998) 28(5):1492–8. doi:10.1002/(SICI)1521-4141(199805)28:05%3C1492::AID-IMMU1492%3E3.0.CO;2-X
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
18. Monleon I, Martinez-Lorenzo MJ, Monteagudo L, Lasierra P, Taules M, Iturralde M, et al. Differential secretion of Fas ligand- or APO2 ligand/TNF-related apoptosis-inducing ligand-carrying microvesicles during activation-induced death of human T cells. J Immunol (2001) 167(12):6736–44. doi:10.4049/jimmunol.167.12.6736
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
19. Pan G, Ni J, Wei YF, Yu G, Gentz R, Dixit VM. An antagonist decoy receptor and a death domain-containing receptor for TRAIL. Science (1997) 277(5327):815–8. doi:10.1126/science.277.5327.815
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
20. Sheridan JP, Marsters SA, Pitti RM, Gurney A, Skubatch M, Baldwin D, et al. Control of TRAIL-induced apoptosis by a family of signaling and decoy receptors. Science (1997) 277(5327):818–21. doi:10.1126/science.277.5327.818
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
21. Walczak H, Degli-Esposti MA, Johnson RS, Smolak PJ, Waugh JY, Boiani N, et al. TRAIL-R2: a novel apoptosis-mediating receptor for TRAIL. EMBO J (1997) 16(17):5386–97. doi:10.1093/emboj/16.17.5386
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
22. Marsters SA, Sheridan JP, Pitti RM, Huang A, Skubatch M, Baldwin D, et al. A novel receptor for Apo2L/TRAIL contains a truncated death domain. Curr Biol (1997) 7(12):1003–6. doi:10.1016/S0960-9822(06)00422-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
23. Degli-Esposti MA, Dougall WC, Smolak PJ, Waugh JY, Smith CA, Goodwin RG. The novel receptor TRAIL-R4 induces NF-kappaB and protects against TRAIL-mediated apoptosis, yet retains an incomplete death domain. Immunity (1997) 7(6):813–20. doi:10.1016/S1074-7613(00)80399-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
24. Emery JG, McDonnell P, Burke MB, Deen KC, Lyn S, Silverman C, et al. Osteoprotegerin is a receptor for the cytotoxic ligand TRAIL. J Biol Chem (1998) 273(23):14363–7. doi:10.1074/jbc.273.23.14363
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
25. Falschlehner C, Emmerich CH, Gerlach B, Walczak H. TRAIL signalling: decisions between life and death. Int J Biochem Cell Biol (2007) 39(7-8):1462–75. doi:10.1016/j.biocel.2007.02.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
26. Corazza N, Kassahn D, Jakob S, Badmann A, Brunner T. TRAIL-induced apoptosis: between tumor therapy and immunopathology. Ann N Y Acad Sci (2009) 1171:50–8. doi:10.1111/j.1749-6632.2009.04905.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
27. Takimoto R, El-Deiry WS. Wild-type p53 transactivates the KILLER/DR5 gene through an intronic sequence-specific DNA-binding site. Oncogene (2000) 19(14):1735–43. doi:10.1038/sj.onc.1203489
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
28. Fulda S. The dark side of TRAIL signaling. Cell Death Differ (2013) 20(7):845–6. doi:10.1038/cdd.2013.36
29. Gonzalvez F, Ashkenazi A. New insights into apoptosis signaling by Apo2L/TRAIL. Oncogene (2013) 29(34):4752–65. doi:10.1038/onc.2010.221
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
30. Picarda G, Trichet V, Teletchea S, Heymann D, Redini F. TRAIL receptor signaling and therapeutic option in bone tumors: the trap of the bone microenvironment. Am J Cancer Res (2012) 2(1):45–64.
31. Wagner KW, Punnoose EA, Januario T, Lawrence DA, Pitti RM, Lancaster K, et al. Death-receptor O-glycosylation controls tumor-cell sensitivity to the proapoptotic ligand Apo2L/TRAIL. Nat Med (2007) 13(9):1070–7. doi:10.1038/nm1627
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
32. Held J, Schulze-Osthoff K. Potential and caveats of TRAIL in cancer therapy. Drug Resist Updat (2001) 4(4):243–52. doi:10.1054/drup.2001.0208
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
33. Wang S. The promise of cancer therapeutics targeting the TNF-related apoptosis-inducing ligand and TRAIL receptor pathway. Oncogene (2008) 27(48):6207–15. doi:10.1038/onc.2008.298
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
34. Igney FH, Krammer PH. Death and anti-death: tumour resistance to apoptosis. Nat Rev Cancer (2002) 2(4):277–88. doi:10.1038/nrc776
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
35. Kischkel FC, Lawrence DA, Chuntharapai A, Schow P, Kim KJ, Ashkenazi A. Apo2L/TRAIL-dependent recruitment of endogenous FADD and caspase-8 to death receptors 4 and 5. Immunity (2000) 12(6):611–20. doi:10.1016/S1074-7613(00)80212-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
36. Li H, Zhu H, Xu CJ, Yuan J. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell (1998) 94(4):491–501. doi:10.1016/S0092-8674(00)81590-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
37. Luo X, Budihardjo I, Zou H, Slaughter C, Wang X. Bid, a Bcl2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell (1998) 94(4):481–90. doi:10.1016/S0092-8674(00)81589-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
38. Du C, Fang M, Li Y, Li L, Wang X. Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell (2000) 102(1):33–42. doi:10.1016/S0092-8674(00)00008-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
39. Golks A, Brenner D, Fritsch C, Krammer PH, Lavrik IN. c-FLIPR, a new regulator of death receptor-induced apoptosis. J Biol Chem (2005) 280(15):14507–13. doi:10.1074/jbc.M414425200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
40. Holoch PA, Griffith TS. TNF-related apoptosis-inducing ligand (TRAIL): a new path to anti-cancer therapies. Eur J Pharmacol (2009) 625(1-3):63–72. doi:10.1016/j.ejphar.2009.06.066
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
41. Dickens LS, Boyd RS, Jukes-Jones R, Hughes MA, Robinson GL, Fairall L, et al. A death effector domain chain DISC model reveals a crucial role for caspase-8 chain assembly in mediating apoptotic cell death. Mol Cell (2012) 47(2):291–305. doi:10.1016/j.molcel.2012.05.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
42. Safa AR, Pollok KE. Targeting the anti-apoptotic protein c-FLIP for cancer therapy. Cancers (Basel) (2011) 3(2):1639–71. doi:10.3390/cancers3021639
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
43. Bagnoli M, Canevari S, Mezzanzanica D. Cellular FLICE-inhibitory protein (c-FLIP) signalling: a key regulator of receptor-mediated apoptosis in physiologic context and in cancer. Int J Biochem Cell Biol (2010) 42(2):210–3. doi:10.1016/j.biocel.2009.11.015
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
45. Chang L, Kamata H, Solinas G, Luo JL, Maeda S, Venuprasad K, et al. The E3 ubiquitin ligase itch couples JNK activation to TNFalpha-induced cell death by inducing c-FLIP(L) turnover. Cell (2006) 124(3):601–13. doi:10.1016/j.cell.2006.01.021
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
46. Pawar PS, Micoli KJ, Ding H, Cook WJ, Kappes JC, Chen Y, et al. Calmodulin binding to cellular FLICE-like inhibitory protein modulates Fas-induced signalling. Biochem J (2008) 412(3):459–68. doi:10.1042/BJ20071507
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
47. McCourt C, Maxwell P, Mazzucchelli R, Montironi R, Scarpelli M, Salto-Tellez M, et al. Elevation of c-FLIP in castrate-resistant prostate cancer antagonizes therapeutic response to androgen receptor-targeted therapy. Clin Cancer Res (2012) 18(14):3822–33. doi:10.1158/1078-0432.CCR-11-3277
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
48. Quintavalle C, Incoronato M, Puca L, Acunzo M, Zanca C, Romano G, et al. c-FLIPL enhances anti-apoptotic Akt functions by modulation of Gsk3beta activity. Cell Death Differ (2010) 17(12):1908–16. doi:10.1038/cdd.2010.65
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
49. Spencer SL, Gaudet S, Albeck JG, Burke JM, Sorger PK. Non-genetic origins of cell-to-cell variability in TRAIL-induced apoptosis. Nature (2009) 459(7245):428–32. doi:10.1038/nature08012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
50. Scheper GC, van der Knaap MS, Proud CG. Translation matters: protein synthesis defects in inherited disease. Nat Rev Genet (2007) 8(9):711–23. doi:10.1038/nrg2142
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
51. Kozak M. Emerging links between initiation of translation and human diseases. Mamm Genome (2002) 13(8):401–10. doi:10.1007/s00335-002-4002-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
52. Barnhart BC, Simon MC. Taking aim at translation for tumor therapy. J Clin Invest (2007) 117(9):2385–8. doi:10.1172/JCI33107
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
53. von der Haar T, Gross JD, Wagner G, McCarthy JE. The mRNA cap-binding protein eIF4E in post-transcriptional gene expression. Nat Struct Mol Biol (2004) 11(6):503–11. doi:10.1038/nsmb779
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
54. Richter JD, Sonenberg N. Regulation of cap-dependent translation by eIF4E inhibitory proteins. Nature (2005) 433(7025):477–80. doi:10.1038/nature03205
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
55. Graff JR, Konicek BW, Carter JH, Marcusson EG. Targeting the eukaryotic translation initiation factor 4E for cancer therapy. Cancer Res (2008) 68(3):631–4. doi:10.1158/0008-5472.CAN-07-5635
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
56. Fan S, Li Y, Yue P, Khuri FR, Sun SY. The eIF4E/eIF4G interaction inhibitor 4EGI-1 augments TRAIL-mediated apoptosis through c-FLIP Down-regulation and DR5 induction independent of inhibition of cap-dependent protein translation. Neoplasia (2010) 12(4):346–56. doi:10.1593/neo.10144
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
57. Herr I, Debatin KM. Cellular stress response and apoptosis in cancer therapy. Blood (2001) 98(9):2603–14. doi:10.1182/blood.V98.9.2603
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
58. Zou W, Liu X, Yue P, Zhou Z, Sporn MB, Lotan R, et al. c-Jun NH2-terminal kinase-mediated up-regulation of death receptor 5 contributes to induction of apoptosis by the novel synthetic triterpenoid methyl-2-cyano-3,12-dioxooleana-1, 9-dien-28-oate in human lung cancer cells. Cancer Res (2004) 64(20):7570–8. doi:10.1158/0008-5472.CAN-04-1238
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
59. Kim I, Xu W, Reed JC. Cell death and endoplasmic reticulum stress: disease relevance and therapeutic opportunities. Nat Rev Drug Discov (2008) 7(12):1013–30. doi:10.1038/nrd2755
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
60. Szegezdi E, Logue SE, Gorman AM, Samali A. Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep (2006) 7(9):880–5. doi:10.1038/sj.embor.7400779
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
61. Soboloff J, Berger SA. Sustained ER Ca2+ depletion suppresses protein synthesis and induces activation-enhanced cell death in mast cells. J Biol Chem (2002) 277(16):13812–20. doi:10.1074/jbc.M112129200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
62. Oslowski CM, Urano F. Measuring ER stress and the unfolded protein response using mammalian tissue culture system. Methods Enzymol (2011) 490:71–92. doi:10.1016/B978-0-12-385114-7.00004-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
63. Masud A, Mohapatra A, Lakhani SA, Ferrandino A, Hakem R, Flavell RA. Endoplasmic reticulum stress-induced death of mouse embryonic fibroblasts requires the intrinsic pathway of apoptosis. J Biol Chem (2007) 282(19):14132–9. doi:10.1074/jbc.M700077200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
64. He Q, Lee DI, Rong R, Yu M, Luo X, Klein M, et al. Endoplasmic reticulum calcium pool depletion-induced apoptosis is coupled with activation of the death receptor 5 pathway. Oncogene (2002) 21(17):2623–33. doi:10.1038/sj.onc.1205345
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
65. Shiraishi T, Yoshida T, Nakata S, Horinaka M, Wakada M, Mizutani Y, et al. Tunicamycin enhances tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis in human prostate cancer cells. Cancer Res (2005) 65(14):6364–70. doi:10.1158/0008-5472.CAN-05-0312
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
66. Liu H, Jiang CC, Lavis CJ, Croft A, Dong L, Tseng HY, et al. 2-Deoxy-D-glucose enhances TRAIL-induced apoptosis in human melanoma cells through XBP-1-mediated up-regulation of TRAIL-R2. Mol Cancer (2009) 8:122. doi:10.1186/1476-4598-8-122
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
67. Teng Y, Gao M, Wang J, Kong Q, Hua H, Luo T, et al. Inhibition of eIF2alpha dephosphorylation enhances TRAIL-induced apoptosis in hepatoma cells. Cell Death Dis (2014) 5:e1060. doi:10.1038/cddis.2014.24
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
68. Burikhanov R, Zhao Y, Goswami A, Qiu S, Schwarze SR, Rangnekar VM. The tumor suppressor Par-4 activates an extrinsic pathway for apoptosis. Cell (2009) 138(2):377–88. doi:10.1016/j.cell.2009.05.022
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
69. Abdelrahim M, Newman K, Vanderlaag K, Samudio I, Safe S. 3,3’-diindolylmethane (DIM) and its derivatives induce apoptosis in pancreatic cancer cells through endoplasmic reticulum stress-dependent upregulation of DR5. Carcinogenesis (2006) 27(4):717–28. doi:10.1093/carcin/bgi270
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
70. Martin-Perez R, Niwa M, Lopez-Rivas A. ER stress sensitizes cells to TRAIL through down-regulation of FLIP and Mcl-1 and PERK-dependent up-regulation of TRAIL-R2. Apoptosis (2012) 17(4):349–63. doi:10.1007/s10495-011-0673-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
71. Tiwary R, Yu W, Li J, Park SK, Sanders BG, Kline K. Role of endoplasmic reticulum stress in alpha-TEA mediated TRAIL/DR5 death receptor dependent apoptosis. PLoS One (2010) 5(7):e11865. doi:10.1371/journal.pone.0011865
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
72. Murai M, Inoue T, Suzuki-Karasaki M, Ochiai T, Ra C, Nishida S, et al. Diallyl trisulfide sensitizes human melanoma cells to TRAIL-induced cell death by promoting endoplasmic reticulum-mediated apoptosis. Int J Oncol (2012) 41(6):2029–37. doi:10.3892/ijo.2012.1656
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
73. Zhang HG, Wang J, Yang X, Hsu HC, Mountz JD. Regulation of apoptosis proteins in cancer cells by ubiquitin. Oncogene (2004) 23(11):2009–15. doi:10.1038/sj.onc.1207373
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
74. Jesenberger V, Jentsch S. Deadly encounter: ubiquitin meets apoptosis. Nat Rev Mol Cell Biol (2002) 3(2):112–21. doi:10.1038/nrm731
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
75. Adams J. The proteasome: a suitable antineoplastic target. Nat Rev Cancer (2004) 4(5):349–60. doi:10.1371/journal.pone.0084248
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
76. Leverkus M, Sprick MR, Wachter T, Mengling T, Baumann B, Serfling E, et al. Proteasome inhibition results in TRAIL sensitization of primary keratinocytes by removing the resistance-mediating block of effector caspase maturation. Mol Cell Biol (2003) 23(3):777–90. doi:10.1128/MCB.23.3.777-790.2003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
77. Johnson TR, Stone K, Nikrad M, Yeh T, Zong WX, Thompson CB, et al. The proteasome inhibitor PS-341 overcomes TRAIL resistance in Bax and caspase 9-negative or Bcl-xL overexpressing cells. Oncogene (2003) 22(32):4953–63. doi:10.1038/sj.onc.1206656
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
78. He Q, Huang Y, Sheikh MS. Proteasome inhibitor MG132 upregulates death receptor 5 and cooperates with Apo2L/TRAIL to induce apoptosis in Bax-proficient and -deficient cells. Oncogene (2004) 23(14):2554–8. doi:10.1038/sj.onc.1207351
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
79. Conticello C, Adamo L, Vicari L, Giuffrida R, Iannolo G, Anastasi G, et al. Antitumor activity of bortezomib alone and in combination with TRAIL in human acute myeloid leukemia. Acta Haematol (2008) 120(1):19–30. doi:10.1159/000151511
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
80. Voortman J, Resende TP, Abou El Hassan MA, Giaccone G, Kruyt FA. TRAIL therapy in non-small cell lung cancer cells: sensitization to death receptor-mediated apoptosis by proteasome inhibitor bortezomib. Mol Cancer Ther (2007) 6(7):2103–12. doi:10.1158/1535-7163.MCT-07-0167
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
81. Sarhan D, Wennerberg E, D’Arcy P, Gurajada D, Linder S, Lundqvist A. A novel inhibitor of proteasome deubiquitinating activity renders tumor cells sensitive to TRAIL-mediated apoptosis by natural killer cells and T cells. Cancer Immunol Immunother (2013) 62(8):1359–68. doi:10.1007/s00262-013-1439-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
82. Baritaki S, Suzuki E, Umezawa K, Spandidos DA, Berenson J, Daniels TR, et al. Inhibition of Yin Yang 1-dependent repressor activity of DR5 transcription and expression by the novel proteasome inhibitor NPI-0052 contributes to its TRAIL-enhanced apoptosis in cancer cells. J Immunol (2008) 180(9):6199–210. doi:10.4049/jimmunol.180.9.6199
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
83. Inoue T, Shiraki K, Fuke H, Yamanaka Y, Miyashita K, Yamaguchi Y, et al. Proteasome inhibition sensitizes hepatocellular carcinoma cells to TRAIL by suppressing caspase inhibitors and AKT pathway. Anticancer Drugs (2006) 17(3):261–8. doi:10.1097/00001813-200603000-00004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
84. Lemke J, von Karstedt S, Zinngrebe J, Walczak H. Getting TRAIL back on track for cancer therapy. Cell Death Differ (2014) 21(9):1350–64. doi:10.1038/cdd.2014.81
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
85. Liston P, Roy N, Tamai K, Lefebvre C, Baird S, Cherton-Horvat G, et al. Suppression of apoptosis in mammalian cells by NAIP and a related family of IAP genes. Nature (1996) 379(6563):349–53. doi:10.1038/379349a0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
86. Yang YL, Li XM. The IAP family: endogenous caspase inhibitors with multiple biological activities. Cell Res (2000) 10(3):169–77. doi:10.1038/sj.cr.7290046
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
87. Suzuki Y, Nakabayashi Y, Takahashi R. Ubiquitin-protein ligase activity of X-linked inhibitor of apoptosis protein promotes proteasomal degradation of caspase-3 and enhances its anti-apoptotic effect in Fas-induced cell death. Proc Natl Acad Sci U S A (2001) 98(15):8662–7. doi:10.1073/pnas.161506698
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
88. Yuan BZ, Chapman J, Ding M, Wang J, Jiang B, Rojanasakul Y, et al. TRAIL and proteasome inhibitors combination induces a robust apoptosis in human malignant pleural mesothelioma cells through Mcl-1 and Akt protein cleavages. BMC Cancer (2013) 13:140. doi:10.1186/1471-2407-13-140
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
89. Bullenkamp J, Raulf N, Ayaz B, Walczak H, Kulms D, Odell E, et al. Bortezomib sensitises TRAIL-resistant HPV-positive head and neck cancer cells to TRAIL through a caspase-dependent, E6-independent mechanism. Cell Death Dis (2014) 5:e1489. doi:10.1038/cddis.2014.455
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
90. Schlegel CR, Fonseca AV, Stocker S, Georgiou ML, Misterek MB, Munro CE, et al. DAPK2 is a novel modulator of TRAIL-induced apoptosis. Cell Death Differ (2014) 21(11):1780–91. doi:10.1038/cdd.2014.93
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
91. Hartl FU, Hayer-Hartl M. Molecular chaperones in the cytosol: from nascent chain to folded protein. Science (2002) 295(5561):1852–8. doi:10.1126/science.1068408
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
92. Morimoto RI, Kline MP, Bimston DN, Cotto JJ. The heat-shock response: regulation and function of heat-shock proteins and molecular chaperones. Essays Biochem (1997) 32:17–29.
93. Panner A, Murray JC, Berger MS, Pieper RO. Heat shock protein 90alpha recruits FLIPS to the death-inducing signaling complex and contributes to TRAIL resistance in human glioma. Cancer Res (2007) 67(19):9482–9. doi:10.1158/0008-5472.CAN-07-0569
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
94. Gething MJ, Sambrook J. Protein folding in the cell. Nature (1992) 355(6355):33–45. doi:10.1038/355033a0
95. Hightower LE. Heat shock, stress proteins, chaperones, and proteotoxicity. Cell (1991) 66(2):191–7. doi:10.1016/0092-8674(91)90611-2
96. Paolini M, Mesirca R, Pozzetti L, Gialluca N, Bauer C, Biagi GL, et al. Cancer chemoprevention: some complications and limitations. Cancer Detect Prev (1998) 22(1):68–74. doi:10.1046/j.1525-1500.1998.00088.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
97. Pratt WB, Toft DO. Regulation of signaling protein function and trafficking by the hsp90/hsp70-based chaperone machinery. Exp Biol Med (Maywood) (2003) 228(2):111–33.
98. Blagosklonny MV, Toretsky J, Bohen S, Neckers L. Mutant conformation of p53 translated in vitro or in vivo requires functional HSP90. Proc Natl Acad Sci U S A (1996) 93(16):8379–83. doi:10.1073/pnas.93.16.8379
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
99. Beere HM, Wolf BB, Cain K, Mosser DD, Mahboubi A, Kuwana T, et al. Heat-shock protein 70 inhibits apoptosis by preventing recruitment of procaspase-9 to the Apaf-1 apoptosome. Nat Cell Biol (2000) 2(8):469–75. doi:10.1038/35019501
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
100. Pandey P, Saleh A, Nakazawa A, Kumar S, Srinivasula SM, Kumar V, et al. Negative regulation of cytochrome c-mediated oligomerization of Apaf-1 and activation of procaspase-9 by heat shock protein 90. EMBO J (2000) 19(16):4310–22. doi:10.1093/emboj/19.16.4310
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
101. Samali A, Cai J, Zhivotovsky B, Jones DP, Orrenius S. Presence of a pre-apoptotic complex of pro-caspase-3, Hsp60 and Hsp10 in the mitochondrial fraction of jurkat cells. EMBO J (1999) 18(8):2040–8. doi:10.1093/emboj/18.8.2040
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
102. Lu X, Xiao L, Wang L, Ruden DM. Hsp90 inhibitors and drug resistance in cancer: the potential benefits of combination therapies of Hsp90 inhibitors and other anti-cancer drugs. Biochem Pharmacol (2012) 83(8):995–1004. doi:10.1016/j.bcp.2011.11.011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
103. Williams CR, Tabios R, Linehan WM, Neckers L. Intratumor injection of the Hsp90 inhibitor 17AAG decreases tumor growth and induces apoptosis in a prostate cancer xenograft model. J Urol (2007) 178(4 Pt 1):1528–32. doi:10.1016/j.juro.2007.05.120
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
104. Ma Y, Lakshmikanthan V, Lewis RW, Kumar MV. Sensitization of TRAIL-resistant cells by inhibition of heat shock protein 90 with low-dose geldanamycin. Mol Cancer Ther (2006) 5(1):170–8. doi:10.1158/1535-7163.MCT-05-0129
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
105. Vasilevskaya IA, O’Dwyer PJ. 17-Allylamino-17-demethoxygeldanamycin overcomes TRAIL resistance in colon cancer cell lines. Biochem Pharmacol (2005) 70(4):580–9. doi:10.1016/j.bcp.2005.05.018
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
106. Georgakis GV, Li Y, Rassidakis GZ, Martinez-Valdez H, Medeiros LJ, Younes A. Inhibition of heat shock protein 90 function by 17-allylamino-17-demethoxy-geldanamycin in Hodgkin’s lymphoma cells down-regulates Akt kinase, dephosphorylates extracellular signal-regulated kinase, and induces cell cycle arrest and cell death. Clin Cancer Res (2006) 12(2):584–90. doi:10.1158/1078-0432.CCR-05-1194
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
107. Sui X, Chen R, Wang Z, Huang Z, Kong N, Zhang M, et al. Autophagy and chemotherapy resistance: a promising therapeutic target for cancer treatment. Cell Death Dis (2013) 4:e838. doi:10.1038/cddis.2013.350
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
108. Yang ZJ, Chee CE, Huang S, Sinicrope FA. The role of autophagy in cancer: therapeutic implications. Mol Cancer Ther (2011) 10(9):1533–41. doi:10.1158/1535-7163.MCT-11-0047
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
109. Garg AD, Nowis D, Golab J, Vandenabeele P, Krysko DV, Agostinis P. Immunogenic cell death, DAMPs and anticancer therapeutics: an emerging amalgamation. Biochim Biophys Acta (2009) 1805(1):53–71. doi:10.1016/j.bbcan.2009.08.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
110. Hou W, Han J, Lu C, Goldstein LA, Rabinowich H. Enhancement of tumor-TRAIL susceptibility by modulation of autophagy. Autophagy (2008) 4(7):940–3. doi:10.4161/auto.6769
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
111. Aguilera DG, Das CM, Sinnappah-Kang ND, Joyce C, Taylor PH, Wen S, et al. Reactivation of death receptor 4 (DR4) expression sensitizes medulloblastoma cell lines to TRAIL. J Neurooncol (2009) 93(3):303–18. doi:10.1007/s11060-008-9788-x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
112. Bolden JE, Peart MJ, Johnstone RW. Anticancer activities of histone deacetylase inhibitors. Nat Rev Drug Discov (2006) 5(9):769–84. doi:10.1038/nrd2133
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
113. Esteller M. Epigenetics in cancer. N Engl J Med (2008) 358(11):1148–59. doi:10.1056/NEJMra072067
114. Pingoud-Meier C, Lang D, Janss AJ, Rorke LB, Phillips PC, Shalaby T, et al. Loss of caspase-8 protein expression correlates with unfavorable survival outcome in childhood medulloblastoma. Clin Cancer Res (2003) 9(17):6401–9.
115. Zuzak TJ, Steinhoff DF, Sutton LN, Phillips PC, Eggert A, Grotzer MA. Loss of caspase-8 mRNA expression is common in childhood primitive neuroectodermal brain tumour/medulloblastoma. Eur J Cancer (2002) 38(1):83–91. doi:10.1016/S0959-8049(01)00355-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
116. Riffkin CD, Gray AZ, Hawkins CJ, Chow CW, Ashley DM. Ex vivo pediatric brain tumors express Fas (CD95) and FasL (CD95L) and are resistant to apoptosis induction. Neuro Oncol (2001) 3(4):229–40. doi:10.1215/15228517-3-4-229
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
117. Jin Z, El-Deiry WS. Overview of cell death signaling pathways. Cancer Biol Ther (2005) 4(2):139–63. doi:10.4161/cbt.4.2.1508
118. Moon DO, Asami Y, Long H, Jang JH, Bae EY, Kim BY, et al. Verrucarin A sensitizes TRAIL-induced apoptosis via the upregulation of DR5 in an eIF2alpha/CHOP-dependent manner. Toxicol In vitro (2013) 27(1):257–63. doi:10.1016/j.tiv.2012.09.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
119. Yoon MJ, Kang YJ, Kim IY, Kim EH, Lee JA, Lim JH, et al. Monensin, a polyether ionophore antibiotic, overcomes TRAIL resistance in glioma cells via endoplasmic reticulum stress, DR5 upregulation and c-FLIP downregulation. Carcinogenesis (2013) 34(8):1918–28. doi:10.1093/carcin/bgt137
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
120. Trivedi R, Maurya R, Mishra DP. Medicarpin, a legume phytoalexin sensitizes myeloid leukemia cells to TRAIL-induced apoptosis through the induction of DR5 and activation of the ROS-JNK-CHOP pathway. Cell Death Dis (2014) 5:e1465. doi:10.1038/cddis.2014.429
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
121. He L, Jang JH, Choi HG, Lee SM, Nan MH, Jeong SJ, et al. Oligomycin A enhances apoptotic effect of TRAIL through CHOP-mediated death receptor 5 expression. Mol Carcinog (2013) 52(2):85–93. doi:10.1002/mc.21831
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
122. Jiang CC, Chen LH, Gillespie S, Kiejda KA, Mhaidat N, Wang YF, et al. Tunicamycin sensitizes human melanoma cells to tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis by up-regulation of TRAIL-R2 via the unfolded protein response. Cancer Res (2007) 67(12):5880–8. doi:10.1158/0008-5472.CAN-07-0213
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
123. Su RY, Chi KH, Huang DY, Tai MH, Lin WW. 15-deoxy-Delta12,14-prostaglandin J2 up-regulates death receptor 5 gene expression in HCT116 cells: involvement of reactive oxygen species and C/EBP homologous transcription factor gene transcription. Mol Cancer Ther (2008) 7(10):3429–40. doi:10.1158/1535-7163.MCT-08-0498
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
124. Prasad S, Yadav VR, Ravindran J, Aggarwal BB. ROS and CHOP are critical for dibenzylideneacetone to sensitize tumor cells to TRAIL through induction of death receptors and downregulation of cell survival proteins. Cancer Res (2011) 71(2):538–49. doi:10.1158/0008-5472.CAN-10-3121
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
125. Yang JF, Cao JG, Tian L, Liu F. 5, 7-Dimethoxyflavone sensitizes TRAIL-induced apoptosis through DR5 upregulation in hepatocellular carcinoma cells. Cancer Chemother Pharmacol (2012) 69(1):195–206. doi:10.1007/s00280-011-1686-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
126. Kim YJ, Choi WI, Ko H, So Y, Kang KS, Kim I, et al. Neobavaisoflavone sensitizes apoptosis via the inhibition of metastasis in TRAIL-resistant human glioma U373MG cells. Life Sci (2014) 95(2):101–7. doi:10.1016/j.lfs.2013.10.035
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
127. Kim HB, Kim MJ, Kim DY, Lee JW, Bae JH, Kim DW, et al. High susceptibility of metastatic cells derived from human prostate and colon cancer cells to TRAIL and sensitization of TRAIL-insensitive primary cells to TRAIL by 4,5-dimethoxy-2-nitrobenzaldehyde. Mol Cancer (2011) 10:46. doi:10.1186/1476-4598-10-46
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
128. Sung ES, Park KJ, Choi HJ, Kim CH, Kim YS. The proteasome inhibitor MG132 potentiates TRAIL receptor agonist-induced apoptosis by stabilizing tBid and Bik in human head and neck squamous cell carcinoma cells. Exp Cell Res (2012) 318(13):1564–76. doi:10.1016/j.yexcr.2012.04.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
129. Koschny R, Boehm C, Sprick MR, Haas TL, Holland H, Xu LX, et al. Bortezomib sensitizes primary meningioma cells to TRAIL-induced apoptosis by enhancing formation of the death-inducing signaling complex. J Neuropathol Exp Neurol (2014) 73(11):1034–46. doi:10.1097/NEN.0000000000000129
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
130. Brooks AD, Jacobsen KM, Li W, Shanker A, Sayers TJ. Bortezomib sensitizes human renal cell carcinomas to TRAIL apoptosis through increased activation of caspase-8 in the death-inducing signaling complex. Mol Cancer Res (2010) 8(5):729–38. doi:10.1158/1541-7786.MCR-10-0022
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
131. Seol DW. p53-Independent up-regulation of a TRAIL receptor DR5 by proteasome inhibitors: a mechanism for proteasome inhibitor-enhanced TRAIL-induced apoptosis. Biochem Biophys Res Commun (2011) 416(1-2):222–5. doi:10.1016/j.bbrc.2011.11.053
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
132. Kahana S, Finniss S, Cazacu S, Xiang C, Lee HK, Brodie S, et al. Proteasome inhibitors sensitize glioma cells and glioma stem cells to TRAIL-induced apoptosis by PKCepsilon-dependent downregulation of AKT and XIAP expressions. Cell Signal (2011) 23(8):1348–57. doi:10.1016/j.cellsig.2011.03.017
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
133. Jane EP, Premkumar DR, Pollack IF. Bortezomib sensitizes malignant human glioma cells to TRAIL, mediated by inhibition of the NF-{kappa}B signaling pathway. Mol Cancer Ther (2011) 10(1):198–208. doi:10.1158/1535-7163.MCT-10-0725
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
134. Seki N, Toh U, Sayers TJ, Fujii T, Miyagi M, Akagi Y, et al. Bortezomib sensitizes human esophageal squamous cell carcinoma cells to TRAIL-mediated apoptosis via activation of both extrinsic and intrinsic apoptosis pathways. Mol Cancer Ther (2010) 9(6):1842–51. doi:10.1158/1535-7163.MCT-09-0918
135. Kandasamy K, Kraft AS. Proteasome inhibitor PS-341 (VELCADE) induces stabilization of the TRAIL receptor DR5 mRNA through the 3’-untranslated region. Mol Cancer Ther (2008) 7(5):1091–100. doi:10.1158/1535-7163.MCT-07-2368
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
136. Liu FT, Agrawal SG, Gribben JG, Ye H, Du MQ, Newland AC, et al. Bortezomib blocks Bax degradation in malignant B cells during treatment with TRAIL. Blood (2008) 111(5):2797–805. doi:10.1182/blood-2007-08-110445
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
137. Conticello C, Adamo L, Giuffrida R, Vicari L, Zeuner A, Eramo A, et al. Proteasome inhibitors synergize with tumor necrosis factor-related apoptosis-induced ligand to induce anaplastic thyroid carcinoma cell death. J Clin Endocrinol Metab (2007) 92(5):1938–42. doi:10.1210/jc.2006-2157
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
138. Siegelin MD, Habel A, Gaiser T. 17-AAG sensitized malignant glioma cells to death-receptor mediated apoptosis. Neurobiol Dis (2009) 33(2):243–9. doi:10.1016/j.nbd.2008.10.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
139. Mellier G, Liu D, Bellot G, Holme AL, Pervaiz S. Small molecule sensitization to TRAIL is mediated via nuclear localization, phosphorylation and inhibition of chaperone activity of Hsp27. Cell Death Dis (2013) 4:e890. doi:10.1038/cddis.2013.413
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
140. Monma H, Harashima N, Inao T, Okano S, Tajima Y, Harada M. The HSP70 and autophagy inhibitor pifithrin-mu enhances the antitumor effects of TRAIL on human pancreatic cancer. Mol Cancer Ther (2013) 12(4):341–51. doi:10.1158/1535-7163.MCT-12-0954
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
141. Singh TR, Shankar S, Srivastava RK. HDAC inhibitors enhance the apoptosis-inducing potential of TRAIL in breast carcinoma. Oncogene (2005) 24(29):4609–23. doi:10.1038/sj.onc.1208585
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
142. Inoue S, MacFarlane M, Harper N, Wheat LM, Dyer MJ, Cohen GM. Histone deacetylase inhibitors potentiate TNF-related apoptosis-inducing ligand (TRAIL)-induced apoptosis in lymphoid malignancies. Cell Death Differ (2004) 11(Suppl 2):S193–206. doi:10.1038/sj.cdd.4401535
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
143. Muhlethaler-Mottet A, Flahaut M, Bourloud KB, Auderset K, Meier R, Joseph JM, et al. Histone deacetylase inhibitors strongly sensitise neuroblastoma cells to TRAIL-induced apoptosis by a caspases-dependent increase of the pro- to anti-apoptotic proteins ratio. BMC Cancer (2006) 6:214. doi:10.1186/1471-2407-6-214
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
144. Nakata S, Yoshida T, Horinaka M, Shiraishi T, Wakada M, Sakai T. Histone deacetylase inhibitors upregulate death receptor 5/TRAIL-R2 and sensitize apoptosis induced by TRAIL/APO2-L in human malignant tumor cells. Oncogene (2004) 23(37):6261–71. doi:10.1038/sj.onc.1207830
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
145. D’Acunto CW, Carratu A, Rodriquez M, Taddei M, Parente L, Petrella A. LGP1, A histone deacetylase inhibitor analogue of FR235222, sensitizes promyelocytic leukaemia U937 cells to TRAIL-mediated apoptosis. Anticancer Res (2010) 30(3):887–94.
146. Kim HB, Kim MJ, Lee SH, Lee JW, Bae JH, Kim DW, et al. Amurensin G, a novel SIRT1 inhibitor, sensitizes TRAIL-resistant human leukemic K562 cells to TRAIL-induced apoptosis. Biochem Pharmacol (2012) 84(3):402–10. doi:10.1016/j.bcp.2012.03.014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
147. Kaminskyy VO, Surova OV, Vaculova A, Zhivotovsky B. Combined inhibition of DNA methyltransferase and histone deacetylase restores caspase-8 expression and sensitizes SCLC cells to TRAIL. Carcinogenesis (2011) 32(10):1450–8. doi:10.1093/carcin/bgr135
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
148. Eramo A, Pallini R, Lotti F, Sette G, Patti M, Bartucci M, et al. Inhibition of DNA methylation sensitizes glioblastoma for tumor necrosis factor-related apoptosis-inducing ligand-mediated destruction. Cancer Res (2005) 65(24):11469–77. doi:10.1158/0008-5472.CAN-05-1724
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
149. Fulda S, Debatin KM. 5-Aza-2’-deoxycytidine and IFN-gamma cooperate to sensitize for TRAIL-induced apoptosis by upregulating caspase-8. Oncogene (2006) 25(37):5125–33. doi:10.1038/sj.onc.1209518
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
150. Fulda S, Kufer MU, Meyer E, van Valen F, Dockhorn-Dworniczak B, Debatin KM. Sensitization for death receptor- or drug-induced apoptosis by re-expression of caspase-8 through demethylation or gene transfer. Oncogene (2001) 20(41):5865–77. doi:10.1038/sj.onc.1204750
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
151. Pradelli LA, Beneteau M, Chauvin C, Jacquin MA, Marchetti S, Munoz-Pinedo C, et al. Glycolysis inhibition sensitizes tumor cells to death receptors-induced apoptosis by AMP kinase activation leading to Mcl-1 block in translation. Oncogene (2010) 29(11):1641–52. doi:10.1038/onc.2009.448
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
152. Taniguchi H, Horinaka M, Yoshida T, Yano K, Goda AE, Yasuda S, et al. Targeting the glyoxalase pathway enhances TRAIL efficacy in cancer cells by downregulating the expression of antiapoptotic molecules. Mol Cancer Ther (2012) 11(10):2294–300. doi:10.1158/1535-7163.MCT-12-0031
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
153. Goda AE, Yoshida T, Horinaka M, Yasuda T, Shiraishi T, Wakada M, et al. Mechanisms of enhancement of TRAIL tumoricidal activity against human cancer cells of different origin by dipyridamole. Oncogene (2008) 27(24):3435–45. doi:10.1038/sj.onc.1211008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
154. Azijli K, van Roosmalen IA, Smit J, Pillai S, Fukushima M, de Jong S, et al. The novel thymidylate synthase inhibitor trifluorothymidine (TFT) and TRAIL synergistically eradicate non-small cell lung cancer cells. Cancer Chemother Pharmacol (2014) 73(6):1273–83. doi:10.1007/s00280-014-2465-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
155. He T, Haapa-Paananen S, Kaminskyy VO, Kohonen P, Fey V, Zhivotovsky B, et al. Inhibition of the mitochondrial pyrimidine biosynthesis enzyme dihydroorotate dehydrogenase by doxorubicin and brequinar sensitizes cancer cells to TRAIL-induced apoptosis. Oncogene (2014) 33(27):3538–49. doi:10.1038/onc.2013.313
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
156. Sah NK, Munshi A, Kurland JF, McDonnell TJ, Su B, Meyn RE. Translation inhibitors sensitize prostate cancer cells to apoptosis induced by tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) by activating c-Jun N-terminal kinase. J Biol Chem (2003) 278(23):20593–602. doi:10.1074/jbc.M211010200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
157. Hernandez A, Wang QD, Schwartz SA, Evers BM. Sensitization of human colon cancer cells to TRAIL-mediated apoptosis. J Gastrointest Surg (2001) 5(1):56–65. doi:10.1016/S1091-255X(01)80014-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
158. Munoz-Pinedo C, Ruiz-Ruiz C, Ruiz de Almodovar C, Palacios C, Lopez-Rivas A. Inhibition of glucose metabolism sensitizes tumor cells to death receptor-triggered apoptosis through enhancement of death-inducing signaling complex formation and apical procaspase-8 processing. J Biol Chem (2003) 278(15):12759–68. doi:10.1074/jbc.M212392200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
159. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science (2009) 324(5930):1029–33. doi:10.1126/science.1160809
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
160. Dang CV. Links between metabolism and cancer. Genes Dev (2012) 26(9):877–90. doi:10.1101/gad.189365.112
161. Xu RH, Pelicano H, Zhou Y, Carew JS, Feng L, Bhalla KN, et al. Inhibition of glycolysis in cancer cells: a novel strategy to overcome drug resistance associated with mitochondrial respiratory defect and hypoxia. Cancer Res (2005) 65(2):613–21.
162. Viale A, De Franco F, Orleth A, Cambiaghi V, Giuliani V, Bossi D, et al. Cell-cycle restriction limits DNA damage and maintains self-renewal of leukaemia stem cells. Nature (2009) 457(7225):51–6. doi:10.1038/nature07618
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
163. Boada J, Roig T, Perez X, Gamez A, Bartrons R, Cascante M, et al. Cells overexpressing fructose-2,6-bisphosphatase showed enhanced pentose phosphate pathway flux and resistance to oxidative stress. FEBS Lett (2000) 480(2-3):261–4. doi:10.1016/S0014-5793(00)01950-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
164. Gatenby RA, Gillies RJ. Why do cancers have high aerobic glycolysis? Nat Rev Cancer (2004) 4(11):891–9. doi:10.1038/nrc1478
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
165. Zhong D, Liu X, Schafer-Hales K, Marcus AI, Khuri FR, Sun SY, et al. 2-Deoxyglucose induces Akt phosphorylation via a mechanism independent of LKB1/AMP-activated protein kinase signaling activation or glycolysis inhibition. Mol Cancer Ther (2008) 7(4):809–17. doi:10.1158/1535-7163.MCT-07-0559
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
166. Maher JC, Krishan A, Lampidis TJ. Greater cell cycle inhibition and cytotoxicity induced by 2-deoxy-D-glucose in tumor cells treated under hypoxic vs aerobic conditions. Cancer Chemother Pharmacol (2004) 53(2):116–22. doi:10.1007/s00280-003-0724-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
167. Bonnet S, Archer SL, Allalunis-Turner J, Haromy A, Beaulieu C, Thompson R, et al. A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell (2007) 11(1):37–51. doi:10.1016/j.ccr.2006.10.020
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
168. Sun RC, Fadia M, Dahlstrom JE, Parish CR, Board PG, Blackburn AC. Reversal of the glycolytic phenotype by dichloroacetate inhibits metastatic breast cancer cell growth in vitro and in vivo. Breast Cancer Res Treat (2010) 120(1):253–60. doi:10.1007/s10549-009-0435-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
169. Zhao Y, Butler EB, Tan M. Targeting cellular metabolism to improve cancer therapeutics. Cell Death Dis (2013) 4:e532. doi:10.1038/cddis.2013.60
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
170. Shi HS, Li D, Zhang J, Wang YS, Yang L, Zhang HL, et al. Silencing of pkm2 increases the efficacy of docetaxel in human lung cancer xenografts in mice. Cancer Sci (2010) 101(6):1447–53. doi:10.1111/j.1349-7006.2010.01562.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
171. Nam SY, Amoscato AA, Lee YJ. Low glucose-enhanced TRAIL cytotoxicity is mediated through the ceramide-Akt-FLIP pathway. Oncogene (2002) 21(3):337–46. doi:10.1038/sj.onc.1205068
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
172. Kaelin WG Jr, Thompson CB. Q&A: cancer: clues from cell metabolism. Nature (2010) 465(7298):562–4. doi:10.1038/465562a
173. Erickson JW, Cerione RA. Glutaminase: a hot spot for regulation of cancer cell metabolism? Oncotarget (2010) 1(8):734–40.
174. Rahman L, Voeller D, Rahman M, Lipkowitz S, Allegra C, Barrett JC, et al. Thymidylate synthase as an oncogene: a novel role for an essential DNA synthesis enzyme. Cancer Cell (2004) 5(4):341–51. doi:10.1016/S1535-6108(04)00080-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
175. Khutornenko AA, Roudko VV, Chernyak BV, Vartapetian AB, Chumakov PM, Evstafieva AG. Pyrimidine biosynthesis links mitochondrial respiration to the p53 pathway. Proc Natl Acad Sci U S A (2010) 107(29):12828–33. doi:10.1073/pnas.0910885107
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
176. Steeg PS. Tumor metastasis: mechanistic insights and clinical challenges. Nat Med (2006) 12(8):895–904. doi:10.1038/nm1469
177. Steeg PS, Theodorescu D. Metastasis: a therapeutic target for cancer. Nat Clin Pract Oncol (2008) 5(4):206–19. doi:10.1038/ncponc1066
178. Srivastava RK, Kurzrock R, Shankar S. MS-275 sensitizes TRAIL-resistant breast cancer cells, inhibits angiogenesis and metastasis, and reverses epithelial-mesenchymal transition in vivo. Mol Cancer Ther (2010) 9(12):3254–66. doi:10.1158/1535-7163.MCT-10-0582
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
179. Tsai JH, Yang J. Epithelial-mesenchymal plasticity in carcinoma metastasis. Genes Dev (2013) 27(20):2192–206. doi:10.1101/gad.225334.113
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
180. Berx G, Raspe E, Christofori G, Thiery JP, Sleeman JP. Pre-EMTing metastasis? Recapitulation of morphogenetic processes in cancer. Clin Exp Metastasis (2007) 24(8):587–97. doi:10.1007/s10585-007-9114-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
181. Jechlinger M, Grunert S, Tamir IH, Janda E, Ludemann S, Waerner T, et al. Expression profiling of epithelial plasticity in tumor progression. Oncogene (2003) 22(46):7155–69. doi:10.1038/sj.onc.1206887
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
182. Hurt EM, Saykally JN, Anose BM, Kalli KR, Sanders MM. Expression of the ZEB1 (deltaEF1) transcription factor in human: additional insights. Mol Cell Biochem (2008) 318(1-2):89–99. doi:10.1007/s11010-008-9860-z
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
183. Spaderna S, Schmalhofer O, Wahlbuhl M, Dimmler A, Bauer K, Sultan A, et al. The transcriptional repressor ZEB1 promotes metastasis and loss of cell polarity in cancer. Cancer Res (2008) 68(2):537–44. doi:10.1158/0008-5472.CAN-07-5682
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
184. Wang H, Xu C, Kong X, Li X, Wang Y, Ding X, et al. Trail resistance induces epithelial-mesenchymal transition and enhances invasiveness by suppressing PTEN via miR-221 in breast cancer. PLoS One (2014) 9(6):e99067. doi:10.1371/journal.pone.0099067
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
185. Grazia G, Vegetti C, Benigni F, Penna I, Perotti V, Tassi E, et al. Synergistic anti-tumor activity and inhibition of angiogenesis by cotargeting of oncogenic and death receptor pathways in human melanoma. Cell Death Dis (2014) 5:e1434. doi:10.1038/cddis.2014.410
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
186. Johnston JB, Kabore AF, Strutinsky J, Hu X, Paul JT, Kropp DM, et al. Role of the TRAIL/APO2-L death receptors in chlorambucil- and fludarabine-induced apoptosis in chronic lymphocytic leukemia. Oncogene (2003) 22(51):8356–69. doi:10.1038/sj.onc.1207004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
187. Mouzakiti A, Packham G. Regulation of tumour necrosis factor-related apoptosis-inducing ligand (TRAIL)-induced apoptosis in Burkitt’s lymphoma cell lines. Br J Haematol (2003) 122(1):61–9. doi:10.1046/j.1365-2141.2003.04424.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
188. Bellail AC, Qi L, Mulligan P, Chhabra V, Hao C. TRAIL agonists on clinical trials for cancer therapy: the promises and the challenges. Rev Recent Clin Trials (2009) 4(1):34–41. doi:10.2174/157488709787047530
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
189. Herbst RS, Eckhardt SG, Kurzrock R, Ebbinghaus S, O’Dwyer PJ, Gordon MS, et al. Phase I dose-escalation study of recombinant human Apo2L/TRAIL, a dual proapoptotic receptor agonist, in patients with advanced cancer. J Clin Oncol (2010) 28(17):2839–46. doi:10.1200/JCO.2009.25.1991
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
190. Almasan A, Ashkenazi A. Apo2L/TRAIL: apoptosis signaling, biology, and potential for cancer therapy. Cytokine Growth Factor Rev (2003) 14(3-4):337–48. doi:10.1016/S1359-6101(03)00029-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
191. Hao C, Song JH, Hsi B, Lewis J, Song DK, Petruk KC, et al. TRAIL inhibits tumor growth but is nontoxic to human hepatocytes in chimeric mice. Cancer Res (2004) 64(23):8502–6. doi:10.1158/0008-5472.CAN-04-2599
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
192. Yada A, Yazawa M, Ishida S, Yoshida H, Ichikawa K, Kurakata S, et al. A novel humanized anti-human death receptor 5 antibody CS-1008 induces apoptosis in tumor cells without toxicity in hepatocytes. Ann Oncol (2008) 19(6):1060–7. doi:10.1093/annonc/mdn015
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
193. Buchsbaum DJ, Zhou T, Lobuglio AF. TRAIL receptor-targeted therapy. Future Oncol (2006) 2(4):493–508. doi:10.2217/14796694.2.4.493
194. Carlo-Stella C, Lavazza C, Locatelli A, Vigano L, Gianni AM, Gianni L. Targeting TRAIL agonistic receptors for cancer therapy. Clin Cancer Res (2007) 13(8):2313–7. doi:10.1158/1078-0432.CCR-06-2774
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
195. Jo M, Kim TH, Seol DW, Esplen JE, Dorko K, Billiar TR, et al. Apoptosis induced in normal human hepatocytes by tumor necrosis factor-related apoptosis-inducing ligand. Nat Med (2000) 6(5):564–7. doi:10.1038/75045
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
196. Qin J, Chaturvedi V, Bonish B, Nickoloff BJ. Avoiding premature apoptosis of normal epidermal cells. Nat Med (2001) 7(4):385–6. doi:10.1038/86401
197. Lawrence D, Shahrokh Z, Marsters S, Achilles K, Shih D, Mounho B, et al. Differential hepatocyte toxicity of recombinant Apo2L/TRAIL versions. Nat Med (2001) 7(4):383–5. doi:10.1038/nm1201-1291
198. Ashkenazi A, Holland P, Eckhardt SG. Ligand-based targeting of apoptosis in cancer: the potential of recombinant human apoptosis ligand 2/tumor necrosis factor-related apoptosis-inducing ligand (rhApo2L/TRAIL). J Clin Oncol (2008) 26(21):3621–30. doi:10.1200/JCO.2007.15.7198
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
199. Stieglmaier J, Bremer E, Kellner C, Liebig TM, Ten Cate B, Peipp M, et al. Selective induction of apoptosis in leukemic B-lymphoid cells by a CD19-specific TRAIL fusion protein. Cancer Immunol Immunother (2008) 57(2):233–46. doi:10.1007/s00262-007-0370-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
200. Bremer E, van Dam GM, de Bruyn M, van Riezen M, Dijkstra M, Kamps G, et al. Potent systemic anticancer activity of adenovirally expressed EGFR-selective TRAIL fusion protein. Mol Ther (2008) 16(12):1919–26. doi:10.1038/mt.2008.203
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
201. Kelley RF, Totpal K, Lindstrom SH, Mathieu M, Billeci K, Deforge L, et al. Receptor-selective mutants of apoptosis-inducing ligand 2/tumor necrosis factor-related apoptosis-inducing ligand reveal a greater contribution of death receptor (DR) 5 than DR4 to apoptosis signaling. J Biol Chem (2005) 280(3):2205–12. doi:10.1074/jbc.M410660200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
202. van der Sloot AM, Tur V, Szegezdi E, Mullally MM, Cool RH, Samali A, et al. Designed tumor necrosis factor-related apoptosis-inducing ligand variants initiating apoptosis exclusively via the DR5 receptor. Proc Natl Acad Sci U S A (2006) 103(23):8634–9. doi:10.1073/pnas.0510187103
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
203. Griffith TS, Rauch CT, Smolak PJ, Waugh JY, Boiani N, Lynch DH, et al. Functional analysis of TRAIL receptors using monoclonal antibodies. J Immunol (1999) 162(5):2597–605.
204. Ichikawa K, Liu W, Zhao L, Wang Z, Liu D, Ohtsuka T, et al. Tumoricidal activity of a novel anti-human DR5 monoclonal antibody without hepatocyte cytotoxicity. Nat Med (2001) 7(8):954–60. doi:10.1038/91000
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
205. Plummer R, Attard G, Pacey S, Li L, Razak A, Perrett R, et al. Phase 1 and pharmacokinetic study of lexatumumab in patients with advanced cancers. Clin Cancer Res (2007) 13(20):6187–94. doi:10.1158/1078-0432.CCR-07-0950
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
206. Ashkenazi A. Targeting the extrinsic apoptosis pathway in cancer. Cytokine Growth Factor Rev (2008) 19(3-4):325–31. doi:10.1016/j.cytogfr.2008.04.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
207. MacFarlane M, Kohlhaas SL, Sutcliffe MJ, Dyer MJ, Cohen GM. TRAIL receptor-selective mutants signal to apoptosis via TRAIL-R1 in primary lymphoid malignancies. Cancer Res (2005) 65(24):11265–70. doi:10.1158/0008-5472.CAN-05-2801
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
208. Chuntharapai A, Dodge K, Grimmer K, Schroeder K, Marsters SA, Koeppen H, et al. Isotype-dependent inhibition of tumor growth in vivo by monoclonal antibodies to death receptor 4. J Immunol (2001) 166(8):4891–8. doi:10.4049/jimmunol.166.8.4891
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
209. Mom CH, Verweij J, Oldenhuis CN, Gietema JA, Fox NL, Miceli R, et al. Mapatumumab, a fully human agonistic monoclonal antibody that targets TRAIL-R1, in combination with gemcitabine and cisplatin: a phase I study. Clin Cancer Res (2009) 15(17):5584–90. doi:10.1158/1078-0432.CCR-09-0996
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
210. Zhang XD, Zhang XY, Gray CP, Nguyen T, Hersey P. Tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis of human melanoma is regulated by smac/DIABLO release from mitochondria. Cancer Res (2001) 61(19):7339–48.
211. Belyanskaya LL, Marti TM, Hopkins-Donaldson S, Kurtz S, Felley-Bosco E, Stahel RA. Human agonistic TRAIL receptor antibodies mapatumumab and lexatumumab induce apoptosis in malignant mesothelioma and act synergistically with cisplatin. Mol Cancer (2007) 6:66. doi:10.1186/1476-4598-6-66
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
212. Amelio I, Gostev M, Knight RA, Willis AE, Melino G, Antonov AV. DRUGSURV: a resource for repositioning of approved and experimental drugs in oncology based on patient survival information. Cell Death Dis (2014) 5:e1051. doi:10.1038/cddis.2014.9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: TRAIL, cancer, apoptosis, TRAIL-resistance, DR4, DR5
Citation: Trivedi R and Mishra DP (2015) Trailing TRAIL resistance: novel targets for TRAIL sensitization in cancer cells. Front. Oncol. 5:69. doi: 10.3389/fonc.2015.00069
Received: 05 December 2014; Accepted: 09 March 2015;
Published online: 02 April 2015.
Edited by:
Gerry Melino, Medical Research Council, UKReviewed by:
Alessandro Rufini, University of Leicester, UKCopyright: © 2015 Trivedi and Mishra. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Durga Prasad Mishra, Scientist Cell Death Research Laboratory, Endocrinology Division, CSIR-Central Drug Research Institute, Life Science South Lab No 106, B.S. 10/1, Sector-10, Jankipuram Extension, Sitapur Road, Lucknow-226031, India e-mail:ZHBtQGNkcmkucmVzLmlu
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.