 Ana Sofia Ribeiro
Ana Sofia Ribeiro Joana Paredes
Joana Paredes
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Oncol. , 05 January 2015
Sec. Molecular and Cellular Oncology
Volume 4 - 2014 | https://doi.org/10.3389/fonc.2014.00371
This article is part of the Research Topic Epithelial mesenchymal interactions and transitions during carcinoma progression View all 7 articles
Epithelial–mesenchymal transition (also known as EMT) is a fundamental mechanism occurring during embryonic development and tissue differentiation, being also crucial for cancer progression. Actually, the EMT program contributes to the dissemination of cancer cells from solid tumors and to the formation of micro-metastasis that subsequently develop into clinically detectable metastases. Besides being a process that is defined by the progressive loss of epithelial cell characteristics and the acquisition of mesenchymal features, EMT has also been implicated in therapy resistance, immune escape, and maintenance of cancer stem cell properties, such as self-renewal capacity. However, the majority of the studies usually neglect the progressive alterations occurring during intermediate EMT states, which imply a range of phenotypic cellular heterogeneity that can potentially generate more metastable and plastic tumor cells. In fact, few studies have tried to identify these transitory states, partly due to the current lack of a detailed understanding of EMT, as well as of reliable readouts for its progression. Herein, a brief review of evidences is presented, showing that P-cadherin expression, which has been already identified as a breast cancer stem cell marker and invasive promoter, is probably able to identify an intermediate EMT state associated with a metastable phenotype. This hypothesis is based on our own work, as well as on the results described by others, which suggest the use of P-cadherin as a promising EMT marker, clearly functioning as an important clinical prognostic factor and putative therapeutic target in breast carcinogenesis.
Epithelial–mesenchymal transition (EMT) is a highly regulated transdifferentiation cellular program, by which static and polarized epithelial cells convert to an invasive and motile mesenchymal morphology. During the EMT process, there is the progressive loss of epithelial characteristics and the acquisition of mesenchymal features, which, in a cancer context, leads to the development of cells that are chemoresistant, able to escape to immune cells and with stem cell properties. Moreover, this phenotypic transformation gives cancer cells the ability to invade locally, to resist apoptosis, and to metastasize (1–4). Interestingly, the phenotypic plasticity afforded by EMT is revealed by the occurrence of the reverse process, a mesenchymal–epithelial transition (or MET) (5–7), which involves the conversion of mesenchymal cells to an epithelial cell phenotype.
Epithelial–mesenchymal transition is a multistep program that involves a series of changes by which epithelial cells lose their epithelial characteristics and acquire properties that are typical of mesenchymal cells. One of the earliest events during EMT involves the decrease in proteins from tight and adherent junctions (8). Epithelial-like cells exhibit an organized apical–basal polarity maintained by the precise arrangement of actin filaments and adhesive structures, such as tight junctions, adherens junctions, and desmosomes (9). Specialized adhesive molecules, such as cadherins, integrins, and other cell-surface proteins, are essential for the maintenance of the epithelial phenotype by stabilizing cell–cell contacts. Thus, a decreased expression of these proteins complex triggers redistribution of key molecules at the cell surface, disruption of the polarity complex, and cytoskeletal reorganization (10). Following loss of junctional complexes and downregulation of E-Cadherin (epithelial cadherin), β−catenin is no longer sequestered in the cytoplasm and translocates to the nucleus to activate β-catenin responsive genes. In the nucleus, a transcriptional shift is observed, where several transcription factors suppress epithelial markers and activate mesenchymal genes, promoting a mesenchymal-like phenotype (11). At this stage, cytoskeletal proteins are upregulated, as is the deposition of the extracellular matrix (ECM) components. All these alterations stimulate integrin signaling and promote the formation of focal adhesion complexes, leading in this way to an increase in cell motility and invasion capacity.
Conversely to epithelial cell phenotype, mesenchymal cells are characterized by a unique spindle morphology defined by a front-back-end polarity and enhanced invasive potential (12, 13). However, not all cells undergo a complete EMT; instead, most cells undergo partial EMT, an intermediate state in which cells retain some cell–cell adhesion characteristic of epithelial cells, but gain some migratory ability, that is a feature of mesenchymal cells (1). This means that cancer cells in this transient EMT phenotype are endowed with special properties, such as collective cell migration and invasion. Moreover, the transient phenotypic changes during EMT have been also associated with the acquisition of stem-like properties. Actually, the intermediate EMT stage has been coined as the metastable phenotype (14), since it describes the simultaneous existence of both epithelial and mesenchymal characteristics, being of great importance for the understanding of the major cellular changes associated with the progression of the EMT program.
Curiously, the identification of EMT transient stages has been neglected, although it is thought to be more prevalent than the pure mesenchymal cell morphology. In fact, it has been accepted by several authors that this hybrid phenotype, reflecting epithelial–mesenchymal plasticity, should be included in these intermediate states of EMT, implying a range of phenotypic cellular heterogeneity that can generate more plastic and metastable tumor cells (1). Interestingly, the metastatic capacity of these cells is consistent with the expression of stem cell markers in colorectal cells undergoing EMT (15), suggesting that such plasticity may be found in progenitor cells in various organs. This plasticity can explain why it is so rare to observe cells undergoing EMT during cancer progression, and also that the acquisition of mesenchymal characteristics may be transitory, undergoing a reverse process during the last stages of tumorigenesis (6).
Due to the tremendous difficulty in capturing cells in the intermediate states of EMT (16), most studies have been focused in finding biomarkers that identify the non-invasive full epithelial state, as well as the aggressive, motile, and invasive mesenchymal state.
The conversion of epithelial-like cells into mesenchymal-like cells requires alterations in cellular morphology, adhesion, and migratory capacity. The different degrees of EMT can occur in carcinomas, being the alterations found in the expression of molecular markers used to assess the EMT status (17).
At the molecular level, the EMT transition is characterized by a series of coordinated changes including downregulation of epithelial markers (e.g., cytokeratin 8, 18, 19, E-cadherin, claudins, occludins) and upregulation of mesenchymal markers (e.g., vimentin, N-cadherin) (18), what results in numerous phenotypic changes, such as the loss of cell–cell adhesion and cell polarity, and the acquisition of migratory and invasive properties (3, 15). The cell intermediate filament status changes from a keratin-rich network, which connects to adherens junctions and hemidesmosomes, to a vimentin-rich network connecting to focal adhesions.
The loss of functional E-cadherin containing junctions with the concomitant upregulation of N-cadherin (19, 20) is termed cadherin switch and is a hallmark of the EMT process. This switch has been already reported for carcinomas in the esophagus, prostate, cervix, and ovary (20–23), being associated with tumor progression and metastatic disease.
Epithelial–mesenchymal transition process is also particularly associated with the expression of zinc-finger transcription factors Snail (SNAI1) and Slug (SNAI2), as well as of ZEB1 (zinc-finger E-box-binding homeobox 1), ZEB2, FoxC2 (forkhead box protein C2), and TWIST (24, 25). Moreover, several evidences show that the expression of the intermediate filament protein vimentin can be upregulated by different EMT transcription factors in a direct or indirect way (26–30). Both mesenchymal proteins, vimentin and fibronectin, contribute to changes in cytoskeletal architecture and migratory potential of cancer cells (27). All these alterations cause cells to change to a mesenchymal morphology and to a functional change toward migration, invasion, and resistance to apoptosis.
Concerning the transient/metastable phenotype, few specific markers have been proposed, due to the lack of studies evaluating the progressive and transitory states of EMT, as well as of reliable readouts for its progression. Knowing that, there is the need to find markers to identify this metastable phenotype, which will be crucial to understand how to clinically prevent metastasis and to give mechanistic insights to be translated into therapeutic opportunities.
Cell–cell adhesion, mediated by E-cadherin at adherens junctions, is essential for the maintenance of epithelial tissue architecture and homeostasis. Altered expression and/or function of E-cadherin play a major role in the acquisition of cell invasive properties and in the induction of EMT, as well as tumor progression. During EMT, epithelial adherens junctions are dynamically regulated, which trigger signaling pathways and alterations in the organization of the actin cytoskeleton that are involved in the induction of cell motility. In fact, the well-known cadherins switch, namely the loss of E-cadherin and the gain of N-cadherin, is described as being characteristic of EMT and is associated with tumor cell invasion. However, there are examples of highly aggressive and invasive tumors, like basal-like breast carcinomas, where E-cadherin expression is rarely lost, and N-cadherin is hardly overexpressed (31). In contrast, these tumors usually overexpress other classical cadherin, named P-cadherin, mainly in a wild-type E-cadherin context, which intriguingly has never been linked to the EMT process. However, since it has been already proven that P-cadherin is able to interfere with epithelial cell–cell adhesion and to promote cancer cell invasion and metastasis (32), it is our belief that P-cadherin can be also used as a new EMT marker, mainly to identify an intermediate and transient EMT state associated with a metastable phenotype.
The role of P-cadherin in breast carcinogenesis has been extensively studied in the last few years. Presently, it is known that this protein is de novo expressed in near 30–40% of invasive breast carcinomas, being reported as a valuable prognostic factor in this disease. P-cadherin-positive carcinomas are significantly associated with tumors of high histological grade, with short-term overall and disease-free survival, as well as with distant and loco-regional relapse-free interval (31, 33–35). P-cadherin expression still shows a strong correlation with invasion of the vascular and soft tissues (36). Moreover, the overexpression of this protein has also been positively associated with well-established markers and biological parameters associated to poor prognosis in breast cancer, such as epidermal growth factor receptor (EGFR), cytokeratin 5 (CK5), vimentin, p53 and HER2, high proliferation rates (MIB-1), mitotic index, and decreased cell differentiation (31, 33, 34, 37). P-cadherin expression was also inversely related with age at diagnosis, hormonal receptors (ER and PgR) and Bcl-2 expression (31, 33–35).
Interestingly, P-cadherin is a marker of triple-negative (which means negative for ER, PgR, and HER2) basal-like breast carcinomas, which comprise a heterogeneous group of tumors that accounts for up to 15% of all breast cancer cases (31). These tumors are highly aggressive, affect younger patients, are more prevalent in African-American women, and often are present as interval cancers. Histologically, the majority of basal-like breast cancers are IDC-NST (invasive ductal carcinomas of no special type), high histological grade, and characterized by exceptionally high mitotic indices, the presence of central necrotic or fibrotic zones, pushing borders, conspicuous lymphocytic infiltrate, and typical/atypical medullary features (38). As the name indicates, these tumors express genes and proteins usually found in basal/myoepithelial cells of the normal breast, including high-molecular-weight cytokeratins (5/6, 14, and 17), P-cadherin, caveolins 1 and 2, nestin, αB crystallin, CD109, and EGFR (39). Since, until today, these tumors do not harbor any therapeutic target usually used to treat breast cancer patients, all these proteins, including P-cadherin, can be putative therapeutic options to be targeted.
In contrast to E-cadherin, which is an important invasion suppressor protein, it has been shown that P-cadherin behaves as an invasion promoter in several cancer models, including breast cancer. It has been often reported that P-cadherin induces increased tumor cell motility and invasiveness when aberrantly overexpressed (40–46). Our group has demonstrated that one of the mechanisms underlying the invasive capacity of P-cadherin overexpression in breast cancer cells is mediated by the secretion of matrix metalloproteases (MMPs), which degrade the ECM during invasion (44, 45) and cleave P-cadherin extracellular domain to produce a soluble P-cadherin fragment (sP-cad). Interestingly, we have demonstrated that this fragment has pro-invasive effects in non-invasive breast cancer cells (45) (Figure 1). Accordingly, it was demonstrated a significant increased shedding of sP-cad in nipple aspirate fluids from women with breast cancer, when compared with healthy subjects or with women with pre-cancer conditions, suggesting its release via proteolytic processing in cancer cells (47).
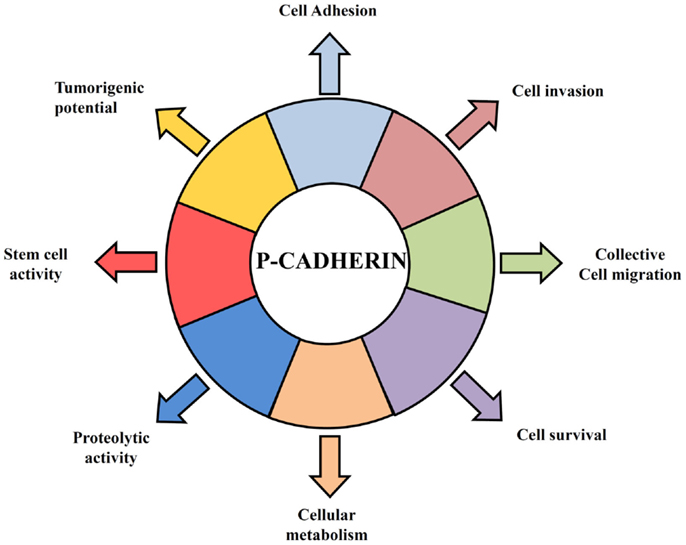
Figure 1. Hallmarks of P-cadherin function in breast cancer cells. P-cadherin overexpressing cells acquire features that give them an advantage to survive in a hostile environment leading to an invasive and tumorigenic phenotype of breast cancer cells. P-cadherin expression affects cell–cell adhesion, since it disrupts the normal suppressor function of E-cadherin, by decreasing the interaction between E-cadherin and intracellular catenins. Overexpression of this protein in breast cancer cells promotes an increase in cell migration and cell invasion, being able to provoke the secretion of pro-invasive factors, such as MMP1 and MMP2, which then lead to P-cadherin ectodomain cleavage (sP-cad) that also has pro-invasive activity by itself. Moreover, P-cadherin expression mediates cancer stem cell properties, conferring resistance to x-ray-induced cell death and being related with a hypoxic, glycolytic, and acid-resistant phenotype in breast cancer cells.
Furthermore, the invasive phenotype mediated by P-cadherin was seemingly dependent on the concomitant expression of wild-type E-cadherin: in cell models where P-cadherin showed an invasion promoter function, E-cadherin was also expressed (44, 46, 48, 49); contrarily, in models, which only express P-cadherin, this protein was described as an invasion suppressor (50–52). This dual functional role of P-cadherin was recently explained by the induction of an aggressive biological cell behavior in cells co-expressing both cadherins compared to cells just expressing one of each cadherin. Indeed, we have found that P-cadherin expression disrupts the normal invasive suppressor function of E-cadherin (32) by destabilizing the normal cadherin/catenin complex (53). The induced-delocalization of β-catenin and p120-catenin from the membrane to the cytoplasm alters the actin cytoskeleton polymerization, and promotes cell migration and motility, as well as an increased invasive and tumorigenic potential (32, 46) (Figure 1).
Breast carcinomas, as well as other solid tumors, contain a small population of cancer cells that have the ability to self-renew and to be tumorigenic, generating all the distinct cancer cells present within the tumor. These cells were called breast cancer stem cells (CSCs), which are the apex of a hierarchy that is comparable to the one established in normal tissues (54). Breast CSCs share a large amount of properties with mammary stem cells, namely the ability to resist to standard cancer therapies, such as radiation and chemotherapy, allowing them to survive and to cause tumor recurrence and metastasis (55–58). Targeting breast CSCs, in combination with current therapies, is the forthcoming goal in breast cancer treatment.
In this context, we have demonstrated that P-cadherin expression is able to identify basal-like breast cancer cells with stem cell properties. Using breast cancer cell lines and primary tumors, we showed that P-cadherin was directly associated with the expression of the breast stem markers CD44, CD49f, and aldehyde dehydrogenase 1 (ALDH1) in the basal-like molecular subtype. Moreover, P-cadherin-enriched cancer cell populations comprised increased in vitro mammosphere-forming efficiency, as well as increased tumorigenicity. Additionally, the expression of this adhesion molecule still conferred resistance to x-ray-induced cell death, sustaining a role for this molecule in another stem cell property (59) (Figure 1).
Importantly, we found that P-cadherin expression was associated with stem-/progenitor-like phenotypes of the breast, including the luminal progenitor population, CD49f+CD24+. Accordingly, Nassour et al. recently demonstrated that the expression of Slug, P-cadherin, and CD49f has a role in the growth dynamics of a subpopulation of cycling progenitor basal cap and duct cells during mammary morphogenesis (60). Moreover, we were able to establish that there is a crosstalk between the expression of P-cadherin and CD49f (α6 integrin). Actually, we have demonstrated that P-cadherin regulates the laminin receptor α6β4 integrin-signaling pathway, which activation explains the stem cell and invasive properties induced by P-cadherin to breast cancer cells (61).
Cancer stem cells are also usually described as hypoxia-resistant, presenting a preponderant glycolytic metabolism. These characteristics are also found enriched in basal-like breast carcinomas, which show increased expression of cancer stem cell markers (62). Interestingly, we demonstrated that cancer cell populations harboring high levels of P-cadherin were the same exhibiting more GLUT1 and CAIX expression. Moreover, its silencing significantly decreased the mammosphere-forming efficiency in the same range as the silencing of HIF-1α, CAIX, or GLUT1, substantiating that all these markers are being expressed by the same breast cancer stem cell population (63) (Figure 1).
All the knowledge that was acquired concerning P-cadherin expression in cancer supported the development of anti-P-cadherin therapeutic strategies. A humanized monoclonal antibody (PF-03732010) was developed to antagonize P-cadherin-regulated cell–cell adhesion and the associated signaling pathway. Actually, a study using this antibody confirmed the role of P-cadherin as a molecule involved in cell invasion, as well as in metastization. The authors observed that PF-03732010 treated cells and tumors showed disrupted P-cadherin signaling and resulted in anti-tumorigenic and anti-metastatic activity (64).
Based on these results and on our own data, we hypothesize that P-cadherin could be a promising marker of the metastable and intermediate EMT phenotype (Figure 2). The rationale for this hypothesis relies on the fact that P-cadherin expression disturbs epithelial cell–cell adhesion and promotes the acquisition of a more undifferentiated cell phenotype, acquiring these cells an intermediate phenotypic state between epithelial and mesenchymal morphology. Additionally, P-cadherin overexpressing cells show increased therapy resistance, stem cell properties, and a more aggressive and invasive behavior. Finally, P-cadherin expression is a factor of poor prognosis in breast cancer, being associated to tumors with high metastatic potential. Thus, all these characteristics points to a transient EMT state, with cancer cells harboring an increased plasticity and metastability.
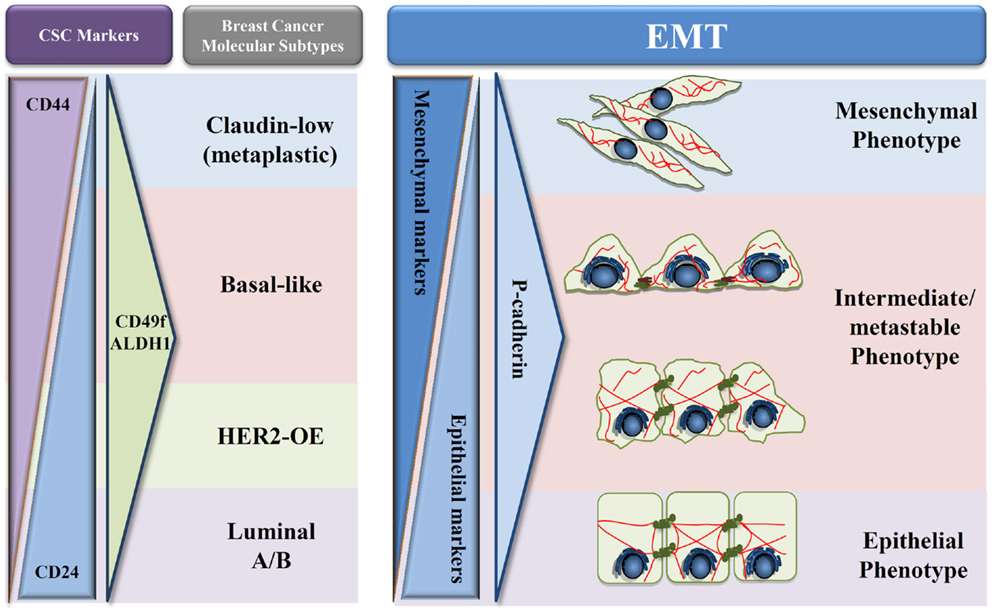
Figure 2. Proposed model of P-cadherin expression in EMT progression. Schematic representation adapted from Schmitt et al. (54) of the different types of breast cancer in what concerns cancer stem cells proteins (CD24, CD44, CD49f, and ALDH1), EMT markers, and P-cadherin expression during EMT progression. A decrease of epithelial proteins with a concomitant increase in mesenchymal markers is observed during the transition from an epithelial to a mesenchymal phenotype. During this process, we hypothesize that P-cadherin expression is in very low levels in both full epithelial and full mesenchymal states; however, an increased expression can be seen in the metastable and intermediate states of EMT.
This premise seems to be corroborated by preliminary data that we have recently produced, where a three-protein EMT signature has been applied to a series of 500 invasive breast carcinomas, using the expression of two classical EMT markers, E-cadherin and vimentin, combined with P-cadherin expression. Our results show that P-cadherin identifies an intermediate state between the epithelial and the mesenchymal phenotypes, associating to a poor prognosis in breast cancer patients, but also with the expression of breast cancer stem cell markers. Although this data need further validation, both in independent breast cancer series and by in vitro and in vivo functional assays, our future aim is to definitely prove the crucial role of P-cadherin in identifying the EMT “intermediate/metastable” phenotype. Proving this hypothesis, we will clarify the role of P-cadherin in the EMT process, identifying it as a putative EMT marker and supporting the use of anti-P-cadherin therapeutics to metastatic breast cancer.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Klymkowsky MW, Savagner P. Epithelial-mesenchymal transition: a cancer researcher’s conceptual friend and foe. Am J Pathol (2009) 174:1588–93. doi:10.2353/ajpath.2009.080545
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
2. Polyak K, Weinberg RA. Transitions between epithelial and mesenchymal states: acquisition of malignant and stem cell traits. Nat Rev Cancer (2009) 9:265–73. doi:10.1038/nrc2620
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
3. Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell (2009) 139:871–90. doi:10.1016/j.cell.2009.11.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
4. Yilmaz M, Christofori G. Emt, the cytoskeleton, and cancer cell invasion. Cancer Metastasis Rev (2009) 28:15–33. doi:10.1007/s10555-008-9169-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
5. Sorlie T, Tibshirani R, Parker J, Hastie T, Marron JS, Nobel A, et al. Repeated observation of breast tumor subtypes in independent gene expression data sets. Proc Natl Acad Sci USA (2003) 100:8418–23. doi:10.1073/pnas.0932692100
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
6. Brabletz T. Emt and met in metastasis: where are the cancer stem cells? Cancer Cell (2012) 22:699–701. doi:10.1016/j.ccr.2012.11.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
7. Carstens JL, Lovisa S, Kalluri R. Microenvironment-dependent cues trigger mirna-regulated feedback loop to facilitate the emt/met switch. J Clin Invest (2014) 124:1458–60. doi:10.1172/JCI75239
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
8. Jeanes A, Gottardi CJ, Yap AS. Cadherins and cancer: how does cadherin dysfunction promote tumor progression? Oncogene (2008) 27:6920–9. doi:10.1038/onc.2008.343
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
9. Han SP, Yap AS. The cytoskeleton and classical cadherin adhesions. Subcell Biochem (2012) 60:111–35. doi:10.1007/978-94-007-4186-7_6
10. Brieher WM, Yap AS. Cadherin junctions and their cytoskeleton(s). Curr Opin Cell Biol (2013) 25:39–46. doi:10.1016/j.ceb.2012.10.010
11. Azmi AS. Unveiling the role of nuclear transport in epithelial-to-mesenchymal transition. Curr Cancer Drug Targets (2013) 13:906–14. doi:10.2174/15680096113136660096
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
12. Hay ED. The mesenchymal cell, its role in the embryo, and the remarkable signaling mechanisms that create it. Dev Dyn (2005) 233:706–20. doi:10.1002/dvdy.20345
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
13. Zavadil J, Haley J, Kalluri R, Muthuswamy SK, Thompson E. Epithelial-mesenchymal transition. Cancer Res (2008) 68:9574–7. doi:10.1158/0008-5472.CAN-08-2316
14. Lee JM, Dedhar S, Kalluri R, Thompson EW. The epithelial-mesenchymal transition: new insights in signaling, development, and disease. J Cell Biol (2006) 172:973–81. doi:10.1083/jcb.200601018
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
15. Ksiazkiewicz M, Markiewicz A, Zaczek AJ. Epithelial-mesenchymal transition: a hallmark in metastasis formation linking circulating tumor cells and cancer stem cells. Pathobiology (2012) 79:195–208. doi:10.1159/000337106
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
16. Krasnapolski MA, Todaro LB, de Kier Joffe EB. Is the epithelial-to-mesenchymal transition clinically relevant for the cancer patient? Curr Pharm Biotechnol (2011) 12:1891–9. doi:10.2174/138920111798377021
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
17. Chaw SY, Majeed AA, Dalley AJ, Chan A, Stein S, Farah CS. Epithelial to mesenchymal transition (emt) biomarkers – e-cadherin, beta-catenin, apc and vimentin – in oral squamous cell carcinogenesis and transformation. Oral Oncol (2012) 48:997–1006. doi:10.1016/j.oraloncology.2012.05.011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
18. Markiewicz A, Ahrends T, Welnicka-Jaskiewicz M, Seroczynska B, Skokowski J, Jaskiewicz J, et al. Expression of epithelial to mesenchymal transition-related markers in lymph node metastases as a surrogate for primary tumor metastatic potential in breast cancer. J Transl Med (2012) 10:226. doi:10.1186/1479-5876-10-226
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
19. Li G, Herlyn M. Dynamics of intercellular communication during melanoma development. Mol Med Today (2000) 6:163–9. doi:10.1016/S1357-4310(00)01692-0
20. Tomita K, van Bokhoven A, van Leenders GJ, Ruijter ET, Jansen CF, Bussemakers MJ, et al. Cadherin switching in human prostate cancer progression. Cancer Res (2000) 60:3650–4.
21. Bailey T, Biddlestone L, Shepherd N, Barr H, Warner P, Jankowski J. Altered cadherin and catenin complexes in the Barrett’s esophagus-dysplasia-adenocarcinoma sequence: correlation with disease progression and dedifferentiation. Am J Pathol (1998) 152:135–44.
22. de Boer CJ, van Dorst E, van Krieken H, Jansen-van Rhijn CM, Warnaar SO, Fleuren GJ, et al. Changing roles of cadherins and catenins during progression of squamous intraepithelial lesions in the uterine cervix. Am J Pathol (1999) 155:505–15. doi:10.1016/S0002-9440(10)65146-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
23. Patel IS, Madan P, Getsios S, Bertrand MA, MacCalman CD. Cadherin switching in ovarian cancer progression. Int J Cancer (2003) 106:172–7. doi:10.1002/ijc.11086
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
24. Lim J, Thiery JP. Epithelial-mesenchymal transitions: insights from development. Development (2012) 139:3471–86. doi:10.1242/dev.071209
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
25. Chu PY, Hu FW, Yu CC, Tsai LL, Yu CH, Wu BC, et al. Epithelial-mesenchymal transition transcription factor zeb1/zeb2 co-expression predicts poor prognosis and maintains tumor-initiating properties in head and neck cancer. Oral Oncol (2013) 49:34–41. doi:10.1016/j.oraloncology.2012.07.012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
26. Guo J, Fu Z, Wei J, Lu W, Feng J, Zhang S. Prrx1 promotes epithelial-mesenchymal transition through the wnt/beta-catenin pathway in gastric cancer. Med Oncol (2015) 32:393. doi:10.1007/s12032-014-0393-x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
27. Ivaska J. Vimentin: central hub in emt induction? Small GTPases (2011) 2:51–3. doi:10.4161/sgtp.2.1.15114
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
28. Ivaska J, Heino J. Cooperation between integrins and growth factor receptors in signaling and endocytosis. Annu Rev Cell Dev Biol (2011) 27:291–320. doi:10.1146/annurev-cellbio-092910-154017
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
29. Kaufhold S, Bonavida B. Central role of snail1 in the regulation of emt and resistance in cancer: a target for therapeutic intervention. J Exp Clin Cancer Res (2014) 33:62. doi:10.1186/s13046-014-0062-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
30. Liu AN, Zhu ZH, Chang SJ, Hang XS. Twist expression associated with the epithelial-mesenchymal transition in gastric cancer. Mol Cell Biochem (2012) 367:195–203. doi:10.1007/s11010-012-1333-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
31. Paredes J, Albergaria A, Oliveira JT, Jeronimo C, Milanezi F, Schmitt FC. P-cadherin overexpression is an indicator of clinical outcome in invasive breast carcinomas and is associated with cdh3 promoter hypomethylation. Clin Cancer Res (2005) 11:5869–77. doi:10.1158/1078-0432.CCR-05-0059
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
32. Ribeiro AS, Sousa B, Carreto L, Mendes N, Nobre AR, Ricardo S, et al. P-cadherin functional role is dependent on e-cadherin cellular context: a proof of concept using the breast cancer model. J Pathol (2013) 229:705–18. doi:10.1002/path.4143
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
33. Gamallo C, Moreno-Bueno G, Sarrio D, Calero F, Hardisson D, Palacios J. The prognostic significance of p-cadherin in infiltrating ductal breast carcinoma. Mod Pathol (2001) 14:650–4. doi:10.1038/modpathol.3880367
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
34. Peralta Soler A, Knudsen KA, Salazar H, Han AC, Keshgegian AA. P-cadherin expression in breast carcinoma indicates poor survival. Cancer (1999) 86:1263–72. doi:10.1002/(SICI)1097-0142(19991001)86:7<1263::AID-CNCR23>3.0.CO;2-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
35. Turashvili G, McKinney SE, Goktepe O, Leung SC, Huntsman DG, Gelmon KA, et al. P-cadherin expression as a prognostic biomarker in a 3992 case tissue microarray series of breast cancer. Mod Pathol (2011) 24:64–81. doi:10.1038/modpathol.2010.189
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
36. Liu N, Yu Q, Liu TJ, Gebreamlak EP, Wang SL, Zhang RJ, et al. P-cadherin expression and basal-like subtype in breast cancers. Med Oncol (2012) 29:2606–12. doi:10.1007/s12032-012-0218-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
37. Sousa B, Paredes J, Milanezi F, Lopes N, Martins D, Dufloth R, et al. P-cadherin, vimentin and ck14 for identification of basal-like phenotype in breast carcinomas: an immunohistochemical study. Histol Histopathol (2010) 25:963–74.
38. Badve S, Dabbs DJ, Schnitt SJ, Baehner FL, Decker T, Eusebi V, et al. Basal-like and triple-negative breast cancers: a critical review with an emphasis on the implications for pathologists and oncologists. Mod Pathol (2011) 24:157–67. doi:10.1038/modpathol.2010.200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
39. Perou CM, Sorlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA, et al. Molecular portraits of human breast tumours. Nature (2000) 406:747–52. doi:10.1038/35021093
40. Cheung LW, Leung PC, Wong AS. Cadherin switching and activation of p120 catenin signaling are mediators of gonadotropin-releasing hormone to promote tumor cell migration and invasion in ovarian cancer. Oncogene (2010) 29:2427–40. doi:10.1038/onc.2009.523
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
41. Kumper S, Ridley AJ. P120ctn and p-cadherin but not e-cadherin regulate cell motility and invasion of du145 prostate cancer cells. PLoS One (2010) 5:e11801. doi:10.1371/journal.pone.0011801
42. Mandeville JA, Silva Neto B, Vanni AJ, Smith GL, Rieger-Christ KM, Zeheb R, et al. P-cadherin as a prognostic indicator and a modulator of migratory behaviour in bladder carcinoma cells. BJU Int (2008) 102:1707–14. doi:10.1111/j.1464-410X.2008.08115.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
43. Paredes J, Correia AL, Ribeiro AS, Albergaria A, Milanezi F, Schmitt FC. P-cadherin expression in breast cancer: a review. Breast Cancer Res (2007) 9:214. doi:10.1186/bcr1774
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
44. Paredes J, Stove C, Stove V, Milanezi F, Van Marck V, Derycke L, et al. P-cadherin is up-regulated by the antiestrogen ici 182,780 and promotes invasion of human breast cancer cells. Cancer Res (2004) 64:8309–17. doi:10.1158/0008-5472.CAN-04-0795
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
45. Ribeiro AS, Albergaria A, Sousa B, Correia AL, Bracke M, Seruca R, et al. Extracellular cleavage and shedding of p-cadherin: a mechanism underlying the invasive behaviour of breast cancer cells. Oncogene (2010) 29:392–402. doi:10.1038/onc.2009.338
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
46. Taniuchi K, Nakagawa H, Hosokawa M, Nakamura T, Eguchi H, Ohigashi H, et al. Overexpressed p-cadherin/cdh3 promotes motility of pancreatic cancer cells by interacting with p120ctn and activating rho-family gtpases. Cancer Res (2005) 65:3092–9. doi:10.1158/0008.5472.CAN-04-3646
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
47. Mannello F, Tonti GA, Medda V, Pederzoli A, Sauter ER. Increased shedding of soluble fragments of p-cadherin in nipple aspirate fluids from women with breast cancer. Cancer Sci (2008) 99:2160–9. doi:10.1111/j.1349-7006.2008.00921.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
48. Ben Hamida A, Labidi IS, Mrad K, Charafe-Jauffret E, Ben Arab S, Esterni B, et al. Markers of subtypes in inflammatory breast cancer studied by immunohistochemistry: prominent expression of p-cadherin. BMC Cancer (2008) 8:28. doi:10.1186/1471-2407-8-28
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
49. Cheung LW, Mak AS, Cheung AN, Ngan HY, Leung PC, Wong AS. P-cadherin cooperates with insulin-like growth factor-1 receptor to promote metastatic signaling of gonadotropin-releasing hormone in ovarian cancer via p120 catenin. Oncogene (2011) 30:2964–74. doi:10.1038/onc.2011.7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
50. Bauer K, Dowejko A, Bosserhoff AK, Reichert TE, Bauer RJ. P-cadherin induces an epithelial-like phenotype in oral squamous cell carcinoma by gsk-3beta-mediated snail phosphorylation. Carcinogenesis (2009) 30:1781–8. doi:10.1093/carcin/bgp175
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
51. Sarrio D, Palacios J, Hergueta-Redondo M, Gomez-Lopez G, Cano A, Moreno-Bueno G. Functional characterization of e- and p-cadherin in invasive breast cancer cells. BMC Cancer (2009) 9:74. doi:10.1186/1471-2407-9-74
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
52. Van Marck V, Stove C, Van Den Bossche K, Stove V, Paredes J, Vander Haeghen Y, et al. P-cadherin promotes cell-cell adhesion and counteracts invasion in human melanoma. Cancer Res (2005) 65:8774–83. doi:10.1158/0008-5472.CAN-04-4414
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
53. Paredes J, Correia AL, Ribeiro AS, Milanezi F, Cameselle-Teijeiro J, Schmitt FC. Breast carcinomas that co-express e- and p-cadherin are associated with p120-catenin cytoplasmic localisation and poor patient survival. J Clin Pathol (2008) 61:856–62. doi:10.1136/jcp.2007.052704
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
54. Schmitt F, Ricardo S, Vieira AF, Dionisio MR, Paredes J. Cancer stem cell markers in breast neoplasias: their relevance and distribution in distinct molecular subtypes. Virchows Arch (2012) 460:545–53. doi:10.1007/s00428-012-1237-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
56. Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell (2008) 133:704–15. doi:10.1016/j.cell.2008.03.027
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
57. Morel AP, Lievre M, Thomas C, Hinkal G, Ansieau S, Puisieux A. Generation of breast cancer stem cells through epithelial-mesenchymal transition. PLoS One (2008) 3:e2888. doi:10.1371/journal.pone.0002888
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
58. Ablett MP, Singh JK, Clarke RB. Stem cells in breast tumours: are they ready for the clinic? Eur J Cancer (2012) 48:2104–16. doi:10.1016/j.ejca.2012.03.019
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
59. Vieira AF, Ricardo S, Ablett MP, Dionisio MR, Mendes N, Albergaria A, et al. P-cadherin is coexpressed with cd44 and cd49f and mediates stem cell properties in basal-like breast cancer. Stem Cells (2012) 30:854–64. doi:10.1002/stem.1075
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
60. Nassour M, Idoux-Gillet Y, Selmi A, Come C, Faraldo ML, Deugnier MA, et al. Slug controls stem/progenitor cell growth dynamics during mammary gland morphogenesis. PLoS One (2012) 7:e53498. doi:10.1371/journal.pone.0053498
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
61. Vieira AF, Ribeiro AS, Dionisio MR, Sousa B, Nobre AR, Albergaria A, et al. P-cadherin signals through the laminin receptor alpha6beta4 integrin to induce stem cell and invasive properties in basal-like breast cancer cells. Oncotarget (2014) 5:679–92.
62. Ricardo S, Vieira AF, Gerhard R, Leitao D, Pinto R, Cameselle-Teijeiro JF, et al. Breast cancer stem cell markers cd44, cd24 and aldh1: expression distribution within intrinsic molecular subtype. J Clin Pathol (2011) 64:937–46. doi:10.1136/jcp.2011.090456
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
63. Sousa B, Ribeiro AS, Nobre AR, Lopes N, Martins D, Pinheiro C, et al. The basal epithelial marker p-cadherin associates with breast cancer cell populations harboring a glycolytic and acid-resistant phenotype. BMC Cancer (2014) 14:734. doi:10.1186/1471-2407-14-734
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
64. Zhang CC, Yan Z, Zhang Q, Kuszpit K, Zasadny K, Qiu M, et al. Pf-03732010: a fully human monoclonal antibody against p-cadherin with antitumor and antimetastatic activity. Clin Cancer Res (2010) 16:5177–88. doi:10.1158/1078-0432.CCR-10-1343
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: P-cadherin, EMT transition, breast cancer, metastasis, metastable phenotype
Citation: Ribeiro AS and Paredes J (2015) P-cadherin linking breast cancer stem cells and invasion: a promising marker to identify an “intermediate/metastable” EMT state. Front. Oncol. 4:371. doi: 10.3389/fonc.2014.00371
Received: 22 October 2014; Paper pending published: 21 November 2014;
Accepted: 11 December 2014; Published online: 05 January 2015.
Edited by:
Pierre Savagner, INSERM, FranceCopyright: © 2015 Ribeiro and Paredes. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Joana Paredes, Institute of Molecular Pathology and Immunology of the University of Porto (IPATIMUP), Rua Dr. Roberto Frias s/n, Porto 4200-465, Portugal e-mail:anBhcmVkZXNAaXBhdGltdXAucHQ=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.