 Virginie Vlaeminck-Guillem
Virginie Vlaeminck-Guillem Germain Gillet1
Germain Gillet1
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol., 18 August 2014
Sec. Genitourinary Oncology
Volume 4 - 2014 | https://doi.org/10.3389/fonc.2014.00222
This article is part of the Research TopicProstate Cancer: what we know and what we would like to knowView all 5 articles
A key question for urologic practitioners is whether an apparently organ-confined prostate cancer (PCa) is actually aggressive or not. The dilemma is to specifically identify among all prostate tumors the very aggressive high-grade cancers that will become life-threatening by developing extra-prostatic invasion and metastatic potential and the indolent cancers that will never modify a patient’s life expectancy. A choice must be made between several therapeutic options to achieve the optimal personalized management of the disease that causes as little harm as possible to patients. Reliable clinical, biological, or pathological markers that would enable distinctions to be made between aggressive and indolent PCas in routine practice at the time of initial diagnosis are still lacking. The molecular mechanisms that explain why a PCa is aggressive or not are also poorly understood. Among the potential markers and/or actors in PCa aggressiveness, Src and other members of the Src kinase family, are valuable candidates. Activation of Src-dependent intracellular pathways is frequently observed in PCa. Indeed, Src is at the cross-roads of several pathways [including androgen receptor (AR), TGFbeta, Bcl-2, Akt/PTEN or MAPK, and ERK …], and is now known to influence some of the cellular and tissular events that accompany tumor progression: cell proliferation, cell motility, invasion, epithelial-to-mesenchymal transition, resistance to apoptosis, angiogenesis, neuroendocrine differentiation, and metastatic spread. Recent work even suggests that Src could also play a part in PCa initiation in coordination with the AR. The aim of this review is to gather data that explore the links between the Src kinase family and PCa progression and aggressiveness.
Prostate cancer (PCa) is the most common non-skin cancer and the second leading cause of cancer deaths in men from Western countries. Diagnosis can be made fortuitously during the pathological examination of prostate tissue removed because of symptomatic benign prostate hyperplasia, but the disease is more often discovered following prostate biopsies performed because of elevated serum prostate-specific antigen (PSA) levels and/or abnormal digital rectal examination. Another diagnostic scenario is the identification of a PCa at a metastatic stage when metastases (most often bone metastases) become clinically significant. At this time, no curative treatment can be expected and the only therapeutic options are androgen deprivation, which is achieved by surgery (resection of the androgen-secreting testicular tissue), LH–RH analogs (that block testicular androgen secretion), and/or antiandrogens (that block androgen action on the nuclear androgen receptor – AR). Although initially effective, androgen deprivation usually results in an escape with the emergence of a lethal castration-resistant PCa (CRPC). Therapeutic options are more diverse for (apparently) localized, prostate-confined cancers; in this situation, treatments (traditionally radical prostatectomy and radiation therapy) are effective and given with a curative intent. These treatments can, however, have a profound impact on a patient’s quality of life, and clinicians have to be sure that they are provided adequately, in particular, clinicians have to face two interconnected risks (Table 1): (1) the risk of over-treating a localized cancer that will never become life-threatening (the so-called indolent PCa), and (2) the risk to ineffectively treating an aggressive cancer (the so-called high-grade PCa). The pregnant problem when facing a PCa is therefore the need to have an appropriate estimation of its aggressiveness in order to manage the patient with the most appropriate treatment. This estimation is currently based on clinical (organ-confined or not on digital rectal examination), biological (PSA and/or derivatives), and pathological (Gleason score or number/proportion of invaded cores at biopsy) criteria, but remains poorly reliable. Research studies have therefore been performed world-wide to identify and evaluate better criteria for assessing PCa aggressiveness. Efforts are usually focused on molecular markers. The hypothesis is that the better characterization of the intrinsic biological properties of the tumor will lead to more personalized treatment. There are well-established genetic alterations associated with PCa, such as the upregulation of AR-signaling, the overexpression of c-Myc, the loss of the tumor-suppressor gene Pten (and subsequent activation of the Akt pathway), and the fusion of Ets genes with upstream AR-regulated promoter sequences (with TMPRSS2-Erg being the most frequently observed fusion gene). Among others, the c-Src tyrosine kinase (TKs) recently received particular attention because of its implication in several aspects of PCa initiation and progression.

Table 1. The therapeutic stakes of prostate cancer.
Tyrosine kinases are known to be involved in several fundamental physiological and pathological processes, and include receptor (usually membrane-located) and non-receptor TKs. Src is the prototypical member of the Src family of kinases (SFK), which is the largest family of non-receptor TKs. The SFK family has nine members (Blk, Fgr, Fyn, Hck, Lck, Lyn, Src, Yes, and Yrk) that are implicated in several signal transduction pathways. All SFK members share a similar structure: four Src homology (SH1 to SH4) domains and a unique amino-terminal domain. Src is the most widely studied SFK member, and high resolution crystallographic studies have revealed the complex mechanisms that allow the switch from an inactive to an active state (Figure 1). Src is essentially locked in an inactive conformation through phosphorylation at the tyrosine Y530, which is located in its negative C-terminal regulatory tail. This phosphorylation is performed by the C-terminal Src kinase (Csk) and homologous enzymes. Phosphorylation of tyrosine Y419 in the SH1 kinase domain is the key event associated with protein unfolding and catalytically active conformation. The transition from the off-conformation to the on-conformation occurs upon stimulation from numerous extracellular and intracellular signals. For example, Src and other SFK members are downstream of multiple membrane receptor TKs, including EGFR-Rc and IGF1-Rc. Other interacting membrane proteins are G-protein coupled receptors and integrins. Src is also known to crosstalk with several intracellular compounds such as other non-receptor TKs, steroid receptors, or components of fundamental kinase pathways (PI3K, MAPK, …). For example, Src is usually found within a complex comprising two other non-receptor TKs: focal adhesion kinases (FAK) and Etk. The three kinases are cross-reactive with each other and therefore share the activation process when one of them is activated by a specific stimulus. Overall, Src can be regarded as a scaffolding adaptor between membrane and/or intracellular proteins and these interactions can result in mutual activation/repression depending on phosphorylation exchanges. Src activation has been observed in several cancers, including PCa (1–3), and several lines of evidence link Src and SFKs to prostate carcinogenesis. Src is highly expressed in PCa cell lines (4–9), as well as in the majority of PCa specimens (2, 5, 8, 10), and has become a new therapeutic target. Src inhibitors have recently reached the clinical development stage in managing patients with metastatic PCa.
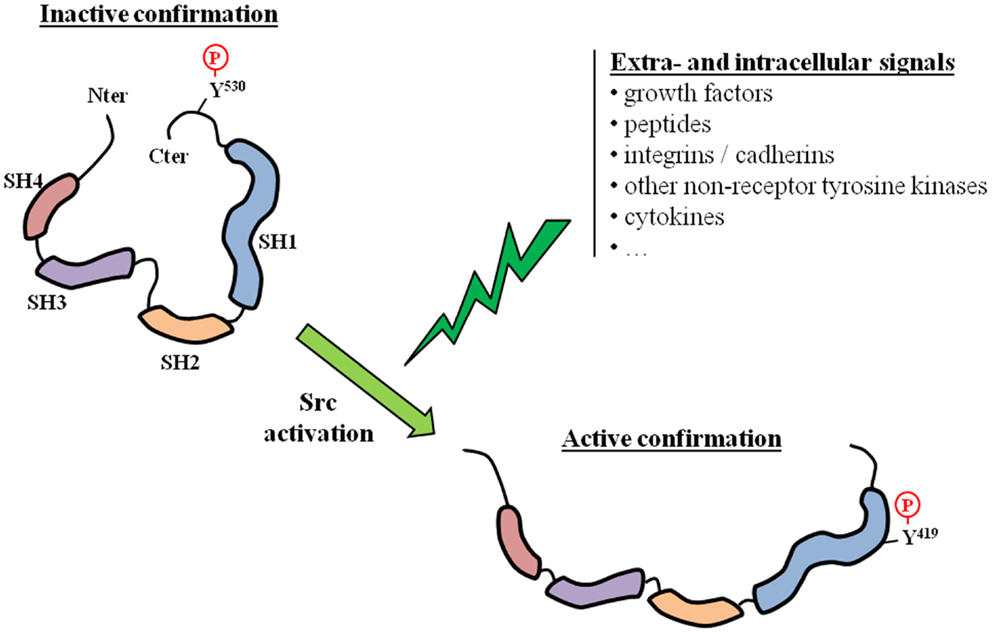
Figure 1. Src activation. The activation of Src requires a switch from an inactive to an active conformation. This corresponds to a phosphorylation switch from a tyrosine residue located in the regulatory C-terminal tail (Y530), which is characteristic of the inactive state, to a tyrosine residue located in the catalytic SH1 domain (Y419). Src activation can result from various extra- and/or intracellular signals.
Prostatic intraepithelial neoplasia (PIN) is the putative precursor lesion of PCa. Due to the difficulty in profiling small lesions, little is known about gene expression and genetic alterations that could account for the transition from PIN to cancer. The initiation of PCa has, however, been linked to the loss or mutation of PTEN and the subsequent activation of AKT-signaling (11), as well as to Ets fusion genes (12). Alterations in AR-signaling have also been advocated, either alone (13) or in combination with the activation of AKT-signaling (14–16). Most of these studies originated from Witte’s laboratory and used an in vivo prostate regeneration system in which prostate tissue is regenerated by combining the embryonic urogenital sinus mesenchyme and the postnatal prostate epithelium (12, 14–16). By specifically over-expressing oncogenes of interest, the influence of extrinsic signals on the initiation and progression of PCa can be evaluated. As a result of Src overexpression in many PCa specimens, the overexpression of Src was induced in this system, either alone or in combination with AR overexpression (17). In these experiments, while the overexpression of either Src alone or the AR alone did not significantly change the prostate tubule structure, the simultaneous overexpression of the AR and Src produced sheets of undifferentiated cells with no glandular organization, which is characteristic of a poorly- or un-differentiated PCa. These results are consistent with those previously obtained in the same regeneration system by chronic exposure to paracrine FGF10 (15): induction of PIN and PCa was achieved (15), probably through Src activation, since Src is known to mediate FGF-signaling, while selective Src loss or inactivation inhibited FGF10-induced PIN and PCa (18). A similar effect was observed for Lyn, but not for Fyn, which are other members of the SFK (18). Of interest is the fact that AR overexpression was necessary for the oncogenic potential of wild-type Src, while a constitutively active mutant Src (Y529F) alone phenocopied the synergistic action of the AR and wild-type Src (17). This suggests that the AR is able to activate Src (19, 20). This is a result that is consistent with the detection of increased levels of activated Src in the tumors induced by the simultaneous overexpression of the AR and Src (17). Indeed, cross-activation between the AR and Src is clearly advocated as a way to explain their synergy (17, 21), which is possibly favored by a physical interaction between both proteins (19, 20, 22) (Figure 2). The AR does indeed contain a proline-rich zone that is affine for the Src SH3 domain and allows the formation of an AR–Src complex (19, 20). The oncogenic properties of this complex are also suggested by the influence of the DOC2/DAB2 (differentially expressed in ovarian cancer 2/disabled 2) protein and its partner DAB2IP. DOC2/DAB2 and DAB2IP are considered to be tumor-suppressor genes and are able to counteract the formation and oncogenic action of the AR–Src complex by physically interacting with Src (23, 24). It is notable that the synergy between the AR and ETS-related gene (ERG) has also been suggested as promoting PCa initiation (12). Since functional interactions between the ERG and Src have also been reported (25), whether mutual and possibly synergistic cross-talks between Src, the ERG, and the AR are involved in PCa initiation should be investigated.
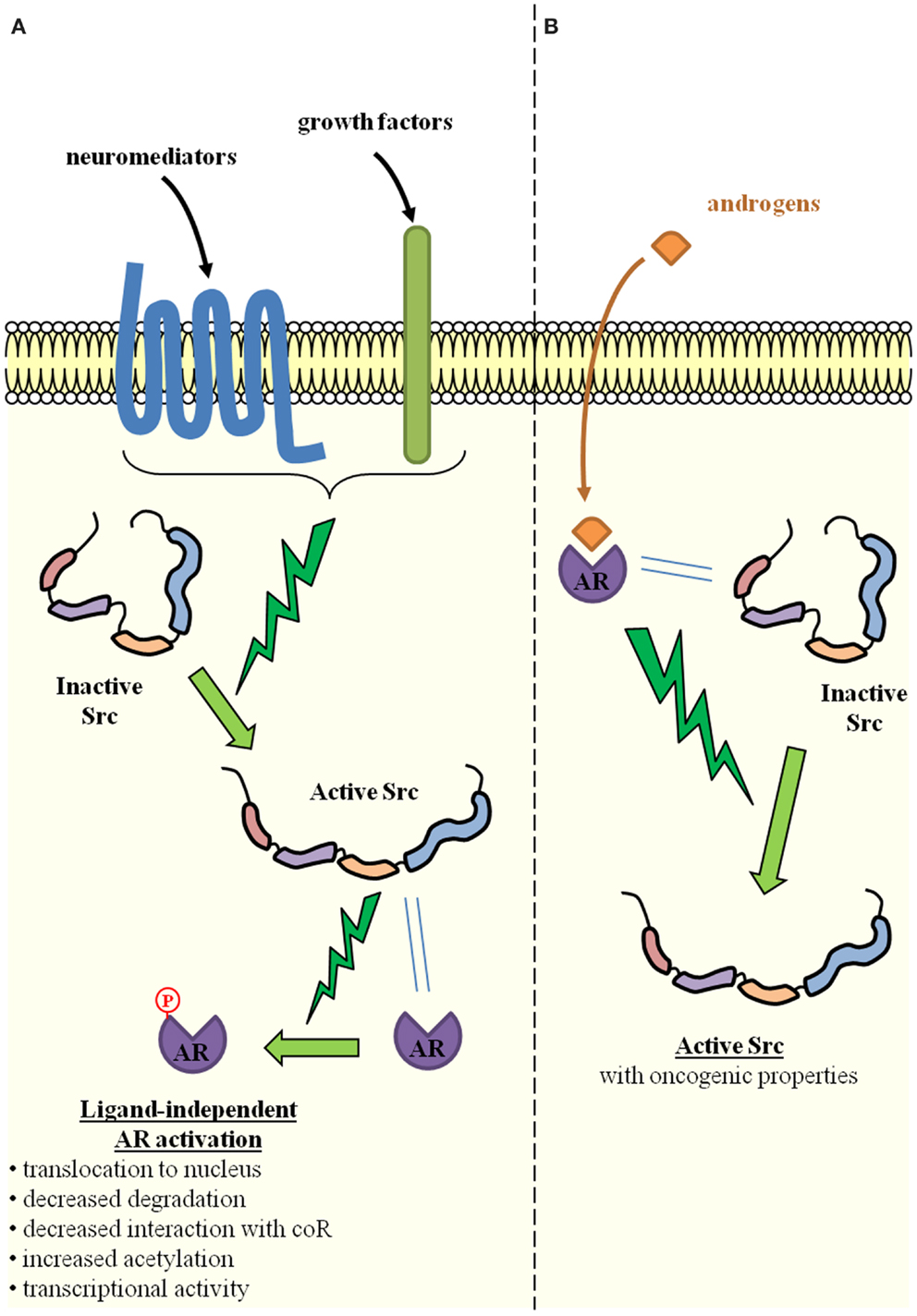
Figure 2. Reciprocal activation of Src and the androgen receptor through direct physical interaction. (A) Src activation is induced by several extracellular signals such as neuromediators produced by neighboring tumor cells with neuroendocrine differentiation, growth factors produced by neighboring tumor or stromal cells. Through direct physical interaction with the AR, Src is able to phosphorylate the AR and thereby induce ligand-independent AR activation (one of the key mechanisms of castration-resistant prostate cancer). Molecular mechanisms include increased AR translocation to the nucleus, decreased proteasomal degradation, decreased interaction with co-repressors (CoR), and/or increased acetylation. The result is the activation of AR-dependent gene expression programs. (B) Conversely, upon ligand binding and direct physical interaction with Src, the AR is able to induce Src activation, which is one of the cellular events associated with oncogenic transformation.
The role of Src and other SFKs in PCa progression has been suggested by a number of demonstrations of their expression in prostate cell lines (4–9) and PCa specimens (5, 8, 10). There are also many reports that SFKs are abnormally activated in PCa cells (1–3), in response to numerous and interconnected stimuli including neuroendocrine ligands (26–28), reactive oxygen species (29), cytokines such as Il-8 (30, 31), growth factors like EGF (27, 29, 32), IGF-1 (28, 33) VEGF (34, 35), or even intracellular activating proteins such as FAK (36). It is notable that all of these molecules are by themselves involved in several basic aspects of cancer progression, including cell proliferation, adhesion, migration, and invasion. SFKs can therefore be regarded as integral components of the signal transduction pathways involved in normal cellular growth, proliferation, migration, and survival, all of which are processes that, if deregulated, promote tumor progression (37–39). With respect to molecular mechanisms, SFKs have been proved to control cell proliferation through the activation of the Ras/ERK/MAPK pathway and to induce specific gene expression programs by affecting the transcriptional activity of several transcription factors like the TGFbeta effectors, the STAT molecules (40). Cell adhesion and migration are also influenced through direct interactions with key partners such as actins, integrins, or kinases such as FAK (40, 41). Indeed, for several years, a great deal of attention has been paid to the roles of Src in these aspects because of the clinical development of Src inhibitors, such as saracatinib (AZD0530), dasatinib, and bosutinib (42). Preclinical in vitro studies demonstrated that these inhibitors, along with others that have not yet reached clinical development play a significant role in controlling cell proliferation, adhesion, and migration, as well as the ability to form tumoral xenografts in immunodeficient mice (Table 2). A direct role for Src has not really been demonstrated, probably because the experimental overexpression of Src – without activation – is unlikely to be sufficient in promoting cancer progression. Correlations have in fact been suggested between Src phosphorylation-mediated activation and cell migration (9, 36). Nevertheless, the direct inactivation of Src through siRNA experiments has been proved to induce reduced migration and growth in PCa cell lines (43, 44).
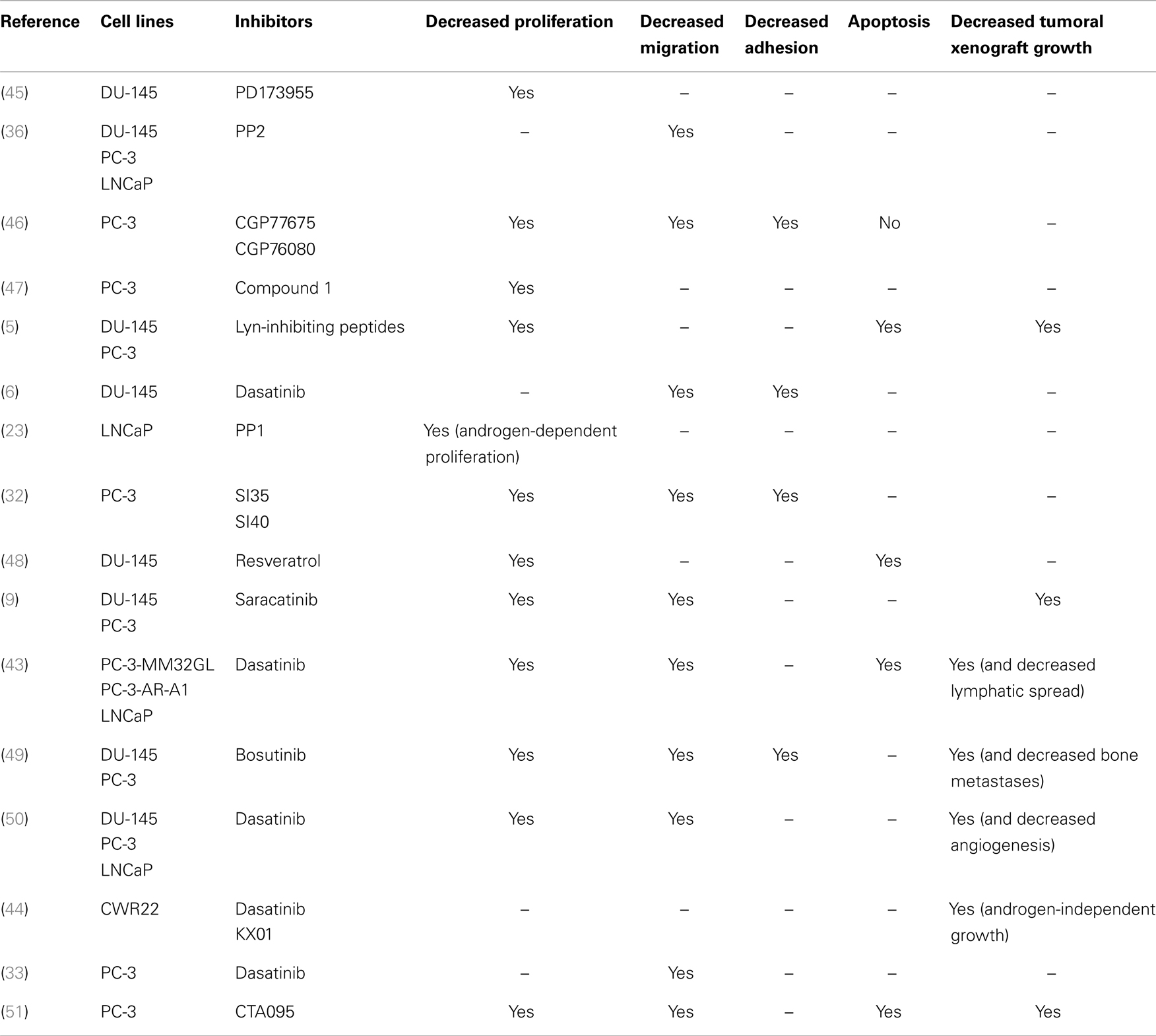
Table 2. Representative studies that evaluated the biological effects of Src inhibitors on basic cellular events associated with prostate cancer progression.
Other studies favor a role of Src in PCa-induced angiogenesis. That Src is a potent actor in angiogenesis has been demonstrated by a substantial amount of evidence. The interplay between Src and VEGF is one of the molecular mechanisms that govern tumor-associated angiogenesis and has also been described with respect to PCa. In prostate cells, hypoxia-induced VEGF expression requires Src activation, which activates Stats3 through phosphorylation and increases HIF1alpha expression (52). Both transcription factors are then able to drive VEGF expression. Il-8 is a known cytokine promoting angiogenesis, and this effect is mediated by Src in prostate cells (31). Src and VEGF cooperation can be targeted by the Src-specific inhibitor dasatinib (50) and several other pharmacological compounds (35, 53–58). Of strong interest for clinical applications, in vivo studies have also demonstrated that Src inhibition is able to provoke decreased tumoral xenograft growth by reducing angiogenesis (50, 56).
Advanced PCa is frequently associated with metastases. The tumor metastatic cascade includes several steps from the detachment of isolated cells with survival capability from the primary tumor, their migration through the vascular endothelium into the blood stream, their extravasation from the blood stream to the receiving tissue, and the local development of the secondary tumor. To migrate through stromal tissue and invade an adjacent blood vessel, the tumor cell has to develop the ability to survive as an isolated cell, devoid of all contact with other cells and the base membrane. This survival depends on the acquisition of resistance to anoikis, i.e., the variety of apoptosis induced by the loss of the close contacts established between an epithelial cell and the base membrane. Mobility, invasion, and resistance to anoikis are an integral part of a general phenomenon known as the epithelium-to-mesenchyme transition (EMT) (59). EMT is a biological process by which a tumor cell loses some of the features of an epithelial cell (e.g., expression of epithelial markers such as E-cadherin) and acquires some of the characteristics of a mesenchymal cell (expression of mesenchymal markers such as N-cadherin). Through interactions with “true” mesenchymal cells, the modified cell migrates more easily through the stroma. EMT is actually a physiological, evolutionary-conserved process that takes place during normal embryonic and fetal development (gastrulation and morphogenesis) and in some adult situations (wound healing and tissue regeneration) (60, 61). EMT is considered to be the first step of metastatic spread (60), and cells engaged in this process are located at the invasion front (62). Within the metastatic site, a mirror process occurs, whereby the secondary tumor develops like the primary one, including with respect to more or less differentiated epithelial cells (mesenchyme-to-epithelium transition) (63). EMT, or at least EMT-like states, has been described in PCa (59).
An archetypal phenomenon associated with EMT, namely, the so-called cadherin switch from the epithelial E-cadherin to the mesenchymal N-cadherin, is of biological importance as the loss of E-cadherin is associated with the loss of cell-cell junctions. E-cadherin expression is therefore tightly regulated and is notably under the control of EMT-specific transcription factors such as Snail, Slug, Twist, and Zeb1/2. Src activation is known as a potent inducer of EMT and has been proved to induce the dissociation of the complex between E-cadherin and beta-catenin through E-cadherin phosphorylation (39, 64). As a first testimony of Src implication in PCa-associated EMT, decreased E-cadherin expression and increased N-cadherin expression have been linked to Src activation in PCa cells (49, 65–67). Although a detailed and direct demonstration of Src as an inducer of EMT in PCa is still lacking, a few studies support the Src implication by showing a correlation between Src activation and the markers of EMT, such as the mesenchymal aspect of epithelial cells (65), the expression of vimentin (67–69), the action of EMT inducers (70), the expression of EMT-specific transcription factors (65) or micro-RNAs (25, 67), or the EMT-associated increase in cell proliferation, invasion, or mobility (65, 69).
Of note is the fact that EMT is one of the aspects of a larger phenomenon called epithelial plasticity (71). Epithelial plasticity refers to the capacity of cells to undergo reversible phenotypic changes during cell invasion or spread. In other words, the epithelial phenotype is partly lost and replaced by another phenotype, for example a mesenchymal one during EMT. The acquisition of stemness characteristics can be regarded as epithelial plasticity. For PCa, other more specific phenotypes are neuroendocrine differentiation (NED) (72, 73) and osteomimicry (74, 75). These various phenotypes represent the Darwinian adaptation of cancer cells to their environment, are usually transient, and are not mutually exclusive. In a recently published model of EMT induction in PC-3 cells by the inactivation of the E-cadherin gene, expression of stemness, NED, and osteomimicry markers was indeed simultaneously observed (69). While NED is usually induced by androgen deprivation (see below), osteomimicry is a means by which metastatic PCa cells adapt to the bone environment (74–77).
Bones are actually the main metastasis site for PCa cells, which have been shown to deregulate the normal remodeling process that assures bone maintenance. They secrete several molecules such as growth factors and cytokines that alter the tight interplay between osteoclasts and osteoblasts. The balance is in favor of osteoblastogenesis, which explains the usual condensing aspect of PCa-derived bone metastases. Src signaling is a key pathway for bone remodeling regulation, and several lines of evidence link it to the bone metastatic process (78). Several cell models of PCa have linked Src activation with the ability to develop distant metastases (43, 49), while various animal studies also demonstrated the efficiency of Src inhibitors when it comes to reducing bone metastases due to PCa (79–82).
Src therefore seems to be implicated in two crucial steps of the PCa metastatic process: EMT and the consequent cell ability to detach from the primary tumor, and local development of the secondary tumor within the metastatic site. It is also worth noting that the initial arrest and adhesion of cancer cells to the vascular endothelium is also an essential step, being intermediary between the two steps referred to, preceding their extravasation from the blood stream. As well as its role in promoting angiogenesis (see above), Src has also been revealed to play a part in the endothelial permeability that permits cancer cell extravasation (83). In PCa cells, the activated transcription factor Stat5 induces decreased E-cadherin expression as well as the increased adhesion of PCa cells to endothelial cells (84). This effect is inhibited by the Src inhibitor PP2, suggesting that Src also mediates endothelial permeability in PCa (84).
The development of the embryonic prostate gland and the maintenance of the adult one are very dependent on androgen stimulation. At least during the initial stages of prostate carcinogenesis, PCa cells are naturally androgen-dependent. Androgen deprivation consequently became the first choice treatment for PCas that could not be physically and locally cured. Resistance to castration is the ability of PCa cells to escape and survive, despite androgen deprivation. As the main cause of PCa-related deaths, resistance to castration has been extensively studied and several mechanisms have been advocated such as AR overexpression, AR activation by non-androgenic ligands (because of specific gene mutations that alter the specificity of the ligand binding), or the activation of AR-bypassing signaling pathways (85). Another significant mechanism is AR activation in the absence of any ligand (85). During this process, the AR is subverted through activation by several soluble factors, essential growth factors such as EGF or IGF-1. These factors act by inducing phosphorylation cascades from their TK membrane receptors. As an integrator of several phosphorylation cascade-inducing extracellular signals, Src has logically been advocated as one of the intermediate effectors that transmit the activation signals from growth factors to the AR pathway.
Indeed, a direct physical interaction has been described between Src and the AR (Figure 2). The AR contains a proline-rich region, which has a clear affinity for the Src SH3 domain (19). This interaction is able to relieve Src folding constraints and therefore induces Src activation. Androgens favor the formation of the AR–Src complex, stimulate the Src/Raf-1/Erk-2 pathway, and consequently lead to the increased proliferation of PCa cells (19, 20). Such an interaction is likely to augment the Src oncogenetic effect in prostate cells (17, 18). It should be noted that AR and Src in fact engage in a ternary complex with the estrogen receptor (ER). Complex formation is therefore favored by either androgens or estrogens and conversely inhibited by antiandrogens or antiestrogens. While the AR interacts with the Src SH3 domain, the ER interacts with the SH2 domain (19). Other partners include TK membrane receptors such as IGF1-R that can be overexpressed and activated by AR-stimulated Src (86).
The interaction between the AR and Src not only favors Src activation but also AR activation (Figure 2). It is well known that AR phosphorylation is implicated in AR translocation from the cytoplasm to the nucleus (8), and its transcriptional activity (8, 87–89). A correlation has been demonstrated between AR expression or activity and Src activation in PCa (8, 21, 90). The tyrosine Y534 in the AR would be the Src target (17, 19, 21, 91) and its phosphorylation would result in ligand-independent AR activation, which is an important mechanism to explain resistance to castration. Activated Src would also inhibit AR interaction with co-repressors, thereby increasing its capacity to be activated without ligand binding (92).
The AR–Src is a target for several signals that thereby influence Src and/or AR activation. The product of the tumor-suppressor gene DOC2/DAB2 is able to repress PCa cell proliferation through direct interaction with Src and the inhibition of the complex formation (23). The EGF/EGFR pathway activates Src in LNCaP cells that are cultured without androgens, probably through the stimulation of the AR–Src complex formation (93, 94). The use of an antiandrogen does indeed block the proliferative effect, underlying the role of the AR as a downstream effector of the EGF/EGFR pathway (93). Similar actions of several other growth factors are likely since many other receptors are expressed in PCa and are able to interact with and activate Src (95). Finally, the AR gene seems to be able to produce a splice variant, called AR8, which lacks the DNA-binding domain, and is instead located at the plasma membrane and favors interactions between the full-length AR, Src, and EGFR (96).
Resistance to hormone castration is often linked to NED, since NED is almost always the result of androgen deprivation (72, 73). NED is characterized by the production of neuropeptides such as chromogranine A, NSE, serotonin, neurophysin, and synaptophysin. All of these products are thought to exert paracrine effects on adjacent (epithelial) cells, keeping them alive despite androgen deprivation (3). Bombesin is another secreted neuropeptide. Bound to its membrane receptor, it activates Src and thus favors AR phosphorylation and its androgen-independent activation (26, 90). The same action is suggested for other neuropeptides such as neurotensin (26, 27), PTHrp (97), and gastrin-releasing peptide (98). It is, however, still unclear whether these neuropeptides directly activate Src or activate it by stimulating the EGF/EGFR pathway and interactions between Src and EGFR (22, 27, 96, 97). Similar complex interactions could also be observed between the neuropeptides, Src, the AR, and the IGF1-R (28). Another molecular mechanism by which bombesin activates the AR has been suggested: the bombesin is able to stimulate, via Src, p300-mediated AR acetylation (99).
As described above, several experimental studies performed in cellulo or in animal models of PCa suggested that molecules able to inhibit Src and/or other members of the SFK could be of clinical interest. Several phase I and phase II clinical trials have therefore been conducted to assess tolerance, side-effects, and optimal dose for three of the several orally bioavailable Src inhibitors: dasatinib (BMSS354825), saracatinib (AZD0530), and KX2-391. The first two act by directly inhibiting the TK activity while the third acts through inhibition of protein–protein interactions. Results were disappointing when saracatinib and KX2-391 were assessed as monotherapy in phase II trials that accrued patients with metastatic CRPC (100, 101). Dasatinib is the most clinically studied SFK inhibitor and has been assessed in several cancers including liquid and solid tumors. It is currently FDA-approved for chronic myelogenous leukemia and, as a second line treatment, for Philadelphia chromosome-positive acute lymphoblastic leukemia. In PCa, dasatinib reached phase II development programs as monotherapy in chemotherapy-naïve men (102, 103), and in men previously treated with one chemotherapy regimen (104), or in association with the cytotoxic docetaxel (105, 106). The relative efficacy of dasatinib (102, 103, 105) and the description of patients with long term survival (104, 106) prompted clinicians to perform phase III trial comparing docetaxel + prednisolone with either dasatinib or placebo (107). Results of this large trial (more than 1500 patients accrued) were disappointing in that the addition of dasatinib did not perform better than docetaxel + prednisolone in terms of overall survival. This failure can be explained by several factors including inadequate study design (108), potential pharmacokinetic interactions between dasatinib and docetaxel (109), a stronger effect on stromal cells than on epithelial cells (despite association with the epithelial-targeted docetaxel), and the too broad specificity of inhibitory effect of dasatinib for numerous receptor and non-receptors TK. Subsequent clinical trials are therefore warranted, which have to be based on an comprehensive knowledge about the optimal timing of a Src inhibitor strategy in PCa progression (before or after chemotherapy? before or after metastatic spreading? …), the intrinsic biology of the patient’s tumor and the value of its association with other new therapeutic agents such as antiangiogenic factors (110).
Overall, Src has been implicated in several steps of prostate carcinogenesis. It is likely that it plays similar, major roles in other cancers. The particularity of the Src oncogenic action in prostate carcinogenesis is its ability to interfere with the androgen pathway. Through both direct and indirect interaction with the AR, Src is able to reinforce the proliferative and antiapoptotic actions of the AR, even in the absence of specific ligands. These molecular mechanisms constitute a solid rationale in favor of the use of Src inhibitors in routinely managing patients with PCa.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Creighton CJ. Multiple oncogenic pathway signatures show coordinate expression patterns in human prostate tumors. PLoS One (2008) 3:e1816. doi: 10.1371/journal.pone.0001816
2. Tatarov O, Mitchell TJ, Seywright M, Leung HY, Brunton VG, Edwards J. SRC family kinase activity is up-regulated in hormone-refractory prostate cancer. Clin Cancer Res (2009) 15:3540–9. doi:10.1158/1078-0432.CCR-08-1857
3. Drake JM, Graham NA, Stoyanova T, Sedghi A, Goldstein AS, Cai H, et al. Oncogene-specific activation of tyrosine kinase networks during prostate cancer progression. Proc Natl Acad Sci U S A (2012) 109:1643–8. doi:10.1073/pnas.1120985109
4. Robinson D, He F, Pretlow T, Kung HJ. A tyrosine kinase profile of prostate carcinoma. Proc Natl Acad Sci U S A (1996) 93:5958–62. doi:10.1073/pnas.93.12.5958
5. Goldenberg-Furmanov M, Stein I, Pikarsky E, Rubin H, Kasem S, Wygoda M, et al. Lyn is a target gene for prostate cancer: sequence-based inhibition induces regression of human tumor xenografts. Cancer Res (2004) 64:1058–66. doi:10.1158/0008-5472.CAN-03-2420
6. Nam S, Kim D, Cheng JQ, Zhang S, Lee JH, Buettner R, et al. Action of the Src family kinase inhibitor, dasatinib (BMS-354825), on human prostate cancer cells. Cancer Res (2005) 65:9185–9. doi:10.1158/0008-5472.CAN-05-1731
7. Chang YM, Kung HJ, Evans CP. Nonreceptor tyrosine kinases in prostate cancer. Neoplasia (2007) 9:90–100. doi:10.1593/neo.06694
8. Asim M, Siddiqui IA, Hafeez BB, Baniahmad A, Mukhtar H. Src kinase potentiates androgen receptor transactivation function and invasion of androgen-independent prostate cancer C4-2 cells. Oncogene (2008) 27:3596–604. doi:10.1038/sj.onc.1211016
9. Chang YM, Bai L, Liu S, Yang JC, Kung HJ, Evans CP. Src family kinase oncogenic potential and pathways in prostate cancer as revealed by AZD0530. Oncogene (2008) 27:6365–75. doi:10.1038/onc.2008.250
10. Derry JJ, Prins GS, Ray V, Tyner AL. Altered localization and activity of the intracellular tyrosine kinase BRK/Sik in prostate tumor cells. Oncogene (2003) 22:4212–20. doi:10.1038/sj.onc.1206465
11. Wang S, Gao J, Lei Q, Rozengurt N, Pritchard C, Jiao J, et al. Prostate-specific deletion of the murine Pten tumor suppressor gene leads to metastatic prostate cancer. Cancer Cell (2003) 4:209–21. doi:10.1016/S1535-6108(03)00215-0
12. Zong Y, Xin L, Goldstein AS, Lawson DA, Teitell MA, Witte ON. ETS family transcription factors collaborate with alternative signaling pathways to induce carcinoma from adult murine prostate cells. Proc Natl Acad Sci U S A (2009) 106:12465–70. doi:10.1073/pnas.0905931106
13. Han G, Buchanan G, Ittmann M, Harris JM, Yu X, Demayo FJ, et al. Mutation of the androgen receptor causes oncogenic transformation of the prostate. Proc Natl Acad Sci U S A (2005) 102:1151–6. doi:10.1073/pnas.0408925102
14. Xin L, Teitell MA, Lawson DA, Kwon A, Mellinghoff IK, Witte ON. Progression of prostate cancer by synergy of AKT with genotropic and nongenotropic actions of the androgen receptor. Proc Natl Acad Sci U S A (2006) 103:7789–94. doi:10.1073/pnas.0602567103
15. Memarzadeh S, Xin L, Mulholland DJ, Mansukhani A, Wu H, Teitell MA, et al. Enhanced paracrine FGF10 expression promotes formation of multifocal prostate adenocarcinoma and an increase in epithelial androgen receptor. Cancer Cell (2007) 12:572–85. doi:10.1016/j.ccr.2007.11.002
16. Memarzadeh S, Cai H, Janzen DM, Xin L, Lukacs R, Riedinger M, et al. Role of autonomous androgen receptor signaling in prostate cancer initiation is dichotomous and depends on the oncogenic signal. Proc Natl Acad Sci U S A (2011) 108:7962–7. doi:10.1073/pnas.1105243108
17. Cai H, Babic I, Wei X, Huang J, Witte ON. Invasive prostate carcinoma driven by c-Src and androgen receptor synergy. Cancer Res (2011) 71:862–72. doi:10.1158/0008-5472.CAN-10-1605
18. Cai H, Smith DA, Memarzadeh S, Lowell CA, Cooper JA, Witte ON. Differential transformation capacity of Src family kinases during the initiation of prostate cancer. Proc Natl Acad Sci U S A (2011) 108:6579–84. doi:10.1073/pnas.1103904108
19. Migliaccio A, Castoria G, Di Domenico M, De Falco A, Bilancio A, Lombardi M, et al. Steroid-induced androgen receptor-oestradiol receptor beta-Src complex triggers prostate cancer cell proliferation. EMBO J (2000) 19:5406–17. doi:10.1093/emboj/19.20.5406
20. Migliaccio A, Castoria G, Di Domenico M, De Falco A, Bilancio A, Lombardi M, et al. Sex steroid hormones act as growth factors. J Steroid Biochem Mol Biol (2002) 83:31–5. doi:10.1016/S0960-0760(02)00264-9
21. Guo Z, Dai B, Jiang T, Xu K, Xie Y, Kim O, et al. Regulation of androgen receptor activity by tyrosine phosphorylation. Cancer Cell (2006) 10:309–19. doi:10.1016/j.ccr.2006.08.021
22. Kraus S, Gioeli D, Vomastek T, Gordon V, Weber MJ. Receptor for activated C kinase 1 (RACK1) and Src regulate the tyrosine phosphorylation and function of the androgen receptor. Cancer Res (2006) 66:11047–54. doi:10.1158/0008-5472.CAN-06-0596
23. Zhoul J, Hernandez G, Tu SW, Huang CL, Tseng CP, Hsieh JT. The role of DOC-2/DAB2 in modulating androgen receptor-mediated cell growth via the nongenomic c-Src-mediated pathway in normal prostatic epithelium and cancer. Cancer Res (2005) 65:9906–13. doi:10.1158/0008-5472.CAN-05-1481
24. Wu K, Liu J, Tseng SF, Gore C, Ning Z, Sharifi N, et al. The role of DAB2IP in androgen receptor activation during prostate cancer progression. Oncogene (2014) 33:1954–63. doi:10.1038/onc.2013.143
25. Kao CJ, Martiniez A, Shi XB, Yang J, Evans CP, Dobi A, et al. miR-30 as a tumor suppressor connects EGF/Src signal to ERG and EMT. Oncogene (2013) 33(19):2495–503. doi:10.1038/onc.2013.200
26. Lee LF, Guan J, Qiu Y, Kung HJ. Neuropeptide-induced androgen independence in prostate cancer cells: roles of nonreceptor tyrosine kinases Etk/Bmx, Src, and focal adhesion kinase. Mol Cell Biol (2001) 21:8385–97. doi:10.1128/MCB.21.24.8385-8397.2001
27. Amorino GP, Deeble PD, Parsons SJ. Neurotensin stimulates mitogenesis of prostate cancer cells through a novel c-Src/Stat5b pathway. Oncogene (2007) 26:745–56. doi:10.1038/sj.onc.1209814
28. DaSilva JO, Amorino GP, Casarez EV, Pemberton B, Parsons SJ. Neuroendocrine-derived peptides promote prostate cancer cell survival through activation of IGF-1R signaling. Prostate (2013) 73:801–12. doi:10.1002/pros.22624
29. Giannoni E, Fiaschi T, Ramponi G, Chiarugi P. Redox regulation of anoikis resistance of metastatic prostate cancer cells: key role for Src and EGFR-mediated pro-survival signals. Oncogene (2009) 28:2074–86. doi:10.1038/onc.2009.77
30. Lee LF, Louie MC, Desai SJ, Yang J, Chen HW, Evans CP, et al. Interleukin-8 confers androgen-independent growth and migration of LNCaP: differential effects of tyrosine kinases Src and FAK. Oncogene (2004) 23:2197–205. doi:10.1038/sj.onc.1207344
31. Araki S, Omori Y, Lyn D, Singh RK, Meinbach DM, Sandman Y, et al. Interleukin-8 is a molecular determinant of androgen independence and progression in prostate cancer. Cancer Res (2007) 67:6854–62. doi:10.1158/0008-5472.CAN-07-1162
32. Angelucci A, Schenone S, Gravina GL, Muzi P, Festuccia C, Vicentini C, et al. Pyrazolo[3,4-d]pyrimidines c-Src inhibitors reduce epidermal growth factor-induced migration in prostate cancer cells. Eur J Cancer (2006) 42:2838–45. doi:10.1016/j.ejca.2006.06.024
33. Varkaris A, Gaur S, Parikh NU, Song JH, Dayyani F, Jin JK, et al. Ligand-independent activation of MET through IGF-1/IGF-1R signaling. Int J Cancer (2013) 133:1536–46. doi:10.1002/ijc.28169
34. Inoue S, Branch CD, Gallick GE, Chada S, Ramesh R. Inhibition of Src kinase activity by Ad-mda7 suppresses vascular endothelial growth factor expression in prostate carcinoma cells. Mol Ther (2005) 12:707–15. doi:10.1016/j.ymthe.2005.05.015
35. Pang X, Zhang L, Lai L, Chen J, Wu Y, Yi Z, et al. 1’-Acetoxychavicol acetate suppresses angiogenesis-mediated human prostate tumor growth by targeting VEGF-mediated Src-FAK-Rho GTPase-signaling pathway. Carcinogenesis (2011) 32:904–12. doi:10.1093/carcin/bgr052
36. Slack JK, Adams RB, Rovin JD, Bissonette EA, Stoker CE, Parsons JT. Alterations in the focal adhesion kinase/Src signal transduction pathway correlate with increased migratory capacity of prostate carcinoma cells. Oncogene (2001) 20:1152–63. doi:10.1038/sj.onc.1204208
37. Summy JM, Gallick GE. Src family kinases in tumor progression and metastasis. Cancer Metastasis Rev (2003) 22:337–58. doi:10.1023/A:1023772912750
38. Kim LC, Song L, Haura EB. Src kinases as therapeutic targets for cancer. Nat Rev Clin Oncol (2009) 6:587–95. doi:10.1038/nrclinonc.2009.129
39. Guarino M. Src signaling in cancer invasion. J Cell Physiol (2010) 223:14–26. doi:10.1002/jcp.22011
40. Ishizawar R, Parsons SJ. c-Src and cooperating partners in human cancer. Cancer Cell (2004) 6:209–14. doi:10.1016/j.ccr.2004.09.001
41. Playford MP, Schaller MD. The interplay between Src and integrins in normal and tumor biology. Oncogene (2004) 23:7928–46. doi:10.1038/sj.onc.1208080
42. Edwards J. Src kinase inhibitors: an emerging therapeutic treatment option for prostate cancer. Expert Opin Investig Drugs (2010) 19:605–14. doi:10.1517/13543781003789388
43. Park SI, Zhang J, Phillips KA, Araujo JC, Najjar AM, Volgin AY, et al. Targeting SRC family kinases inhibits growth and lymph node metastases of prostate cancer in an orthotopic nude mouse model. Cancer Res (2008) 68:3323–33. doi:10.1158/0008-5472.CAN-07-2997
44. Su B, Gillard B, Gao L, Eng KH, Gelman IH. Src controls castration recurrence of CWR22 prostate cancer xenografts. Cancer Med (2013) 2:784–92. doi:10.1002/cam4.144
45. Moasser MM, Srethapakdi M, Sachar KS, Kraker AJ, Rosen N. Inhibition of Src kinases by a selective tyrosine kinase inhibitor causes mitotic arrest. Cancer Res (1999) 59:6145–52.
46. Recchia I, Rucci N, Festuccia C, Bologna M, Mackay AR, Migliaccio S, et al. Pyrrolopyrimidine c-Src inhibitors reduce growth, adhesion, motility and invasion of prostate cancer cells in vitro. Eur J Cancer (2003) 39:1927–35. doi:10.1016/S0959-8049(03)00394-0
47. Lombardo LJ, Lee FY, Chen P, Norris D, Barrish JC, Behnia K, et al. Discovery of N-(2-chloro-6-methyl- phenyl)-2-(6-(4-(2-hydroxyethyl)- piperazin-1-yl)-2-methylpyrimidin-4- ylamino)thiazole-5-carboxamide (BMS-354825), a dual Src/Abl kinase inhibitor with potent antitumor activity in preclinical assays. J Med Chem (2004) 47:6658–61. doi:10.1021/jm049486a
48. Kotha A, Sekharam M, Cilenti L, Siddiquee K, Khaled A, Zervos AS, et al. Resveratrol inhibits Src and Stat3 signaling and induces the apoptosis of malignant cells containing activated Stat3 protein. Mol Cancer Ther (2006) 5:621–9. doi:10.1158/1535-7163.MCT-05-0268
49. Rabbani SA, Valentino ML, Arakelian A, Ali S, Boschelli F. SKI-606 (bosutinib) blocks prostate cancer invasion, growth, and metastasis in vitro and in vivo through regulation of genes involved in cancer growth and skeletal metastasis. Mol Cancer Ther (2010) 9:1147–57. doi:10.1158/1535-7163.MCT-09-0962
50. Rice L, Lepler S, Pampo C, Siemann DW. Impact of the SRC inhibitor dasatinib on the metastatic phenotype of human prostate cancer cells. Clin Exp Metastasis (2012) 29:133–42. doi:10.1007/s10585-011-9436-2
51. Guo W, Liu R, Bhardwaj G, Ma AH, Changou C, Yang JC, et al. CTA095, a novel Etk and Src dual inhibitor, induces apoptosis in prostate cancer cells and overcomes resistance to Src inhibitors. PLoS One (2013) 8:e70910. doi:10.1371/journal.pone.0070910
52. Gray MJ, Zhang J, Ellis LM, Semenza GL, Evans DB, Watowich SS, et al. HIF-1alpha, STAT3, CBP/p300 and Ref-1/APE are components of a transcriptional complex that regulates Src-dependent hypoxia-induced expression of VEGF in pancreatic and prostate carcinomas. Oncogene (2005) 24:3110–20. doi:10.1038/sj.onc.1208513
53. Yi T, Yi Z, Cho SG, Luo J, Pandey MK, Aggarwal BB, et al. Gambogic acid inhibits angiogenesis and prostate tumor growth by suppressing vascular endothelial growth factor receptor 2 signaling. Cancer Res (2008) 68:1843–50. doi:10.1158/0008-5472.CAN-07-5944
54. Pang X, Yi Z, Zhang X, Sung B, Qu W, Lian X, et al. Acetyl-11-keto-beta-boswellic acid inhibits prostate tumor growth by suppressing vascular endothelial growth factor receptor 2-mediated angiogenesis. Cancer Res (2009) 69:5893–900. doi:10.1158/0008-5472.CAN-09-0755
55. Pang X, Wu Y, Wu Y, Lu B, Chen J, Wang J, et al. (-)-Gossypol suppresses the growth of human prostate cancer xenografts via modulating VEGF signaling-mediated angiogenesis. Mol Cancer Ther (2011) 10:795–805. doi:10.1158/1535-7163.MCT-10-0936
56. Wu Y, He L, Zhang L, Chen J, Yi Z, Zhang J, et al. Anacardic acid (6-pentadecylsalicylic acid) inhibits tumor angiogenesis by targeting Src/FAK/Rho GTPases signaling pathway. J Pharmacol Exp Ther (2011) 339:403–11. doi:10.1124/jpet.111.181891
57. Mirzoeva S, Franzen CA, Pelling JC. Apigenin inhibits TGF-beta-induced VEGF expression in human prostate carcinoma cells via a Smad2/3- and Src-dependent mechanism. Mol Carcinog (2013) 53(8):598–609. doi:10.1002/mc.22005
58. Saraswati S, Kumar S, Alhaider AA. Alpha-santalol inhibits the angiogenesis and growth of human prostate tumor growth by targeting vascular endothelial growth factor receptor 2-mediated AKT/mTOR/P70S6K signaling pathway. Mol Cancer (2013) 12:147. doi:10.1186/1476-4598-12-147
59. Nauseef JT, Henry MD. Epithelial-to-mesenchymal transition in prostate cancer: paradigm or puzzle? Nat Rev Urol (2011) 8:428–39. doi:10.1038/nrurol.2011.85
60. Yang J, Weinberg RA. Epithelial-mesenchymal transition: at the crossroads of development and tumor metastasis. Dev Cell (2008) 14:818–29. doi:10.1016/j.devcel.2008.05.009
61. Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell (2009) 139:871–90. doi:10.1016/j.cell.2009.11.007
62. Brabletz T, Jung A, Reu S, Porzner M, Hlubek F, Kunz-Schughart LA, et al. Variable beta-catenin expression in colorectal cancers indicates tumor progression driven by the tumor environment. Proc Natl Acad Sci U S A (2001) 98:10356–61. doi:10.1073/pnas.171610498
63. Fodde R, Brabletz T. Wnt/beta-catenin signaling in cancer stemness and malignant behavior. Curr Opin Cell Biol (2007) 19:150–8. doi:10.1016/j.ceb.2007.02.007
64. Fujita Y, Krause G, Scheffner M, Zechner D, Leddy HE, Behrens J, et al. Hakai, a c-Cbl-like protein, ubiquitinates and induces endocytosis of the E-cadherin complex. Nat Cell Biol (2002) 4:222–31. doi:10.1038/ncb758
65. Deep G, Gangar SC, Agarwal C, Agarwal R. Role of E-cadherin in antimigratory and antiinvasive efficacy of silibinin in prostate cancer cells. Cancer Prev Res (Phila) (2011) 4:1222–32. doi:10.1158/1940-6207.CAPR-10-0370
66. Putzke AP, Ventura AP, Bailey AM, Akture C, Opoku-Ansah J, Celiktas M, et al. Metastatic progression of prostate cancer and e-cadherin regulation by zeb1 and SRC family kinases. Am J Pathol (2011) 179:400–10. doi:10.1016/j.ajpath.2011.03.028
67. Majid S, Dar AA, Saini S, Arora S, Shahryari V, Zaman MS, et al. miR-23b represses proto-oncogene Src kinase and functions as methylation-silenced tumor suppressor with diagnostic and prognostic significance in prostate cancer. Cancer Res (2012) 72:6435–46. doi:10.1158/0008-5472.CAN-12-2181
68. Wei J, Xu G, Wu M, Zhang Y, Li Q, Liu P, et al. Overexpression of vimentin contributes to prostate cancer invasion and metastasis via src regulation. Anticancer Res (2008) 28:327–34.
69. Deep G, Jain AK, Ramteke A, Ting H, Vijendra KC, Gangar SC, et al. SNAI1 is critical for the aggressiveness of prostate cancer cells with low E-cadherin. Mol Cancer (2014) 13:37. doi:10.1186/1476-4598-13-37
70. Diamantopoulou Z, Kitsou P, Menashi S, Courty J, Katsoris P. Loss of receptor protein tyrosine phosphatase beta/zeta (RPTPbeta/zeta) promotes prostate cancer metastasis. J Biol Chem (2012) 287:40339–49. doi:10.1074/jbc.M112.405852
71. Bitting RL, Schaeffer D, Somarelli JA, Garcia-Blanco MA, Armstrong AJ. The role of epithelial plasticity in prostate cancer dissemination and treatment resistance. Cancer Metastasis Rev (2014) 33(2–3):441–68. doi:10.1007/s10555-013-9483-z
72. Yuan TC, Veeramani S, Lin MF. Neuroendocrine-like prostate cancer cells: neuroendocrine transdifferentiation of prostate adenocarcinoma cells. Endocr Relat Cancer (2007) 14:531–47. doi:10.1677/ERC-07-0061
73. Sun Y, Niu J, Huang J. Neuroendocrine differentiation in prostate cancer. Am J Transl Res (2009) 1:148–62.
74. Koeneman KS, Yeung F, Chung LW. Osteomimetic properties of prostate cancer cells: a hypothesis supporting the predilection of prostate cancer metastasis and growth in the bone environment. Prostate (1999) 39:246–61. doi:10.1002/(SICI)1097-0045(19990601)39:4<246::AID-PROS5>3.0.CO;2-U
75. Hagberg Thulin M, Jennbacken K, Damber JE, Welen K. Osteoblasts stimulate the osteogenic and metastatic progression of castration-resistant prostate cancer in a novel model for in vitro and in vivo studies. Clin Exp Metastasis (2014) 31:269–83. doi:10.1007/s10585-013-9626-1
76. Odero-Marah VA, Wang R, Chu G, Zayzafoon M, Xu J, Shi C, et al. Receptor activator of NF-kappaB Ligand (RANKL) expression is associated with epithelial to mesenchymal transition in human prostate cancer cells. Cell Res (2008) 18:858–70. doi:10.1038/cr.2008.84
77. Zhau HE, Odero-Marah V, Lue HW, Nomura T, Wang R, Chu G, et al. Epithelial to mesenchymal transition (EMT) in human prostate cancer: lessons learned from ARCaP model. Clin Exp Metastasis (2008) 25:601–10. doi:10.1007/s10585-008-9183-1
78. Saad F, Lipton A. SRC kinase inhibition: targeting bone metastases and tumor growth in prostate and breast cancer. Cancer Treat Rev (2010) 36:177–84. doi:10.1016/j.ctrv.2009.11.005
79. Araujo JC, Poblenz A, Corn P, Parikh NU, Starbuck MW, Thompson JT, et al. Dasatinib inhibits both osteoclast activation and prostate cancer PC-3-cell-induced osteoclast formation. Cancer Biol Ther (2009) 8:2153–9. doi:10.4161/cbt.8.22.9770
80. Koreckij T, Nguyen H, Brown LG, Yu EY, Vessella RL, Corey E. Dasatinib inhibits the growth of prostate cancer in bone and provides additional protection from osteolysis. Br J Cancer (2009) 101:263–8. doi:10.1038/sj.bjc.6605178
81. Lee YC, Huang CF, Murshed M, Chu K, Araujo JC, Ye X, et al. Src family kinase/abl inhibitor dasatinib suppresses proliferation and enhances differentiation of osteoblasts. Oncogene (2010) 29:3196–207. doi:10.1038/onc.2010.73
82. Yang JC, Bai L, Yap S, Gao AC, Kung HJ, Evans CP. Effect of the specific Src family kinase inhibitor saracatinib on osteolytic lesions using the PC-3 bone model. Mol Cancer Ther (2010) 9:1629–37. doi:10.1158/1535-7163.MCT-09-1058
83. Kim MP, Park SI, Kopetz S, Gallick GE. Src family kinases as mediators of endothelial permeability: effects on inflammation and metastasis. Cell Tissue Res (2009) 335:249–59. doi:10.1007/s00441-008-0682-9
84. Gu L, Vogiatzi P, Puhr M, Dagvadorj A, Lutz J, Ryder A, et al. Stat5 promotes metastatic behavior of human prostate cancer cells in vitro and in vivo. Endocr Relat Cancer (2010) 17:481–93. doi:10.1677/ERC-09-0328
85. Feldman BJ, Feldman D. The development of androgen-independent prostate cancer. Nat Rev Cancer (2001) 1:34–45. doi:10.1038/35094009
86. Pandini G, Mineo R, Frasca F, Roberts CT Jr, Marcelli M, Vigneri R, et al. Androgens up-regulate the insulin-like growth factor-I receptor in prostate cancer cells. Cancer Res (2005) 65:1849–57. doi:10.1158/0008-5472.CAN-04-1837
87. Culig Z, Hobisch A, Cronauer MV, Radmayr C, Trapman J, Hittmair A, et al. Androgen receptor activation in prostatic tumor cell lines by insulin-like growth factor-I, keratinocyte growth factor, and epidermal growth factor. Cancer Res (1994) 54:5474–8.
88. Gioeli D, Ficarro SB, Kwiek JJ, Aaronson D, Hancock M, Catling AD, et al. Androgen receptor phosphorylation. Regulation and identification of the phosphorylation sites. J Biol Chem (2002) 277:29304–14. doi:10.1074/jbc.M204131200
89. Heinlein CA, Chang C. Androgen receptor in prostate cancer. Endocr Rev (2004) 25:276–308. doi:10.1210/er.2002-0032
90. Desai SJ, Ma AH, Tepper CG, Chen HW, Kung HJ. Inappropriate activation of the androgen receptor by nonsteroids: involvement of the Src kinase pathway and its therapeutic implications. Cancer Res (2006) 66:10449–59. doi:10.1158/0008-5472.CAN-06-2582
91. Liu Y, Karaca M, Zhang Z, Gioeli D, Earp HS, Whang YE. Dasatinib inhibits site-specific tyrosine phosphorylation of androgen receptor by Ack1 and Src kinases. Oncogene (2010) 29:3208–16. doi:10.1038/onc.2010.103
92. Asim M, Hafeez BB, Siddiqui IA, Gerlach C, Patz M, Mukhtar H, et al. Ligand-dependent corepressor acts as a novel androgen receptor corepressor, inhibits prostate cancer growth, and is functionally inactivated by the Src protein kinase. J Biol Chem (2011) 286:37108–17. doi:10.1074/jbc.M111.292771
93. Migliaccio A, Di Domenico M, Castoria G, Nanayakkara M, Lombardi M, De Falco A, et al. Steroid receptor regulation of epidermal growth factor signaling through Src in breast and prostate cancer cells: steroid antagonist action. Cancer Res (2005) 65:10585–93. doi:10.1158/0008-5472.CAN-05-0912
94. Hitosugi T, Sasaki K, Sato M, Suzuki Y, Umezawa Y. Epidermal growth factor directs sex-specific steroid signaling through Src activation. J Biol Chem (2007) 282:10697–706. doi:10.1074/jbc.M610444200
95. Tatarov O, Edwards J. The role of SRC family kinases in prostate cancer. Transl Oncogenomics (2007) 2:67–77.
96. Yang X, Guo Z, Sun F, Li W, Alfano A, Shimelis H, et al. Novel membrane-associated androgen receptor splice variant potentiates proliferative and survival responses in prostate cancer cells. J Biol Chem (2011) 286:36152–60. doi:10.1074/jbc.M111.265124
97. DaSilva J, Gioeli D, Weber MJ, Parsons SJ. The neuroendocrine-derived peptide parathyroid hormone-related protein promotes prostate cancer cell growth by stabilizing the androgen receptor. Cancer Res (2009) 69:7402–11. doi:10.1158/0008-5472.CAN-08-4687
98. Yang JC, Ok JH, Busby JE, Borowsky AD, Kung HJ, Evans CP. Aberrant activation of androgen receptor in a new neuropeptide-autocrine model of androgen-insensitive prostate cancer. Cancer Res (2009) 69:151–60. doi:10.1158/0008-5472.CAN-08-0442
99. Gong J, Zhu J, Goodman OB Jr, Pestell RG, Schlegel PN, Nanus DM, et al. Activation of p300 histone acetyltransferase activity and acetylation of the androgen receptor by bombesin in prostate cancer cells. Oncogene (2006) 25:2011–21. doi:10.1038/sj.onc.1209231
100. Lara PN Jr, Longmate J, Evans CP, Quinn DI, Twardowski P, Chatta G, et al. A phase II trial of the Src-kinase inhibitor AZD0530 in patients with advanced castration-resistant prostate cancer: a California Cancer Consortium Study. Anticancer Drugs (2009) 20:179–84. doi:10.1097/CAD.0b013e328325a867
101. Antonarakis ES, Heath EI, Posadas EM, Yu EY, Harrison MR, Bruce JY, et al. A phase 2 study of KX2-391, an oral inhibitor of Src kinase and tubulin polymerization, in men with bone-metastatic castration-resistant prostate cancer. Cancer Chemother Pharmacol (2013) 71:883–92. doi:10.1007/s00280-013-2079-z
102. Yu EY, Wilding G, Posadas E, Gross M, Culine S, Massard C, et al. Phase II study of dasatinib in patients with metastatic castration-resistant prostate cancer. Clin Cancer Res (2009) 15:7421–8. doi:10.1158/1078-0432.CCR-09-1691
103. Yu EY, Massard C, Gross ME, Carducci MA, Culine S, Hudes G, et al. Once-daily dasatinib: expansion of phase II study evaluating safety and efficacy of dasatinib in patients with metastatic castration-resistant prostate cancer. Urology (2011) 77:1166–71. doi:10.1016/j.urology.2011.01.006
104. Twardowski PW, Beumer JH, Chen CS, Kraft AS, Chatta GS, Mitsuhashi M, et al. A phase II trial of dasatinib in patients with metastatic castration-resistant prostate cancer treated previously with chemotherapy. Anticancer Drugs (2013) 24:743–53. doi:10.1097/CAD.0b013e328361feb0
105. Araujo JC, Mathew P, Armstrong AJ, Braud EL, Posadas E, Lonberg M, et al. Dasatinib combined with docetaxel for castration-resistant prostate cancer: results from a phase 1-2 study. Cancer (2012) 118:63–71. doi:10.1002/cncr.26204
106. Araujo JC, Trudel GC, Paliwal P. Long-term use of dasatinib in patients with metastatic castration-resistant prostate cancer after receiving the combination of dasatinib and docetaxel. Cancer Manag Res (2013) 6:25–30. doi:10.2147/CMAR.S41667
107. Araujo JC, Trudel GC, Saad F, Armstrong AJ, Yu EY, Bellmunt J, et al. Docetaxel and dasatinib or placebo in men with metastatic castration-resistant prostate cancer (READY): a randomised, double-blind phase 3 trial. Lancet Oncol (2013) 14:1307–16. doi:10.1016/S1470-2045(13)70479-0
108. Lara PN Jr, Evans CP. Dasatinib and docetaxel in advanced prostate cancer. Lancet Oncol (2013) 14:1248–9. doi:10.1016/S1470-2045(13)70500-X
109. Leveque D. Pharmacokinetic interactions of dasatinib and docetaxel. Lancet Oncol (2014) 15:e51. doi:10.1016/S1470-2045(14)70004-X
Keywords: prostate cancer, c-Src, SFK family, tyrosine-kinase, aggressiveness, prognosis, epithelial-to-mesenchymal transition, neuroendocrine differentiation
Citation: Vlaeminck-Guillem V, Gillet G and Rimokh R (2014) Src: marker or actor in prostate cancer aggressiveness. Front. Oncol. 4:222. doi: 10.3389/fonc.2014.00222
Received: 16 June 2014; Paper pending published: 15 July 2014;
Accepted: 02 August 2014; Published online: 18 August 2014.
Edited by:
Fabio Grizzi, Humanitas Clinical and Research Center, ItalyReviewed by:
Scott T. Tagawa, Weill Cornell Medical College, USACopyright: © 2014 Vlaeminck-Guillem, Gillet and Rimokh. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Virginie Vlaeminck-Guillem, U1052 INSERM, UMR CNRS 5286, Lyon 1 University, Cancer Research Centre of Lyon, Centre Léon Bérard, 28 rue Laennec, 69008 Lyon, France e-mail:dmlyZ2luaWUudmxhZW1pbmNrLWd1aWxsZW1AdW5pdi1seW9uMS5mcg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.