
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 20 May 2014
Sec. Women's Cancer
Volume 4 - 2014 | https://doi.org/10.3389/fonc.2014.00111
This article is part of the Research Topic Epigenetic and Transcriptional Mechanisms in Gynecologic Cancers Development, Progression, and Resistance to Treatment View all 14 articles
 Dineo Khabele1,2*
Dineo Khabele1,2*Epithelial ovarian cancer remains the deadliest gynecologic malignancy. Despite advances in treatment, new approaches are needed. Histone deacetylases (HDACs) are a family of enzymes that regulate gene expression by removing acetyl groups from lysine residues on histones and non-histone proteins. Inhibition of HDACs with small molecules has led to the development of histone deacetylase inhibitors (HDACi) that are in clinical use, primarily for hematologic malignancies. Although clinical trials with HDACi as single agents in solid tumors have been disappointing, data from independent labs and recent work by our group show that class I selective HDACi have potent anti-tumor effects in pre-clinical models of ovarian cancer. This review summarizes the role of HDACs in ovarian cancer and the potential niche for selective class I HDACi, particularly HDAC3 in ovarian cancer therapy.
Ovarian cancer is the deadliest gynecological malignancy in the United States, with 21,980 new cases and 14,270 deaths estimated for 2014 (1). Epithelial ovarian cancer is classified into two broad subtypes based on biological, histological, and molecular features (2–4). Type I low-grade serous, low-grade endometrioid, clear cell, and mucinous tumors are typically indolent and relatively chemotherapy resistant. Somatic mutations in KRAS, BRAF, PIK3CA, PTEN, CTNNB1, and ARID1A genes are common in Type I tumors. In contrast, Type II high-grade serous, high-grade endometrioid, carcinosarcoma, and undifferentiated tumors are aggressive, highly proliferative tumors that are relatively chemotherapy sensitive. Type II tumors are genomically unstable with a high degree of copy number alterations, mutations in TP53, and alterations in the homologous recombination (HR) DNA damage repair pathway (2–4). HR deficiency confers relative sensitivity to DNA damaging agents such as cisplatin, carboplatin, and more recently PARP inhibitors (2, 5–7). Approximately 70% of epithelial ovarian cancers are Type II high-grade serous tumors (2).
Current treatment for epithelial ovarian cancer entails a combination of cytoreductive surgery and platinum-based chemotherapy (8–12). Platinum-based chemotherapy has extended survival significantly in patients who undergo optimal tumor debulking surgery (13, 14). Despite optimal initial therapy, however, most tumors recur and options for recurrent disease are restricted by few effective drugs and frequent dose-limiting toxicities of traditional cytotoxic drugs (8, 9). Extending the disease-free interval (initial response to platinum therapy) and re-sensitizing tumors to platinum-based drugs (overcoming platinum resistance), while minimizing toxic side effects is an ongoing and urgent clinical dilemma, and new treatment approaches are urgently needed. This review summarizes the role of histone deacetylase inhibitors (HDACi) as epigenetic anti-cancer therapy and evidence that class I selective HDACi, particularly those biased to HDAC3 may be a promising therapeutic strategy for ovarian cancer.
Histone deacetylases are a large family of enzymes that deacetylate lysine residues on histones and non-histone proteins (15, 16). Deacetylation of lysine residues of histone tails allows tighter binding of the nucleosome to negatively charged DNA, which results in chromatin compaction. Chromatin compaction is associated with silencing of gene transcription and other functions of genome maintenance such as DNA replication and DNA damage response and repair (16–20). Deacetylation of histones represses the transcription of tumor suppressor genes such as the cyclin-dependent kinase inhibitor, p21 p21(WAF1/CIP1), and the DNA damage repair gene BRCA1, and directly or indirectly promotes the expression, activity, or downstream effects of known oncogenes such as c-MYC (21), RAS (22, 23), and AKT (24). Direct deacetylation of non-histone proteins p53, STAT3, c-MYC, α-tubulin, and Hsp90 is implicated in tumorigenesis (15, 25–27).
The first mammalian HDAC was discovered by using the small chemical molecule trapoxin as a probe. Trapoxin is a microbially derived cyclotetrapeptide that inhibits histone deacetylation in vivo and causes cell cycle arrest in mammalian cells (28). The protein HD1 (HDAC1), similar to the yeast transcriptional regulator Rpd3p/Hda1, was subsequently isolated and cloned. Since then, 18 mammalian HDACs have been identified and are grouped into four classes based on homology to yeast deacetylases. The family of Rpd3/Hda1 are class I (HDAC1, 2, 3, and 8); class IIa (HDAC4, 5, 7, 9); class IIb (HDAC6 and 10); and class IV: HDAC11 (15, 17, 29) (Figure 1). Class I HDACs are ubiquitously expressed, whereas class II and IV HDACs have tissue specificity for smooth muscle, heart, brain, liver, and colon (29). Class III HDACs, related to yeast sirtuins are nicotinamide-dependent enzymes and will not be discussed in this review.
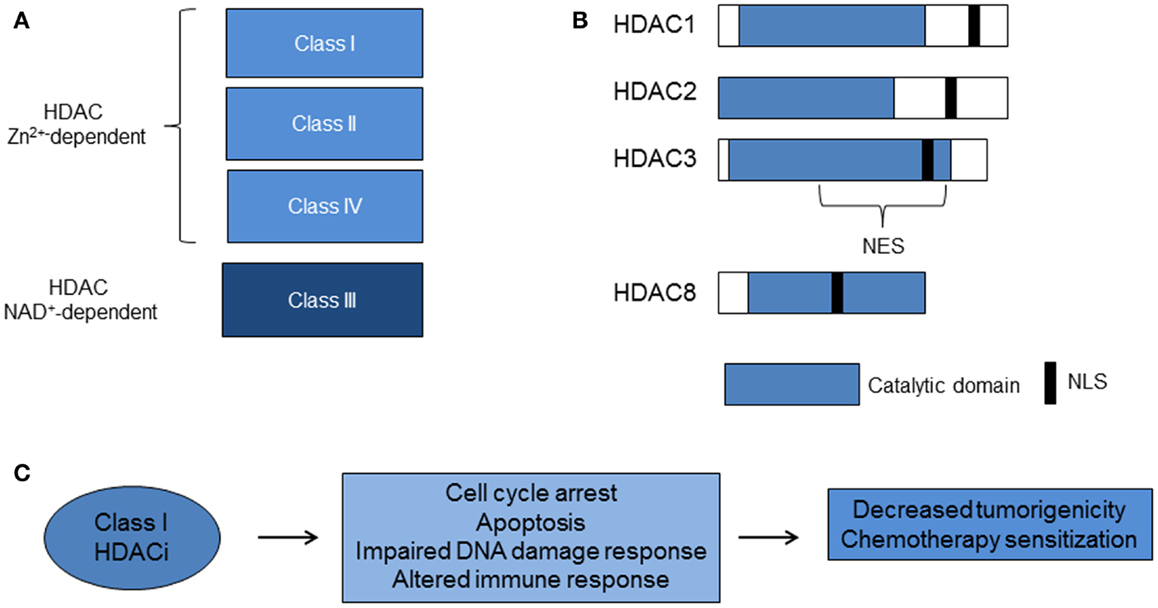
Figure 1. Histone deacetylases and class I HDACi. (A) Zinc (Zn2+)-dependent classes of HDACs. The Class III HDACs are nicotinamide adenine dinucleotide (NAD+)-dependent. (B) Class I HDACs share more than 50% homology, particularly in the catalytic domain. (C) Class I HDACi alter biological pathways that lead to decreased tumorigenicity and chemotherapy sensitization (HDAC, histone deacetylase; HDACi, histone deacetylase inhibitors; NES, nuclear export signal; NLS, nuclear localization signal).
Class I HDACs 1–3 share more than 50% homology, but have distinct structures and cellular functions (15) (Figure 1). HDAC3 lacks the N terminus regions of the other class I HDACs, exists in a distinct multi-protein complex from HDACs 1 and 2, and is associated with N-COR/SMRT co-repressors (15, 16, 30–32). Furthermore, the C terminus of HDAC3 has a unique nuclear export sequence and both nuclear and cytoplasmic localization, which suggests differential function from the other class I HDACs that are confined to the nucleus (33). Knockout mouse models of HDAC1 and HDAC3 enzymes are embryonic lethal and knockout of HDAC2 leads to perinatal death (17). Studies of in vitro silencing of HDACs show HDAC1 and 3 siRNA inhibit cell growth and HDAC3 siRNA causes histone hyperacetylation and apoptosis (34–36). These studies point to a critical role for class I HDACs 1–3 in cell growth. HDAC8 does not have known co-repressors and its function remains under investigation.
Aberrant expression of HDACs is implicated in the pathogenesis of malignancies, including solid tumors such as ovarian cancer (35–38). Our group has published that class I HDACs are highly expressed in ovarian cancers (36), and recent work shows elevated class I HDAC expression is associated with poorer survival in certain subtypes of ovarian cancer (37). Because of the pleiotropic pro-tumorigenic effects on cellular proliferation, apoptosis, and DNA damage and aberrant expression of class I HDACs in ovarian cancer, class I HDACi are potentially effective agents for the treatment of ovarian cancer.
Histone deacetylase inhibitors are a structurally diverse set of chemical compounds traditionally classified into four major categories: hydroxamic acids (e.g., vorinostat formerly SAHA); benzamides (e.g., MS-275); short aliphatic acids (e.g., valproic acid, VPA); and cyclic peptides (e.g., romidepsin or depsipeptide (FK228). Approximately 11 HDACi, including SAHA, MS-275, VPA, and FK228, are in use clinically or are in clinical trials (15, 16, 29, 39, 40). Vorinostat and romidepsin are the only FDA-approved HDACi and are indicated for the treatment of cutaneous T-cell lymphoma (41–44). However, ongoing clinical trials of HDACi in solid tumors, including ovarian cancer (Table 1) (45–48) are underway.
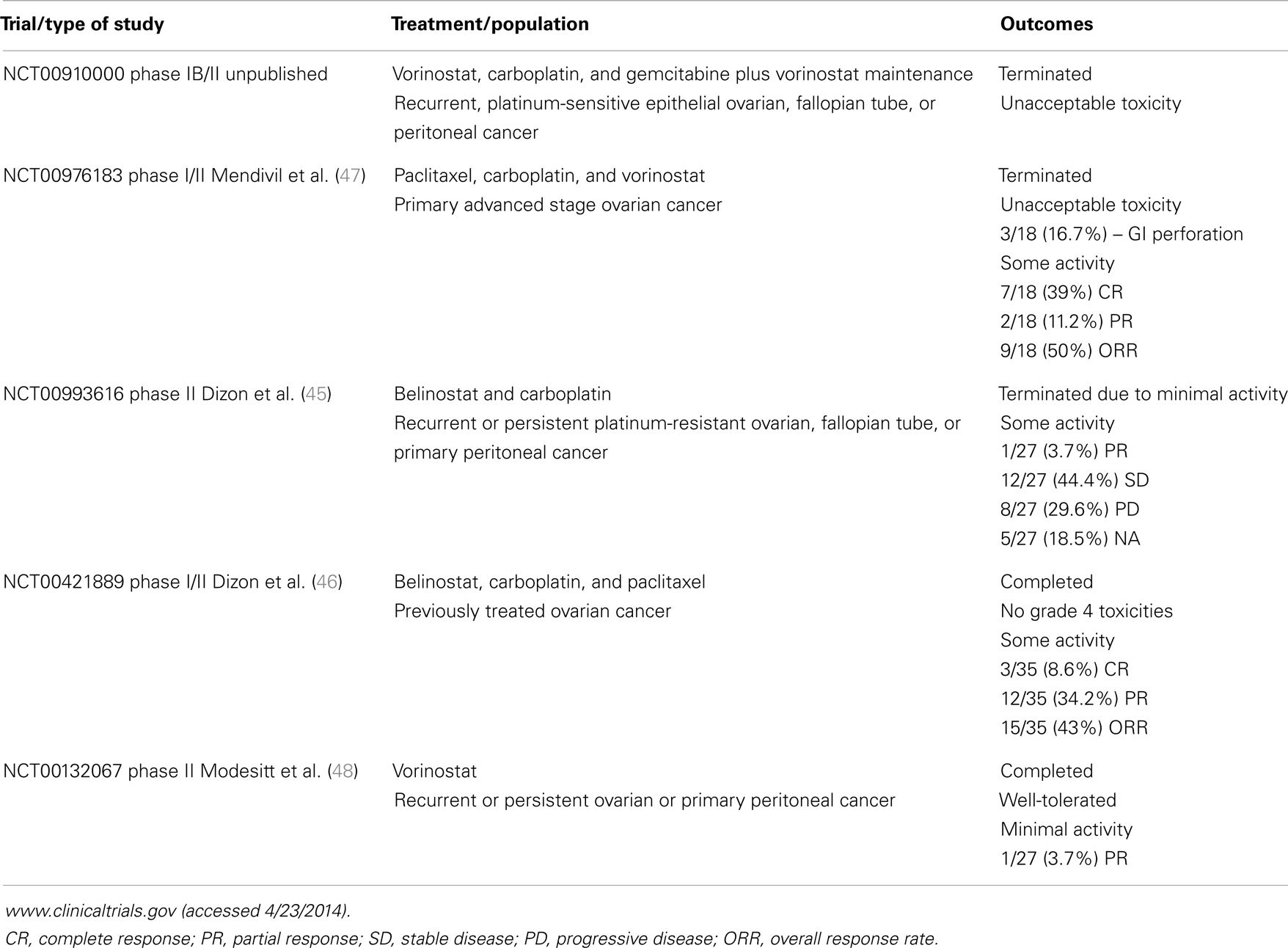
Table 1. Clinical trials of histone deacetylase inhibitors for the treatment of ovarian cancer in the United States.
Although some HDACi are thought to be non-selective inhibitors, many including FK228 have selective bias toward class I HDACs (49). Our group performed a high-throughput study of a diverse group of HDACi in a panel of ovarian cancer cell lines represented in the NCI 60 panel. We demonstrated that the FK228 is the most potent in reducing cell growth (50). FK228 induced cytotoxic effects, measured by induction of the DNA damage response mark [phosphorylation of histone H2AX (pHAX)], inhibition of cell proliferation and increased cell death. FK228 was isolated from Chromobacterium violaceum no. 968, a rare Gram negative bacterium, and approved for the treatment of cutaneous and peripheral T-cell lymphomas (43, 44). The primary mechanism of action of FK228 requires reduction of a characteristic disulfide bond that creates a “warhead” thiol group. The thiol binds to zinc in the catalytic center of both class I and class II HDACs and inhibits HDAC enzymatic activity (51). Based on in vitro binding assays, FK228 preferentially inhibits class I HDACs over class II HDACs, with potent biochemical activity against HDAC3 (51). We have gone on to show that class I biased HDACi with similar bicyclic depsipeptide structures, thailandepsin A (TDP-A) and thailandepsin B (TDP-B) discovered from Burkholderia thailandensis (52), are as potent as FK228 in ovarian cancer cells (53).
Our group has shown HDACi have potent anti-tumor effects in other ovarian cancer cells with relative resistance to cisplatin (36). These findings suggest a role for HDACi in the treatment of platinum-resistant ovarian tumors. For example, NCI/ADR-Res, an ovarian cancer cell line that is resistant to common cytotoxic agents including cisplatin, is the most sensitive to SAHA in the entire set of ovarian cancer cells represented in the NCI 60 panel (50). HDAC proteins play an important role in DNA damage response and repair, and HDACi are known to reduce the expression of HR associated genes such as BRCA1 and RAD51 (54–57). We have recently shown that SAHA inhibits both BRCA1 and RAD51 in response to DNA damage in ovarian cancer cells (58). This implies a role for HDACi altering HR efficiency as a mechanism for sensitizing ovarian cancer cells to DNA damaging drugs. Whether targeting selective class I HDACs indirectly increases DNA damage, impairs DNA repair, or both is an area of active investigation and has potential therapeutic implications for Type II high-grade serous ovarian cancers.
Despite being highly effective in vitro and generally well-tolerated in vivo, clinical responses to HDACi in solid tumors, including ovarian cancers have been disappointing compared to hematologic malignancies (16, 48). Furthermore, evidence from clinical trials and in vitro studies suggest that HDACi are more effective when combined with other anti-tumor agents (16). Table 1 summarizes completed HDACi clinical trials specifically for ovarian cancer in the United States (45–48). In ovarian cancer, single agent trials with HDACi have been disappointing. A Phase II study of single agent vorinostat in platinum refractory recurrent or persistent ovarian cancer showed minimal responses, although it was well-tolerated (48). A Phase II trial of belinostat in women with ovarian cancer, including platinum-resistant disease, showed moderate responses with >50% of the patients with stable disease (59). Interestingly, the best responses were seen in patients with platinum-resistant Type II ovarian cancers in that study. This trend toward improved response in platinum-resistant disease was also observed in a study of belinostat combined with carboplatin and paclitaxel in recurrent ovarian cancer (46). However, in a study specifically for recurrent or persistent ovarian cancer, the combination of belinostat and carboplatin was terminated due to minimal activity (45). The diversity of responses to HDACi in different cell types is not fully understood, but supports observations from our group and others that certain types of cells (e.g., rapidly proliferating cells) are more sensitive than others (e.g., “normal” epithelial cells) to these agents (18, 60). Challenges remain in defining the most appropriate HDACi to combine with other anti-tumor agents.
Histone deacetylase inhibitors have been shown be synergistic with DNA damaging radiation (18, 61–65), suggesting a role for HDACi with DNA damaging chemotherapeutic agents. Combining HDACi with chemotherapeutic drugs that specifically target DNA, such as topoiomerase II inhibitors and cisplatin, enhance the efficacy of these drugs in cancer cells (66–68). Cisplatin or cis-diamminedichloroplatinum (II) is one of the first-line standard chemotherapy agents in the treatment of ovarian cancer (9–12). Cisplatin forms covalent platinum-DNA adducts that lead to double strand breaks, DNA damage, and eventual cell death (69, 70). A multi-drug combination of the pan-HDACi belinostat with a DNA methylation inhibitor, decitabine, enhances sensitivity to cisplatin in cisplatin-resistant ovarian cancer cells (71). The potential mechanism of synergy with DNA damaging agents is suggested by published results that selective inhibition of class I HDACs, specifically HDAC3, leads to a defective response to DNA damage, and aberrant histone deposition on chromatin (18, 66). Conditional knock-down of HDAC3 decreases S phase and causes inefficient repair of double strand DNA breaks induced by radiation (18). Because HDAC3 knock-down suppresses cell viability and contributes to DNA damage and disruption of DNA repair (18, 36, 66, 72), we hypothesized that HDACi compounds with HDAC3 bias will be synergistic with DNA damaging agents in ovarian cancer cells and found that FK228, a class I HDACi that potently inhibits HDAC3, enhances the effects of cisplatin in vitro and in vivo (73).
Selective class I HDACi targeting HDAC3 is an attractive therapeutic strategy. Our group and others have shown that targeting class I HDACs, particularly HDAC3, inhibits cellular proliferation and directly represses transcription of p21 (35, 36, 74, 75). In acute leukemia, the HDAC3-dependent N-CoR complex is recruited by the oncogenic fusion proteins (76–78). Conditional knock-down of HDAC3 in the liver induces DNA damage, chromosomal instability, and changes in metabolism (18, 20, 72). Inactivation of HDAC3 is sufficient to trigger apoptosis in cycling, non-quiescent murine embryonic fibroblasts, suggesting that HDAC3 could be a therapeutic target in highly proliferative cancer cells (18). HDAC3 is also required for efficient DNA replication in hematopoietic stem and progenitor cells, and required for the passage of hematopoietic stem/progenitor cells through the S phase, for stem cell functions, and for lymphopoiesis (79). The HDAC3 selective inhibitor, RGFP966, causes impaired S phase progression, decreased cell growth, and increased DNA damage associated apoptosis via disruptions in DNA replication in refractory cutaneous T-cell lymphoma (CTCL) (80). These results suggest that HDAC3 and other class I HDACi that enhance DNA damage are effective anti-cancer drugs, but should be used at the lowest doses possible over short periods of time.
Observations in ovarian cancer cells and other cancer cell types indicate that selective targeting of HDAC3 may be an attractive therapeutic strategy. Our group and others have shown that inhibiting class I HDACs, particularly HDAC3, inhibits cellular growth and survival, and de-represses p21 transcription leading to increased protein expression in cancer cell lines of diverse origin (35, 36, 74, 75). Class I HDAC inhibition does not induce similar anti-tumor effects in normal ovarian epithelial cell lines (36), consistent with data showing normal, non-transformed cells are spared cytotoxic effects from short-term HDACi treatment (16, 81).
Several studies have indicated that HDAC3 may contribute to inflammatory processes in macrophages during ovarian tumorigenesis, although its role is complex and remains incompletely understood. Ovarian tumorigenesis in the peritoneal cavity involves a complex interplay of signaling and responses between tumor cells and inflammatory cells such as macrophages, T-cells, and dendritic cells (82, 83). The contribution of peritoneal macrophages to the extensive peritoneal tumor implants and malignant ascites characteristic of ovarian cancer is well-recognized (84–86). Thus, therapies designed to alter macrophage function in the ovarian tumor microenvironment are a promising emerging concept reviewed elsewhere (87–89). Our group and others have shown that macrophages are abundant in peritoneal ascites fluid and that ovarian tumors have the ability to polarize macrophages to display tumor-promoting characteristics in a nuclear factor-kappaB (NF-κB)-dependent manner (84, 90), but can be “re-educated” toward an anti-tumor phenotype (84) and thus are a potential target for therapy.
Conditional HDAC3 knock-down in macrophages contributes to the regulation of inflammatory gene expression and function and appears to activate pro-tumorigenic macrophage phenotypes (91, 92). On the other hand, a possible mechanism for an anti-tumor function of HDAC3 in macrophages is direct deacetylation of NF-κB (p65/relA), which is associated with overall termination of the NF-κB transcriptional response (93), but specific activation of transcription of the anti-tumor macrophage cytokine, interleukin-1 (IL-1) (94). Further evidence for the context-dependent role played by HDAC3 in inflammation is that HDAC3 knock-down in macrophages reduces expression of almost half of lipopolysaccharide-induced inflammatory genes (92), but also contributes to transcriptional repression of toll-like receptor activation by deacetylation of p50 (95). These studies indicate that systemic inhibition of HDAC3 may, at least in a specific context, reduce the ability of peritoneal macrophages to mount an anti-tumor response. Therefore, this may represent a possible mechanism by which the efficacy of HDACi in clinical trials is limited. More research is needed to more fully understand HDAC functions in macrophages and inflammatory cells in the peritoneal cavity.
The development of class selective HDACi is ongoing (96). Novel combinations of HDACi with other targeted drugs such as aurora kinase inhibitors are underway (97) and using specific HDACi as hybrid compounds with other anti-cancer drugs is a novel approach (96). For example, targeting HDACs with PI3K inhibitors (96) may have particular relevance for both Type I and Type II ovarian tumors with alterations in the PIK3CA/AKT oncogenic pathway. Finally, hybrid ester-HDACi that selectively trap HDACi in monocytes and macrophages are in development for inflammatory diseases (98) and could be used to target the tumor microenvironment in ovarian cancer.
There are potential limitations to selective HDACi therapy. The effects of class I HDACi on DNA damage and repair pathways suggest that prolonged exposure to these drugs could lead to unacceptable toxicities (47) and secondary malignancies (20). Class I HDACs may also play an oncogenic role depending on the context (20, 99, 100). Further, predictive and prognostic biomarkers of response and toxicity, including potential immune-related toxicities and mechanisms of drug resistance are not known. Finally, the type of combinatory regimen using class I HDACi, dose and sequence of drugs are important considerations that are poorly understood and worthy of further study.
The limited clinical benefit previously seen with HDACi in ovarian cancer could be explained by a variety of factors including the: (1) phenotypic and molecular features of the tumors; (2) strength and selectivity of HDAC inhibition; and (3) type of combinatory treatment. Substantial pre-clinical evidence shows that class I biased HDACi decrease cell proliferation and increase apoptosis, likely through enhanced DNA damage and decreased DNA repair, in molecularly vulnerable ovarian cancer cells. Combination therapy with other epigenetic drugs such as DNA methyl transferase inhibitors, DNA damaging agents (platinum drugs), and small molecule inhibitors of oncogenic pathways such as the PIK3CA/AKT and NF-κB signaling pathways are potential strategic approaches. Targeting the tumor microenvironment with HDAC3 selective inhibitors is another potentially innovative strategy. A better understanding of the most susceptible ovarian cancer subtypes to target and the most effective HDACi to use in rational combinations with other cancer drugs has the potential to drive novel applications of HDACi in ovarian cancer therapy. Questions about the long-term toxicity of class I HDACi, particularly HDAC3-biased compounds and specific drug combinations remain rich areas for investigation. An ongoing debate in the field is the specificity and selectivity of HDACi as anti-cancer agents. If class I selective HDACi biased to HDAC3 can be designed to improve efficacy in subtypes of ovarian cancer such as in HR deficient ovarian tumors without significantly increasing toxicity, the therapeutic impact could be high.
Dr. Dineo Khabele discloses that she received a research grant from the Celgene Corporation and is a consultant to Genentech. However, the author is solely responsible for the content.
Grant Support from NIH/NCI (K08CA148887) and Celgene Corporation. Andrew J. Wilson Ph.D., Ms. Jeanette Saskowski, and Ms. Lynne Black for their editorial contributions.
1. Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin (2014) 64(1):9–29. doi: 10.3322/caac.21208
2. Kurman RJ, Shih Ie M. Molecular pathogenesis and extraovarian origin of epithelial ovarian cancer – shifting the paradigm. Hum Pathol (2011) 42(7):918–31. doi:10.1016/j.humpath.2011.03.003
3. Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature (2011) 474(7353):609–15. doi:10.1038/nature10166
4. Alsop K, Fereday S, Meldrum C, Defazio A, Emmanuel C, George J, et al. BRCA mutation frequency and patterns of treatment response in BRCA mutation-positive women with ovarian cancer: a report from the Australian Ovarian Cancer Study Group. J Clin Oncol (2012) 30(21):2654–63. doi:10.1200/JCO.2011.39.8545
5. Audeh MW, Carmichael J, Penson RT, Friedlander M, Powell B, Bell-McGuinn KM, et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer: a proof-of-concept trial. Lancet (2010) 376(9737):245–51. doi:10.1016/S0140-6736(10)60893-8
6. Fong PC, Boss DS, Yap TA, Tutt A, Wu P, Mergui-Roelvink M, et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med (2009) 361(2):123–34. doi:10.1056/NEJMoa0900212
7. Fong PC, Yap TA, Boss DS, Carden CP, Mergui-Roelvink M, Gourley C, et al. Poly(ADP)-ribose polymerase inhibition: frequent durable responses in BRCA carrier ovarian cancer correlating with platinum-free interval. J Clin Oncol (2010) 28(15):2512–9. doi:10.1200/JCO.2009.26.9589
8. Muggia F. Platinum compounds 30 years after the introduction of cisplatin: implications for the treatment of ovarian cancer. Gynecol Oncol (2009) 112(1):275–81. doi:10.1016/j.ygyno.2008.09.034
9. Yap TA, Carden CP, Kaye SB. Beyond chemotherapy: targeted therapies in ovarian cancer. Nat Rev Cancer (2009) 9(3):167–81. doi:10.1038/nrc2583
10. Cannistra SA. Cancer of the ovary. N Engl J Med (2004) 351(24):2519–29. doi:10.1056/NEJMra041842
11. Ozols RF. Update of the NCCN ovarian cancer practice guidelines. Oncology (Williston Park) (1997) 11(11A):95–105.
12. Covens A, Carey M, Bryson P, Verma S, Fung Kee Fung M, Johnston M. Systematic review of first-line chemotherapy for newly diagnosed postoperative patients with stage II, III, or IV epithelial ovarian cancer. Gynecol Oncol (2002) 85(1):71–80. doi:10.1006/gyno.2001.6552
13. Katsumata N, Yasuda M, Isonishi S, Takahashi F, Michimae H, Kimura E, et al. Long-term results of dose-dense paclitaxel and carboplatin versus conventional paclitaxel and carboplatin for treatment of advanced epithelial ovarian, fallopian tube, or primary peritoneal cancer (JGOG 3016): a randomised, controlled, open-label trial. Lancet Oncol (2013) 14(10):1020–6. doi:10.1016/S1470-2045(13)70363-2
14. Armstrong DK, Bundy B, Wenzel L, Huang HQ, Baergen R, Lele S, et al. Intraperitoneal cisplatin and paclitaxel in ovarian cancer. N Engl J Med (2006) 354(1):34–43. doi:10.1056/NEJMoa052985
15. West AC, Johnstone RW. New and emerging HDAC inhibitors for cancer treatment. J Clin Invest (2014) 124(1):30–9. doi:10.1172/JCI69738
16. Frew AJ, Johnstone RW, Bolden JE. Enhancing the apoptotic and therapeutic effects of HDAC inhibitors. Cancer Lett (2009) 280(2):125–33. doi:10.1016/j.canlet.2009.02.042
17. Haberland M, Montgomery RL, Olson EN. The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat Rev Genet (2009) 10(1):32–42. doi:10.1038/nrg2485
18. Bhaskara S, Chyla BJ, Amann JM, Knutson SK, Cortez D, Sun ZW, et al. Deletion of histone deacetylase 3 reveals critical roles in S phase progression and DNA damage control. Mol Cell (2008) 30(1):61–72. doi:10.1016/j.molcel.2008.02.030
19. Conti C, Leo E, Eichler GS, Sordet O, Martin MM, Fan A, et al. Inhibition of histone deacetylase in cancer cells slows down replication forks, activates dormant origins, and induces DNA damage. Cancer Res (2010) 70(11):4470–80. doi:10.1158/0008-5472.CAN-09-3028
20. Bhaskara S, Knutson SK, Jiang G, Chandrasekharan MB, Wilson AJ, Zheng S, et al. Hdac3 is essential for the maintenance of chromatin structure and genome stability. Cancer Cell (2010) 18(5):436–47. doi:10.1016/j.ccr.2010.10.022
21. Wilson AJ, Velcich A, Arango D, Kurland AR, Shenoy SM, Pezo RC, et al. Novel detection and differential utilization of a c-myc transcriptional block in colon cancer chemoprevention. Cancer Res (2002) 62(21):6006–10.
22. Kobayashi Y, Ohtsuki M, Murakami T, Kobayashi T, Sutheesophon K, Kitayama H, et al. Histone deacetylase inhibitor FK228 suppresses the Ras-MAP kinase signaling pathway by upregulating Rap1 and induces apoptosis in malignant melanoma. Oncogene (2006) 25(4):512–24.
23. Chang HC, Liu LT, Hung WC. Involvement of histone deacetylation in ras-induced down-regulation of the metastasis suppressor RECK. Cell Signal (2004) 16(6):675–9. doi:10.1016/j.cellsig.2003.11.001
24. Pan L, Lu J, Wang X, Han L, Zhang Y, Han S, et al. Histone deacetylase inhibitor trichostatin a potentiates doxorubicin-induced apoptosis by up-regulating PTEN expression. Cancer (2007) 109(8):1676–88. doi:10.1002/cncr.22585
25. Spange S, Wagner T, Heinzel T, Kramer OH. Acetylation of non-histone proteins modulates cellular signalling at multiple levels. Int J Biochem Cell Biol (2009) 41(1):185–98. doi:10.1016/j.biocel.2008.08.027
26. Eot-Houllier G, Fulcrand G, Magnaghi-Jaulin L, Jaulin C. Histone deacetylase inhibitors and genomic instability. Cancer Lett (2009) 274(2):169–76. doi:10.1016/j.canlet.2008.06.005
27. Glozak MA, Sengupta N, Zhang X, Seto E. Acetylation and deacetylation of non-histone proteins. Gene (2005) 363:15–23. doi:10.1016/j.gene.2005.09.010
28. Taunton J, Hassig CA, Schreiber SL. A mammalian histone deacetylase related to the yeast transcriptional regulator Rpd3p. Science (1996) 272(5260):408–11. doi:10.1126/science.272.5260.408
29. Bolden JE, Peart MJ, Johnstone RW. Anticancer activities of histone deacetylase inhibitors. Nat Rev Drug Discov (2006) 5(9):769–84. doi:10.1038/nrd2133
30. Guenther MG, Yu J, Kao GD, Yen TJ, Lazar MA. Assembly of the SMRT-histone deacetylase 3 repression complex requires the TCP-1 ring complex. Genes Dev (2002) 16(24):3130–5. doi:10.1101/gad.1037502
31. Guenther MG, Barak O, Lazar MA. The SMRT and N-CoR corepressors are activating cofactors for histone deacetylase 3. Mol Cell Biol (2001) 21(18):6091–101. doi:10.1128/MCB.21.18.6091-6101.2001
32. Yang WM, Tsai SC, Wen YD, Fejer G, Seto E. Functional domains of histone deacetylase-3. J Biol Chem (2002) 277(11):9447–54. doi:10.1074/jbc.M105993200
33. Escaffit F, Vaute O, Chevillard-Briet M, Segui B, Takami Y, Nakayama T, et al. Cleavage and cytoplasmic relocalization of histone deacetylase 3 are important for apoptosis progression. Mol Cell Biol (2007) 27(2):554–67. doi:10.1128/MCB.00869-06
34. Glaser KB, Li J, Staver MJ, Wei RQ, Albert DH, Davidsen SK. Role of class I and class II histone deacetylases in carcinoma cells using siRNA. Biochem Biophys Res Commun (2003) 310(2):529–36. doi:10.1016/j.bbrc.2003.09.043
35. Wilson AJ, Byun DS, Popova N, Murray LB, L’Italien K, Sowa Y, et al. Histone deacetylase 3 (HDAC3) and other class I HDACs regulate colon cell maturation and p21 expression and are deregulated in human colon cancer. J Biol Chem (2006) 281(19):13548–58. doi:10.1074/jbc.M510023200
36. Khabele D, Son DS, Parl AK, Goldberg GL, Augenlicht LH, Mariadason JM, et al. Drug-induced inactivation or gene silencing of class I histone deacetylases suppresses ovarian cancer cell growth: implications for therapy. Cancer Biol Ther (2007) 6(5):795–801. doi:10.4161/cbt.6.5.4007
37. Weichert W, Denkert C, Noske A, Darb-Esfahani S, Dietel M, Kalloger SE, et al. Expression of class I histone deacetylases indicates poor prognosis in endometrioid subtypes of ovarian and endometrial carcinomas. Neoplasia (2008) 10(9):1021–7.
38. Mariadason JM. HDACs and HDAC inhibitors in colon cancer. Epigenetics (2008) 3(1):28–37. doi:10.4161/epi.3.1.5736
39. Marsoni S, Damia G, Camboni G. A work in progress: the clinical development of histone deacetylase inhibitors. Epigenetics (2008) 3(3):164–71. doi:10.4161/epi.3.3.6253
40. Xu WS, Parmigiani RB, Marks PA. Histone deacetylase inhibitors: molecular mechanisms of action. Oncogene (2007) 26(37):5541–52. doi:10.1038/sj.onc.1210620
41. Mann BS, Johnson JR, Cohen MH, Justice R, Pazdur R. FDA approval summary: vorinostat for treatment of advanced primary cutaneous T-cell lymphoma. Oncologist (2007) 12(10):1247–52. doi:10.1634/theoncologist.12-10-1247
42. Mann BS, Johnson JR, He K, Sridhara R, Abraham S, Booth BP, et al. Vorinostat for treatment of cutaneous manifestations of advanced primary cutaneous T-cell lymphoma. Clin Cancer Res (2007) 13(8):2318–22. doi:10.1158/1078-0432.CCR-06-2672
43. Piekarz RL, Frye R, Turner M, Wright JJ, Allen SL, Kirschbaum MH, et al. Phase II multi-institutional trial of the histone deacetylase inhibitor romidepsin as monotherapy for patients with cutaneous T-cell lymphoma. J Clin Oncol (2009) 27(32):5410–7. doi:10.1200/JCO.2008.21.6150
44. Piekarz RL, Frye R, Prince HM, Kirschbaum MH, Zain J, Allen SL, et al. Phase 2 trial of romidepsin in patients with peripheral T-cell lymphoma. Blood (2011) 117(22):5827–34. doi:10.1182/blood-2010-10-312603
45. Dizon DS, Blessing JA, Penson RT, Drake RD, Walker JL, Johnston CM, et al. A phase II evaluation of belinostat and carboplatin in the treatment of recurrent or persistent platinum-resistant ovarian, fallopian tube, or primary peritoneal carcinoma: a Gynecologic Oncology Group study. Gynecol Oncol (2012) 125(2):367–71. doi:10.1016/j.ygyno.2012.02.019
46. Dizon DS, Damstrup L, Finkler NJ, Lassen U, Celano P, Glasspool R, et al. Phase II activity of belinostat (PXD-101), carboplatin, and paclitaxel in women with previously treated ovarian cancer. Int J Gynecol Cancer (2012) 22(6):979–86. doi:10.1097/IGC.0b013e31825736fd
47. Mendivil AA, Micha JP, Brown JV III, Rettenmaier MA, Abaid LN, Lopez KL, et al. Increased incidence of severe gastrointestinal events with first-line paclitaxel, carboplatin, and vorinostat chemotherapy for advanced-stage epithelial ovarian, primary peritoneal, and fallopian tube cancer. Int J Gynecol Cancer (2013) 23(3):533–9. doi:10.1097/IGC.0b013e31828566f1
48. Modesitt SC, Sill M, Hoffman JS, Bender DP. A phase II study of vorinostat in the treatment of persistent or recurrent epithelial ovarian or primary peritoneal carcinoma: a Gynecologic Oncology Group study. Gynecol Oncol (2008) 109(2):182–6. doi:10.1016/j.ygyno.2008.01.009
49. Bradner JE, West N, Grachan ML, Greenberg EF, Haggarty SJ, Warnow T, et al. Chemical phylogenetics of histone deacetylases. Nat Chem Biol (2010) 6(3):238–43. doi:10.1038/nchembio.313
50. Wilson AJ, Holson E, Wagner F, Zhang YL, Fass DM, Haggarty SJ, et al. The DNA damage mark pH2AX differentiates the cytotoxic effects of small molecule HDAC inhibitors in ovarian cancer cells. Cancer Biol Ther (2011) 12(6):484–93. doi:10.4161/cbt.12.6.15956
51. Furumai R, Matsuyama A, Kobashi N, Lee KH, Nishiyama M, Nakajima H, et al. FK228 (depsipeptide) as a natural prodrug that inhibits class I histone deacetylases. Cancer Res (2002) 62(17):4916–21.
52. Wang C, Henkes LM, Doughty LB, He M, Wang D, Meyer-Almes FJ, et al. Thailandepsins: bacterial products with potent histone deacetylase inhibitory activities and broad-spectrum antiproliferative activities. J Nat Prod (2011) 74(10):2031–8. doi:10.1021/np200324x
53. Wilson AJ, Cheng YQ, Khabele D. Thailandepsins are new small molecule class I HDAC inhibitors with potent cytotoxic activity in ovarian cancer cells: a preclinical study of epigenetic ovarian cancer therapy. J Ovarian Res (2012) 5(1):12. doi:10.1186/1757-2215-5-12
54. Gregoretti IV, Lee YM, Goodson HV. Molecular evolution of the histone deacetylase family: functional implications of phylogenetic analysis. J Mol Biol (2004) 338(1):17–31. doi:10.1016/j.jmb.2004.02.006
55. Koprinarova M, Botev P, Russev G. Histone deacetylase inhibitor sodium butyrate enhances cellular radiosensitivity by inhibiting both DNA nonhomologous end joining and homologous recombination. DNA Repair (Amst) (2011) 10(9):970–7. doi:10.1016/j.dnarep.2011.07.003
56. Adimoolam S, Sirisawad M, Chen J, Thiemann P, Ford JM, Buggy JJ. HDAC inhibitor PCI-24781 decreases RAD51 expression and inhibits homologous recombination. Proc Natl Acad Sci U S A (2007) 104(49):19482–7. doi:10.1073/pnas.0707828104
57. Ladd B, Ackroyd JJ, Hicks JK, Canman CE, Flanagan SA, Shewach DS. Inhibition of homologous recombination with vorinostat synergistically enhances ganciclovir cytotoxicity. DNA Repair (Amst) (2013) 12(12):1114–21. doi:10.1016/j.dnarep.2013.10.008
58. Konstantinopoulos PA, Wilson AJ, Saskowski J, Wass E, Khabele D. Suberoylanilide hydroxamic acid (SAHA) enhances olaparib activity by targeting homologous recombination DNA repair in ovarian cancer. Gynecol Oncol (2014). doi:10.1016/j.ygyno.2014.03.007
59. Mackay HJ, Hirte H, Colgan T, Covens A, MacAlpine K, Grenci P, et al. Phase II trial of the histone deacetylase inhibitor belinostat in women with platinum resistant epithelial ovarian cancer and micropapillary (LMP) ovarian tumours. Eur J Cancer (2010) 46(9):1573–9. doi:10.1016/j.ejca.2010.02.047
60. Son DS, Wilson AJ, Parl AK, Khabele D. The effects of the histone deacetylase inhibitor romidepsin (FK228) are enhanced by aspirin (ASA) in COX-1 positive ovarian cancer cells through augmentation of p21. Cancer Biol Ther (2010) 9(11):928–35. doi:10.4161/cbt.9.11.11873
61. Batty N, Malouf GG, Issa JP. Histone deacetylase inhibitors as anti-neoplastic agents. Cancer Lett (2009) 280(2):192–200. doi:10.1016/j.canlet.2009.03.013
62. Kim IA, No M, Lee JM, Shin JH, Oh JS, Choi EJ, et al. Epigenetic modulation of radiation response in human cancer cells with activated EGFR or HER-2 signaling: potential role of histone deacetylase 6. Radiother Oncol (2009) 92(1):125–32. doi:10.1016/j.radonc.2009.03.008
63. Zhang F, Zhang T, Teng ZH, Zhang R, Wang JB, Mei QB. Sensitization to gamma-irradiation-induced cell cycle arrest and apoptosis by the histone deacetylase inhibitor trichostatin A in non-small cell lung cancer (NSCLC) cells. Cancer Biol Ther (2009) 8:9. doi:10.4161/cbt.8.9.8143
64. Cuneo KC, Fu A, Osusky K, Huamani J, Hallahan DE, Geng L. Histone deacetylase inhibitor NVP-LAQ824 sensitizes human nonsmall cell lung cancer to the cytotoxic effects of ionizing radiation. Anticancer Drugs (2007) 18(7):793–800. doi:10.1097/CAD.0b013e3280b10d57
65. Geng L, Cuneo KC, Fu A, Tu T, Atadja PW, Hallahan DE. Histone deacetylase (HDAC) inhibitor LBH589 increases duration of gamma-H2AX foci and confines HDAC4 to the cytoplasm in irradiated non-small cell lung cancer. Cancer Res (2006) 66(23):11298–304. doi:10.1158/0008-5472.CAN-06-0049
66. Marchion DC, Bicaku E, Turner JG, Schmitt ML, Morelli DR, Munster PN. HDAC2 regulates chromatin plasticity and enhances DNA vulnerability. Mol Cancer Ther (2009) 8(4):794–801. doi:10.1158/1535-7163.MCT-08-0985
67. Ozaki K, Kishikawa F, Tanaka M, Sakamoto T, Tanimura S, Kohno M. Histone deacetylase inhibitors enhance the chemosensitivity of tumor cells with cross-resistance to a wide range of DNA-damaging drugs. Cancer Sci (2008) 99(2):376–84. doi:10.1111/j.1349-7006.2007.00669.x
68. Kim DH, Kim M, Kwon HJ. Histone deacetylase in carcinogenesis and its inhibitors as anti-cancer agents. J Biochem Mol Biol (2003) 36(1):110–9. doi:10.5483/BMBRep.2003.36.1.110
69. Wang D, Lippard SJ. Cisplatin-induced post-translational modification of histones H3 and H4. J Biol Chem (2004) 279(20):20622–5. doi:10.1074/jbc.M402547200
70. Cohen SM, Lippard SJ. Cisplatin: from DNA damage to cancer chemotherapy. Prog Nucleic Acid Res Mol Biol (2001) 67:93–130. doi:10.1016/S0079-6603(01)67026-0
71. Steele N, Finn P, Brown R, Plumb JA. Combined inhibition of DNA methylation and histone acetylation enhances gene re-expression and drug sensitivity in vivo. Br J Cancer (2009) 100(5):758–63. doi:10.1038/sj.bjc.6604932
72. Knutson SK, Chyla BJ, Amann JM, Bhaskara S, Huppert SS, Hiebert SW. Liver-specific deletion of histone deacetylase 3 disrupts metabolic transcriptional networks. EMBO J (2008) 27(7):1017–28. doi:10.1038/emboj.2008.51
73. Wilson AJ, Lalani AS, Wass E, Saskowski J, Khabele D. Romidepsin (FK228) combined with cisplatin stimulates DNA damage-induced cell death in ovarian cancer. Gynecol Oncol (2012) 127(3):579–86. doi:10.1016/j.ygyno.2012.09.016
74. Wilson AJ, Byun DS, Nasser S, Murray LB, Ayyanar K, Arango D, et al. HDAC4 promotes growth of colon cancer cells via repression of p21. Mol Biol Cell (2008) 19(10):4062–75. doi:10.1091/mbc.E08-02-0139
75. Huang W, Tan D, Wang X, Han S, Tan J, Zhao Y, et al. Histone deacetylase 3 represses p15(INK4b) and p21(WAF1/cip1) transcription by interacting with Sp1. Biochem Biophys Res Commun (2006) 339(1):165–71. doi:10.1016/j.bbrc.2005.11.010
76. Gelmetti V, Zhang J, Fanelli M, Minucci S, Pelicci PG, Lazar MA. Aberrant recruitment of the nuclear receptor corepressor-histone deacetylase complex by the acute myeloid leukemia fusion partner ETO. Mol Cell Biol (1998) 18(12):7185–91.
77. Grignani F, De Matteis S, Nervi C, Tomassoni L, Gelmetti V, Cioce M, et al. Fusion proteins of the retinoic acid receptor-alpha recruit histone deacetylase in promyelocytic leukaemia. Nature (1998) 391(6669):815–8. doi:10.1038/35901
78. Atsumi A, Tomita A, Kiyoi H, Naoe T. Histone deacetylase 3 (HDAC3) is recruited to target promoters by PML-RARalpha as a component of the N-CoR co-repressor complex to repress transcription in vivo. Biochem Biophys Res Commun (2006) 345(4):1471–80. doi:10.1016/j.bbrc.2006.05.047
79. Summers AR, Fischer MA, Stengel KR, Zhao Y, Kaiser JF, Wells CE, et al. HDAC3 is essential for DNA replication in hematopoietic progenitor cells. J Clin Invest (2013) 123(7):3112–23. doi:10.1172/JCI60806
80. Wells CE, Bhaskara S, Stengel KR, Zhao Y, Sirbu B, Chagot B, et al. Inhibition of histone deacetylase 3 causes replication stress in cutaneous T cell lymphoma. PLoS One (2013) 8(7):e68915. doi:10.1371/journal.pone.0068915
81. Bolden JE, Shi W, Jankowski K, Kan CY, Cluse L, Martin BP, et al. HDAC inhibitors induce tumor-cell-selective pro-apoptotic transcriptional responses. Cell Death Dis (2013) 4:e519. doi:10.1038/cddis.2013.9
82. Alvero AB, Montagna MK, Craveiro V, Liu L, Mor G. Distinct subpopulations of epithelial ovarian cancer cells can differentially induce macrophages and T regulatory cells toward a pro-tumor phenotype. Am J Reprod Immunol (2012) 67(3):256–65. doi:10.1111/j.1600-0897.2011.01068.x
83. Krempski J, Karyampudi L, Behrens MD, Erskine CL, Hartmann L, Dong H, et al. Tumor-infiltrating programmed death receptor-1+ dendritic cells mediate immune suppression in ovarian cancer. J Immunol (2011) 186(12):6905–13. doi:10.4049/jimmunol.1100274
84. Hagemann T, Lawrence T, McNeish I, Charles KA, Kulbe H, Thompson RG, et al. “Re-educating” tumor-associated macrophages by targeting NF-kappaB. J Exp Med (2008) 205(6):1261–8. doi:10.1084/jem.20080108
85. Hagemann T, Robinson SC, Thompson RG, Charles K, Kulbe H, Balkwill FR. Ovarian cancer cell-derived migration inhibitory factor enhances tumor growth, progression, and angiogenesis. Mol Cancer Ther (2007) 6(7):1993–2002. doi:10.1158/1535-7163.MCT-07-0118
86. Robinson-Smith TM, Isaacsohn I, Mercer CA, Zhou M, Van Rooijen N, Husseinzadeh N, et al. Macrophages mediate inflammation-enhanced metastasis of ovarian tumors in mice. Cancer Res (2007) 67(12):5708–16. doi:10.1158/0008-5472.CAN-06-4375
87. Coussens LM, Zitvogel L, Palucka AK. Neutralizing tumor-promoting chronic inflammation: a magic bullet? Science (2013) 339(6117):286–91. doi:10.1126/science.1232227
88. Hagemann T, Wilson J, Kulbe H, Li NF, Leinster DA, Charles K, et al. Macrophages induce invasiveness of epithelial cancer cells via NF-kappa B and JNK. J Immunol (2005) 175(2):1197–205. doi:10.4049/jimmunol.175.2.1197
89. Kulbe H, Chakravarty P, Leinster DA, Charles KA, Kwong J, Thompson RG, et al. A dynamic inflammatory cytokine network in the human ovarian cancer microenvironment. Cancer Res (2012) 72(1):66–75. doi:10.1158/0008-5472.CAN-11-2178
90. Hagemann T, Wilson J, Burke F, Kulbe H, Li NF, Pluddemann A, et al. Ovarian cancer cells polarize macrophages toward a tumor-associated phenotype. J Immunol (2006) 176(8):5023–32. doi:10.4049/jimmunol.176.8.5023
91. Mullican SE, Gaddis CA, Alenghat T, Nair MG, Giacomin PR, Everett LJ, et al. Histone deacetylase 3 is an epigenomic brake in macrophage alternative activation. Genes Dev (2011) 25(23):2480–8. doi:10.1101/gad.175950.111
92. Chen X, Barozzi I, Termanini A, Prosperini E, Recchiuti A, Dalli J, et al. Requirement for the histone deacetylase Hdac3 for the inflammatory gene expression program in macrophages. Proc Natl Acad Sci U S A (2012) 109(42):E2865–74. doi:10.1073/pnas.1121131109
93. Chen L, Fischle W, Verdin E, Greene WC. Duration of nuclear NF-kappaB action regulated by reversible acetylation. Science (2001) 293(5535):1653–7. doi:10.1126/science.1062374
94. Ziesche E, Kettner-Buhrow D, Weber A, Wittwer T, Jurida L, Soelch J, et al. The coactivator role of histone deacetylase 3 in IL-1-signaling involves deacetylation of p65 NF-kappaB. Nucleic Acids Res (2013) 41(1):90–109. doi:10.1093/nar/gks916
95. Yan Q, Carmody RJ, Qu Z, Ruan Q, Jager J, Mullican SE, et al. Nuclear factor-kappaB binding motifs specify toll-like receptor-induced gene repression through an inducible repressosome. Proc Natl Acad Sci U S A (2012) 109(35):14140–5. doi:10.1073/pnas.1119842109
96. Delcuve GP, Khan DH, Davie JR. Targeting class I histone deacetylases in cancer therapy. Expert Opin Ther Targets (2013) 17(1):29–41. doi:10.1517/14728222.2013.729042
97. Li Y, Liu T, Ivan C, Huang J, Shen DY, Kavanagh JJ, et al. Enhanced cytotoxic effects of combined valproic acid and the aurora kinase inhibitor VE465 on gynecologic cancer cells. Front Oncol (2013) 3:58. doi:10.3389/fonc.2013.00058
98. Needham LA, Davidson AH, Bawden LJ, Belfield A, Bone EA, Brotherton DH, et al. Drug targeting to monocytes and macrophages using esterase-sensitive chemical motifs. J Pharmacol Exp Ther (2011) 339(1):132–42. doi:10.1124/jpet.111.183640
99. Santoro F, Botrugno OA, Dal Zuffo R, Pallavicini I, Matthews GM, Cluse L, et al. A dual role for Hdac1: oncosuppressor in tumorigenesis, oncogene in tumor maintenance. Blood (2013) 121(17):3459–68. doi:10.1182/blood-2012-10-461988
Keywords: histone deacetylases, histone deacetylase inhibitors, epigenetic therapy, ovarian cancer, targeted therapy
Citation: Khabele D (2014) The therapeutic potential of class I selective histone deacetylase inhibitors in ovarian cancer. Front. Oncol. 4:111. doi: 10.3389/fonc.2014.00111
Received: 14 February 2014; Accepted: 30 April 2014;
Published online: 20 May 2014.
Edited by:
Angeles Alvarez Secord, Duke University Medical Center, USAReviewed by:
Angeles Alvarez Secord, Duke University Medical Center, USACopyright: © 2014 Khabele. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dineo Khabele, Department of Obstetrics and Gynecology, Division of Gynecologic Oncology, Vanderbilt University, 1161 21st Avenue South, B-1100 Medical Center North, Nashville, TN 37232, USA e-mail:ZGluZW8ua2hhYmVsZUB2YW5kZXJiaWx0LmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.