 Clotilde Costa
Clotilde Costa Jesús M. Paramio
Jesús M. Paramio Mirentxu Santos
Mirentxu Santos
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol., 17 December 2013
Sec. Cancer Genetics
Volume 3 - 2013 | https://doi.org/10.3389/fonc.2013.00307
This article is part of the Research TopicGenetically modified mouse models of cancerView all 7 articles
The Rb1 gene was the first bona fide tumor suppressor identified and cloned more than 25 years ago. Since then, a plethora of studies have revealed the functions of pRb and the existence of a sophisticated and strictly regulated pathway that modulates such functional roles. An emerging paradox affecting Rb1 in cancer connects the relatively low number of mutations affecting Rb1 gene in specific human tumors, compared with the widely functional inactivation of pRb in most, if not in all, human cancers. The existence of a retinoblastoma family of proteins pRb, p107, and p130 and their potential unique and overlapping functions as master regulators of cell cycle progression and transcriptional modulation by similar processes, may provide potential clues to explain such conundrum. Here, we will review the development of different genetically engineered mouse models, in particular those affecting stratified epithelia, and how they have offered new avenues to understand the roles of the Rb family members and their targets in the context of tumor development and progression.
Mouse models are essential tools to analyze the molecular mechanisms underlying any physiological event that take place in the organism. They provide information about gene function, allow the analysis of specific genetic changes and, under appropriate circumstances, might mimic human diseases. This characteristic enables the close analysis of pathology and the identification and validation of candidate therapeutic targets.
Several approaches are used to generate genetically engineered mouse models (GEMMs). The traditional methodology, based on the elimination of the gene of interest in the whole animal (knockout mice) (1), allows the functional characterization of the gene product during the organism development. However, this may result in embryonic lethality, precluding the study in adult animal tissues. This problem can be circumvented by the use of conditional transgenic gene knockout (e.g., involving cre-loxP system), which allows studies of gene function in specific cell and tissue types, including adult ones (2). Additionally, both experimental systems also provide valuable information about potential compensatory mechanisms, since the possible functions of the specific ablated gene can be carried out by other genes. Such compensation could occur directly, if “compensatory” genes are up-regulated or down-regulated as a direct result of the loss of the gene under study. The existence of such overlapping roles may also help to determine possible cooperative events in disease onset or progression. The Rb and E2F families represent a paradigm of direct compensatory mechanisms revealed using conventional and conditional knockouts (3).
The conditional knockout mice are engineered in such way that genes can be inactivated in a tissue-specific manner. This approach requires the use of specific gene promoters, which in some circumstances may represent a problematic issue due to ectopic expression or poorly characterized control elements. Nonetheless, these GEMMs enable much more sophisticated pathology modeling, as they provide essential information of the potential role of a particular gene or group of genes in a determined cell type. This is of a particular relevance in cancer research where it is possible to determine whether the loss of a particular gene is involved in tumor susceptibility, initiation, or progression to malignancy. On the other hand, in sporadic cancer, initiating mutations probably occur in a unique or very few cells in a certain tissue. These initiated cells acquire proliferative or pro-survival advantages through subsequent genetic alterations and by a cross-talk with the microenvironment lead to tumor development and progression. Accordingly, most of the conventional or tissue-specific mouse models reproduce familial forms of cancer but not sporadic tumors, as mutations are present in every cell of the body or in vast majority of cells in a certain tissue (3). An interesting progress in the field was the development of a ligand-dependent Cre recombinase that can be activated by tamoxifen administration (4). These inducible mouse models, besides allowing the space-temporal control of recombination, also provide a better suited model of human sporadic cancer, as the recombination can be only limited to few cells in a given tissue.
Skin is an essential organ that forms a protective barrier against the environment. This function is primarily exerted by the epidermis, a stratified epithelium in the outermost layer of the skin, and relies on a finely tuned process of coupled differentiation and proliferation. Both processes are compartmentalized in this tissue, and can be characterized by the sequential expression of highly specific markers. The proliferative cells are confined to a single cell basal layer and the non-proliferative differentiating cells are located in the suprabasal layers. This represents one of the main characteristics that make epidermis an ideal model system. In particular, the expression of highly specific proteins in these compartments has allowed the characterization of specific promoters for different epidermal layers. In this context, it is remarkable the predominant use of the basal cell specific keratin K5 (5–7) and keratin K14 promoters (8), including the inducible specific forms (9).
Another attractive biologic characteristic of the epidermis resides in the cell replenishment and renewal processes occurring throughout the individual entire life. Terminally differentiated or damaged epidermal cells are shed from the skin, requiring a permanent tissue renewal fulfilled by epidermal stem cells. In mouse epidermis, these stem cells lie mostly in the hair follicle bulge, display a low proliferative rate and upon specific stress conditions (i.e., wound healing) can give rise to all the epidermal cell subtypes (10–12). Importantly, the hair follicle stem cells are also responsible for the hair cycling, which is coordinated by extremely relevant signaling pathways such as Wnt, BMP, Notch, etc. and they are considered the cells of origin in non-melanoma skin cancer (13–17). Epidermal stem cells can be isolated and characterized using different cell surface markers (18, 19) and they also express K5 and K14 genes. Accordingly, the use of the K5 and K14 control elements may also cause genetic alterations in these long-lived cells.
Besides the above commented attributes, epidermis is also perfectly suited to allow in vitro studies. Keratinocytes can be obtained and cultured in vitro as monolayer cell cultures. In these, differentiation can be achieved in response to external signals in a manner resembling that observed in vivo (20, 21). In humans, cultured keratinocytes, which can also be genetically modified, can be used to engineer skin equivalents (22–24), which can be grafted onto different receptors allowing the evolution of the skin transplant. In mice, skin transplants can avoid specifically complex technical approaches associated with compromised viability (25).
Finally, for the cancer field, the two-stage mouse skin carcinogenesis is perhaps the best characterized experimental carcinogenesis protocol and represents a suitable model for the understanding the multistage nature of tumorigenesis (26). This approach, besides allowing to define the relevance of multiple oncogenic and/or tumor suppressor activities, has also led to the establishment of fundamental molecular aspects of cancer, and has contributed in an essential manner to an ideal conceptual framework to understand many molecular aspects of tumorigenesis such as tumor angiogenesis, epithelial-mesenchymal transition and metastasis, and the role of adult stem cells (27).
The retinoblastoma gene (Rb1), localized in chromosome 13q14.2, was the first tumor suppressor identified more than 25 years ago (28). Years later, other similar genes were discovered encompassing Rb family. They are the retinoblastoma-like 1 (Rbl1) and retinoblastoma-like 2 (Rbl-2) genes, localized in 20q11.2 and 16q12.2, and encoding p107 and p130 proteins, respectively (29–31). pRb, p107, and p130 share structural homology. These proteins are defined by a conserved pocket domain which serves as a binding site and provides the family name “pocket proteins.” The pocket domain consists of A and B domains separated by a spacer region, and is a region where viral oncoproteins bind (Ad-E1A, SV40 LT-antigen, or HPV-E7) (32, 33). Rb family proteins interact with proteins containing a LeuXCysXGlu (LXCXE) motif, found in several viral transforming proteins, such as HPV E7, and in cellular proteins (34). The structure of pRb (and probably of other members of the family) is altered by phosphorylation events, changing its binding affinities (35, 36). Despite the well conserved pocket protein domain, p107 and p130 are closer to each other than either is to pRb (37). They share a motive in the spacer region, not present in pRb, which binds cyclin A-CDK2 and cyclin E-CDK2 complexes. Moreover, they also share a sequence, absent in pRb, next to the N-terminal region (38–40).
The most relevant function of Rb family is to control cell cycle progression (34). This role, which depends on phosphorylation, is determined by the interaction with other proteins, including different transcription factors and nuclear matrix domains. Phosphorylation is carried out by cyclin-dependent kinases (41) which, in turn, are regulated by cyclins and cyclin-dependent kinase inhibitors (42). Hypophosphorylated Rb family members bind to distinct E2F transcription factor family members, thus promoting their inhibition. Moreover, the pocket protein-E2F complexes can act as an active mechanism for transcriptional repression (43, 44). Sequential phosphorylation of the pocket proteins leads to E2F release, allowing gene expression and cell cycle progression (43, 44).
E2Fs transcription factors form a superfamily consisting of E2F and DP (dimerization partner) proteins (45). DP family proteins are cofactors of E2F factors and DP-E2F dimerization increases DNA affinity and gets efficient regulation of the transcription. There are 8 E2F gene members that produce 10 different proteins (E2F3 and E2F7 have two isoforms). They display different affinity to pocket proteins. pRb binds principally E2F1–4; p107 binds E2F4 and 5, mostly in cycling cells, whereas p130 preferentially binds E2F5. E2F family members have unique and redundant functions in cell cycle regulation and also show cell cycle-independent roles (46). Functionally, E2Fs are divided in activators (E2F1–E2F3a) and repressors (E2F3b–8) based on structurally and affinity differences. E2F1–E2F6 factors have a domain to bind DP and DNA, E2F1–5 have a pocket protein binding domain, and E2F7 and 8 can bind DNA without DP proteins. E2Fs have transactivation and repression domains. When retinoblastoma proteins are bound to E2F factors mediate transcriptional repression.
During cell cycle, the three Rb family members can interact with E2Fs in a distinct, specific fashion. Activator E2Fs associate exclusively with pRb, whereas p107 or p130 bind mainly to repressors E2Fs. Importantly, pocket proteins are differentially expressed along the cell cycle, probably reflecting specific functions for each protein during the different cell cycle phases. In general, pRb and p107 are expressed in cycling cells, while p130 is preferentially expressed in quiescent cells, but this aspect is also cell type specific (43, 47, 48).
Besides their cell cycle functions, pocket proteins are involved in other cellular process such as differentiation, apoptosis, angiogenesis, or senescence modulating gene transcription (49–53). Some of these functions are also mediated by interaction with specific E2F family members (54, 55). However, they can do it both directly and indirectly, and in a very broad scale, recruiting co-repressors/activators to specific transcription factors. Recent studies propose Rb proteins to bind co-repressors: histones deacetylases (HDAC1, HDAC2) (56–58), histone de-methylases (RBP2) (59), DNA methyl transferases (DNMT1) (60), helicases (Brg1, Brm) (61, 62), histone methyl transferases (Suv39h1, RIZ, suv4-20h1/h2) (63–65), and histone binding proteins, like HP1, regulating chromatin structure and transcription (63, 66). In this context, the interaction with E2F directs pocket proteins to DNA domains leads to transcriptional repression. Finally, a recent work suggests a role for Rb family in influencing the chromatin structure of larger genomic regions, and also in genome stability maintenance (67).
Rb family functions in multiple processes may exemplify their role as potential tumor suppressors. Most, if not all, of the so called hallmarks of cancer (68, 69) are regulated by this family. Accordingly, the Rb/E2F pathway is disrupted in probably all tumors (34). This subversion occurs mainly by overexpression or mutation of cyclin-dependent kinases, inactivation of CKIs, increased expression of cyclins and, in some cases, amplification and increased expression of specific E2F members. Deletion and inactivating mutations of pRb are restricted to very few specific cancer types, whereas alterations of p107 and/or p130 in human cancers is still a matter of debate, being rarely mutated in human tumors (70, 71), probably due to pRb controls E2F activity in a broad manner (72). The reason why different tumors preferentially display one or another alteration of the Rb pathway is still unknown. However, the most frequent alterations tend to the functional inactivation of the three Rb family members, indicating their potential overlapping function as tumor suppressors. In spite of this, due to the specific functions of pRb in cell differentiation and senescence, it appears to exert specific tumor suppressor activities over p107 and p130. This has been highlighted by the findings in GEMMs (Table 1).
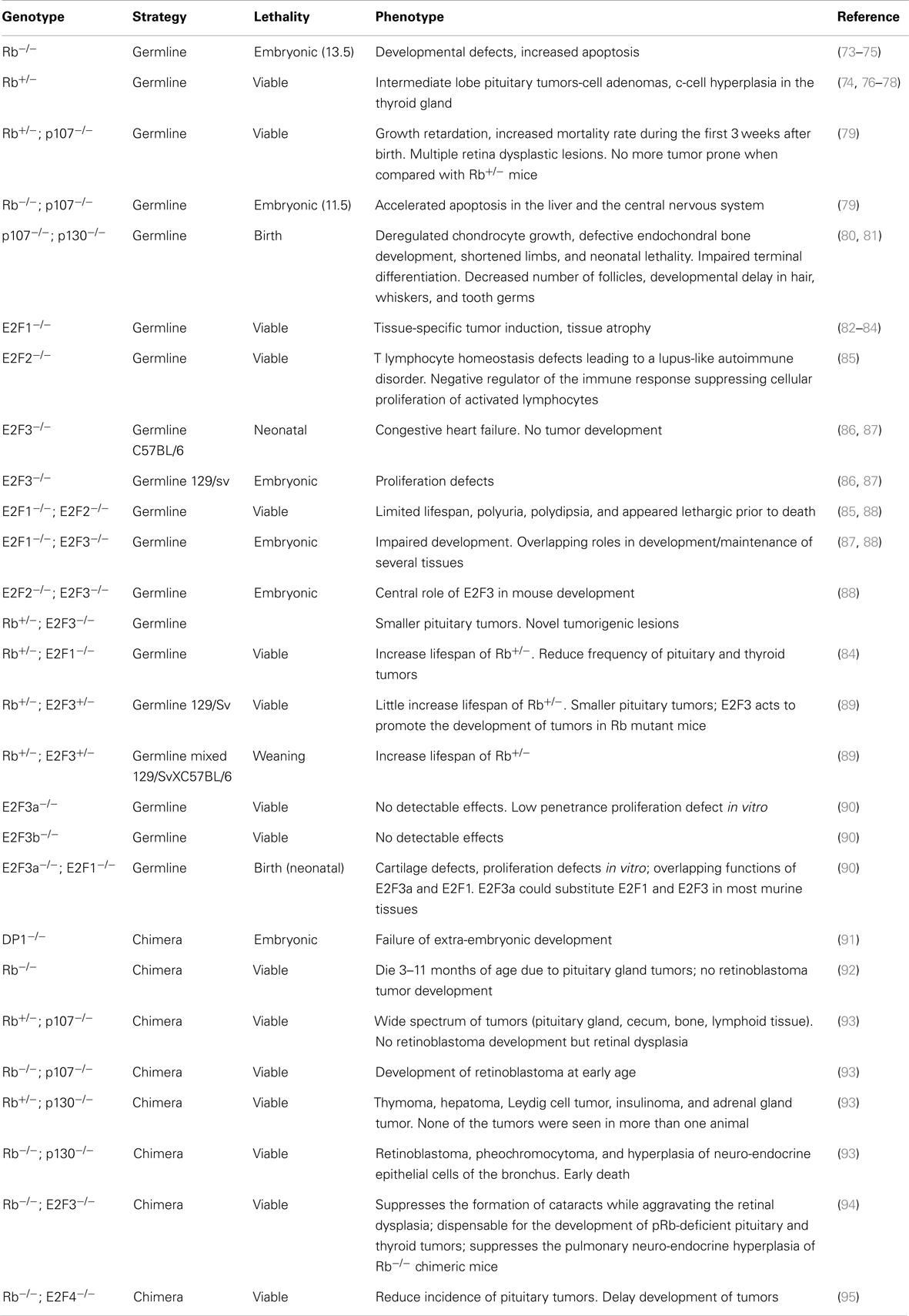
Table 1. Phenotypic abnormalities observed in germline-ablated mouse models affecting retinoblastoma family members.
Retinoblastoma was the first tumor suppressor gene eliminated by targeted deletion in mice (73–75). Rb-null mice die during embryonic development due to multiple embryonary and extraembryonary tissue defects. However, their progress until late stages of development, indicating that pRb is not essential during early normal mouse development. This aspect was particularly emphasized by the generation of chimeric mice with a very high component of Rb-null cells (Table 1 and references therein).
Nevertheless, the early lethality precludes the analysis of the Rb1 gene roles in adult mice. This is in contrast with the targeted deletion of any of the other pocket protein family members, as p107 or p130 deficient mice do not show any obvious phenotype and no tumor predisposition has been observed in any mutant animal (79, 80). In contrast, mice having simultaneous inactivation of p130 and p107 genes show neonatal lethality and limb development defects (80). These findings demonstrate the existence of compensatory roles among the pocket protein family members (i.e., p130 fulfills p107 functions in its absence or vice versa) and also that, in certain tissues and cellular settings, p107 and p130 perform shared growth-regulatory functions that are not fulfilled by pRb. In agreement, the embryonic lethality of Rb-null mice occurs earlier when is accompanied by the deletion of any other pocket protein (79, 96).
Genetically deficient cells for all the three Rb family members (TKO mouse embryonic fibroblasts) are resistant to G1 arrest (97, 98). Chimeric embryos composed of TKO cells develop until day 9 of gestation and some cells are able to arrest in G0/G1, to exit cell cycle and to differentiate (in teratomas and in culture), pointing to a cell type dependent mechanism and illustrating the robustness of cell cycle regulatory networks (99). Regarding the role of Rb family in cancer, KO mice have also provided important clues. To circumvent the embryonic lethality due to Rb1 deficiency, heterozygous mice have been widely employed. Different studies gave evidences of compensatory roles between retinoblastoma family members (see Table 1). Lee and colleagues provided in vivo evidence that p107 and pRb have overlapping functions in development and adult tissues of mutant mice for the first time (79). pRb and p107 interact with E2Fs in a different manner, which could explain specific and related functions (100). As the Rb+/− and Rb−/− chimeric mice do not develop retinoblastoma, additional mutations besides loss of pRb function are needed to induce this type of tumor. p107 plays a tumor suppressor function in absence of pRb (101), and is responsible for a limited tumor spectrum observed in mice which have lost pRb in a variety of tissues (93). In Rb−/− mice, the lack of E2F4 suppresses pituitary and thyroid tumors formation, with enhancement of p107 and p130 levels associated with the activator E2Fs. This may indicate that Retinoblastoma family acts as a tumor suppressor possibly regulating activator E2Fs rather than establishing repressive E2F complex. Accordingly, in those tissues with very low p107 and p130 expression levels, pRb compensation is unlikely, making those tissues more susceptible to tumor development as a consequence of Rb1 loss (102). In addition, the different phenotypes observed could be related to the diverse capacity of the Rb family members to interact with target proteins (103).
Although numerous indirect evidences have suggested a role of pRb pathway in epidermal homeostasis (104–108) and in skin carcinogenesis (109–115), the early lethality of Rb-null mice precluded the study of actual Rb1 functions in skin. Nonetheless, conventional knockout models allowed us to demonstrate that the simultaneous absence of p107 and p130 produces severe skin abnormalities affecting terminal differentiation and the expression of several morphogens involved in the inductive signals between epithelium and underlying mesenchyme (81, 116). Importantly, these signals are essential in epidermal stem cell homeostasis and have been also involved in epidermal tumor development (13).
Long-lived adult stem cells have more chances to accumulate the number of genetic hits essential for tumor development (13), and many pathways implicated in stem cell quiescence are deregulated in tumor progression. Remarkably, Rb family activity also plays a critical role to balance proliferation and cell survival in human embryonic stem cells (117). In epidermis, tissue homeostasis is strictly dependent on the functionality of epidermal stem cells. Whole transcriptome analysis and bioinformatic approaches using these purified epidermal stem cells revealed that the Rb-E2F axis is also an essential mediator of stem cell quiescence (118). Moreover, transgenic mouse models showed that the aberrant accumulation of altered stem cells in hair follicles and their subsequent migration to the interfollicular epidermis contribute to HPV-induced tumor development (17), which is also mediated by Rb family inactivation (24).
The development of an epidermal-specific Rb-deficient mouse model and its combination with several other GEMMs has allowed us to establish a comprehensive framework for a better understanding of the functions of the Rb-dependent signaling in different aspects of epidermal homeostasis, carcinogenesis, and metastatic development (Table 2).
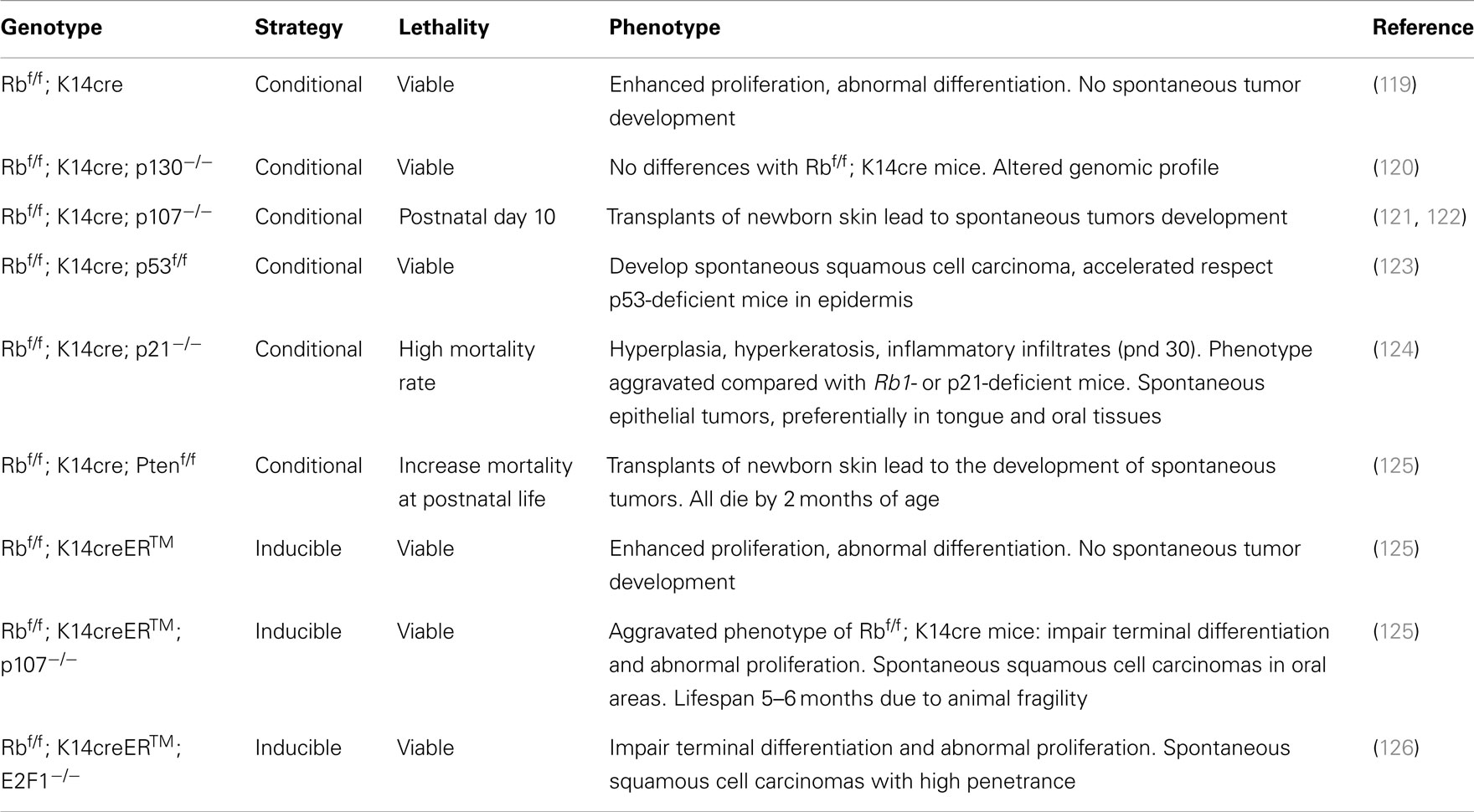
Table 2. Phenotypic Skin abnormalities observed in mouse models lacking pRb in epidermis.
We and others demonstrated that pRb absence in epidermis is characterized by moderate hyperplasia and hyperkeratosis associated with increased proliferation and altered differentiation (121, 127). Although these characteristics might suggest a cancer-prone phenotype, no spontaneous tumor development was observed even after a long latency (121, 127). This might indicate the possible overlapping roles of the other retinoblastoma family members, p107 and p130, in suppressing skin tumorigenesis in the absence of pRb. In support of this, the phenotypic changes observed in pRb-deficient skin are aggravated by concomitant HPV E7 oncogene expression (127), indicating the involvement of other E7 targets (such as p107 and/or p130). Importantly, similar findings suggesting this compensatory or/and overlapping mechanisms have been also suggested for multiple tissues including retina, mammary gland, muscle, bone, etc. (128).
Loss of p130 did not further aggravate the phenotypic consequences of pRb ablation in skin, suggesting a potential absence of functional compensation between these two proteins in skin (120). In agreement, multiple cell cycle and proliferation genes showed a similar pattern between keratinocytes lacking pRb alone or both pRb and p130 (120). In spite of this, gene profiling analyses demonstrated that the combined absence of pRb and p130 generated changes in a large number of genes compared to pRb, indicating a primary role of p130 in modulating transcription through the specific interaction with particular E2F proteins in the absence of pRb (120). Further studies also showed a potential link between p130, specific E2Fs and chromatin remodeling machinery through the cyclin-cdk inhibitor p27 (129).
In contrast with the moderate epidermal phenotype produced by the absence of pRb or combined pRb and p130 epidermal loss, the lack of p107 and pRb in epidermis produced a severe phenotype consisting of dramatic growth retardation, no hair, and death by postnatal day 10 (121). Importantly, whilst the phenotype was aggravated with the progressive loss of one or both alleles of p107, a single functional copy of pRb was sufficient to rescue all epidermal defects (121). These data demonstrated a functional overlap between pRb and p107 in epidermis and illustrated a dose dependent effect of p107 in vivo in the context of Rb1 deficiency, suggestive of a potential tumor suppressor role for p107 in the absence of pRb. This was further supported by the findings obtained with skin grafts (used to circumvent the perinatal lethality), which invariably evolved to differentiated SCC (122). Furthermore, primary double deficient keratinocytes displayed a high sensitivity to Ha Ras-mediated transformation and a particular resistance to oncogene-induced senescence (122). Importantly, biochemical and whole transcriptome analysis suggested a possible impaired p53 function in these double deficient keratinocytes (122).
Two-stage chemical carcinogenesis protocols in epidermal-specific Rb-deficient mice showed that the absence of pRb lead to the generation of fewer and smaller tumors than in control animals (119), but with increased malignant conversion to SCCs. Biochemical analyses indicated that, in the absence of pRb, multiple pathways, including the aberrant p53 induction mediated by E2F/p19ARF, are activated, leading to increased tumor apoptosis (119). Importantly, the increased expression and activity of p53 generates a selective pressure leading to premature p53 loss and then increased malignant conversion (119, 130). This hypothesis was also reinforced by the observation of restored susceptibility to tumor development obtained in mice lacking pRb and a single p107 allele, upon similar carcinogenesis protocols (131).
The above commented findings pointed to a primordial role of p53 in suppressing skin tumorigenesis, as suggested previously (8, 132, 133), and also indicated a possible cooperative roles of both tumor suppressors in skin, in agreement with the findings in other tissues (76, 134–139). To demonstrate such hypothesis, we generated mice lacking both Rb1 and Trp53 genes in epidermis (123). We observed that the spontaneous SCC development due to epidermal loss of p53 (8) is severely accelerated in mice lacking pRb and p53 (123), whereas the epidermal proliferation and differentiation phenotype due to pRb loss is not enhanced by the simultaneous inactivation of pRb and p53 (123). Detailed analyses indicated that tumorigenesis due to p53 loss was associated to early chromosome instability, and under these settings the increased proliferation promoted by the absence of Rb1 resulted in an accelerated process (140). Of note, the transcriptome analysis of tumors arising in these deficient mice revealed a highly significant overlap with human tumors. These were characterized by p53 mutation, poor prognosis and/or very high predisposition to develop distant metastasis (141), regardless the tissue of origin, and including clinically relevant human cancers such as breast and lung tumors (141, 142). In agreement, squamous cell carcinomas generated in these mouse models are highly metastatic and displayed early signs of epithelial-mesenchymal transition and deregulated expression of specific miRNAs (143). The possible link between Rb family and deregulated miRNA expression has also been reported in other tissues, such as muscle and retina (144, 145), and also upon HPV E7 expression in human keratinocytes (24). Remarkably, the specific upregulation of miR-21 dependent in p53-deficient metastatic tumor cells, which appears to be mediated by increased mTOR and Stat3 activity (143), was also found in human metastatic lung tumors bearing mutated p53 gene (143), thus supporting a potential mechanism of metastatic spreading in human tumors. Nonetheless, the possible relative contribution of Rb1 and Trp53 tumor suppressor genes to this process remains unsolved.
The cyclin-cdk inhibitor p21 is a bona fide transcriptional target of p53 (146, 147). It has been reported as responsible for the potential mechanism mediating cell cycle arrest in the absence of pRb (148–150), and it has been also involved in epidermal homeostasis and carcinogenesis (151–157). Consequently, we have recently generated mice lacking p21 in the absence of epidermal pRb. Remarkably these mice developed skin inflammatory processes followed by spontaneous tumor development, with strong resemblance to human head and neck SCCs by histopathology and transcriptome characteristics (124). Further, these mice also showed aberrant Stat3 signaling, thus reinforcing our previous findings connecting pRb, p53, and Stat3 (143).
Most sporadic cancers are originated by several mutations, including loss of function of tumor suppressors, occurring only in very few cells of adult tissues. To mimic such situation Sage and co-workers developed an experimental approach that allows Rb1 elimination in an acute manner in primary mouse embryonic fibroblasts (158). This revealed a significant functional difference between such acute loss of pRb and that achieved by inactivation during embryonic development (158). Importantly, one of the main molecular bases of such difference relies on the acclimatization of the tissues to a permanent loss of pRb that allows the induction of compensatory genes, such as p107 (158). Remarkably, the proliferative arrest mediated by differentiation stimuli in keratinocytes in vitro also displayed similar characteristics upon acute or chronic Rb1 loss (121). These results might have important implications for the interpretation of Rb-loss in GEMMs, and in particular in the context of epidermal tumor development.
To find out possible differences between acute and chronic Rb1 loss in adult mice, we have recently generated a new mouse model lacking pRb in epidermis in an inducible manner by topical tamoxifen administration (125, 126). Acute pRb deletion in epidermis in vivo caused moderate hyperplasia due to changes in epidermal proliferation and differentiation similar to those observed after chronic pRb loss (116). This phenotype is durable along mice lifespan (125), indicative of efficient targeting in adult stem cells. However, no tumor development was observed even 1 year after recombination induction (125). These observations indicated that, although the acute loss of pRb is sufficient to overcome some processes associated to prolonged pRb ablation, the extended time required for the development of spontaneous tumors allows the induction of compensatory genes and thus tumor suppression.
In order to analyze whether p107 is responsible for this tumor suppression, we generated a mouse model susceptible of acute pRb loss in the absence of p107 (125). These mice displayed abnormal epidermal differentiation, hair loss, frail appearance, and lesions in the cheek, neck, eyelids, and snout, leading to lifespan reduction up to 6 months after recombination induction. Moreover, all mice developed spontaneous tumors starting from 2 months after recombination and affecting preferentially perioral areas (125). Biochemical and genomic analyses of these tumors and primary keratinocytes demonstrated that combined pRb and p107 absence limits the transcriptional tumor suppressive functions of p53, leading to a reduced Pten gene expression. Consequently, this mouse model confirmed the existence of an in vivo novel functional connection between the three major suppressor genes TP53, Pten, and Rb1, previously suggested from in vitro experiments (159). Similarly, Lambert and colleagues have reported that loss of pRb and p107 can predispose to oral tumors in mice after chemical carcinogenesis (160). However, the inactivation of all three family members was unable to induce cervical cancer (161). These findings support a tissue context role for the Rb family in tumor suppression in stratified epithelia, and the different results observed in distinct mouse strains may also point to possible genetic and/or epigenetic factors affecting their tumor suppressor functions.
The loss of pRb is accompanied by increased E2F activity and deregulated expression of several E2F members, including increased E2F1 expression (116). E2F1 is the best described E2F family member. Although initially considered as a potential oncogene due to its ability to drive S-phase progression in quiescent cells (162, 163), data obtained in E2F1-null mice (82, 83) revealed a potential tumor suppressor role associated with impaired apoptosis induction (82, 83). Such apoptosis takes place through p53-dependent and -independent mechanisms (119, 164, 165). Such functional difference is also emphasized in epidermal carcinogenesis. E2F1 overexpression in transgenic mouse epidermis leads to spontaneous tumor development, which is accelerated by p53 loss (166, 167). However, such transgenic expression of E2F1 was found to inhibit ras-driven skin carcinogenesis in part through a p53/p19arf dependent process (114, 168, 169). This process is also reminiscent to that observed upon chemical carcinogenesis protocols in mice lacking epidermal pRb (119). Importantly, loss of E2F1 reduces tumorigenesis and extends the lifespan of Rb1 heterozygous mice indicating that an important part of the several tumor suppressor activities of pRb depend on its ability to repress E2F1 (170, 171). Of note, E2F1 is dispensable for the normal skin development and homeostasis, but plays important roles during wound healing in vivo (107). As the elimination of Rb1 in epidermis leads to increased E2F1 expression and activity (116), which also partially explain the abnormal susceptibility to chemical carcinogenesis protocols (119), we obtained a mouse model of epidermal inducible Rb1 loss in an E2F1-deficient background (126). The epidermis of mice lacking Rb1 and E2F1 was characterized by generalized hyperplasia and abnormal differentiation similar to that observed in mice under acute or chronic epidermal Rb1 loss (126). This suggests that some of the functions of Rb1 in epidermis are E2F1-independent. Nonetheless, the combined deficiency in Rb1 and E2F1 results in spontaneous epidermal carcinomas with high penetrance (126). These tumors displayed a hair follicle origin and a functional p19/p53 axis (126). Whole transcriptome analysis of these tumors revealed a potential involvement of Wnt signaling, as well as a significant overlap with human tumors (126). These results demonstrated, for the first time, that tumor suppressor functions of pRb in vivo are partially E2F1-dependent in specific tissues.
The retinoblastoma pathway is an essential mechanism allowing proper cell cycle progression. Therefore, not surprisingly, most human tumors show aberrant disruption of this pathway rather than mutation in Rb1 per se. Nonetheless, it is composed by a large number of different proteins with specific and diverse functions, and the different tumor types usually show distinct type of alterations. The analysis of mouse models bearing different combinations of genetic alterations in a tissue-specific manner, and in an acute or chronic mode, will continue to provide invaluable information on the retinoblastoma pathway and its involvement in the control of different cancer hallmarks. Such studies may highlight the significant events and the molecular entities involved in initiation and progression of the disease in different tissue contexts. In skin, the findings on the retinoblastoma family proteins and the Rb-dependent pathway support this type of studies for specific squamous cancers. Nonetheless, similar or different molecular events may take place in other tissues, and it is essential to consider that tumor development may differ in mice or humans. These factors require extremely refined comparative studies to validate the models in order to identify potential biomarkers and/or molecular targets for new therapies. Nonetheless, these mouse models could provide a highly valuable tool for preclinical research.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We are indebted to all members (past and present) of the Molecular Oncology Unit of the CIEMAT for their work, suggestions, comments, and helpful discussions and to the multiple excellent external collaborators that have allowed our research throughout the recent years. This work has been partially supported by Grants: MINECO grants SAF2011-26122-C02-01 and SAF2012-34378, CAM Oncocycle Program Grants S2010/BMD-2470, ISCIII-RETIC grants RD06/0020/0029 and RD12/0036/0009 to Jesús M. Paramio and ISCIII Grant PI12/01959 to Mirentxu Santos.
1. Capecchi MR. Targeted gene replacement. Sci Am (1994) 270(3):52–9. doi: 10.1038/scientificamerican0394-52
2. Nagy A. Cre recombinase: the universal reagent for genome tailoring. Genesis (2000) 26(2):99–109. doi:10.1002/(SICI)1526-968X(200002)26:2<99::AID-GENE1>3.3.CO;2-2
3. Berns A. Tumour suppressors: timing will tell. Nature (2003) 424(6945):140–1. doi:10.1038/424140a
4. Littlewood TD, Hancock DC, Danielian PS, Parker MG, Evan GI. A modified oestrogen receptor ligand-binding domain as an improved switch for the regulation of heterologous proteins. Nucleic Acids Res (1995) 23(10):1686–90. doi:10.1093/nar/23.10.1686
5. Ramirez A, Bravo A, Jorcano JL, Vidal M. Sequences 5′ of the bovine keratin 5 gene direct tissue- and cell-type-specific expression of a lacZ gene in the adult and during development. Differentiation (1994) 58(1):53–64. doi:10.1007/s002580050065
6. Ramirez A, Vidal M, Bravo A, Larcher F, Jorcano JL. A 5′-upstream region of a bovine keratin 6 gene confers tissue-specific expression and hyperproliferation-related induction in transgenic mice. Proc Natl Acad Sci U S A (1995) 92(11):4783–7. doi:10.1073/pnas.92.11.4783
7. Sinha S, Fuchs E. Identification and dissection of an enhancer controlling epithelial gene expression in skin. Proc Natl Acad Sci U S A (2001) 98(5):2455–60. doi:10.1073/pnas.051633598
8. Jonkers J, Meuwissen R, van der Gulden H, Peterse H, van der Valk M, Berns A. Synergistic tumor suppressor activity of BRCA2 and p53 in a conditional mouse model for breast cancer. Nat Genet (2001) 29(4):418–25. doi:10.1038/ng747
9. Vasioukhin V, Degenstein L, Wise B, Fuchs E. The magical touch: genome targeting in epidermal stem cells induced by tamoxifen application to mouse skin. Proc Natl Acad Sci U S A (1999) 96(15):8551–6. doi:10.1073/pnas.96.15.8551
10. Fuchs E, Merrill BJ, Jamora C, DasGupta R. At the roots of a never-ending cycle. Dev Cell (2001) 1(1):13–25. doi:10.1016/S1534-5807(01)00022-3
11. Watt FM. Stem cell fate and patterning in mammalian epidermis. Curr Opin Genet Dev (2001) 11(4):410–7.
13. Lorz C, Segrelles C, Paramio JM. On the origin of epidermal cancers. Curr Mol Med (2009) 9(3):353–64. doi:10.2174/156652409787847227
14. Beck B, Blanpain C. Mechanisms regulating epidermal stem cells. EMBO J (2012) 31(9):2067–75. doi:10.1038/emboj.2012.67
15. Blanpain C, Fuchs E. Epidermal stem cells of the skin. Annu Rev Cell Dev Biol (2006) 22:339–73. doi:10.1146/annurev.cellbio.22.010305.104357
16. Lapouge G, Youssef KK, Vokaer B, Achouri Y, Michaux C, Sotiropoulou PA, et al. Identifying the cellular origin of squamous skin tumors. Proc Natl Acad Sci U S A (2012) 108(18):7431–6. doi:10.1073/pnas.1012720108
17. da Silva-Diz V, Sole-Sanchez S, Valdes-Gutierrez A, Urpi M, Riba-Artes D, Penin RM, et al. Progeny of Lgr5-expressing hair follicle stem cell contributes to papillomavirus-induced tumor development in epidermis. Oncogene (2013) 32(32):3732–43. doi:10.1038/onc.2012.375
18. Blanpain C, Lowry WE, Geoghegan A, Polak L, Fuchs E. Self-renewal, multipotency, and the existence of two cell populations within an epithelial stem cell niche. Cell (2004) 118(5):635–48. doi:10.1016/j.cell.2004.08.012
19. Tumbar T, Guasch G, Greco V, Blanpain C, Lowry WE, Rendl M, et al. Defining the epithelial stem cell niche in skin. Science (2004) 303(5656):359–63. doi:10.1126/science.1092436
20. Hennings H, Michael D, Cheng C, Steinert P, Holbrook K, Yuspa SH. Calcium regulation of growth and differentiation of mouse epidermal cells in culture. Cell (1980) 19(1):245–54. doi:10.1016/0092-8674(80)90406-7
21. Yuspa SH, Kilkenny AE, Steinert PM, Roop DR. Expression of murine epidermal differentiation markers is tightly regulated by restricted extracellular calcium concentrations in vitro. J Cell Biol (1989) 109(3):1207–17. doi:10.1083/jcb.109.3.1207
22. Llames SG, Del Rio M, Larcher F, Garcia E, Garcia M, Escamez MJ, et al. Human plasma as a dermal scaffold for the generation of a completely autologous bioengineered skin. Transplantation (2004) 77(3):350–5. doi:10.1097/01.TP.0000112381.80964.85
23. Del Rio M, Larcher F, Serrano F, Meana A, Munoz M, Garcia M, et al. A preclinical model for the analysis of genetically modified human skin in vivo. Hum Gene Ther (2002) 13(8):959–68. doi:10.1089/10430340252939069
24. Buitrago-Perez A, Hachimi M, Duenas M, Lloveras B, Santos A, Holguin A, et al. A humanized mouse model of HPV-associated pathology driven by E7 expression. PLoS One (2012) 7(7):e41743. doi:10.1371/journal.pone.0041743
25. Martinez-Cruz AB, Costa C, Saiz C, Paramio JM, Santos M. In vivo transplantation of genetically modified mouse embryonic epidermis. Methods Mol Biol (2010) 585:361–7. doi:10.1007/978-1-60761-380-0_25
26. Slaga TJ, Budunova IV, Gimenez-Conti IB, Aldaz CM. The mouse skin carcinogenesis model. J Investig Dermatol Symp Proc (1996) 1(2):151–6.
27. Schwarz M, Munzel PA, Braeuning A. Non-melanoma skin cancer in mouse and man. Arch Toxicol (2012) 87(5):783–98. doi:10.1007/s00204-012-0998-9
28. Friend SH, Bernards R, Rogelj S, Weinberg RA, Rapaport JM, Albert DM, et al. A human DNA segment with properties of the gene that predisposes to retinoblastoma and osteosarcoma. Nature (1986) 323(6089):643–6. doi:10.1038/323643a0
29. Ewen ME, Xing YG, Lawrence JB, Livingston DM. Molecular cloning, chromosomal mapping, and expression of the cDNA for p107, a retinoblastoma gene product-related protein. Cell (1991) 66(6):1155–64. doi:10.1016/0092-8674(91)90038-Z
30. Yeung RS, Bell DW, Testa JR, Mayol X, Baldi A, Grana X, et al. The retinoblastoma-related gene, RB2, maps to human chromosome 16q12 and rat chromosome 19. Oncogene (1993) 8(12):3465–8.
31. Mayol X, Grana X, Baldi A, Sang N, Hu Q, Giordano A. Cloning of a new member of the retinoblastoma gene family (pRb2) which binds to the E1A transforming domain. Oncogene (1993) 8(9):2561–6.
33. Chan HM, Smith L, La Thangue NB. Role of LXCXE motif-dependent interactions in the activity of the retinoblastoma protein. Oncogene (2001) 20(43):6152–63. doi:10.1038/sj.onc.1204793
34. Weinberg RA. The retinoblastoma protein and cell cycle control. Cell (1995) 81(3):323–30. doi:10.1016/0092-8674(95)90385-2
35. Lamber EP, Beuron F, Morris EP, Svergun DI, Mittnacht S. Structural insights into the mechanism of phosphoregulation of the retinoblastoma protein. PLoS One (2013) 8(3):e58463. doi:10.1371/journal.pone.0058463
36. Burke JR, Hura GL, Rubin SM. Structures of inactive retinoblastoma protein reveal multiple mechanisms for cell cycle control. Genes Dev (2012) 26(11):1156–66. doi:10.1101/gad.189837.112
37. Classon M, Dyson N. p107 and p130: versatile proteins with interesting pockets. Exp Cell Res (2001) 264(1):135–47. doi:10.1006/excr.2000.5135
38. Ewen ME, Faha B, Harlow E, Livingston DM. Interaction of p107 with cyclin A independent of complex formation with viral oncoproteins. Science (1992) 255(5040):85–7. doi:10.1126/science.1532457
39. Faha B, Ewen ME, Tsai LH, Livingston DM, Harlow E. Interaction between human cyclin A and adenovirus E1A-associated p107 protein. Science (1992) 255(5040):87–90. doi:10.1126/science.1532458
40. Hannon GJ, Demetrick D, Beach D. Isolation of the Rb-related p130 through its interaction with CDK2 and cyclins. Genes Dev (1993) 7(12A):2378–91. doi:10.1101/gad.7.12a.2378
41. Malumbres M, Barbacid M. Cell cycle, CDKs and cancer: a changing paradigm. Nat Rev Cancer (2009) 9(3):153–66. doi:10.1038/nrc2602
42. Harper JW, Elledge SJ. Cdk inhibitors in development and cancer. Curr Opin Genet Dev (1996) 6(1):56–64.
43. Henley SA, Dick FA. The retinoblastoma family of proteins and their regulatory functions in the mammalian cell division cycle. Cell Div (2013) 7(1):10. doi:10.1186/1747-1028-7-10
44. Dick FA, Rubin SM. Molecular mechanisms underlying RB protein function. Nat Rev Mol Cell Biol (2013) 14(5):297–306. doi:10.1038/nrm3567
45. Frolov MV, Dyson NJ. Molecular mechanisms of E2F-dependent activation and pRB-mediated repression. J Cell Sci (2004) 117(Pt 11):2173–81. doi:10.1242/jcs.01227
46. McClellan KA, Slack RS. Specific in vivo roles for E2Fs in differentiation and development. Cell Cycle (2007) 6(23):2917–27. doi:10.4161/cc.6.23.4997
47. Moberg K, Starz MA, Lees JA. E2F-4 switches from p130 to p107 and pRB in response to cell cycle reentry. Mol Cell Biol (1996) 16(4):1436–49.
48. Hurford RK Jr, Cobrinik D, Lee MH, Dyson N. pRB and p107/p130 are required for the regulated expression of different sets of E2F responsive genes. Genes Dev (1997) 11(11):1447–63. doi:10.1101/gad.11.11.1447
49. Condorelli GL, Testa U, Valtieri M, Vitelli L, De Luca A, Barberi T, et al. Modulation of retinoblastoma gene in normal adult hematopoiesis: peak expression and functional role in advanced erythroid differentiation. Proc Natl Acad Sci U S A (1995) 92(11):4808–12. doi:10.1073/pnas.92.11.4808
50. Khidr L, Chen PL. RB, the conductor that orchestrates life, death and differentiation. Oncogene (2006) 25(38):5210–9. doi:10.1038/sj.onc.1209612
51. Hilgendorf KI, Leshchiner ES, Nedelcu S, Maynard MA, Calo E, Ianari A, et al. The retinoblastoma protein induces apoptosis directly at the mitochondria. Genes Dev (2013) 27(9):1003–15. doi:10.1101/gad.211326.112
52. Gabellini C, Del Bufalo D, Zupi G. Involvement of RB gene family in tumor angiogenesis. Oncogene (2006) 25(38):5326–32. doi:10.1038/sj.onc.1209631
53. Chicas A, Wang X, Zhang C, McCurrach M, Zhao Z, Mert O, et al. Dissecting the unique role of the retinoblastoma tumor suppressor during cellular senescence. Cancer Cell (2010) 17(4):376–87. doi:10.1016/j.ccr.2010.01.023
54. Claudio PP, Stiegler P, Howard CM, Bellan C, Minimo C, Tosi GM, et al. RB2/p130 gene-enhanced expression down-regulates vascular endothelial growth factor expression and inhibits angiogenesis in vivo. Cancer Res (2001) 61(2):462–8.
55. Trimarchi JM, Lees JA. Sibling rivalry in the E2F family. Nat Rev Mol Cell Biol (2002) 3(1):11–20. doi:10.1038/nrm714
56. Luo RX, Postigo AA, Dean DC. Rb interacts with histone deacetylase to repress transcription. Cell (1998) 92(4):463–73. doi:10.1016/S0092-8674(00)80940-X
57. Magnaghi-Jaulin L, Groisman R, Naguibneva I, Robin P, Lorain S, Le Villain JP, et al. Retinoblastoma protein represses transcription by recruiting a histone deacetylase. Nature (1998) 391(6667):601–5. doi:10.1038/35410
58. Brehm A, Miska EA, McCance DJ, Reid JL, Bannister AJ, Kouzarides T. Retinoblastoma protein recruits histone deacetylase to repress transcription. Nature (1998) 391(6667):597–601. doi:10.1038/35404
59. Benevolenskaya EV, Murray HL, Branton P, Young RA, Kaelin WG Jr. Binding of pRB to the PHD protein RBP2 promotes cellular differentiation. Mol Cell (2005) 18(6):623–35. doi:10.1016/j.molcel.2005.05.012
60. Robertson KD, Ait-Si-Ali S, Yokochi T, Wade PA, Jones PL, Wolffe AP. DNMT1 forms a complex with Rb, E2F1 and HDAC1 and represses transcription from E2F-responsive promoters. Nat Genet (2000) 25(3):338–42. doi:10.1038/77124
61. Dunaief JL, Strober BE, Guha S, Khavari PA, Alin K, Luban J, et al. The retinoblastoma protein and BRG1 form a complex and cooperate to induce cell cycle arrest. Cell (1994) 79(1):119–30. doi:10.1016/0092-8674(94)90405-7
62. Singh P, Coe J, Hong W. A role for retinoblastoma protein in potentiating transcriptional activation by the glucocorticoid receptor. Nature (1995) 374(6522):562–5. doi:10.1038/374562a0
63. Nielsen SJ, Schneider R, Bauer UM, Bannister AJ, Morrison A, O’Carroll D, et al. Rb targets histone H3 methylation and HP1 to promoters. Nature (2001) 412(6846):561–5. doi:10.1038/35087620
64. Gonzalo S, Blasco MA. Role of Rb family in the epigenetic definition of chromatin. Cell Cycle (2005) 4(6):752–5. doi:10.4161/cc.4.6.1720
65. Steele-Perkins G, Fang W, Yang XH, Van Gele M, Carling T, Gu J, et al. Tumor formation and inactivation of RIZ1, an Rb-binding member of a nuclear protein-methyltransferase superfamily. Genes Dev (2001) 15(17):2250–62. doi:10.1101/gad.870101
66. Vandel L, Nicolas E, Vaute O, Ferreira R, Ait-Si-Ali S, Trouche D. Transcriptional repression by the retinoblastoma protein through the recruitment of a histone methyltransferase. Mol Cell Biol (2001) 21(19):6484–94. doi:10.1128/MCB.21.19.6484-6494.2001
67. Talluri S, Dick FA. Regulation of transcription and chromatin structure by pRB: here, there and everywhere. Cell Cycle (2012) 11(17):3189–98. doi:10.4161/cc.21263
68. Hanahan D, Weinberg RA. The hallmarks of cancer. Cell (2000) 100(1):57–70. doi:10.1016/S0092-8674(00)81683-9
69. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell (2011) 144(5):646–74. doi:10.1016/j.cell.2011.02.013
70. Helin K, Holm K, Niebuhr A, Eiberg H, Tommerup N, Hougaard S, et al. Loss of the retinoblastoma protein-related p130 protein in small cell lung carcinoma. Proc Natl Acad Sci U S A (1997) 94(13):6933–8. doi:10.1073/pnas.94.13.6933
71. Claudio PP, Howard CM, Pacilio C, Cinti C, Romano G, Minimo C, et al. Mutations in the retinoblastoma-related gene RB2/p130 in lung tumors and suppression of tumor growth in vivo by retrovirus-mediated gene transfer. Cancer Res (2000) 60(2):372–82.
72. Ikeda MA, Jakoi L, Nevins JR. A unique role for the Rb protein in controlling E2F accumulation during cell growth and differentiation. Proc Natl Acad Sci U S A (1996) 93(8):3215–20. doi:10.1073/pnas.93.8.3215
73. Clarke AR, Maandag ER, van Roon M, van der Lugt NM, van der Valk M, Hooper ML, et al. Requirement for a functional Rb-1 gene in murine development. Nature (1992) 359(6393):328–30. doi:10.1038/359328a0
74. Jacks T, Fazeli A, Schmitt EM, Bronson RT, Goodell MA, Weinberg RA. Effects of an Rb mutation in the mouse. Nature (1992) 359(6393):295–300. doi:10.1038/359295a0
75. Lee EY, Chang CY, Hu N, Wang YC, Lai CC, Herrup K, et al. Mice deficient for Rb are nonviable and show defects in neurogenesis and haematopoiesis. Nature (1992) 359(6393):288–94. doi:10.1038/359288a0
76. Williams BO, Remington L, Albert DM, Mukai S, Bronson RT, Jacks T. Cooperative tumorigenic effects of germline mutations in Rb and p53. Nat Genet (1994) 7(4):480–4. doi:10.1038/ng0894-480
77. Hu N, Gutsmann A, Herbert DC, Bradley A, Lee WH, Lee EY. Heterozygous Rb-1 delta 20/+mice are predisposed to tumors of the pituitary gland with a nearly complete penetrance. Oncogene (1994) 9(4):1021–7.
78. Harrison DJ, Hooper ML, Armstrong JF, Clarke AR. Effects of heterozygosity for the Rb-1t19neo allele in the mouse. Oncogene (1995) 10(8):1615–20.
79. Lee MH, Williams BO, Mulligan G, Mukai S, Bronson RT, Dyson N, et al. Targeted disruption of p107: functional overlap between p107 and Rb. Genes Dev (1996) 10(13):1621–32. doi:10.1101/gad.10.13.1621
80. Cobrinik D, Lee MH, Hannon G, Mulligan G, Bronson RT, Dyson N, et al. Shared role of the pRB-related p130 and p107 proteins in limb development. Genes Dev (1996) 10(13):1633–44. doi:10.1101/gad.10.13.1633
81. Ruiz S, Segrelles C, Bravo A, Santos M, Perez P, Leis H, et al. Abnormal epidermal differentiation and impaired epithelial-mesenchymal tissue interactions in mice lacking the retinoblastoma relatives p107 and p130. Development (2003) 130(11):2341–53. doi:10.1242/dev.00453
82. Field SJ, Tsai FY, Kuo F, Zubiaga AM, Kaelin WG Jr, Livingston DM, et al. E2F-1 functions in mice to promote apoptosis and suppress proliferation. Cell (1996) 85(4):549–61. doi:10.1016/S0092-8674(00)81255-6
83. Yamasaki L, Jacks T, Bronson R, Goillot E, Harlow E, Dyson NJ. Tumor induction and tissue atrophy in mice lacking E2F-1. Cell (1996) 85(4):537–48. doi:10.1016/S0092-8674(00)81254-4
84. Yamasaki L. Growth regulation by the E2F and DP transcription factor families. Results Probl Cell Differ (1998) 22:199–227.
85. Murga M, Fernandez-Capetillo O, Field SJ, Moreno B, Borlado LR, Fujiwara Y, et al. Mutation of E2F2 in mice causes enhanced T lymphocyte proliferation, leading to the development of autoimmunity. Immunity (2001) 15(6):959–70. doi:10.1016/S1074-7613(01)00254-0
86. Humbert PO, Verona R, Trimarchi JM, Rogers C, Dandapani S, Lees JA. E2f3 is critical for normal cellular proliferation. Genes Dev (2000) 14(6):690–703.
87. Cloud JE, Rogers C, Reza TL, Ziebold U, Stone JR, Picard MH, et al. Mutant mouse models reveal the relative roles of E2F1 and E2F3 in vivo. Mol Cell Biol (2002) 22(8):2663–72. doi:10.1128/MCB.22.8.2663-2672.2002
88. Wu L, Timmers C, Maiti B, Saavedra HI, Sang L, Chong GT, et al. The E2F1-3 transcription factors are essential for cellular proliferation. Nature (2001) 414(6862):457–62. doi:10.1038/35106593
89. Ziebold U, Lee EY, Bronson RT, Lees JA. E2F3 loss has opposing effects on different pRB-deficient tumors, resulting in suppression of pituitary tumors but metastasis of medullary thyroid carcinomas. Mol Cell Biol (2003) 23(18):6542–52. doi:10.1128/MCB.23.18.6542-6552.2003
90. Danielian PS, Friesenhahn LB, Faust AM, West JC, Caron AM, Bronson RT, et al. E2f3a and E2f3b make overlapping but different contributions to total E2f3 activity. Oncogene (2008) 27(51):6561–70. doi:10.1038/onc.2008.253
91. Kohn MJ, Bronson RT, Harlow E, Dyson NJ, Yamasaki L. Dp1 is required for extra-embryonic development. Development (2003) 130(7):1295–305. doi:10.1242/dev.00355
92. Maandag EC, van der Valk M, Vlaar M, Feltkamp C, O’Brien J, van Roon M, et al. Developmental rescue of an embryonic-lethal mutation in the retinoblastoma gene in chimeric mice. EMBO J (1994) 13(18):4260–8.
93. Dannenberg JH, Schuijff L, Dekker M, van der Valk M, Riele HT. Tissue-specific tumor suppressor activity of retinoblastoma gene homologs p107 and p130. Genes Dev (2004) 18(23):2952–62. doi:10.1101/gad.322004
94. Parisi T, Yuan TL, Faust AM, Caron AM, Bronson R, Lees JA. Selective requirements for E2f3 in the development and tumorigenicity of Rb-deficient chimeric tissues. Mol Cell Biol (2007) 27(6):2283–93. doi:10.1128/MCB.01854-06
95. Parisi T, Bronson RT, Lees JA. Inhibition of pituitary tumors in Rb mutant chimeras through E2f4 loss reveals a key suppressive role for the pRB/E2F pathway in urothelium and ganglionic carcinogenesis. Oncogene (2009) 28(4):500–8. doi:10.1038/onc.2008.406
96. Mulligan G, Jacks T. The retinoblastoma gene family: cousins with overlapping interests. Trends Genet (1998) 14(6):223–9. doi:10.1016/S0168-9525(98)01470-X
97. Sage J, Mulligan GJ, Attardi LD, Miller A, Chen S, Williams B, et al. Targeted disruption of the three Rb-related genes leads to loss of G(1) control and immortalization. Genes Dev (2000) 14(23):3037–50. doi:10.1101/gad.843200
98. Dannenberg JH, van Rossum A, Schuijff L, te Riele H. Ablation of the retinoblastoma gene family deregulates G(1) control causing immortalization and increased cell turnover under growth-restricting conditions. Genes Dev (2000) 14(23):3051–64. doi:10.1101/gad.847700
99. Wirt SE, Sage J. p107 in the public eye: an Rb understudy and more. Cell Div (2010) 5:9. doi:10.1186/1747-1028-5-9
100. Cobrinik D. Regulatory interactions among E2Fs and cell cycle control proteins. Curr Top Microbiol Immunol (1996) 208:31–61.
101. Robanus-Maandag E, Dekker M, van der Valk M, Carrozza ML, Jeanny JC, Dannenberg JH, et al. p107 is a suppressor of retinoblastoma development in pRb-deficient mice. Genes Dev (1998) 12(11):1599–609. doi:10.1101/gad.12.11.1599
102. Lee EY, Cam H, Ziebold U, Rayman JB, Lees JA, Dynlacht BD. E2F4 loss suppresses tumorigenesis in Rb mutant mice. Cancer Cell (2002) 2(6):463–72. doi:10.1016/S1535-6108(02)00207-6
103. Mulligan GJ, Wong J, Jacks T. p130 is dispensable in peripheral T lymphocytes: evidence for functional compensation by p107 and pRB. Mol Cell Biol (1998) 18(1):206–20.
104. Martinez LA, Chen Y, Fischer SM, Conti CJ. Coordinated changes in cell cycle machinery occur during keratinocyte terminal differentiation. Oncogene (1999) 18(2):397–406. doi:10.1038/sj.onc.1202300
105. Paramio JM, Lain S, Segrelles C, Lane EB, Jorcano JL. Differential expression and functionally co-operative roles for the retinoblastoma family of proteins in epidermal differentiation. Oncogene (1998) 17(8):949–57. doi:10.1038/sj.onc.1202031
106. D’Souza SJ, Pajak A, Balazsi K, Dagnino L. Ca2+ and BMP-6 signaling regulate E2F during epidermal keratinocyte differentiation. J Biol Chem (2001) 276(26):23531–8. doi:10.1074/jbc.M100780200
107. D’Souza SJ, Vespa A, Murkherjee S, Maher A, Pajak A, Dagnino L. E2F-1 is essential for normal epidermal wound repair. J Biol Chem (2002) 277(12):10626–32. doi:10.1074/jbc.M111956200
108. Paramio JM, Segrelles C, Casanova ML, Jorcano JL. Opposite functions for E2F1 and E2F4 in human epidermal keratinocyte differentiation. J Biol Chem (2000) 275(52):41219–26. doi:10.1074/jbc.M004973200
109. Rodriguez-Puebla ML, de Marval PL, LaCava M, Moons DS, Kiyokawa H, Conti CJ. Cdk4 deficiency inhibits skin tumor development but does not affect normal keratinocyte proliferation. Am J Pathol (2002) 161(2):405–11. doi:10.1016/S0002-9440(10)64196-X
110. Miliani de Marval PL, Macias E, Conti CJ, Rodriguez-Puebla ML. Enhanced malignant tumorigenesis in Cdk4 transgenic mice. Oncogene (2004) 23(10):1863–73. doi:10.1038/sj.onc.1207309
111. Sotillo R, Garcia JF, Ortega S, Martin J, Dubus P, Barbacid M, et al. Invasive melanoma in Cdk4-targeted mice. Proc Natl Acad Sci U S A (2001) 98(23):13312–7. doi:10.1073/pnas.241338598
112. Paramio JM, Segrelles C, Ruiz S, Martin-Caballero J, Page A, Martinez J, et al. The ink4a/arf tumor suppressors cooperate with p21cip1/waf in the processes of mouse epidermal differentiation, senescence, and carcinogenesis. J Biol Chem (2001) 276(47):44203–11. doi:10.1074/jbc.M105650200
113. Wang D, Russell J, Xu H, Johnson DG. Deregulated expression of DP1 induces epidermal proliferation and enhances skin carcinogenesis. Mol Carcinog (2001) 31(2):90–100. doi:10.1002/mc.1044
114. Pierce AM, Schneider-Broussard R, Gimenez-Conti IB, Russell JL, Conti CJ, Johnson DG. E2F1 has both oncogenic and tumor-suppressive properties in a transgenic model. Mol Cell Biol (1999) 19(9):6408–14.
115. Chu C, Qu K, Zhong FL, Artandi SE, Chang HY. Genomic maps of long noncoding RNA occupancy reveal principles of RNA-chromatin interactions. Mol Cell (2011) 44(4):667–78. doi:10.1016/j.molcel.2011.08.027
116. Ruiz S, Segrelles C, Santos M, Lara MF, Paramio JM. Functional link between retinoblastoma family of proteins and the Wnt signaling pathway in mouse epidermis. Dev Dyn (2004) 230(3):410–8. doi:10.1002/dvdy.20065
117. Conklin JF, Baker J, Sage J. The RB family is required for the self-renewal and survival of human embryonic stem cells. Nat Commun (2012) 3:1244. doi:10.1038/ncomms2254
118. Lorz C, Garcia-Escudero R, Segrelles C, Garin MI, Ariza JM, Santos M, et al. A functional role of RB-dependent pathway in the control of quiescence in adult epidermal stem cells revealed by genomic profiling. Stem Cell Rev (2010) 6(2):162–77. doi:10.1007/s12015-010-9139-0
119. Ruiz S, Santos M, Lara MF, Segrelles C, Ballestin C, Paramio JM. Unexpected roles for pRb in mouse skin carcinogenesis. Cancer Res (2005) 65(21):9678–86. doi:10.1158/0008-5472.CAN-05-1853
120. Lara MF, Garcia-Escudero R, Ruiz S, Santos M, Moral M, Martinez-Cruz AB, et al. Gene profiling approaches help to define the specific functions of retinoblastoma family in epidermis. Mol Carcinog (2008) 47(3):209–21. doi:10.1002/mc.20376
121. Ruiz S, Santos M, Segrelles C, Leis H, Jorcano JL, Berns A, et al. Unique and overlapping functions of pRb and p107 in the control of proliferation and differentiation in epidermis. Development (2004) 131(11):2737–48. doi:10.1242/dev.01148
122. Lara MF, Santos M, Ruiz S, Segrelles C, Moral M, Martinez-Cruz AB, et al. p107 acts as a tumor suppressor in pRb-deficient epidermis. Mol Carcinog (2008) 47(2):105–13. doi:10.1002/mc.20367
123. Martinez-Cruz AB, Santos M, Lara MF, Segrelles C, Ruiz S, Moral M, et al. Spontaneous squamous cell carcinoma induced by the somatic inactivation of retinoblastoma and Trp53 tumor suppressors. Cancer Res (2008) 68(3):683–92. doi:10.1158/0008-5472.CAN-07-3049
124. Saiz-Ladera C, Lara MF, Garin M, Ruiz S, Santos M, Lorz C, et al. p21 suppresses inflammation and tumorigenesis on pRB-deficient stratified epithelia. Oncogene (2013). doi:10.1038/onc.2013.417
125. Costa C, Santos M, Segrelles C, Duenas M, Lara MF, Agirre X, et al. A novel tumor suppressor network in squamous malignancies. Sci Rep (2012) 2:828. doi:10.1038/srep00828
126. Costa C, Santos M, Martinez-Fernandez M, Duenas M, Lorz C, Garcia-Escudero R, et al. E2F1 loss induces spontaneous tumour development in Rb-deficient epidermis. Oncogene (2013) 32(24):2937–51. doi:10.1038/onc.2012.316
127. Balsitis SJ, Sage J, Duensing S, Munger K, Jacks T, Lambert PF. Recapitulation of the effects of the human papillomavirus type 16 E7 oncogene on mouse epithelium by somatic Rb deletion and detection of pRb-independent effects of E7 in vivo. Mol Cell Biol (2003) 23(24):9094–103. doi:10.1128/MCB.23.24.9094-9103.2003
128. Black EP, Huang E, Dressman H, Rempel R, Laakso N, Asa SL, et al. Distinct gene expression phenotypes of cells lacking Rb and Rb family members. Cancer Res (2003) 63(13):3716–23.
129. Pippa R, Espinosa L, Gundem G, Garcia-Escudero R, Dominguez A, Orlando S, et al. p27Kip1 represses transcription by direct interaction with p130/E2F4 at the promoters of target genes. Oncogene (2012) 31(38):4207–20. doi:10.1038/onc.2011.582
130. Ruiz S, Santos M, Paramio JM. Is the loss of pRb essential for the mouse skin carcinogenesis? Cell Cycle (2006) 5(6):625–9. doi:10.4161/cc.5.6.2580
131. Santos M, Ruiz S, Lara MF, Segrelles C, Moral M, Martinez-Cruz AB, et al. Susceptibility of pRb-deficient epidermis to chemical skin carcinogenesis is dependent on the p107 allele dosage. Mol Carcinog (2008) 47(11):815–21. doi:10.1002/mc.20426
132. Kemp CJ, Wheldon T, Balmain A. p53-deficient mice are extremely susceptible to radiation-induced tumorigenesis. Nat Genet (1994) 8(1):66–9. doi:10.1038/ng0994-66
133. Kemp CJ, Donehower LA, Bradley A, Balmain A. Reduction of p53 gene dosage does not increase initiation or promotion but enhances malignant progression of chemically induced skin tumors. Cell (1993) 74(5):813–22. doi:10.1016/0092-8674(93)90461-X
134. Wang NP, To H, Lee WH, Lee EY. Tumor suppressor activity of RB and p53 genes in human breast carcinoma cells. Oncogene (1993) 8(2):279–88.
135. Morgenbesser SD, Williams BO, Jacks T, DePinho RA. p53-dependent apoptosis produced by Rb-deficiency in the developing mouse lens. Nature (1994) 371(6492):72–4. doi:10.1038/371072a0
136. Harvey M, Vogel H, Lee EY, Bradley A, Donehower LA. Mice deficient in both p53 and Rb develop tumors primarily of endocrine origin. Cancer Res (1995) 55(5):1146–51.
137. Marino S, Vooijs M, van Der Gulden H, Jonkers J, Berns A. Induction of medulloblastomas in p53-null mutant mice by somatic inactivation of Rb in the external granular layer cells of the cerebellum. Genes Dev (2000) 14(8):994–1004.
138. Bardeesy N, Bastian BC, Hezel A, Pinkel D, DePinho RA, Chin L. Dual inactivation of RB and p53 pathways in RAS-induced melanomas. Mol Cell Biol (2001) 21(6):2144–53. doi:10.1128/MCB.21.6.2144-2153.2001
139. Rubio R, Garcia-Castro J, Gutierrez-Aranda I, Paramio J, Santos M, Catalina P, et al. Deficiency in p53 but not retinoblastoma induces the transformation of mesenchymal stem cells in vitro and initiates leiomyosarcoma in vivo. Cancer Res (2010) 70(10):4185–94. doi:10.1158/0008-5472.CAN-09-4640
140. Martinez-Cruz AB, Santos M, Garcia-Escudero R, Moral M, Segrelles C, Lorz C, et al. Spontaneous tumor formation in Trp53-deficient epidermis mediated by chromosomal instability and inflammation. Anticancer Res (2009) 29(8):3035–42.
141. Garcia-Escudero R, Martinez-Cruz AB, Santos M, Lorz C, Segrelles C, Garaulet G, et al. Gene expression profiling of mouse p53-deficient epidermal carcinoma defines molecular determinants of human cancer malignancy. Mol Cancer (2010) 9:193. doi:10.1186/1476-4598-9-193
142. Duenas M, Santos M, Aranda JF, Bielza C, Martinez-Cruz AB, Lorz C, et al. Mouse p53-deficient cancer models as platforms for obtaining genomic predictors of human cancer clinical outcomes. PLoS One (2012) 7(8):e42494. doi:10.1371/journal.pone.0042494
143. Bornachea O, Santos M, Martinez-Cruz AB, Garcia-Escudero R, Duenas M, Costa C, et al. EMT and induction of miR-21 mediate metastasis development in Trp53-deficient tumours. Sci Rep (2012) 2:434. doi:10.1038/srep00434
144. Conkrite K, Sundby M, Mukai S, Thomson JM, Mu D, Hammond SM, et al. miR-17 ∼92 cooperates with RB pathway mutations to promote retinoblastoma. Genes Dev (2011) 25(16):1734–45. doi:10.1101/gad.17027411
145. Marzi MJ, Puggioni EM, Dall’Olio V, Bucci G, Bernard L, Bianchi F, et al. Differentiation-associated microRNAs antagonize the Rb-E2F pathway to restrict proliferation. J Cell Biol (2012) 199(1):77–95. doi:10.1083/jcb.201206033
146. el-Deiry WS, Tokino T, Velculescu VE, Levy DB, Parsons R, Trent JM, et al. WAF1, a potential mediator of p53 tumor suppression. Cell (1993) 75(4):817–25. doi:10.1016/0092-8674(93)90500-P
147. Xiong Y, Hannon GJ, Zhang H, Casso D, Kobayashi R, Beach D. p21 is a universal inhibitor of cyclin kinases. Nature (1993) 366(6456):701–4. doi:10.1038/366701a0
148. Brugarolas J, Bronson RT, Jacks T. p21 is a critical CDK2 regulator essential for proliferation control in Rb-deficient cells. J Cell Biol (1998) 141(2):503–14. doi:10.1083/jcb.141.2.503
149. Brugarolas J, Moberg K, Boyd SD, Taya Y, Jacks T, Lees JA. Inhibition of cyclin-dependent kinase 2 by p21 is necessary for retinoblastoma protein-mediated G1 arrest after gamma-irradiation. Proc Natl Acad Sci U S A (1999) 96(3):1002–7. doi:10.1073/pnas.96.3.1002
150. Dimri GP, Nakanishi M, Desprez PY, Smith JR, Campisi J. Inhibition of E2F activity by the cyclin-dependent protein kinase inhibitor p21 in cells expressing or lacking a functional retinoblastoma protein. Mol Cell Biol (1996) 16(6):2987–97.
151. Philipp J, Vo K, Gurley KE, Seidel K, Kemp CJ. Tumor suppression by p27Kip1 and p21Cip1 during chemically induced skin carcinogenesis. Oncogene (1999) 18(33):4689–98. doi:10.1038/sj.onc.1202840
152. Topley GI, Okuyama R, Gonzales JG, Conti C, Dotto GP. p21(WAF1/Cip1) functions as a suppressor of malignant skin tumor formation and a determinant of keratinocyte stem-cell potential. Proc Natl Acad Sci U S A (1999) 96(16):9089–94. doi:10.1073/pnas.96.16.9089
153. Weinberg WC, Fernandez-Salas E, Morgan DL, Shalizi A, Mirosh E, Stanulis E, et al. Genetic deletion of p21WAF1 enhances papilloma formation but not malignant conversion in experimental mouse skin carcinogenesis. Cancer Res (1999) 59(9):2050–4.
154. Missero C, Di Cunto F, Kiyokawa H, Koff A, Dotto GP. The absence of p21Cip1/WAF1 alters keratinocyte growth and differentiation and promotes ras-tumor progression. Genes Dev (1996) 10(23):3065–75. doi:10.1101/gad.10.23.3065
155. Dotto GP. p21(WAF1/Cip1): more than a break to the cell cycle? Biochim Biophys Acta (2000) 1471(1):M43–56.
156. Devgan V, Nguyen BC, Oh H, Dotto GP. p21Waf1/Cip1 suppresses keratinocyte differentiation independently of the cell cycle through transcriptional upregulation of the IGF-1 gene. J Biol Chem (2006) 281(41):30463–70. doi:10.1074/jbc.M604684200
157. Di Cunto F, Topley G, Calautti E, Hsiao J, Ong L, Seth PK, et al. Inhibitory function of p21Cip1/WAF1 in differentiation of primary mouse keratinocytes independent of cell cycle control. Science (1998) 280(5366):1069–72. doi:10.1126/science.280.5366.1069
158. Sage J, Miller AL, Perez-Mancera PA, Wysocki JM, Jacks T. Acute mutation of retinoblastoma gene function is sufficient for cell cycle re-entry. Nature (2003) 424(6945):223–8. doi:10.1038/nature01764
159. Paramio JM, Navarro M, Segrelles C, Gomez-Casero E, Jorcano JL. PTEN tumour suppressor is linked to the cell cycle control through the retinoblastoma protein. Oncogene (1999) 18(52):7462–8. doi:10.1038/sj.onc.1203151
160. Shin MK, Pitot HC, Lambert PF. Pocket proteins suppress head and neck cancer. Cancer Res (2012) 72(5):1280–9. doi:10.1158/0008-5472.CAN-11-2833
161. Shin MK, Sage J, Lambert PF. Inactivating all three Rb family pocket proteins is insufficient to initiate cervical cancer. Cancer Res (2012) 72(20):5418–27. doi:10.1158/0008-5472.CAN-12-2083
162. Johnson DG, Cress WD, Jakoi L, Nevins JR. Oncogenic capacity of the E2F1 gene. Proc Natl Acad Sci U S A (1994) 91(26):12823–7. doi:10.1073/pnas.91.26.12823
163. Johnson DG, Schwarz JK, Cress WD, Nevins JR. Expression of transcription factor E2F1 induces quiescent cells to enter S phase. Nature (1993) 365(6444):349–52. doi:10.1038/365349a0
164. Powers JT, Hong S, Mayhew CN, Rogers PM, Knudsen ES, Johnson DG. E2F1 uses the ATM signaling pathway to induce p53 and Chk2 phosphorylation and apoptosis. Mol Cancer Res (2004) 2(4):203–14.
165. Nahle Z, Polakoff J, Davuluri RV, McCurrach ME, Jacobson MD, Narita M, et al. Direct coupling of the cell cycle and cell death machinery by E2F. Nat Cell Biol (2002) 4(11):859–64. doi:10.1038/ncb868
166. Pierce AM, Fisher SM, Conti CJ, Johnson DG. Deregulated expression of E2F1 induces hyperplasia and cooperates with ras in skin tumor development. Oncogene (1998) 16(10):1267–76. doi:10.1038/sj.onc.1201666
167. Pierce AM, Gimenez-Conti IB, Schneider-Broussard R, Martinez LA, Conti CJ, Johnson DG. Increased E2F1 activity induces skin tumors in mice heterozygous and nullizygous for p53. Proc Natl Acad Sci U S A (1998) 95(15):8858–63. doi:10.1073/pnas.95.15.8858
168. Wikonkal NM, Remenyik E, Knezevic D, Zhang W, Liu M, Zhao H, et al. Inactivating E2f1 reverts apoptosis resistance and cancer sensitivity in Trp53-deficient mice. Nat Cell Biol (2003) 5(7):655–60. doi:10.1038/ncb1001
169. Russell JL, Weaks RL, Berton TR, Johnson DG. E2F1 suppresses skin carcinogenesis via the ARF-p53 pathway. Oncogene (2006) 25(6):867–76. doi:10.1038/sj.onc.1209120
170. Yamasaki L, Bronson R, Williams BO, Dyson NJ, Harlow E, Jacks T. Loss of E2F-1 reduces tumorigenesis and extends the lifespan of Rb1(±)mice. Nat Genet (1998) 18(4):360–4. doi:10.1038/ng0498-360
Keywords: pRb, p107, p130, epidermis, E2F, p53, SCC, transgenic mice
Citation: Costa C, Paramio JM and Santos M (2013) Skin tumors Rb(eing) uncovered. Front. Oncol. 3:307. doi: 10.3389/fonc.2013.00307
Received: 30 August 2013; Accepted: 04 December 2013;
Published online: 17 December 2013.
Edited by:
Amancio Carnero, Instituto de Biomedicina de Sevilla, SpainReviewed by:
Philipp Kaldis, Agency for Science, Technology and Research, SingaporeCopyright: © 2013 Costa, Paramio and Santos. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jesús M. Paramio and Mirentxu Santos, Molecular Oncology Unit, Department of Basic Research, Centro de Investigaciones Energéticas Medioambientales y Teconológicas (ed70A), Ave. Complutense 40, E-28040 Madrid, Spain e-mail:amVzdXNtLnBhcmFtaW9AY2llbWF0LmVz;bWlyZW50eHUuc2FudG9zQGNpZW1hdC5lcw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.