


94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 04 June 2012
Sec. Molecular and Cellular Oncology
Volume 2 - 2012 | https://doi.org/10.3389/fonc.2012.00058
This article is part of the Research Topic Radiation-induced effects and the immune system View all 14 articles
Chronic inflammation has emerged as one of the hallmarks of cancer. Inflammation also plays a pivotal role in modulating radiation responsiveness of tumors. As discussed in this review, ionizing radiation (IR) leads to activation of several transcription factors modulating the expression of numerous mediators in tumor cells and cells of the microenvironment promoting cancer development. Novel therapeutic approaches thus aim to interfere with the activity or expression of these factors, either in single-agent or combinatorial treatment or as supplements of the existing therapeutic concepts. Among them, NF-κB, STAT-3, and HIF-1 play a crucial role in radiation-induced inflammatory responses embedded in a complex inflammatory network. A great variety of classical or novel drugs including nutraceuticals such as plant phytochemicals have the capacity to interfere with the inflammatory network in cancer and are considered as putative radiosensitizers. Thus, targeting the inflammatory signaling pathways induced by IR offers the opportunity to improve the clinical outcome of radiation therapy by enhancing radiosensitivity and decreasing putative metabolic effects. Since inflammation and sex steroids also impact tumorigenesis, a therapeutic approach targeting glucocorticoid receptors and radiation-induced production of tumorigenic factors might be effective in sensitizing certain tumors to IR.
Chronic inflammation has emerged as one of the hallmarks of cancer impacting any stage of tumorigenesis (Colotta et al., 2009; Grivennikov and Karin, 2010). The persistent expression of inflammatory mediators exert pleiotropic effects on the malignant process. On the one hand, they affect carcinogenesis and malignant transformation, tumor growth, invasion, and metastasis, on the other hand they activate immune effector mechanisms limiting tumor growth. The link between cancer and inflammation can be viewed as consisting of two pathways: an intrinsic and an extrinsic pathway (Mantovani et al., 2008). At the level of the tumor cell, both pathways converge and induce the activation of several transcription factors culminating in the formation of numerous pro-inflammatory molecules that recruit and activate various leukocyte populations into the tumor microenvironment (for a review, see Multhoff et al., 2012). The tumor cell-derived pro-inflammatory molecules now activate the same transcription factors within the cells of the microenvironment and the tumor cells themselves resulting in a more pronounced generation of inflammatory mediators driving a tumor-promoting amplification loop. This amplification mechanism further enhances the impact of inflammatory stimuli within the tumor environment and triggers the manifestation of a cancer-related inflammatory milieu contributing to tumor growth and invasiveness.
NF-κB provides a mechanistic link between inflammation, carcinogenesis, and tumor radioresistance (Magne et al., 2006; Ben-Neriah and Karin, 2011). NF-κB is regarded as the key orchestrator controlling the ability of both, preneoplastic and malignant cells, to resist apoptosis-based tumor surveillance mechanisms activated by DNA damage and chromosomal rearrangement or anti-cancer drugs and radiation, respectively. NF-κB might also regulate tumor angiogenesis and invasiveness (Karin, 2006), and may contribute to the characteristic radio-/chemoresistance of tumor cells (Fahy et al., 2004; Singh and Khar, 2006; Antoon et al., 2011; Chaturvedi et al., 2011). In conjunction with STAT-3 and HIF-1, NF-κB serves as a modulator of the expression of several factors promoting cancer development. Novel therapeutic approaches thus aim to interfere with the activity or expression of these factors, either in single-agent or combinatorial treatment or as supplements of the existing therapeutic concepts. Noteworthy, targeting the pro-inflammatory signaling pathways for tumor radiosensitization represents a promising novel therapeutical approach in cancer. A great variety of classical or novel drugs including nutraceuticals have the capacity to interfere with the inflammatory network in cancer and are progressively tested for tumor radiosensitization. Accumulating evidence over the last few years indicate that most chemotherapeutic agents and radiation therapy activate NF-κB (Wang et al., 1996; Sandur et al., 2009; Li and Sethi, 2010). Thus, NF-κB blockage has been recognized as a promising tool in increasing radiosensitivity of tumors. Apart from NF-κB, STAT-3, HIF-1, and PGHS-2 are further inflammatory factors crucially involved in radioresistance of tumors. Thus, interrupting the inflammatory network in cancer by targeting these molecules may be a promising radiosensitization approach in cancer therapy. Table 1 provides an overview on natural and (semi-)synthetic compounds that are considered as putative radiosensitizers.
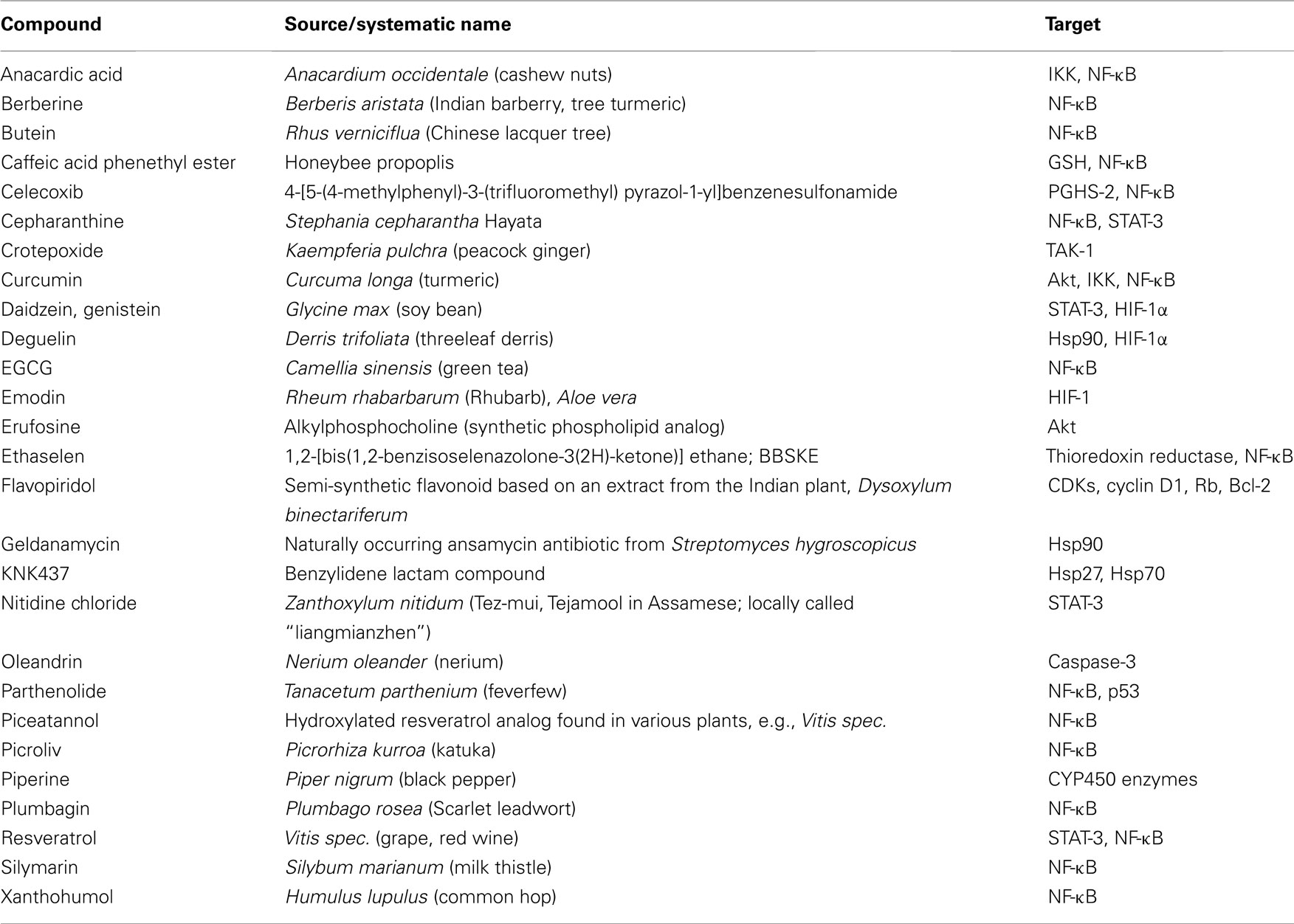
Table 1. Natural and (semi-)synthetic compounds as putative radiosensitizers und their targets.
Recent observations allow a deeper insight into the molecular and cellular mechanisms linking inflammation and tumorigenesis. Emerging data suggest that genetic destabilization of tumor cells is regarded as a further hallmark of most human cancers contributing to tumor initiation and progression (Colotta et al., 2009). Apart from the production of cytokines, chemokines, proteases and prostanoids, inflammatory cells are able to produce reactive oxygen species (ROS) and reactive nitrogen species (RNS). Leukocytes are the main source of RNS and ROS acting as chemical effectors in inflammation-driven carcinogenesis (Kundu and Surh, 2008). All of these mediators act together in perpetuating and amplifying the inflammatory cascade. As outlined in Figure 1, they suppress DNA repair mechanisms leading to an increase in genetic instability termed microsatellite instability (MSI) as a result of mutations or epigenetic alterations of members of the mismatch repair (MMR) family (Hakem, 2008). The MMR system is strongly affected by inflammatory conditions. It has been shown previously that the transcription factor HIF-1 is induced in tumor cells not only by different cytokines and prostaglandins (Jung et al., 2003) but also by ROS and RNS (Sandau et al., 2000). HIF-1 is a heterodimeric transcription factor consisting of a constitutively expressed β-subunit and an oxygen-regulated α-subunit Kaelin Jr. and Ratcliffe 2008). It was proved that HIF-1 plays a pivotal role in hypoxia-induced tumor radioresistance (Moeller and Dewhirst, 2006; Harada, 2011). In this context, our own investigations revealed no correlation between basal HIF-1α levels and the survival fraction in irradiated tumor cell lines implying that basal HIF-1α levels in human tumor cell lines obviously do not predict their radiosensitivity under normoxia (Schilling et al., 2012a). Moreover, HIF-1α has been found as being responsible for genetic instability to down-regulated MMR proteins by inhibiting the MMR proteins MSH-2 and MSH-6, thereby decreasing levels of the MSH-2/MSH-6 complex, MutSα, which recognizes base mismatches. HIF-1α displaces the transcriptional activator c-Myc from Sp1 binding to repress MutSα expression in a p53-dependent manner (Koshiji et al., 2005). Chang et al. (2002 observed that hydrogen peroxide inactivates members of the MMR family at the protein level. From this observation the authors speculate that inactivation of the MMR function in response to oxidative stress may be responsible for the low-frequency MSI (MSI-L) seen in non-neoplastic and cancer tissues associated with chronic inflammation.
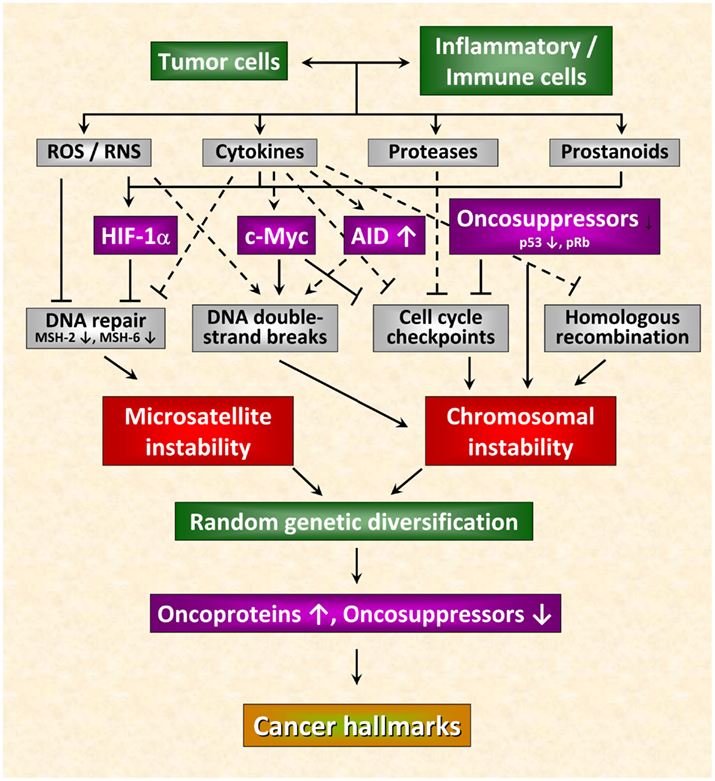
Figure 1. Inflammation-induced molecular pathways causing genetic instability in cancer cells. Genetic destabilization of tumor cells is regarded as a further hallmark of most human cancers contributing to tumor initiation and progression. Apart from the production of cytokines, chemokines, proteases, and prostanoids, inflammatory cells are able to produce reactive oxygen (ROS) and nitrogen species (RNS). All of these mediators act together in perpetuating and amplifying the inflammatory cascade. On the one hand, they suppress DNA repair mechanisms leading to microsatellite instability. On the other hand, they can cause chromosomal instability culminating in abnormal chromosomal segregation and aneuploidy. These inflammatory mediators induce DNA double-strand breaks, affect function of mitotic checkpoint molecules and dysregulate homologous recombination of DNA double-strand break repair leading to random genetic diversification of tumor cells. Cancer cells harboring the optimal combination of activated oncoproteins and inactivated oncosuppressor proteins will develop the malignant phenotype (figure adapted from Colotta et al., 2009; for details see text).
Chromosomal instability can also be the result of the deleterious action of inflammatory mediators culminating in abnormal chromosomal segregation and aneuploidy. The deleterious actions of inflammatory mediators include direct or indirect induction of DNA double-strand breaks (Karanjawala et al., 2002; Mills et al., 2003), defective mitotic checkpoints (Rajagopalan et al., 2003; Menssen et al., 2007), and dysregulated homologous recombination of DNA double-strand break repair (Saintigny et al., 2001; Hakem, 2008; Plo et al., 2008). Further critical molecules that affect genetic stability comprise activation-induced cytidine deaminase (AID; Endo et al., 2008), c-Myc (Vafa et al., 2002), phospho-retinoblastoma protein pRb (Pickering and Kowalik, 2006), and p53 (Tomasini et al., 2008). By causing microsatellite as well as chromosomal instability these molecules induce random genetic diversification of tumor cells. As already discussed by Colotta et al. (2009), cancer clones harboring the optimal combination of activated oncoproteins and inactivated oncosuppressors will develop the malignant phenotype (Figure 1).
In the tumor microenvironment, an intensive interaction between tumor cells and infiltrating immune cells occurs. The latter comprise macrophages, dendritic cells (DC), T cells as well as NK cells with macrophages and T cells as being the most frequent ones (Luster et al., 2005). Inflammatory mediators secreted by tumor and immune cells have been found to play a dual role in tumor development. On the one hand, they promote tumor development and survival of tumor cells, on the other hand they exert surveillance mechanisms against tumor cells (Ben Baruch, 2006; Kim et al., 2006b). In case of a predominance in anti-tumor immunity, tumor cells are eradicated whereas a predominance in surveillance mechanisms provides cancer cells with an immunosuppressive network extending the immune evasion and promoting tumor progression and metastasis (Hadden, 2003). Among the inflammatory mediators secreted by tumor and immune cells TNF, IL-6, and IL-17 act as crucial players in developing chronic inflammation resulting in immune escape and acceleration of tumor progression and metastasis. Upon activation, myeloid cells produce pro- and anti-inflammatory mediators not only affecting growth, survival, and invasiveness of tumor cells but also controlling functional activities of Th1, NK, Treg, and Th17 cells (Lin and Karin, 2007). TRAIL, a member of the TNF superfamily and the product of activated T and NK cells, directly induces apoptosis in numerous cancer cells (LeBlanc and Ashkenazi, 2003) thus playing a crucial role in tumor surveillance mechanisms. Regulatory T cells (Treg) function as key components for regulating anti-tumor immunity (Yamaguchi and Sakaguchi, 2006). Treg specifically suppress the cytotoxicity of expanded CD8+ cytotoxic T cells (Chen et al., 2005) and induce release of IL-17 from Th17 cells acting as key players in chronic inflammation (Mangan et al., 2006). IL-17-mediated effects on the inflammatory response involve recruitment of immune cells (Park et al., 2005), induction of pro-inflammatory factors (IL-1, IL-6, TNF) as well as promotion of angiogenesis and tumor growth (Numasaki et al., 2003). Development of Th17 cells is stimulated by IL-6, IL-23, TGF-β, and TNF released from activated myeloid cells. Anti-inflammatory IL-10 inhibits tumor progression and development by blocking synthesis of IL-6, IL-12, and TNF via NF-κB inhibition (Moore et al., 2001). Furthermore, the anti-tumor activity of Treg is mediated by IL-10 released from Treg themselves (Erdman et al., 2003). IL-23 belonging to the IL-12 family of cytokines enhances the production of IFN-γ and IL-12 by activated T cells, induces IL-17 release from Th17 cells and promotes inflammation at its final stage (Cho et al., 2006). Moreover, IL-23 has been found to up-regulate expression of MMP-9, to increase angiogenesis and to decrease CD8+ T cell recruitment to tumors possibly providing the basis for the development of a tumor-promoting environment (Langowski et al., 2006). IL-12, also released from antigen-presenting cells (APC) after stimulation with IL-23, is a further member of the IL-12 family of cytokines and harbors anti-tumor activities in particular via stimulation of Th1- and CTL-mediated immune responses (Trinchieri, 2003; Langowski et al., 2006). Table 2 summarizes the cellular and molecular outcomes based on interactions between various cell types in the tumor microenvironment.

Table 2. Effects of cell–cell interactions in the tumor microenvironment.
An increasing number of data currently reveal the close relationship between the two classical pathways in tumor progression: inflammation and gonadal hormones. Since the discovery of the hormone dependency of mammary carcinoma in the 1890s, it has become clear that gonadal steroids play a crucial role in the pathogenesis of breast and prostate cancer. The Scottish surgeon George Thomas Beatson was about the first who showed that oophorectomy in a premenopausal woman with breast cancer led to a complete remission (Beatson, 1896a,b) highlighting the role of sex steroids in tumor pathogenesis. However, more recent investigations revealed an astonishing effect of gonadal hormones on tumorigenesis. Female somatic cells including tumor cells express receptors for sex steroid hormones such as estrogen and progesterone affecting growth of hormone-dependent breast cancer cells (Henderson and Canellos, 1980a,b). Women are well known as being less susceptible to tumors at organ sites not representing classical targets for gonadal hormones including the liver. Thus, hepatocellular carcinoma (HCC), the most common liver cancer, occurs mainly in men. The same gender disparity is seen in mice given a chemical carcinogen, diethylnitrosamine (DEN) resulting in liver parenchymal damage followed by activation of Kupffer cells (KC; Naugler et al., 2007). As demonstrated in this study, DEN induces NF-κB-dependent production of pro-inflammatory and growth-promoting IL-6 in KC via IL-1 and TLR signaling cascades, respectively, finally leading to tumor development. Estrogen inhibits activation of NF-κB and blocks secretion of IL-6 from KC thus protecting against liver cancer in females.
Prostate cancer is an androgen-dependent cancer whose susceptibility to gonadal hormones is regulated by selective androgen-receptor modulators (SARM) attenuating the proliferative properties of androgens on tumor cells. Pro-inflammatory IL-1 derived from tumor cells or cells of the microenvironment reverses the properties of SARM from being inhibitory to activatory (Zhu et al., 2006). This de-repression effect requires TGF-β-activated kinase 1 (TAK-1)-binding protein (TAB) 2 (=TAB-2; Takaesu et al., 2000). At the molecular level, IL-1 signaling induces phosphorylation of TAB-2. TAB-2 acts as a sensor for inflammatory signals by serving as a molecular beacon for recruitment of MEKK1, which in turn mediates dismissal of the nuclear receptor co-repressor (N-CoR) holoco-repressor complex from the androgen-receptor and permits de-repression of androgen and estrogen receptor target genes. According to Zhu et al. (2006), this strategy might have come into notice in order to trigger reversal of gonadal hormone-dependent repression of a limited cohort of target genes in response to inflammatory signals linking inflammatory, and nuclear receptor ligand responses to essential reproductive functions. Treatment of prostate cancer by androgen deprivation either by suppression of testicular androgen production or by the use of pharmacological SARM such as flutamide or bicalutamide remains the standard systemic therapy. It has been shown previously that bicalutamide does not function as an androgen-receptor (AR) antagonist by preventing AR binding to DNA but instead stimulates the assembly of a transcriptionally inactive receptor on DNA (Masiello et al., 2002). Recent reports demonstrate that AR can also bind to co-repressor proteins, including N-CoR, and that this binding is enhanced in the presence of bicalutamide indicating that co-repressor binding could further contribute to the in vivo antagonist activity of bicalutamide (Cheng et al., 2002; Shang et al., 2002; Yoon and Wong, 2006). Interestingly, the group of Hollenberg clearly demonstrated that the AR/N-CoR interaction is not enhanced by AR antagonists used currently for the treatment of prostate cancer, but can be markedly enhanced by mifepristone (RU486) in vitro (Hodgson et al., 2005). RU486 can thus be considered as a novel AR antago-nist that will likely have novel activities in vivo. However, clinical trials of RU486 or related drugs are needed to determine whether these may be more efficacious than currently available AR antagonists in the treatment of prostate cancer, particularly advanced androgen-independent prostate cancer. A recent phase II study was conducted to assess the efficacy of mifepristone as an AR antagonist in patients with castration-resistant prostate cancer (CRPC). In this study, RU486 showed only limited activity in patients with CRPC, but stimulated a marked increase in adrenal androgens (Taplin et al., 2008). From these findings the authors hypothesized that inhibition of glucocorticoid receptors by mifepristone might lead to an increase in adrenocorticotropic hormone followed by an increase in adrenal androgens, and that their conversion by tumor cells to testosterone and DHT might have limited the efficacy of mifepristone. As stated by Taplin et al. (2008), a therapeutic approach that combines mifepristone with a second drug harboring a complementary mechanism might be effective in blocking the compensatory rise in adrenal androgens seen in patients with CRPC. These data clearly demonstrate the impact of inflammation and gonadal hormones in tumor progression thus consolidating the fundamental work of Rudolf Virchow and George Thomas Beatson in the field of tumor-associated inflammation.
Several lines of evidence indicate that inflammation plays a pivotal role in modulating radiation responsiveness of tumors. Radiation treatment is obviously a two-edged sword. On the one hand, sublethal doses of ionizing radiation (IR) induces a nuclear DNA damage response. On the other hand, they trigger a cellular damage response in tumors by inducing pro-inflammatory pathways predominantly mediated via activation of NF-κB, the central linker between inflammation, carcinogenesis, and radioresistance. Apart from NF-κB activation, radiation activates/up-regulates the expression of immediate early genes encoding for, e.g., c-Fos, c-Myc, c-Jun (Hong et al., 1997) as well as TNF (Zhou et al., 2001), GM-CSF (Akashi et al., 1992), PGHS-2 (Steinauer et al., 2000), and ICAM-1 (Son et al., 2006). Radiation also induces activation of receptor tyrosine kinase pathways (Fedrigo et al., 2011) and mitochondria-associated responses (Aykin-Burns et al., 2011). As demonstrated by Valerie et al. (2007), radiation-induced activation of plasma membrane receptors occurs via generation of ionizing events in the liquid phase of the cytosol that are amplified, possibly via mitochondria, generating large amounts of ROS and RNS that inhibit protein tyrosine phosphatase (PTPase) activities. Moreover, radiation activates acidic sphingomyelinase and increases the production of ceramide. Inhibition of PTPases leads to activation of non-receptor and receptor tyrosine kinases (RTK) including epidermal growth factor receptor (EGFR) and the activation of down-stream signal transduction pathways (Goldkorn et al., 1997; Szumiel, 2008). Radiation-induced ceramide was found to promote membrane-associated receptor activation by facilitating the clustering of receptors within lipid rafts (Maziere et al., 2001; Galabova-Kovacs et al., 2006). As a consequence, activated RTK induce down-stream pro-survival pathways (e.g., Akt) that might act as promising targets in enhancing radiosensitivity of tumors.
Furthermore, various inflammatory mediators are reported as being up-regulated during radiation responses. In human glioblastoma cells, exposure to gamma-irradiation stimulated release of IL-6 and IL-8 into culture supernatants (Pasi et al., 2010). Radiation and chemotherapy led to a remarkable increase in the production of these cytokines in human oral carcinoma cells (Tamatani et al., 2004). IR induced a tremendous increase in IL-1, IL-6, and GM-CSF production by human lung cancer cells (Zhang et al., 1994). A similar effect was observed in patients with head and neck cancer where increased IL-6 and IL-8 levels can be detected after chemoradiotherapy (Meirovitz et al., 2010). Elevated IL-6 serum levels are also reported in patients with locally advanced non-small cell lung cancer undergoing concurrent chemoradiation therapy (Wang et al., 2010).
Some concern had been raised about the effect of IR on the expression of the pleiotropic cytokine TNF. The majority of the studies points to an activatory action of IR. For instance, Chendil et al. (2004) reported on a radiation-induced up-regulated TNF protein in prostate cancer cells PC-3 leading to an increase in NF-κB activity followed by an induction of Bcl-2 protein. Furthermore, IR persistently induced NF-κB DNA-binding activity and NF-κB-dependent TNF transactivation and secretion (Veeraraghavan et al., 2011a). In contrast, three-dimensional conformal blocking radiation therapy did not alter serum levels of TNF in patients with prostate cancer (Lopes and Callera, 2011). It is interesting to note that in an immunocompetent animal model of pancreatic cancer the combination of the radio inducible TNF-expressing adenovector Ad.Egr-TNF with IR resulted in significant anti-tumor effects mediated by the immune system (Meng et al., 2010). As demonstrated in this study, Ad.Egr-TNF/IR therapy contributed to local tumor control through TNF production in the tumor microenvironment. TNF induced expression of the known potent immune regulator IFN-β that, in turn, stimulated the production of chemokines leading to the recruitment of CD8+ T cells to the tumor. Several clinical and preclinical studies with Ad.Egr-TNF/IR have suggested that this local approach suppresses the growth of distant metastases (Moral and Tomillero, 2008). However, a Phase III trial comparing Ad.Egr-TNF (TNFerade™) along with standard of care therapy (defined as infusion 5-FU and radiation therapy, followed by gemcitabine or gemcitabine/erlotinib maintenance therapy) versus standard of care therapy in the treatment of locally advanced, unresectable pancreatic cancer failed. Since the interim analysis did not provide sufficient evidence of the clinical effectiveness of TNFerade, the supplying company, GenVec, announced the discontinuation of the trial in 2010. From these observations one can hypothesize that up-regulated TNF probably enhances the impact of tumorigenic stimuli within the tumor or the tumor microenvironment thereby forcing a critical amplification mechanism in tumor-associated inflammation triggered by pro-inflammatory mediators.
It should be kept in mind that radiation therapy represents an efficient local anti-cancer approach leading to elimination of both, tumor cells and cells of the tumor microenvironment such as endothelial cells and tumor-induced suppressor T cells (North, 1984). IR also affects function of immune cells culminating in homing of APC and effector T cells (Ganss et al., 2002). According to the study of Ganss et al. (2002), the combination of irradiation and adoptive tumor-specific T cell therapy ensures antigen-driven tumor cell eradication with anti-angiogenic effects on tumor endothelium. It has been shown previously that sublethal doses of IR stimulates anti-tumor T cell responses and up-regulates MHC class I/II expression in melanoma cells rendering the cells more sensitive to T cell recognition (Abdel-Wahab et al., 1996), obviously through tumor-specific antigen presentation by DC (Ciernik et al., 1999). Clinical phase I/II trials are ongoing and will shed light on the efficiency of low-dose single phase fraction radiotherapy on tumor-infiltrating T cells responses in patients with liver metastasis derived from colorectal caner (Reissfelder et al., 2011) and primarily operable pancreatic cancer (Timke et al., 2011).
PGHS-2, the rate-limiting enzyme involved in converting arachidonic acid to prostanoids, has emerged as another crucial NF-κB-dependent pro-inflammatory mediator in tumorigenesis. Aberrant up-regulation of PGHS-2 is frequently observed in various pre-cancerous and malignant tissues. Because most of the PGHS-2-induced effects are mediated by its product PGE2, down-regulation of prostaglandins in tumor tissues by PGHS-2 inhibition blocks several neoplastic pathways restricting tumor growth. Radiation is known to induce inflammation and NF-κB consequently up-regulating/activating PGHS-2. In this context, PGHS-2 inhibitors have been tested for their anti-tumor efficiency in combination with radiation or chemotherapy. It was found that these inhibitors exert promising anti-cancer effects in a variety of human tumor cells and increase the sensitivity of tumor cells toward chemotherapy and/or radiation therapy. For instance, the PGHS-2 inhibitor NS398 enhanced the radiosensitivity of radioresistant esophageal cancer cells CSC-like Eca109R50Gy most likely by down-regulating the expression of β-catenin as well as inhibiting activation of Akt and inducing apoptosis (Che et al., 2010, 2011). NS398 was also found to radiosensitize human melanoma cells through G2/M arrest of the cell cycle, predominantly via necrotic mechanisms (Johnson et al., 2008). A further PGHS-2 inhibitor, celecoxib, increased radiation-induced cell death, and clonogenic kill of prostate cancer cells in vitro providing a rationale for clinical evaluation of celecoxib in combination with irradiation in prostate cancer patients (Handrick et al., 2009). Celecoxib also enhanced radiosensitivity of bronchial and colon carcinoma cells by inhibiting EGFR-mediated mechanisms of radioresistance independent of PGHS-2 activity implying that PGHS-2 inhibition might ameliorate the therapeutic outcome of radiation therapy even in patients with PGHS-2-independent tumor radioresistance (Dittmann et al., 2008). In line with this observation, targeting PGHS-2 by different pharmacological inhibitors led to radio enhancement of human glioma cells in the absence of the PGHS-2 protein (Kuipers et al., 2007). Nimesulide is another PGHS-2-selective inhibitor that has been found to increase the efficacy of radiation therapy in non-small cell lung cancer cells possibly via suppression of NF-κB-mediated, radiation-induced cytoprotective genes (Grimes et al., 2006).
However, selective PGHS-2 inhibitors have come under scrutiny because of reports suggesting an increased cardiovascular risk associated with their use (Solomon et al., 2008). The thitherto used high therapeutic concentrations of these drugs may contribute to a pro-thrombotic state in patients with higher risk for serious cardiovascular events. A novel approach to over-come the limitations associated with the toxicity of PGHS-2 inhibitors might be the combination of pharmacological PGHS-2 inhibitors at low doses with naturally occurring compounds such as the catechin EGCG which is a promising chemopreventive agent derived from green tea (Cerella et al., 2010; Härdtner et al., 2012). Of note, nutraceuticals such as plant-derived polyphenols have been studied intensively for their potential chemopreventive properties and have been found as being pharmacologically safe. These compounds comprise genistein, curcumin, resveratrol, silymarin, caffeic acid phenethyl ester, flavopiridol, emodin, green tea polyphenols (e.g., EGCG), piperine, oleandrin, ursolic acid, and betulinic acid. These phytochemicals sensitize tumor cells to chemotherapeutic agents and radiation therapy by inhibiting pathways responsible for treatment resistance (Garg et al., 2005; Nambiar et al., 2011). Among them, curcumin derived from the rhizomes of Curcuma longa has been identified to improve the anti-tumor effects of IR by blocking NF-κB pathways, down-regulating anti-apoptotic Bcl-XL and survivin as well as increasing G2/M phase arrest in the cell cycle distribution in Burkitt’s lymphoma cells (Qiao et al., 2012). Curcumin also potentiates radiation therapy-induced cell death by targeting radiation therapy-induced NF-κB activation in pancreatic cancer cells (Veeraraghavan et al., 2011b).
Apart from NF-κB and PGHS-2, STAT-3 is a further inflammatory molecule crucially involved in radioresistance of tumors. Since enhanced radioresistance of cancer cells is additionally related to radiation-induced activation of the JAK/STAT pathway, inhibition of STAT by, e.g., phytochemicals might sensitize tumors to radiation therapy. It has been shown previously that STAT-3-mediated radiosensitization obviously occurs via down-regulation of anti-apoptotic survivin (Kim et al., 2006a). In this context, resveratrol, a polyphenolic phytoalexin, selectively targets numerous cell signaling pathways and decreases clonogenic survival primarily via an apoptotic mechanism. In melanoma cells, resveratrol inhibits STAT-3 and NF-κB-dependent transcription, culminating in suppression of c-FLIP and Bcl-XL expression, while activating the MAPK and the ATM-Chk2-p53 pathways (Johnson et al., 2008). Resveratrol also up-regulates TRAIL promoter activity and induces TRAIL surface expression in some melanoma cell lines, resulting in a rapid apoptosis development (Johnson et al., 2008). As also demonstrated in this study, sequential treatment of melanoma cells, first with gamma-irradiation to up-regulate TRAIL receptor surface expression, and then with resveratrol to suppress anti-apoptotic proteins c-FLIP and Bcl-XL and induce TRAIL surface expression, dramatically up-regulated apoptosis in some melanoma cell lines. Nitidine chloride, a natural phytochemical alkaloid derived from Zanthoxylum nitidum, was identified as a potent STAT-3 signaling inhibitor suppressing angiogenesis and growth of human gastric cancer (Chen et al., 2012a). From these data one can hypothesize that phytochemicals in combination with IR may play a significant role in enhancing the therapeutic efficacy of cancer treatment.
As already mentioned, HIF-1, the key mediator in hypoxia signaling pathways, is crucially involved in hypoxia-induced tumor radioresistance. This obviously includes radiation-induced activation of HIF-1 (Moeller et al., 2004; Harada et al., 2009a,b), HIF-1-dependent induction of VEGF and protection of endothelial cells from radiation-induced cytotoxicity by VEGF (Gorski et al., 1999) as well as delivery of oxygen and nutrients to tumor cells by radioprotected tumor blood vessels (Zeng et al., 2008). N-Myc down-stream-regulated gene 2 (NDRG2) was recently identified in cervical cancer cells as a new HIF-1 target gene acting down-stream of HIF-1 to promote radioresistance via suppression of radiation-induced Bax expression (Liu et al., 2010). Therefore, it would be reasonable to study the efficacy of HIF-1 and NDRG2 blockage as radiosensitizer for tumor therapy. Strategies to over-come radioresistance of hypoxic tumor cells comprise, among others, hyperbaric oxygenation, gene therapy approaches, fractionated radiotherapy, radiosensitization by mimicking the effect of molecular oxygen using nitroimidazole derivates as well as suppression of hypoxic tumor cell radioresistance by HIF-1 inhibitors (for a review, see Harada, 2011). As described previously, administration of the HIF-1 inhibitor YC-1 to hypoxic cobalt-treated cells derived from squamous-cell carcinoma of the larynx effectively inhibited HIF-1α expression, and enhanced the sensitivity of cells to radiation, decreasing the surviving fraction to that of normoxic cells (Moon et al., 2009). YC-1 was found to reduce the number of tumor lesions after tumor cell inoculation in nude mice (Shin et al., 2007). Compared to radiation therapy alone, inhibition of radiation-induced HIF-1 activation by YC-1 led to a significant reduction in tumor cell growth (Harada et al., 2009b).
Another HIF-1 inhibitor, acriflavine, was found to inhibit tumor growth and angiogenesis in a xenograft tumor model for human prostate cancer through blockage of HIF-1 dimerization (Lee et al., 2009). Since the same agent blocked lung metastasis in an orthotopic breast cancer model (Wong et al., 2012), it would be of interest to test the efficacy of this HIF-1 inhibitor in increasing radioresistance of certain tumors. Interestingly, inhibition of Hsp90 function by 17-allylamino-17-demethoxygeldanamycin or deguelin, a novel natural inhibitor of Hsp90, suppressed increases in HIF-1α/Hsp90 interaction and HIF-1α expression in radioresistant lung cancer cells (Kim et al., 2009). Hsp90 interacts with HIF-1α in competition with receptor of activated protein C kinase 1 (RACK-1) and inhibits oxygen-independent degradation of HIF-1α (Semenza, 2007). The study by Kim et al. (2009) also demonstrated that the combined treatment of radiation with deguelin significantly decreased the survival and angiogenic potential of radioresistant lung cancer cells in vitro and inhibited tumor growth and angiogenesis in vivo.
Even phytochemicals have the capacity to inhibit radiation-induced HIF-1 activation. Pre-treatment of prostate cancer cells with soy isoflavones inhibited Src/STAT-3/HIF-1α activation by radiation and nuclear translocation of HIF-1α. These findings correlated with decreased expression of APE1/Ref-1 and DNA-binding activity of HIF-1α and NF-κB (Singh-Gupta et al., 2009). Apurinic/apyrimidinic (AP) endonuclease 1/redox factor-1 (APE1/Ref-1) is a multifunctional protein involved in DNA repair that also functions as a redox activator of cellular transcription factors. Emodin, a natural anthraquinone enriched in the traditional Chinese herbal medicines and novel small HIF-1 inhibitor, was found to improve efficacy of chemotherapeutic drugs by inhibiting transactivation of HIF-1 without impairing mRNA expression and stability of HIF-1α protein in prostate cancer cells (Huang et al., 2008). Another approach to HIF-1 blockage might be the use of cell-permeable HIF-1 antagonists (Shi et al., 2007). As mentioned before, HIF-1 contributes to tumor radioresistance by up-regulating survivin expression under hypoxic conditions. Moreover, in hypoxic tumor cells the HIF-1 signaling pathway is activated and could be further enhanced by radiation, thereby providing survival signals to adjacent vascular endothelial cells by up-regulation of VEGF and basic fibroblast growth factor (bFGF) and resulting in tumor radioresistance through vascular radioprotection. Thus, HIF-1 antagonists might decrease tumor angiogenesis and sensitize tumor cells to radiotherapy.
Targeting HIF-1 for radiosensitization can also be achieved by inhibiting its up-stream mediators. In this respect, the PI3K/Akt/mTOR pathway can be considered as the most relevant target in sensitizing tumors to radiation therapy. Mutations in this pathway can be found in various human cancers and have been found to up-regulate HIF-1α expression (Zhong et al., 2000). The PI3K/Akt/mTOR pathway also affects the NF-κB-mediated expression of PGHS-2 (Yang et al., 2009) and radiation-induced MMP-9 expression (Cheng et al., 2006). Several studies demonstrated that specific blockage of this pathway by certain inhibitors such as erufosine (Rudner et al., 2010), LY294002 (Nakamura et al., 2005; Kao et al., 2007), RAD001 (Albert et al., 2006), and rapamycin (Majumder et al., 2004) ensures efficient radiosensitization of distinct tumors cells.
In recent years NF-κB inhibition by synthetic compounds as well as nutraceuticals of different sources has been approved for tumor radiosensitization. In particular the use of nutraceuticals became a popular approach due to the broad anti-tumor and anti-inflammatory properties in conjunction with low toxicity risks of these compounds (Deorukhkar et al., 2007; Deorukhkar and Krishnan, 2010). How these drugs block NF-κB activation is becoming increasingly apparent. Among them, curcumin has emerged as one of the best studied plant-derived polyphenols. In a phase II clinical trial curcumin showed beneficial effects in patients with advanced pancreatic cancer (Dhillon et al., 2008). Curcumin down-regulated expression of NF-κB, PGHS-2, and phosphorylated STAT-3 in peripheral blood mononuclear cells from these patients. Furthermore, curcumin has been shown to suppress TNF-mediated NF-κB activation by inhibiting inhibitor of kappaB alpha (IκBα) in human myeloid ML-1a cells (Singh and Aggarwal, 1995). Curcumin also confers radiosensitizing effects in prostate cancer cells by inhibiting TNF-mediated NF-κB activation resulting in Bcl-2 protein down-regulation and concomitant activation of cytochrome c and caspase-9 and -3 (Chendil et al., 2004). More recently, curcumin was found to sensitize colorectal cancer cells to radiotherapy by suppressing radiation-induced NF-κB activation via inhibition of radiation-induced phosphorylation and degradation of IκBα, inhibition of inhibitor of kappaB kinase (IKK) activity, and inhibition of Akt phosphorylation (Sandur et al., 2009) consequently leading to down-regulation of several tumorigenic factors in colorectal cancer xenografts in nude mice (Kunnumakkara et al., 2008). Apart from its IKK-inhibitory capacity, curcumin also blocks p65 phosphorylation and acetylation and represses the p300/CREB-binding protein (CBP) HAT activity-dependent transcriptional activation from chromatin (Balasubramanyam et al., 2004). Similarly, anacardic acid (6-pentadecylsalicylic acid) derived from traditional medicinal plants, such as cashew nuts, has been identified to inhibit NF-κB activation, to suppress the activation of IκBα kinase that led to abrogation of phosphorylation and degradation of IκBα and to inhibit acetylation and nuclear translocation of p65 (Sung et al., 2008). The same study demonstrated that down-regulation of the p300 HAT gene by RNA interference abrogated the effect of anacardic acid on NF-κB suppression. Further nutraceuticals including the soy isoflavone genistein (Raffoul et al., 2006), the isoquinoline alkaloid berberine from medicinal plants such as Berberis aristata, Coptis chinensis, Coptis japonica, Coscinium fenestratum, and Hydrastis canadensis (Pandey et al., 2008), piceatannol (3,3′,4,5′-trans-trihydroxystilbene), a naturally occurring hydroxylated analog of resveratrol found in various plants (Son et al., 2010), the principal prenylated flavonoid xanthohumol from Humulus lupulus (Harikumar et al., 2009), and the polyphenol butein (3,4,2′,4′-tetrahydroxychalcone) from Rhus verniciflua Stokes (Pandey et al., 2007) have been identified as blocking NF-κB by direct interaction with IKKβ on cysteine 179 residue. NF-κB inhibition by direct interaction with one of its subunits occurs in the presence of numerous phytochemicals including sesquiterpene lactones (Garcia-Pineres et al., 2001). In radiation-resistant human CGL1 cells, parthenolide, a major active component of the herbal medicine feverfew (Tanacetum parthenium), enhanced radiosensitivity through NF-κB inhibition and apoptosis induction via p53 stabilization, induction of pro-apoptotic Bax, and phosphorylation of pro-apoptotic Bid (Mendonca et al., 2007). The radiosensitization effect of parthenolide is enhanced in the presence of tumor suppressor protein PTEN (phosphatase and tensin homolog deleted on chromosome 10), in part, by suppressing the absolute amount of activated p-Akt in human prostate cancer cells (Sun et al., 2007). The group of Aggarwal recently found out that crotepoxide (a substituted cyclohexane diepoxide), isolated from Kaempferia pulchra (peacock ginger), inhibited activation of TGF-β-activated kinase (TAK)-1, which led to suppression of IκBα kinase, abrogation of IκBα phosphorylation and degradation, nuclear translocation of p65, and suppression of NF-κB-dependent reporter genes encoding for anti-apoptotic (Bcl-2, Bcl-XL, IAP1/2, Mcl-1, survivin, TRAF-1), pro-apoptotic (Bax, Bid), pro-inflammatory (PGHS-2), proliferation- (cyclin D1, c-Myc), invasion- (ICAM-1, MMP-9), and angiogenesis-promoting (VEGF) factors (Prasad et al., 2010). Moreover, Tamatani et al. (2007) analyzed the effects of radiation therapy in combination with cepharanthine on NF-κB activation and expression of its down-stream effector molecules in human oral squamous-cell carcinoma cells. Cepharanthine is a biscoclaurine alkaloid extracted from the roots of Stephania cepharantha Hayata, and is widely used in Japan for the treatment of patients with leucopenia, nasal allergy, and venomous snakebites. The authors could show that treatment of cancer cells with cepharanthine combined with exposure to IR enhanced radiosensitivity via NF-κB inhibition and concomitant down-regulation of IL-6, IL-8, and anti-apoptotic proteins such as cellular inhibitor of apoptosis protein (cIAP)-1 and -2. Moreover, the pleiotropic effects of cepharanthine also includes inhibition of STAT-3 in the human osteosarcoma cell line SaOS2 (Chen et al., 2012b). These examples highlight the crucial role of naturally occurring compounds in targeting inflammatory signaling pathways for sensitizing tumors to radiation therapy. Beside them, a variety of further compounds have been identified to enhance radiosensitivity via inhibition of NF-κB including celecoxib (Raju et al., 2005), pitavastatin (Tsuboi et al., 2009), docosahexaenoic acid (Zand et al., 2008), and the novel organoselenium thioredoxin reductase inhibitor ethaselen (Wang et al., 2011).
It is commonly accepted that aggressive radio-/chemotherapy often results in a negative selection toward highly aggressive tumor clones. In recent days, the induction of radiation-induced apoptosis, the principal purpose of radiation therapy, has come under scrutiny due to reports suggesting that dying tumor cells use the apoptotic pathway to stimulate the re-population of tumors subjected to radiation therapy. The process of re-population was originally discovered in the second half of the twentieth century and is commonly accepted as playing a crucial role in radio-/chemotherapy (Hermens and Barendsen, 1969; Stephens et al., 1978). Interestingly, the group of Li from Aurora (USA) demonstrated that radiation-induced apoptosis comprises caspase-3 cleavage and concomitant activation of Ca2+-independent phospholipase A2 (iPLA2) followed by PGE2 release promoting tumor cell re-population in vitro and in vivo (Huang et al., 2011). The relevance of this mechanism for the caspase-mediated tumor cell re-population was confirmed by caspase-3 determination in different cancer patients. Herein, elevated expression levels of activated caspase-3 in tumor tissues correlated with poor clinical outcome in patients with head and neck cancer as well as advanced stage breast cancer. However, the contribution of macrophages and the subsequently generated clearance-related anti-inflammatory milieu to radiation-induced tumor cell re-population and poor therapeutic outcome should be taken into consideration. As discussed by Tauber and co-workers, apoptotic manifestations such as externalization of phosphatidylserine, bleb formation, and DNA fragmentation are crucially involved in macrophage activation and depend on caspase-3 activation (Jänicke et al., 1998; Coleman et al., 2001; Sebbagh et al., 2001). The authors conclude that caspase-3-positive apoptotic cells can recruit more macrophages and are more efficiently internalized by the phagocytes (“silent clearance”) culminating in a strong anti-inflammatory and growth-promoting phagocyte response via release of clearance-associated cytokines, e.g., PGE2 than their caspase-3-negative counterparts. However, further preclinical and clinical studies using certain caspase inhibitors are required to strengthen this hypothesis. In this context, the novel caspase inhibitor GS-9450 was found to down-regulate caspase-3 expression on peripheral T cells from chronically HCV-infected patients in a phase II clinical trial (Arends et al., 2011). It would therefore be of interest to study a putative beneficial effect of a combinatorial treatment with caspase inhibitors and radiation therapy on the clinical outcome of patients with various advanced cancer types.
It is well known that chronic inflammation induced by non-infectious agents can also contribute to carcinogenesis and act as a driving force in tumorigenesis. Several factors such as growth factors, oncoproteins, and toxins can affect the host via an activation of pattern recognition receptors (PRR) interacting with exogenous pathogen-associated molecular patterns (PAMP; Kawai and Akira, 2011). Apart from PAMP, the same receptor superfamily also recognizes endogenous “alarmins” both of them comprising the group of danger-associated molecular patterns (DAMP; Bianchi, 2007). Receptor ligation leads to activation of inflammatory cells and initiation of host responses that tend to eradicate invading microorganisms (Akira et al., 2006; Karin et al., 2006). Not surprisingly, inadequate pathogen elimination, recurring tissue injury, prolonged inflammatory signaling, and failure of anti-inflammatory mechanisms can all culminate in chronic inflammation promoting cancer development.
Signaling via PRR also makes an impact on radiation responses. It has been shown previously that both, low (0.075 Gy) and high (2 Gy) doses of IR causes sustained stimulation of IL-12 and IL-18 secretion by mouse macrophages (Shan et al., 2007) with concomitant activation of NF-κB accompanied by elevated cytoplasmic MyD88 levels and an up-regulated surface expression of CD14 and TLR-4/MD-2 (Shan et al., 2007), the latter acting as LPS sensor. From these findings the authors hypothesized that IR can stimulate the secretion of IL-12 and IL-18 presumably via activation of the Toll signaling pathway in macrophages. A detailed description of the TLR-4 signaling pathway is visualized schematically in Figure 2.
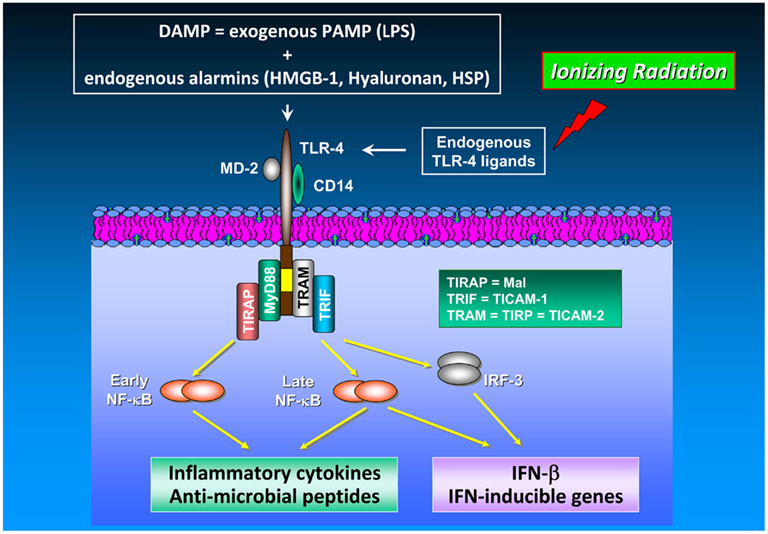
Figure 2. TLR-4 signaling in cancer. The TLR-4/MD-2 receptor complex recognizes and binds exogenous PAMP (e.g., endotoxins such as LPS) as well as endogenous alarmins (HMGB-1, hyaluronan, heat shock proteins). Release of DAMP into the extracellular space is achieved by a number of different mechanisms including (i) leakage from necrotic cells, (ii) increased synthesis and post-translational modification in response to inflammation, and (iii) degradation of inactive precursors into TLR-mimetic degradation products in inflammatory environments (Mencin et al., 2009). TLR-4 induces two distinct signaling pathways controlled by the TIRAP/MyD88 and TRAM/TRIF pairs of adaptor proteins, which elicit the production of pro-inflammatory cytokines and type I interferons, respectively. The cytosolic adapter molecules mentioned above comprise myeloid differentiation protein 88 (MyD88), Toll/IL-1R resistance domain-containing adapter inducing IFN-β (TRIF), TIR domain-containing adapter protein (TIRAP), and TRIF-related adaptor molecule (TRAM). TIRAP is also termed Mal (MyD88 adaptor-like), TRIF is also known as Toll/IL-1R homology domain-containing adaptor molecule 1 (TICAM-1), whereas TRAM is alternatively entitled TIR-containing protein (TIRP) and TICAM-2, respectively. TLR-4-mediated signal transduction occurs via MyD88-dependent and MyD88-independent (i.e., TRAM/TRIF-dependent) pathways. Both, MyD88-dependent and MyD88-independent pathways induce expression of genes involved in pro-inflammatory and anti-microbial responses (Akira and Takeda, 2004). In TLR-4 signaling, MyD88 up-regulates inflammatory cytokines via NF-κB activation. Moreover, the MyD88-independent pathway does not only induce inflammatory gene expression in an NF-κB-dependent manner but also up-regulates type I interferon expression via the transcription factor IRF-3. NF-κB activation and subsequent inflammatory cytokine production are mediated by different mechanisms and kinetics in the MyD88-dependent and the MyD88-independent pathway: NF-κB activation in the MyD88-dependent pathway is an early event occurring with fast kinetics whereas NF-κB activation via the MyD88-independent pathway represents a late event occurring with slower kinetics. Unlike TLR-4 signaling in immune cells which has been found to enhance anti-tumor immunity by, e.g., IL-12/IFN-γ up-regulation and promotion of DC maturation and function, TLR-4 signaling in cancer cells increases their tumorigenic capacity under certain circumstances (Oblak and Jerala, 2011). Noteworthy, HMGB-1 which is released from irradiated tumor cells functions as an endogenous TLR-4 ligand leading to the development of a tumor antigen-specific T cell immunity mediated by an interaction of HMGB-1 with TLR-4 on DC.
Interestingly, RP105 (radioprotective 105 kDa), a TLR-related molecule, was recently identified on human B cells and DC (Fugier-Vivier et al., 1997) and characterized as being similar to TLR-4 in that the extracellular leucine-rich repeats associate with MD-1, an MD-2-like molecule (Miyake et al., 1995; Fugier-Vivier et al., 1997). MD-2 directly binds to lipid A, the active center of lipopolysaccharide (LPS), leading to dimerization of TLR-4/MD-2 (Ohto et al., 2011). An antibody raised against surface-bound RP105 was found to drive B cell proliferation and protection from subsequent radiation- or dexamethasone-induced apoptosis (Miyake et al., 1994). Studies by Divanovic et al. (2005) demonstrated that: (a) RP105 is a specific inhibitor of TLR-4 signaling in human embryonal kidney cells HEK293; (b) RP105/MD-1 interacts directly with TLR-4/MD-2, thus abolishing the LPS-binding capacity of the complex; (c) RP105 regulates TLR-4 signaling in DC and macrophages; and (d) RP105 regulates in vivo responses to LPS supporting the assumption that RP105 acts as a physiological negative regulator of TLR-4 responses. This brief overview makes it clear that, on the one hand, pro-inflammatory responses to radiation and TLR signaling enhance the impact of tumorigenic factors in the tumor and the tumor microenvironment and, on the other hand, might represent pivotal target structures in radiation therapy. As discussed by Schaue and McBride 2010), radiation-induced DAMP signaling via TLR-2/-4 has emerged as a critical component in affecting the outcome of anti-cancer therapies. In this context, radiation was found to induce secretion of the prototypical DAMP, the high-mobility-group box 1 (HMGB-1) “alarmin” protein from dying tumor cells as a prerequisite for the development of a tumor antigen-specific T cell immunity mediated by an interaction of HMGB-1 with TLR-4 on DC (Apetoh et al., 2007). The same study revealed that patients with breast cancer who carry a TLR-4 loss-of-function allele relapsed more quickly after radiotherapy and chemotherapy than those carrying the normal TLR-4 allele implying a clinically relevant immunoadjuvant pathway triggered by tumor cell death. An intriguing novel finding comprises the reduction of metastatic ability and MMP-9 expression in MGC-803 gastric cancer cells by silencing of the HMGB-1 expression using an HMGB-1-specific RNAi lentiviral vector (Song et al., 2012). As also shown in this study, HMGB-1 silencing decreased cell proliferation and sensitized cells to apoptosis implying HMGB-1 as being a potential target for the therapeutic intervention of certain cancers such as gastric cancer.
A promising approach in cancer therapy also might be targeting heat shock proteins (HSP), a class of proteins which are induced under physiologic stress to promote cell survival in the face of endogenous or exogenous injury. Compared to normal cells, tumors frequently have elevated basal Hsp70 levels which are further enhanced in response to a number of pathological and environmental stresses such as nutrient deficiency, hypoxia, heavy metals, irradiation, and/or chemotherapeutic agents. Also normal cells show an increase in the synthesis of Hsp70 following stress in order to mediate protection against lethal damage and to maintain protein homeostasis. Screening of nearly 1,000 primary human tumor biopsies and the corresponding normal tissues revealed that human carcinomas, but none of the tested normal tissues, frequently present Hsp70 on their cell surface (Multhoff et al., 1995; Multhoff, 2007). A membrane Hsp70-positive tumor phenotype has been found to be associated with a significantly decreased overall survival in tumor patients. Therefore, the expression of this molecule could serve as a negative prognostic marker (Pfister et al., 2007).
Apart from their intracellular localization, Hightower and Guidon Jr. (1989) reported on an ER/Golgi-independent release of Hsp70 from viable cells with intact cell membranes already in the late 1980s. Extracellular HSP are considered as molecules with immunomodulatory functions (Pockley and Multhoff, 2008; Pockley et al., 2008) either as cross-presenters of immunogenic peptides (Srivastava, 1997; Asea et al., 2000) or in a peptide-free version as chaperokines (Asea et al., 2002) or stimulators of innate immune responses (Multhoff et al., 1995). Despite these well-documented immunological functions, the mechanisms of HSP export are still controversially discussed since cytosolic HSP lack a consensus signal for secretion. However, apart from Hsp70, other molecules lacking a secretory signal such as IL-1α, IL-1β, and HMBG-1 are also found outside of cells (Nickel and Seedorf, 2008; Eder, 2009). Hsp70 also has been found to be located on the cell surface although lacking a transmembrane domain (Multhoff and Hightower, 1996). Membrane Hsp70 might help to maintain stability of tumor cells and thus might protect tumors from lethal damage induced by environmental stress (Horvath et al., 2008; Horvath and Vigh, 2010). The fundamental work of the group of Antonio De Maio has demonstrated an interaction of members of the HSP70 family with artificial membranes containing phosphatidylserine (PS; Arispe and De Maio, 2000; Arispe et al., 2002; Vega et al., 2008; De Maio, 2011). Our group reported on an interaction of Hsp70 with the sphingolipid globotriaosylceramide (Gb3) in the plasma membrane of non-stressed human gastrointestinal stromal tumors (Gehrmann et al., 2008). Gb3 is found in cholesterol-rich microdomains, also termed as lipid rafts, which serve as signal transduction platforms. Following irradiation or hypoxia-induced stress Hsp70 was found to be associated predominantly with PS outside of lipid rafts in the plasma membrane of tumor cells (Schilling et al., 2009). These data indicate that environmental stress might result in a re-organization of the lipid bilayer and might modulate the interaction of Hsp70 with lipid components. Surprisingly, only tumors but not the corresponding normal tissues were found as being membrane Hsp70-positive using the IgG1 mouse monoclonal antibody cmHsp70.1. In contrast, other Hsp70-specific antibodies failed to bind to membrane Hsp70 on viable tumor cells (Stangl et al., 2011). The discovery that neither high/low salt concentrations nor changes in the pH were able to release Hsp70 from the plasma membrane of tumor cells (Gehrmann et al., 2008; Vega et al., 2008) confirmed our hypothesis that in tumor cells Hsp70 is an integral membrane protein which can associate with raft (Gb3) and non-raft (PS) lipid components.
We recently observed an increased surface expression of Hsp70 in colorectal tumor cells after IR alone or in combination with hyperthermia (HT) while the amount of extracellular Hsp70 was only increased when HT was given additionally (Schildkopf et al., 2011). Moreover, a high up-regulation of the co-stimulation molecule CD80 and the chemokine receptor CCR7 on DC was measured after contact with supernatants of X-ray plus HT-treated cells. This was dependent on extracellular Hsp70. Combined treatments further led to significantly increased phagocytosis rates of macrophages and DC and increased pro-inflammatory cytokine (IL-8, IL-12) secretion. From these findings we conclude that X-ray combined with HT induces Hsp70-dependent activation of immune cells and might generate a tumor microenvironment beneficial for cure.
HSP over-expression in tumor cells plays a pivotal role in tumorigenesis by inhibiting apoptosis and senescence. Recent studies indicate an involvement of HSP such as Hsp70/Hsp72 and Hsp90 in the recognition of PAMP by binding to TLR-4 within lipid rafts (Triantafilou and Triantafilou, 2004; Wheeler et al., 2009). Since extracellular residing Hsp70 acts as a danger signal for the immune system (Matzinger, 1998), this stress protein has been added to the list of “alarmins” comprising the group of DAMP together with PAMP, hyaluronan, and other HSP members. Consequently, developing means of abrogating HSP expression may provide a way to render cancer cells more susceptible to radiation or chemotherapy. Various attempts are underway to target these proteins, particularly small HSP, in developing potent radiation and chemotherapy sensitizers (Guttmann and Koumenis, 2011). For instance, Hsp27 has been found as being implicated in the resistance to chemotherapy in several types of cancers. The group of Moriwaki analyzed the effects of a gemcitabine treatment in pancreatic cancer cells (Nakashima et al., 2011). Gemcitabine is an anti-tumor drug and currently considered to be the standard of care for the treatment of advanced pancreatic cancer, but the clinical outcome is still not satisfactory. It was shown that gemcitabine suppressed growth of pancreatic cancer cells by inducing apoptosis. Gemcitabine also caused activation of p38 mitogen-activated protein kinase (MAPK), MAPK-activated protein kinase 2 (MAPKAPK-2) with concomitant serine phosphorylation of Hsp27 at position 15, 78, and 82 without affecting total Hsp27 levels. From these results the authors conclude that the phosphorylation status of Hsp27 obviously plays a pivotal role in gemcitabine-induced growth suppression of pancreatic cancer. Of Note, the HSP inhibitor KNK437, a benzylidene lactam compound, was observed to dramatically reduce expression of Hsp27 in gemcitabine-resistant pancreatic cancer cells KLM1-R and to enhance the in vitro anti-tumor cytotoxic effect of gemcitabine on KLM1-R compared to single-agent gemcitabine (Taba et al., 2011). KNK437 also sensitizes prostate cancer cells to the apoptotic effect of hyperthermia by down-regulating heat-induced Hsp70 mRNA expression (Sahin et al., 2011). Moreover, in patients with locally advanced squamous-cell esophageal cancer neoadjuvant radiochemotherapy (NRCT) led to a decreased expression of Hsp16.2, Hsp90, and heme-binding protein 2 (SOUL), and an increased Bax/Bcl-2 ratio was found in the responding tumors (Farkas et al., 2011).
Also Hsp90 may represent a potentially attractive target for specific molecular anti-cancer agents, because Hsp90 expression is up-regulated in tumors as compared with normal tissues, which implies that tumor cells might be preferentially affected by Hsp90-targeted therapies (Ferrarini et al., 1992). In this context, geldanamycin (GA), a naturally occurring ansamycin antibiotic, along with its clinically used analogs such as 17-allylamino-17-demethoxygeldanamycin (17-AAG), has been evaluated in preclinical and clinical trials for its significant anti-tumor properties. These agents disrupt Hsp90 association with client proteins by occupying the nucleotide-binding site of Hsp90 (Grenert et al., 1997; Prodromou et al., 1997; Stebbins et al., 1997), thereby preventing binding of Hsp90 with ATP and profoundly affecting the composition of Hsp90-containing multimolecular chaperone complexes (Obermann et al., 1998; Maloney and Workman, 2002). As demonstrated by the group of Gius, treatment of two human cervical carcinoma cell lines (HeLa, SiHa) with geldanamycin and 17-AAG resulted in cytotoxicity and, when combined with IR, enhanced the radiation response. In addition, mouse in vivo models using 17-AAG at clinically achievable concentrations yielded results that paralleled the in vitro radiosensitization studies of both single and fractioned courses of irradiation (Bisht et al., 2003). We recently analyzed the effects of the novel Hsp90 inhibitor NVP-AUY922 compared to 17-AAG on the HIF-1α/HIF-2α expression in combination with radiosensitivity in lung cancer cell lines under normoxic and hypoxic conditions (Schilling et al., 2012b). NVP-AUY922 is a synthetic, isoxazole/resorcinol-based second generation Hsp90 inhibitor exhibiting an enhanced metabolic stability and a tighter binding to Hsp90 compared to 17-AAG (Brough et al., 2008). As given in our study, both inhibitors reduced basal and hypoxia-induced HIF-1α levels in EPLC-272H lung carcinoma cells. However, despite a down-regulation of HIF-1α upon Hsp90 inhibition, sensitivity toward irradiation remained unaltered in EPLC-272H cells under normoxic and hypoxic conditions. In contrast, treatment of H1339 lung carcinoma cells with NVP-AUY922 and 17-AAG resulted in a significant up-regulation of their initially high HIF-1α levels and a concomitant increase in radiosensitivity indicating the ability of an HIF-1α-independent radiosensitization of normoxic and hypoxic H1339 lung cancer cells via Hsp90 inhibition. From these observations it can be concluded that treatment strategies combining HSP targeting and radiochemotherapy appear to be a high potential therapeutic benefits for cancer patients.
Although radiation therapy, alone or in combination with chemotherapy, is the primary treatment for several tumors, radioresistance dramatically attenuates radiocurability. Several lines of evidence indicate that inflammation plays a pivotal role in modulating radiation responsiveness of tumors. As discussed in this review, exposure to IR leads to activation of several transcription factors modulating the expression of numerous factors promoting cancer development. Novel therapeutic approaches thus aim to interfere with the activity or expression of these factors, either in single-agent or combinatorial treatment or as supplements of the existing therapeutic concepts. Among them, NF-κB, STAT-3, and HIF-1 play a crucial role in radiation-induced inflammatory responses. A great variety of classical or novel drugs including nutraceuticals have the capacity to interfere with the inflammatory network in cancer and are considered to function as putative radiosensitizers (Figure 3). Thus, targeting the inflammatory signaling pathways induced by IR offers the opportunity to improve the clinical outcome of radiation therapy by enhancing radiosensitivity and decreasing putative metabolic effects. Since inflammation and sex steroids also impact tumorigenesis, a therapeutic approach targeting glucocorticoid receptors, and radiation-induced production of tumorigenic factors might be effective in sensitizing tumor cells to IR in certain cases.
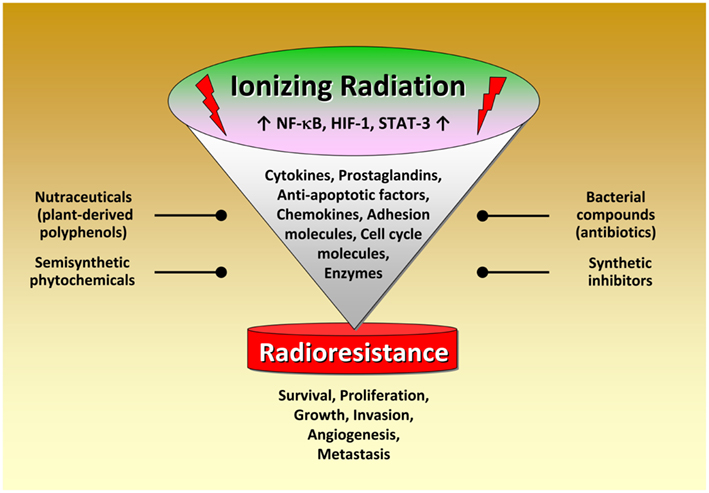
Figure 3. Radiation-induced activation of inflammatory pathways in tumor cells. Schematical simplified representation of the complex intracellular mechanisms leading to radioresistance. Exposure to ionizing radiation leads to activation of several transcription factors modulating the expression of numerous factors promoting cancer development. Novel therapeutic approaches thus aim to interfere with the activity or expression of these factors, either in single-agent or combinatorial treatment or as supplements of the existing therapeutic concepts. Noteworthy, targeting the pro-inflammatory signaling pathways for tumor radiosensitization represents a promising novel therapeutical approach in cancer. A great variety of classical or novel drugs including nutraceuticals have the capacity to interfere with the inflammatory network in cancer and are considered as putative radiosensitizers.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was supported by the Deutsche Forschungsgemeinschaft (DFG) SFB-824, DFG-Cluster of Excellence: Munich Advanced Photonics, Bundesministerium für Bildung und Forschung (BMBF) MOBITUM (01EZ0826) Innovative Therapies (01GU0823), m4 – Personalized Medicine and Targeted Therapies (01EX1021C), Kompetenzverbund Strahlenforschung (03NUK007E), and multimmune GmbH.
Abdel-Wahab, Z., Dar, M. M., Hester, D., Vervaert, C., Gangavalli, R., Barber, J., Darrow, T. L., and Seigler, H. F. (1996). Effect of irradiation on cytokine production, MHC antigen expression, and vaccine potential of interleukin-2 and interferon-gamma gene-modified melanoma cells. Cell. Immunol. 171, 246–254.
Akashi, M., Hachiya, M., Koeffler, H. P., and Suzuki, G. (1992). Irradiation increases levels of GM-CSF through RNA stabilization which requires an AU-rich region in cancer cells. Biochem. Biophys. Res. Commun. 189, 986–993.
Akira, S., Uematsu, S., and Takeuchi, O. (2006). Pathogen recognition and innate immunity. Cell 124, 783–801.
Albert, J. M., Kim, K. W., Cao, C., and Lu, B. (2006). Targeting the Akt/mammalian target of rapamycin pathway for radiosensitization of breast cancer. Mol. Cancer Ther. 5, 1183–1189.
Antoon, J. W., White, M. D., Slaughter, E. M., Driver, J. L., Khalili, H. S., Elliott, S., Smith, C. D., Burow, M. E., and Beckman, B. S. (2011). Targeting NFκB mediated breast cancer chemoresistance through selective inhibition of sphingosine kinase-2. Cancer Biol. Ther. 11, 678–689.
Apetoh, L., Ghiringhelli, F., Tesniere, A., Obeid, M., Ortiz, C., Criollo, A., Mignot, G., Maiuri, M. C., Ullrich, E., Saulnier, P., Yang, H., Amigorena, S., Ryffel, B., Barrat, F. J., Saftig, P., Levi, F., Lidereau, R., Nogues, C., Mira, J. P., Chompret, A., Joulin, V., Clavel-Chapelon, F., Bourhis, J., Andre, F., Delaloge, S., Tursz, T., Kroemer, G., and Zitvogel, L. (2007). Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat. Med. 13, 1050–1059.
Arends, J. E., Hoepelman, A. I., Nanlohy, N. M., Höppener, F. J., Hirsch, K. R., Park, J. G., and van Baarle, D. (2011). Low doses of the novel caspase-inhibitor GS-9450 leads to lower caspase-3 and -8 expression on peripheral CD4+ and CD8+ T-cells. Apoptosis 16, 959–966.
Arispe, N., and De Maio, A. (2000). ATP and ADP modulate a cation channel formed by Hsc70 in acidic phospholipid membranes. J. Biol. Chem. 275, 30839–30843.
Arispe, N., Doh, M., and De Maio, A. (2002). Lipid interaction differentiates the constitutive and stress-induced heat shock proteins Hsc70 and Hsp70. Cell Stress Chaperones 7, 330–338.
Asea, A., Kraeft, S. K., Kurt-Jones, E. A., Stevenson, M. A., Chen, L. B., Finberg, R. W., Koo, G. C., and Calderwood, S. K. (2000). HSP70 stimulates cytokine production through a CD14-dependant pathway, demonstrating its dual role as a chaperone and cytokine. Nat. Med. 6, 435–442.
Asea, A., Rehli, M., Kabingu, E., Boch, J. A., Bare, O., Auron, P. E., Stevenson, M. A., and Calderwood, S. K. (2002). Novel signal transduction pathway utilized by extracellular HSP70: role of toll-like receptor (TLR) 2 and TLR4. J. Biol. Chem. 277, 15028–15034.
Aykin-Burns, N., Slane, B. G., Liu, A. T., Owens, K. M., O’Malley, M. S., Smith, B. J., Domann, F. E., and Spitz, D. R. (2011). Sensitivity to low-dose/low-LET ionizing radiation in mammalian cells harboring mutations in succinate dehydrogenase subunit C is governed by mitochondria-derived reactive oxygen species. Radiat. Res. 175, 150–158.
Balasubramanyam, K., Varier, R. A., Altaf, M., Swaminathan, V., Siddappa, N. B., Ranga, U., and Kundu, T. K. (2004). Curcumin, a novel p300/CREB-binding protein-specific inhibitor of acetyltransferase, represses the acetylation of histone/nonhistone proteins and histone acetyltransferase-dependent chromatin transcription. J. Biol. Chem. 279, 51163–51171.
Beatson, G. T. (1896a). On the treatment of inoperable cases of carcinoma of the mamma: suggestions for a new method of treatment, with illustrative cases. Lancet 2, 104–107.
Beatson, G. T. (1896b). On the treatment of inoperable cases of carcinoma of the mamma: suggestions for a new method of treatment, with illustrative cases. Lancet 2, 162–165.
Ben Baruch, A. (2006). Inflammation-associated immune suppression in cancer: the roles played by cytokines, chemokines and additional mediators. Semin. Cancer Biol. 16, 38–52.
Ben-Neriah, Y., and Karin, M. (2011). Inflammation meets cancer, with NF-κB as the matchmaker. Nat. Immunol. 12, 715–723.
Bianchi, M. E. (2007). DAMPs, PAMPs and alarmins: all we need to know about danger. J. Leukoc. Biol. 81, 1–5.
Bisht, K. S., Bradbury, C. M., Mattson, D., Kaushal, A., Sowers, A., Markovina, S., Ortiz, K. L., Sieck, L. K., Isaacs, J. S., Brechbiel, M. W., Mitchell, J. B., Neckers, L. M., and Gius, D. (2003). Geldanamycin and 17-allylamino-17-demethoxygeldanamycin potentiate the in vitro and in vivo radiation response of cervical tumor cells via the heat shock protein 90-mediated intracellular signaling and cytotoxicity. Cancer Res. 63, 8984–8995.
Brough, P. A., Aherne, W., Barril, X., Borgognoni, J., Boxall, K., Cansfield, J. E., Cheung, K. M., Collins, I., Davies, N. G., Drysdale, M. J., Dymock, B., Eccles, S. A., Finch, H., Fink, A., Hayes, A., Howes, R., Hubbard, R. E., James, K., Jordan, A. M., Lockie, A., Martins, V., Massey, A., Matthews, T. P., McDonald, E., Northfield, C. J., Pearl, L. H., Prodromou, C., Ray, S., Raynaud, F. I., Roughley, S. D., Sharp, S. Y., Surgenor, A., Walmsley, D. L., Webb, P., Wood, M., Workman, P., and Wright, L. (2008). 4,5-diarylisoxazole Hsp90 chaperone inhibitors: potential therapeutic agents for the treatment of cancer. J. Med. Chem. 51, 196–218.
Cerella, C., Sobolewski, C., Dicato, M., and Diederich, M. (2010). Targeting COX-2 expression by natural compounds: a promising alternative strategy to synthetic COX-2 inhibitors for cancer chemoprevention and therapy. Biochem. Pharmacol. 80, 1801–1815.
Chang, C. L., Marra, G., Chauhan, D. P., Ha, H. T., Chang, D. K., Ricciardiello, L., Randolph, A., Carethers, J. M., and Boland, C. R. (2002). Oxidative stress inactivates the human DNA mismatch repair system. Am. J. Physiol. Cell Physiol. 283, C148–C154.
Chaturvedi, M. M., Sung, B., Yadav, V. R., Kannappan, R., and Aggarwal, B. B. (2011). NF-κB addiction and its role in cancer: “one size does not fit all.” Oncogene 30, 1615–1630.
Che, S. M., Zhang, X. Z., Hou, L., and Song, T. B. (2010). Cyclooxygenase-2 inhibitor NS398 enhances radiosensitivity of radioresistant esophageal cancer cells by inhibiting AKT activation and inducing apoptosis. Cancer Invest. 28, 679–688.
Che, S. M., Zhang, X. Z., Liu, X. L., Chen, X., and Hou, L. (2011). The radiosensitization effect of NS398 on esophageal cancer stem cell-like radioresistant cells. Dis. Esophagus 24, 265–273.
Chen, J., Wang, J., Lin, L., He, L., Wu, Y., Zhang, L., Yi, Z., Chen, Y., Pang, X., and Liu, M. (2012a). Inhibition of STAT3 signaling pathway by nitidine chloride suppressed the angiogenesis and growth of human gastric cancer. Mol. Cancer Ther. 11, 277–287.
Chen, Z., Huang, C., Yang, Y. L., Ding, Y., Ou-Yang, H. Q., Zhang, Y. Y., and Xu, M. (2012b). Inhibition of the STAT3 signaling pathway is involved in the antitumor activity of cepharanthine in SaOS2 cells. Acta Pharmacol. Sin. 33, 101–108.
Chen, M. L., Pittet, M. J., Gorelik, L., Flavell, R. A., Weissleder, R., von Boehmer, H., and Khazaie, K. (2005). Regulatory T cells suppress tumor-specific CD8 T cell cytotoxicity through TGF-β signals in vivo. Proc. Natl. Acad. Sci. U.S.A. 102, 419–424.
Chendil, D., Ranga, R. S., Meigooni, D., Sathishkumar, S., and Ahmed, M. M. (2004). Curcumin confers radiosensitizing effect in prostate cancer cell line PC-3. Oncogene 23, 1599–1607.
Cheng, J. C., Chou, C. H., Kuo, M. L., and Hsieh, C. Y. (2006). Radiation-enhanced hepatocellular carcinoma cell invasion with MMP-9 expression through PI3K/Akt/NF-κB signal transduction pathway. Oncogene 25, 7009–7018.
Cheng, S., Brzostek, S., Lee, S. R., Hollenberg, A. N., and Balk, S. P. (2002). Inhibition of the dihydrotestosterone-activated androgen receptor by nuclear receptor corepressor. Mol. Endocrinol. 16, 1492–1501.
Cho, M. L., Kang, J. W., Moon, Y. M., Nam, H. J., Jhun, J. Y., Heo, S. B., Jin, H. T., Min, S. Y., Ju, J. H., Park, K. S., Cho, Y. G., Yoon, C. H., Park, S. H., Sung, Y. C., and Kim, H. Y. (2006). STAT3 and NF-κB signal pathway is required for IL-23-mediated IL-17 production in spontaneous arthritis animal model IL-1 receptor antagonist-deficient mice. J. Immunol. 176, 5652–5661.
Ciernik, I. F., Romero, P., Berzofsky, J. A., and Carbone, D. P. (1999). Ionizing radiation enhances immunogenicity of cells expressing a tumor-specific T-cell epitope. Int. J. Radiat. Oncol. Biol. Phys. 45, 735–741.
Coleman, M. L., Sahai, E. A., Yeo, M., Bosch, M., Dewar, A., and Olson, M. F. (2001). Membrane blebbing during apoptosis results from caspase-mediated activation of ROCK I. Nat. Cell Biol. 3, 339–345.
Colotta, F., Allavena, P., Sica, A., Garlanda, C., and Mantovani, A. (2009). Cancer-related inflammation, the seventh hallmark of cancer: links to genetic instability. Carcinogenesis 30, 1073–1081.
De Maio, A. (2011). Extracellular heat shock proteins, cellular export vesicles, and the stress observation system: a form of communication during injury, infection, and cell damage. It is never known how far a controversial finding will go! Dedicated to Ferruccio Ritossa. Cell Stress Chaperones 16, 235–249.
Deorukhkar, A., and Krishnan, S. (2010). Targeting inflammatory pathways for tumor radiosensitization. Biochem. Pharmacol. 80, 1904–1914.
Deorukhkar, A., Krishnan, S., Sethi, G., and Aggarwal, B. B. (2007). Back to basics: how natural products can provide the basis for new therapeutics. Expert. Opin. Investig. Drugs 16, 1753–1773.
Dhillon, N., Aggarwal, B. B., Newman, R. A., Wolff, R. A., Kunnumakkara, A. B., Abbruzzese, J. L., Ng, C. S., Badmaev, V., and Kurzrock, R. (2008). Phase II trial of curcumin in patients with advanced pancreatic cancer. Clin. Cancer Res. 14, 4491–4499.
Dittmann, K. H., Mayer, C., Ohneseit, P. A., Raju, U., Andratschke, N. H., Milas, L., and Rodemann, H. P. (2008). Celecoxib induced tumor cell radiosensitization by inhibiting radiation induced nuclear EGFR transport and DNA-repair: a COX-2 independent mechanism. Int. J. Radiat. Oncol. Biol. Phys. 70, 203–212.
Divanovic, S., Trompette, A., Atabani, S. F., Madan, R., Golenbock, D. T., Visintin, A., Finberg, R. W., Tarakhovsky, A., Vogel, S. N., Belkaid, Y., Kurt-Jones, E. A., and Karp, C. L. (2005). Negative regulation of Toll-like receptor 4 signaling by the Toll-like receptor homolog RP105. Nat. Immunol. 6, 571–578.
Endo, Y., Marusawa, H., Kou, T., Nakase, H., Fujii, S., Fujimori, T., Kinoshita, K., Honjo, T., and Chiba, T. (2008). Activation-induced cytidine deaminase links between inflammation and the development of colitis-associated colorectal cancers. Gastroenterology 135, 889–898.
Erdman, S. E., Rao, V. P., Poutahidis, T., Ihrig, M. M., Ge, Z., Feng, Y., Tomczak, M., Rogers, A. B., Horwitz, B. H., and Fox, J. G. (2003). CD4(+)CD25(+) regulatory lymphocytes require interleukin 10 to interrupt colon carcinogenesis in mice. Cancer Res. 63, 6042–6050.
Fahy, B. N., Schlieman, M. G., Virudachalam, S., and Bold, R. J. (2004). Inhibition of AKT abrogates chemotherapy-induced NF-κB survival mechanisms: implications for therapy in pancreatic cancer. J. Am. Coll. Surg. 198, 591–599.
Farkas, R., Pozsgai, E., Bellyei, S., Cseke, L., Szigeti, A., Vereczkei, A., Marton, S., Mangel, L., Horvath, O. P., and Papp, A. (2011). Correlation between tumor-associated proteins and response to neoadjuvant treatment in patients with advanced squamous-cell esophageal cancer. Anticancer Res. 31, 1769–1775.
Fedrigo, C. A., Grivicich, I., Schunemann, D. P., Chemale, I. M., dos, S. D., Jacovas, T., Boschetti, P. S., Jotz, G. P., Braga, F. A., and da Rocha, A. B. (2011). Radioresistance of human glioma spheroids and expression of HSP70, p53 and EGFr. Radiat. Oncol. 6, 156.
Ferrarini, M., Heltai, S., Zocchi, M. R., and Rugarli, C. (1992). Unusual expression and localization of heat-shock proteins in human tumor cells. Int. J. Cancer 51, 613–619.
Fugier-Vivier, I., de Bouteiller, O., Guret, C., Fossiez, F., Banchereau, J., Mattei, M. G., Ait-Yahia, S., Garcia, E., Lebecque, S., and Liu, Y. J. (1997). Molecular cloning of human RP105. Eur. J. Immunol. 27, 1824–1827.
Galabova-Kovacs, G., Kolbus, A., Matzen, D., Meissl, K., Piazzolla, D., Rubiolo, C., Steinitz, K., and Baccarini, M. (2006). ERK and beyond: insights from B-Raf and Raf-1 conditional knockouts. Cell Cycle 5, 1514–1518.
Ganss, R., Ryschich, E., Klar, E., Arnold, B., and Hammerling, G. J. (2002). Combination of T-cell therapy and trigger of inflammation induces remodeling of the vasculature and tumor eradication. Cancer Res. 62, 1462–1470.
Garcia-Pineres, A. J., Castro, V., Mora, G., Schmidt, T. J., Strunck, E., Pahl, H. L., and Merfort, I. (2001). Cysteine 38 in p65/NF-κB plays a crucial role in DNA binding inhibition by sesquiterpene lactones. J. Biol. Chem. 276, 39713–39720.
Garg, A. K., Buchholz, T. A., and Aggarwal, B. B. (2005). Chemosensitization and radiosensitization of tumors by plant polyphenols. Antioxid. Redox Signal. 7, 1630–1647.
Gehrmann, M., Liebisch, G., Schmitz, G., Anderson, R., Steinem, C., De Maio, A., Pockley, G., and Multhoff, G. (2008). Tumor-specific Hsp70 plasma membrane localization is enabled by the glycosphingolipid Gb3. PLoS ONE 3, e1925. doi:10.1371/journal.pone.0001925
Goldkorn, T., Balaban, N., Shannon, M., and Matsukuma, K. (1997). EGF receptor phosphorylation is affected by ionizing radiation. Biochim. Biophys. Acta 1358, 289–299.
Gorski, D. H., Beckett, M. A., Jaskowiak, N. T., Calvin, D. P., Mauceri, H. J., Salloum, R. M., Seetharam, S., Koons, A., Hari, D. M., Kufe, D. W., and Weichselbaum, R. R. (1999). Blockage of the vascular endothelial growth factor stress response increases the antitumor effects of ionizing radiation. Cancer Res. 59, 3374–3378.
Grenert, J. P., Sullivan, W. P., Fadden, P., Haystead, T. A., Clark, J., Mimnaugh, E., Krutzsch, H., Ochel, H. J., Schulte, T. W., Sausville, E., Neckers, L. M., and Toft, D. O. (1997). The amino-terminal domain of heat shock protein 90 (hsp90) that binds geldanamycin is an ATP/ADP switch domain that regulates hsp90 conformation. J. Biol. Chem. 272, 23843–23850.
Grimes, K. R., Warren, G. W., Fang, F., Xu, Y., and St. Clair, W. H. (2006). Cyclooxygenase-2 inhibitor, nimesulide, improves radiation treatment against non-small cell lung cancer both in vitro and in vivo. Oncol. Rep. 16, 771–776.
Grivennikov, S. I., and Karin, M. (2010). Inflammation and oncogenesis: a vicious connection. Curr. Opin. Genet. Dev. 20, 65–71.
Guttmann, D. M., and Koumenis, C. (2011). The heat shock proteins as targets for radiosensitization and chemosensitization in cancer. Cancer Biol. Ther. 12, 1023–1031.
Hadden, J. W. (2003). Immunodeficiency and cancer: prospects for correction. Int. Immunopharmacol. 3, 1061–1071.
Handrick, R., Ganswindt, U., Faltin, H., Goecke, B., Daniel, P. T., Budach, W., Belka, C., and Jendrossek, V. (2009). Combined action of celecoxib and ionizing radiation in prostate cancer cells is independent of pro-apoptotic Bax. Radiother. Oncol. 90, 413–421.
Harada, H. (2011). How can we overcome tumor hypoxia in radiation therapy? J. Radiat. Res. 52, 545–556.
Harada, H., Itasaka, S., Kizaka-Kondoh, S., Shibuya, K., Morinibu, A., Shinomiya, K., and Hiraoka, M. (2009a). The Akt/mTOR pathway assures the synthesis of HIF-1α protein in a glucose- and reoxygenation-dependent manner in irradiated tumors. J. Biol. Chem. 284, 5332–5342.
Harada, H., Itasaka, S., Zhu, Y., Zeng, L., Xie, X., Morinibu, A., Shinomiya, K., and Hiraoka, M. (2009b). Treatment regimen determines whether an HIF-1 inhibitor enhances or inhibits the effect of radiation therapy. Br. J. Cancer 100, 747–757.
Härdtner, C., Multhoff, G., Falk, W., and Radons, J. (2012). (-)-Epigallocatechin-3-gallate, a green tea-derived catechin, synergizes with celecoxib to inhibit IL-1-induced tumorigenic mediators by human pancreatic adenocarcinoma cells Colo357. Eur. J. Pharmacol. 684, 36–43.
Harikumar, K. B., Kunnumakkara, A. B., Ahn, K. S., Anand, P., Krishnan, S., Guha, S., and Aggarwal, B. B. (2009). Modification of the cysteine residues in IκBα kinase and NF-κB (p65) by xanthohumol leads to suppression of NF-κB-regulated gene products and potentiation of apoptosis in leukemia cells. Blood 113, 2003–2013.
Henderson, I. C., and Canellos, G. P. (1980a). Cancer of the breast: the past decade (first of two parts). N. Engl. J. Med. 302, 17–30.
Henderson, I. C., and Canellos, G. P. (1980b). Cancer of the breast: the past decade (second of two parts). N. Engl. J. Med. 302, 78–90.
Hermens, A. F., and Barendsen, G. W. (1969). Changes of cell proliferation characteristics in a rat rhabdomyosarcoma before and after x-irradiation. Eur. J. Cancer 5, 173–189.
Hightower, L. E., and Guidon, P. T. Jr. (1989). Selective release from cultured mammalian cells of heat-shock (stress) proteins that resemble glia-axon transfer proteins. J. Cell. Physiol. 138, 257–266.
Hodgson, M. C., Astapova, I., Cheng, S., Lee, L. J., Verhoeven, M. C., Choi, E., Balk, S. P., and Hollenberg, A. N. (2005). The androgen receptor recruits nuclear receptor CoRepressor (N-CoR) in the presence of mifepristone via its N and C termini revealing a novel molecular mechanism for androgen receptor antagonists. J. Biol. Chem. 280, 6511–6519.
Hong, J. H., Chiang, C. S., Sun, J. R., Withers, H. R., and McBride, W. H. (1997). Induction of c-fos and junB mRNA following in vivo brain irradiation. Brain Res. Mol. Brain Res. 48, 223–228.
Horvath, I., Multhoff, G., Sonnleitner, A., and Vigh, L. (2008). Membrane-associated stress proteins: more than simply chaperones. Biochim. Biophys. Acta 1778, 1653–1664.
Huang, Q., Li, F., Liu, X., Li, W., Shi, W., Liu, F. F., O’Sullivan, B., He, Z., Peng, Y., Tan, A. C., Zhou, L., Shen, J., Han, G., Wang, X. J., Thorburn, J., Thorburn, A., Jimeno, A., Raben, D., Bedford, J. S., and Li, C. Y. (2011). Caspase 3-mediated stimulation of tumor cell repopulation during cancer radiotherapy. Nat. Med. 17, 860–866.
Huang, X. Z., Wang, J., Huang, C., Chen, Y. Y., Shi, G. Y., Hu, Q. S., and Yi, J. (2008). Emodin enhances cytotoxicity of chemotherapeutic drugs in prostate cancer cells: the mechanisms involve ROS-mediated suppression of multidrug resistance and hypoxia inducible factor-1. Cancer Biol. Ther. 7, 468–475.
Jänicke, R. U., Sprengart, M. L., Wati, M. R., and Porter, A. G. (1998). Caspase-3 is required for DNA fragmentation and morphological changes associated with apoptosis. J. Biol. Chem. 273, 9357–9360.
Johnson, G. E., Ivanov, V. N., and Hei, T. K. (2008). Radiosensitization of melanoma cells through combined inhibition of protein regulators of cell survival. Apoptosis 13, 790–802.
Jung, Y. J., Isaacs, J. S., Lee, S., Trepel, J., and Neckers, L. (2003). IL-1β-mediated up-regulation of HIF-1α via an NFκB/COX-2 pathway identifies HIF-1 as a critical link between inflammation and oncogenesis. FASEB J. 17, 2115–2117.
Kaelin, W. G. Jr., and Ratcliffe, P. J. (2008). Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Mol. Cell 30, 393–402.
Kao, G. D., Jiang, Z., Fernandes, A. M., Gupta, A. K., and Maity, A. (2007). Inhibition of phosphatidylinositol-3-OH kinase/Akt signaling impairs DNA repair in glioblastoma cells following ionizing radiation. J. Biol. Chem. 282, 21206–21212.
Karanjawala, Z. E., Murphy, N., Hinton, D. R., Hsieh, C. L., and Lieber, M. R. (2002). Oxygen metabolism causes chromosome breaks and is associated with the neuronal apoptosis observed in DNA double-strand break repair mutants. Curr. Biol. 12, 397–402.
Karin, M., Lawrence, T., and Nizet, V. (2006). Innate immunity gone awry: linking microbial infections to chronic inflammation and cancer. Cell 124, 823–835.
Kawai, T., and Akira, S. (2011). Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity 34, 637–650.
Kim, K. W., Mutter, R. W., Cao, C., Albert, J. M., Shinohara, E. T., Sekhar, K. R., and Lu, B. (2006a). Inhibition of signal transducer and activator of transcription 3 activity results in down-regulation of survivin following irradiation. Mol. Cancer Ther. 5, 2659–2665.
Kim, R., Emi, M., Tanabe, K., and Arihiro, K. (2006b). Tumor-driven evolution of immunosuppressive networks during malignant progression. Cancer Res. 66, 5527–5536.
Kim, W. Y., Oh, S. H., Woo, J. K., Hong, W. K., and Lee, H. Y. (2009). Targeting heat shock protein 90 overrides the resistance of lung cancer cells by blocking radiation-induced stabilization of hypoxia-inducible factor-1α. Cancer Res. 69, 1624–1632.
Koshiji, M., To, K. K., Hammer, S., Kumamoto, K., Harris, A. L., Modrich, P., and Huang, L. E. (2005). HIF-1α induces genetic instability by transcriptionally downregulating MutSalpha expression. Mol. Cell 17, 793–803.
Kuipers, G. K., Slotman, B. J., Wedekind, L. E., Stoter, T. R., Berg, J., Sminia, P., and Lafleur, M. V. (2007). Radiosensitization of human glioma cells by cyclooxygenase-2 (COX-2) inhibition: independent on COX-2 expression and dependent on the COX-2 inhibitor and sequence of administration. Int. J. Radiat. Biol. 83, 677–685.
Kundu, J. K., and Surh, Y. J. (2008). Inflammation: gearing the journey to cancer. Mutat. Res. 659, 15–30.
Kunnumakkara, A. B., Diagaradjane, P., Guha, S., Deorukhkar, A., Shentu, S., Aggarwal, B. B., and Krishnan, S. (2008). Curcumin sensitizes human colorectal cancer xenografts in nude mice to gamma-radiation by targeting nuclear factor-κB-regulated gene products. Clin. Cancer Res. 14, 2128–2136.
Langowski, J. L., Zhang, X., Wu, L., Mattson, J. D., Chen, T., Smith, K., Basham, B., McClanahan, T., Kastelein, R. A., and Oft, M. (2006). IL-23 promotes tumour incidence and growth. Nature 442, 461–465.
LeBlanc, H. N., and Ashkenazi, A. (2003). Apo2L/TRAIL and its death and decoy receptors. Cell Death Differ. 10, 66–75.
Lee, K., Zhang, H., Qian, D. Z., Rey, S., Liu, J. O., and Semenza, G. L. (2009). Acriflavine inhibits HIF-1 dimerization, tumor growth, and vascularization. Proc. Natl. Acad. Sci. U.S.A. 106, 17910–17915.
Li, F., and Sethi, G. (2010). Targeting transcription factor NF-κB to overcome chemoresistance and radioresistance in cancer therapy. Biochim. Biophys. Acta 1805, 167–180.
Lin, W. W., and Karin, M. (2007). A cytokine-mediated link between innate immunity, inflammation, and cancer. J. Clin. Invest. 117, 1175–1183.
Liu, J., Zhang, J., Wang, X., Li, Y., Chen, Y., Li, K., Zhang, J., Yao, L., and Guo, G. (2010). HIF-1 and NDRG2 contribute to hypoxia-induced radioresistance of cervical cancer Hela cells. Exp. Cell Res. 316, 1985–1993.
Lopes, C. O., and Callera, F. (2011). Three-dimensional conformal radiotherapy in prostate cancer patients: rise in interleukin 6 (IL-6) but not IL-2, IL-4, IL-5, tumor necrosis factor-α, MIP-1-α, and LIF levels. Int. J. Radiat. Oncol. Biol. Phys. 82, 1385–1388.
Luster, A. D., Alon, R., and von Andrian, U. H. (2005). Immune cell migration in inflammation: present and future therapeutic targets. Nat. Immunol. 6, 1182–1190.
Magne, N., Toillon, R. A., Bottero, V., Didelot, C., Houtte, P. V., Gerard, J. P., and Peyron, J. F. (2006). NF-κB modulation and ionizing radiation: mechanisms and future directions for cancer treatment. Cancer Lett. 231, 158–168.
Majumder, P. K., Febbo, P. G., Bikoff, R., Berger, R., Xue, Q., McMahon, L. M., Manola, J., Brugarolas, J., McDonnell, T. J., Golub, T. R., Loda, M., Lane, H. A., and Sellers, W. R. (2004). mTOR inhibition reverses Akt-dependent prostate intraepithelial neoplasia through regulation of apoptotic and HIF-1-dependent pathways. Nat. Med. 10, 594–601.
Maloney, A., and Workman, P. (2002). HSP90 as a new therapeutic target for cancer therapy: the story unfolds. Expert. Opin. Biol. Ther. 2, 3–24.
Mangan, P. R., Harrington, L. E., O’Quinn, D. B., Helms, W. S., Bullard, D. C., Elson, C. O., Hatton, R. D., Wahl, S. M., Schoeb, T. R., and Weaver, C. T. (2006). Transforming growth factor-β induces development of the T(H)17 lineage. Nature 441, 231–234.
Mantovani, A., Allavena, P., Sica, A., and Balkwill, F. (2008). Cancer-related inflammation. Nature 454, 436–444.
Masiello, D., Cheng, S., Bubley, G. J., Lu, M. L., and Balk, S. P. (2002). Bicalutamide functions as an androgen receptor antagonist by assembly of a transcriptionally inactive receptor. J. Biol. Chem. 277, 26321–26326.
Maziere, C., Conte, M. A., Leborgne, L., Levade, T., Hornebeck, W., Santus, R., and Maziere, J. C. (2001). UVA radiation stimulates ceramide production: relationship to oxidative stress and potential role in ERK, JNK, and p38 activation. Biochem. Biophys. Res. Commun. 281, 289–294.
Meirovitz, A., Kuten, M., Billan, S., Abdah-Bortnyak, R., Sharon, A., Peretz, T., Sela, M., Schaffer, M., and Barak, V. (2010). Cytokines levels, severity of acute mucositis and the need of PEG tube installation during chemo-radiation for head and neck cancer – a prospective pilot study. Radiat. Oncol. 5, 16.
Mencin, A., Kluwe, J., and Schwabe, R. F. (2009). Toll-like receptors as targets in chronic liver diseases. Gut 58, 704–720.
Mendonca, M. S., Chin-Sinex, H., Gomez-Millan, J., Datzman, N., Hardacre, M., Comerford, K., Nakshatri, H., Nye, M., Benjamin, L., Mehta, S., Patino, F., and Sweeney, C. (2007). Parthenolide sensitizes cells to X-ray-induced cell killing through inhibition of NF-kappaB and split-dose repair. Radiat. Res. 168, 689–697.
Meng, Y., Mauceri, H. J., Khodarev, N. N., Darga, T. E., Pitroda, S. P., Beckett, M. A., Kufe, D. W., and Weichselbaum, R. R. (2010). Ad.Egr-TNF and local ionizing radiation suppress metastases by interferon-β-dependent activation of antigen-specific CD8+ T cells. Mol. Ther. 18, 912–920.
Menssen, A., Epanchintsev, A., Lodygin, D., Rezaei, N., Jung, P., Verdoodt, B., Diebold, J., and Hermeking, H. (2007). c-MYC delays prometaphase by direct transactivation of MAD2 and BubR1: identification of mechanisms underlying c-MYC-induced DNA damage and chromosomal instability. Cell Cycle 6, 339–352.
Mills, K. D., Ferguson, D. O., and Alt, F. W. (2003). The role of DNA breaks in genomic instability and tumorigenesis. Immunol. Rev. 194, 77–95.
Miyake, K., Yamashita, Y., Hitoshi, Y., Takatsu, K., and Kimoto, M. (1994). Murine B cell proliferation and protection from apoptosis with an antibody against a 105-kD molecule: unresponsiveness of X-linked immunodeficient B cells. J. Exp. Med. 180, 1217–1224.
Miyake, K., Yamashita, Y., Ogata, M., Sudo, T., and Kimoto, M. (1995). RP105, a novel B cell surface molecule implicated in B cell activation, is a member of the leucine-rich repeat protein family. J. Immunol. 154, 3333–3340.
Moeller, B. J., Cao, Y., Li, C. Y., and Dewhirst, M. W. (2004). Radiation activates HIF-1 to regulate vascular radiosensitivity in tumors: role of reoxygenation, free radicals, and stress granules. Cancer Cell 5, 429–441.
Moeller, B. J., and Dewhirst, M. W. (2006). HIF-1 and tumour radiosensitivity. Br. J. Cancer 95, 1–5.
Moon, S. Y., Chang, H. W., Roh, J. L., Kim, G. C., Choi, S. H., Lee, S. W., Cho, K. J., Nam, S. Y., and Kim, S. Y. (2009). Using YC-1 to overcome the radioresistance of hypoxic cancer cells. Oral Oncol. 45, 915–919.
Moore, K. W., de Waal, M. R., Coffman, R. L., and O’Garra, A. (2001). Interleukin-10 and the interleukin-10 receptor. Annu. Rev. Immunol. 19, 683–765.
Moral, M. A., and Tomillero, A. (2008). Gateways to clinical trials. Methods Find. Exp. Clin. Pharmacol. 30, 149–171.
Multhoff, G. (2007). Heat shock protein 70 (Hsp70): membrane location, export and immunological relevance. Methods 43, 229–237.
Multhoff, G., Botzler, C., Wiesnet, M., Müller, E., Meier, T., Wilmanns, W., and Issels, R. D. (1995). A stress-inducible 72-kDa heat-shock protein (HSP72) is expressed on the surface of human tumor cells, but not on normal cells. Int. J. Cancer 61, 272–279.
Multhoff, G., and Hightower, L. E. (1996). Cell surface expression of heat shock proteins and the immune response. Cell Stress Chaperones 1, 167–176.
Multhoff, G., Molls, M., and Radons, J. (2012). Chronic inflammation in cancer development. Front. Immunol. 2:98. doi:10.3389/fimmu.2011.00098
Nakamura, J. L., Karlsson, A., Arvold, N. D., Gottschalk, A. R., Pieper, R. O., Stokoe, D., and Haas-Kogan, D. A. (2005). PKB/Akt mediates radiosensitization by the signaling inhibitor LY294002 in human malignant gliomas. J. Neurooncol. 71, 215–222.
Nakashima, M., Adachi, S., Yasuda, I., Yamauchi, T., Kawaguchi, J., Itani, M., Yoshioka, T., Matsushima-Nishiwaki, R., Hirose, Y., Kozawa, O., and Moriwaki, H. (2011). Phosphorylation status of heat shock protein 27 plays a key role in gemcitabine-induced apoptosis of pancreatic cancer cells. Cancer Lett. 313, 218–225.
Nambiar, D., Rajamani, P., and Singh, R. P. (2011). Effects of phytochemicals on ionization radiation-mediated carcinogenesis and cancer therapy. Mutat. Res. 728, 139–157.
Naugler, W. E., Sakurai, T., Kim, S., Maeda, S., Kim, K., Elsharkawy, A. M., and Karin, M. (2007). Gender disparity in liver cancer due to sex differences in MyD88-dependent IL-6 production. Science 317, 121–124.
Nickel, W., and Seedorf, M. (2008). Unconventional mechanisms of protein transport to the cell surface of eukaryotic cells. Annu. Rev. Cell Dev. Biol. 24, 287–308.
North, R. J. (1984). Gamma-irradiation facilitates the expression of adoptive immunity against established tumors by eliminating suppressor T cells. Cancer Immunol. Immunother. 16, 175–181.
Numasaki, M., Fukushi, J., Ono, M., Narula, S. K., Zavodny, P. J., Kudo, T., Robbins, P. D., Tahara, H., and Lotze, M. T. (2003). Interleukin-17 promotes angiogenesis and tumor growth. Blood 101, 2620–2627.
Obermann, W. M., Sondermann, H., Russo, A. A., Pavletich, N. P., and Hartl, F. U. (1998). In vivo function of Hsp90 is dependent on ATP binding and ATP hydrolysis. J. Cell Biol. 143, 901–910.
Oblak, A., and Jerala, R. (2011). Toll-like receptor 4 activation in cancer progression and therapy. Clin. Dev. Immunol. 2011, 609579.
Ohto, U., Miyake, K., and Shimizu, T. (2011). Crystal structures of mouse and human RP105/MD-1 complexes reveal unique dimer organization of the toll-like receptor family. J. Mol. Biol. 413, 815–825.
Pandey, M. K., Sandur, S. K., Sung, B., Sethi, G., Kunnumakkara, A. B., and Aggarwal, B. B. (2007). Butein, a tetrahydroxychalcone, inhibits nuclear factor (NF)-κB and NF-κB-regulated gene expression through direct inhibition of IκBα kinase β on cysteine 179 residue. J. Biol. Chem. 282, 17340–17350.
Pandey, M. K., Sung, B., Kunnumakkara, A. B., Sethi, G., Chaturvedi, M. M., and Aggarwal, B. B. (2008). Berberine modifies cysteine 179 of IκBα kinase, suppresses nuclear factor-κB-regulated antiapoptotic gene products, and potentiates apoptosis. Cancer Res. 68, 5370–5379.
Park, H., Li, Z., Yang, X. O., Chang, S. H., Nurieva, R., Wang, Y. H., Wang, Y., Hood, L., Zhu, Z., Tian, Q., and Dong, C. (2005). A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat. Immunol. 6, 1133–1141.
Pasi, F., Facoetti, A., and Nano, R. (2010). IL-8 and IL-6 bystander signalling in human glioblastoma cells exposed to gamma radiation. Anticancer Res. 30, 2769–2772.
Pfister, K., Radons, J., Busch, R., Tidball, J. G., Pfeifer, M., Freitag, L., Feldmann, H. J., Milani, V., Issels, R., and Multhoff, G. (2007). Patient survival by Hsp70 membrane phenotype: association with different routes of metastasis. Cancer 110, 926–935.
Pickering, M. T., and Kowalik, T. F. (2006). Rb inactivation leads to E2F1-mediated DNA double-strand break accumulation. Oncogene 25, 746–755.
Plo, I., Nakatake, M., Malivert, L., de Villartay, J. P., Giraudier, S., Villeval, J. L., Wiesmuller, L., and Vainchenker, W. (2008). JAK2 stimulates homologous recombination and genetic instability: potential implication in the heterogeneity of myeloproliferative disorders. Blood 112, 1402–1412.
Pockley, A. G., and Multhoff, G. (2008). Cell stress proteins in extracellular fluids: friend or foe? Novartis Found. Symp. 291, 86–95.
Pockley, A. G., Muthana, M., and Calderwood, S. K. (2008). The dual immunoregulatory roles of stress proteins. Trends Biochem. Sci. 33, 71–79.
Prasad, S., Yadav, V. R., Sundaram, C., Reuter, S., Hema, P. S., Nair, M. S., Chaturvedi, M. M., and Aggarwal, B. B. (2010). Crotepoxide chemosensitizes tumor cells through inhibition of expression of proliferation, invasion, and angiogenic proteins linked to proinflammatory pathway. J. Biol. Chem. 285, 26987–26997.
Prodromou, C., Roe, S. M., O’Brien, R., Ladbury, J. E., Piper, P. W., and Pearl, L. H. (1997). Identification and structural characterization of the ATP/ADP-binding site in the Hsp90 molecular chaperone. Cell 90, 65–75.
Qiao, Q., Jiang, Y., and Li, G. (2012). Curcumin improves the antitumor effect of X-ray irradiation by blocking the NF-κB pathway: an in-vitro study of lymphoma. Anticancer Drugs. doi:10.1097/CAD.0b013e3283503fbc. [Epub ahead of print].
Raffoul, J. J., Wang, Y., Kucuk, O., Forman, J. D., Sarkar, F. H., and Hillman, G. G. (2006). Genistein inhibits radiation-induced activation of NF-κB in prostate cancer cells promoting apoptosis and G2/M cell cycle arrest. BMC Cancer 6, 107. doi:10.1186/1471-2407-6-107
Rajagopalan, H., Nowak, M. A., Vogelstein, B., and Lengauer, C. (2003). The significance of unstable chromosomes in colorectal cancer. Nat. Rev. Cancer 3, 695–701.
Raju, U., Ariga, H., Dittmann, K., Nakata, E., Ang, K. K., and Milas, L. (2005). Inhibition of DNA repair as a mechanism of enhanced radioresponse of head and neck carcinoma cells by a selective cyclooxygenase-2 inhibitor, celecoxib. Int. J. Radiat. Oncol. Biol. Phys. 63, 520–528.
Reissfelder, C., Timke, C., Schmitz-Winnenthal, H., Rahbari, N. N., Koch, M., Klug, F., Roeder, F., Edler, L., Debus, J., Buchler, M. W., Beckhove, P., Huber, P. E., and Weitz, J. (2011). A randomized controlled trial to investigate the influence of low dose radiotherapy on immune stimulatory effects in liver metastases of colorectal cancer. BMC Cancer 11, 419. doi:10.1186/1471-2407-11-419
Rudner, J., Ruiner, C. E., Handrick, R., Eibl, H. J., Belka, C., and Jendrossek, V. (2010). The Akt-inhibitor Erufosine induces apoptotic cell death in prostate cancer cells and increases the short term effects of ionizing radiation. Radiat. Oncol. 5, 108.
Sahin, E., Sahin, M., Sanlioglu, A. D., and Gumuslu, S. (2011). KNK437, a benzylidene lactam compound, sensitises prostate cancer cells to the apoptotic effect of hyperthermia. Int. J. Hyperthermia 27, 63–73.
Saintigny, Y., Dumay, A., Lambert, S., and Lopez, B. S. (2001). A novel role for the Bcl-2 protein family: specific suppression of the RAD51 recombination pathway. EMBO J. 20, 2596–2607.
Sandau, K. B., Faus, H. G., and Brune, B. (2000). Induction of hypoxia-inducible-factor 1 by nitric oxide is mediated via the PI 3K pathway. Biochem. Biophys. Res. Commun. 278, 263–267.
Sandur, S. K., Deorukhkar, A., Pandey, M. K., Pabon, A. M., Shentu, S., Guha, S., Aggarwal, B. B., and Krishnan, S. (2009). Curcumin modulates the radiosensitivity of colorectal cancer cells by suppressing constitutive and inducible NF-κB activity. Int. J. Radiat. Oncol. Biol. Phys. 75, 534–542.
Schaue, D., and McBride, W. H. (2010). Links between innate immunity and normal tissue radiobiology. Radiat. Res. 173, 406–417.
Schildkopf, P., Frey, B., Ott, O. J., Rubner, Y., Multhoff, G., Sauer, R., Fietkau, R., and Gaipl, U. S. (2011). Radiation combined with hyperthermia induces HSP70-dependent maturation of dendritic cells and release of pro-inflammatory cytokines by dendritic cells and macrophages. Radiother. Oncol. 101, 109–115.
Schilling, D., Bayer, C., Emmerich, K., Molls, M., Vaupel, P., Huber, R. M., and Multhoff, G. (2012a). Basal HIF-1α expression levels are not predictive for radiosensitivity of human cancer cell lines. Strahlenther. Onkol. 188, 353–358.
Schilling, D., Bayer, C., Emmerich, K., Molls, M., Vaupel, P., Huber, R. M., and Multhoff, G. (2012b). Radiosensitization of normoxic and hypoxic H1339 lung tumor cells by heat shock protein 90 inhibition is independent of hypoxia inducible factor-1α. PLoS ONE 7, e31110. doi:10.1371/journal.pone.0031110
Schilling, D., Gehrmann, M., Steinem, C., De Maio, A., Pockley, A. G., Abend, M., Molls, M., and Multhoff, G. (2009). Binding of heat shock protein 70 to extracellular phosphatidylserine promotes killing of normoxic and hypoxic tumor cells. FASEB J. 23, 2467–2477.
Sebbagh, M., Renvoize, C., Hamelin, J., Riche, N., Bertoglio, J., and Breard, J. (2001). Caspase-3-mediated cleavage of ROCK I induces MLC phosphorylation and apoptotic membrane blebbing. Nat. Cell Biol. 3, 346–352.
Shan, Y. X., Jin, S. Z., Liu, X. D., Liu, Y., and Liu, S. Z. (2007). Ionizing radiation stimulates secretion of pro-inflammatory cytokines: dose-response relationship, mechanisms and implications. Radiat. Environ. Biophys. 46, 21–29.
Shang, Y., Myers, M., and Brown, M. (2002). Formation of the androgen receptor transcription complex. Mol. Cell 9, 601–610.
Shi, M., Guo, X. T., Shu, M. G., Chen, F. L., and Li, L. W. (2007). Cell-permeable hypoxia-inducible factor-1 (HIF-1) antagonists function as tumor radiosensitizers. Med. Hypotheses 69, 33–35.
Shin, D. H., Kim, J. H., Jung, Y. J., Kim, K. E., Jeong, J. M., Chun, Y. S., and Park, J. W. (2007). Preclinical evaluation of YC-1, a HIF inhibitor, for the prevention of tumor spreading. Cancer Lett. 255, 107–116.
Singh, S., and Aggarwal, B. B. (1995). Activation of transcription factor NF-κB is suppressed by curcumin (diferuloylmethane) [corrected]. J. Biol. Chem. 270, 24995–25000.
Singh, S., and Khar, A. (2006). Biological effects of curcumin and its role in cancer chemoprevention and therapy. Anticancer Agents Med. Chem. 6, 259–270.
Singh-Gupta, V., Zhang, H., Banerjee, S., Kong, D., Raffoul, J. J., Sarkar, F. H., and Hillman, G. G. (2009). Radiation-induced HIF-1α cell survival pathway is inhibited by soy isoflavones in prostate cancer cells. Int. J. Cancer 124, 1675–1684.
Solomon, S. D., Wittes, J., Finn, P. V., Fowler, R., Viner, J., Bertagnolli, M. M., Arber, N., Levin, B., Meinert, C. L., Martin, B., Pater, J. L., Goss, P. E., Lance, P., Obara, S., Chew, E. Y., Kim, J., Arndt, G., and Hawk, E. (2008). Cardiovascular risk of celecoxib in 6 randomized placebo-controlled trials: the cross trial safety analysis. Circulation 117, 2104–2113.
Son, E. W., Rhee, D. K., and Pyo, S. (2006). Gamma-irradiation-induced intercellular adhesion molecule-1 (ICAM-1) expression is associated with catalase: activation of Ap-1 and JNK. J. Toxicol. Environ. Health Part A 69, 2137–2155.
Son, P. S., Park, S. A., Na, H. K., Jue, D. M., Kim, S., and Surh, Y. J. (2010). Piceatannol, a catechol-type polyphenol, inhibits phorbol ester-induced NF-κB activation and cyclooxygenase-2 expression in human breast epithelial cells: cysteine 179 of IKKβ as a potential target. Carcinogenesis 31, 1442–1449.
Song, B., Song, W. G., Li, Z. J., Xu, Z. F., Wang, X. W., Wang, C. X., and Liu, J. (2012). Effect of HMGB1 silencing on cell proliferation, invasion and apoptosis of MGC-803 gastric cancer cells. Cell Biochem. Funct. 30, 11–17.
Stangl, S., Gehrmann, M., Riegger, J., Kuhs, K., Riederer, I., Sievert, W., Hube, K., Mocikat, R., Dressel, R., Kremmer, E., Pockley, A. G., Friedrich, L., Vigh, L., Skerra, A., and Multhoff, G. (2011). Targeting membrane heat-shock protein 70 (Hsp70) on tumors by cmHsp70.1 antibody. Proc. Natl. Acad. Sci. U.S.A. 108, 733–738.
Stebbins, C. E., Russo, A. A., Schneider, C., Rosen, N., Hartl, F. U., and Pavletich, N. P. (1997). Crystal structure of an Hsp90-geldanamycin complex: targeting of a protein chaperone by an antitumor agent. Cell 89, 239–250.
Steinauer, K. K., Gibbs, I., Ning, S., French, J. N., Armstrong, J., and Knox, S. J. (2000). Radiation induces upregulation of cyclooxygenase-2 (COX-2) protein in PC-3 cells. Int. J. Radiat. Oncol. Biol. Phys. 48, 325–328.
Stephens, T. C., Currie, G. A., and Peacock, J. H. (1978). Repopulation of gamma-irradiated Lewis lung carcinoma by malignant cells and host macrophage progenitors. Br. J. Cancer 38, 573–582.
Sun, Y., St. Clair, D. K., Fang, F., Warren, G. W., Rangnekar, V. M., Crooks, P. A., and St. Clair, W. H. (2007). The radiosensitization effect of parthenolide in prostate cancer cells is mediated by nuclear factor-kappaB inhibition and enhanced by the presence of PTEN. Mol. Cancer Ther. 6, 2477–2486.
Sung, B., Pandey, M. K., Ahn, K. S., Yi, T., Chaturvedi, M. M., Liu, M., and Aggarwal, B. B. (2008). Anacardic acid (6-nonadecyl salicylic acid), an inhibitor of histone acetyltransferase, suppresses expression of nuclear factor-κB-regulated gene products involved in cell survival, proliferation, invasion, and inflammation through inhibition of the inhibitory subunit of nuclear factor-κBα kinase, leading to potentiation of apoptosis. Blood 111, 4880–4891.
Szumiel, I. (2008). Intrinsic radiation sensitivity: cellular signaling is the key. Radiat. Res. 169, 249–258.
Taba, K., Kuramitsu, Y., Ryozawa, S., Yoshida, K., Tanaka, T., Mori-Iwamoto, S., Maehara, S., Maehara, Y., Sakaida, I., and Nakamura, K. (2011). KNK437 downregulates heat shock protein 27 of pancreatic cancer cells and enhances the cytotoxic effect of gemcitabine. Chemotherapy 57, 12–16.
Takaesu, G., Kishida, S., Hiyama, A., Yamaguchi, K., Shibuya, H., Irie, K., Ninomiya-Tsuji, J., and Matsumoto, K. (2000). TAB2, a novel adaptor protein, mediates activation of TAK1 MAPKKK by linking TAK1 to TRAF6 in the IL-1 signal transduction pathway. Mol. Cell 5, 649–658.
Tamatani, T., Azuma, M., Ashida, Y., Motegi, K., Takashima, R., Harada, K., Kawaguchi, S., and Sato, M. (2004). Enhanced radiosensitization and chemosensitization in NF-κB-suppressed human oral cancer cells via the inhibition of γ-irradiation- and 5-FU-induced production of IL-6 and IL-8. Int. J. Cancer 108, 912–921.
Tamatani, T., Azuma, M., Motegi, K., Takamaru, N., Kawashima, Y., and Bando, T. (2007). Cepharanthin-enhanced radiosensitivity through the inhibition of radiation-induced nuclear factor-κB activity in human oral squamous cell carcinoma cells. Int. J. Oncol. 31, 761–768.
Taplin, M. E., Manola, J., Oh, W. K., Kantoff, P. W., Bubley, G. J., Smith, M., Barb, D., Mantzoros, C., Gelmann, E. P., and Balk, S. P. (2008). A phase II study of mifepristone (RU-486) in castration-resistant prostate cancer, with a correlative assessment of androgen-related hormones. BJU Int. 101, 1084–1089.
Timke, C., Winnenthal, H. S., Klug, F., Roeder, F. F., Bonertz, A., Reissfelder, C., Rochet, N., Koch, M., Tjaden, C., Buechler, M. W., Debus, J., Werner, J., Beckhove, P., Weitz, J., and Huber, P. E. (2011). Randomized controlled phase I/II study to investigate immune stimulatory effects by low dose radiotherapy in primarily operable pancreatic cancer. BMC Cancer 11, 134. doi:10.1186/1471-2407-11-134
Tomasini, R., Mak, T. W., and Melino, G. (2008). The impact of p53 and p73 on aneuploidy and cancer. Trends Cell Biol. 18, 244–252.
Triantafilou, M., and Triantafilou, K. (2004). Heat-shock protein 70 and heat-shock protein 90 associate with Toll-like receptor 4 in response to bacterial lipopolysaccharide. Biochem. Soc. Trans. 32, 636–639.
Trinchieri, G. (2003). Interleukin-12 and the regulation of innate resistance and adaptive immunity. Nat. Rev. Immunol. 3, 133–146.
Tsuboi, Y., Kurimoto, M., Nagai, S., Hayakawa, Y., Kamiyama, H., Hayashi, N., Kitajima, I., and Endo, S. (2009). Induction of autophagic cell death and radiosensitization by the pharmacological inhibition of nuclear factor-kappa B activation in human glioma cell lines. J. Neurosurg. 110, 594–604.
Vafa, O., Wade, M., Kern, S., Beeche, M., Pandita, T. K., Hampton, G. M., and Wahl, G. M. (2002). c-Myc can induce DNA damage, increase reactive oxygen species, and mitigate p53 function: a mechanism for oncogene-induced genetic instability. Mol. Cell 9, 1031–1044.
Valerie, K., Yacoub, A., Hagan, M. P., Curiel, D. T., Fisher, P. B., Grant, S., and Dent, P. (2007). Radiation-induced cell signaling: inside-out and outside-in. Mol. Cancer Ther. 6, 789–801.
Veeraraghavan, J., Natarajan, M., Aravindan, S., Herman, T. S., and Aravindan, N. (2011a). Radiation-triggered tumor necrosis factor (TNF) α-NFκB cross-signaling favors survival advantage in human neuroblastoma cells. J. Biol. Chem. 286, 21588–21600.
Veeraraghavan, J., Natarajan, M., Lagisetty, P., Awasthi, V., Herman, T. S., and Aravindan, N. (2011b). Impact of curcumin, raspberry extract, and neem leaf extract on rel protein-regulated cell death/radiosensitization in pancreatic cancer cells. Pancreas 40, 1107–1119.
Vega, V. L., Rodriguez-Silva, M., Frey, T., Gehrmann, M., Diaz, J. C., Steinem, C., Multhoff, G., Arispe, N., and De Maio, A. (2008). Hsp70 translocates into the plasma membrane after stress and is released into the extracellular environment in a membrane-associated form that activates macrophages. J. Immunol. 180, 4299–4307.
Wang, C. Y., Mayo, M. W., and Baldwin, A. S. Jr. (1996). TNF- and cancer therapy-induced apoptosis: potentiation by inhibition of NF-κB. Science 274, 784–787.
Wang, L., Fu, J. N., Wang, J. Y., Jin, C. J., Ren, X. Y., Tan, Q., Li, J., Yin, H. W., Xiong, K., Wang, T. Y., Liu, X. M., and Zeng, H. H. (2011). Selenium-containing thioredoxin reductase inhibitor ethaselen sensitizes non-small cell lung cancer to radiotherapy. Anticancer Drugs 22, 732–740.
Wang, X. S., Shi, Q., Williams, L. A., Mao, L., Cleeland, C. S., Komaki, R. R., Mobley, G. M., and Liao, Z. (2010). Inflammatory cytokines are associated with the development of symptom burden in patients with NSCLC undergoing concurrent chemoradiation therapy. Brain Behav. Immun. 24, 968–974.
Wheeler, D. S., Chase, M. A., Senft, A. P., Poynter, S. E., Wong, H. R., and Page, K. (2009). Extracellular Hsp72, an endogenous DAMP, is released by virally infected airway epithelial cells and activates neutrophils via Toll-like receptor (TLR)-4. Respir. Res. 10, 31.
Wong, C. C., Zhang, H., Gilkes, D. M., Chen, J., Wei, H., Chaturvedi, P., Hubbi, M. E., and Semenza, G. L. (2012). Inhibitors of hypoxia-inducible factor 1 block breast cancer metastatic niche formation and lung metastasis. J. Mol. Med. doi:10.1007/s00109-011-0855-y. [Epub ahead of print].
Yamaguchi, T., and Sakaguchi, S. (2006). Regulatory T cells in immune surveillance and treatment of cancer. Semin. Cancer Biol. 16, 115–123.
Yang, C. M., Lee, I. T., Lin, C. C., Yang, Y. L., Luo, S. F., Kou, Y. R., and Hsiao, L. D. (2009). Cigarette smoke extract induces COX-2 expression via a PKCα/c-Src/EGFR, PDGFR/PI3K/Akt/NF-κB pathway and p300 in tracheal smooth muscle cells. Am. J. Physiol. Lung Cell Mol. Physiol. 297, L892–L902.
Yoon, H. G., and Wong, J. (2006). The corepressors silencing mediator of retinoid and thyroid hormone receptor and nuclear receptor corepressor are involved in agonist- and antagonist-regulated transcription by androgen receptor. Mol. Endocrinol. 20, 1048–1060.
Zand, H., Rahimipour, A., Salimi, S., and Shafiee, S. M. (2008). Docosahexaenoic acid sensitizes Ramos cells to Gamma-irradiation-induced apoptosis through involvement of PPAR-γ activation and NF-κB suppression. Mol. Cell. Biochem. 317, 113–120.
Zeng, L., Ou, G., Itasaka, S., Harada, H., Xie, X., Shibuya, K., Kizaka-Kondoh, S., Morinibu, A., Shinomiya, K., and Hiraoka, M. (2008). TS-1 enhances the effect of radiotherapy by suppressing radiation-induced hypoxia-inducible factor-1 activation and inducing endothelial cell apoptosis. Cancer Sci. 99, 2327–2335.
Zhang, J. S., Nakatsugawa, S., Niwa, O., Ju, G. Z., and Liu, S. Z. (1994). Ionizing radiation-induced IL-1 alpha, IL-6 and GM-CSF production by human lung cancer cells. Chin. Med. J. 107, 653–657.
Zhong, H., Chiles, K., Feldser, D., Laughner, E., Hanrahan, C., Georgescu, M. M., Simons, J. W., and Semenza, G. L. (2000). Modulation of hypoxia-inducible factor 1α expression by the epidermal growth factor/phosphatidylinositol 3-kinase/PTEN/AKT/FRAP pathway in human prostate cancer cells: implications for tumor angiogenesis and therapeutics. Cancer Res. 60, 1541–1545.
Zhou, D., Yu, T., Chen, G., Brown, S. A., Yu, Z., Mattson, M. P., and Thompson, J. S. (2001). Effects of NF-κB1 (p50) targeted gene disruption on ionizing radiation-induced NF-κB activation and TNFα, IL-1α, IL-1β and IL-6 mRNA expression in vivo. Int. J. Radiat. Biol. 77, 763–772.
Keywords: radiation, inflammation, radioresistance, PGHS-2, heat shock proteins, NF-κB, STAT-3, HIF-1
Citation: Multhoff G and Radons J (2012) Radiation, inflammation, and immune responses in cancer. Front. Oncol. 2:58. doi: 10.3389/fonc.2012.00058
Received: 15 March 2012; Paper pending published: 25 April 2012;
Accepted: 18 May 2012; Published online: 04 June 2012.
Edited by:
Udo S. Gaipl, University Hospital Erlangen, GermanyReviewed by:
Stephen Tait, University of Glasgow, UKCopyright: © 2012 Multhoff and Radons. This is an openaccess article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Jürgen Radons, multimmune GmbH, Arnulfstr. 197, D-80634 Munich, Germany. e-mail:cmFqMTAwNjJAd2ViLmRl
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.