

95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 08 February 2012
Sec. Cancer Molecular Targets and Therapeutics
Volume 2 - 2012 | https://doi.org/10.3389/fonc.2012.00004
Cancer researchers have been looking for ways to harness the immune system and to reinstate immune surveillance, to kill cancer cells without collateral damage. Here we scan current approaches to targeting the immune system against cancer, and emphasize our own approach. We are using chemical vectors attached to a specific ligand, to introduce synthetic dsRNA, polyinosine/cytosine (polyIC), into tumors. The ligand binds to a receptor protein that is overexpressed on the surface of the tumor cells. Upon ligand binding, the receptor complex is internalized, introducing the polyIC into the cell. In this fashion a large amount of synthetic dsRNA can be internalized, leading to the activation of dsRNA-binding proteins, such as dsRNA dependent protein kinase (PKR), Toll-like receptor 3 (TLR3), retinoic acid-inducible gene I (RIG-1), and melanoma differentiation-associated gene 5 (MDA5). The simultaneous activation of these signaling proteins leads to the rapid demise of the targeted cell and to cytokine secretion. The cytokines lead to a strong bystander effect and to the recruitment of immune cells that converge upon the targeted cells. The bystander effects lead to the destruction of neighboring tumor cells not targeted themselves by the vector. Normal cells, being more robust than tumor cells, survive. This strategy has several advantages: (1) recruitment of the immune system is localized to the tumor. (2) The response is rapid, leading to fast tumor eradication. (3) The bystander effects lead to the eradication of tumor cells not harboring the target. (4) The multiplicity of pro-death signaling pathways elicited by PolyIC minimizes the likelihood of the emergence of resistance. In this chapter we focus on EGFR as the targeted receptor, which is overexpressed in many tumors. In principle, the strategy can be extended to other tumors that overexpress a protein that can be internalized by a ligand, which can be a small molecule, a single chain antibody, or an affibody.
A great boost for cancer research was rendered in 1971, when President Nixon signed the National Cancer Act. This historic moment was a turning point in the history of cancer research. Other countries soon followed the lead of the United States, increasing funding for cancer research. The massive investment created great expectations, but progress toward a cancer cure has been slow.
The concept of targeted cancer therapy has led to the development of many new agents, but these are unable to contend with the complexity of cancer. In fact cancer comprises more than 100 diseases, with numerous variants. The tumor does not standalone, but rather interacts intimately with its microenvironment as well as with the immune network, generating an organ-like entity. The built-in genomic instability of the tumor makes its treatment much like aiming at a moving target, since the wiring of the cancer “organ” changes continuously. We have come to realize that the notions “Achilles’ heel” and “oncogene addiction” are reductionist notions, applying to a very narrow spectrum of diseases, such as early stage chronic myelogenous leukemia (CML) and a subclass of gastrointestinal stromal tumor (GIST), a subclass of lung cancer and a few other cancers. In all these cases there is a prominent oncogene coding for chimeric protein such as Bcr–Abl in early CML, or mutated kinases like mutant forms of EGFR or the chimeric protein EML4–ALK in subsets of non-small cell lung carcinoma patients, and mutated BRaf in metastatic melanoma. In all these cases the cell is “addicted” to the oncogenes and therefore blocking its protein product brings about dramatic responses. However, within months or even less the disease recurs except early CML, where the remission is long-term. Almost invariably, the disease eventually breaks away from dependence on a single oncogene, and the initially dominant target is demoted to one of many. Long ago, we asserted the need for “smart cocktails” (Levitzki, 1992) of targeted and non-targeted therapies. These “smart cocktails” have to be modified continuously during the course of treatment, as the cancer and its intimately associated microenvironment undergo continuous evolution. This is indeed the current modality of treatment, and it is showing incremental but steady improvement in patient outcome. Dealing with a moving target, which actually consists a continuously changing oncogenic network poses a tremendous challenge to the therapist and cancer researcher.
Cancer is a genetic disease, rooted in the gradual acquisition of somatic mutations that eventually overcome the barriers to unregulated growth. The surveillance mechanisms that prevent cancer include a variety of DNA damage control mechanisms, which often trigger cellular senescence. The death of the emerging cancer cell may also be induced by stress pathways that are turned on in the mutated cells and/or by the p53 pathway. The immune system also participates in the action. The immune system identifies newly emerging cancer cells, eliminating them. When this immune surveillance is breached, cancer can develop.
It is now recognized that the immune system interacts in a complex fashion with the tumor and can even cooperate with the tumor. Thus, one speaks today about immunoediting rather than immune surveillance. Immunoediting comprises three steps: elimination, equilibrium, and escape. Initially, the innate and adaptive immune systems eliminate emerging nascent tumor cells. Cancer cells that are not eliminated attain equilibrium with the immune network and become dormant. When tumor dormancy is compromised due to changes in the tumor, the tumor microenvironment or the immune system, cancer cells escape the equilibrium, and the tumor thrives. Factors that contribute to this continuous evolution include the built-in genomic instability of the tumor cell and changes that may occur in the immune system, for example as the result of viral or bacterial infection. The complex interaction between the emerging tumor and its microenvironment, which includes the complex immune network, has been the subject of intense research over the past few years. The newly revised classic article “The Hallmarks of Cancer” now discusses the microenvironment as well as the immune system (Hanahan and Weinberg, 2011). Since researchers and clinicians alike recognize the dynamic involvement of the immune system at all stages of cancer initiation, development, and progression, attention is now being devoted to finding ways to compel the immune system to fight cancer rather than cooperate with it. A comprehensive review on the interrelationships between cancer and the immune system has been published recently (Vesely et al., 2011).
Killing cancer cells by the immune system has been on the agenda for many years, but only in the last decade has real progress been made (Blattman and Greenberg, 2004). Five approaches are being adopted: (1) development of antibodies to attack cancer cells by homing to cancer antigens. These anti-cancer antibodies are sometimes armed with cytotoxic chemicals, bacterial toxins, or labeled with a radioactive element; (2) enhancing the action of anti-tumor T cells, by means of antibodies or other methods; (3) development of therapeutic vaccines; (4) development of prophylactic vaccines; (5) development of strategies to target the anti-tumor action of the immune system. In all cases attempts are being made to combine these strategies with each other, as well as with other types of targeted therapies and cytotoxic therapies.
Since these are emerging fields I shall not attempt to review all the details but rather will refer the reader to key reviews that cover various aspects of immune therapy. I will devote more space to our own attempts to harness the immune system and target its action to the tumor. Our chemical biological approach complements current efforts by the scientific community to harness the immune system against cancer. It differs from the other approaches in that it is highly targeted to the tumor and does not evoke a systemic immune response.
Although monoclonal antibodies were discovered and developed by Köhler and Milstein (1975), only in the 1990s did anti-cancer antibodies start to move into the clinic. Chimeric monoclonal antibodies, containing human constant domains and mouse variable domains, have been developed for human therapy. Rituximab/Rituxan/MabThera was the first monoclonal antibody to be developed for clinical use. Its success in the late 1990s catalyzed the development of other anti-cancer antibodies, such as Herceptin/trastuzumab targeting Her-2, Cetuximab/Erbitux targeting EGFR, and alemtuzumab targeting CD52 for the treatment of chronic lymphocytic leukemia (CLL), cutaneous T-cell lymphoma (CTCL), and T-cell lymphoma. These are all partly “humanized” mouse monoclonal antibodies, in which the only mouse sequences are the ones responsible for specificity. More recently fully humanized antibodies, such as the anti-EGFR antibody, panitumumab, have begun to enter the clinic. Humanization of antibodies reduces their immunogenicity, increases their stability, and enhances their ability to induce antibody-dependent cellular cytotoxicity (ADCC).
Rituximab/Rituxan has revolutionized the treatment of CD20-positive, non-Hodgkin lymphomas (NHL). This antibody purges leukemic cells as well as normal B cells. The anti-leukemic effect is impressive and toxic effects slight, mainly due to the ability of humans to live with no B cells, a fact that was not really appreciated early on and is actually quite astonishing. Herceptin/Trastuzumab is effective in the treatment of breast cancer patients whose tumors strongly overexpress Her-2, especially when given early. The Herceptin treatment is always given in combination with cytotoxic therapy or the dual EGFR/Her-2 kinase inhibitor lapatinib. A significant number of patients with Her-2 positive breast cancer, who are initially sensitive to Herceptin, later become refractory to Herceptin treatment and succumb to the disease. Avastin, the anti-vascular endothelial growth factor (VEGF) inhibitor, is used in the treatment of metastatic colon cancer in combination with chemotherapy. Similarly, Erbitux/Cetuximab, an anti-EGFR antibody, is used in combination with chemotherapy or radiation for the treatment of head and neck cancer, as well as other cancers in which EGFR is overexpressed. Historically, the combination of antibodies with other targeted agents against EGFR was already demonstrated in 1991, when an EGFR-directed tyrosine phosphorylation inhibitor (tyrphostin) was combined with an EGFR-directed antibody (Yoneda et al., 1991). Interestingly, patients who do not express/overexpress EGFR on their tumors were also reported to respond to Erbitux. In view of the success of anti-tumor antibodies it is not surprising that many more are being developed and are on the way to the clinic.
Monoclonal antibodies operate by two mechanisms: first, direct inhibition of signal transduction by the receptor or the growth factor at which the antibody is aimed and second, ADCC and complement-dependent cytotoxicity (CDC). These two bystander effects are extra benefits provided by antibodies that are not provided by small molecular weight signaling inhibitors like tyrosine phosphorylation inhibitors (tyrphostins; Levitzki, 1992). In fact, combinations of antibodies with low molecular weight signaling inhibitors are emerging as standard therapies. Combinations of antibodies are also useful, even if they are against the same molecular target, as long as they recognize different epitopes (see below).
Antibody-dependent cellular cytotoxicity is mediated by the Fc domain of an antibody. The humanized Fc domains on the monoclonal antibodies bind to the Fc receptors on immune accessory cells, mainly natural killer (NK) cells, but also neutrophils, mononuclear phagocytes, and dendritic cells (DC). This interaction activates the immune cells, which then engulf and destroy the bound tumor cells. NK cells also release cytokines and chemokines that inhibit cell proliferation and tumor-induced angiogenesis, and induce the expression of major histocompatibility (MHC) antigens on the tumor, enhancing tumor immunogenicity. Antibodies that mediate ADCC also activate the complement system. CDC leads to the direct killing of tumor cells through the formation of a “membrane attack complex” (MAC). MAC generates a pore at the target cell membrane, leading to tumor cell destruction. Various experiments demonstrate that the efficacy of Herceptin and Rituxan is due, to a significant degree, to these bystander effects. The intensity of the bystander effects depends on the quality of the Fc domain. The absence of Fc receptors or their diminished affinities to the antibodies compromise their efficacy. For example it was shown, in pre-clinical experiments, that experimental animals lacking Fc receptors on their immune effector cells do not respond well to Herceptin since ADCC is absent. It was also shown that polymorphisms in the IgG receptors, CD16 (FcγRIIIa), and CD32a (FcγRIIa), predict how well follicular lymphoma patients respond to rituximab. Also, in animals that lack elements of the complement pathway, rituximab is not effective. Since the role of the Fc–Fc-receptor interaction in the efficacy of the antibody was recognized, second generation antibodies against CD20 with stronger affinities for the CD20 antigen have been generated, by mutating the Fc portion selected, to enhance ADCC and CDC. These examples underscore the key role that bystander effects play in the anti-tumor activities of antibodies. It is therefore likely that other anti-cancer antibodies that are being developed will also be engineered to enhance the bystander anti-tumor effects.
Recently it was demonstrated that antibodies against different epitopes of EGFR synergize to down-regulate EGFR and exhibit strong anti-tumor effects in pre-clinical models. In the case of Her-2, a combination of antibodies has been examined in patients. Pertuzumab, which potently inhibits Her-2 dimerization and Her-2 mediated signaling, was combined with the Her-2-targeted monoclonal antibody trastuzumab/Herceptin. These antibodies have complementary mechanisms of action and result in enhanced anti-tumor activity when combined, in pre-clinical models. Phase II studies in humans have shown a good toxicity profile and good efficacy. In view of the development of fast and effective means to produce an array of antibodies targeted to a particular protein, it is likely that using a mixture of antibodies against a single cancer antigen will emerge as an effective strategy.
The immune system not only induces an immune response to a specific target, but also involves a complex regulatory network that attenuates the immune response in order to avoid autoimmune reactions. Recognizing the self-restraining action of the immune response, strategies to down-regulate the attenuation of the immune response, thus elevating the innate anti-tumor immune response are being developed (Korman et al., 2006); (Peggs et al., 2008). The most advanced and dramatic example of this strategy is the development of ipilimumab and its recent approval for clinical use.
Ipilimumab is an antibody against cytotoxic T-lymphocyte-associated 4 (CTLA-4). CTLA-4 raises the signal threshold for T-cell activation, leading to the attenuation of T-cell activation. Clinical trials have shown efficacy for CTLA-4 blocking antibodies (Hodi et al., 2010) in melanoma, and renal cell carcinoma. Ipilimumab received FDA approval in 2011 for the treatment of metastatic melanoma. The use of antiCTLA-4/ipilimumab is now being evaluated for the treatment of other tumors. Using a similar approach attempts are being made to manipulate immune checkpoints regulated by B7H1, B7H4, OX-40, CD137, CD40, and PD1, enhancing anti-tumor immune responses.
The development of agents that temper with immune checkpoints is attractive, since in principle such agents can be utilized for many types of tumors. Clearly, dosing and combination with other agents will be different for every type of cancer and will have to be individualized. An important caveat is that such general agents can cause severe autoimmune toxic effects, as has already been observed. Better understanding of the complex regulation of the immune response and more clinical experience is likely to bring a gradual improvement of this approach. It is likely that for every cancer the therapeutic window will be defined for each individual patient. A certain amount of toxicity (e.g., rash) is an indication of efficacy and may be used as a possible early generation biomarker. Manipulation of the immune checkpoints on T cells, NK cells, macrophages, and DC, can in principle amplify the effects of ADCC and vaccines, leading to clinically useful host-generated anti-tumor effects. It is therefore likely that antibodies that inhibit the moderating signals of the immune response will be combined with other anti-tumor therapeutic modalities.
Antibodies can be used to bring non-specific therapy directly to the tumor site, thus reducing toxic side effects induced by the systemic application of the cytotoxic agent.
Attempts have been made for many years to target cytotoxic agents to cancer cells, using antibodies. Recently such an antibody conjugate, Brentuximab vedotin, was approved for clinical use. Brentuximab vedotin is a chimeric monoclonal antibody, which targets CD30 and possesses three to five units of the antimitotic agent monomethyl auristat (MMAE) bound to the protein. MMAE is responsible for the anti-tumor activity. In a clinical trial, 34% of patients with refractory Hodgkin lymphoma achieved complete remission and another 40% had partial remission with tumor reductions in 94% of patients. In anaplastic large cell lymphoma (ALCL), 87% of patients had tumors shrink at least 50%, and 97% of patients had various extents of tumor shrinkage.
Antibodies can be used to target radioisotopes to the tumor site. This form of therapy is known as radioimmunotherapy (RIT). A few radioactively labeled antibodies are already in the clinic, such as 90Y-labeled Rituxan (Zevalin) and 131I-labeled labeled Rituxan (Bexxar). Both radioactively labeled anti-CD20 antibodies are used in patients with B-cell NHL who do not respond to the regular anti-CD20 antibody rituximab. In principle, radiolabeled antibodies can be also used to treat metastases of solid tumors to the bone marrow and lymph nodes, which are accessible to circulating mAbs. A number of radiolabeled antibodies are in development to treat prostate cancer, for example. However, many solid tumors are radio-resistant or develop radioresistance and therefore are not as accessible as hematological malignancies to RIT.
Antibodies and ligands can be used to target toxins to the tumor site. The first immunotoxins were made by chemically conjugating antibody to toxin, but today recombinant fusion proteins are used. Pseudomonas exotoxin and Diptheria toxin have been successfully incorporated into recombinant immunotoxins (Pastan et al., 2006). Both of these toxins are ADP ribosylases that covalently modify elongation factor 2 (EF2), inhibiting protein synthesis, and leading to cell death. The fusion protein is molecularly engineered such that the natural binding domains of these bacterial toxins are deleted, to prevent binding to normal cells. The recombinant protein consists of a tumor-targeting ligand or antibody, and the translocation and catalytic domains of the toxin. Following internalization into the tumor cell, the immunotoxin is cleaved within the translocation domain, releasing the catalytic domain. RFB4 (dsFv)-PE38 (BL22), a recombinant immunotoxin containing an anti-CD22 variable domain (Fv) fused to truncated pseudomonas exotoxin, was found to be successful in treating drug resistant (purine analog treatment) hairy cell leukemia patients. One problem that limits the use of fusion proteins for therapy is the development of neutralizing antibodies. Although most patients develop anti-denileukin diftitox antibodies, even after one cycle of treatment, some patients continue to respond after re-treatment. The Pastan–Bigner groups also reported a positive clinical outcome with a recombinant Pseudomonas exotoxin targeted to the EGFR by fusion with TGFα (Sampson et al., 2005).
The immune system is educated and trained to identify, seek, and destroy invading pathogens, and does it with great efficiency. It is however much less effective at recognizing tumor antigens and destroying them, partly because tumors express antigens common to normal cells such as EGFR or Her-2, only at higher levels. Furthermore, many tumors evade the immune system by failing to express antigens that can be recognized by the immune system, and/or they develop various strategies that interfere with the execution of the anti-tumor immune response.
Although vaccines have a strong track record in fighting serious infections, such as polio, mumps, and measles, they have so far had limited success in the treatment of cancer. Vaccines work best with soluble antigens, and are much less efficacious against bulky, solid tumors. Moreover, cancer cells change continuously, due to their genomic instability and therefore change their antigen presentation and/or HLA markers. Furthermore, cancer cells frequently secrete immunosuppressive cytokines, such as TGFβ and IL-10 (see Stat3 as a Target). In spite of these caveats, a number of therapeutic vaccines are at various stages of development, but the results so far are not too encouraging. Numerous vaccines against peptides that represent various cancer antigens have been tested, with rather limited success. Several cellular vaccines, which present multiple antigens, are also in advanced trials, with the hope that multiple tumor antigens will boost the anti-tumor activity of emerging vaccines. So far the success of vaccines has been limited but success may be emerging. For example, Sipuleucel-T is an active cellular immunotherapy against castration-resistant prostate cancer (Kantoff et al., 2010). The vaccine consists of autologous peripheral blood mononuclear cells (PBMCs), which include antigen-presenting cells (APCs) that have been activated ex vivo with a recombinant fusion protein. The activating protein, called PA2024, consists of a prostate antigen, prostatic acid phosphatase, fused to granulocyte–macrophage colony-stimulating factor, an immune cell activator. This vaccine showed clinical efficacy and therefore obtained FDA approval in 2010. It should be stressed however that the survival benefit reported for this vaccine is of a few months only.
A recent report claims that women with HPV-16-positive, grade 3 vulvar intraepithelial neoplasia respond to vaccination by a synthetic long-peptide vaccine against the HPV-16 oncoproteins E6 and E7 with a few complete responses (Kenter et al., 2009).
We now understand that one of the main reasons vaccines have not been successful is that the tumor microenvironment is not conducive to the migration and optimal activation of DCs, so the ensuing immune response is weak and ineffective. Thus in order to stimulate an anti-tumor immune response one has to channel the tumor antigens into the appropriate DC subset and induce the DCs to develop into potent immunostimulatory APCs. In order to enhance the anti-tumor immune response, attempts are being made to stimulate the immune system by the activation of Toll-like receptor 9 (TLR9; Hennessy et al., 2010). TLR9 recognizes unmethylated CpG in DNA molecules, common in bacteria, and rare in vertebrates. TLR9 is expressed intracellularly, within the endosomal compartments of DCs, B lymphocytes, monocytes, and NK cells. TLR9 signals initiate pro-inflammatory reactions that result in the production of cytokines such as interferon type I and IL-12. Arousal of the innate immune system is likely to tilt the tumor microenvironment toward preventing the tumor from evading therapeutic vaccines. Another approach is to prepare DCs for anti-cancer vaccines, in the presence of interferon α (for review see Moschella et al., 2010).
Several cancers are strongly associated with viral infection. Therefore, one strategy for cancer prevention is to vaccinate against viruses that cause cancer. Indeed, the vaccine against hepatitis B virus (HBV; Blumberg, 2010) is expected to reduce the incidence of HBV-related hepatocellular carcinoma. Intensive attempts are also being made to develop a vaccine against hepatitis C virus. In 1984, large-scale hepatitis B vaccination of newborns began in Taiwan. During the years 1984–1999 the incidence of childhood liver carcinoma decreased: a 62–70% decline in mortality due to liver carcinoma among children has been reported. In the western world cervical cancer is the fourth leading cause of cancer-related death in women, after breast cancer, lung cancer, and colon cancer. In developing countries, where routine Pap smears are less widespread, cervical cancer is the leading cancer among women. The recent introduction of vaccines against the most common high-risk strains of human papilloma virus (HPV) is expected to reduce mortality from cervical cancer, which is caused by this virus and is the major cause of cancer-related deaths among women in the developing world.
This is actually a form of individualized immune therapy. In this approach one isolates CD8+ cytotoxic T cells directed at the tumor from a patient’s own tumor (tumor infiltrating lymphocytes, TILs), and expands the cells in culture ex vivo. One can even sort out the T cells that recognize a specific tumor antigen. While ex vivo, the T cells can also be genetically engineered to home more effectively to the tumor and even enhance their tumor killing efficacy.
Genetically modified T cells can originate from the tumor itself or from normal T cells of the patient. One engineers the T-cell receptor such that when reintroduced into the patient the T cells will home to the tumor and kill it. The advantage of using normal T cells over TILs is that one is not limited by the relatively small number of TILs and more importantly, one does not depend on the necessary access to the tumor to get TILs, which is not always possible. It is hoped that the identification of specific tumor antigens for each patients will enable individualized T-cell therapy.
Before reintroduction of the T cells, the patient is treated with systemic chemotherapy or irradiation to deplete him of lymphocytes, and then the anti-tumor T cells are reintroduced into the patient. The approach has been used with a measure of success in the treatment of metastatic melanoma (Dudley et al., 2008; Rosenberg et al., 2008) and in CLL. One problem is the tendency of the T cells to undergo apoptosis, once they encounter the tumor. This can be addressed and even overcome by the co-administration of IL-2, known to enhance the survival of T cells and to promote their proliferation. Cancer patients also suffer from lowered numbers of CD4+ T helper cells, which support the survival of cytotoxic T cells. CD4+cells are harder to expand ex vivo than CD8+ cells, and adoptive transfer of CD4+ cells is so far less advanced than that of CD8+ cells. Since the immune system is conducive of tumor development and maintenance, when diagnosed it is important to break immune tolerance in order to make cellular therapy more effective. This can be achieved by using cyclophosphamide that was found to enhance the efficacy of TILs (for reviews see Moschella et al., 2010; Sistigu et al., 2011).
Cytotoxic T cells can be induced to attack a tumor by means of bispecific antibodies, which have specificity both for T cells and for the tumor (Wolf et al., 2005). These antibodies are also known as bispecific T-cell engagers (BiTEs). One such antibody is Blinatumomab/MT103, a dual specificity antibody. Blinatumomab combines two binding sites: a CD3 site for T cells and a CD19 site for the target B cells. The drug works by linking these two cell types and triggering T-cell activation, which exerts cytotoxic effects on the target malignant B cells. In a phase I clinical trial of blinatumomab, patients with NHL showed tumor regression and even complete remission. At present the bispecific antibody is being tested on acute lymphoblastic lymphoma (ALL).
Future experiments will assess the use of bispecific antibodies against solid tumors. A bispecific antibody, MT110, directed against an epithelial cell adhesion molecule (Ep-CAM/CD326) that is overexpressed on most human carcinomas and against CD3, is being assessed for therapeutic potential for the treatment of cancers of the colon, breast, pancreas, and prostate. Pre-clinical experiments using animal models are highly promising, but only clinical trials, which are still ongoing, will establish efficacy. In order to minimize autoimmune effects, MT110 was designed to have only moderate affinity for Ep-CAM (Amann et al., 2009).
The tumor microenvironment is not conducive to attack by the immune system. Tumors that are in equilibrium with the immune system remain dormant, but tumors that escape immune control run wild. Tumors utilize a number of strategies to evade the immune system. One important mechanism, common to many different cancers, is the secretion of cytokines, such as IL-10, TGFβ, and VEGF. Other mechanisms, triggered by biochemical pathways emanating from the tumor cell, include the expression of decoy receptors such as a non-signaling Fas ligand, disabling apoptotic pathways, and the down-regulation of MHC type I molecules, leading to the evasion of recognition by CD8+ CTL. The systemic anti-tumor immune response can be enhanced by using ligands such as CpG to enhance TLR9 (see Therapeutic Vaccines). Activation of TLR9 is not targeted specifically to the tumor, but rather enhances overall immune responsiveness. As indicated above, the activation of TLR9 sensitizes a tumor to all tumor-targeted immune therapies. Therefore, it is likely that TLR9 agonists will find their place in immunotherapeutic regimens as well as other therapeutic modalities that can be augmented by the utilization of TLR9 agonists.
We decided to develop an immunotherapeutic strategy that would be highly focused on the tumor and have limited general immune effects. Recognizing that antibodies induce a bystander effect at the tumor site and therefore are more efficacious than tyrphostins, we sought a strategy to enhance the bystander effect at the tumor site, whilst still using the receptor tyrosine kinase as the tumor-specific target. We sought a simple and inexpensive approach.
The synthetic long chain dsRNA, PolyIC, has strong anti-tumor activity and has been used as an immune adjuvant for many years (Talmadge et al., 1985; Salem et al., 2005, 2006; Kumar et al., 2008; Navabi et al., 2009; Okada et al., 2011). We therefore considered this molecule for our efforts. In a recent trial, type1 polarized DCs were loaded with synthetic peptides for glioma-associated antigen plus polyIC complexed with polylysine. There was a positive immune response in 58% of the 22 recipients, and sustained complete remission was reported in one patient (Okada et al., 2011). In another clinical situation, poly AU showed an improvement in 20-year survival rates in a subset of breast cancer patients with tumor cells that expressed TLR3(Lacour et al., 1991). There are more than 24,000 entries in Google Scholar when one inserts “PolyIC Adjuvant Cancer.” The response of metazoan cells to dsRNA is a mechanism utilized mostly, but not exclusively, against dsRNA viruses and ssRNA viruses that generated sRNA during replication. Long chain dsRNA induces strong anti-apoptotic effects within the cell. The activation of PKR by the dsRNA leads to the phosphorylation of eIF2α and a halt in protein translation. dsRNA also stimulates RNAaseL, contributing to the inhibition of translation. In addition to the direct intracellular pro-apoptotic effects of long chain dsRNA, there are strong bystander effects on neighboring cells, mostly mediated by TLR3, retinoic acid-inducible gene I (RIG-1), and melanoma differentiation-associated gene 5 (MDA5) (Figure 1).
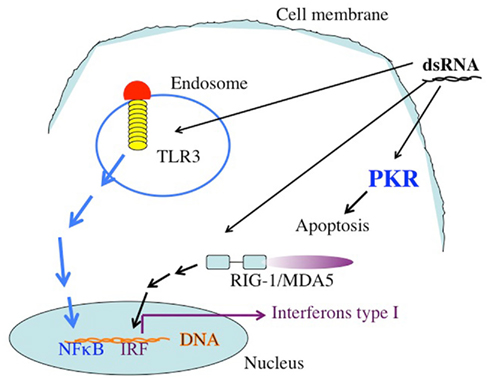
Figure 1. The targets of dsRNA. TLR3 recognizes dsRNA extacellularly or in endosomal compartments. The TLR3 TIR domain is associated with the adaptor molecule TRIF, which upon ligation activates the protein kinases TBK-1 and IKKe. IRF-3 is phosphorylated by TBK-1 and IKKe on C-terminal serines, leading to its dimerization and translocation into the nucleus. Active IRF-3 induces transcription from the IFN-β promoter. Intracellular dsRNA is recognized by the RNA helicase RIG-I (or MDA5), which activates TBK-1 and IKKe via its CARD domain, leading to IRF-3 activation as well. Both pathways also activate NFκB by mechanisms that are not fully understood yet. Secreted IFN-β binds to the IFN receptor (IFNR) leading to transcriptional activation of ISGs (interferon stimulated genes), such as IRF-7. IRF-7 further stimulates transcription from the IFN-α and -β promoters in a positive feedback loop. In addition to these targets, which are responsible for the activation of the immune system dsRNA binds to the enzyme dsRNA dependent protein kinase, PKR. The activation of PKR leads to the phosphorylation of the eIF2a, leading to the inhibition of protein translation.
The tumor cells that internalize the dsRNA are induced to secrete interferons alpha and beta and other cytokines that attract T cells and NK cells – which in turn secrete additional cytokines – to the locality. The activation of PKR and of the innate immune network is rapid and localized to the tumor. We therefore expected that activation of innate immunity would be focused on the tumor, with minimal activation of the overall immune system.
The systemic application of long chain dsRNA such as PolyIC is toxic, mainly due to strong systemic immune reactions. This is why PolyIC and Ampligen (a complex of PolyIC with polylysine) are not used systemically but only in the adjuvant setting. Still, we were highly impressed by the profound effects of PolyIC and therefore decided to formulate it in a way that it would be targeted to the tumor. It was clear to us that we needed to develop PolyIC-carrying vectors that would home selectively to the tumor and be internalized quickly into the tumor, to induce strong anti-tumor effects and avoid systemic toxicity. To this end, the vector needed to possess a ligand that would guide PolyIC to a specific receptor on the tumor, such that receptor-mediated endocytosis would rapidly lead to internalization of the PolyIC. Thus, the ligand would act as a targeted Trojan Horse. Another pre-requisite was that the receptor should be expressed at high levels on the tumor and much less on normal tissues, opening a therapeutic window.
A suitable receptor is the EGFR, for which the group of Ernst Wagner (Ludwig Maximilian University, Munich) has developed synthetic chemical vectors bound to EGF, and they use these to introduce genes into EGFR-overexpressing tumor cells. Using their vectors, we have been developing the Trojan horse approach to eradicate tumors, using polyethyleneimine–polyethyleneglycol–EGF based vectors (Figure 2). Remembering that tumor cells are much more vulnerable than normal cells to all stresses (Benhar et al., 2002), we hypothesized that we could define a wide enough therapeutic window to affect tumor cells almost exclusively, sparing the normal cells surrounding the tumors. The strategy, if successful, would have an advantage over other immunotherapeutic approaches, since both the innate immune system and the adaptive immune system would be activated and directed to the tumor. Moreover, this strategy can, in principle, overcome immunoinhibitory mechanisms developed by many cancers, due to the overwhelming immune activating effects of polyIC as well as to the fast-acting nature of polyIC.
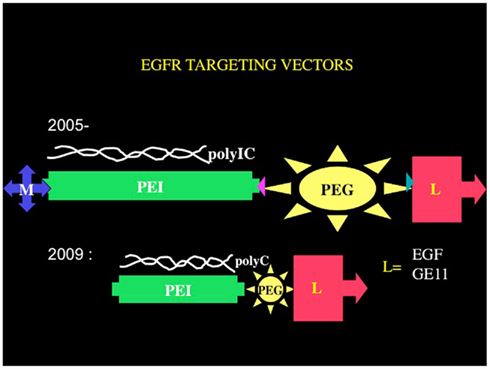
Figure 2. Chemical vectors targeting the EGFR. The core structural elements of the chemical vectors targeting the EGFR consists of branched polyethyleneimine tethered by polyethyleneglycol to an EGFR homing ligand EGF or a peptide GE11. The tetraconjugate possesses a melittin moiety that facilitates endosomal release. The triconjugate is composed of a linear and shorter polyethyleneimine molecule tethered to the EGFR homing ligand via a short polyethyleneglycol.
Initially we utilized PolyIC complexed with branched-polyethyleneimine–polyethyleneglycol–EGF (PolyIC/brPEI–PEG–EGF) mixed with brPEI–melittin (Shir et al., 2006). These two entities mix in nanoparticles that home to EGFR and internalize into the cell. Melittin was used to enhance endosomal release, to enable the PolyIC to bind to and activate PKR, which resides in the cytoplasm. Other targets for dsRNA, such as TLR3, RIG-1, and MDA5, should be activated in the endosome (Figure 1). Using this nanoparticle we demonstrated the complete eradication of intracranial tumors generated in mice from glioblastoma cell lines that overexpressed EGFR. The particles were applied locally to the tumor site, using Alzat pumps. Even tumors 15mm3 in size were completely eradicated by a 3-day treatment.
The vector did not kill glioblastoma cells that overexpressed mutated Δ(2–7) EGFR (EGFRvIII) receptors, which do not bind and therefore do not internalize the PolyIC-loaded EGFR homing vector. Strikingly, when cells overexpressing wild type EGFR were mixed with cells overexpressing Δ(2–7) EGFR the latter were killed: intracranial tumors comprising 50% cells overexpressing EGFR and 50% Δ(2–7) EGFR, were eradicated. The cells that overexpressed wild type EGFR and internalized the polyIC initiated strong bystander effects (Figure 3), which led to the demise of the mutant EGFR-expressing cells. Notably, the nude mice used in these experiments lacked T cells, yet they mounted a partial bystander response, sufficient to cure the experimental animals. These results show that the bystander effect is effective and allows the complete cure of animals that harbor large intracranial tumors, by a 3-day treatment. At the same time we did not observe any damage to the normal brain tissue during treatment. Furthermore, the necrotic tissue bordering the tumor in the brain also disappeared.
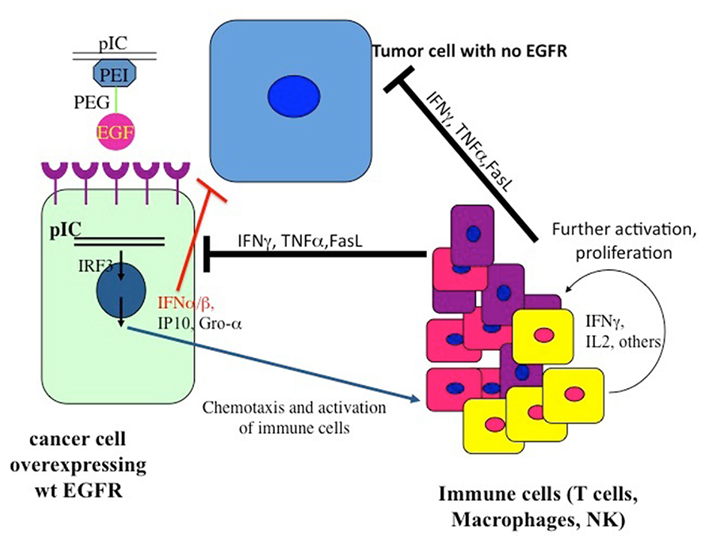
Figure 3. The pattern of the recruitment of the immune system to the tumor. The dsRNA internalized into the tumor cells binds to dsRNA-binding signaling proteins, which then activate a plethora of signaling pathways, leading to the recruitment of immune cells to the tumor as well as to apoptotic programs (Figure 1).
At this point an improved version of the vector was introduced: a tetraconjugate, Melittin–brPEI–PEG–EGF (MPPE), in which the melittin was incorporated into the EGF homing vector (Figure 2; Shir et al., 2006). The tetraconjugate proved to be highly effective and quickly replaced the mixture of the triconjugate with Mel-PEI. The rapid eradication of intracranial tumors by local application of the EGFR-targeted PolyIC convinced us that the strategy could be moved to clinical development for the local treatment of EGFR-overexpressing tumors, not only in the brain. It should be noted that in human GBM one finds a heterogeneous mixture of cells, including many types of pluripotent cells/cancer stem cells that may or may not express high levels of EGFR (Mazzoleni et al., 2010). Elimination of the EGFR-overexpressing cells, using AG 1478, a selective EGFR tyrphostin, enables the EGFR-negative cells to survive and thrive (Mazzoleni et al., 2010). It is likely that treatment with the EGFR-targeted PolyIC, will also eliminate the EGFR-negative GBM cells, limiting or even preventing tumor recurrence (Figure 3).
We next decided to adapt the EGFR-targeted dsRNA killing strategy to eradicate disseminated EGFR-overexpressing tumors, since humans usually succumb to disseminated tumors. Toward this end we established a model for disseminated tumors: we injected two million cells intravenously to SCID mice and waited for tumors to develop in various organs. Treatment with the vector PolyIC/MPPE began 5, 10, or 15 days after intravenous cell injection. Complete eradication – defined as survival for more than 1 year – of 5- or 10-day-old disseminated tumors derived from A431 cells or MD-MB468 was achieved. The animals were treated for 10 days with two 1-day intermissions, thus avoiding toxicity. Large (∼200 μm) 15-day-old tumors were only partially inhibited: 60% of animals with A431 tumors survived over 1 year; animals with disseminated MD-MB468 tumors showed long-term survival but no complete tumor eradication (Shir et al., 2011).
The SCID mouse model does not enable us to examine the full capacity of our dsRNA therapy, because SCID mice have extremely deficient immune systems. Our next step, therefore, was to reconstitute the SCID mouse immune system with human PBMCs and use these mice for the study of targeted PolyIC therapy of disseminated cancers. When SCID mice harboring 15-day EGFR-overexpressing tumors were treated with EGFR-targeted PolyIC, and PBMC were introduced, the mice were completely cured by a short treatment (4 days; Shir et al., 2011). The mice survived for over a year, with no tumor recurrence. Furthermore, tumors not expressing EGFR were oblivious to the treatment and animals with these tumors succumbed to the disease. Interferon gamma, TNF-alpha and IL-2 were generated within the tumors themselves during the treatment, but few cytokines were detected in the blood stream. We also demonstrated that immune cells infiltrated the tumor. These findings validated the targeted nature of the innate immunity aroused by the EGFR homing vector armed with PolyIC (Shir et al., 2011) (Figure 3).
In cellular studies we found that the PolyIC synergizes with immune cells recruited to PolyIC-treated tumor cells, to induce tumor cell death. In the disseminated tumor model, the direct killing effect of PolyIC combined with the effect of the PBMC-derived immune cells attracted to the PolyIC-treated tumors, led to the complete elimination of the disseminated tumors. Activated immune cells converging on the tumor enhanced the bystander effect, killing even those cancer cells that were not attacked directly by the PolyIC.
The fast immune response that we observed strongly suggests that innate immunity is the primary player in the animal model we have constructed. Infiltration of APCs, such as macrophages, into the tumor and induction of class I and class II MHC by IFNs eventually should lead also to an adaptive immune response against the tumor. In our experiments, we did not observe systemic immunotoxic reactions, most likely because the tumor acted as a “sink,” quickly eliminating the vector from the blood stream, and because the treatment was short (4 days).
In principle, EGFR-targeted polyIC can be combined with other cancer immunotherapies available today. These include cancer vaccines and cancer-targeted (engineered or extracted) T cells. To mediate anti-tumor effects in vivo, cancer-targeted T cells must travel to the tumor site, extravasate from the circulation, and then mediate effector functions to cause destruction of cancer cells. IP-10 and Gro-α are strongly induced by targeted polyIC selectively in tumor cells, which should facilitate both traffic to the tumor and extravasation of T cells. Similarly, the polyIC treatment induces IFNs, which should enhance T-cell-mediated cancer killing. The absence of an immunocompetent mouse model of EGFR-overexpressing cancer currently precludes a study of the activation of the mouse’s own immune system by polyIC/MPPE. Nonetheless, our data suggest that systemic treatment of EGFR-overexpressing metastatic tumors with EGFR-targeted polyIC may lead to a complete cure, in patients with a functional immune system.
In its latest version, the PolyIC homing vector has been re-engineered to eliminate the melittin. The branched polyethyleneimine (brPEI) was replaced by a linear version of PEI and the PEG shortened, producing the triconjugate L-PEI (22 kDa)-PEG (2kDa)-EGF (Figure 2) (PPE; Schaffert et al., 2011).
The re-engineered vector was as good as the melittin-containing tetraconjugate MPPE. The PolyIC/L-PEI–PEG–EGF (PolyIC/PPE) shows excellent efficacy and is now replacing the tetraconjugate in our experiments and has been recently adopted for advanced pre-clinical development by Bioline Rx. We have also shown recently that EGF can be replaced by an EGFR-binding peptide, GE 11, identified by phage display (Li et al., 2005). The vector PEI–PEG–GE11 (Figure 2) showed enhanced transfection into EGFR-overexpressing tumor cells, including glioblastoma and hepatoma cells, but without causing EGFR activation (Schafer et al., 2011). PolyIC/PEI–PEG–GE11 was less selective, however, when compared with PolyIC/PPE as an anti-tumor agent (Abourbeh et al., 2012). The absence of EGFR activation by GE1 (Schafer et al., 2011) may be advantageous, but it is not clear whether the very short exposure of a patient to an EGFR-activating vector is actually a problem. The presence of TGFα in the recombinant pseudomonas toxin-TGFα did not prevent the FDA from allowing its successful use in treating GBM patients, in clinical trials (Pastan et al., 2006). We therefore believe that PolyIC/PPE is suitable for clinical development, while we are continuing to evaluate PPGE11 (Abourbeh et al., 2012) in pre-clinical models.
The chemical vector we utilize is actually built like Lego, such that the homing ligand can be replaced, with the other elements remaining constant. Any tumor that overexpresses a surface protein that can be internalized upon ligand binding is a candidate for targeted PolyIC therapy, once the coupling conditions have been optimized for the relevant ligand. Table 1 depicts tumors that overexpress receptor proteins that can in principle be targeted by a vector similar to the ones used for targeting EGFR-overexpressing tumors.

Table 1. Examples of cancers with overexpressed growth factor receptors.
Stat3 plays a crucial role in tumor development at multiple levels. Stat3 functions as an oncoprotein in tumor cells and inhibits the anti-tumor effects of immune cells, tilting the immune system toward cooperation with the tumor cells. A comprehensive discussion of the role of Stat3 in the interaction between the tumor and the immune system is given in (Yu et al., 2007, 2009). We shall describe briefly the action of Stat3, since we believe that Stat3 inhibition can be combined effectively with targeted PolyIC. Stat3 mediates crosstalk between tumor cells and immune cells, forming an immunosuppressive network in many tumors, including GBM. Stat3 is persistently activated in many kinds of cancer, in both the tumor cells and the tumor-associated stromal cells, including diverse immune cells. Activated Stat3 promotes the expression of numerous immunosuppressive factors, such as VEGF, interleukin-10 (IL-10), IL-6, and IL-23. The receptors for many Stat3-regulated factors in turn are Stat3 activators and are expressed by various cells of hematopoietic origin. This feed-forward loop of Stat3 activation underlies the crosstalk among tumor cells and diverse immune cells in the tumor microenvironment (Yu et al., 2007, 2009). Although Stat3 is a well-known transcription factor, more recent studies have demonstrated its ability to inhibit the expression of a large number of Th1 immunostimulatory genes. For example, Stat3 inhibits the expression of the MHC class II molecules, CD80, CD86, and IL-12, by DCs. This prevents DC maturation, which is required to stimulate the anti-tumor effects of CD8+ T cells and NK cells. Consequently, persistent activation of Stat3 in tumor cells and in tumor stromal immune cells is associated with profound immunosuppression. Furthermore, immature myeloid cells that accumulate in the tumor produce VEGF, bFGF, and other angiogenic factors in a Stat3-dependent manner, promoting tumor vascularization, or angiogenesis. Tumor vascularization, in turn, protects the tumor cells from apoptosis and counteracts the interferon-γ-dependent effects of CD8+ T cells. Thus, inhibition of Stat3 should be effective in treating many forms of cancer.
A number of strategies for Stat3 inhibition are being followed, including low molecular weight inhibitors that target Stat3 dimerization, inhibiting Stat3 binding to DNA, and siRNA. Stat3 siRNA linked to CpG oligonucleotide, the ligand for TLR9 receptor, was effective at inhibiting Stat3 in myeloid cells and B cells in vivo, activating the immune system to exert its anti-tumor activity (Kortylewski et al., 2009). It is likely that a combination of targeted dsRNA such as PolyIC with emerging Stat3 inhibitors will have additive or even synergistic effects. Stat3 inhibition is expected to disarm the shield of the tumor and its microenvironment, sensitizing the tumor to other forms of therapy, including targeted PolyIC therapy.
In the last few years the utilization of monoclonal antibodies to treat various malignancies has accelerated at a much higher pace than the development of new medicinal compounds. This trend reflects the recognition that harnessing the immune system is an effective way to attack cancer. An important feature of antibody action is the induction of ADCC, which brings about the demise of tumor cells neighboring the targeted tumor cell. This feature is missing from small molecular weight signal transduction inhibitors. In this review we argue that one could upgrade the bystander effects induced by antibodies if one were able to target the immune stimulator PolyIC to the tumor. This strategy would bring about strong anti-tumor activities, allowing a short and effective treatment. The strategy can be utilized to treat tumors that overexpress proteins that internalize upon ligand binding. The prevalence of such tumors is very wide, so this strategy should be applicable to the treatment of many types of cancer.
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
I would like to thank Dr. Shoshana Klein from our unit for her comments and editing. The experimental work described in the review was supported by grants from The Association for International Cancer Research (AICR), UK, a grant from the National Cancer Institute (NIH), USA 1R01CA125500 – 01A2, and an ERC a grant from the European Research Council under the European Union’s Seventh Framework Programme (FP7/2007-2013)/ERC grant agreement no. 249898.
Abourbeh, G., Shir, A., Mishani, E., Ogris, M., Roedl, W., Wagner, E., and Levitzki, A. (2012). PolyIC GE11polyplex inhibits EGFR overexpressing tumors. IUBMB Life (in press).
Amann, M., D’Argouges, S., Lorenczewski, G., Brischwein, K., Kischel, R., Lutterbuese, R., Mangold, S., Rau, D., Volkland, J., Pflanz, S., Raum, T., Munz, M., Kufer, P., Schlereth, B., Baeuerle, P. A., and Friedrich, M. (2009). Antitumor activity of an EpCAM/CD3-bispecific BiTE antibody during long-term treatment of mice in the absence of T-cell anergy and sustained cytokine release. J. Immunother. 32, 452–464.
Benhar, M., Engelberg, D., and Levitzki, A. (2002). ROS, stress-activated kinases and stress signaling in cancer. EMBO Rep. 3, 420–425.
Blattman, J. N., and Greenberg, P. D. (2004). Cancer immunotherapy: a treatment for the masses. Science 305, 200–205.
Blumberg, B. S. (2010). Primary and secondary prevention of liver cancer by HBV. Front. Biosci. 2, 756–763.
Dudley, M. E., Yang, J. C., Sherry, R., Hughes, M. S., Royal, R., Kammula, U., Robbins, P. F., Huang, J., Citrin, D. E., Leitman, S. F., Wunderlich, J., Restifo, N. P., Thomasian, A., Downey, S. G., Smith, F. O., Klapper, J., Morton, K., Laurencot, C., White, D. E., and Rosenberg, S. A. (2008). Adoptive cell therapy for patients with metastatic melanoma: evaluation of intensive myeloablative chemoradiation preparative regimens. J. Clin. Oncol. 26, 5233–5239.
Hanahan, D., and Weinberg, R. A. (2011). Hallmarks of cancer: the next generation. Cell 144, 646–674.
Hennessy, E. J., Parker, A. E., and O’Neill, L. A. (2010). Targeting Toll-like receptors: emerging therapeutics? Nat. Rev. Drug Discov. 9, 293–307.
Hodi, F. S., O’Day, S. J., Mcdermott, D. F., Weber, R. W., Sosman, J. A., Haanen, J. B., Gonzalez, R., Robert, C., Schadendorf, D., Hassel, J. C., Akerley, W., Van Den Eertwegh, A. J., Lutzky, J., Lorigan, P., Vaubel, J. M., Linette, G. P., Hogg, D., Ottensmeier, C. H., Lebbe, C., Peschel, C., Quirt, I., Clark, J. I., Wolchok, J. D., Weber, J. S., Tian, J., Yellin, M. J., Nichol, G. M., Hoos, A., and Urba, W. J. (2010). Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 363, 711–723.
Kantoff, P. W., Higano, C. S., Shore, N. D., Berger, E. R., Small, E. J., Penson, D. F., Redfern, C. H., Ferrari, A. C., Dreicer, R., Sims, R. B., Xu, Y., Frohlich, M. W., and Schellhammer, P. F. (2010). Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N. Engl. J. Med. 363, 411–422.
Kenter, G. G., Welters, M. J., Valentijn, A. R., Lowik, M. J., Berends-Van Der Meer, D. M., Vloon, A. P., Essahsah, F., Fathers, L. M., Offringa, R., Drijfhout, J. W., Wafelman, A. R., Oostendorp, J., Fleuren, G. J., Van Der Burg, S. H., and Melief, C. J. (2009). Vaccination against HPV-16 oncoproteins for vulvar intraepithelial neoplasia. N. Engl. J. Med. 361, 1838–1847.
Kohler, G., and Milstein, C. (1975). Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 256, 495–497.
Korman, A. J., Peggs, K. S., and Allison, J. P. (2006). Checkpoint blockade in cancer immunotherapy. Adv. Immunol. 90, 297–339.
Kortylewski, M., Swiderski, P., Herrmann, A., Wang, L., Kowolik, C., Kujawski, M., Lee, H., Scuto, A., Liu, Y., Yang, C., Deng, J., Soifer, H. S., Raubitschek, A., Forman, S., Rossi, J. J., Pardoll, D. M., Jove, R., and Yu, H. (2009). In vivo delivery of siRNA to immune cells by conjugation to a TLR9 agonist enhances antitumor immune responses. Nat. Biotechnol. 27, 925–932.
Kumar, H., Koyama, S., Ishii, K. J., Kawai, T., and Akira, S. (2008). Cutting edge: cooperation of IPS-1- and TRIF-dependent pathways in poly IC-enhanced antibody production and cytotoxic T cell responses. J. Immunol. 180, 683–687.
Lacour, J., Laplanche, A., Delozier, T., Berlie, J., Mourali, N., Julien, J. P., De Gislain, C., Namer, M., Petit, J. C., Denis, V., et al. (1991). Polyadenylic-polyuridylic acid plus locoregional and pelvic radiotherapy versus chemotherapy with CMF as adjuvants in operable breast cancer. A 6 1/2 year follow-up analysis of a randomized trial of the French federation of cancer centers (F.F.C.C.). Breast Cancer Res. Treat. 19, 15–21.
Levitzki, A. (1992). Tyrphostins: tyrosine kinase blockers as novel antiproliferative agents and dissectors of signal transduction. FASEB J. 14, 3275–3282.
Li, Z., Zhao, R., Wu, X., Sun, Y., Yao, M., Li, J., Xu, Y., and Gu, J. (2005). Identification and characterization of a novel peptide ligand of epidermal growth factor receptor for targeted delivery of therapeutics. FASEB J. 19, 1978–1985.
Mazzoleni, S., Politi, L. S., Pala, M., Cominelli, M., Franzin, A., Sergi Sergi, L., Falini, A., De Palma, M., Bulfone, A., Poliani, P. L., and Galli, R. (2010). Epidermal growth factor receptor expression identifies functionally and molecularly distinct tumor-initiating cells in human glioblastoma multiforme and is required for gliomagenesis. Cancer Res. 70, 7500–7513.
Moschella, F., Proietti, E., Capone, I., and Belardelli, F. (2010). Combination strategies for enhancing the efficacy of immunotherapy in cancer patients. Ann. N. Y. Acad. Sci. 1194, 169–178.
Navabi, H., Jasani, B., Reece, A., Clayton, A., Tabi, Z., Donninger, C., Mason, M., and Adams, M. (2009). A clinical grade poly I:C-analogue (Ampligen) promotes optimal DC maturation and Th1-type T cell responses of healthy donors and cancer patients in vitro. Vaccine 27, 107–115.
Okada, H., Kalinski, P., Ueda, R., Hoji, A., Kohanbash, G., Donegan, T. E., Mintz, A. H., Engh, J. A., Bartlett, D. L., Brown, C. K., Zeh, H., Holtzman, M. P., Reinhart, T. A., Whiteside, T. L., Butterfield, L. H., Hamilton, R. L., Potter, D. M., Pollack, I. F., Salazar, A. M., and Lieberman, F. S. (2011). Induction of CD8+ T-cell responses against novel glioma-associated antigen peptides and clinical activity by vaccinations with {alpha}-type 1 polarized dendritic cells and polyinosinic-polycytidylic acid stabilized by lysine and carboxymethyl cellulose in patients with recurrent malignant glioma. J. Clin. Oncol. 29, 330–336.
Pastan, I., Hassan, R., Fitzgerald, D. J., and Kreitman, R. J. (2006). Immunotoxin therapy of cancer. Nat. Rev. Cancer 6, 559–565.
Peggs, K. S., Quezada, S. A., and Allison, J. P. (2008). Cell intrinsic mechanisms of T-cell inhibition and application to cancer therapy. Immunol. Rev. 224, 141–165.
Rosenberg, S. A., Restifo, N. P., Yang, J. C., Morgan, R. A., and Dudley, M. E. (2008). Adoptive cell transfer: a clinical path to effective cancer immunotherapy. Nat. Rev. Cancer 8, 299–308.
Salem, M. L., El-Naggar, S. A., Kadima, A., Gillanders, W. E., and Cole, D. J. (2006). The adjuvant effects of the toll-like receptor 3 ligand polyinosinic-cytidylic acid poly (I:C) on antigen-specific CD8+ T cell responses are partially dependent on NK cells with the induction of a beneficial cytokine milieu. Vaccine 24, 5119–5132.
Salem, M. L., Kadima, A. N., Cole, D. J., and Gillanders, W. E. (2005). Defining the antigen-specific T-cell response to vaccination and poly(I:C)/TLR3 signaling: evidence of enhanced primary and memory CD8 T-cell responses and antitumor immunity. J. Immunother. 28, 220–228.
Sampson, J. H., Reardon, D. A., Friedman, A. H., Friedman, H. S., Coleman, R. E., Mclendon, R. E., Pastan, I., and Bigner, D. D. (2005). Sustained radiographic and clinical response in patient with bifrontal recurrent glioblastoma multiforme with intracerebral infusion of the recombinant targeted toxin TP-38: case study. Neuro-oncology 7, 90–96.
Schafer, A., Pahnke, A., Schaffert, D., Van Weerden, W. M., De Ridder, C. M., Rodl, W., Vetter, A., Spitzweg, C., Kraaij, R., Wagner, E., and Ogris, M. (2011). Disconnecting the Yin and Yang relation of epidermal growth factor receptor (EGFR)-mediated delivery: a fully synthetic, EGFR-targeted gene transfer system avoiding receptor activation. Hum. Gene Ther. 12, 1463–1473.
Schaffert, D., Kiss, M., Rodl, W., Shir, A., Levitzki, A., Ogris, M., and Wagner, E. (2011). Poly(I:C)-mediated tumor growth suppression in EGF-receptor overexpressing tumors using EGF-polyethylene glycol-linear polyethylenimine as carrier. Pharm. Res. 28, 731–741.
Shir, A., Ogris, M., Roedl, W., Wagner, E., and Levitzki, A. (2011). EGFR-homing dsRNA activates cancer-targeted immune response and eliminates disseminated EGFR-overexpressing tumors in mice. Clin. Cancer Res. 17, 1033–1043.
Shir, A., Ogris, M., Wagner, E., and Levitzki, A. (2006). EGF receptor-targeted synthetic double-stranded RNA eliminates glioblastoma, breast cancer, and adenocarcinoma tumors in mice. PLoS Med. 3, e6. doi: 10.1371/journal.pmed.0030006
Sistigu, A., Viaud, S., Chaput, N., Bracci, L., Proietti, E., and Zitvogel, L. (2011). Immunomodulatory effects of cyclophosphamide and implementations for vaccine design. Semin. Immunopathol. 33, 369–383.
Talmadge, J. E., Adams, J., Phillips, H., Collins, M., Lenz, B., Schneider, M., Schlick, E., Ruffmann, R., Wiltrout, R. H., and Chirigos, M. A. (1985). Immunomodulatory effects in mice of polyinosinic-polycytidylic acid complexed with poly-L-lysine and carboxymethylcellulose. Cancer Res. 45, 1058–1065.
Vesely, M. D., Kershaw, M. H., Schreiber, R. D., and Smyth, M. J. (2011). Natural innate and adaptive immunity to cancer. Annu. Rev. Immunol. 29, 235–271.
Wolf, E., Hofmeister, R., Kufer, P., Schlereth, B., and Baeuerle, P. A. (2005). BiTEs: bispecific antibody constructs with unique anti-tumor activity. Drug Discov. Today 10, 1237–1244.
Yoneda, T., Lyall, R. M., Alsina, M. M., Persons, P. E., Spada, A. P., Levitzki, A., Zilberstein, A., and Mundy, G. R. (1991). The antiproliferative effects of tyrosine kinase inhibitors tyrphostins on a human squamous cell carcinoma in vitro and in nude mice. Cancer Res. 51, 4430–4435.
Yu, H., Kortylewski, M., and Pardoll, D. (2007). Crosstalk between cancer and immune cells: role of STAT3 in the tumour microenvironment. Nat. Rev. Immunol. 7, 41–51.
Keywords: PolyIC, bystander effect, cancer
Citation: Levitzki A (2012) Targeting the immune system to fight cancer using chemical receptor homing vectors carrying polyinosine/cytosine (PolyIC). Front. Oncol. 2:4. doi: 10.3389/fonc.2012.00004
Received: 31 October 2011; Accepted: 05 January 2012;
Published online: 08 February 2012.
Edited by:
Ruggero De Maria, Istituto Superiore di Sanità, ItalyReviewed by:
Paola Nisticò, Regina Elena National Cancer Institute, ItalyCopyright: © 2012 Levitzki. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Alexander Levitzki, Unit of Cellular Signaling, Department of Biological Chemistry, The Alexander Silberman Institute of Life Sciences, The Hebrew University of Jerusalem, Givat Ram, Jerusalem 91904, Israel. e-mail:YWxleC5sZXZpdHpraUBtYWlsLmh1amkuYWMuaWw=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.