
- 1 The Department of Oncological Sciences, University of Utah School of Medicine, Salt Lake City, UT, USA
- 2 The Center for Children’s Cancer Research, Huntsman Cancer Institute, University of Utah, Salt Lake City, UT, USA
- 3 The Division of Pediatric Hematology/Oncology, University of Utah School of Medicine, Salt Lake City, UT, USA
The glutathione S-transferases (GSTs) are a family of ubiquitously expressed polymorphic enzymes important for detoxifying endogenous and exogenous compounds. In addition to their classic activity of detoxification by conjugation of compounds with glutathione, many other functions are now found to be associated with GSTs. The associations between GST polymorphisms/functions and human disease susceptibility or treatment outcome, mostly in adults, have been extensively studied and reviewed. This mini review focuses on studies related to GST epidemiology and functions related to pediatric cancer. Opportunities to exploit GST in pediatric cancer therapy are also discussed.
General Information on Glutathione S-Transferases
The GST Family
Glutathione S-transferase (GST) isozymes were originally discovered in rat liver cytosol during the early 1960s (Booth et al., 1961; Coombes and Stakelum, 1961). GSTs are drug-metabolizing enzymes that catalyze conjugation of glutathione with carcinogens, drugs, toxins, or products of oxidative stress. Two distinct superfamilies of GST isozymes exist (Table 1). One superfamily comprises cytosolic, soluble, dimeric enzymes. At present, eight distinct classes have been identified in this superfamily: alpha, kappa, mu, omega, pi, sigma, theta, and zeta. The other superfamily is composed of microsomal, membrane bound, trimeric proteins. This family of proteins is also called MAPEG (membrane-associated proteins in eicosanoid and glutathione metabolism; Hayes and Strange, 2000). While GSTs are expressed in all tissues, specific isozyme distribution across different tissues in mammals is variable and complex (Hiley et al., 1989; Strange et al., 1989).
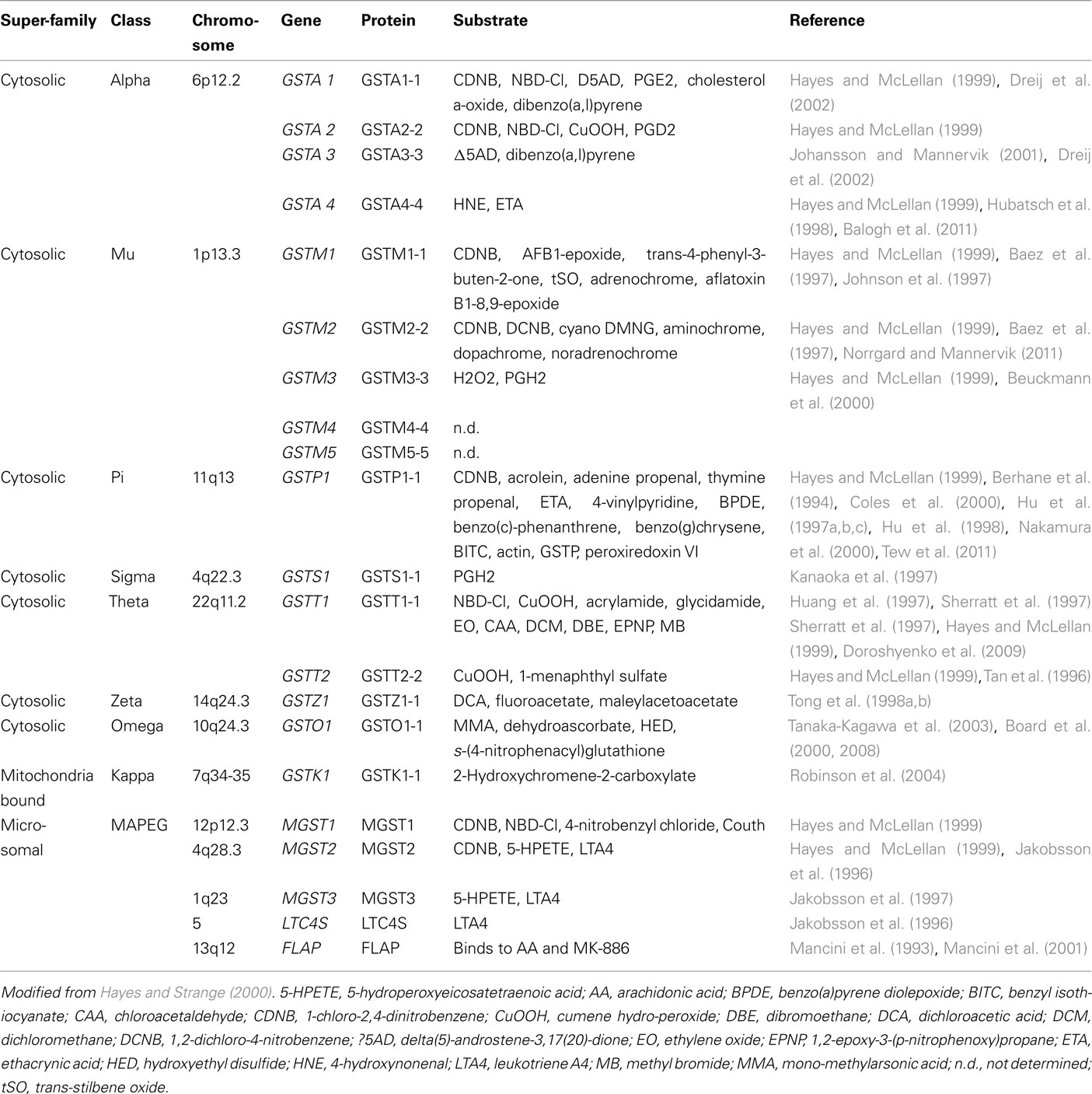
Table 1. Human GSTs and their biochemical properties.
GST Substrates
Crystal structures have been determined for many soluble GSTs, often with bound substrates or products (Oakley, 2011). The “canonical fold” of a soluble GST subunit reveals an N-terminal α/β domain forming the GSH-binding site (G-site) and a second, α-helical domain forming most of the H-site that binds the electrophilic substrate (Figure 1). CDNB (1-chloro-2,4-dinitrobenzene) was initially adopted by biochemists as a “universal” GST substrate. However, it was soon realized that, although different transferases may exhibit overlapping substrate specificities, no common substrate exists that is metabolized by all GST isozymes (Table 1). Different amino acids in the substrate-binding site (H-site) of GST isozymes can account for substrate specificities. In view of the separate evolutionary histories of the cytosolic and MAPEG superfamilies, it is not surprising that they display marked differences in catalytic activities (Armstrong, 1997). Because its binding pocket for electrophiles is hydrophobic, Microsomal GST1 (MGST1) is uniquely suited among the GSTs to detoxify reactive intermediates of a more hydrophobic nature (Schaffert, 2011). The substrate specificity of individual GST isozymes suggests that each of them play a unique role in biotransformation of drugs. Genetic variations in these enzymes will therefore influence the response of individuals to environmental agents.
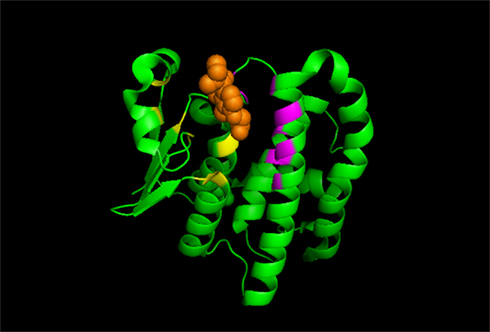
Figure 1. A cartoon diagram of GSTP, a representative of a soluble GST enzyme. The N-terminal GSH-binding site (G-site, in yellow) and the C-terminal H-site (in purple) that binds the electrophilic substrate (in orange) are shown.
Polymorphism and Epidemiology
Genetic polymorphisms in the GST genes arise from nucleotide alterations that may change codons to generate unique alleles or even null genotypes. These amino acid substitutions cause steric changes in the substrate-binding site of the enzyme. As a consequence, the enzymatic activities of GST are significantly affected. Because different GST proteins differ in their ability to catalyze specific detoxification reactions, polymorphisms in GST will likely impact response to specific therapies. Many molecular epidemiological studies have tested associations between polymorphisms of GST genes and disease susceptibility. The results of such studies have often been conflicting. As a consequence, a number of meta-analyses have been carried out as part of the Human Genome Epidemiology (HuGE) Network, which provided evidence that GST polymorphisms can result in a small but significant increase in risk of some types of cancers or diseases (Hayes and Strange, 2000; Bolt and Their, 2006; Di Pietro et al., 2010; Economopoulos and Sergentanis, 2010; Josephy, 2010; McMahon et al., 2010). However, most meta-analyses suffered from a serious limitation: fail to distinguish between heterozygous and homozygous genotypes, which resulted in heterogeneity between studies. Now boundaries of the deletion polymorphisms have been cloned and analytical methods that assess copy number such as real-time PCR are now available (Timofeeva et al., 2009). These will be helpful in dissolving some of the heterogeneity observed in clinical evaluations.
Functions of GST
The classic activity of the GSTs is to detoxify reactive electrophiles by conjugation to glutathione (GSH), thereby reducing the likelihood of deleterious interactions between such reactive species and essential cellular components like proteins and nucleic acids. Knockout and transgenic mouse models were generated for many GST family members which helped to reveal the physiological function of GST isozymes (Elsby et al., 2003; Henderson and Wolf, 2011). Based on these and other model systems, many other activities are now associated with GSTs, including regulation of signaling pathways and anti-apoptotic activity by GSTP (Tew et al., 2011), anti- and pro-inflammatory functions of sigma-class GSTs (Flanagan and Smythe, 2011), activities of MGST1 related to mitochondria (Aniya and Imaizumi, 2011), regulation of the cardiac muscle ryanodine receptor (Dulhunty et al., 2011), and functions associated with asthma (Minelli et al., 2010).
GSTs and signaling pathway regulation
It is becoming apparent that GSTP family members participate in the maintenance of cellular redox homeostasis through a variety of mechanisms (Tew et al., 2011). GSTP1, for example, displays an anti-apoptotic activity based on protein–protein interactions with c-Jun N-terminal kinase (JNK; Adler et al., 1999; Figure 2). GSTP1 was implicated in mediating S-glutathionylation of specific clusters of target proteins and in reactions that play a negative regulatory role in some kinase pathways through other protein–protein interactions. GSTP1 has also been implicated in regulating tumor necrosis factor-alpha (TNF-alpha) signaling primarily through a physical association with tumor necrosis factor receptor-associated factor 2 (TRAF2; Wu et al., 2006). In addition, physical interactions between the HPV16 E7 viral factor and GSTP1 were found to improve survival capabilities of host cells (Mileo et al., 2009). GSTA1 is also capable of suppressing JNK signaling caused by inflammatory cytokines or oxidative stress, likely because of the homology between GST A and P family members, (Romero et al., 2006). It was also reported that GSTM1 binds and inhibits the activity of Apoptosis Signaling Kinase-1 (ASK1; Cho et al., 2001). The GSTM1:ASK1 interaction dissociates under oxidative stress or heat shock, leading to activation of ASK1 and apoptosis (Dorion et al., 2002).
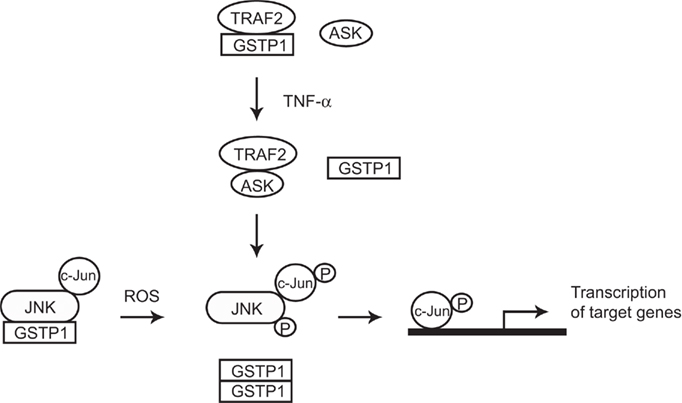
Figure 2. Cartoon model of how GSTP1–JNK and GSTP1–TRAF2 interactions modulate JNK and TNF-α signaling pathways. In non-stressed cells, GSTP1 form complexes with JNK and TRAF2 respectively. GSTP1–JNK interaction keeps JNK at low activity, while GSTP1–TRAF2 interaction competes against TRAF2–ASK1 binding which also leads to low JNK activity. In cells exposed to ROS or TNF-α, GSTP1 dissociates from the complexes and accumulates as oligomers, with resultant activation of JNK and downstream signaling pathways.
MGST1 and its dual function in mitochondria
Microsomal GST1 protects cells (and mitochondria) from oxidative stress by both conjugation and glutathione peroxidase functions (Johansson et al., 2010; Aniya and Imaizumi, 2011; Schaffert, 2011). MCF7 cells overexpressing MGST1 showed protection against agents that induce peroxidation. Mitochondria in these cells were shown to be protected from oxidative insult as measured by calcium loading capacity and respiration. MGST1 also induced cellular resistance against cisplatin in these cells (Johansson et al., 2010). In addition to these protective effects, MGST1 also has deleterious effects that contribute to oxidative stress-induced liver injury. Preliminary studies indicated that mitochondrial MGST1 was activated by reactive oxygen species (ROS). This activation led to its aggregation and induction of non-classical mitochondrial permeability transition (MPT). MPT induces mitochondrial dysfunction which results in apoptosis and necrosis. Therefore, depending on the context, MGST1 activation can either exhibit a protective or toxic effect on the liver and possibly other tissues (Schaffert, 2011).
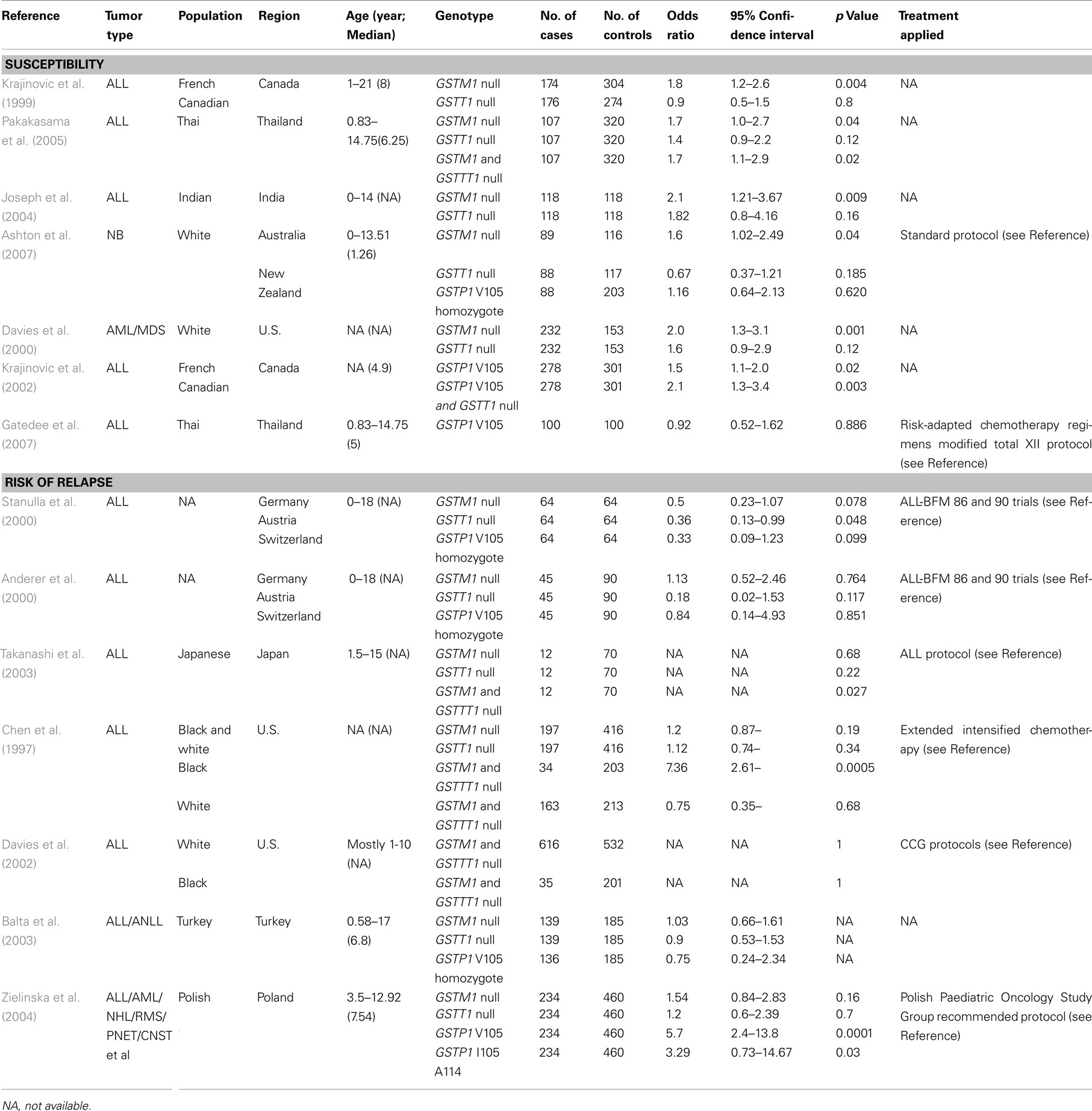
Table 2. Case–control studies on association of GST polymorphism with susceptibility or risk of relapse of childhood cancers.
GST in Pediatric Cancer
Epidemiology
As in adult cancers, a large number of epidemiological studies have tested possible associations between GST polymorphic variants as well as deletions with susceptibility of pediatric cancers (Table 2). GSTM1 is subject to a deletion polymorphism that is found in as many as 75% of Caucasians (Zhong et al., 1991; Huang et al., 2009). Multiple case-controlled studies have indicated statistically significant associations of the GSTM1 null genotype with increased risk of childhood cancers. Groups from different regions in the world found that GSTM1 null genotype related to increased risk of acute leukemia (ALL and AML) in various populations (Krajinovic et al., 1999; Davies et al., 2000; Joseph et al., 2004; Pakakasama et al., 2005; Marino et al., 2009). Ashton et al. (2007) found that GSTM1 null genotype is associated with high risk of neuroblastoma in Australia and New Zealand. GSTP1 V105 was found to relate to increased susceptibility to childhood leukemia in Canada (Krajinovic et al., 2002). However, another group found that there was no statistically significant association between GSTP1 polymorphism and susceptibility to ALL in Thai children (Gatedee et al., 2007). Interestingly, one group reported that GSTO1 A140D and GSTO2 N142D polymorphism were both significantly associated with susceptibility to ALL in Thailand (Pongstaporn et al., 2009). These conflicting results were thought to be related to differences in ethnicity and age of the patients, treatment, and follow-up periods included in these studies.
GST and Drug Resistance
Glutathione S-transferases have been implicated in the development of resistance toward chemotherapeutic agents which lead to relapse in pediatric cancers (Table 2). GSTT1 null genotype has been shown to confer a reduced risk of relapse in childhood ALL in several case studies (Anderer et al., 2000; Stanulla et al., 2000). However, the GSTT1 null genotype was also shown to be associated with an increased risk of death after chemotherapy in childhood AML (Davies et al., 2001). As to the GSTM1–GSTT1 double-null genotype, one group reported an increased risk of early relapse of ALL with double-null genotype (Takanashi et al., 2003) while a subsequent study reported the opposite result (Kham et al., 2004). Yet other studies showed that the double-null genotype was not associated with risk of relapse or treatment outcome in ALL (Chen et al., 1997; Davies et al., 2002; Balta et al., 2003; Jazbec et al., 2003). GSTP1 expression was significantly increased in relapsed AML, and GSTP1 V105 was found to be related to increased relapse rate of childhood leukemia (Sauerbrey et al., 1994; Beck et al., 1996; Zielinska et al., 2004).
Several studies using microarray technology have identified glutathione metabolism pathway to reflect tumor resistance to chemotherapy in Ewing’s sarcoma. The authors found that the expression of MGST1 clearly predict Ewing’s sarcoma prognosis and to be associated with doxorubicin chemosensitivity (Townsend and Tew, 2003; Schaefer et al., 2008; Scotlandi et al., 2009). In a separate microarray analysis for target genes of EWS/FLI, the master regulator of Ewing’s sarcoma, GSTM4 was found to be upregulated by EWS/FLI. Reduction of GSTM4 levels resulted in an increased sensitivity of Ewing’s sarcoma cells to chemotherapeutic agents (etoposide and fenretinide). This suggested a role for GSTM4 in drug resistance. In support of this hypothesis, patients with Ewing’s sarcoma whose tumors had higher levels of GSTM4 expression had worse outcomes than those with lower expression levels (Luo et al., 2009).
GSTM1 has been reported to be a significant risk factor for hematologic relapse in childhood ALL. Transduction of GSTM1 into T-acute lymphoblastic leukemia cells selectively decreased cellular sensitivity to dexamethasone in a manner that was independent of glutathione conjugation, but was due to apoptosis inhibition. Interestingly, p38-MAPK and Bim activation were suppressed, and NF-kappaB p50 was activated, in these GSTM1 expressing cells. The authors proposed that GSTM1 is a novel regulator of dexamethasone-induced apoptosis, and causes dexamethasone resistance by suppression of Bim through dual mechanisms of downregulation of p38-MAPK and up-regulation of NF-kappaB p50 (Hosono et al., 2010). Consistently, GSTM1 null genotype was associated with a reduced risk of relapse in ALL (Stanulla et al., 2000). Association of GSTM1 with resistance to adriamycin and cisplatin was also found in childhood hepatoblastoma (Bader et al., 1998).
Drug resistance is a common problem in the treatment of childhood rhabdomyosarcoma (RMS). To identify causes of drug resistance in this disease, Seitz et al. (2010) performed gene expression analysis of tumors from mice transplanted with embryonal or alveolar RMS cells and treated with vincristine. The authors found 2314 differentially expressed genes between the groups in alveolar RMS and 1387 in embryonal RMS. Pathway analysis revealed a cluster of five overexpressed genes of the GST family in animals treated with vincristine, suggesting a cause for drug resistance. In vitro experiments confirmed up-regulation of GST activity following incubation with doxorubicin and topotecan in RMS cell lines. Incubation with GST inhibitors resulted in a decreased cell viability. The authors concluded that reversal of drug resistance in childhood RMS may be achieved by GST inhibitors, at least in part. Thus, the GST family represents a promising target for further treatment strategies in childhood RMS (Seitz et al., 2010).
The association of GSTs with risk of relapse and drug resistance may not be a straightforward reflection of their ability to participate in detoxification reactions. Greater understanding of the numerous factors affecting GST expression and activity, as well as GST functions, may reveal further connections between GST and individual responses to disease and drugs.
Regulation of GST
Most cases of Ewing’s sarcoma express the EWS/FLI fusion oncoprotein (Turc-Carel et al., 1988). EWS/FLI functions as an aberrant transcription factor that mediates the transformed phenotype through the deregulation of several key target genes (Kinsey et al., 2006; Owen and Lessnick, 2006; Smith et al., 2006; Tirado et al., 2006; Luo et al., 2009). Furthermore, EWS/FLI has been shown to transcriptionally activate some of its gene targets through GGAA-containing microsatellite promoter elements (Gangwal et al., 2008). Interestingly, GSTM4 contains a GGAA-microsatellite in its promoter. In vitro and in vivo studies revealed that EWS/FLI binds to the microsatellite and up-regulates GSTM4 via this element. Other work has shown that the ability of EWS/FLI to activate gene expression through GGAA-microsatellite response elements is proportional to the length of the microsatellite, suggesting that microsatellite polymorphisms might affect target gene expression (Gangwal et al., 2010). Indeed, this hypothesis was supported by the finding that the number of GGAA repeats present in the NR0B1 promoter positively correlated with the level of NR0B1 mRNA expression in Ewing’s cells (Garcia-Aragoncillo et al., 2008). Further work will be needed to determine if a similar relationship exists for the GSTM4 gene, and if such a relationship correlates with drug resistance in Ewing’s sarcoma.
The GGAA-microsatellite is not shared by other GST family members, however. Other GST promoters contain response elements such as an antioxidant response element and a xenobiotic response element, as well as putative binding sites for transcription factors such as AP-1, MAF, Nrf1, Jun, Fos, and NF-kappaB. Such complex response elements suggest a mechanism for differential regulation of GST isozymes across tumor types in response to differing toxic insults.
GST as Targets in Pediatric Cancer Treatment
The design and discovery of compounds that bind GST isozymes and modulate their biological activity has become an important aim in cancer research because GST isozymes are overexpressed in many cancer cell lines (Tew et al., 1996), and induce drug resistance by inactivating many chemotherapeutic compounds via GSH conjugation (Tew et al., 1997). There are a number of candidate GST-targeted drugs at various stages of preclinical development (Tew et al., 1997; Ruzza and Quintieri, 2009; Wondrak, 2009; Sau et al., 2010). Ethacrynic acid, an inhibitor that lacks class specificity for GSTs, represented a first attempt in this direction; however, its low affinity and deleterious side effects have discouraged its use in clinical practice (Tew et al., 1997). More recently, GSH peptidomimetic compounds have been designed, including TER 199 (Figure 3). However, many GSH derivatives are actively extruded from cancer cells by specific export pumps, such as the multidrug resistance protein, and so are unlikely to be highly efficacious (Muller et al., 1994; Morgan et al., 1996).
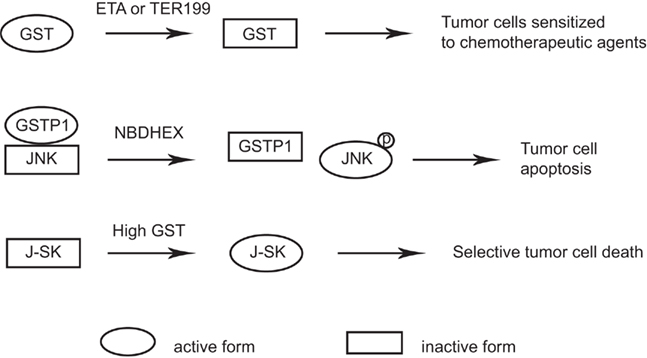
Figure 3. GST as targets in pediatric cancer treatment. GST non-specific inhibitors such as ETA or TER199 decrease the GST activity in tumors and sensitize tumor cells to chemotherapeutic agents. NBDHEX treatment of tumor cells interrupts GSTP1–JNK interaction which releases and activates JNK and results in tumor cell apoptosis. Prodrugs such as J-SK is activated specifically in tumors with high GST activity and selectively kills tumor cells.
A new class of GST inhibitors, called NBD derivatives, has been designed recently (Ricci et al., 2005). A representative of this class is NBDHEX, which interacts with GSTP1 and triggers apoptosis in human tumor cells through dissociation of the JNK–GSTP1 complex (Turella et al., 2005; Figure 3). Osteosarcoma, Ewing’s sarcoma, and rhabdomyosarcoma cell lines were all found to be sensitive to NBDHEX in vitro and in xenograft models (Scotlandi et al., 2009; Pasello et al., 2011). Importantly, NBDHEX was not extruded from tumor cells by multidrug resistance protein pumps (Ricci et al., 2005; Turella et al., 2005, 2006). NBDHEX had synergistic effect with doxorubicin, vincristine, cisplatin in an in vitro study. In vivo studies confirmed the cytostatic efficacy of NBDHEX and its synergy with vincristine in Ewing’s sarcoma cells, and also its effect against the metastatic spread of osteosarcoma cells (Pasello et al., 2011). Although NBDHEX is still under preclinical in vivo evaluation, it may be an interesting new therapeutic option for patients who are not highly responsive to conventional regimens.
Another approach is to design prodrugs that exploit the high GST expression levels found in drug resistant tumors and cells. Prodrugs would be preferentially activated by GST in malignant cells, thus sparing normal tissues and enhancing the therapeutic index. JS-K is a member of the O2-aryl diazeniumdiolate compound family which was designed to release nitric oxide (NO) when activated by GSTs (Shami et al., 2006). JS-K has shown promise as a novel cancer therapeutic agent in a number of studies. For instance, JS-K selectively induces programmed cell death in breast cancer cells while sparing normal mammary epithelial cells under the same conditions (McMurtry et al., 2011; Figure 3). The selective anti-tumor activity of JS-K warrants its further investigation in pediatric cancers.
Conclusion
Many epidemiological studies have investigated possible associations between GST polymorphisms with risk of pediatric cancers. Some studies have indicated increased risk for specific genotypes, such as the GSTT1 and GSTM1 deletions, while other studies have not confirmed this association. These conflicting results need to be interpreted with caution. Many GST activities other than detoxification have been discovered and may contribute to different GST activities among individuals. GSTs have also been implicated in the development of resistance toward chemotherapeutic drugs. Although further studies are required to reveal the underlying mechanisms, drugs targeting GSTs have been designed to overcome resistance to conventional therapeutic agents. Little progress has been made in understanding how GST expression and activity is regulated. Such studies will allow an understanding of the upstream players in the glutathione metabolism pathway and provide potential new approaches for the treatment of pediatric cancers.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Stephen L. Lessnick is supported by the NIH (R21 CA138295, R01 CA140394), the Terri Anna Perine Sarcoma Fund, and the University of Utah Department of Pediatrics and Huntsman Cancer Institute/Huntsman Cancer Foundation. Joshua D. Schiffman is supported by Damon Runyon Clinical Investigator Award, Alex’s Lemonade Stand Foundation Epidemiology Award, and the Eunice Kennedy Shriver Children’s Health Research Career Development Award NICHD 5K12HD001410. Wen Luo is supported by the Alex’s Lemonade Stand Foundation and the Primary Children’s Medical Center Foundation. We also acknowledge NIH support to the Huntsman Cancer Institute (P30 CA042014).
References
Adler, V., Yin, Z., Fuchs, S. Y., Benezra, M., Rosario, L., Tew, K. D., Pincus, M. R., Sardana, M., Henderson, C. J., Wolf, C. R., Davis, R. J., and Ronai, Z. (1999). Regulation of JNK signaling by GSTp. EMBO J. 18, 1321–1334.
Anderer, G., Schrappe, M., Brechlin, A. M., Lauten, M., Muti, P., Welte, K., and Stanulla, M. (2000). Polymorphisms within glutathione S-transferase genes and initial response to glucocorticoids in childhood acute lymphoblastic leukaemia. Pharmacogenetics 10, 715–726.
Aniya, Y., and Imaizumi, N. (2011). Mitochondrial glutathione transferases involving a new function for membrane permeability transition pore regulation. Drug Metab. Rev. 43, 292–299.
Armstrong, R. N. (1997). Structure, catalytic mechanism, and evolution of the glutathione transferases. Chem. Res. Toxicol. 10, 2–18.
Ashton, L. J., Murray, J. E., Haber, M., Marshall, G. M., Ashley, D. M., and Norris, M. D. (2007). Polymorphisms in genes encoding drug metabolizing enzymes and their influence on the outcome of children with neuroblastoma. Pharmacogenet. Genomics 17, 709–717.
Bader, P., Fuchs, J., Wenderoth, M., von Schweinitz, D., Niethammer, D., and Beck, J. F. (1998). Altered expression of resistance associated genes in hepatoblastoma xenografts incorporated into mice following treatment with adriamycin or cisplatin. Anticancer Res. 18, 3127–3132.
Baez, S., Segura-Aguilar, J., Widersten, M., Johansson, A. S., and Mannervik, B. (1997). Glutathione transferases catalyse the detoxication of oxidized metabolites (?-quinones) of catecholamines and may serve as an antioxidant system preventing degenerative cellular processes. Biochem. J. 324, 25–28.
Balogh, L. M., Le Trong, I., Kripps, K. A., Shireman, L. M., Stenkamp, R. E., Zhang, W., Mannervik, B., and Atkins, W. M. (2011). Substrate specificity combined with stereopromiscuity in glutathione transferase A4-4-dependent metabolism of 4-hydroxynonenal. Biochemistry 49, 1541–1548.
Balta, G., Yuksek, N., Ozyurek, E., Ertem, U., Hicsonmez, G., Altay, C., and Gurgey, A. (2003). Characterization of MTHFR, GSTM1, GSTT1, GSTP1, and CYP1A1 genotypes in childhood acute leukemia. Am. J. Hematol. 73, 154–160.
Beck, J., Handgretinger, R., Klingebiel, T., Dopfer, R., Schaich, M., Ehninger, G., Niethammer, D., and Gekeler, V. (1996). Expression of PKC isozyme and MDR-associated genes in primary and relapsed state AML. Leukemia 10, 426–433.
Berhane, K., Widersten, M., Engstrom, A., Kozarich, J. W., and Mannervik, B. (1994). Detoxication of base propenals and other α,β-unsaturated aldehyde products of radical reactions and lipid peroxidation by human glutathione transferase. Proc. Natl. Acad. Sci. U.S.A. 91, 1480–1484.
Beuckmann, C. T., Fujimori, K., Urade, Y., and Hayaishi, O. (2000). Identification of mu-class glutathione transferases M2-2 and M3-3 as cytosolic prostaglandin E synthases in the human brain. Neurochem. Res. 25, 733–738.
Board, P. G., Coggan, M., Cappello, J., Zhou, H., Oakley, A. J., and Anders, M. W. (2008). S-(4-nitrophenacyl)glutathione is a specific substrate for glutathione transferase omega 1-1. Anal. Biochem. 374, 25–30.
Board, P. G., Coggan, M., Chelvanavagam, G., Easteal, S., Jermjin, L. S., Schulte, G. K., Danley, D. E., Hoth, L. R., Griffor, M. C., Kamath, A. V., Rosner, M. H., Chrunvk, B. A., Perregaux, D. E., Gabel, C. A., Geoghegan, K. F., and Pandit, J. (2000). Identification, characterization, and crystal structure of the Omega class glutathione transferases. J. Biol. Chem. 275, 24798–24806.
Bolt, H. M., and Their, R. (2006). Relevance of the deletion polymorphisms of the glutathione S-transferases GSTT1 and GSTM1 in pharmacology and toxicology. Curr. Drug Metab. 7, 613–628.
Booth, J., Boyland, E., and Sims, P. (1961). An enzyme from rat liver catalyzing conjugations with glutathione. Biochem. J. 79, 516–524.
Chen, C. L., Liu, Q., Pui, C. H., Rivera, G. K., Sandlund, J. T., Ribeiro, R., Evans, W. E., and Relling, M. V. (1997). Higher frequency of glutathione S-transferase deletions in black children with acute lymphoblastic leukemia. Blood 89, 1701–1707.
Cho, S. G., Lee, Y. H., Park, H. S., Ryoo, K., Kang, K. W., Park, J., Eom, S. J., Kim, M. J., Chang, T. S., Choi, S. Y., Shim, J., Kim, Y., Dong, M. S., Lee, M. J., Kim, S. G., Ichijo, H., and Choi, E. J. (2001). Glutathione S-transferase mu modulates the stress-activated signals by suppressing apoptosis signal-regulating kinase 1. J. Biol. Chem. 276, 12749–12755.
Coles, B., Yang, M., Lang, N. P., and Kadlubar, F. F. (2000). Expression of hGSTP1 alleles in human lung and catalytic activity of the native protein variants towards 1-chloro-2,4-dinitrobenzene, 4-vinylpyridine and (+)-anti benzo[a]pyrene-7,8-diol-9,10-oxide. Cancer Lett. 156, 167–175.
Coombes, B., and Stakelum, G. S. (1961). A liver enzyme that conjugates sulfobromophthalein with glutathione. J. Clin. Invest. 40, 981–988.
Davies, S. M., Bhatia, S., Ross, J. A., Kiffmeyer, W. R., Gaynon, P. S., Radloff, G. A., Robison, L. L., and Perentesis, J. P. (2002). Glutathione S-transferase genotypes, genetic susceptibility, and outcome of therapy in childhood acute lymphoblastic leukemia. Blood 100, 67–71.
Davies, S. M., Robison, L. L., Buckley, J. D., Radloff, G. A., Ross, J. A., and Perentesis, J. P. (2000). Glutathione S-transferase polymorphisms in children with myeloid leukemia: a Children’s Cancer Group study. Cancer Epidemiol. Biomarkers Prev. 9, 563–566.
Davies, S. M., Robison, L. L., Buckley, J. D., Tjoa, T., Woods, W. G., Radloff, G. A., Ross, J. A., and Perentesis, J. P. (2001). Glutathione S-transferase polymorphisms and outcome of chemotherapy in childhood acute myeloid leukemia. J. Clin. Oncol. 19, 1279–1287.
Di Pietro, G., Magno, L. A., and Rios-Santos, F. (2010). Glutathione S-transferases: an overview in cancer research. Expert Opin. Drug Metab. Toxicol. 6, 153–170.
Dorion, S., Lambert, H., and Landry, J. (2002). Activation of the p38 signaling pathway by heat shock involves the dissociation of glutathione s-transferase Mu from Ask1. J. Biol. Chem. 277, 30792–30797.
Doroshyenko, O., Fuhr, U., Kunz, D., Frank, D., Kinzig, M., Jetter, A., Reith, Y., Lazar, A., Taubert, D., Kirchheiner, J., Baum, M., Eisenbrand, G., Berger, F. I., Bertow, D., Berkessel, A., Sorgel, F., Schomig, E., and Tomalik-Scharte, D. (2009). In vivo role of cytochrome P450 2E1 and glutathione-S-transferase activity for acrylamide toxicokinetics in humans. Cancer Epidemiol. Biomarkers Prev. 18, 433–443.
Dreij, K., Sundberg, K., Johansson, A. S., Nordling, E., Seidel, A., Persson, B., Mannervik, B., and Jernstrom, B. (2002). Catalytic activities of human alpha class glutathione transferases toward carcinogenic dibenzo[a,l]pyrene diol epoxides. Chem. Res. Toxicol. 15, 825–831.
Dulhunty, A. F., Hewawasam, R., Liu, D., Casarotto, M. G., and Board, P. G. (2011). Regulation of the cardiac muscle ryanodine receptor by glutathione transferases. Drug Metab. Rev. 43, 236–252.
Economopoulos, K. P., and Sergentanis, T. N. (2010). GSTM1, GSTT1, GSTP1, GSTA1 and colorectal cancer risk: a comprehensive meta-analysis. Eur. J. Cancer 46, 1617–1631.
Elsby, R., Kitteringham, N. R., Goldring, C. E., Lovatt, C. A., Chamberlain, M., Henderson, C. J., Wolf, C. R., and Park, B. K. (2003). Increased constitutive c-Jun N-terminal kinase signaling in mice lacking glutathione S-transferase Pi. J. Biol. Chem. 278, 22243–22249.
Flanagan, J. U., and Smythe, M. L. (2011). Sigma-class glutathione transferases. Drug Metab. Rev. 43, 194–214.
Gangwal, K., Close, D., Enriquez, C. A., Hill, C. P., and Lessnick, S. L. (2010). Emergent properties of EWS/FLI regulation via GGAA microsatellites in Ewing’s sarcoma. Genes Cancer 1, 177–187.
Gangwal, K., Sankar, S., Hollenhorst, P. C., Kinsey, M., Haroldsen, S. C., Shah, A. A., Boucher, K. M., Watkins, W. S., Jorde, L. B., Graves, B. J., and Lessnick, S. L. (2008). Microsatellites as EWS/FLI response elements in Ewing’s sarcoma. Proc. Natl. Acad. Sci. U.S.A. 105, 10149–10154.
Garcia-Aragoncillo, E., Carillo, J., Lalli, E., Agra, N., Gómez-López, G., Pestaña, A., and Alonso, J. (2008). DAX1, a direct target of EWS/FLI oncoprotein, is a principle regulator of cell-cycle progression in Ewing’s tumor cells. Oncogene 27, 6034–6043.
Gatedee, J., Pakakassama, S., Muangman, S., and Pongstaporn, W. (2007). Glutathione S-transferase P1 genotypes, genetic susceptibility and outcome of therapy in thai childhood acute lymphoblastic leukemia. Asian Pac. J. Cancer Prev. 8, 294–296.
Hayes, J. D., and McLellan, L. I. (1999). Glutathione and glutathione-dependent enzymes represent a co-ordinately regulated defence against oxidative stress. Free Radic. Res. 31, 273–300.
Hayes, J. D., and Strange, R. C. (2000). Glutathione s-transferase polymorphisms and their biological consequences. Pharmacology 61, 154–166.
Henderson, C. J., and Wolf, C. R. (2011). Knockout and transgenic mice in glutathione transferase research. Drug Metab. Rev. 43, 152–164.
Hiley, C., Bell, J., Hume, R., and Strange, R. (1989). Differential expression of alpha and pi isoenzymes of glutathione s-transferase in developing human kidney. Biochim. Biophys. Acta 990, 321–324.
Hosono, N., Kishi, S., Iho, S., Urasaki, Y., Yoshida, A., Kurooka, H., Yokota, Y., and Ueda, T. (2010). Glutathione s-transferase M1 inhibits dexamethasone-induced apoptosis in association with the suppression of Bim through dual mechanisms in a lymphoblastic leukemia cell line. Cancer Sci. 101, 767–773.
Hu, X., Ji, X., Srivastava, S. K., Xia, H., Awasthi, S., Nanduri, B., Awasthi, Y. C., Zimniak, P., and Singh, S. V. (1997a). Mechanism of differential catalytic efficiency of two polymorphic forms of human glutathione S-transferase P1-1 in the glutathione conjugation of carcinogenic diol epoxides of chrysene. Arch. Biochem. Biophys. 345, 32–38.
Hu, X., O’Donnell, R., Srivastava, S. K., Xia, H., Zimniak, P., Nanduri, B., Bleicher, R. J., Awasthi, S., Awasthi, Y. C., Ji, X., and Singh, S. V. (1997b). Active site architecture of polymorphic forms of human glutathione S-transferase P1-1 accounts for their enantioselectivity and disparate activity in the glutathione conjugation of 7β,8α-dihydroxy-9α,10α-oxy-7,8,9,10-tetrahydrobenzo(α)pyrene. Biochem. Biophys. Res. Commun. 235, 424–428.
Hu, X., Xia, H., Srivastava, S. K., Herzog, C., Awasthi, Y. C., Ji, X., Zimniak, P., and Singh, S. V. (1997c). Activity of four allelic forms of glutathione S-transferase hGSTP1-1 for diol epoxides of polycyclic aromatic hydrocarbons. Biochem. Biophys. Res. Commun. 238, 397–402.
Hu, X., Xia, H., Srivastava, S. K., Pal, A., Awasthi, Y. C., Zimniak, P., and Singh, S. V. (1998). Catalytic efficiencies of allelic variants of human glutathione –transferase P1-1 toward carcinogenic anti-diol epoxides of benzo[c]phenanthrene and benzo[g]chrysene. Cancer Res. 58, 5340–5343.
Huang, C. Y., Huang, K. L., Cheng, T. J., Wang, J. D., and Hsieh, L. L. (1997). The GST T1 and CYP2E1 genotypes are possible factors causing vinyl chloride induced abnormal liver function. Arch. Toxicol. 71, 482–488.
Huang, J. X., Li, F. Y., Xiao, W., Song, Z. X., Qian, R. Y., Chen, P., and Salminen, E. (2009). Expression of thymidylate synthase and glutathione-s-transferase pi in patients with esophageal squamous cell carcinoma. World J. Gastroenterol. 15, 4316–4321.
Hubatsch, I., Ridderstrom, M., and Mannervik, B. (1998). Human glutathione transferase A4-4: an alpha class enzyme with high catalytic efficiency in the conjugation of 4-hydroxynonenal and other genotoxic products of lipid peroxidation. Biochem. J. 330, 175–179.
Jakobsson, P. J., Mancini, J. A., and Ford-Hutchinson, A. W. (1996). Identification and characterization of a novel human microsomal glutathione S-transferase with leukotrience C4 synthase activity and significant sequence identity to 5-lipoxygenase-activating protein and leukotriene C4 synthase. J. Biol. Chem. 271, 22203–22210.
Jakobsson, P. J., Mancini, J. A., Riendeau, D., and Ford-Hutchinson, A. W. (1997). Identification and characterization of a novel microsomal enzyme with glutathione-dependent transferase and peroxidase activities. J. Biol. Chem. 272, 22934–22939.
Jazbec, J., Aplenc, R., Dolzan, V., Debeljak, M., and Jereb, B. (2003). GST polymorphisms and occurrence of second neoplasms after treatment of childhood leukemia. Leukemia 17, 2540–2542.
Johansson, A. S., and Mannervik, B. (2001). Human glutathione transferase A3-3, a highly efficient catalyst of double-bond isomerization in the biosynthetic pathway of steroid hormones. J. Biol. Chem. 276, 33061–33065.
Johansson, K., Jarvliden, J., Gogvadze, V., and Morgenstern, R. (2010). Multiple roles of microsomal glutathione transferase 1 in cellular protection: a mechanistic study. Free Radic. Biol. Med. 49, 1638–1645.
Johnson, W. W., Ueng, Y. F., Widersten, W., Mannervik, B., Hayes, J. D., Sherratt, P. J., Ketterer, B., and Guengerich, F. P. (1997). Conjugation of highly reactive aflatoxin B1 exo-8,9-epoxide catalysed by rat and human glutathione transferases: estimation of kinetic parameters. Biochemistry 36, 3056–3060.
Joseph, T., Kusumakumary, P., Chacko, P., Abraham, A., and Pillai, M. R. (2004). Genetic polymorphism of CYP1A1, CYP2D6, GSTM1 and GSTT1 and susceptibility to acute lymphoblastic leukaemia in Indian children. Pediatr. Blood Cancer 43, 560–567.
Josephy, P. D. (2010). Genetic variations in human glutathione transferase enzymes: significance for pharmacology and toxicology. Hum. Genomics Proteomics 2010, 876940–876953.
Kanaoka, Y., Ago, H., Inagaki, E., Nanayama, T., Miyano, M., Kikuno, R., Fujii, Y., Eguchi, N., Toh, H., Urade, Y., and Hayaishi, O. (1997). Cloning and crystal structure of hematopoietic prostaglandin D synthase. Cell 90, 1085–1095.
Kham, S. K., Soh, C. K., Heng, C. K., Ariffin, H., Quah, T. C., and Yeoh, A. E. (2004). Double deletions of glutathione S-transferase genes (GSTM1 and GSTT1) reduce the risk of early relapse in childhood B-lineage acute lymphoblastic leukaemia (ALL). Ann. Acad. Med. Singapore 33, S83–S85.
Kinsey, M., Smith, R., and Lessnick, S. L. (2006). NR0B1 is required for the oncogenic phenotype mediated by EWS/FLI in Ewing’s sarcoma. Mol. Cancer Res. 4, 851–859.
Krajinovic, M., Labuda, D., Richer, C., Karimi, S., and Sinnett, D. (1999). Susceptibility to childhood acute lymphoblastic leukemia: influence of CYP1A1, CYP2D6, GSTM1, and GSTT1 genetic polymorphisms. Blood 93, 1496–1501.
Krajinovic, M., Labuda, D., and Sinnett, D. (2002). Glutathione S-transferase P1 genetic polymorphisms and susceptibility to childhood acute lymphoblastic leukaemia. Pharmacogenetics 12, 655–658.
Luo, W., Gangwal, K., Sankar, S., Boucher, K. M., Thomas, D., and Lessnick, S. L. (2009). GSTM4 is a microsatellite-containing EWS/FLI target involved in Ewing’s sarcoma oncogenesis and therapeutic resistance. Oncogene 28, 4126–4132.
Mancini, J. A., Abramovitz, M., Cox, M. E., Wong, E., Charleson, S., Perrier, H., Wang, Z., Prasit, P., and Vickers, P. J. (1993). 5-lipoxygenase-activating protein is an arachidonate binding protein. FEBS Lett. 318, 277–281.
Mancini, J. A., Blood, K., Guay, J., Gordon, R., Claveau, D., Chan, C. C., and Riendeau, D. (2001). Cloning, expression, and up-regulation of inducible rat prostaglandin e synthase during lipopolysaccharide-induced pyresis and adjuvant-induced arthritis. J. Biol. Chem. 279, 4469–4475.
Marino, S., Verzegnassi, F., Tamaro, P., Stocco, G., Bartoli, F., Decorti, G., and Rabusin, M. (2009). Response to glucocorticoids and toxicity in childhood acute lymphoblastic leukemia: role of polymorphisms of genes involved in glucocorticoid response. Pediatr. Blood Cancer 53, 984–991.
McMahon, B. A., Koyner, J. L., and Murray, P. T. (2010). Urinary glutathione S-transferases in the pathogenesis and diagnostic evaluation of acute kidney injury following cardiac surgery: a critical review. Curr. Opin. Crit. Care 16, 550–555.
McMurtry, V., Saavedra, J. E., Nieves-Alicea, R., Simeone, A. M., Keefer, L. K., and Tari, A. M. (2011). JS-K, a nitric oxide-releasing prodrug, induces breast cancer cell death while sparing normal mammary epithelial cells. Int. J. Oncol. 38, 963–971.
Mileo, A. M., Abbruzzese, C., Mattarocci, S., Bellacchio, E., Pisano, P., Federico, A., Maresca, V., Picardo, M., Giorgi, A., Maras, B., Schininà, M. E., and Paggi, M. G. (2009). Human papillomavirus-16 E7 interacts with glutathione S-transferase P1 and enhances its role in cell survival. PLoS ONE 4, e7254. doi: 10.1371/journal.pone.0007254
Minelli, C., Granell, R., Newson, R., Rose-Zerilli, M. J., Torrent, M., Ring, S. M., Holloway, J. W., Shaheen, S. O., and Henderson, J. A. (2010). Glutathione-S-transferase genes and asthma phenotypes: a human genome epidemiology (HuGE) systematic review and meta-analysis including unpublished data. Int. J. Epidemiol. 39, 539–562.
Morgan, A. S., Ciaccio, P. J., Tew, K. D., and Kauvar, L. M. (1996). Isozyme-specific glutathione S-transferase inhibitors potentiate drug sensitivity in cultured human tumor cell lines. Cancer Chemother. Pharmacol. 37, 363–370.
Muller, M., Meijer, C., Zaman, G. J., Borst, P., Scheper, R. J., Mulder, N. H., de Vries, E. G., and Jansen, P. L. (1994). Proc. Natl. Acad. Sci. U.S.A. 91, 13033–13037.
Nakamura, Y., Ohigashi, H., Masuda, S., Murakami, A., Morimitsu, Y., Kawamoto, Y., Osawa, T., Imagawa, M., and Uchida, K. (2000). Redox regulation of glutathione S-transferase induction by benzyl isothiocyanate: correlation of enzyme induction with the formation of reactive oxygen intermediates. Cancer Res. 60, 219–225.
Norrgard, M. A., and Mannervik, B. (2011). Engineering GST M2-2 for high activity with indene 1,2-oxide and indication of an H-site residue sustaining catalytic promiscuity. J. Mol. Biol. 412, 111–120.
Oakley, A. (2011). Glutathione transferases: a structural perspective. Drug Metab. Rev. 43, 138–151.
Owen, L. A., and Lessnick, S. L. (2006). Identification of target genes in their native cellular context: an analysis of EWS/FLI in Ewing’s sarcoma. Cell Cycle 5, 2049–2053.
Pakakasama, S., Mukda, E., Sasanakul, W., Kadegasem, P., Udomsubpayakul, U., Thithapandha, A., and Hongeng, S. (2005). Polymorphisms of drug-metabolizing enzymes and risk of childhood acute lymphoblastic leukemia. Am. J. Hematol. 79, 202–205.
Pasello, M., Manara, M. C., Michelacci, F., Fanelli, M., Hattinger, C. M., Nicoletti, G., Landuzzi, L., Lollini, P. L., Caccuri, A., Picci, P., Scotlandi, K., and Serra, M. (2011). Targeting glutathione-s transferase enzymes in musculoskeletal sarcomas: a promising therapeutic strategy. Anal. Cell. Pathol. (Amst.) 34, 131–145.
Pongstaporn, W., Pakakasama, S., Sanguansin, S., Hongeng, S., and Petmitr, S. (2009). Polymorphism of glutathione S-transferase Omega gene: association with risk of childhood acute lymphoblastic leukemia. J. Cancer Res. Clin. Oncol. 135, 673–678.
Ricci, G., De Maria, F., Antonini, G., Turella, P., Bullo, A., Stella, L., Filomeni, G., and Caccuri, A. M. (2005). 7-Nitro-2,1,3-benzoxadiazole derivatives, a new class of suicide inhibitors for glutathione s-transferases. J. Biol. Chem. 280, 26397–26405.
Robinson, A., Huttley, G. A., Booth, H. S., and Board, P. G. (2004). Modelling and bioinformatics studies of the human Kappa-class glutathione transferase predict a novel third glutathione transferase family with similarity to prokaryotic 2-hydroxychromene-2-carboxylate isomerases. Biochem. J. 379, 541–552.
Romero, L., Andrews, K., Ng, L., O’Rourke, K., Maslen, A., and Kirby, G. (2006). Human GSTA-1 reduces c-Jun N-terminal kinase signaling and apoptosis in Caco-2 cells. Biochem. J. 400, 135–141.
Ruzza, P., and Quintieri, L. (2009). Enzymes as useful tools and potential targets in cancer chemotherapy. Anticancer Agents Med. Chem. 9, 716.
Sau, A., Pellizzari Tregno, F., Valentino, F., Federici, G., and Caccuri, A. M. (2010). Glutathione transferases and development of new principles to overcome drug resistance. Arch. Biochem. Biophys. 500, 116–122.
Sauerbrey, A., Zintl, F., and Volm, M. (1994). P-glycoprotein and glutathione S-transferase pi in childhood acute lymphoblastic leukaemia. Br. J. Cancer 70, 1144–1149.
Schaefer, K. L., Eisenacher, M., Braun, Y., Brachwitz, K., Wai, D. H., Dirksen, U., Lanvers-Kaminsky, C., Juergens, H., Herrero, D., Stegmaier, S., Koscielniak, E., Eggert, A., Nathrath, M., Gosheger, G., Schneider, D. T., Bury, C., Diallo-Danebrock, R., Ottaviano, L., Gabbert, H. E., and Poremba, C. (2008). Microarray analysis of Ewing’s sarcoma family of tumors reveals characteristic gene expression signatures associated with metastasis and resistance to chemotherapy. Eur. J. Cancer 44, 699–709.
Schaffert, C. S. (2011). Role of MGST1 in reactive intermediate-induced injury. World J. Gastroenterol. 17, 2552–2557.
Scotlandi, K., Remondini, D., Castellani, G., Manara, M. C., Nardi, F., Cantiani, L., Francesconi, M., Mercuri, M., Caccuri, A. M., Serra, M., Knuutila, S., and Picci, P. (2009). Overcoming resistance to conventional drugs in Ewing sarcoma and identification of molecular predictors of outcome. J. Clin. Oncol. 27, 2209–2216.
Seitz, G., Bonin, M., Fuchs, J., Poths, S., Ruck, P., Warmann, S. W., and Armeanu-Ebinger, S. (2010). Inhibition of glutathione-s-transferase as a treatment strategy for multidrug resistance in childhood rhabdomyosarcoma. Int. J. Oncol. 36, 491–500.
Shami, P. J., Saavedra, J. E., Bonifant, C. L., Chu, J., Udupi, V., Malaviya, S., Carr, B. I., Kar, S., Wang, M., Jia, L., Ji, X., and Keefer, L. K. (2006). Antitumor activity of JS-K [O2-(2,4-dinitrophenyl) 1-[(4-ethoxycarbonyl)piperazin-1-yl]diazen-1-ium-1,2-diolate] and related O2-aryl diazeniumdiolates in vitro and in vivo. J. Med. Chem. 49, 4356–4366.
Sherratt, P. J., Pulford, D. J., Harrison, D. J., Green, T., and Hayes, J. D. (1997). Evidence that human class theta glutathione S-transferase T1-1 can catalyse the activation of dichloromethane, a liver and lung carcinogen in the mouse. Comparison of the tissue distribution of GSTT1 with that of classes alpha, mu and pi GST in human. Biochem. J. 326, 837–846.
Smith, R., Owen, L. A., Trem, D. J., Wong, J. S., Whangbo, J. S., Golub, T. R., and Lessnick, S. L. (2006). Expression profiling of EWS/FLI identifies NKX2.2 as a critical target gene in Ewing’s sarcoma. Cancer Cell 9, 405–416.
Stanulla, M., Schrappe, M., Brechlin, A. M., Zimmermann, M., and Welte, K. (2000). Polymorphisms within glutathione S-transferase genes (GSTM1, GSTT1, GSTP1) and risk of relapse in childhood B-cell precursor acute lymphoblastic leukemia: a case-control study. Blood 95, 1222–1228.
Strange, R. C., Howie, A. F., Hume, R., Matharoo, B., Bell, J., Hiley, C., Jones, P., and Beckett, G. J. (1989). The development expression of alpha-, mu- and pi-class glutathione s-transferases in human liver. Biochem. Biophys. Acta 993, 186–190.
Takanashi, M., Morimoto, A., Yagi, T., Kuriyama, K., Kano, G., Imamura, T., Hibi, S., Todo, S., and Imashuku, S. (2003). Impact of glutathione S-transferase gene deletion on early relapse in childhood B-precursor acute lymphoblastic leukemia. Haematologica 88, 1238–1244.
Tan, K. L., Chelyanayagam, G., Parker, M. W., and Board, P. G. (1996). Mutagenesis of the active site of the human Theta-class glutathione transferase GSTT2-2: catalysis with different substrates involves different residues. Biochem. J. 319, 315–321.
Tanaka-Kagawa, T., Jinno, H., Hasegawa, T., Makino, Y., Seko, Y., Hanjoka, N., and Ando, M. (2003). Functional characterization of two variant human GSTO1-1s (Ala40Asp and Thr217Asn). Biochem. Biophys. Res. Commun. 301, 516–520.
Tew, K. D., Dutta, S., and Schultz, M. (1997). Inhibitors of glutathione S-transferases as therapeutic agents. Adv. Drug Deliv. Rev. 26, 91–104.
Tew, K. D., Manevich, Y., Grek, C., Xiong, Y., Uys, J., and Townsend, D. M. (2011). The role of glutathione s-transferase P in signaling pathways and s-glutathionylation in cancer. Free Radic. Biol. Med. 51, 299–313.
Tew, K. D., Monks, A., Barone, L., Rosser, D., Akerman, G., Montali, J. A., Wheatley, J. B., and Schmidt, D. E. (1996). Glutathione-associated enzymes in the human cell lines of the National Cancer Institute Drug Screening Program. Mol. Pharmacol. 50, 149–159.
Timofeeva, M., Jager, B., Rosenberger, A., Sauter, W., Wichmann, H. E., KORA Study Group, Bickeboller, H., and Risch, A. (2009). A multiplex real-time PCR method for detection of GSTM1 and GSTT1 copy numbers. Clin. Biochem. 42, 500–509.
Tirado, O. M., Mateo-Lozano, S., Villar, J., Dettin, L. E., Llort, A., Gallego, S., Ban, J., Kovar, H., and Notario, V. (2006). Caveolin-1 (CAV1) is a target of EWS/FLI-1 and a key determinant of the oncogenic phenotype and tumorigenicity of Ewing’s sarcoma cells. Cancer Res. 66, 9937–9947.
Tong, Z., Board, P. G., and Anders, M. W. (1998a). Glutathione transferase zeta catalyses the oxygenation of the carcinogen dichloroacetic acid to glyoxylic acid. Biochem. J. 331, 371–374.
Tong, Z., Board, P. G., and Anders, M. W. (1998b). Glutathione transferase zeta-catalyzed biotransformation of dichloroacetic acid and other α-haloacids. Chem. Res. Toxicol. 11, 1332–1338.
Townsend, D. M., and Tew, K. D. (2003). The role of glutathione-s-transferase in anti-cancer drug resistance. Oncogene 22, 7369–7375.
Turc-Carel, C., Aurias, A., Mugneret, F., Lizard, S., Sidaner, I., Volk, C., Thiery, J. P., Olschwang, S., Philip, I., Berger, M. P., Philip, T., Lenoir, G. M., and Mazabraud, A. (1988). Chromosomes in Ewing’s sarcoma. I. An evaluation of 85 cases of remarkable consistency of t(11;22)(q24;q12). Cancer Genet. Cytogenet. 32, 229–238.
Turella, P., Cerella, C., Filomeni, G., Bullo, A., De Maria, F., Ghibelli, L., Ciriolo, M. R., Cianfriglia, M., Mattei, M., Federici, G., Ricci, G., and Caccuri, A. M. (2005). Proapoptotic activity of new glutathione S-transferase inhibitors. Cancer Res. 65, 3751–3761.
Turella, P., Filomeni, G., Dupuis, M. L., Ciriolo, M. R., Molinari, A., De Maria, F., Tombesi, M., Cianfriglia, M., Federici, G., Ricci, G., and Caccuri, A. M. (2006). A strong glutathione s-transferase inhibitor overcomes the P-glycoprotein-mediated resistance in tumor cells: NBDHEX triggers a caspase-dependent apoptosis in MDR1-expressing leukemia cells. J. Biol. Chem. 281, 23725–23732.
Wondrak, G. T. (2009). Redox-directed cancer therapeutics: molecular mechanisms and opportunities. Antioxid. Redox Signal. 11, 3013–3069.
Wu, Y., Fan, Y., Xue, B., Luo, L., Shen, J., Zhang, S., Jiang, Y., and Yin, Z. (2006). Human glutathion s-transferase P1-1 interacts with TRAF2 and regulates TRAF2-ASK1 signals. Oncogene 25, 5787–5800.
Zhong, S., Howie, A. F., Ketterer, B., Taylor, J., Hayes, J. D., Beckett, G. J., Wathen, C. G., Wolf, C. R., and Spurr, N. K. (1991). Glutathione S-transferase mu locus: use of genotyping and phenotyping assays to assess association with lung cancer susceptibility. Carcinogenesis 12, 1533–1537.
Keywords: glutathione S-transferase, pediatric cancer, epidemiology, drug resistance, therapeutic target, microsatellite
Citation: Luo W, Kinsey M, Schiffman JD and Lessnick SL (2011) Glutathione S-transferases in pediatric cancer. Front. Oncol. 1:39. doi: 10.3389/fonc.2011.00039
Received: 22 July 2011; Accepted: 03 October 2011;
Published online: 24 October 2011.
Edited by:
Mignon Lee-Cheun Loh, University of California, USAReviewed by:
Martin Stanulla, University Hospital Schleswig-Holstein, GermanyKenneth Tew, Medical University of South Carolina, USA
Copyright: © 2011 Luo, Kinsey, Schiffman and Lessnick. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Stephen L. Lessnick, Huntsman Cancer Institute, 2000 Circle of Hope, Salt Lake City, UT 84112, USA. e-mail: stephen.lessnick@hci.utah.edu