 Meng Ma
Meng Ma Xifeng Fei
Xifeng Fei Dongyi Jiang2
Dongyi Jiang2 Xiangtong Xie
Xiangtong Xie Qiang Huang
Qiang Huang- 1Department of Neurosurgery, Dushu Lake Hospital Affiliated to Soochow University, Suzhou, China
- 2Department of Neurosurgery, Suzhou Kowloon Hospital, Shanghai Jiaotong University School of Medicine, Suzhou, China
- 3Department of Neurosurgery, The Second Affiliated Hospital of Soochow University, Suzhou, China
Glioma is the most prevalent primary malignant tumor of the central nervous system. While traditional treatment modalities such as surgical resection, radiotherapy, and chemotherapy have made significant advancements in glioma treatment, the prognosis for glioma patients remains often unsatisfactory. Ferroptosis, a novel form of programmed cell death, plays a crucial role in glioma and is considered to be the most functionally rich programmed cell death process. Histone deacetylases have emerged as a key focus in regulating ferroptosis in glioma. By inhibiting the activity of histone deacetylases, histone deacetylase inhibitors elevate acetylation levels of both histones and non-histone proteins, thereby influencing various cellular processes. Numerous studies have demonstrated that histone deacetylases are implicated in the development of glioma and hold promise for its treatment. This article provides an overview of research progress on the mechanism by which histone deacetylases contribute to ferroptosis in glioma.
Introduction
Glioma is the most prevalent primary malignant tumor of the central nervous system. While traditional treatment modalities such as surgical resection, radiotherapy, and chemotherapy have shown progress in glioma management, patient prognosis remains unsatisfactory due to the extensive phenotypic heterogeneity and plasticity of the tumor [1, 2]. The heterogeneity emerges as a consequence of the genetic diversity of glioma cells, and its plasticity is manifested in the tumor cells’ capacity to adapt to environmental alterations [2]. Although immune treatment strategies such as checkpoint blockade (ICB), immune modulators, and CAR-T cell therapy have shown potential in other tumors, the immune characteristic of glioma being a “cold tumor” limits the effect of immune therapy. Therefore there is an urgent need to find ways to enhance the effectiveness of glioma immunotherapy [3].
Ferroptosis, as a novel form of programmed cell death, is the most functionally diverse process in glioma and is involved in the immunological microenvironment of glioma, offering new research avenues for glioma treatment [4, 5]. Acetylation, a crucial post-translational modification, orchestrates cellular growth and development through modulating gene transcription, with the fine balance of histone acetylation levels being precisely regulated by the coordinated actions of histone acetyltransferases (HATs) and histone deacetylases (HDACs) [6, 7]. Histone deacetylase inhibitors (HDACi) are currently being used alone or in combination to treat certain tumors [8–12]. The acetylation status of histones affects the process of ferroptosis in tumor cells. By using HDACi to decrease HDAC activity, the induction of ferroptosis in adrenocortical cancer cells can be accelerated, inhibiting tumor growth and proliferation. However, the specific mechanism underlying this process is not yet fully understood [13]. Therefore, this article primarily reviews research progress on the mechanism of HDACs in the process of ferroptosis in glioma.
A New Perspective in Glioma Research: Ferroptosis and Glioma
Ferroptosis
Iron, an essential trace element in the human body, plays a crucial role in vital activities such as DNA synthesis, ATP production, and mitochondrial metabolism [14]. Maintaining iron homeostasis is important for the normal progression of life processes. Disruption of iron homeostasis can affect normal physiological processes and lead to various diseases such as tumors, aging, and infections. Excessive iron within cells can lead to the production of reactive oxygen species (ROS) through the Fenton reaction, triggering oxidative stress responses [15, 16]. Conversely, iron deficiency can impact cell structure in the body and result in diseases such as anemia, weakened immunity, and digestive disorders and so on [17–19].
Cells, as the fundamental units of life, are confronted with the challenge of iron overload during their growth and development. This involves how cells respond to various oxidative stress responses, a process that ultimately determines the fate of the cell. Dixon et al. have identified a novel form of programmed cell death known as ferroptosis. Ferroptosis is characterized by lipid peroxidation on the cell membrane, leading to an accumulation of iron within the cell as a primary manifestation [20]. Unlike traditional forms of apoptosis (such as apoptosis, pyroptosis, etc.), ferroptosis is distinguished by mitochondrial shrinkage and a reduction in the number of mitochondrial cristae [21]. Additionally, certain conditional factors such as synthesis and peroxidation of polyunsaturated fatty acid phospholipids (PUFA-PLs), disorder in iron metabolism, and mitochondrial dysfunction are necessary for the occurrence of ferroptosis [22]. Cells possess four defense systems against ferroptosis: GPX4-GSH system, FSP1-CoQH2 system, DHODH-CoQH2 system, and GCH1-BH4 system [21]. When these defense systems fail to effectively buffer against ferroptosis, it results in cellular demise. These mechanisms offer potential pathways for intervening in ferroptosis.
Ferroptosis is a form of cell death associated with various pathological processes, particularly in tumors, neurodegenerative, and inflammatory diseases [23–30]. It holds great potential in tumor therapy. Tumor cells have a higher metabolic rate and require more oxygen, leading to the production of reactive oxygen species. This characteristic makes tumor cells more susceptible to ferroptosis, making it an important mechanism for tumor suppression. However, tumor cells can counteract ferroptosis by limiting the synthesis and peroxidation of PUFA-PLs [31], restricting the availability of unstable iron [32], and upregulating cellular defense systems against ferroptosis [33]. These regulatory responses play a key role in the survival of tumor cells and may pose as obstacles to treatment. The induction of ferroptosis not only inhibits tumor growth but may also damage non-tumor cells. Therefore, precise control over the induction of ferroptosis is currently a focus in research efforts aimed at primarily affecting tumor cells while protecting normal cells, especially immune system-related cells. In this way, ferroptosis is expected to be developed as a new therapeutic strategy that effectively inhibits tumors while preserving the body’s immune function—a significant development in treating various diseases.
The Impact of Ferroptosis on the Blood-Brain Barrier
The blood-brain barrier (BBB) is a critical protective mechanism of the central nervous system, regulating the passage of substances into and out of the brain while maintaining its stability. Composed of endothelial cells, pericytes, astrocytes, microglia, and neurons among others, the interaction between these cells is essential for BBB function [34]. The unique characteristics of BBB endothelial cells include a flat appearance, tight junctions, and a higher number of mitochondria which contribute to regulating and maintaining BBB function [35]. Lipids are a vital component of cell membranes and are particularly abundant in the central nervous system [36]. Lipid peroxidation can alter cell membrane fluidity and permeability, impacting cell structure and function. Cyclooxygenase (COX), cytochrome P450 (CYP), and lipoxygenase (LOXs) are the three primary lipid oxidases involved in iron-dependent lipid peroxidation [37]. Iron accumulation and lipid peroxidation are linked to BBB dysfunction; understanding the relationship between ferroptosis and BBB dysfunction offers new avenues for treating central nervous system diseases.
The p53 protein is responsible for maintaining the permeability and integrity of BBB cells by reducing the level of lipid peroxidation [38]. Dysfunction of the BBB is among the early pathophysiological changes observed in neurodegenerative diseases and brain injuries, such as stroke, traumatic brain injury (TBI), Alzheimer’s disease (AD), and Parkinson’s disease (PD) [39–41]. The BBB serves as the primary barrier for iron to enter the brain and plays a critical role in maintaining brain iron balance. Research has indicated that iron chelators can protect brain microvascular endothelial cells from toxicity and functional damage, thereby preserving endothelial cell stability [42]. Excessive accumulation of iron can induce the expression of matrix metalloproteinases (MMPs), which in turn leads to degradation of vascular basement membrane components, causing damage to the BBB [43, 44]. Additionally, reactive oxygen species (ROS) can impact BBB function by altering intracranial vascular tension, increasing platelet aggregation, and endothelial cell permeability; ultimately resulting in focal lesions on the endothelial cell membrane [45]. The interaction between ROS and MMPs may ultimately lead to dysfunction of the BBB [46]. Although initial progress has been achieved in comprehending the connection between BBB dysfunction and ferroptosis, the current literature is still inadequate in uncovering the molecular mechanism of their interaction, and there is a shortage of conclusive experimental data to substantiate it. Additionally, existing studies have not comprehensively expounded the universality and applicability of BBB dysfunction in various pathological conditions. In light of this, the research on the BBB is particularly crucial in the process of ferroptosis. This not only enriches our understanding of the pathological mechanism of central nervous system diseases but also holds significant guiding significance for the development of new treatment strategies and the improvement of treatment efficacy.
The Role of Ferroptosis in Glioma
In recent years, ferroptosis has garnered widespread attention in the field of tumor research, particularly demonstrating potentially significant effects in glioma and the tumor microenvironment [5, 47]. Glioma is a prevalent malignant tumor of the central nervous system, and its treatment difficulty stems from the resistance of tumor cells to traditional radiotherapy and chemotherapy, as well as the limitation of drug delivery imposed by the blood-brain barrier. The discovery of ferroptosis offers a new perspective and potential treatment strategy for glioma.
Due to their rapid proliferation and high metabolic activity, glioma cells have a high demand for iron, making them potentially vulnerable in terms of iron metabolism. The induction of ferroptosis is closely related to the disorder of iron metabolism. Therefore, regulating iron metabolism or directly inducing ferroptosis may become a new approach to the treatment of glioma. Studies have shown that certain iron chelators and ferroptosis inducers can increase the sensitivity of glioma cells to radiotherapy and chemotherapy, thereby enhancing the therapeutic effect [48, 49]. ATF3 contributes to brucine-triggered glioma cell ferroptosis via promotion of hydrogen peroxide and iron [50]. High levels of NRF2 sensitize temozolomide-resistant glioblastoma cells to ferroptosis via ABCC1/MRP1 upregulation [51]. PRMT1 driven PTX3 regulates ferritinophagy in glioma [52]. Gastrodin Inhibits H2O2-Induced Ferroptosis through Its Antioxidative Effect in Rat Glioma Cell Line C6 [53]. Ibuprofen induces ferroptosis of glioblastoma cells via downregulation of nuclear factor erythroid 2-related factor 2 signaling pathway. Anticancer Drugs [54]. Pseudolaric acid B triggers ferroptosis in glioma cells via activation of Nox4 and inhibition of xCT [55]. TRIM7 modulates NCOA4-mediated ferritinophagy and ferroptosis in glioblastoma cells [56]. Targeting NQO1/GPX4-mediated ferroptosis by plumbagin suppresses in vitro and in vivo glioma growth [57]. In addition, the induction of ferroptosis not only involves inducing ferroptosis in glioma cells but may also affect the iron metabolism of related cells in the tumor microenvironment. The microenvironment of glioma is usually rich in factors that promote tumor growth, and promoting or inhibiting ferroptosis may change the balance of these factors, thus affecting the growth and invasion of the tumor. For instance, SLC1A5 enhances malignant phenotypes through modulating ferroptosis status and immune microenvironment in glioma [58]. Within the glioma microenvironment, the ferroptosis inducer erastin has been shown to enhance the polarization of macrophages towards the M2 phenotype. Conversely, the ferroptosis inhibitor ferrostatin-1 has been found to increase the number of M1-like macrophages and decrease the number of M2-like macrophages in the glioma microenvironment, thereby exerting an inhibitory effect on glioma development [4]. Additionally, an innovative NRF2 nano-modulator has demonstrated its ability to induce ferroptosis in lung cancer, leading to a shift in M2 macrophage polarization towards M1 and enhancing anti-tumor immunity [59]. The critical factors influencing ferroptosis in glioma are presented in Table 1. However, there are challenges associated with utilizing ferroptosis for treating glioma. Firstly, a key issue is how to precisely induce ferroptosis in glioma cells without causing damage to normal cells. Secondly, effective drug delivery to the tumor site is hindered by the presence of the blood-brain barrier. As a result, researchers are exploring various strategies such as developing new ferroptosis inducers, leveraging nanotechnology to improve drug penetration, and combining other treatment methods for comprehensive care.
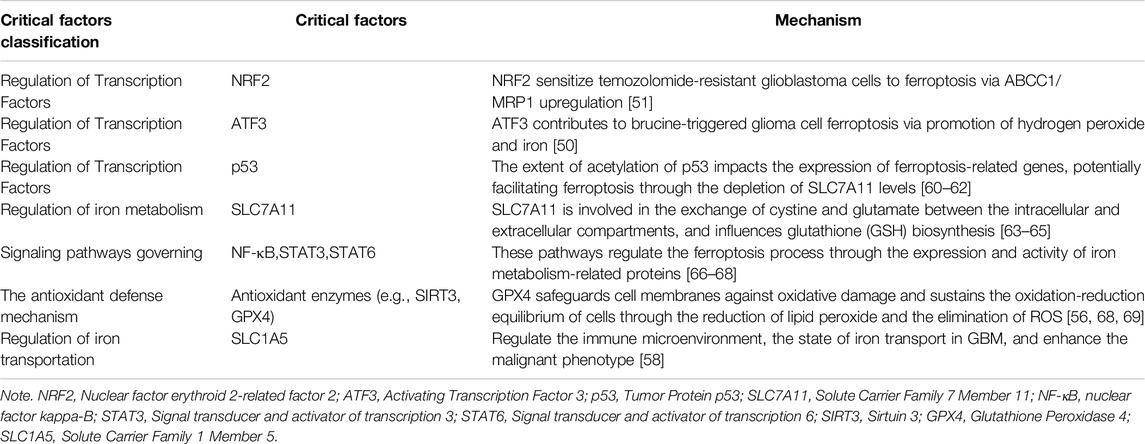
Table 1. Critical factors of ferroptosis in glioma.
A New Target for Glioma Treatment: Histone Deacetylases
The level of histone acetylation is jointly regulated by HATs and HDACs, which influence the structure of chromatin and gene transcription. There are five families of HATs, including p300/CBP, GNAT, SRC, MYST, and TAFII250. These enzymes catalyze the transfer of acetyl-coenzyme A to lysine residues, promoting chromatin relaxation and gene activation [70, 71]. The function of HDACs is opposite to that of HATs; HDACs catalyze deacetylation by hydrolyzing the acetyl groups on lysine residues, leading to a decrease in histone acetylation levels, which enhances the interaction between histones and DNA. This results in a more condensed chromatin state and suppresses gene transcription [72]. The HDAC family is diverse, with 18 members divided into four classes. Class I HDACs (HDAC1, HDAC2, HDAC3, and HDAC8) are related to the yeast RPD3 deacetylase and are primarily involved in cell proliferation, differentiation, DNA damage response, and tissue development in the nucleus. Class II HDACs can be further divided into two subclasses: Class IIa (HDAC4, HDAC5, HDAC7, and HDAC9) and Class IIb (HDAC6 and HDAC10), which are homologous to the yeast Hda1 deacetylase. Class III HDACs (SIRT1, SIRT2, SIRT3, SIRT4, SIRT5, SIRT6, and SIRT7) share sequence similarity with the yeast Sir2 protein. Class IV HDAC (HDAC11) has sequence similarity with Class I and Class II proteins [73]. The balance of these enzymes is crucial for maintaining cellular functions.
In glioma, the abnormal activity of HDACs is associated with tumor growth, invasiveness, and drug resistance [74–77]. HDACs regulate the expression of related genes by modulating the acetylation level of histones. These genes may involve iron metabolism and antioxidant defense systems, thereby indirectly affecting the process of ferroptosis. Therefore, HDAC inhibitors (HDACIs), as potential therapeutic agents, have become a new direction in the treatment of glioma. Understanding the role and expression patterns of HDACs in different subtypes of glioma will help develop more precise and personalized treatment plans.
The Impact of Acetylation on Ferroptosis
Acetylation, as an epigenetic modification, orchestrates cellular processes by influencing the intricate structure of chromatin, thereby altering protein function, stability, and subcellular localization. The impact of acetylation on ferroptosis primarily manifests through its modulation of the antioxidant defense mechanism, regulation of transcription factors, and control over iron metabolism-related proteins and signaling pathways.
Acetylation Affects the Antioxidant Defense Mechanism
Antioxidant enzymes are a critical component of the cellular antioxidant defense system, protecting cells from oxidative damage by neutralizing reactive oxygen species (ROS). Acetylation has been shown to modulate the activity of these antioxidant enzymes. For example, SIRT3 enhances the activity and stability of antioxidant enzymes through deacetylation, thereby enhancing the cell’s antioxidant capacity [69]. The cystine/glutamate antiporter SLC7A11 (also known as xCT) plays a significant role in ferroptosis by facilitating the exchange of cystine and glutamate across the cell membrane. This process imports cystine for glutathione (GSH) biosynthesis and supports antioxidant defense. SLC7A11 is also overexpressed in various human cancers [63]. Recent studies indicate that overexpression of SLC7A11 partially promotes tumor growth by inhibiting ferroptosis. Additionally, it has been observed that knockdown of the TP53 gene increases transcriptional levels of SLC7A11; conversely, overexpression of TP53 downregulates SLC7A11 expression and cystine release. These findings suggest a negative correlation between p53 expression and SLC7A11 expression, indicating that p53 may suppress the activity of SLC7A11 directly or indirectly promote ferroptosis to inhibit glioma cell growth [60]. However, high overexpression of SLC7A11 can unexpectedly inhibit tumor metastasis [78], highlighting the dynamic nature of ferroptosis. Although there is currently no direct evidence supporting acetylation’s regulation on SLC7A11, it is possible that acetylation may influence its gene expression by affecting transcription factors such as p53. This underscores the need for further comprehensive study into understanding how different regulatory mechanisms impact ferroptosis dynamics.
Acetylation Regulates the Activity of Transcription Factors
Acetylation plays a critical role in regulating the activity and DNA-binding capacity of transcription factors. An example of this is the impact of acetylation on p53, which in turn affects the expression of genes related to ferroptosis. As a crucial tumor suppressor, p53 regulates various biological processes such as cell proliferation, DNA repair, apoptosis, and autophagy [79]. The pioneering work of researchers like Wei Gu et al. has confirmed that the p53 protein can undergo acetylation modification through a process involving multiple proteins and complex regulation [80]. While there is functional redundancy in the acetylation sites of p53, meaning that loss of one or more sites can be compensated by acetylation at other sites, it has been observed that loss of the eight primary acetylation sites in human p53 (the 8 KR mutant) results in diminished transcriptional activity and hinders its ability to induce cell cycle arrest and/or apoptosis [61]. This finding challenges previous notions about functional redundancy and has sparked significant interest in understanding the functional role of p53 acetylation sites.
The acetylation modification also plays an important role in regulating not only the overall transcriptional activity but also the site-specific transcriptional selectivity of p53. This precise control allows for regulation of key biological processes such as cell cycle arrest, apoptosis, senescence, autophagy, and metabolism [81–83]. Furthermore, p53 has been shown to inhibit glioma growth by inducing ferroptosis [84], while a novel histone deacetylase inhibitor called MPT0B291 has demonstrated efficacy in suppressing glioma growth both in vitro and in vivo by increasing the level of p53 acetylation [62]. In conclusion, it is evident that the acetylation state of p53 is crucial for its function in inhibiting tumor metabolism and inducing ferroptosis.
Acetylation Regulates Iron Metabolism-Related Proteins and Cellular Signaling Pathways
Acetylation plays a significant role in the regulation of iron metabolism-related proteins. Hepcidin, a small peptide secreted by the liver, is crucial for maintaining systemic iron homeostasis. The downregulation of hepcidin expression is associated with the epigenetic regulation of HDAC3. Hepcidin controls systemic iron levels by inhibiting intestinal iron absorption and recycling [85]. Acetylation modification can participate in the regulation of various signaling pathways, which may impact the process of ferroptosis. STAT6 has been found to inhibit ferroptosis and alleviate acute lung injury by competitively binding to CREB-binding protein, a key acetyltransferase that regulates the p53/SLC7A11 pathway [66]. The NF-κB and STAT3 pathways also play important roles in regulating ferroptosis. Activation of the NF-κB pathway can increase the expression of LCN2, an iron-sequestering cytokine that may be related to tumor resistance to ferroptosis-inducing drugs [67]. Additionally, STAT3 can promote GPX4 transcription by binding to its promoter region. GPX4 is an essential antioxidant enzyme that protects cells from ferroptosis. Therefore, inhibition of STAT3 may reduce GPX4 expression and promote ferroptosis [68]. In summary, acetylation can influence the process of ferroptosis through multiple mechanisms, which may play a pivotal role in tumor development and treatment. Future research needs to delve deeper into these mechanisms and explore the specific role of acetylation in ferroptosis to develop more effective therapeutic strategies.
The Role of HDAC Inhibitors in Ferroptosis in Glioma
HDACi influence various cellular processes by suppressing HDAC activity, leading to increased acetylation levels of histones and non-histone proteins within the cell. This ultimately exerts anti-tumor effects by inducing histone acetylation and regulating gene transcription [86]. While research on HDACi in the context of ferroptosis is still relatively scarce, as our understanding of the role of acetylation in ferroptosis deepens, researchers are beginning to focus on the role of HDACi in this process.
As epigenetic regulatory molecules, HDACi can modulate the expression of approximately 5%–20% of genes [87]. For instance, treatment with quisinostat (a HDACi) in tongue squamous cell carcinoma cells resulted in an increase in ROS levels, a decrease in GPX4 protein expression, an increase in p53 protein expression, and changes in mitochondrial morphology and function were observed. These findings suggest the occurrence of ferroptosis [88]. Additionally, a study by Li F et al. demonstrated that quisinostat could activate p53 and promote ferroptosis by upregulating the acetylation of p53 [89]. Ines M L Wolf et al. found that the HDAC inhibitor SAHA specifically inhibits the expression of SLC7A11 transporter protein, which leads to increased ROS activity within glioma cells [64]. Furthermore, research conducted by Zhang T et al. revealed that vorinostat can reverse tumor cell resistance to ferroptosis by downregulating the expression of SLC7A11 [65].
Although HDAC inhibitors such as vorinostat, romidepsin, belinostat, panobinostat, and chidamide have been approved for the treatment of certain diseases, such as cutaneous T-cell lymphoma and multiple myeloma, these drugs have issues with low target specificity and low sensitivity to solid tumors. Therefore, researchers are developing the next-generation of HDAC inhibitors and exploring combination therapy strategies. Curcumin is a natural pan-HDAC inhibitor that has shown anti-tumor potential. However, its specific mechanism still requires further research. Curcumin has been found to reduce the vitality, GSH, and MMP levels in breast cancer cells while increasing ROS levels, apoptosis rates, and DNA damage. New drugs based on curcumin also show potential for the treatment of glioblastoma [90, 91]. In addition to exploring new HDAC inhibitors, researchers are also investigating combination therapies. For example, the combined use of HDAC inhibitors with other drugs like SASP can enhance the effect of ferroptosis. SASP itself is a known inducer of ferroptosis; when used in combination with vorinostat it can further promote this process by targeting SLC7A11 expression which is related to cancer cell insensitivity to HDAC inhibitors [92]. Furthermore Endri Karaj et al. have designed a new class of dual-mechanism hybrid molecules that can induce ferroptosis while inhibiting HDAC activity. These novel compounds are expected to become new types of anti-cancer agents that may reduce toxic side effects caused by ferroptosis [93]. Zille M et al and Paganoni S et al discovered that HDACi could induce ferroptosis in GBM without causing neurotoxic side effects [94, 95]. In summary, while research on the role of HDAC inhibitors in promoting ferroptosis has made progress, there remains a significant need to explore their complex mechanisms of action and to conduct a thorough assessment of the potential side effects in clinical applications.
Conclusion
In recent years, ferroptosis has emerged as a novel mechanism of cell death and has become a prominent topic in the field of tumor research. Numerous researchers are dedicated to elucidating the mechanisms of ferroptosis and its association with various diseases, particularly its potential applications in tumor treatment. Despite offering new strategies for tumor treatment, there is currently a lack of clinical drug trials targeting ferroptosis, which constrains our ability to assess the safety and efficacy of ferroptosis-inducing agents in a clinical context [96]. Acetylation, an important post-translational modification, has been shown to impact the process of ferroptosis in studies. However, our current understanding of how acetylation affects ferroptosis is still limited, primarily focusing on individual key protein acetylation changes, and there is a need for further research to understand how acetylation influences protein interactions and its overall role in ferroptosis. This article reviews the role of histone deacetylases (HDACs) and their inhibitors in regulating the process of ferroptosis and anticipates future research directions. It is important to acknowledge the limitations inherent in this review, including potential publication bias towards positive findings, a possible overreliance on in vitro studies, and a lack of comprehensive clinical data to support the translational potential of HDAC inhibitors in ferroptosis. Additionally, the current body of research may not fully account for the heterogeneity of tumor types and stages, the individual variability among patients, or the long-term effects and safety profiles of HDAC inhibitors. Future studies should emphasize the role of acetylation in ferroptosis and explore its impact on tumor development and other diseases to establish a theoretical basis and experimental evidence for developing new treatment strategies. It is also crucial to address these limitations by employing rigorous study designs, seeking diverse patient populations, and conducting long-term follow-up studies to better understand the mechanisms and implications of ferroptosis in disease pathology. With continued deepening research efforts, we aim to gain a better understanding of the complex regulatory network involved in ferroptosis and make new breakthroughs in clinical treatment, while being mindful of the potential biases and gaps in the current literature.
Author Contributions
Conception: QH and ZW. Interpretation or analysis of data: MM, XF, DJ, HC, and XX. Preparation of the manuscript: MM and XF. Revision for important intellectual content: QH and ZW. Supervision: QH, ZW, DJ, HC, and XX. All authors contributed to the article and approved the submitted version.
Funding
The authors declare that financial support was received for the research, authorship, and/or publication of this article. This work was funded by grants from the Youth medical talent Foundation of Jiangsu (QNRC2016217), the Health Talent Training Project of Gusu (GSWS2020122), the Health Talent Training Project of Gusu (GSWS2021061), Suzhou Medical, Health Technology Innovation Project (SKY2021028), Science and technology planning project of Suzhou (SZM2022004).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Rong, L, Li, N, and Zhang, Z. Emerging Therapies for Glioblastoma: Current State and Future Directions. J Exp Clin Cancer Res (2022) 41(1):142. doi:10.1186/s13046-022-02349-7
2. Yabo, YA, Niclou, SP, and Golebiewska, A. Cancer Cell Heterogeneity and Plasticity: A Paradigm Shift in Glioblastoma. Neuro-Oncology (2022) 24(5):669–82. doi:10.1093/neuonc/noab269
3. Wang, H, Xu, T, Huang, Q, Jin, W, and Chen, J. Immunotherapy for Malignant Glioma: Current Status and Future Directions. Trends Pharmacol Sci (2020) 41(2):123–38. doi:10.1016/j.tips.2019.12.003
4. Liu, T, Zhu, C, Chen, X, Guan, G, Zou, C, Shen, S, et al. Ferroptosis, as the Most Enriched Programmed Cell Death Process in Glioma, Induces Immunosuppression and Immunotherapy Resistance. Neuro-Oncology (2022) 24(7):1113–25. doi:10.1093/neuonc/noac033
5. Wang, K, Wang, J, Zhang, J, Zhang, A, Liu, Y, Zhou, J, et al. Ferroptosis in Glioma Immune Microenvironment: Opportunity and Challenge. Front Oncol (2022) 12:917634. doi:10.3389/fonc.2022.917634
6. Zaib, S, Rana, N, and Khan, I. Histone Modifications and Their Role in Epigenetics of Cancer. Curr Med Chem (2022) 29(14):2399–411. doi:10.2174/0929867328666211108105214
7. Neganova, ME, Klochkov, SG, Aleksandrova, YR, and Aliev, G. Histone Modifications in Epigenetic Regulation of Cancer: Perspectives and Achieved Progress. Semin Cancer Biol (2022) 83:452–71. doi:10.1016/j.semcancer.2020.07.015
8. Parveen, R, Harihar, D, and Chatterji, BP. Recent Histone Deacetylase Inhibitors in Cancer Therapy. Cancer (2023) 129(21):3372–80. doi:10.1002/cncr.34974
9. Schelker, C, Nowak-Sliwinska, P, and Borchard, G. HDACIs and TKIs Combinations and Their Liposomal Delivery for Cancer Treatment. J Controlled Release (2023) 358:59–77. doi:10.1016/j.jconrel.2023.04.006
10. Chang, HH, Chang, YY, Tsai, BC, Chen, LJ, Chang, AC, Chuang, JY, et al. A Selective Histone Deacetylase Inhibitor Induces Autophagy and Cell Death via SCNN1A Downregulation in Glioblastoma Cells. Cancers (Basel) (2022) 14(18):4537. doi:10.3390/cancers14184537
11. Spurgeon, SE, Sharma, K, Claxton, DF, Ehmann, C, Pu, J, Shimko, S, et al. Phase 1-2 Study of Vorinostat (SAHA), Cladribine and Rituximab (SCR) in Relapsed B-Cell Non-Hodgkin Lymphoma and Previously Untreated Mantle Cell Lymphoma. Br J Haematol (2019) 186(6):845–54. doi:10.1111/bjh.16008
12. Gusyatiner, O, Bady, P, Pham, MDT, Lei, Y, Park, J, Daniel, RT, et al. BET Inhibitors Repress Expression of Interferon-Stimulated Genes and Synergize With HDAC Inhibitors in Glioblastoma. Neuro-Oncology (2021) 23(10):1680–92. doi:10.1093/neuonc/noab115
13. Oliveira, T, Hermann, E, Lin, D, Chowanadisai, W, Hull, E, and Montgomery, M. HDAC Inhibition Induces EMT and Alterations in Cellular Iron Homeostasis to Augment Ferroptosis Sensitivity in SW13 Cells. Redox Biol (2021) 47:102149. doi:10.1016/j.redox.2021.102149
14. Aisen, P, Enns, C, and Wessling-Resnick, M. Chemistry and Biology of Eukaryotic Iron Metabolism. Int J Biochem Cell Biol (2001) 33(10):940–59. doi:10.1016/s1357-2725(01)00063-2
15. Kosman, DJ. Redox Cycling in Iron Uptake, Efflux, and Trafficking. J Biol Chem (2010) 285(35):26729–35. doi:10.1074/jbc.R110.113217
16. Lambeth, JD, and Neish, AS. Nox Enzymes and New Thinking on Reactive Oxygen: A Double-Edged Sword Revisited. Annu Rev Pathol Mech Dis (2014) 9:119–45. doi:10.1146/annurev-pathol-012513-104651
17. Wegmüller, R, Bah, A, Kendall, L, Goheen, MM, Sanyang, S, Danso, E, et al. Hepcidin-Guided Screen-And-Treat Interventions for Young Children With Iron-Deficiency Anaemia in the Gambia: An Individually Randomised, Three-Arm, Double-Blind, Controlled, Proof-of-Concept, Non-Inferiority Trial. Lancet Glob Health (2023) 11(1):e105–e116. doi:10.1016/S2214-109X(22)00449-1
18. Auerbach, M, Henry, D, Derman, RJ, Achebe, MM, Thomsen, LL, and Glaspy, J. A Prospective, Multi-Center, Randomized Comparison of Iron Isomaltoside 1000 versus Iron Sucrose in Patients With Iron Deficiency Anemia; the FERWON-IDA Trial. Am J Hematol (2019) 94(9):1007–14. doi:10.1002/ajh.25564
19. Savarese, G, von Haehling, S, Butler, J, Cleland, JGF, Ponikowski, P, and Anker, SD. Iron Deficiency and Cardiovascular Disease [Published Correction Appears in Eur Heart J. 2023 May 7;44(18):1607]. Eur Heart J (2023) 44(1):14–27. doi:10.1093/eurheartj/ehac569
20. Dixon, SJ, Lemberg, KM, Lamprecht, MR, Skouta, R, Zaitsev, EM, Gleason, CE, et al. Ferroptosis: An Iron-Dependent Form of Non-Apoptotic Cell Death. Cell (2012) 149(5):1060–72. doi:10.1016/j.cell.2012.03.042
21. Lei, G, Zhuang, L, and Gan, B. Targeting Ferroptosis as a Vulnerability in Cancer. Nat Rev Cancer (2022) 22(7):381–96. doi:10.1038/s41568-022-00459-0
22. Jiang, X, Stockwell, BR, and Conrad, M. Ferroptosis: Mechanisms, Biology and Role in Disease. Nat Rev Mol Cel Biol (2021) 22(4):266–82. doi:10.1038/s41580-020-00324-8
23. Shen, C, Liu, J, Liu, H, Li, G, Wang, H, Tian, H, et al. Timosaponin AIII Induces Lipid Peroxidation and Ferroptosis by Enhancing Rab7-Mediated Lipophagy in Colorectal Cancer Cells. Phytomedicine (2024) 122:155079. doi:10.1016/j.phymed.2023.155079
24. Dong, X, Li, Y, Sheng, X, Zhou, W, Sun, A, and Dai, H. Mitochondria-Related Signaling Pathways Involved in Breast Cancer Regulate Ferroptosis. Genes Dis (2024) 11(1):358–66. doi:10.1016/j.gendis.2023.03.019
25. Chen, X, Wang, Z, Li, C, Zhang, Z, Lu, S, Wang, X, et al. SIRT1 Activated by AROS Sensitizes Glioma Cells to Ferroptosis via Induction of NAD+ Depletion-Dependent Activation of ATF3. Redox Biol (2024) 69:103030. doi:10.1016/j.redox.2024.103030
26. Yang, YH, Li, W, Ren, LW, Yang, H, Zhang, Y, Zhang, S, et al. S670, an Amide Derivative of 3-O-Acetyl-11-Keto-β-Boswellic Acid, Induces Ferroptosis in Human Glioblastoma Cells by Generating ROS and Inhibiting STX17-Mediated Fusion of Autophagosome and Lysosome. Acta Pharmacol Sin (2024) 45(1):209–22. doi:10.1038/s41401-023-01157-9
27. Feng, L, Sun, J, Xia, L, Shi, Q, Hou, Y, Zhang, L, et al. Ferroptosis Mechanism and Alzheimer's Disease. Neural Regen Res (2024) 19(8):1741–50. doi:10.4103/1673-5374.389362
28. Xiang, Y, Song, X, and Long, D. Ferroptosis Regulation Through Nrf2 and Implications for Neurodegenerative Diseases. Arch Toxicol (2024) 98:579–615. doi:10.1007/s00204-023-03660-8
29. Deng, L, He, S, Guo, N, Tian, W, Zhang, W, and Luo, L. Molecular Mechanisms of Ferroptosis and Relevance to Inflammation. Inflamm Res (2023) 72(2):281–99. doi:10.1007/s00011-022-01672-1
30. Wang, F, He, J, Xing, R, Sha, T, and Sun, B. Molecular Mechanisms of Ferroptosis and Their Role in Inflammation. Int Rev Immunol (2023) 42(1):71–81. doi:10.1080/08830185.2021.2016739
31. Ubellacker, JM, Tasdogan, A, Ramesh, V, Shen, B, Mitchell, EC, Martin-Sandoval, MS, et al. Lymph Protects Metastasizing Melanoma Cells From Ferroptosis. Nature (2020) 585(7823):113–8. doi:10.1038/s41586-020-2623-z
32. Alvarez, SW, Sviderskiy, VO, Terzi, EM, Papagiannakopoulos, T, Moreira, AL, Adams, S, et al. NFS1 Undergoes Positive Selection in Lung Tumours and Protects Cells From Ferroptosis. Nature (2017) 551(7682):639–43. doi:10.1038/nature24637
33. Anandhan, A, Dodson, M, Schmidlin, CJ, Liu, P, and Zhang, DD. Breakdown of an Ironclad Defense System: The Critical Role of NRF2 in Mediating Ferroptosis. Cel Chem Biol (2020) 27(4):436–47. doi:10.1016/j.chembiol.2020.03.011
34. Sprowls, SA, Arsiwala, TA, Bumgarner, JR, Shah, N, Lateef, SS, Kielkowski, BN, et al. Improving CNS Delivery to Brain Metastases by Blood-Tumor Barrier Disruption. Trends Cancer (2019) 5(8):495–505. doi:10.1016/j.trecan.2019.06.003
35. Liebner, S, and Engelhardt, B. Development of the Blood–Brain Barrier. In: The Blood Brain Barrier and Its Microenvironment: Basic Physiology to Neurological Disease. New York: Taylor & Francis (2005). p. 1–25.
36. Lacombe, RJS, Chouinard-Watkins, R, and Bazinet, RP. Brain Docosahexaenoic Acid Uptake and Metabolism. Mol Aspects Med (2018) 64:109–34. doi:10.1016/j.mam.2017.12.004
37. Chauhan, G, Roy, K, Kumar, G, Kumari, P, Alam, S, Kishore, K, et al. Distinct Influence of COX-1 and COX-2 on Neuroinflammatory Response and Associated Cognitive Deficits During High Altitude Hypoxia. Neuropharmacology (2019) 146:138–48. doi:10.1016/j.neuropharm.2018.11.026
38. Akhter, MS, Uddin, MA, Kubra, KT, and Barabutis, N. P53-Induced Reduction of Lipid Peroxidation Supports Brain Microvascular Endothelium Integrity. J Pharmacol Sci (2019) 141(1):83–5. doi:10.1016/j.jphs.2019.09.008
39. Zlokovic, BV. The Blood-Brain Barrier in Health and Chronic Neurodegenerative Disorders. Neuron (2008) 57(2):178–201. doi:10.1016/j.neuron.2008.01.003
40. Yang, C, Hawkins, KE, Doré, S, and Candelario-Jalil, E. Neuroinflammatory Mechanisms of Blood-Brain Barrier Damage in Ischemic Stroke. Am J Physiology-Cell Physiol (2019) 316(2):C135–C153. doi:10.1152/ajpcell.00136.2018
41. Cash, A, and Theus, MH. Mechanisms of Blood-Brain Barrier Dysfunction in Traumatic Brain Injury. Int J Mol Sci (2020) 21(9):3344. doi:10.3390/ijms21093344
42. Rand, D, Ravid, O, Atrakchi, D, Israelov, H, Bresler, Y, Shemesh, C, et al. Endothelial Iron Homeostasis Regulates Blood-Brain Barrier Integrity via the HIF2α-Ve-Cadherin Pathway. Pharmaceutics (2021) 13(3):311. doi:10.3390/pharmaceutics13030311
43. Woo, JH, Choi, YS, and Choi, JH. Iron-Storage Protein Ferritin Is Upregulated in Endometriosis and Iron Overload Contributes to a Migratory Phenotype. Biomedicines (2020) 8(11):454. doi:10.3390/biomedicines8110454
44. Jin, Z, Liang, J, Li, J, and Kolattukudy, PE. Absence of MCP-Induced Protein 1 Enhances Blood-Brain Barrier Breakdown After Experimental Stroke in Mice. Int J Mol Sci (2019) 20(13):3214. doi:10.3390/ijms20133214
45. Correale, J, and Villa, A. The Blood-Brain-Barrier in Multiple Sclerosis: Functional Roles and Therapeutic Targeting. Autoimmunity (2007) 40(2):148–60. doi:10.1080/08916930601183522
46. Lin, M, Sun, W, Gong, W, Zhou, Z, Ding, Y, and Hou, Q. Methylophiopogonanone A Protects Against Cerebral Ischemia/Reperfusion Injury and Attenuates Blood-Brain Barrier Disruption In Vitro. PLoS One (2015) 10(4):e0124558. doi:10.1371/journal.pone.0124558
47. Chi, H, Li, B, Wang, Q, Gao, Z, Feng, B, Xue, H, et al. Opportunities and Challenges Related to Ferroptosis in Glioma and Neuroblastoma. Front Oncol (2023) 13:1065994. doi:10.3389/fonc.2023.1065994
48. Ye, LF, Chaudhary, KR, Zandkarimi, F, Harken, AD, Kinslow, CJ, Upadhyayula, PS, et al. Radiation-Induced Lipid Peroxidation Triggers Ferroptosis and Synergizes with Ferroptosis Inducers. ACS Chem Biol (2020) 15(2):469–84. doi:10.1021/acschembio.9b00939
49. Alexiou, GA, Gerogianni, P, Vartholomatos, E, and Kyritsis, AP. Deferiprone Enhances Temozolomide Cytotoxicity in Glioma Cells. Cancer Invest (2016) 34(10):489–95. doi:10.1080/07357907.2016.1233424
50. Lu, S, Wang, XZ, He, C, Wang, L, Liang, S, Wang, C, et al. ATF3 Contributes to Brucine-Triggered Glioma Cell Ferroptosis via Promotion of Hydrogen Peroxide and Iron. Acta Pharmacol Sin (2021) 42(10):1690–702. doi:10.1038/s41401-021-00700-w
51. de Souza, I, Monteiro, LKS, Guedes, CB, Silva, MM, Andrade-Tomaz, M, Contieri, B, et al. High Levels of NRF2 Sensitize Temozolomide-Resistant Glioblastoma Cells to Ferroptosis via ABCC1/MRP1 Upregulation. Cell Death Dis (2022) 13(7):591. doi:10.1038/s41419-022-05044-9
52. Lathoria, K, Gowda, P, Umdor, SB, Patrick, S, Suri, V, and Sen, E. PRMT1 Driven PTX3 Regulates Ferritinophagy in Glioma. Autophagy (2023) 19(7):1997–2014. doi:10.1080/15548627.2023.2165757
53. Jiang, T, Chu, J, Chen, H, Cheng, H, Su, J, Wang, X, et al. Gastrodin Inhibits H2O2-Induced Ferroptosis through its Antioxidative Effect in Rat Glioma Cell Line C6. Biol Pharm Bull (2020) 43(3):480–7. doi:10.1248/bpb.b19-00824
54. Gao, X, Guo, N, Xu, H, Pan, T, lei, H, Yan, A, et al. Ibuprofen Induces Ferroptosis of Glioblastoma Cells via Downregulation of Nuclear Factor Erythroid 2-Related Factor 2 Signaling Pathway. Anticancer Drugs (2020) 31(1):27–34. doi:10.1097/CAD.0000000000000825
55. Wang, Z, Ding, Y, Wang, X, Lu, S, Wang, C, He, C, et al. Pseudolaric Acid B Triggers Ferroptosis in Glioma Cells via Activation of Nox4 and Inhibition of xCT. Cancer Lett (2018) 428:21–33. doi:10.1016/j.canlet.2018.04.021
56. Li, K, Chen, B, Xu, A, Shen, J, Li, K, Hao, K, et al. TRIM7 Modulates NCOA4-Mediated Ferritinophagy and Ferroptosis in Glioblastoma Cells. Redox Biol (2022) 56:102451. doi:10.1016/j.redox.2022.102451
57. Zhan, S, Lu, L, Pan, SS, Wei, X, Miao, R, Liu, X, et al. Targeting NQO1/GPX4-Mediated Ferroptosis by Plumbagin Suppresses In Vitro and In Vivo Glioma Growth. Br J Cancer (2022) 127(2):364–76. doi:10.1038/s41416-022-01800-y
58. Han, L, Zhou, J, Li, L, Wu, X, Shi, Y, Cui, W, et al. SLC1A5 Enhances Malignant Phenotypes Through Modulating Ferroptosis Status and Immune Microenvironment in Glioma. Cel Death Dis (2022) 13(12):1071. doi:10.1038/s41419-022-05526-w
59. Hsieh, CH, Hsieh, HC, Shih, FH, Wang, PW, Yang, LX, Shieh, DB, et al. An Innovative NRF2 Nano-Modulator Induces Lung Cancer Ferroptosis and Elicits an Immunostimulatory Tumor Microenvironment. Theranostics (2021) 11(14):7072–91. doi:10.7150/thno.57803
60. Umans, RA, Martin, J, Harrigan, ME, Patel, DC, Chaunsali, L, Roshandel, A, et al. Transcriptional Regulation of Amino Acid Transport in Glioblastoma Multiforme. Cancers (Basel) (2021) 13(24):6169. doi:10.3390/cancers13246169
61. Kang, R, Kroemer, G, and Tang, D. The Tumor Suppressor Protein P53 and the Ferroptosis Network. Free Radic Biol Med (2019) 133:162–8. doi:10.1016/j.freeradbiomed.2018.05.074
62. Buyandelger, B, Bar, EE, Hung, KS, Chen, RM, Chiang, YH, Liou, JP, et al. Histone Deacetylase Inhibitor MPT0B291 Suppresses Glioma Growth In Vitro and In Vivo Partially Through Acetylation of P53. Int J Biol Sci (2020) 16(16):3184–99. doi:10.7150/ijbs.45505
63. Koppula, P, Zhuang, L, and Gan, B. Cystine Transporter SLC7A11/xCT in Cancer: Ferroptosis, Nutrient Dependency, and Cancer Therapy. Protein Cell (2021) 12(8):599–620. doi:10.1007/s13238-020-00789-5
64. Wolf, IM, Fan, Z, Rauh, M, Seufert, S, Hore, N, Buchfelder, M, et al. Histone Deacetylases Inhibition by SAHA/Vorinostat Normalizes the Glioma Microenvironment via xCT Equilibration. Sci Rep (2014) 4:6226. doi:10.1038/srep06226
65. Zhang, T, Sun, B, Zhong, C, Xu, K, Wang, Z, Hofman, P, et al. Targeting Histone Deacetylase Enhances the Therapeutic Effect of Erastin-Induced Ferroptosis in EGFR-Activating Mutant Lung Adenocarcinoma. Transl Lung Cancer Res (2021) 10(4):1857–72. doi:10.21037/tlcr-21-303
66. Yang, Y, Ma, Y, Li, Q, Ling, Y, Zhou, Y, Chu, K, et al. STAT6 Inhibits Ferroptosis and Alleviates Acute Lung Injury via Regulating P53/SLC7A11 Pathway. Cel Death Dis (2022) 13(6):530. doi:10.1038/s41419-022-04971-x
67. Lan, Y, Yang, T, Yue, Q, Wang, Z, Zhong, X, Luo, X, et al. IRP1 Mediated Ferroptosis Reverses Temozolomide Resistance in Glioblastoma via Affecting LCN2/FPN1 Signaling Axis Depended on NFKB2. iScience (2023) 26(8):107377. doi:10.1016/j.isci.2023.107377
68. Huang, Q, Li, J, Ma, M, Lv, M, Hu, R, Sun, J, et al. High-Throughput Screening Identification of a Small-Molecule Compound That Induces Ferroptosis and Attenuates the Invasion and Migration of Hepatocellular Carcinoma Cells by Targeting the STAT3/GPX4 Axis. Int J Oncol (2023) 62(3):42. doi:10.3892/ijo.2023.5490
69. Wang, Z, Sun, R, Wang, G, Chen, Z, Li, Y, Zhao, Y, et al. SIRT3-Mediated Deacetylation of PRDX3 Alleviates Mitochondrial Oxidative Damage and Apoptosis Induced by Intestinal Ischemia/reperfusion Injury. Redox Biol (2020) 28:101343. doi:10.1016/j.redox.2019.101343
70. Gajer, JM, Furdas, SD, Gründer, A, Gothwal, M, Heinicke, U, Keller, K, et al. Histone Acetyltransferase Inhibitors Block Neuroblastoma Cell Growth In Vivo. Oncogenesis (2015) 4(2):e137. doi:10.1038/oncsis.2014.51
71. Karsli-Ceppioglu, S, Dagdemir, A, Judes, G, Ngollo, M, Penault-Llorca, F, Pajon, A, et al. Epigenetic Mechanisms of Breast Cancer: An Update of the Current Knowledge. Epigenomics (2014) 6(6):651–64. doi:10.2217/epi.14.59
72. Schneider, A, Chatterjee, S, Bousiges, O, Selvi, BR, Swaminathan, A, Cassel, R, et al. Acetyltransferases (HATs) as Targets for Neurological Therapeutics. Neurotherapeutics (2013) 10(4):568–88. doi:10.1007/s13311-013-0204-7
73. Seto, E, and Yoshida, M. Erasers of Histone Acetylation: The Histone Deacetylase Enzymes. Cold Spring Harbor Perspect Biol (2014) 6(4):a018713. doi:10.1101/cshperspect.a018713
74. Cheng, Z, Li, S, Yuan, J, Li, Y, Cheng, S, Huang, S, et al. HDAC1 Mediates Epithelial-Mesenchymal Transition and Promotes Cancer Cell Invasion in Glioblastoma. Pathol - Res Pract (2023) 246:154481. doi:10.1016/j.prp.2023.154481
75. Kwak, S, Park, SH, Kim, SH, Sung, GJ, Song, JH, Jeong, JH, et al. miR-3189-Targeted GLUT3 Repression by HDAC2 Knockdown Inhibits Glioblastoma Tumorigenesis Through Regulating Glucose Metabolism and Proliferation. J Exp Clin Cancer Res (2022) 41(1):87. doi:10.1186/s13046-022-02305-5
76. Wu, AC, Yang, WB, Chang, KY, Lee, JS, Liou, JP, Su, RY, et al. HDAC6 Involves in Regulating the LncRNA-MicroRNA-mRNA Network to Promote the Proliferation of Glioblastoma Cells. J Exp Clin Cancer Res (2022) 41(1):47. doi:10.1186/s13046-022-02257-w
77. Li, X, Zhang, W, Xing, Z, Hu, S, Zhang, G, Wang, T, et al. Targeting SIRT3 Sensitizes Glioblastoma to Ferroptosis by Promoting Mitophagy and Inhibiting SLC7A11. Cel Death Dis (2024) 15(2):168. doi:10.1038/s41419-024-06558-0
78. Yan, Y, Teng, H, Hang, Q, Kondiparthi, L, Lei, G, Horbath, A, et al. SLC7A11 Expression Level Dictates Differential Responses to Oxidative Stress in Cancer Cells. Nat Commun (2023) 14(1):3673. doi:10.1038/s41467-023-39401-9
79. Fojo, T. p53 as a Therapeutic Target: Unresolved Issues on the Road to Cancer Therapy Targeting Mutant P53. Drug Resist Updates (2002) 5(5):209–16. doi:10.1016/s1368-7646(02)00119-x
80. Gu, W, and Roeder, RG. Activation of P53 Sequence-Specific DNA Binding by Acetylation of the P53 C-Terminal Domain. Cell (1997) 90(4):595–606. doi:10.1016/s0092-8674(00)80521-8
81. Ito, A, Lai, CH, Zhao, X, Saito, S, Hamilton, MH, Appella, E, et al. p300/CBP-Mediated P53 Acetylation Is Commonly Induced by P53-Activating Agents and Inhibited by MDM2. EMBO J (2001) 20(6):1331–40. doi:10.1093/emboj/20.6.1331
82. Tang, Y, Zhao, W, Chen, Y, Zhao, Y, and Gu, W. Acetylation Is Indispensable for P53 Activation [Published Correction Appears in Cell. 2008 Jun 27;133(7):1290]. Cell (2008) 133(4):612–26. doi:10.1016/j.cell.2008.03.025
83. Muñoz-Fontela, C, González, D, Marcos-Villar, L, Campagna, M, Gallego, P, González-Santamaría, J, et al. Acetylation Is Indispensable for P53 Antiviral Activity. Cell Cycle (2011) 10(21):3701–5. doi:10.4161/cc.10.21.17899
84. Zhang, Y, Dube, C, Gibert, M, Cruickshanks, N, Wang, B, Coughlan, M, et al. The P53 Pathway in Glioblastoma. Cancers (Basel) (2018) 10(9):297. doi:10.3390/cancers10090297
85. Pasricha, SR, Lim, PJ, Duarte, TL, Casu, C, Oosterhuis, D, Mleczko-Sanecka, K, et al. Hepcidin Is Regulated by Promoter-Associated Histone Acetylation and HDAC3. Nat Commun (2017) 8(1):403. doi:10.1038/s41467-017-00500-z
86. Cheshmazar, N, Hamzeh-Mivehroud, M, Nozad Charoudeh, H, Hemmati, S, Melesina, J, and Dastmalchi, S. Current Trends in Development of HDAC-Based Chemotherapeutics. Life Sci (2022) 308:120946. doi:10.1016/j.lfs.2022.120946
87. Smith, KT, and Workman, JL. Histone Deacetylase Inhibitors: Anticancer Compounds. Int J Biochem & Cel Biol (2009) 41(1):21–5. doi:10.1016/j.biocel.2008.09.008
88. Wang, X, Liu, K, Gong, H, Li, D, Chu, W, Zhao, D, et al. Death by Histone Deacetylase Inhibitor Quisinostat in Tongue Squamous Cell Carcinoma via Apoptosis, Pyroptosis, and Ferroptosis. Toxicol Appl Pharmacol (2021) 410:115363. doi:10.1016/j.taap.2020.115363
89. Li, F, Wang, T, Wang, Z, Chen, X, and Liu, R. Histone Deacetylase Inhibitor Quisinostat Activates Caspase Signaling and Upregulates P53 Acetylation to Inhibit the Proliferation of HepG2 Cells. Mol Med Rep (2017) 16(5):6094–101. doi:10.3892/mmr.2017.7355
90. Shi, L, Wang, Z, and Sun, G. Curcumin Induces Glioma Stem-Like Cell Formation. Neuroreport (2015) 26(3):167–72. doi:10.1097/WNR.0000000000000320
91. Wang, H, Shi, L, and Wang, Z. A Novel Hydroxamic Acid-Based Curcumin Derivative as Potent Histone Deacetylase Inhibitor for the Treatment of Glioblastoma. Front Oncol (2021) 11:756817. doi:10.3389/fonc.2021.756817
92. Miyamoto, K, Watanabe, M, Boku, S, Sukeno, M, Morita, M, Kondo, H, et al. xCT Inhibition Increases Sensitivity to Vorinostat in a ROS-Dependent Manner. Cancers (Basel) (2020) 12(4):827. doi:10.3390/cancers12040827
93. Karaj, E, Sindi, SH, Kuganesan, N, Koranne, RA, Knoff, JR, James, AW, et al. First-in-Class Dual Mechanism Ferroptosis-HDAC Inhibitor Hybrids. J Med Chem (2022) 65(21):14764–91. doi:10.1021/acs.jmedchem.2c01276
94. Zille, M, Kumar, A, Kundu, N, Bourassa, MW, Wong, VSC, Willis, D, et al. Ferroptosis in Neurons and Cancer Cells Is Similar But Differentially Regulated by Histone Deacetylase Inhibitors. eNeuro (2019) 6(1):0263–18.2019. doi:10.1523/ENEURO.0263-18.2019
95. Paganoni, S, Macklin, EA, Hendrix, S, Berry, JD, Elliott, MA, Maiser, S, et al. Trial of Sodium Phenylbutyrate-Taurursodiol for Amyotrophic Lateral Sclerosis. N Engl J Med (2020) 383(10):919–30. doi:10.1056/NEJMoa1916945
Keywords: ferroptosis, histone deacetylases, histone deacetylase inhibitors, glioma, targeted therapy
Citation: Ma M, Fei X, Jiang D, Chen H, Xie X, Wang Z and Huang Q (2024) Research Progress on the Mechanism of Histone Deacetylases in Ferroptosis of Glioma. Oncol. Rev. 18:1432131. doi: 10.3389/or.2024.1432131
Received: 13 May 2024; Accepted: 02 August 2024;
Published: 12 August 2024.
Edited by:
Eduard Yakubov, Paracelsus Medical Private University, Nuremberg, GermanyReviewed by:
Alka Singh, The University of Chicago, United StatesAayushi Mahajan, Columbia University, United States
Copyright © 2024 Ma, Fei, Jiang, Chen, Xie, Wang and Huang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhimin Wang, d2FuZ3ptMjAxN0AxMjYuY29t
†These authors have contributed equally to this work