 Yonwaba Mzizi1,2,†Saidon Mbambara1,2,3,†Boitumelo Moetlhoa2,4Johncy Mahapane5Sipho Mdanda1,2
Yonwaba Mzizi1,2,†Saidon Mbambara1,2,3,†Boitumelo Moetlhoa2,4Johncy Mahapane5Sipho Mdanda1,2 Mike Sathekge1,2
Mike Sathekge1,2 Mankgopo Kgatle1,2,6*
Mankgopo Kgatle1,2,6*
- 1Department of Nuclear Medicine, University of Pretoria and Steve Biko Academic Hospital, Pretoria, South Africa
- 2Basic and Translational Research, Nuclear Medicine Research Infrastructure (NuMeRI), Steve Biko Academic Hospital, Pretoria, South Africa
- 3Department of Biomedical Sciences, Tropical Diseases Research Centre, Ndola, Zambia
- 4School of Health Systems and Public Health, Faculty of Health Sciences, University of Pretoria, Pretoria, South Africa
- 5Department of Radiography, University of Pretoria, Pretoria, South Africa
- 6Department of Medicine, University of Cape Town and Groote Schuur Hospital, Cape Town, South Africa
Ionising radiation (IR) is a form of energy that travels as electromagnetic waves or particles. While it is vital in medical and occupational health settings, IR can also damage DNA, leading to mutations, chromosomal aberrations, and transcriptional changes that disrupt the functions of certain cell regulators, genes, and transcription factors. These disruptions can alter functions critical for cancer development, progression, and treatment response. Additionally, IR can affect various cellular proteins and their regulators within different cell signalling pathways, resulting in physiological changes that may promote cancer development, progression, and resistance to treatment. Understanding these impacts is crucial for developing strategies to mitigate the harmful effects of IR exposure and improve cancer treatment outcomes. This review focuses on specific genes and protein biomarkers regulated in response to chronic IR exposure, and how their regulation impacts disease onset, progression, and treatment response.
Introduction
Ionising radiation (IR) is energy emitted as electromagnetic waves or particles, measured in electron volts (eV) (1). It originates from natural sources like water, soil, and vegetation, as well as artificial sources such as x-rays, gamma rays or particles, and contains higher energy than non-IR (1). Exposure to x-rays and gamma rays is measured in roentgen (R), with one R producing 0.008771 Gray (Gy), defined as the amount of radiation needed to produce ions resulting in a charge of 0.000258 coulombs per kilogram of air under standard conditions (1).
The use of IR is becoming more prevalent in medical and occupational environments. Although it has significantly improved cancer treatment, IR and radiation therapy-prolonged exposure based on absorbed dose and dose rate can damage DNA. This may cause base and sugar damage, as well as single and double strand breaks (SSBs and DSBs). x-rays and gamma rays have low linear energy transfer (LET) and are less densely ionising, while carbon ions have high LET and are more densely ionising. LET levels influence the type of DNA damage, with low LET radiation typically causing about 1,000 SSBs and 40 DSBs. These damages can result in DNA lesions, leading to the loss and rearrangement of genomic sequences. This may alter phenotypic effects, potentially causing malignancy and impacting the effectiveness of cancer treatments (2).
Radiation effects are classified as deterministic or stochastic. Deterministic effects depend on the dose, with higher doses causing more severe outcomes like skin reddening and radiation burns. Stochastic effects are dose-independent and include DNA damage and radiation-induced cancer (3). Diagnostic x-rays, CT scans and radiation therapy can lead to such effects, and potentially leading cancer development, progression and resistance (4). The risk of leukaemia and other secondary cancers increases in adults exposed to radiation from sources like nuclear power plants (5). The first case of radiation-induced cancer was reported in 1902 by Frieben, seven years after the discovery of x-rays (6, 7). Following the Chernobyl disaster in 1986, there were excess cases of cancers, including leukaemia, thyroid cancers, and lymphomas among survivors (8). It is clear that IR causes DNA damage, which can potentially lead to cancer and affect treatment outcomes. Therefore, it is crucial to understand the clinical implications of IR-induced DNA damage and the associated molecular targets to devise more effective preventative and therapeutic strategies. This paper will explore gene and protein biomarkers that are regulated or altered in response to IR-induced DNA damage and their clinical significance in cancer and treatment (also outlined in Table 1).
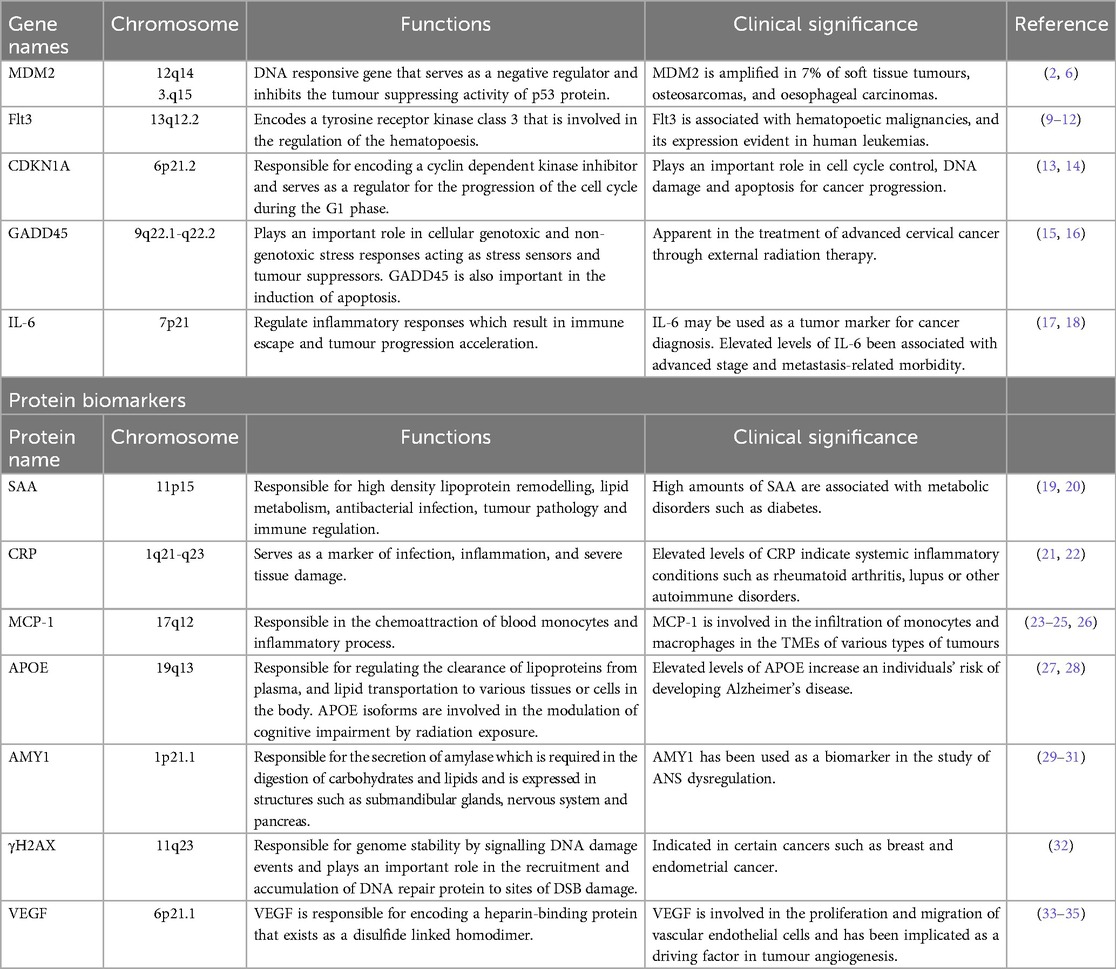
Table 1. Summary of gene and protein-based markers associated with ionising radiation exposure, their functions and clinical relevance.
IR-based DNA damage and repair mechanisms
Stress from internal or external factors can cause DNA damage, including base pair changes, replication errors, and breaks in the DNA double helix. Cells counteract these effects through the DNA damage response (DDR), which signals damage and recruits repair factors (1). DNA repair mechanisms like nonhomologous end-joining (NHEJ) and homologous recombination (HR) are crucial for maintaining genomic stability. NHEJ joins damaged DNA ends with little or no homology, which may cause deletions or insertions, while HR uses the undamaged sister chromatid to accurately repair the DNA.
The DDR system detects DNA damage using kinases such as ATM and ATR proteins as described in Figure 1. ATM senses double strand breaks (DSBs) caused by IR, while ATR responds to single strand breaks (SSBs) and replication fork stalling due to IR exposure (14, 17, 36).
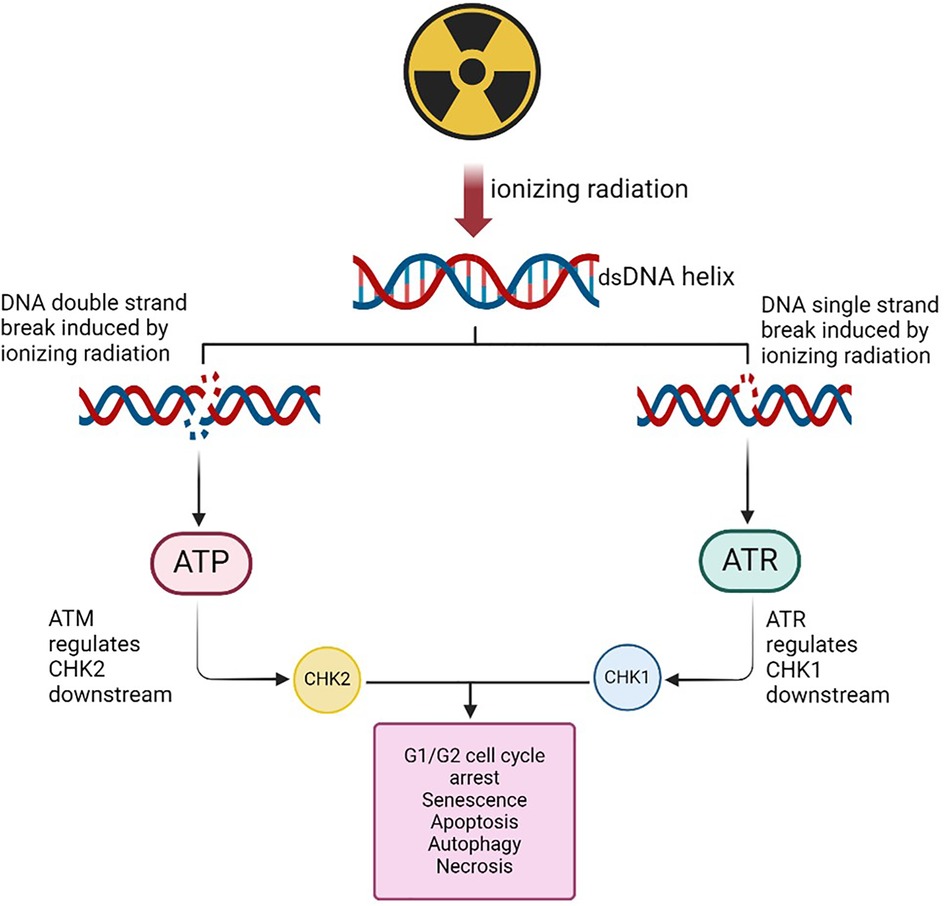
Figure 1. DNA strand breaks due to irradiation may result in the activation of the ATM and the ATR proteins. ATR in turn upregulates the CHK-1 gene that may be implicated in the cell cycle process in the G1/G2 phase. The ATM is involved in the recruitment of the CHK-2 and affect cell cycle process leading to apoptosis, senescence or autophagy. (Created with BioRender.com, Agreement No: LB27EKK6WL).
DNA damage and repair are crucial indicators of the body's response to IR. IR exposure generates reactive oxygen species (ROS), causing damage at specific sites. Repair mechanisms then activate, halting the cell cycle to fix the damage. Successful repair makes cells less radiosensitive, aiding their survival and replication (37). Cells that survive radiotherapy become more resistant to further radiation due to repair mechanisms (38, 39). Radiosensitivity is highest during the G2/M phases and lowest during the late S phase (39, 40). Successful repairs increase resistance to future radiation damage (38). DNA repair involves enzymes like DNA ligase, which mends strand breaks. These processes can lead to genetic changes, potentially contributing to tumorigenesis (41). Epigenetic modifiers, such as DNA methylation and histone modifications, also play a role in regulating DNA repair by affecting nucleosome and chromatin structure (37).
Gene biomarkers
IR-induced DNA damage can lead to somatic mutations that disrupt cell regulation and potentially cause cancer, leaving a mutational signature on the cancer cell genome (41). Genes involved in cell cycle regulation, DNA repair, and oxidative stress response are implicated in IR-induced DNA damage, influencing tumour development and response to radiation therapy (41). IR can alter gene expression, with some genes like murine double minute 2 (MDM2), growth arrest and DNA damage inducible alpha (GADD45), Flt3 Ligand (Flt3l), and cyclin dependent kinase inhibitor 1A (CDKN1A) are consistently up regulated, affecting DNA repair and cell cycle control (Figure 2) (41). Long-term exposure can also increase blood biomarkers such as interleukins that are associated with inflammation and cancer risk. Understanding these genes can help develop personalized radiotherapy plans, improving treatment efficacy and minimizing side effects.
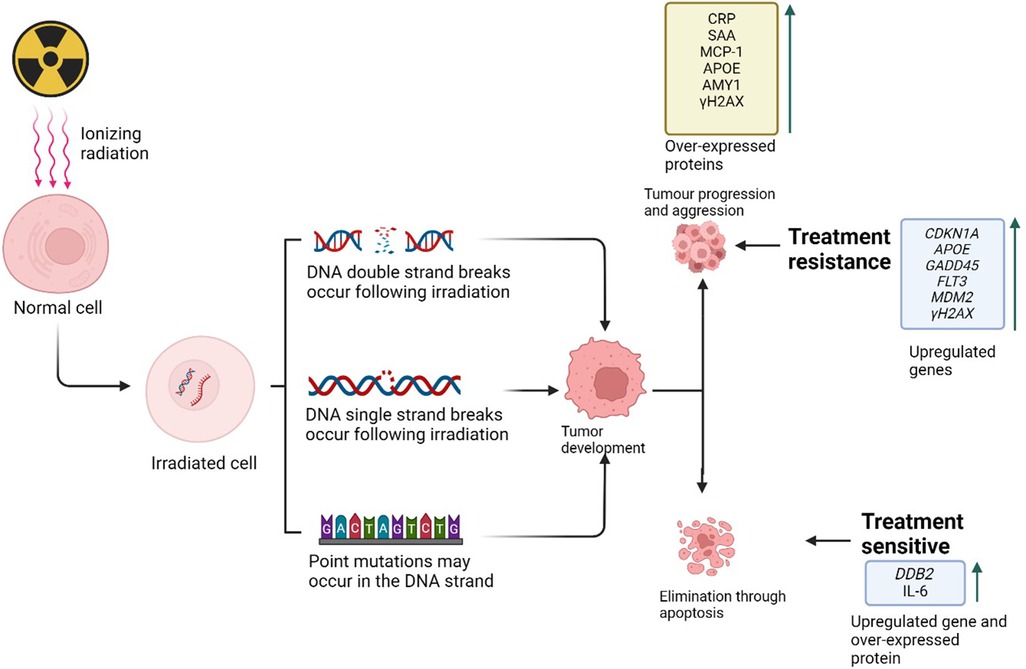
Figure 2. Ionizing radiation-induced DNA damage plays a crucial role in cancer progression and treatment response. This damage includes double-strand breaks (DSBs) and single-strand breaks (SSBs) in irradiated cells, which can lead to the development and progression of cancerous cells. These cells may differentiate and proliferate through the upregulation of various pathways, resulting in either apoptosis or tumor formation, thereby influencing treatment response (Created with BioRender.com, Agreement No: AP27B1MHP8).
Murine double minute 2
MDM2 is a gene with four conserved domains that negatively regulates the tumor-suppressing activity of p53 (6). It is a proto-oncogene amplified in 7% of all cancers, particularly in soft tissue tumours, osteosarcomas, and oesophageal carcinomas. Elevated MDM2 transcript levels are also found in other cancers without gene amplification (2, 6).
MDM2 is a transcriptional target of p53 that regulates E3 ligase activity by binding to, inhibiting, and ubiquitinating p53 gene, leading to its degradation via the 26S proteasome (2, 4). In the absence of IR, MDM2 keeps p53 levels low by preventing its transcriptional activity and tagging it for degradation (2, 4). Upon IR exposure, p53 is activated, leading to the accumulation and transcription of p53-regulated genes through DNA damage response pathways (5).
Elevated p53 levels and activity in response to ionizing radiation (IR) stimulate MDM2 overexpression, which helps control cell death (42–44). This was shown in both humanized and non-humanized mouse models irradiated with 4 and 8 Gy for 24 h (45). Another study found increased p53 levels after 200 rad irradiation in DA-1 murine lymphoma and ML-1 myeloid leukaemia cell lines, leading to MDM2 release (46). Peak p53 levels were seen within an hour of radiation exposure, with peak MDM2 levels at 1.5–2 h. A second wave of p53 was observed hours later, further stimulating MDM2 expression and counteracting p53 activities in cells recovering from DNA damage (47). Upregulation of MDM2 gene was also noted in both p53-positive and p53-negative mouse tissues before and after γ radiation exposure (46).
In cancer cells with wild type p53, a direct correlation between p53 and MDM2 levels and the extent of DNA damage from IR was observed (47). A study on cervical cancer treated with radiation therapy found that remaining cancer cells showed contributions from p53 and MDM2, leading to radioresistance (47). Targeting the p53-MDM2 interaction is seen as a potential cancer treatment strategy. Combining MDM2 inhibitors with IR could be effective, as supported by a clinical trial using Nutlin-3, which showed increased efficacy and improved outcomes in treating both solid and haematological cancers, particularly enhancing survival in glioblastoma patients when combined with radiation therapy (48).
In experimental mouse models, knocking out MDM2 gene in embryonic mice was lethal, causing death at 3.5 days post-coitum due to p53-dependent apoptosis, which was prevented by deleting the p53 gene. Hemizygous MDM2± mice were normal under homeostatic conditions but more sensitive to ionizing radiation compared to wild-type animals (2, 6). This suggests that MDM2 gene is a crucial regulator of the p53 protein, which controls cell cycle arrest and apoptosis. Without MDM2 gene, p53 activity becomes unregulated, leading to excessive apoptosis and embryonic death.
Flt3 Ligand
Flt3l is a crucial ligand for the Flt3 receptor tyrosine kinase encoded by Flt3 gene, playing a vital role in blood cell formation by stimulating the growth and differentiation of hematopoietic stem cells (9). It collaborates with other growth factors like G-CSF, GM-CSF, SCF, and IL-3 to develop various blood cell types and promote the proliferation of stem cells and dendritic (10), making it significant in cancer immunotherapy.
The Flt3 gene encodes the Flt3 ligand, which plays a significant role in response to IR exposure. IR can induce mutations in the Flt3 gene, particularly internal tandem duplications (ITD) and point mutations (11). These mutations lead to ligand-independent dimerization and constitutive activation of the Flt3 receptor, triggering downstream signalling pathways that promote uncontrolled cell growth (11). This is associated with the development and poor prognosis of acute myeloid leukaemia (AML). Such mutations are found in about 30% of AML patients and a smaller number of patients with acute lymphocytic leukaemia (ALL) or myelodysplastic syndrome (11).
Abnormal plasma Flt3l levels indicate radiation exposure-induced damage, making them useful for clinical and emergency assessments (12). A deeper understanding of ionizing radiation-induced damage and Flt3 gene mutations can aid in developing targeted therapies and improving outcomes for patients with radiation-induced cancers. In cancer therapy, Flt3l is being studied to enhance the effectiveness of radiation treatment by boosting the immune response and reducing metastases.
Growth arrest and DNA damage inducible alpha
GADD45 is a gene induced by DNA damage and regulated by the p53 protein during the G1 phase of the cell cycle (18, 49). The GADD45α, GADD45β, and GADD45γ genes encoding the GADD45 protein family, are upregulated in response to DNA damage caused by ionizing radiation, playing a significant role in cancer development and treatment (50, 51). For example, lymphoblastoid cells exposed to 3Gy and 10Gy γ radiation showed increased GADD45 gene expression from 1 to 24 h post-exposure (15). When cells are exposed to ionizing radiation, the upregulation of GADD45 gene aids in DNA repair, cell cycle arrest, and apoptosis, preventing the propagation of damaged cells that could lead to cancer (50, 51).
GADD45 proteins also function as tumour suppressors, maintaining genomic integrity by resolving DNA damage induced by ionizing radiation (52, 53). However, their expression can be altered in response to IR exposure. For instance, a study by Snyder et al. (51) demonstrated that peripheral blood from healthy subjects showed altered expression of various DNA-repair genes, including GADD45, following exposure to 0 and 2Gy of x-ray radiation. Similar results were observed in a study by Smirnov et al. (54), where B cells irradiated with 10Gy showed changes in GADD45 expression at 2- and 6-h post-irradiation. Reduced expression of GADD45 tumour suppressors is associated with genomic instability and increased mutation rates, potentially leading to cancer progression.
Aberrant expression of GADD45 tumour suppressors can also affect how cancer cells respond to radiation therapy (55). For instance, upregulation of GADD45A has been shown to enhance the effectiveness of radiotherapy by increasing the sensitivity of human tongue squamous carcinoma cell lines to IR. Conversely, inactivation of GADD45A can make cancer cells more resistant to radiation, impacting treatment outcomes. Overexpression of GADD45 gene has been linked to increased lethality in cervical cancer in response to radiation therapy. This finding is supported by Asuthkar et al. (16), who demonstrated that GADD45 induction by IR sensitises medulloblastoma cells to radiation treatment. Understanding the role of GADD45 can help develop better therapeutic strategies to improve cancer treatment outcomes.
Cyclin dependent kinase inhibitor 1A
The CDKN1A gene is responsible for encoding a cyclin dependent kinase inhibitor and serves as a regulator for the progression of the cell cycle during the G1 phase. The expression of CDKN1A gene is governed by the p53 protein and plays a critical role in the cellular response to DNA damage, including damage caused by IR. P53 becomes activated in response to ionizing radiation exposure. This leads to overexpression of CDKN1A that triggers cell cycle arrest, particularly at the G1 phase, preventing cells with damaged DNA from proliferating. If the damage has not been resolved or repaired, CDKN1A gene promotes apoptosis and cellular senescence to eliminate damaged cells that could potentially cause cancer (56). The transcription of the CDKN1A gene reaches its peak 4 h after radiation exposure, while the DDB2 gene, also crucial for DNA damage repair, peaks at 24 h post-irradiation (18).
DDB2 works in synergy with CDKN1A, and usually becomes overexpressed following DDB2 in response to IR induced DNA damage to enhance nucleotide excision repair pathway to initiate the repair of DNA lesions (57). Elevated levels of CDKN1 and functional p53 are linked to increased cell cycle arrest and apoptosis, which help eliminate cancer cells and result in greater sensitivity to radiation therapy. On the other hand, upregulation in DDB2 gene may facilitate the repair of radiation-induced DNA damage, leading to resistance in treatment response and aggressiveness in melanoma cancer (58). Amundson et al. (41) demonstrated that CDKN1A mRNA levels, along with mRNA for discoidin domain receptor tyrosine kinase 2 (DDR2), xeroderma pigmentosum C (XPC), tumour necrosis factor- related apoptosis-inducing ligand (TRAIL receptor 2), four and a half LIM Domain protein 2 (FHL2), cyclin G and other cyclin proteins, peak between 12 and 24 h post-irradiation (59–62). This suggests that CDKN1A may work in conjunction with other cell cycle regulators in response to IR. Understanding the interactions among these genes can help develop more effective therapeutic strategies.
Checkpoint proteins such as CHK1 and CHK2 are crucial for the cellular response to IR, impacting DNA repair, cell cycle regulation, and cancer treatment outcomes. They transmit signals from ATM and ATR to facilitate DNA repair (Figure 2). CHK1 is typically active during the synthesis (S) and G2 phases of the cell cycle in response to DNA damage caused by IR (63). CHK1 and CHK2 contribute to the intrinsic resistance mechanisms against radiotherapies that damage DNA. Inhibiting their activities can help develop more effective therapeutic strategies by sensitizing tumours to radiation and preventing the repair of radiation-induced DNA damage.
Protein biomarkers
Proteins, which are polymers of amino acids linked by peptide bonds, can serve as biomarkers indicating DNA damage from environmental factors like IR. These protein biomarkers operate at the post-translational level, involving modifications that occur after protein synthesis. Such post-translational modifications (PTMs) can greatly influence protein function, localisation, stability, and interactions. Proteins can become overexpressed in response to IR, influencing oncogenes and tumor suppressors, and affecting cell growth and survival pathways. These proteins can serve as biomarkers for predicting cancer development, progression, and treatment sensitivity or resistance Figure 2. Identifying and understanding these biomarkers in the context of IR and cancer can enhance our knowledge of how IR impacts cancer and aid in predicting radiosensitivity, optimizing radiation therapy, and improving treatment outcomes.
C-reactive protein
C-reactive protein (CRP) is a protein marker produced by the liver in response to inflammation and severe tissue damage (64). CRP levels increase with both the dose and duration of radiation exposure (64). For instance, blood CRP levels rose in 30 rhesus monkeys exposed to 1–8.5 Gy of gamma cobalt-60 radiation (19). A borderline significant increase in CRP levels was observed in radiological technologists who had undergone prior radiation therapy, depending on the dose (63). Similarly, CRP levels in peripheral blood mononuclear cells of mice increased progressively with the dose and time after total body irradiation with 1–7 Gy of gamma radiation (64).
Studies have shown that radiotherapy, which involves high doses of IR, is associated with increased CRP levels and a higher risk of inflammatory diseases (28). Radiation pneumonitis, an inflammatory clinical outcome and a dose-limiting toxicity of radiation therapy, is a side effect of both chemotherapy and radiation therapy. In a separate study, elevated CRP levels were detected in association with inflammatory conditions, including autoimmune diseases (such as hypothyroidism, hyperthyroidism, rheumatoid arthritis, type-1 diabetes, and Crohn's disease) and non-autoimmune diseases (such as type-2 diabetes and osteoarthritis), suggesting that CRP is a marker of the risk of developing an inflammatory condition following radiation therapy (28).
Research has also demonstrated that elevated CRP levels are observed in response to inflammation resulting from IR exposure. This suggests an activation of inflammatory responses and chronic inflammation that could lead to carcinogenesis (65). For instance, a strong correlation between abnormal CRP levels and IR was reported in both breast and prostate cancers (21, 22). Furthermore, high CRP levels have been linked to increased risks of breast, lung, and colorectal cancers (66).
CRP contributes to cancer development by fostering an inflammatory environment, promoting tumour growth and survival, and modulating immune responses. Elevated CRP levels can indicate cancer risk and progression. Chronic inflammation from high CRP levels supports tumour growth, angiogenesis, and metastasis (66, 67). CRP also enhances tumour cell proliferation and survival by protecting them from therapy-induced apoptosis (67). In its monomeric form (mCRP), CRP activates inflammatory mechanisms by interacting with cell membranes and immune cells, aiding initial defence against tumours and creating a carcinogenic environment (68). High baseline CRP levels in healthy individuals are linked to increased future cancer risk, making CRP a potential biomarker for cancer risk and progression (69).
Monocyte chemotactic protein 1
Monocyte chemotactic protein 1 (MCP-1), is a chemokine essential for the immune system, recruiting monocytes to areas of damage, inflammation, and tumours (23). When the body is exposed to ionizing radiation, MCP-1 is activated to help repair the damage by attracting immune cells to the affected site, contributing to a pro-inflammatory environment that supports tumour growth, progression, and metastasis (23, 24). MCP-1 initiates a proinflammatory response by activating signalling pathways like NF-κB, which are involved in inflammation and can drive tumour progression (25). It has a dual role, capable of either promoting or inhibiting tumour growth depending on the tumour microenvironment. While MCP-1 can support tumour progression by recruiting TAMs and encouraging angiogenesis, it can also boost anti-tumour immune responses under certain conditions (23).
Production of MCP-1 can also be induced in response to radiation therapy for cancer treatment, potentially leading to an inflammatory environment that supports tumour survival and resistance to therapy (23). For example, radiation-induced MCP-1 has been shown to correlate with lung toxicity and inflammation, complicating treatment outcomes in patients with non-small cell lung cancer (NSCLC) (70). Additionally, MCP-1 has been implicated in breast cancer metastasis, particularly to the lungs and brain (26).
To mitigate the effects of MCP-1 during radiation therapy, several strategies can be employed. Anti-inflammatory drugs like corticosteroids can reduce inflammation and MCP-1 levels, minimising the pro-inflammatory environment that supports tumour growth. Specific chemokine inhibitors targeting MCP-1 or its receptor, CCR2, can block its activity and reduce immune cell recruitment to the tumour site (71). Radiation techniques such as fractionated radiation therapy (delivering smaller, frequent doses) and targeted techniques like stereotactic radiosurgery (SRS) and intensity-modulated radiation therapy (IMRT) can minimise MCP-1 production and spare healthy tissue (72). Combining radiation with immunotherapy or chemotherapy agents that have anti-inflammatory properties can enhance anti-tumour responses and mitigate MCP-1's effects. Additionally, maintaining a balanced diet rich in anti-inflammatory foods and engaging in regular, moderate exercise can help manage inflammation and improve overall immune function during radiation therapy.
Serum amyloid A
Serum amyloid A (SAA) is a vital biomarker for both x-ray and γ-ray radiation exposure, as well as cancer progression. Its levels can reflect the extent of radiation-induced inflammation and provide insights into cancer prognosis and treatment responses. SAA levels in the blood can significantly increase in response to IR, such as x-rays and γ-rays (73). As part of the body's acute phase response to IR damage, SAA triggers inflammation. Studies have shown that SAA levels can rise 10–100 times shortly after IR exposure, with levels remaining elevated depending on the severity of inflammation (61). In a study by Huang et al. (20), the expression of SAA during early and late radiation-induced inflammation was assessed in mice exposed to various doses of radiation (1, 2, 4, 8, and 12 Gy) at different time points. It was found that SAA levels moderately increased at 6 h post-irradiation, peaked at 12 h across all doses, and further increased between days 5 and 7 (20). Additionally, significant increases in SAA levels were observed 24 h after total body irradiation with 1–8 Gy of x-rays. These findings suggest that SAA levels increase in a dose-dependent manner following radiation exposure, indicating its potential as a biomarker for assessing radiation exposure and the resulting inflammatory response (73).
SAA plays roles in high-density lipoprotein remodelling, lipid metabolism, antibacterial infection, tumour pathology, and immune regulation (20). Elevated SAA levels often correlate with poor clinical outcomes in various cancers, including renal cell, lung, breast, ovarian, and prostate cancers (19, 74). Aberrant SAA levels drive inflammation, leading to cell proliferation, cancer progression, angiogenesis, and metastasis. While the clinical significance of radiation-induced protein markers in cancer is generally limited, SAA is gaining recognition in the context of lung cancer treatment with radiation therapy. In lung cancer, high SAA levels have been identified as a predictive biomarker for patients at risk of developing radiation pneumonitis following radiation therapy (27).
Interleukins
Interleukin (IL) proteins are part of the cytokine superfamily, consisting of 38 different types of ILs, and they facilitate interactions between cells (17). These cytokines interact with various elements, including cancer stem cells, epithelial-mesenchymal transition (EMT), and miRNAs, during tumorigenesis. Among them, IL-6 is the most well-recognized for its role in cellular functions. IL-6 is a multifunctional, pleiotropic cytokine in the IL-6 family, involved in the growth and differentiation of B and T lymphocytes. It has been shown to transform human mammospheres and pre-malignant mammary epithelial cells in vitro, making them tumorigenic in vivo (17).
In the context of IR, IL-6 has been implicated in cancer progression and resistance to treatment by reducing oxidative stress and DNA damage (75). Breummer et al. (18) reported a significant increase in IL-6 following radiation exposure of myeloid cells at doses below 20 Gy. Upregulation of IL-6 enhances the mobility and tumorigenesis of breast cancer epithelial cells in response to IR. In mouse mammary glands, IL-6 is frequently expressed after IR exposure and is produced by IR-senescent fibroblasts. Additionally, epithelial cells, primary human mammospheres, and pre-malignant mammary epithelial cell lines show increased IL-6 expression following IR exposure. Elevated IL-6 levels are linked to the radiation response in prostate cancer, glioblastoma, liver cancer, and lung cancer, highlighting its significant role in cancer therapy (76, 77).
Other serum-based protein markers may include interleukin-22, insulin-like growth factor binding protein-1 (IGFBP-1), IGFBP-3, insulin-like growth factor 1 (IGF-1) and leukaemia inhibitor factor (LIF) (32). For instance, Wei et al. (32) demonstrated an aberrant accumulation of interleukin-22, IGFBP-1, IGFBP-3, IGF-1 and LIF in the sera of 2-month-old mice irradiated through exposure with carbon atoms which contain an energy of 80 MeV at a rate of 0.25 Gy per min (32), indicating their potential roles as biomarkers in IR exposure.
Salivary Alpha Amylase
Salivary Alpha Amylase (AMY1) is crucial in the context of radiation exposure, with abnormal levels linked to cancer and its treatment. Stress typically activates the sympathetic nervous system, releasing stress hormones like adrenaline and cortisol, which in turn stimulate the production and release of AMY1 in saliva (29).
AMY1 responds to stress in different ways depending on the stress level. During acute stress, an immediate “fight-or-flight” response occurs, leading to a rapid increase in AMY1 levels in saliva as part of the first line of immune defence (29). Conversely, prolonged or chronic stress, which affects digestion and immune function, results in sustained elevated levels of AMY1 (29).
Recognised as a stress marker, AMY1 levels can become irregular due to radiation therapy. For instance, radiation therapy for head and neck cancer has been shown to alter AMY1 levels and salivary gland function. Enzyme activity assays in patients undergoing radiation therapy for neck and head cancer demonstrated that radiation-induced alterations in AMY1 lead to changes in saliva production (30). Elevated levels of circulating AMY1 have been linked to a higher risk of lung, breast, ovarian, and gastric cancers, significantly associated with increased cell proliferation and metastasis (31). The simultaneous activation of the phosphatidylinositol 3-kinase (PI3 K)/protein kinase B (AKT) and mitogen-activated protein kinases (MAPK) signalling pathways by AMY1 fosters a robust environment for tumour growth, resistance to apoptosis, and metastasis (78, 79). In certain cancers, AMY1 is thought to have antiproliferative effects that inhibit tumour cell growth by regulating various pathways. It can block signalling pathways that promote cancer cell proliferation, thereby slowing or halting their growth and inducing apoptosis (79). AMY1 also modifies the tumour microenvironment, making it less favourable for cancer cell survival by altering cytokine and growth factor levels (79). Furthermore, AMY1 activates pathways such as PI3K/Akt and MAPK, which help suppress tumour growth and metastasis (79). Therefore, AMY1 could serve as a potential biomarker for evaluating the impact of radiation therapy on salivary glands and overall stress levels in patients. Understanding AMY1's role in cancer and radiation exposure might lead to new therapeutic strategies to alleviate side effects and improve patient outcomes.
Murine double minute 2
At the post-translational level, MDM2 also serves as a crucial protein biomarker involved in the cellular response to IR exposure, potentially contributing to cancer development and metastasis. When cells are exposed to IR, MDM2 expression increases in a p53-dependent manner, helping manage the cell's response to DNA damage. MDM2 promotes cell survival by limiting p53's apoptotic function during IR exposure.
As the primary negative regulator of the p53 tumour suppressor protein, MDM2 inhibits p53's activity by promoting its ubiquitination and degradation. This inhibition prevents p53 from inducing cell cycle arrest and apoptosis in response to IR, leading to tumour formation through the proliferation of damaged cells. Inhibiting MDM2 can enhance the sensitivity of cancer cells to radiation, improving the effectiveness of radiotherapy.
Additionally, MDM2 acts as an oncogene, with its overexpression driving cancer progression by promoting cell proliferation and survival. It also contributes to metastasis and tumour growth by enhancing the invasive capabilities of cancer cells and their spread to other parts of the body.
Furthermore, MDM2 can alter the tumour immune microenvironment, helping cancer cells evade immune detection and destruction, complicating the immune system's ability to combat cancer. High levels of MDM2 are linked to resistance to therapies such as chemotherapy and immunotherapy. Targeting MDM2 with specific inhibitors is being explored to overcome this resistance and improve treatment outcomes.
Apolipoprotein-E
The apolipoprotein-E (APOE) protein, encoded by the APOE gene, is crucial for lipid metabolism, regulating the clearance of lipoproteins from plasma and transporting lipids to various tissues and cells (64). APOE also plays a role in the body's response to IR, managing the inflammatory response and aiding in the repair and recovery of neural injuries in the central nervous system (80–83). Studies have shown that APOE can influence behavioural impairment following radiation exposure (82, 83). For example, a study by Higuchi et al. (82) found that both APOE knockout and wild-type mice experienced impaired motor coordination and stamina after receiving 2 Gy of total body irradiation. While these effects resolved in wild-type mice by 60 days post-irradiation, they persisted in knockout mice (82). Additionally, knockout mice showed reduced exploratory activity up to 186 days post-treatment, unlike wild-type mice. These findings highlight the role of APOE in the recovery and repair of radiation-induced injury in the central nervous system (82).
Overexpression of APOE has been observed in brain regions like the prefrontal cortex, amygdala, and hippocampus of rhesus macaques in response to radiation therapy, suggesting a role in the brain's response to radiation (83). APOE expression varies among tumor types and cancer cell lines, and it has been shown to have prognostic value and influence treatment outcomes in cancers such as lower-grade glioma, kidney renal clear cell carcinoma, and kidney renal papillary cell carcinoma (84).
Phosphorylated H2A Histone Family Member X
Phosphorylated H2A Histone Family Member X (γH2AX) is a modified protein, and a recognized marker of DNA double-strand break (DSB) damage caused by radiation, playing a crucial role in cancer research and treatment (21). Indirect ionisation of oxide species generates free radicals, often leading to cell death from IR exposure by breaking down the DNA backbone and creating DSBs. These breaks result in the phosphorylation of histone H2A (H2AX) on serine 139, forming γH2AX, an early indicator of DNA damage post-IR exposure (21).
γH2AX acts as a docking site for various DNA repair proteins at DSB sites in cells exposed to IR. Proteins such as MDC1, 53BP1, and BRCA1 are crucial for making DNA DSBs accessible to the repair machinery. The formation of IR-induced γH2AX activates ATM, ATR, and DNA-PK, which phosphorylate H2AX to enhance the DNA damage signal and coordinate the repair process (21, 22, 85). In this context, ATM is the primary mediator, activated by autophosphorylation at Serine 1981. ATR phosphorylates H2AX during SSBs and DNA replication, while DNA-PK does so during DNA fragmentation and under hypertonic conditions (86). Additionally, the DNA damage response mechanisms are triggered, leading to cell cycle arrest, allowing the cell to repair the damage, survive, and maintain genomic integrity before division.
Abnormal levels of γH2AX may be linked to genomic instability, potentially leading to genetic mutations implicated in cancer development, progression, and treatment outcomes. Human skin cells exposed to radiation (4 Gy of 6 MeV electrons) showed elevated γH2AX levels and increased 53BP1 foci, indicating DNA damage. This damage, including single and double-strand breaks, was observed in various skin cell types and persisted for weeks, leading to fibrosis, a hallmark of cancer development. Additionally, higher γH2AX levels were noted in triple-negative breast cancer cell lines compared to non-triple-negative ones when irradiated with 10 Gy, with γH2AX present in all 54 breast cancer cell lines tested (87, 88). Mice exposed to gamma radiation for 24 h showed elevated γH2AX levels, strongly correlating with unrepaired DNA DSBs observed via γH2AX foci and radiogenic lung cancer. While increased γH2AX may act as a tumour suppressor and promote cell cycle arrest and senescence in premalignant lesions, it is often associated with poor prognosis and tumour aggressiveness (33). Nonetheless, γH2AX is a valuable biomarker for detecting and assessing IR-induced DNA damage and response to cancer radiation therapy.
Vascular endothelial growth factor (VEGF)
VEGF is a vital protein that promotes the formation of new blood vessels, especially in response to IR to repair damaged vessels. IR generates oxidative stress by producing reactive oxygen species (ROS) and free radicals, which activate signalling pathways such as the epidermal growth factor receptor (EGFR). This activation triggers downstream pathways, including MAPK, resulting in increased VEGF production.
Although VEGF itself does not directly cause cancer, its induction by IR can contribute to the progression and aggressiveness of existing tumours by providing them with essential nutrients and oxygen (34). For instance, radiation-induced hypoxia, caused by damaged blood vessels within tumours, can lead to increased VEGF production through the activation of hypoxia-inducible factor 1-alpha (HIF-1α) (35). HIF-1α is a transcription factor that stabilizes and activates under hypoxic conditions, promoting VEGF production to form new blood vessels necessary for supplying the tumour with oxygen and nutrients (35). VEGF can also bind to VEGF receptors 1 and 2 on the surface of newly formed blood vessels, activating several additional signalling pathways that stimulate cell proliferation, migration, and survival. This process can potentially lead to tumour progression and treatment resistance by allowing new blood vessels to repair radiation-induced damage and support tumour regrowth (89).
However, pharmacologically blocking VEGF has been shown to improve patient outcomes by reducing angiogenesis and enhancing tumour oxygenation. This involves using anti-VEGF agents like bevacizumab in combination with radiation therapies. This approach reduces VEGF activity, leading to decreased angiogenesis and improved effectiveness of radiation therapy against tumour cells (90).
In summary, VEGF plays a dual role in cancer treatment with IR. It promotes blood vessel formation and can lead to tumour resistance. However, using anti-VEGF therapies can enhance radiation treatment effectiveness and improve cancer control.
Limitations
Identifying biomarkers for radiation exposure that are applicable across various scenarios and types of radiation is challenging. Despite numerous studies over the years, no biomarkers have been specifically validated for radiation exposure alone. Additionally, the lack of data from past nuclear attacks or accidents, which needs to be collected over a specific period following radiation exposure, has significantly hindered progress in researching potential biomarkers. Most studies to date have focused on the effects of whole-body radiation exposure, with very few examining the irradiation of specific body parts. Therefore, the effects of partial body exposure also need to be investigated. Extensive and innovative research is required to develop effective biomarkers for radiation exposure.
Conclusion and future research opportunities
IR seems to influence various genes and transcriptional regulators that control the cell cycle in ways that are linked to cancer development, progression and resistance to treatment. While there are safety measures in place to manage occupational IR exposure, it is crucial to understand the genomic effects that may contribute to diseases, particularly cancer and treatment response. This understanding will help ensure that additional safety and therapeutic measures can be implemented if needed.
Assessing biomarkers after IR exposure has significant potential for cancer treatment and prognosis. These biomarkers can help design compounds that enhance radiosensitivity, support DNA repair, and provide radioprotection. They can also evaluate the risk of cancer development in organs and tissues at different IR doses. Advanced technologies like next-generation sequencing are needed to identify new biomarkers on a genome-wide scale, aiding in cancer prevention and therapy. Understanding IR-induced organ and tissue injury is crucial for managing cancer patients, and developing models to measure IR exposure is essential for monitoring its long-term effects in clinical research.
Author contributions
YM: Writing – original draft, Writing – review & editing. SaM: Writing – original draft, Writing – review & editing. BM: Writing – review & editing. JM: Writing – review & editing. SiM: Writing – review & editing. MS: Writing – review & editing. MK: Conceptualization, Supervision, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Acknowledgments
Figures were created with BioRender.com (Agreement numbers: LB27EKK6WL and AP27B1MHP8).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Kron T, Lehmann J, Greer P. Dosimetry of IR in modern radiation oncology. Phys Med Biol. (2016) 61(14):167. doi: 10.1088/0031-9155/61/14/R167
2. Stephens P, Campbell P. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell. (2011) 144(1):27–40. doi: 10.1016/j.cell.2010.11.055
3. IRCP. IRCP Publication 103: The Recommendations of the International Committee on Radiological Protection (2007).
4. Preston-Martin S, Thomas D, Yu M, Henderson B. Diagnostic radiography as a risk factor for chronic myeloid and monocytic leukaemia. Cancer. (1989) 59(1):639–44. doi: 10.1038/bjc.1989.130
5. Shino P, Chung C, Myrianthopoulos N. Pre-conception radiation, intrauterine diagnostic radiation and childhood neoplasia. Natl Cancer Inst. (1980) 65(1):681–6. doi: 10.1093/jnci/65.4.681
6. Merrifield M, Kovalchuk O. Epigenetics in radiation biology: a new research frontier. Front Genet. (2013) 4:40. doi: 10.3389/fgene.2013.00040
7. Little JB. Radiation carcinogenesis. Carcinogenesis. (2000) 21(3):397–404. doi: 10.1093/carcin/21.3.397
8. Gluzman D, Imamura N, Sklyarenko L, Nadgornaya V, Zavelevich M, Machilo V. Malignant diseases of hematopoietic and lymphoid tissues in chernobyl clean-up workers. Haematol J. (2005) 5(1):565–71. doi: 10.1038/sj.thj.6200568
9. Tsapogas P, Mooney C, Brown G, Rolink A. The cytokine Flt3-ligand in normal and malignant hematopoeis. Int J Mol Sci. (2017) 18(1):1115. doi: 10.3390/ijms18061115
10. Gilliland D, Griffin J. The role of FLT3 in haematopoeisis and leukaemia. Blood. (2002) 100(5):1532–42. doi: 10.1182/blood-2002-02-0492
11. Finnon R, Brown N, Moodie J, Badie C, Olme C, Huiskamp R, et al. Flt3 ITD mutations in a mouse model of radiation-induced acute myeloid leukaemia. Leukaemia. (2012) 26(6):1445–6. doi: 10.1038/leu.2011.377
12. Ge S, Shen X, Wu T, Du J, Dai H, Qiu Q, et al. Mutation spectrum of FLT3 and significance of non-canonical FLT3 mutations in haematological malignancy. Br J Haematol. (2023) 202(3):539–49. doi: 10.1111/bjh.18877
13. Perry M. Mdm2 in the response to radiation. Mol Cancer Res. (2004) 2(1):9–19. doi: 10.1158/1541-7786.9.2.1
14. Maier P, Hartmann L, Wenz F, Herskind C. Cellular pathways in response to ionising radiation and their targetability for tumour radiosensitization. Int J Mol Sci. (2016) 17(1):102. doi: 10.3390/ijms17010102
15. Jen K, Cheung V. Transcriptional response of lymphoblastoid cells to ionizing radiation. Genome Res. (2003) 13(9):2092–100. doi: 10.1101/gr.1240103
16. Asuthkar S, Nalla A, Gondi C, Dinh D, Gujrati M, Mohanam S, et al. Gadd45a sensitizes medulloblastoma cells to irradiation and suppresses MMP-9-mediated EMT. J Neuro-Oncol. (2011) 13(10):1059–73. doi: 10.1093/neuonc/nor109
17. Anstrom G. Techniques to measure DNA single-strand breaks in cells: a review. Int J Radiat Biol. (1988) 54(5):695–707. doi: 10.1080/09553008814552151
18. Bruemmer D, Yin F, Liu J, Berger JP, Sakai T, Blaschke F, et al. Regulation of the growth arrest and DNA damage-inducible gene 45 (GADD45) by peroxisome proliferator-activated receptor gamma in vascular smooth muscle cells. Circ Res. (2003) 93(4):e38–47. doi: 10.1161/01.RES.0000088344
19. Ryu J, Jung I, Park E, Kim K, Yeom J. Radiation-induced c-reactive protein triggers apoptosis of vascular smooth muscle cells through ros interfering with the stat3/ref-1 complex. J Cell Mol Med. (2022) 26(7):2104–18. doi: 10.1111/jcmm.17233
20. Huang J, Qi Z, Chen M, Xiao T, Guan J, Zhou M. Serum amyloid a1 as a biomarker for radiation dose estimation and lethality prediction in irradiated mouse. Ann Transl Med. (2019) 7(23):715. doi: 10.21037/atm.2019.12.27
21. Pernot E, Hall J, Baatout S, Benotmane M, Blanchardon E, Bouffler S, et al. IR biomarkers for potential use in epidemiological studies. Mutat Res. (2012) 751(1):258–86. doi: 10.1016/j.mrrev.2012.05.003
22. Podhorecka M, Skladanowski A, Bozko P. H2AX phosphorylation: its role in DNA damage response and cancer therapy. J Nucleic Acids. (2020) 2010(1):1–9. doi: 10.4061/2010/920161
23. Yoshimura T. The chemokine MCP-1 (CCL2) in the host interaction with cancer: a foe or ally? Cell Mol Immunol. (2018) 15(4):335–45. doi: 10.1038/cmi.2017.135
24. Yoshimura T, Li C, Wangm Y, Matsukawa A. The chemokine monocyte chemoattractant protein-1/CCL2 is a promoter of breast cancer metastases. Cell Mol Immunol. (2023) 20(7):714–38. doi: 10.1038/s41423-023-01013-0
25. Schiller K, Zilhardt M, Alley J. Secretion of MCP-1 and other pancrine factors in a novel tumour-bone coculture model. BMC Cancer. (2009) 9(1):45. doi: 10.1186/1471-2407-9-45
26. Lhuillier C, Rudqvist N, Elemento O. Radiation therapy and anti-tumour immunity: exposing immunogenic mutations to the immune system. Genome Med. (2019) 11(1):40. doi: 10.1186/s13073-019-0653-7
27. Wang Y, Chang H, Chang Y, Huang S, Ko H, Chang C, et al. Serum amyloid a as a predictive marker for radiation pneumonitis in lung cancer patients. Int J Radiat Oncol Biol Phys. (2012) 85(3):791–7. doi: 10.1016/j.ijrobp.2012.06.018
28. Lee E, Nelson O, Puyana C, Takita C, Wright J, Zhao W, et al. Association between c-reactive protein and radiotherapy-related pain in a tri-racial/ethnic population of breast cancer patients: a prospective cohort study. Breast Cancer Res. (2019) 21(1):1–11. doi: 10.1186/s13058-018-1086-8
29. Ali N, Nater UM. Salivary alpha-amylase as a biomarker of stress in behavioral medicine. Int J Behav Med. (2020) 27(3):337–42. doi: 10.1007/s12529-019-09843-x
30. Carpenter D, Mitchell L, Armour J. Copy number variation of human AMY1 is a minor contributor to variation in salivary amylase expression and activity. Hum Genomics. (2017) 11(1):2. doi: 10.1186/s40246-017-0097-3
31. Zhou J, Sheng J, Fan Y, Zhu X, Tao Q, He Y, et al. Association between serum amyloid A levels and cancers: a systematic review and meta-analysis. Postgrad Med J. (2018) 49(1115):499–507. doi: 10.1136/postgradmedj-2018-136004
32. Wei W, Bai H, Feng X, Hua J, Long K, He J, et al. Serum proteins as new biomarkers for whole-body exposure to high- and low-LET ionizing radiation. Dose Response. (2020) 18(1):1559325820914172. doi: 10.1177/1559325820914172
33. Ochola D, Sharif R, Bedfordd J, Keefe T, Kato T, Fallgreen C, et al. Persistence of gamma-H2AX foci in bronchial cells correlates with susceptibility to radiation associated lung cancer in mice. Radiat Res. (2019) 191(1):67–75. doi: 10.1667/RR14979.1
34. Patel SA, Nilsson MB, Le X, Cascone T, Jain RK, Heymach JV. Molecular mechanisms and future implications of VEGF/VEGFR in cancer therapy. Clin Cancer Res. (2023) 29(1):30–9. doi: 10.1158/1078-0432.CCR-22-1366
35. Bertout JA, Patel SA, Simon MC. The impact of O2 availability on human cancer. Nat Rev Cancer. (2008) 8(12):967–75. doi: 10.1038/nrc2540
36. Marechal A, Zou L. DNA damage sensing by the ATM and ATR kinases. Cold Spring Harbor Perspect Biol. (2013) 5(9):1–17. doi: 10.1101/cshperspect.a012716
37. Clay D, Fox D. DNA damage responses during the cell cycle: insights from model organisms and beyond. Genes (Basel). (2021) 12(12):1882. doi: 10.3390/genes12121882
38. Wu Y, Song Y, Wang R, Wang T. Molecular mechanisms of tumour resistance to radiotherapy. Mol Cancer. (2023) 22(1):96. doi: 10.1186/s12943-023-01801-2
39. Xing J, Stea B. Molecular mechanisms of radiosensitivity and resistance to radiotherapy. Clin Exp Metastasis. (2024) 41(1):517–24. doi: 10.1007/s10585-023-10260-4
40. Syljuåsen RG. Cell cycle effects in radiation oncology. In: Wenz F, editor. Radiation Oncology. Cham: Springer (2019). p. 1–8. doi: 10.1007/978-3-319-52619-5_101-1
41. Amundson S, Do K, Shahab S, Bittner M, Meltzer P, Trent J, et al. Identification of potential mrna biomarkers in peripheral blood lymphocytes for human exposure to IR. Radiat Res. (2000) 154(1):342–6. doi: 10.1667/0033-7587(2000)154[0342:IOPMBI]2.0.CO;2
42. Mendrysa S, McElwee M, Michalowski J, O'Leary K, Young K, Perry M. Mdm2 is critical for inhibition of p53 during lymphopoiesis and the response to ionizing irradiation. Mol Cell Biol. (2003) 23(2):462–72. doi: 10.1128/MCB.23.2.462-473.2003
43. Ozaki T, Nakagawara A. Role of p53 in cell death and human cancers. Cancers (Basel). (2011) 3(1):994–1013. doi: 10.3390/cancers3010994
44. Ghandhi S, Smilenov L, Shuryak I, Pujol-Canadell M, Amundson S. Discordant gene responses to radiation in humans and mice and the role of hematopoietically humanized mice in the search for radiation biomarkers. Sci Rep. (2019) 9(1):19434. doi: 10.1038/s41598-019-55982-2
45. Haupt Y, Maya R, Kazaz A, Oren M. Mdm2 promotes the rapid degradation of p53. Nature. (1997) 387(6630):296–9. doi: 10.1038/387296a0
46. Bouvard V, Zaitchouk T, Vacher M, Duthu A, Canivet M, Choisy-Rossi C. Tissue and cell-specific expression of the p53-target genes: bax, fas, mdm2 and waf1/p21, before and following ionising irradiation in mice. Oncogene. (2000) 19(5):649–60. doi: 10.1038/sj.onc.1203366
47. Okazaki R. Role of p53 in regulating radiation responses. Life. (2022) 12(7):1099–104. doi: 10.3390/life12071099
48. Miles X, Vandevoorde C, Hunter A, Bolcaen J. MDM2/X inhibitors as radiosensitizers for glioblastoma targeted therapy. Front Oncol. (2021) 11:703442. doi: 10.3389/fonc.2021.703442
49. Tamura R, de Vasconcellos J, Sarkar D, Libermann T, Fisher P, Zerbini L. Gadd45 proteins: central players in tumorigenesis. Curr Mol Med. (2012) 12(5):634–51. doi: 10.2174/156652412800619978
50. Budworth H, Snijders A, Marchetti F, Mannion B, Bhatnagar S, Kwoh E. DNA repair and cell cycle biomarkers of radiation exposure and inflammation stress in human blood. PLoS One. (2012) 7(11):48619. doi: 10.1371/journal.pone.0048619
51. Snyder A, Morgan W. Gene expression profiling after irradiation: clues to understanding acute and persistent responses? Cancer Metastasis Rev. (2004) 23(3-4):259–68. doi: 10.1023/B:CANC.0000031765.17886.fa
52. Hildesheim J, Fornace AJ Jr. Gadd45a: an elusive yet attractive candidate gene in pancreatic cancer. Clin Cancer Res. (2002) 8(8):2475–9.12171872
53. Yamasawa K, Nio Y, Dong M, Yamaguchi K, Itakura M. Clinicopathological significance of abnormalities in Gadd45 expression and its relationship to p53 in human pancreatic cancer. Clin Cancer Res. (2002) 8(8):2563–9.12171884
54. Smirnov D, Brady L, Halasa K, Morley M, Solomon S, Cheung V. Genetic variation in radiation-induced cell death. Genome Res. (2012) 22(2):332–9. doi: 10.1101/gr.122044.111
55. Zhang X, Wang C, Zhou C, Liu G, Wei F, Sun Z. Overexpression of Gadd45a enhances radiotherapy efficacy in humans Tca8113 cell line. Acta Pharmacol Sin. (2011) 32(2):253–8. doi: 10.1038/aps.2010.208
56. Oulette S, Vigneault F, Lessard M, Leclerc S, Drouin R, Guerin S. Transcriptional regulation of the cyclin-dependent kinase inhibitor 1A (p21) gene by NFI in proliferating human cells. Nucleic Acids Res. (2006) 34(22):6472–87. doi: 10.1093/nar/gkl861
57. Bao N, Han J, Zhou H. A protein with broad functions: damage-specific DNA-binding protein. Mol Biol Rep. (2022) 49(1):12181–92. doi: 10.1007/s11033-022-07963-4
58. Al Hmada Y, Brodell R, Kharouf N, Flanagan T, Alamodi A, Hassan SL, et al. Mechanisms of melanoma progression and treatment resistance: role of cancer stem-like cells. Cancers (Basel). (2024) 16(2):470. doi: 10.3390/cancers16020470
59. Kang C, Park K, Song J, Jeoung D, Cho C, Kim T. Possible biomarkers for IR exposure in human peripheral blood lymphocytes. Radiat Res. (2003) 159(1):312–9. doi: 10.1667/0033-7587(2003)159[0312:PBFIRE]2.0.CO;2
60. Jeng Y, Hsu H. Mutation of the DR5/TRAIL receptor 2 gene is infrequent in hepatocellular carcinoma. Cancer Lett. (2002) 181(2):205–8. doi: 10.1016/S0304-3835(02)00051-4
61. Cao C, Mok S, Cheng V, Tsui S. The FHL2 regulation in the transcriptional circuitry of human cancers. Gene. (2015) 572(1):1–7. doi: 10.1016/j.gene.2015.07.043
62. Dupont CA, Dardalhon-Cuménal D, Kyba M, Brock HW, Randsholt NB, Peronnet F. Drosophila Cyclin G and epigenetic maintenance of gene expression during development. Epigenetics Chromatin. (2015) 8:18. doi: 10.1186/s13072-015-0008-6
63. Bartek J, Lukas J. Chk1 and Chk2 kinases in checkpoint control and cancer. Cancer Cell. (2003) 3(5):421–9. doi: 10.1016/S1535-6108(03)00110-7
64. Ossetrova NI, Blakely WF. Multiple blood-proteins approach for early-response exposure assessment using an in vivo murine radiation model. Int J Radiat Biol. (2009) 85(10):837–50.19863200
65. Sproston N, Ashworth J. Role of c-reactive protein at sites of inflammation and infection. Front Immunol. (2018) 9(1):754. doi: 10.3389/fimmu.2018.00754
66. Zhu M, Ma Z, Zhang X. C-reactive protein and cancer risk: a pan-cancer study of prospective cohort and Mendelian randomisation analysis. BMC Med. (2022) 20(1):301. doi: 10.1186/s12916-022-02506-x
67. Aarstad H, Guobrandsdottir G, Hjelle K, Bostad L, Bruserud O, Tvedt T, et al. The biological context of c-reactive protein as a prognostic marker in renal cell carcinoma: studies on the acute phase cytokine profile. Cancers (Basel). (2020) 12(7):1961. doi: 10.3390/cancers12071961
68. Hart P, Rajab I, Alebraheem M, Potempa L. C- reactive protein and cancer: diagnostic and therapeutic insights. Front Immunol. (2020) 11(1):595835. doi: 10.3389/fimmu.2020.595835
69. O’Brian D, Prunty M, Hill A, Shoag J. The role of c-reactive protein in kidney, bladder and prostate cancer. Front Immunol. (2021) 12(1):721989. doi: 10.3389/fimmu.2021.721989
70. Siva S, MacManus M, Kron T, Best N, Smith J, Lobachevsky P. A pattern of early radiation induced inflammatory cytokine expression is associated with lung toxicity in patients with non-small cell lung cancer. PLoS One. (2014) 9(10):e109560. doi: 10.1371/journal.pone.0109560
71. Moraes F, Shultz D, Murphy E, Khuntia D, Chao S, Suh J. Strategies to mitigate the effects of whole-brain radiation therapy on neurocognitive function in patients with brain metastases. Appl Radiat Oncol. (2017) 6(1):6–10. doi: 10.37549/ARO1119
72. Chaulin A. The essential strategies to mitigate cardiotoxicity caused by docorubicin. Life. (2023) 13(1):2148. doi: 10.3390/life13112148
73. Chen R, Chen O, Zheng J, Zeng Z, Chen M, Li L, et al. Serum amyloid protein A in inflammatory bowel disease: from bench to bedside. Cell Death Discov. (2023) 9(1):154. doi: 10.1038/s41420-023-01455-5
74. Liu C. Serum amyloid A protein in clinical cancer diagnosis. Pathol Oncol Res. (2012) 18(2):117–21. doi: 10.1007/s12253-011-9459-7
75. Yuki T, Genro K, Hiromu M. Acquisition of radioresistance by IL-6 treatment caused by suppressive of oxidative stress derived from mitochondria after gamma radiation. J Radiat Res. (2017) 58(4):412–20. doi: 10.1093/jrr/rrw084
76. Matsouka Y, Nakayama H, Yoshida R, Hirosue A, Nagata M, Tanaka T, et al. IL-6 controls resistance to radiation by suppressing oxidative stress via the Nrf2-antioxidant pathway in oral squamous cell carcinoma. Br J Cancer. (2016) 115(1):1234–44. doi: 10.1038/bjc.2016.327
77. Wu C, Chen M, Chen W. The role of IL-6 in the radiation research of prostate cancer. Radiat Oncol. (2013) 8(1):159. doi: 10.1186/1748-717X-8-159
78. Du Plessis M, Davis T, Olivier D, De Villiers W, Engelbrecht A. A functional role for serum amyloid A in the molecular regulation of autophagy in breast cancer. Front Oncol. (2022) 12(1):1000925. doi: 10.3389/fonc.2022.1000925
79. Anesini CA, Alonso MR, Martino RF. Antiproliferative and cytotoxic activities. In: Sülsen V, Martino V, editors. Sesquiterpene Lactones. Cham: Springer (2018). p. 303–23. doi: 10.1007/978-3-319-78274-4_13
80. Huang Y, Mahley R. Apolipoprotein e: structure and function in lipid metabolism, neurobiology, and Alzheimer’s diseases. Neurobiol Dis. (2014) 72(1):3–12. doi: 10.1016/j.nbd.2014.08.025
81. Liu C, Kanekiyo T, Xu H, Bu G. Apolipoprotein e and Alzheimer disease: risk, mechanisms and therapy. Nat Rev Neurol. (2013) 9(2):106–18. doi: 10.1038/nrneurol.2012.263
82. Higuchi Y, Nelson G, Vazquez M, Laskowitz D, Slater J, Pearlstein R. Apolipoprotein e expression and behavioral toxicity of high charge, high energy (hze) particle radiation. J Radiat Res. (2002) 43(Suppl):S219–S24. doi: 10.1269/jrr.43.S219
83. Kundu P, Zimmerman B, Perez R, Whitlow C, Cline J, Olson J. Apolipoprotein e levels in the amygdala and prefrontal cortex predict relative regional brain volumes in irradiated rhesus macaques. Sci Rep. (2021) 11(1):22130. doi: 10.1038/s41598-021-01480-3
84. Leng X, Liu J, Jin A. Multi-omics analyses reveal function of apolipoprotein E in alterative splicing and tumour immune microenvironment in kidney renal clear cell carcinoma via pan-cancer analysis. Cell Biochem Biophys. (2024) 82(1):1–13. doi: 10.1007/s12013-023-01211-7
85. Banuelos C, Banath J, Kim J, Aquino-Parsons C, Olive P. γH2AX expression in tumours exposed to cisplatin and fractionated irradiation. Clin Cancer Res. (2009) 15(10):3344–53. doi: 10.1158/1078-0432.CCR-08-3114
86. Mah LJ, Orlowski C, Ververis K, Vasireddy RS, El-Osta A, Karagiannis TC. Evaluation of the efficacy of radiation-modifying compounds using γH2AX as a molecular marker of DNA double-strand breaks. Genome Integr. (2011) 2(1):3. doi: 10.1186/2041-9414-2-3
87. Takano S, Shibamoto Y, Wang Z, Kondo T, Hashimoto S, Kawai T, et al. Optimal timing of γH2AX analysis to predict cellular lethal damage in cultured tumour cell lines after exposure to diagnostic and therapeutic radiation doses. J Radiat Res. (2023) 64(2):317–27. doi: 10.1093/jrr/rrac096
88. He Y, Gong Y, Lin J, Chang DW, Gu J, Roth JA, et al. Ionizing radiation-induced γ-H2AX activity in whole blood culture and the risk of lung cancer. Cancer Epidemiol Biomarkers Prev. (2013) 22(3):443–51. doi: 10.1158/1055-9965.EPI-12-0794
89. Kil W, Toflion P, Camphausen K. Post-radiation increase in VEGF enhances glioma cell mortility in vitro. Radiat Oncol. (2012) 7(1):25. doi: 10.1186/1748-717X-7-25
90. Kim Y, Lee H, Kim T, Eisinger-Mathason T, Zhang A, Schmidt B, et al. Overcoming evasive resistance from vascular endothelial growth factor a inhibition in sarcomas by genetic or pharmacologic targeting of hypoxia-inducible factor 1 alpha. Int J Cancer. (2013) 132(1):29–41. doi: 10.1002/ijc.27666
Keywords: alteration, biomarkers, cancer, genes, ionising radiation, proteins, radiation, treatment resistance
Citation: Mzizi Y, Mbambara S, Moetlhoa B, Mahapane J, Mdanda S, Sathekge M and Kgatle M (2024) Ionising radiation exposure-induced regulation of selected biomarkers and their impact in cancer and treatment. Front. Nucl. Med. 4:1469897. doi: 10.3389/fnume.2024.1469897
Received: 24 July 2024; Accepted: 30 September 2024;
Published: 21 October 2024.
Edited by:
Shams A. M. Issa, Al-Azhar University, EgyptReviewed by:
Hesham M. H. Zakaly, Ural Federal University, RussiaJohan K. E. Spetz, University of Gothenburg, Sweden
Copyright: © 2024 Mzizi, Mbambara, Moetlhoa, Mahapane, Mdanda, Sathekge and Kgatle. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mankgopo Kgatle, a2dhdGxlLm1hbmtnb3BvQGdtYWlsLmNvbQ==
†These authors have contributed equally to this work and share first authorship