Department of Psychiatry and Centre for High-Throughput Biology, University of British Columbia, Vancouver, BC, Canada
Numerous studies have been performed to examine gene expression patterns in the rodent hippocampus in the kindling model of epilepsy. However, recent reviews of this literature have revealed limited agreement among studies. Because this conclusion was based on retrospective comparison of reported “hit lists” from individual studies, we hypothesized that re-analysis of the original expression data would help address this concern. In this paper, we reanalyzed four genome-wide expression studies of excitotoxin-induced kindling in rat and performed a statistical meta-analysis. The meta-analysis revealed over 800 genes which show significant change in expression 24 h after initial seizure induction, and 59 genes altered after 10 days. To evaluate our results in light of previous work, we assembled a reference list of genes formed from a consensus of the published literature. Our profiles include most of the genes in this reference list, and most of the additional genes are from pathways or biological processes previously recognized to be altered in kindling. In addition our results emphasized expression changes in lipid metabolism and protein degradation pathways. We conclude that a cautious re-analysis of published expression data can help illuminate genes and pathways underling kindling. Supplementary Material is available at http://www.chibi.ubc.ca/faculty/pavlidis/meta-analysis-of-brain-kindling/
Epilepsy is a common disorder characterized by uncontrolled neuronal activity and behavioral seizures. Because millions of individuals are affected by epilepsy, there is intense interest in understanding how epilepsy develops. For many years researchers have used animal models as a tool for studying the basic mechanisms of epilepsy. In the kindling model, a brief exposure to a neurochemical or other insult results in development of epilepsy with a delay of several days to weeks (McNamara et al., 2006
). As is the case for many areas of neurology, these models have been investigated using high-throughput molecular methods such as gene expression microarrays, often using the hippocampus as the brain region of study (Zagulska-Szymczak et al., 2001
). The main topic of this paper is the combined or meta-analysis of such data sets, in an effort to increase our understanding of the molecular changes occurring in the hippocampus of rodents undergoing kindling protocols.
Recently, Wang et al. (2009)
reviewed 18 expression studies of kindling, using comparisons of the differentially expressed gene lists reported in those studies. They identified 72 genes that were reported as differentially expressed in more than one study, in contrast to nearly 2000 reported in any study. A wide range of biological processes were implicated, including cellular metabolism, immune response and synaptic transmission. Wang et al. (2009)
concluded that this apparent poor agreement between studies was a cause of some confusion in the field, rather than resulting in substantial progress, and recommend that experimental designs be improved. As pointed out by Wang et al. (2009)
, one limitation of such a review was that each study used different criteria for selecting genes. Some of the approaches used in individual studies do not permit accurate estimation of error rates (e.g., genes were selected by “fold-change” alone).
An alternative approach to surveying author-reported “hit lists” is to perform a more formal meta-analysis, in which the results are combined statistically to evaluate changes in expression in each gene (Borenstein et al., 2009
). This would be appropriate for cases where p-values or effect sizes are reported for each gene. Because in general researchers only report such values for positive findings, performing a meta-analysis of gene expression microarray data typically involves reanalyzing the expression data, rather than simply using the published summary statistics. Once one is contemplating reanalyzing all the data, we might ask if the data can be directly combined in a single analysis. Such an analysis should be more powerful, if inter-laboratory variation can be corrected or accounted for. Meta-analysis approaches have been successfully applied to cancer data in the past (Rhodes et al., 2004
; Grützmann et al., 2005
; Chan et al., 2008
), but so far have not been utilized for brain injury data.
In this paper, we describe two different types of combined analysis of kindling in rat hippocampus. We used both a “traditional” meta-analysis method as well as a method for directly combining the data using analysis of variance (ANOVA). We show that in addition to confirming many essential features of the effect of kindling in agreement with Wang et al. (2009) and other review studies, meta-analysis uncovers many differentially expressed genes which were not reported in the original studies (or review articles summarizing expression studies), and which are not statistically significantly expressed in any individual study. We conclude that combined data analysis is a useful tool for making efficient use of accumulated expression data, and these methods can be fruitfully applied to the analysis of epilepsy.
Microarray Datasets
We searched two microarray data repositories, Gene Expression Omnibus (GEO) (Barrett et al., 2007
) and ArrayExpress (Parkinson et al., 2009
), for datasets related to chemically induced seizures/brain-injuries. Focusing our search on rat as experimental animal, we identified only two publicly available datasets. We further performed an exhaustive literature search and identified a number of papers presenting relevant data not in the public repositories, and were able to obtain data directly from the authors for two additional studies. In total we identified two datasets on chemically-induced kindling we considered highly comparable for studying the effects after 24 h, and three studies for examining effects 10 days following treatment. The studies are summarized in Table 1
, and some details about the datasets are given below.
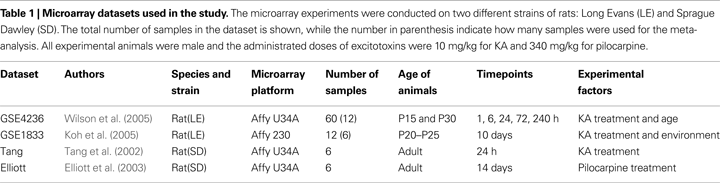
Dataset GSE4236 was described by Wilson et al. (2005)
. The purpose of the study was to compare differences in gene expression after kainic acid (KA) treatment between mature (P30) and immature (P15) rats. The brain samples were taken from hippocampus at five different timepoints after KA administration: 1, 6, 24, 72, and 240 h. There are three biological replicates for each age group, timepoint and treatment (KA and control), resulting in 60 samples all together. For the purpose of our analysis we used only data for P30 rats and for two timepoints, 24 and 240 h.
Dataset GSE1833 (Koh et al., 2005
) was produced for the purpose of studying an effect of the environment on KA-treated rats. Hippocampus samples of P20–P25 old rats were extracted 10 days after KA treatment. There are three biological replicates for each combination of treatment (KA, control) and environment (enriched, control), resulting in 12 samples. We used only samples corresponding to control environment.
Dataset “Tang” was obtained directly from the authors. Tang et al. (2002)
studied gene expression changes after various brain insults: ischemic stroke, intracerebral haemorrhage, kainate-induced seizures, insulin-induced hypoglycemia, and hypoxia. For KA-treated and control animals parietal cortex was used for microarray experiments, which were performed 24 h after the treatment. The dataset contains 27 samples in total of which we used only kainate treated (3) and controls (3).
The final dataset, “Elliott”, was described by Elliott et al. (2003) and was obtained directly from the authors. The study aimed to find genes that are similarly regulated during normal nervous system development and epileptogenesis. The seizures were induced using pilocarpine injection, while control animals were treated with saline. The samples were taken from dentate gyrus 14 days after pilocarpine treatment. The total number of samples is 12 of which we used only pilocarpine treated (3) and controls (3).
The datasets were combined according to matching timepoints: GSE4236 (24 h) and Tang datasets were used for the meta-analysis of gene expression 24 h after KA-treatment and GSE4236 (240 h), GSE1833 and Elliott datasets were combined for a 240+-h timepoint. The probesets for each array design (Affymetrix U34A and 230) were mapped to known and predicted genes using the UCSC GoldenPath annotation database (Hinrichs et al., 2006
) essentially as described by Barnes et al. (2005)
. The 24-h meta-datasets contained expression values from GSE4236 and Tang datasets for all the genes that had specific Affymetrix U34A probe sets (i.e., aligned to only one place in the genome). The 240+-h dataset, which combined datasets from two different platforms, contained the expression values from GSE4236, GSE1833 and Elliott datasets for all the genes that were present on both platforms.
For genes represented by more than one probe set, a final gene expression value was computed by averaging the expression values of all the probesets that mapped to that gene. The original datasets were pre-processed by the authors and we did not do any further processing before assembling the meta-datasets except for log2 transform for the datasets where the expression values were on the linear scale. Finally, the meta-datasets were quantile normalized across all samples to help correct for any scale differences between datasets.
Reference Gene List
For evaluation purposes, we compiled a list of genes that have been previously linked to epileptogenesis or excitotoxic-brain injury by studies of gene expression. We used four review articles (Zagulska-Szymczak et al., 2001
; Lukasiuk and Pitkänen, 2004
; Lukasiuk et al., 2006
; Wang et al., 2009
) that provide lists of relevant genes based on the existing literature. Zagulska-Szymczak et al. (2001)
reviewed the spatio-temporal patterns of gene expression in hippocampus and listed genes found to be induced in all hippocampal subfields after KA injection in previous studies. The list is focused on genes up-regulated within early to immediate time interval after KA injection (usually the first 24 h). Lukasiuk and Pitkänen (2004)
reviewed nine articles that describe use of microarray experiments for studying temporal lobe epilepsy in humans or in animal models. The gene list they assembled contains the genes that are identified as differentially expressed in at least two of the studies. The follow-up study reviewed 15 papers describing global analysis of gene expression in two models of epileptogenic brain insult: status epilepticus and traumatic brain injury (Lukasiuk et al., 2006
). The gene list contains genes that were reported to have altered expression in at least three studies. Finally, the most recent review article (Wang et al., 2009
) addressed global expression studies of mesial temporal epilepsy that arises in hippocampus, parahippocampal gyrus and amygdala. The authors compared results published in 18 different studies that used various epileptogenesis models and methodologies for gene expression analysis and assembled a list of genes reported to be regulated in two or more studies.
Our reference gene list was created by taking a union of these four published gene lists. The number of genes in each original list was 43, 48, 70 and 69, in the order they were described above. There were a number of genes that were present on more than one gene list, partially due to fact that the same papers were reviewed multiple times. A large number of gene symbols in the original lists were outdated and we had to consult sources such as the Rat Genome Database (Twigger et al., 2007
) to resolve identifiers. For gene symbols and names that were ambiguous (referred to more than one gene) we checked the cited primary publications for a more specific reference. There were 14 gene names or symbols which we were not able to resolve.
The final list of genes contained 183 rat genes that have been found to be differentially expressed during epileptogenesis in more than one study. The complete list is given as Supplementary Material. Out of 182 genes, there are 42 that do not have probes on the Affymetrix U34A platform, 40 that do not have probes on the Affymetrix 230 platform, 4 that are mapped by nonspecific probes (which have multiple alignments in the genome) on the Affymetrix U34A platform, and 3 genes for which expression was below background level on the Affymetrix 230 platform. After removing these, there are 136 and 130 genes from the reference list that could be tested for altered gene expression at 24- and 240+-h timepoint, respectively, in the present study.
Meta-Analysis
We used two approaches to analyze data for each timepoint. The first approach, Fisher’s meta-analysis (Fisher, 1925
, 1948
), is a standard, commonly used method that combines results from multiple independent tests all having the same null hypothesis (H0). The method combines p-values resulting from the statistical analysis of individual datasets into one test statistics (F) in the following way:

An important characteristic of Fisher’s test statistic is that under the null hypothesis it has a χ2 distribution and thus p-values for F can be computed from that distribution using 2k degrees of freedom, where k is the number of tests being combined. As usual, H0 is rejected if p-value is less than the selected significance level α, which in our case is 0.05.
For the purposes of our analysis p-values that go into computation of F statistic are computed for each individual dataset using Welch’s two-sided t-test for unequal variance. The null hypothesis tested for each gene in the dataset is that the mean for treated samples is the same as the mean for control samples. Significant p-values (p-value <0.05) indicate that there is differential expression between the two treatments, however, the direction of expression change is not considered. In order to compute meaningful F statistics we can combine p-values only if their corresponding t-statistics, which indicate direction of expression change, are of the same sign. If the t-statistics for a gene are not of the same sign for all of the tests we assign value of 0.1 to F deeming it insignificant in the meta-analysis. The resulting p-values were further adjusted for multiple testing using the q-value method (Storey and Tibshirani, 2003
) to control the false discovery rate.
The other meta-analytical approach we used was analysis of variance (ANOVA) using a fixed-effect model (FEM). FEM attempt to partition the observed variance between samples into components due to different explanatory variables (factors). The effects are considered “fixed” if they are associated with certain, specifically selected, repeatable levels of experimental factor (e.g., treatment with levels “kainate” and “control”). In this study we treated “laboratory” as a fixed effect because we only had two studies per condition. Were more studies used, a random effects model would likely be more appropriate for modeling variation among laboratories. Our FEM model can be expressed as follows:
yij = αi + βj + εij, i = 1,…,n, j = 1,…,m, εij ∼ N (0,σ2)
where yij is the expression level of a gene for the treatment’s level i in dataset j, βi is the fixed effect of the treatment level i, bj is the fixed effect of the laboratories j, and εij is a random residual error, assumed to be normally distributed for the purposes of hypothesis testing. In our current study n = 2 for two levels of experimental factor treatment, “excitotoxin” and “control”, and m, which is the number of datasets, is 2 and 3 for 24- and 240+-h timepoint, respectively.
We chose to use an additive model because we are not interested in the interaction between the experimental treatment factor and the laboratory factor. The model implies that the difference in expression levels between two treatments will be the same for each laboratory (but the absolute expression levels for each treatment might be different) and if it is not the average difference between the laboratories is considered as treatment effect.
For each gene in a meta-dataset, we computed FEM parameters and p-values associated with fixed effects using the expression values. Small p-values (p-value <0.05) for treatment effect indicate differential expression between two treatments. To control for multiple testing, we also computed q-values. The direction of expression change was computed from FEM parameter estimates.
Functional Similarity of Gene Sets
To assess the functional similarity between the significant genes obtained by meta-analysis and reference gene set we employed Gene Ontology (GO) term overlap method developed by Mistry and Pavlidis (2008)
. If annotg is the set of all direct GO term annotations for gene g including all associated parent terms, the GO term overlap score for two genes is calculated as the number of terms that occur in both gene annotation sets:
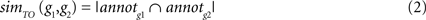
We used GO term overlap as a measure of functional similarity between genes (Mistry and Pavlidis, 2008
). For comparison with 100,000 random pairs we computed the overlap scores between all possible pairs g1 and g2, where g1 is from the meta-analysis gene set and g2 is from the reference gene set. For Cytoscape’s illustration g1 and g2 are all possible pairs from the union of meta-analysis and reference gene set.
Both meta-analyses and all associated computations were conducted using the R statistical computing environment (R Development Core Team, 2007
). Computation of q-values was performed using q-value (Storey and Tibshirani, 2003
). As suggested in qvalue manual
1
, instead of default “smoother” setting for parameter pi0.meth, we used the “bootstrap” setting for estimating π0 because the Fisher’s p-value distribution had an unusual shape (second peak at 1) due to our modification of the method as described above.
Analysis of Individual Datasets
We analyzed four microarray datasets relating to the excitotoxic brain insult in rats (Materials and Methods). We first conducted differential expression analysis on the individual datasets using Welch’s two-sided t-test for unequal variance. The results are shown in Table 2
. The table shows the number of significant genes for each type of analysis after multiple-test correction (q-value <0.05). It can be seen that for individual datasets, with the exception of GSE4326 for the 24-h timepoint, there is almost no evidence of differential expression after treatment. The same statistical test and criterion of significance were used in two of the original publications (Koh et al., 2005
; Wilson et al., 2005
), however the first study focused on the analysis of neuropeptides and the second on the comparison between two different environments and thus the results cannot be directly compared. The other two datasets were analyzed for changes in gene expression after excitotoxic insult using different statistical approaches and the number of differentially expressed genes reported in the original publications was 144 (276) for stringent (less stringent) criteria for Tang dataset (Tang et al., 2002
) and 129 for the Elliott dataset (Elliott et al., 2003
).

Meta-Analysis
Previous studies have shown that alterations in gene expression after excitotoxic insult are transient and time-specific (Zagulska-Szymczak et al., 2001
; Lukasiuk and Pitkänen, 2004
; Lukasiuk et al., 2006
). This can also be observed for GSE4236 dataset (Figure 1
). The number of genes for which expression levels after the insult are significantly different than in control animals increases up to 24 h, when it is the highest, and then it drops to 0 after 3 days. While the genes that have altered expression after 1 h remained differentially expressed at 6 h, there is a number of genes that are specific for the 6- and 24-h timepoints.
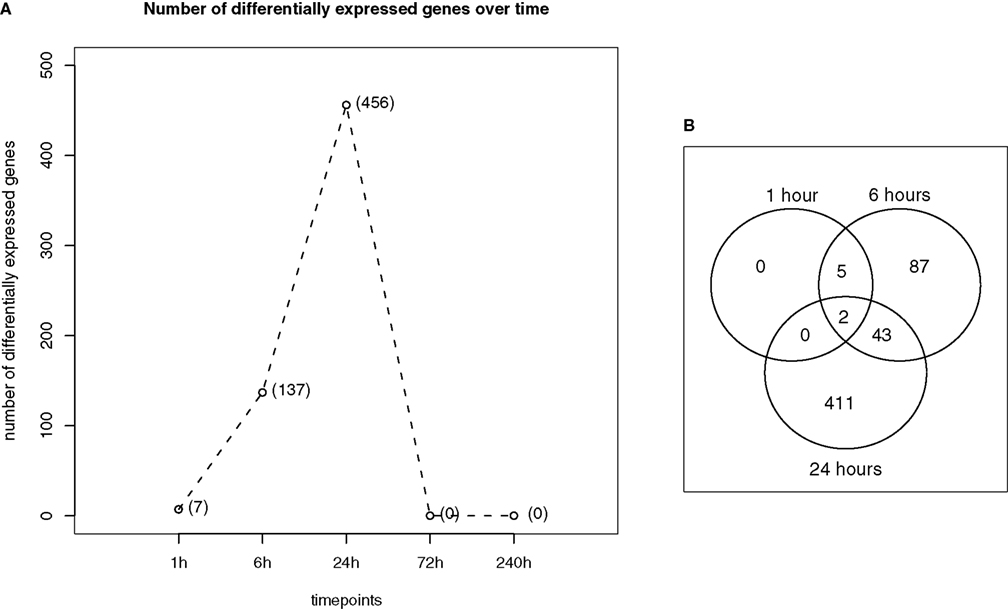
Figure 1. Gene expression over time after KA-treatment for GSE4236 dataset. (A) Changes in the number of differentially expressed genes between different timepoints after treatment. The number in parenthesis represent the number of significant genes for that timepoint (q-value <0.05). (B) Overlap in gene expression between different timepoints. A gene is significant for two timepoints if q-value <0.05 for both timepoints and the direction of change is the same.
Based on these observations and previous findings it is clear that meta-analysis of differential expression during epileptogenesis needs to be time-specific. Therefore, we grouped the available datasets according to the timepoints as described in Section “Materials and Methods”.
Fisher’s method, described in Materials and Methods, identified 293 significant genes for the 24-h timepoint and 8 genes for the later timepoint (see Table 2
). Out of 293 genes, 174 were up-regulated and 119 down-regulated 24 h after the excitotoxic insult. For the 240+-h timepoint, all of the significant genes were up-regulated.
As expected, the more powerful FEM approach identified more genes than Fisher’s approach (Table 2
). For the 24-h timepoint FEM identified 827 genes as differentially expressed, 381 with increased expression and 446 with decreased expression after the insult when compared to gene expression of control animals.
The agreement between the two methods is relatively high: the overlap between the identified genes was 285 (8 unique to Fisher’s and 542 unique to FEM method) for 24-h timepoint and 7 (1 unique to Fisher’s and 52 unique to FEM method) for 240+-h timepoint. We also computed the overall agreement between the methods, by first sorting all the genes by their q-values in each of the meta-datasets and than computing a Pearson correlation coefficient between the ranks. The rank correlations between the methods were r = 0.87 and r = 0.43 for 24- and 240+-h timepoints, respectively. These results indicate that FEM method identifies a large number of genes in addition to the ones identified by Fisher’s approach and both methods are more powerful than the simple overlap approach (Table 2
), which has been used in the literature in the field so far (Lukasiuk and Pitkänen, 2004
; Lukasiuk et al., 2006
; Wang et al., 2009
).
Importantly, we identified a number of genes that were not identified as significant in the individual datasets’ analyses. In other words, by combining data sets we were able to detect more subtle changes in expression. For the 24-h timepoint there are 454 that are found to be significant by the FEM analysis, but not found by t-test analyses of the GSE4236 or Tang datasets. The 240+-h timepoint individual datasets do not yield any significant genes by t-tests, while FEM identifies 59. The apparent reason for this is that FEM, through careful modelling of different sources of variance in the data is utilizing the samples from all the included studies thus increasing the statistical power of the test.
Usually the genes that are found to be significant have consistent patterns of expression across all the studies, as shown in Figure 2
. The heatmaps show the expression levels of top 50 up-regulated and down-regulated genes ranked by their q-values for 24-h timepoint and of all the significant genes for 240+-h timepoint. The difference between expression values for control and treatment samples is more evident for the earlier timepoint (Figure 1
). Differences in expression for the later timepoint are less numerous and more subtle, but FEM is still able to detect them.
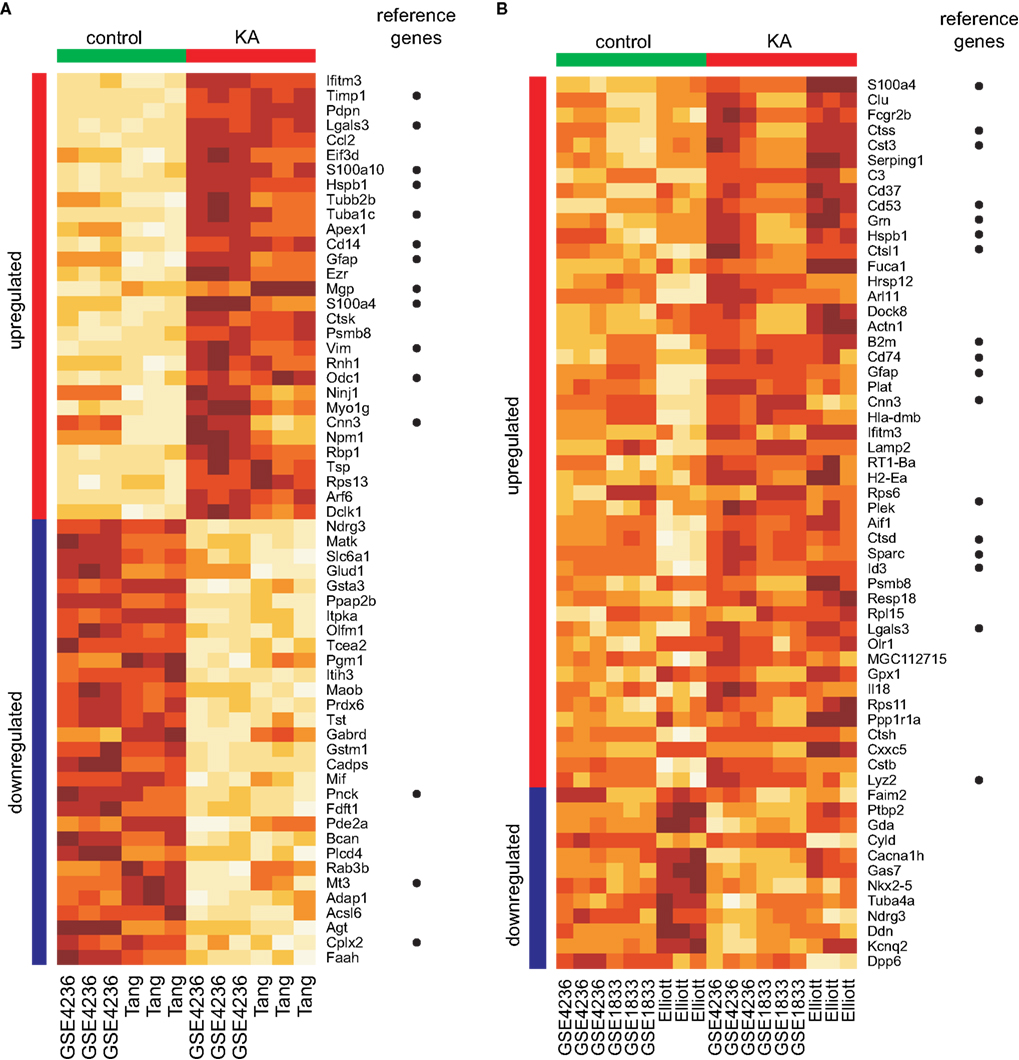
Figure 2. Visualization of expression values for some of the significant genes identified by FEM analysis. (A) Top 50 up-regulated genes and top 50 down-regulated genes for 24-h timepoint. (B) Significant genes for the 240+-h timepoint. In each heatmap the up-regulated and down-regulated genes are ranked separately by their q-values. The direction of change is indicated by the colour bars on the left of the heatmaps (red, up-regulated; blue, down-regulated). Colour bars on the top indicate control (green) and treated (red) samples. The columns correspond to samples and are labelled by the name of the dataset they are coming from. The colours in the heatmap indicate the relative level of expression (light brown for low expression and dark brown for high expression). The expression values for each gene were normalized to have mean 0 and variance 1 to facilitate comparison between genes.
Consistency with Previous Findings
We compared the genes identified to be significant by two meta-analysis approaches with a reference list of genes previously associated with epileptogenesis (see Materials and Methods). For the 24-h timepoint, Fisher’s and FEM methods identify 47 and 90 out of 136 genes on the reference list (46 genes did not have probes, see Materials and Methods), respectively. The majority of genes (30) that the FEM method failed to identify as significant have been reported to have altered expression at earlier timepoints, 6 were reported only for traumatic brain injury models and for three genes we were not able to find any reference in the original studies. This leaves only 7 genes missed by the FEM method. For 240+-h timepoint the numbers of identified reference genes are 7 and 19 out of 130 genes. The reason why there are very few reference genes identified as significant for 240+-h timepoint is the fact that most of the genes on the reference list were implicated to be differentially expressed in the earlier timepoints after excitotoxic insult (Zagulska-Szymczak et al., 2001
; Lukasiuk and Pitkänen, 2004
; Lukasiuk et al., 2006
; Wang et al., 2009
). Another evident discrepancy is between Fisher’s and FEM’s genes overlap with reference list and as noted earlier, FEM’s performance is superior.
In addition, we compared our FEM results with CarpeDB
2
, an epilepsy genetics database that currently contains 431 genes or loci genetically associated with epilepsy. After updating the official gene symbols from CarpeDB and identifying rat homologues of human and mouse genes, we had a list of 255 rat genes. Of these 255, 43 are genes from our 24-h gene list. This enrichment is statistically significant (p < 0.001, computed from the hypergeometric distribution). Only 9 of these 43 genes were on the reference list. For the 240+ timepoint the overlap was only four genes, but still statistically significant (p < 0.05).
Functional Analysis
We used Ingenuity Pathway Analysis (IPA; Ingenuity® Systems
3
) for the functional analysis of genes identified to be differentially expressed using FEM and for comparison between these genes and reference genes (see Materials and Methods). The functional analysis identified the biological functions that were most significantly enriched in the selected genes. IPA uses Fisher’s exact test to calculate a p-value representing the probability that each biological function assigned to the dataset is due to chance alone. In the next two sections we present analyses of IPA “Biological functions”, which are often broad groupings of genes akin to GO categories; and “Canonical pathways”, which are manually-curated pathways.
Biological functions
The most significant biological functions associated with genes found to be differentially expressed 24 h after excitotoxic insult were related to cell death, nervous system development and function, immunity, development and growth, signalling and basic cellular processes, such as transcription and protein synthesis. For 240+-h timepoint, the most significant biological functions were similarly related to immunity, cell death, development and growth and cell signalling. This is general agreement to what was previously reported in the literature. The proportion of genes associated with neurological disease is 48.3% and 54.2% for 24- and 240+-h gene set, respectively. For immunological disease these numbers are 27.2% and 50.8%.
When directly compared to reference genes in terms of biological functions, the 24-h gene set is generally enriched in the same functions as the reference set but with significantly more genes in each category, supporting the biological relevance of the 24-h gene set and suggesting that it is more complete than the reference set. One exception is that there are 105 genes in the 24-h gene set associated with protein synthesis while there are none for the reference set. Table 3
shows a comparison between functional categories identified in our analysis of the 24-h gene set and in reference gene set.
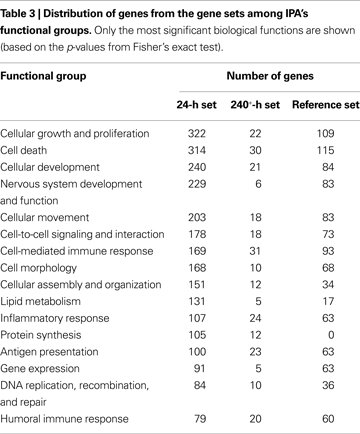
The 240+-h gene set is mostly enriched in functions that relate to immunity, cell death and development. The biological functions “organ development”, “protein synthesis” and “protein degradation” are present for 240+-h gene set but not for the reference set. More details about functional grouping of 240+-h genes and the comparison with 24-h and reference gene sets are given in Table 3
.
Canonical pathways
We also used IPA’s “canonical pathways” to look at the representation of genes from the three different gene lists. In each canonical pathway relating to neurological function or disease, there were significantly more 24-h genes than the reference genes, which reinforces the finding that our 24-h gene set contains more biologically relevant genes than the reference set. Table 4
lists these pathways and gives the total number of genes in the pathway, the number of 24-h genes and the number of reference genes in the pathway. One specific illustrated example is given for reelin signalling in neurons pathway in Figure 3
.

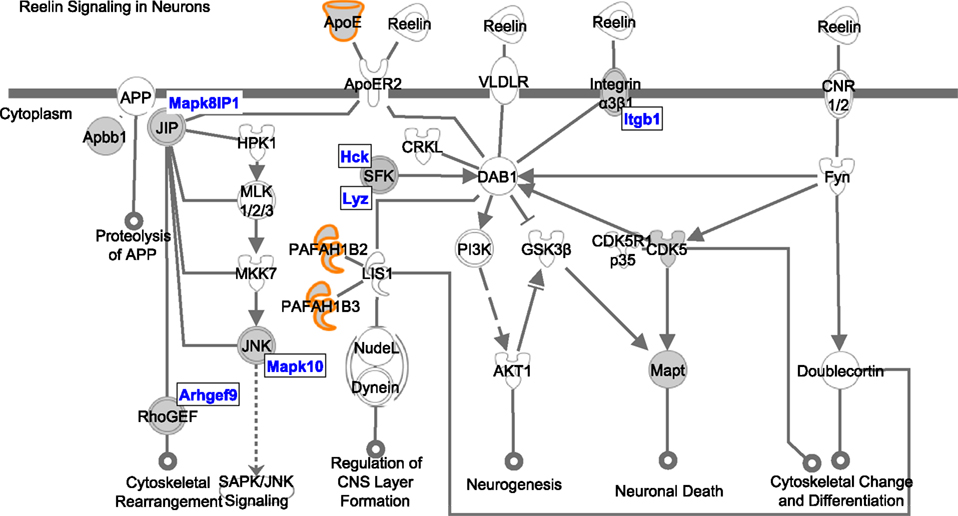
Figure 3. Depiction of the “Reelin signalling in neurons” pathway generated by Ingenuity. The genes and complexes (double lines) highlighted in grey are form 24-h gene set and the genes outlined in orange are also in the reference set. The genes from the 24-h list that are components of complexes are highlighted in blue.
For 240+-h gene set there are just a few significantly enriched canonical pathways, the most significant being antigen presentation pathway with 6 out of 39 genes belonging to the pathway present on the 240+-h gene list (3 genes from the reference set).
GO term similarity
In order to investigate the degree of functional similarity between the meta-analysis gene sets and the reference set more directly, we computed the GO term overlap between all possible gene pairs from our gene set and reference gene set, as described in Section “Materials and Methods”. This was done separately for 24-h (135,043 gene pairs) and 240+-h gene sets (7461 gene pairs). We then compared the distributions of these scores with the distribution of GO term similarity scores for 100,000 random rat gene pairs. The mean (median) values of the GO term similarity scores are 10.20 (8), 8.49 (7) and 3.79 (2) for 24-h – reference gene pairs, 240+-h – reference gene pairs and 100,000 random gene pairs, respectively. The differences between medians are highly significant when our meta-analysis – reference gene pairs are compared to random gene pairs: p-value from Wilcoxon rank sum test is <2.2 × 10−16 for both timepoints. This indicates that the GO term similarity is significantly higher between meta-analysis and reference genes than expected at random. An illustration of level and degree of similarity between 24-h and reference genes is given in Figure 4
, which is GO similarity network created in Cytoscape (Shannon et al., 2003
) for gene pairs between and within 24-h and reference gene sets that share at least 60 GO terms (this aribtrary threshold was chosen to facilitate visualization).
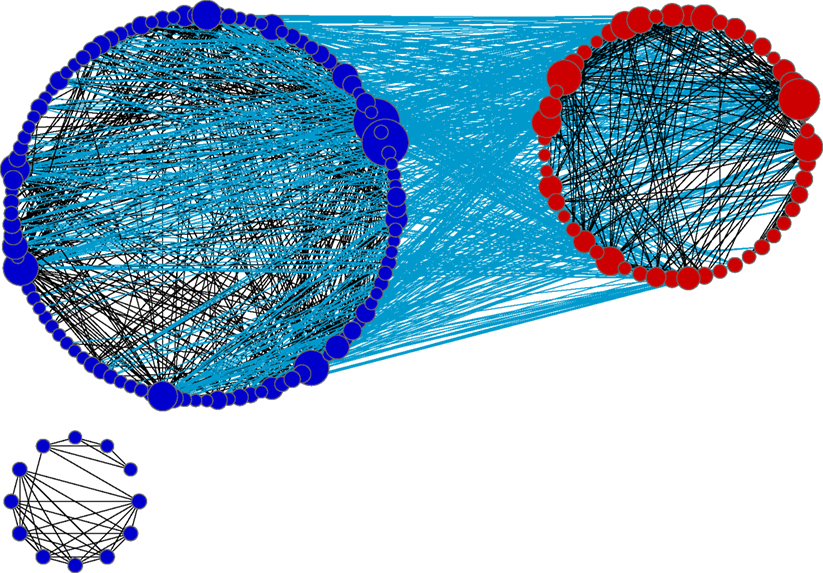
Figure 4. GO similarity network created in Cytoscape. Blue nodes represent genes that are only on our 24-h gene list; red nodes represent reference genes. The two types of nodes were grouped separately so that the interactions between two groups are easily visible. An edge between two nodes signifies that there are at least 60 GO terms that are common for that pair of genes. Edges between reference and 24-h genes are shown in light blue, while grey nodes are between reference genes or between 24 h genes. The size of a node correspond to its degree (its number of edges). Genes which lack GO similarity of at least 60 to another gene are not displayed.
Our contribution in this report is twofold. First, we have reanalyzed four expression studies of epileptogenesis using meta-analytical techniques, providing a much more extensive characterization of expression changes. Second, from a methodological perspective, we show some advantages of combining data sets for reanalysis, as opposed to comparing “hit lists” or performing meta-analyses based on p-values.
A striking finding of previous reviews (Zagulska-Szymczak et al., 2001
; Lukasiuk and Pitkänen, 2004
; Lukasiuk et al., 2006
; Wang et al., 2009
) was that most of the genes found to be differentially expressed in epileptogenesis are not reproduced by other studies (upwards of 1500 genes). The consensus list of affected genes was quite small: fewer than 100 genes. A potential explanation is that the gene selection procedures used in individual studies are suboptimal; many studies are also underpowered. In addition, simply taking overlaps between gene lists is a stringent way of comparing studies; its main advantage is simplicity, not sensitivity. Together these problems lead to an apparent poor reproducibility of findings across studies (very few genes are found by more than one study). In our view, reproducibility (or lack thereof) is a function of study design (small sample sizes being a major culprit), ad hoc gene selection methods (e.g., fold-change), and the method used to assess reproducibility.
Combining studies across laboratories, while not always possible or appropriate, ameliorates all of these issues. Thus our results paint a somewhat different picture of the situation than the simple overlapping hit-list approach. Despite using only four small expression studies, we recaptured a large fraction of the genes identified as reliable by a review of 18 studies. This suggests that these four studies (representing two timepoints after induction) share reliable biological signals, but were underpowered to pick up all of those changes by themselves.
Importantly, our analysis picks up many additional genes which were (1) not identified by the original authors and (2) not found in surveys of 18 studies. It might be tempting to think these additional genes represent false positives from the meta-analysis, but our functional analysis shows that the vast majority of the genes are highly functionally related to genes deemed “reliable” by less sensitive methods (Figure 4
). In other words, our reanalysis shows that the impact of kindling on pathways previously implicated is far more extensive than appreciated using a single study or the reference list. Rather than a random grab-bag of 2000 genes identified by pooling noisy hit lists across studies, the combined analysis yields a more focused list that is largely easily interpreted in light of previous reviews of the literature. For example, while three genes on the reference list are in the Reelin signalling pathway (Figure 3
), our analysis shows that at least 12 additional genes are implicated, suggesting a widespread change in regulation of the pathway.
We found that our 24-h gene list was significantly enriched for genes listed by CarpeDB as being implicated in the genetics of epilepsy. This was surprising, as we did not necessarily expect that genes which change expression in kindling are susceptibility genes for epilepsy. To our knowledge this has not previously been examined systematically. A simple (but speculative) hypothesis to explain this result is that many of the susceptibility genes we found are in some sense protective in the face of a excitotoxic injury. This model would predict that failure to change expression after the insult increases the likelihood of developing spontaneous seizures.
We hypothesized that in addition to “fleshing out” expression changes in pathways already known to be affected by epileptogenesis, we might identify novel genes and/or pathways that were not detected previously. As shown by the small cluster of blue nodes in Figure 4
, there are genes which have documented functions that are less similar in function to the others. The small cluster in Figure 4
is predominantly made up of up-regulated proteosomal protein genes, which to our knowledge has not been noted in expression studies of epileptogenesis. This could reflect a response to oxidative stress (Jung et al., 2007
). Importantly, Figure 4
does not reflect many other genes we found which are not as well “connected” (Figure 4
is heavily filtered for display purposes). On closer inspection of such genes, we found plausible relationships between epilepsy and many of the genes we checked manually with literature searches. For example, fatty acid amide hydrolase (Faah), which is down-regulated 24 h after kainate injection, is involved in metabolism of endocannabinoids, and inhibitors of Faah have been shown to reduce the damaging effects of kainate, if given within a few hours (Karanian et al., 2007
). The expression results may reflect one endogenous mechanism for increasing endocannabanoids after kainate injury, which has been shown to occur at shorter time scales (Marsicano et al., 2003
). Another example is SLC4A3 (a 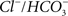 exchanger), mutations in which are associated with idiopathic generalized epilepsy (Vilas et al., 2009
).
exchanger), mutations in which are associated with idiopathic generalized epilepsy (Vilas et al., 2009
).
exchanger), mutations in which are associated with idiopathic generalized epilepsy (Vilas et al., 2009
).These results give us confidence that the genes identified by careful reanalysis of RNA patterns can yield additional results of relevance to understanding the brain’s response to excitotoxic insult. In addition to the proteasome expression changes noted above, numerous genes associated with lipid metabolism were altered in expression (Table 3
). While a few genes in this group were on the reference list, lipid metabolism was not noted as a biological process of interest in any of the reviews used as sources for the list (Zagulska-Szymczak et al., 2001
; Lukasiuk and Pitkänen, 2004
; Lukasiuk et al., 2006
; Wang et al., 2009
). While the meaning of these patterns is not clear (“lipid metabolism” is a broad category of genes), it illustrates the ability of reanalysis and meta-analysis of expression data to help uncover biologically relevant patterns.
In conclusion, we have shown that combining even small numbers of data sets can greatly increase power to detect expression changes. Applied to excitotoxin models of epileptogenesis in the rodent hippocampus, we show that changes in RNA levels in several pathways, rather than being focused on a few genes, are much more widespread than seems to be appreciated. We suspect that expanding the scope of our meta-analysis to include additional data sets and models will yield additional insights.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supported by NIH Grant GM076990, the Canadian Foundation for Innovation, and salary awards to PP from the Michael Smith Foundation for Health Research and the Canadian Institutes for Health Research. We thank Dr Daniel Goldowitz for helpful discussions and encouragement. We thank Drs Frank Sharp and Robert Elliott for generously sharing their microarray data.
The Supplementary Material for this article can be found online at http://www.chibi.ubc.ca/faculty/pavlidis/meta-analysis-of-brain-kindling/
Hinrichs, A. S., Karolchik, D., Baertsch, R., Barber, G. P., Bejerano, G., Clawson, H., Diekhans, M., Furey, T. S., Harte, R. A., Hsu, F., Hillman-Jackson, J., Kuhn, R. M., Pedersen, J. S., Pohl, A., Raney, B. J., Rosenbloom, K. R., Siepel, A., Smith, K. E., Sugnet, C. W., Sultan-Qurraie, A., Thomas, D. J., Trumbower, H., Weber, R. J., Weirauch, M., Zweig, A. S., Haussler, D., and Kent, W. J. (2006). The UCSC Genome browser database: update 2006. Nucleic Acids Res. 34, 590–598.
Marsicano, G., Goodenough, S., Monory, K., Hermann, H., Eder, M., Cannich, A., Azad, S. C., Cascio, M. G., Gutiérrez, S. O., van der Stelt, M., López-Rodriguez, M. L., Casanova, E., Schütz, G., Zieglgänsberger, W., Di Marzo, V., Behl, C., and Lutz, B. (2003). Cb1 cannabinoid receptors and on-demand defense against excitotoxicity. Science 302, 84–88.
Parkinson, H., Kapushesky, M., Kolesnikov, N., Rustici, G., Shojatalab, M., Abeygunawardena, N., Berube, H., Dylag, M., Emam, I., Farne, A., Holloway, E., Lukk, M., Malone, J., Mani, R., Pilicheva, E., Rayner, T. F., Rezwan, F., Sharma, A., Williams, E., Bradley, X. Z., Adamusiak, T., Brandizi, M., Burdett, T., Coulson, R., Krestyaninova, M., Kurnosov, P., Maguire, E., Neogi, S. G., Rocca-Serra, P., Sansone, S. A., Sklyar, N., Zhao, M., Sarkans, U., and Brazma, A. (2009). Arrayexpress update – from an archive of functional genomics experiments to the atlas of gene expression. Nucleic Acids Res. 37, 868–872.
Rhodes, D. R., Yu, J., Shanker, K., Deshpande, N., Varambally, R., Ghosh, D., Barrette, T., Pandey, A., Chinnaiyan, A. M. (2004). Large-scale meta-analysis of cancer microarray data identifies common transcriptional profiles of neoplastic transformation and progression. Proc. Natl. Acad. Sci. U.S.A. 101, 9309–9314.