
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
BRIEF RESEARCH REPORT article
Front. Neurosci. , 26 March 2025
Sec. Neurodevelopment
Volume 19 - 2025 | https://doi.org/10.3389/fnins.2025.1554093
This article is part of the Research Topic Advances in DYRK1A Syndrome: Underlying Mechanisms, Disease Models, and Novel Therapeutic Approaches View all 4 articles
 Nicolas Le May1
Nicolas Le May1 Jérémie Courraud2Imène Boujelbène2,3Cathy Obringer1
Jérémie Courraud2Imène Boujelbène2,3Cathy Obringer1 Tomoo Ogi4
Tomoo Ogi4 Alan R. Lehmann5Fanny Laffargue6Daphné Lehalle7
Alan R. Lehmann5Fanny Laffargue6Daphné Lehalle7 Seiji Mizuno8Shehla Mohammed9Clothilde Ormières10
Seiji Mizuno8Shehla Mohammed9Clothilde Ormières10 Marjolaine Willems11
Marjolaine Willems11 Vincent Laugel1,12†
Vincent Laugel1,12† Nadège Calmels1,3*†
Nadège Calmels1,3*†Nucleotide excision repair (NER) disorders are genetic conditions caused by defects in the pathway responsible for repairing DNA lesions due to UV radiation. These defects lead to a variety of heterogeneous disorders, including Cockayne syndrome (CS) and trichothiodystrophy (TTD). In this study, we report 11 patients initially suspected of having CS or TTD who were ultimately diagnosed with DYRK1A haploinsufficiency syndrome using high-throughput sequencing. Comparing clinical presentations, we observed that DYRK1A symptoms overlapped with CS, with shared features such as intellectual disability and microcephaly, systematically present in both disorders and other common symptoms including feeding difficulties, abnormal brain imaging, ataxic gait, hypertonia, and deep-set eyes. However, distinctive features of DYRK1A syndrome, such as severely impaired language, febrile seizures, and autistic behavior or anxiety, helped differentiate it from CS, which typically manifests with severe growth delay, bilateral cataracts, and pigmentary retinopathy. Among the cohort, three patients carried novel DYRK1A variants, including two truncating and one in-frame variant p.Val237_Leu241delinsGlu whose pathogenicity have been confirmed through functional analysis of DYRK1A protein. While previous research has implicated DYRK1A in DNA repair, with DYRK1A being one of the most downregulated genes in CS cells, our study found that DYRK1A patient-derived cell lines did not exhibit NER defects and did not share the CS transcriptomic signature. These findings suggest that if clinical symptoms overlap stems from common molecular disruptions, DYRK1A is involved downstream of the CS genes. This research highlights the importance of considering DYRK1A haploinsufficiency syndrome in the differential diagnoses for NER disorders.
Nucleotide excision repair (NER) is a critical DNA repair mechanism that protects cells from a variety of DNA damages, such as those caused by ultraviolet (UV) radiation and chemical mutagens (Nieto Moreno et al., 2023). Beyond their role in DNA repair, NER proteins also influence the dynamic regulation of transcription and chromatin structure, which are essential for maintaining genomic stability and proper gene expression. Defects in the NER pathway lead to a variety of distinct, heterogeneous disorders, such as xeroderma pigmentosum (XP), Cockayne Syndrome (CS), and Trichothiodystrophy (TTD), all with autosomal recessive inheritance. Whereas XP is a genodermatosis mainly characterized by a predisposition to sunlight-induced skin pigmentation changes and skin cancers, as well as neurological problems in 30–50% of cases, CS and TTD are neurodevelopmental and subsequently neurodegenerative disorders leading to progressive microcephaly, growth retardation, developmental delay, neurological and neurosensory impairments and skin and/or hair abnormalities. CS encompasses a wide spectrum of severity, with the classic form, known as type I, onset occurring before the age of 2. The genetic abnormalities underlying these diseases are multiple and complex, with mutations in the same gene leading to different conditions (Ferri et al., 2020). Moreover, the existence of combined forms, such as XP/CS, further complicates the diagnosis. Historically, the diagnosis was confirmed by DNA repair activity testing on primary fibroblasts, such as recovery of RNA synthesis (RRS) and Unscheduled DNA synthesis (UDS) assays (Mayne and Lehmann, 1982). More recently, the diagnosis of these heterogeneous conditions has significantly benefited from the use of high-throughput sequencing techniques, as formerly demonstrated by sequencing a panel of NER genes (Calmels et al., 2016). At the end of these investigations, however, some patients with suspected DNA repair disease remain without any identified molecular cause. These patients may carry variants in unexplored regions of NER genes such as promoter or deep intronic regions (Schalk et al., 2018). They may also be clinical phenocopies (so called NER-like patients) whose molecular origin lies in other genes than those of the NER pathway.
DYRK1A haploinsufficiency syndrome (OMIM# 614104) is a neurodevelopmental disorder caused by mutations in the DYRK1A gene, which encodes a protein kinase essential for brain development. It is characterized by a range of cognitive and developmental impairments, including microcephaly, intellectual disability, motor delay, speech difficulties, feeding problems, autism spectrum traits, seizures, and dysmorphic features. Studies have reported that a majority of patients exhibit atopic skin or skin that appears thin and translucent (Courraud et al., 2021). Some individuals may also present with fine hair texture. Interestingly, DYRK1A has been previously reported as one of the most down-regulated ATF3-dependent genes in CS fibroblasts, suggesting a potential functional link between NER proteins and DYRK1A (Epanchintsev et al., 2020).
Here we report on 11 patients, initially suspected of NER disorders, who were finally diagnosed with DYRK1A haploinsufficiency syndrome through further genetic investigations. Extended genetic investigations in NER-like patients enabled us to identify the first four patients. In a second phase, we identified an extra DYRK1A patient by a retrospective analysis of our in-house cohort of NER-like patients. Finally, six additional NER-like patients with a DYRK1A variant were enrolled through an international collaboration with British and Japanese teams involved in diagnosing NER disorders. We will discuss the phenotypic overlap between NER disorders and DYRK1A haploinsufficiency syndrome and attempt to explore the potential underlying molecular mechanisms of these common features.
All patients included in this study gave written consent for genetic testing. DYRK1A coding sequence was analyzed through Sanger sequencing, gene panel sequencing or exome sequencing. DYRK1A variants are named according to GRCh38 version of genome assembly, NM_001396.5 transcript and NP_001387.2 protein. New variants have been registered in ClinVar Database.
Primary fibroblasts from four individuals with DYRK1A variant (patients 1, 2, 4 and 5) were available for DNA repair functional assays and were compared to fibroblasts from a healthy control, from a Cockayne individual (carrying pathogenic variants in the ERCC8/CSA gene) for RRS assay and from a xeroderma pigmentosum individual (carrying pathogenic variants in the ERCC4/XPF gene) for UDS assay. Cells were stored in the registered biobank DC-2014-222 CPP 09/40. Recovery of RNA synthesis (RRS) and unscheduled DNA synthesis (UDS) following UVC irradiation of the primary fibroblasts were performed using fluorescent non-radioactive assays as previously described (Jia et al., 2015; Calmels et al., 2016).
Expression profiles were studied on primary fibroblast cultures from two controls, three CSB patients with ERCC6 mutations, three CSA patients with ERCC8 mutations and from patients 1, 2, 4 and 5. Fibroblasts were seeded on 10 cm plates using 1×106 cells. Twenty-four hours later, cells were UV-irradiated (20 J/m2) and then incubated for 24 h in DMEM (1 g/L glucose) with GLUTAMAX (Life Technologies, Inc., Rockville, MD) supplemented with 10% of Fetal Calf Serum (FCS) and Penicillin (100 UI/mL) + Streptomycin (100 μg/mL). Total RNA was extracted from the fibroblasts using the GenElute Mammalian Total RNA Miniprep kit (Sigma-Aldrich) and reverse transcribed with SuperScript IV reverse transcriptase (Invitrogen). Quantitative PCR was performed using SYBR Green Master Mix and the CFX384 Real-time system (Biorad). The primer sequences for genes analyzed by qPCR are listed in Supplementary Table S1. mRNA expression of the various genes was calculated as the ratio of values obtained from irradiated versus non-irradiated cells, normalized to the housekeeping GAPDH mRNA. Biological triplicates and technical duplicates were analyzed. T-test was performed to compare the expression level of each gene in CSA, CSB and DYRK1A patients to the expression level in control cell lines, applying Welch’s correction.
Level, cellular localization, and kinase activity of DYRK1A mutant proteins were studied as previously reported by our team (Courraud et al., 2021). Briefly, wild-type (WT) and mutant DYRK1A proteins were overexpressed in three different cell lines (HEK293, HeLa, COS1) using DYRK1A expression plasmids. Protein level was studied in the three cell types by Western blot. One-way ANOVA with multiple comparison test was performed to compare the level of mutant DYRK1A proteins to the level of WT DYRK1A protein, applying Bonferroni’s correction. Cellular localization was studied in HeLa cells by immunofluorescence. Chi-square test was performed to compare localization of mutants and wild-type DYRK1A proteins. DYRK1A autophosphorylation activity was evaluated in HEK293 cells using the ratio between Tyr321 DYRK1A and total DYRK1A protein. One-way ANOVA test was performed to compare mutants to wild-type DYRK1A proteins.
We set up a cohort of 11 patients initially suspected of having an NER disorder (CS or TTD) but carrying a pathogenic variant in the DYRK1A gene (NM_001396.5) (Table 1).
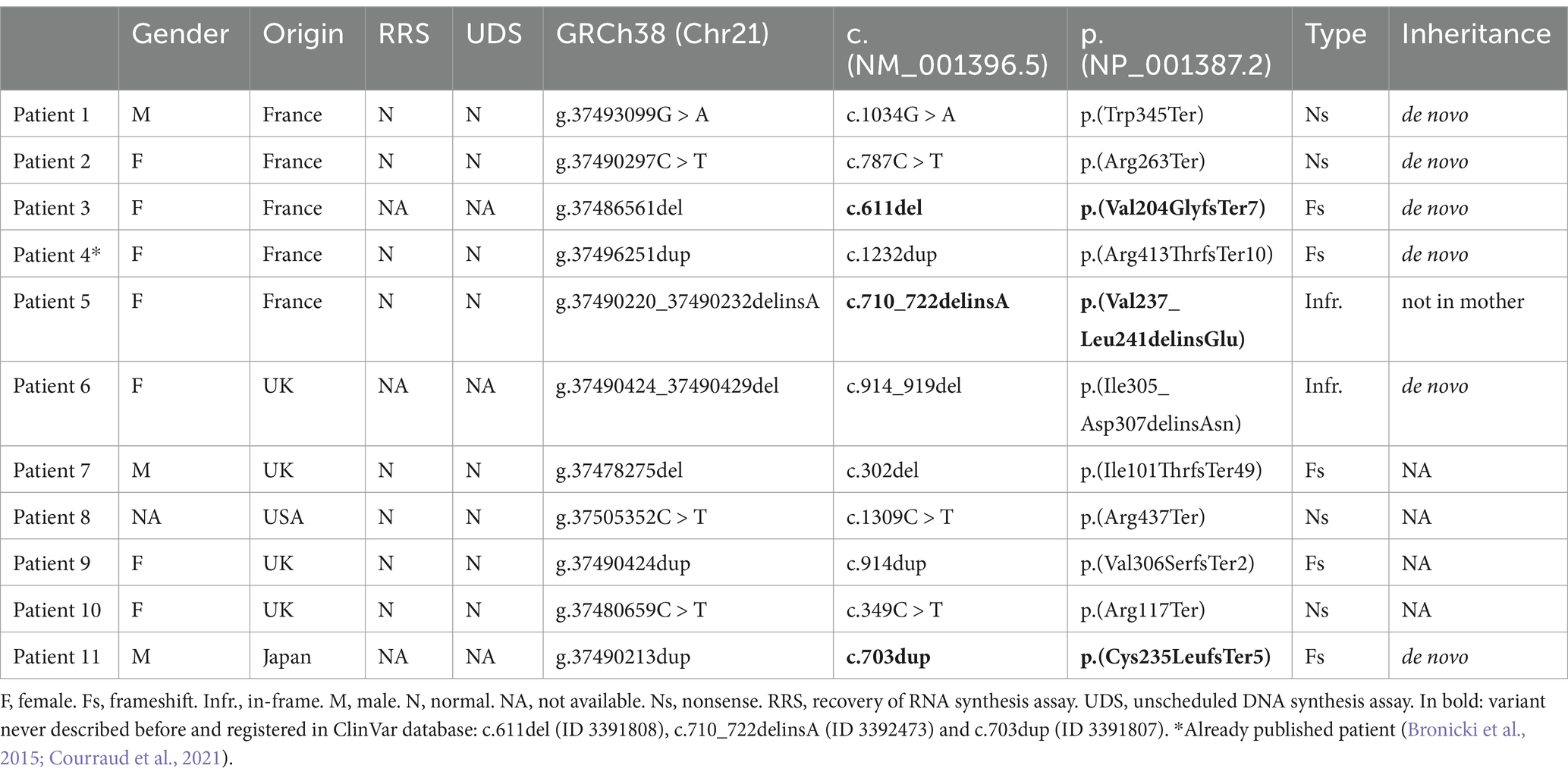
Table 1. List of variants identified in DYRK1A in individuals initially suspected of nucleotide excision repair (NER) disorder.
Patients 1 to 4 are French patients initially addressed for an NER disorder (CS or TTD). Sanger sequencing of the 2 main CS genes, ERCC6 and ERCC8, or high-throughput sequencing (HTS) of a panel of 16 genes involved in the NER pathway (Calmels et al., 2016), did not identify any pathogenic variant. Extended genetic analyses using HTS of a large panel of genes involved in neurodevelopmental disorders have revealed the presence of de novo pathogenic variants in the DYRK1A gene.
In order to identify additional cases, we analyzed a posteriori our in-house cohort of 233 “NER-like” patients. Based on clinical records, we selected individuals with microcephaly, intellectual disability, global development delay and impaired language. Patients presenting with photosensitivity or pigmentary retinopathy, symptoms never described in DYRK1A patients, were excluded. Among the three selected patients, DYRK1A sequencing using high-throughput or Sanger sequencing, revealed an in-frame deletion/insertion variant, p.(Val237_Leu241delinsGlu) in one patient (patient 5). The variant was not maternally inherited but father’s DNA was not available for testing.
Finally, through an international collaboration with British and Japanese diagnosis teams involved in NER disorders, six additional NER-like patients carrying a loss-of-function DYRK1A variant identified by exome sequencing were recruited (patients 6 to 11). The variant occurred de novo in two of them, while parental DNA was not available for the remaining cases.
We reviewed the clinical manifestations of the patients (Table 2; Supplementary Table S2). Consistent with previous reports (Bronicki et al., 2015; Ji et al., 2015; van Bon et al., 2016; Courraud et al., 2021) recurrent features include psychomotor delay evolving to intellectual disability, prenatal and postnatal moderate growth delay (median height: −2.6 ± 1.4 DS), microcephaly and major speech impairment, feeding difficulties, seizures, autistic traits and anxiety, delayed gross motor development with unstable gait, brain magnetic resonance imaging (MRI) abnormalities including enlarged ventricles, cerebral atrophy and cerebellar hypoplasia, and distinctive facial features. We found skin and hair anomalies especially atopic dermatitis and thinning hair. When clinical data were available, we measured the clinical DYRK1A score (Courraud et al., 2021). The range and mean scores (from 14 to 18.5, mean = 16.2) is similar to those of the published cohort (from 13 to 18.5, mean = 15.5) and all evaluated patients have a score above the discriminant threshold of 13 (Supplementary Table S3).
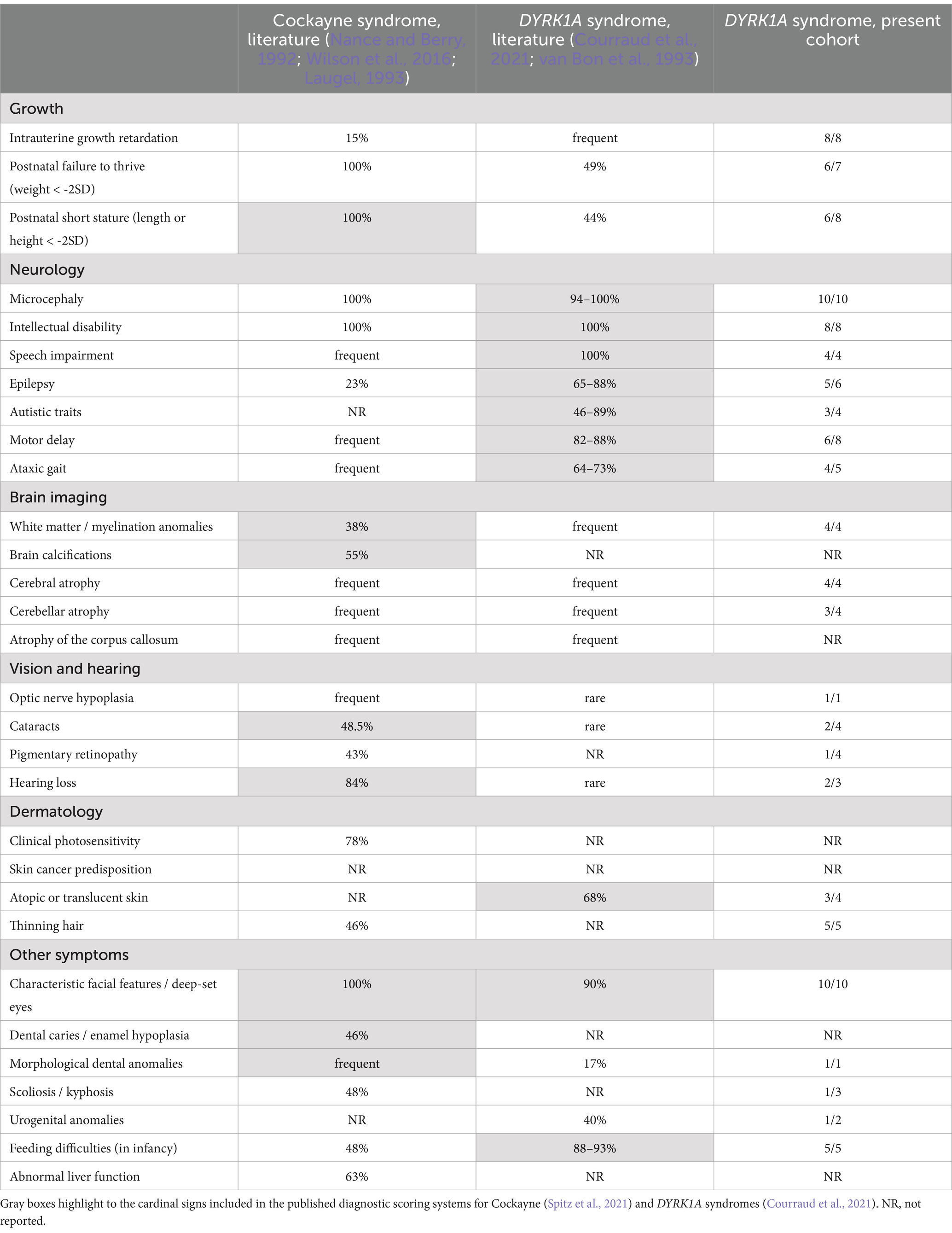
Table 2. Comparative clinical characteristics between classical descriptions of Cockayne and DYRK1A syndromes (percentages are derived from existing literature) and this DYRK1A cohort (numbers reflect occurrences of each symptom among the patients for whom the information was available).
Among the 11 DYRK1A variants described in this cohort, nine were loss of function (five frameshift variants and four nonsense variants) distributed throughout the protein, while the remaining two were in-frame deletion/insertion variants affecting amino acids located within the kinase domain (Table 1). Interestingly, only three of these variants have not been previously described, whereas the others have already been reported in individuals with DYRK1A syndrome or unspecified neurodevelopmental delays. Notably, two of the variants, p.(Arg437Ter) and p.(Arg117Ter), are the most recurrent variants associated with DYRK1A syndrome, with at least eight submissions in ClinVar and likely more cases identified but not reported. The DYRK1A patients in our cohort, selected based on their similarity to NER patients, exhibit clinical and molecular characteristics closely resembling those of previously published DYRK1A cases.
To confirm the loss-of-function consequences of the p.(Val237_Leu241delinsGlu) (also called Val237delins) in-frame variant, we investigated the consequences of this variant in vitro. We overexpressed wild-type (WT) and variant DYRK1A proteins in three different cell lines (HEK293, HeLa, COS1) including a known pathogenic missense variant (p.Ser311Pro) as a positive control as well as a benign missense variant (p.Ala341Ser) as negative control. A significant reduction in DYRK1A protein level was observed for the Val237delins variant (Figure 1A). While the wild-type DYRK1A protein is predominantly localized in the nucleus, the mutant variant Val237delins exhibited a significant relocalization to the cytoplasm in a substantial proportion of cells. (Figure 1B). DYRK1A activation requires autophosphorylation at Tyrosine 321 (Himpel et al., 2001). To assess the level of active DYRK1A protein, we detected phospho-DYRK1A (Tyr321) via immunoprecipitation followed by immunoblot using anti-phospho-HIPK2. We demonstrated that the Val237delins variant prevents autophosphorylation (Figure 1C). The effects of the second in-frame deletion/insertion variant of the cohort, p.Ile305_Asp307delinsAsn, was not investigated in vitro even when it was first reported (Méjécase et al., 2021). However, the deleterious consequences of a missense variant involving the same amino-acid, Ile305Arg, have been demonstrated previously associating a reduced protein level and an absence of autophosphorylation activity (Courraud et al., 2021).
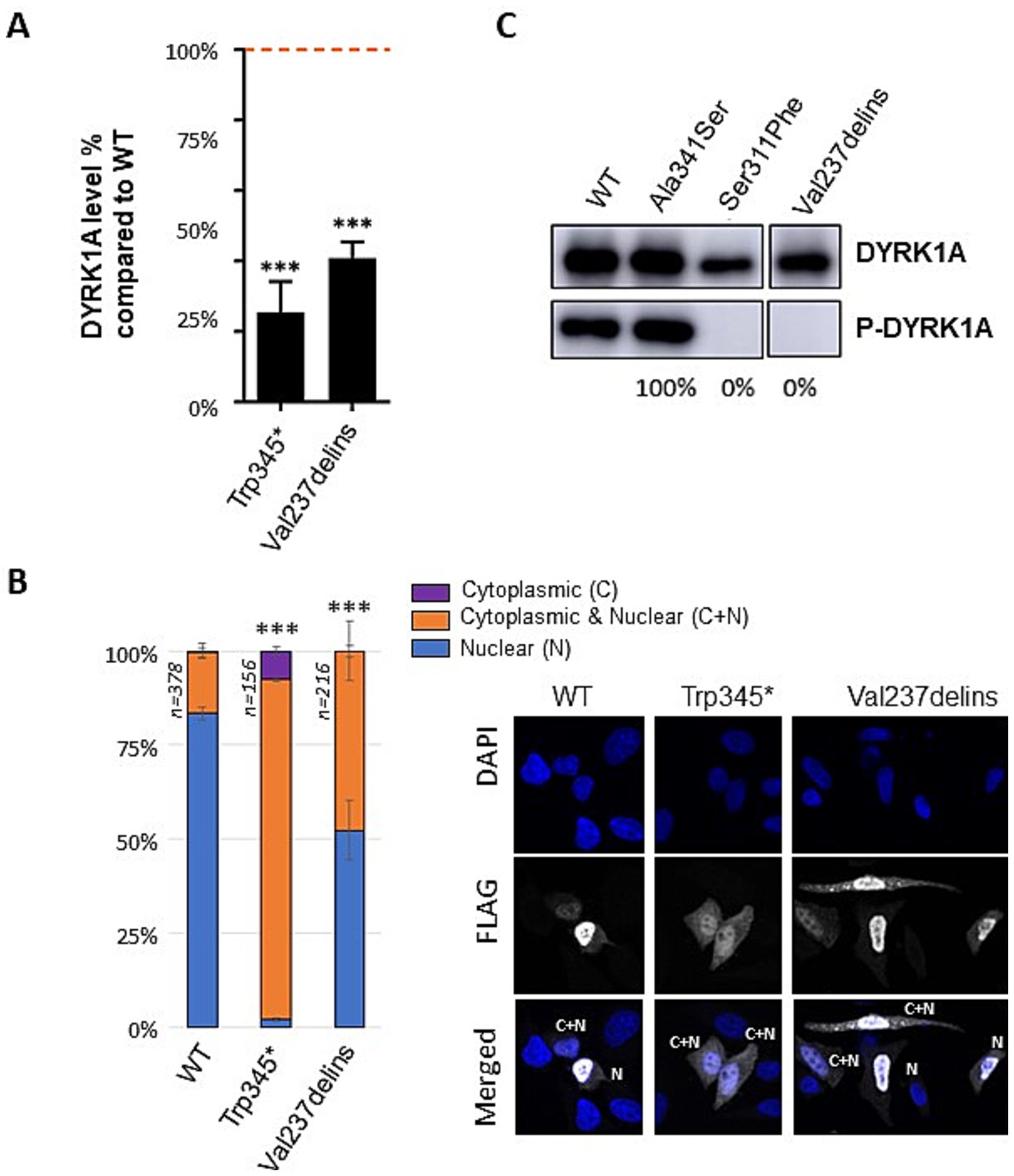
Figure 1. Effect of the p.(Val237_Leu231delinsGlu) (Val237delins); Val237delins) variant on DYRK1A protein level, cellular localization and autophosphorylation. (A) Level of mutant DYRK1A proteins expressed in HeLa, HEK293 and COS cells transiently transfected with DYRK1A constructs containing wild-type (WT), Trp345Ter or Val237delins cDNA sequences. Protein levels were normalized on the level of GFP protein (expressed from a cotransfected pEGFP plasmid). Quantifications were performed on a total of n = 9 series of cells (n = 3 Hela cells, n = 3 HEK293 and n = 3 COS-7 cells) using ImageJ software. One-way ANOVA with multiple comparison test was performed to compare the level of mutant DYRK1A proteins to the level of WT DYRK1A protein, applying Bonferroni’s correction: ns: not significant; ***p < 0.001; error bars represent SEM, standard error of the mean. (B) Cellular localization of DYRK1A WT and mutant proteins observed in HeLa cells after overexpression of FLAG-tagged wild-type and mutant DYRK1A constructs (n = 3 series of Hela cells; 50 cells minimum counted per series; the total number of cells counted per condition is indicated in the graph; scale bars of illustrative images correspond to 10 μm). Three types of cellular localization of DYRK1A protein have been observed: DYRK1A located mostly in the nucleus (blue on the graph), in both nucleus and cytoplasm (orange), or mostly in the cytoplasm (purple). Scale bars: 50 μm. Chi-square test was performed to compare localization of mutants and wild-type DYRK1A proteins, ns: not significant; ***p < 0.001; error bars represent SEM, standard error of the mean. Immunofluorescence images illustrate the data (DAPI-labeled nucleus, flag-labeled DYRK1A and merge). (C) Measure of DYRK1A autophosphorylation on its Tyr321 tested in HEK293 cells (n = 3) transfected with constructs containing WT and Val237delins cDNA sequences, as well two other variants, Ala341Ser which is a benign variant from gnomAD not supposed to affect DYRK1A kinase activity and Ser311Phe which is a kinase-dead variant. An immunoprecipitation with anti-DYRK1A antibody was followed by an immunoblot using anti-phospho-HIPK2 as described in Widowati et al. and Courraud et al. DYRK1A phospho-Tyr321 levels were normalized on DYRK1A total level. The percentage of phosphorylated form for the different mutants compared to the WT proteins is indicated under the blot. One-way ANOVA test was performed to compare mutants to wild-type DYRK1A proteins. ns, not significant; ***p < 0.001; error bars represent SEM, standard error of the mean.
DYRK1A is known to play a major role in DNA repair, especially homologous recombination (Guard et al., 2019; Menon et al., 2019; Roewenstrunk et al., 2019). To explore the mechanisms underlying the clinical overlap between NER and DYRK1A syndrome patients, we evaluated DNA repair assays as well as the CS expression signature on four fibroblastic cell lines from patients with DYRK1A pathogenic variants. The Recovery of RNA Synthesis (RRS) test is an assay measuring the ability of cells to restore normal levels of transcription, following ultraviolet (UV) irradiation. This recovery accompanies the elimination of DNA lesions on the transcribed DNA strand by the NER sub-pathway transcription-coupled repair (TCR) and is deficient in CS cells. The unscheduled DNA Synthesis (UDS) assay evaluates the global genome repair ability of the cells after UVC irradiation. All available cell lines of patients with DYRK1A variants (patients 1, 2, 4 and 5) exhibited normal DNA repair assays after UV irradiation (Figures 2A,B).
Patients with Cockayne syndrome exhibit a distinct transcriptional signature 24 h post-UV irradiation marked by widespread changes in gene expression, including notable upregulation of transcriptional repressor ATF3 and downregulation of ATF3-dependent genes, DYRK1A being one of the most downregulated ATF3-targeted genes (Epanchintsev et al., 2020). We therefore analyzed by RT-qPCR the expression of CSB, ATF3 and four ATF3-targeted genes (CDK5RAP2, NRG1, NIPBL and DYRK1A, which have been described as a signature expression profile for CS) in two control cell lines, 6 CS cell lines (3 CSA linked to ERCC8 variants and 3 CSB linked to ERCC6 variants) and the four available DYRK1A cell lines. Whereas we confirmed the expected expression signature in CS patients, there was no upregulation of ATF3 nor downregulation of ATF3-dependent genes in DYRK1A syndrome cell lines (Figures 2C,D).
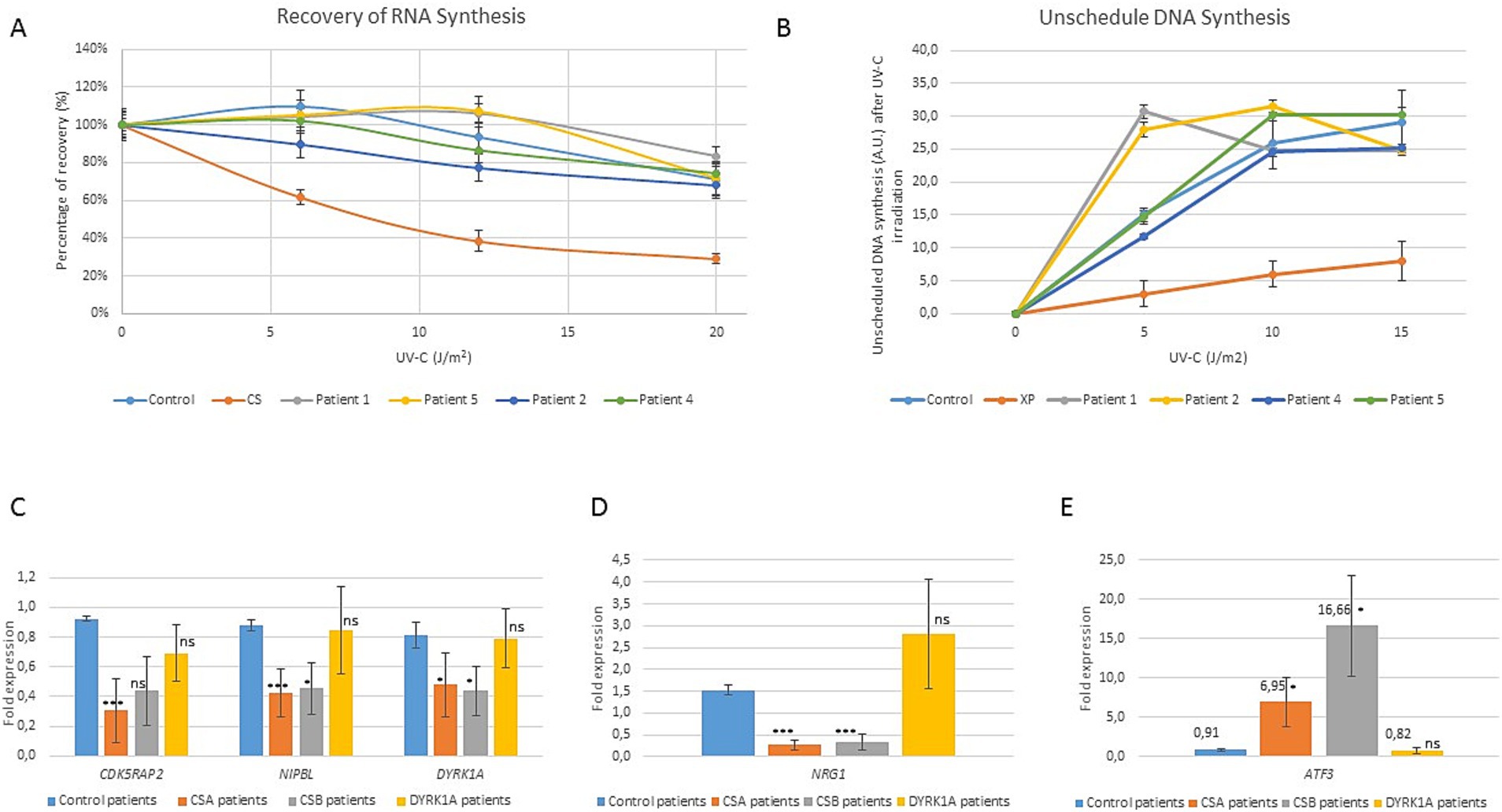
Figure 2. Molecular characterization of DYRK1A patients cell lines compared to Cockayne syndrome patients cell line. (A) Recovery of RNA synthesis (RRS) measured 23 h after UV irradiation in fibroblasts from patients 1, 2, 4 and 5, compared to control cell line (individual non affected with CS) and CSA-mutated cell line (CS). Error bars represent SEM, standard error of the mean. (B) Unscheduled DNA synthesis (UDS) measured 23 h after UV irradiation in fibroblasts from patients 1, 2, 4 and 5, compared to control cell line (individual non affected with CS) and XPF-mutated cell line (XP). A.U.: arbitrary units. (C–E) Study of CS transcriptomic signature and ATF3 expression in DYRK1A patients 1, 2, 4 and 5 compared to controls, CSA and CSB patients. Expression level of CDK5RAP2, NIPBL, DYRK1A, NRG1and ATF3 genes was studied by quantitative RT-PCR in the different cell lines 24 h hours after treatment with 20 J/m2 UV-C. Gene expression at 24 h was normalized to 0 h and to GAPDH expression. Error bars represent standard deviation. T-test was performed to compare the expression level of each gene in CSA, CSB and DYRK1A patients to the expression level in control cell lines, applying Welch’s correction: ns: not significant; *p < 0.005;***p < 0.001.
Here we report on a cohort of 11 DYRK1A patients initially suspected to be affected by nucleotide excision repair disorders. This illustrates the clinical overlap between these disorders that may be connected by disturbance of common molecular pathways.
The DYRK1A gene, located on chromosome 21, codes for a serine/threonine dual specificity kinase. Due to the multiplicity of functions performed by its target proteins, DYRK1A is involved in many cellular functions such as cell cycle regulation, apoptosis, synaptic functions, cytoskeletal maintenance and metabolism. DYRK1A is also thought to play a role in the regulation of splicing and transcription. This dosage sensitive protein plays a critical role in neuronal differentiation and brain development (Ananthapadmanabhan et al., 2023).
Haploinsufficiency of the DYRK1A gene causes a rare autosomal-dominant intellectual developmental disorder (MRD-7 or DYRK1A syndrome, OMIM #614104). This syndrome manifests with characteristic features such as microcephaly, facial dysmorphism, severe intellectual disability, autism spectrum disorder (ASD), speech impairment and seizures (Bronicki et al., 2015; Ji et al., 2015; van Bon et al., 2016; Evers et al., 2017; Courraud et al., 2021). This condition is now recognized as one of the most frequent genetic causes of intellectual disability (Deciphering Developmental Disorders Study, 2017).
Among the 11 DYRK1A variants described in this cohort, four have never been described before. These are 3 truncating variants (p.(Trp345Ter), p.(Val204GlyfsTer7), p.(Val237_Leu241delinsGlu) and p.(Cys235LeufsTer5)) and one in-frame variant p.Val237_Leu241delinsGlu whose pathogenicity has been confirmed by reduced DYRK1A protein expression, reduced nuclear localization and autophosphorylation inability.
Of the 11 variants identified, de novo occurrence was confirmed in six cases. The remaining variants, for which segregation analysis was not possible, include four previously reported pathogenic variants and one novel variant (p.(Val237_Leu241delinsGlu)), whose pathogenicity has been validated through functional studies.
Although initially suspected of NER disease, the 11 DYRK1A patients of the present cohort are indistinguishable from previously reported DYRK1A cases by comparing the clinical DYRK1A score measured on a 20-point scale. Two of the main symptoms observed in DYRK1A patients in general and in this cohort in particular, intellectual disability and microcephaly, are also systematically observed in CS patients. Other frequently observed symptoms in this cohort such as feeding troubles and ataxic gait/hypertonia are also commonly found in CS (Supplementary Table S2). The main facial feature shared by the two syndromes is deep-set eyes (Supplementary Figure S1). Despite this striking clinical overlap, some symptoms distinguish the two conditions. DYRK1A patients can be distinguished to CS type I patients by a specifically severely impaired language, more frequent seizures and autistic behavior or anxiety. Whereas growth delay is a common feature in both disorders, it is more severe in CS patients (median height −6.62 ± 3.09 SD in a large CS patients cohort (Wilson et al., 2016) compared to −2.6 ± 1.4 DS in the present DYRK1A cohort). Moreover, bilateral cataracts and pigmentary retinopathy are more frequent in CS patients. Hearing loss is described for 2 patients in our cohort of 11 patients, while to our knowledge, sensorineural hearing loss had only been reported in one DYRK1A patient previously in the literature (Meissner et al., 2020) and is a common feature in CS. Hair anomalies can be observed in DYRK1A syndrome and in TTD (Stefanini et al., 2010; Courraud et al., 2021). Cutaneous photosensitivity is specific to NER disorders (Wilson et al., 2016). Cerebellar and cerebral atrophy together with white matter anomalies and myelination anomalies may be seen in both DYRK1A and NER disorders whereas cerebral calcifications typical of CS have not been observed in DYRK1A patients (Koob et al., 2016; Courraud et al., 2021).
Other patients initially suspected of having Cockayne syndrome have been reported in the literature. One such case involved a patient with growth delay, psychomotor impairment, photosensitivity, and brain calcifications, who was ultimately diagnosed with Aicardi-Goutières syndrome due to mutations in the SAMHD1 gene (Senju et al., 2022). More recently, four patients with a Cockayne syndrome-like phenotype, but with a normal response in recovery of RNA synthesis after UV irradiation, were diagnosed with MORC2-related disorders (Stafki et al., 2023). Our report highlights that DYRK1A syndrome should now be considered a new differential diagnosis for Cockayne syndrome and included in diagnostic panels for repair disorders.
Transcriptomic signature studied in both CSB and CSA Cockayne patients upon genotoxic (UV) stress has revealed an upregulation of transcriptional repressor ATF3 and downregulation of ATF3-dependent genes (Kristensen et al., 2013; Epanchintsev et al., 2017, 2020). Interestingly, DYRK1A was reported as one of the most down-regulated ATF3-dependent genes in CS fibroblasts. Moreover, several DYRK1A interactome studies have resulted in the discovery of a novel role of DYRK1A in DNA repair (Guard et al., 2019; Menon et al., 2019; Roewenstrunk et al., 2019). We therefore wanted to test whether the cells from our DYRK1A patients had CS cell characteristics. DYRK1A fibroblasts, available for patients 1, 2, 4 and 5, did not exhibit any NER defect (normal RRS and UDS assay), nor ATF3 upregulation or ATF3-gene downregulation after UV exposure (Figure 2). These results are in accordance with normal UDS assay recently found by Bélanger et al in post-UV HeLa cells treated with siRNA against Dyrk1A (Bélanger et al., 2023). Nevertheless the authors do not exclude a non-canonical role of DYRK1A in S-phase global-genome repair (Bélanger et al., 2023). An indirect link between DYRK1A and DNA repair has also been demonstrated via phosphorylation regulation of the DREAM complex, a repressor of the various DNA repair systems in C. elegans somatic cells. (Bujarrabal-Dueso et al., 2023). Finally, the identification of NER-independent pathways for the resolution of DNA damage on actively transcribed strands suggests new pathophysiological mechanisms to explore, in which the pleiotropic DYRK1A protein could play a role (Bujarrabal-Dueso et al., 2023). Altogether, these results points the fact that DYRK1A is not involved in canonical transcription-coupled repair, the NER sub-pathway impaired in CS. Even if a CS deficit leads to downregulation of DYRK1A, the reciprocal is not the case. DYRK1A is probably involved downstream of CS in the molecular cascade and previously published interactome studies revealed no protein interactions between DYRK1A and CSA nor CSB (Guard et al., 2019; Roewenstrunk et al., 2019). Nevertheless, these common molecular pathways can perhaps explain some similar clinical symptoms that we previously discussed between the two diseases.
To conclude, we reported 11 DYRK1A patients initially suspected to be affected by nucleotide excision repair disorders. These two disorders share many symptoms, leading to identified DYRK1A haploinsufficiency syndrome as a main differential diagnosis of Cockayne syndrome. We showed that these DYRK1A patients were clinically indistinguishable from other DYRK1A patients, that DYRK1A patients’ cell lines did not exhibit any NER defect and did not share the CS transcriptomic signature. Nevertheless, DYRK1A having been identified as a down-regulated gene in CS cells, it could be hypothesized that the clinical overlap relies on the dysregulation of a common molecular pathway, DYRK1A acting downstream of CSA/CSB.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article.
The studies involving humans were approved by French ethics committee (Comité de protection des personnes (CPP) Alsace n°1, France). The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin. Written informed consent was obtained from the individual(s), and minor(s)’ legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
NM: Methodology, Writing – original draft, Writing – review & editing, Conceptualization, Investigation. JC: Formal analysis, Writing – review & editing. IB: Writing – original draft, Investigation. CaO: Writing – original draft, Investigation. TO: Resources, Writing – review & editing. AL: Resources, Writing – review & editing. FL: Resources, Writing – original draft. DL: Resources, Writing – original draft. SeM: Resources, Writing – original draft. ShM: Resources, Writing – original draft. ClO: Resources, Writing – original draft. MW: Resources, Writing – original draft. VL: Methodology, Writing – review & editing, Writing – original draft, Conceptualization, Resources. NC: Conceptualization, Writing – original draft, Writing – review & editing, Methodology.
The author(s) declare that financial support was received for the research and/or publication of this article. This research was supported by financial funding from the Jérôme Lejeune Foundation and Agence de la Biomédecine.
We sincerely thank Amélie Piton for her contributions to this project, including scientific advices and support in facilitating publication. Her expertise was essential in advancing this work. The authors are also deeply grateful to all participating patients and their families.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors declare that no Gen AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnins.2025.1554093/full#supplementary-material
SUPPLEMENTARY FIGURE S1 | Comparison of facial dysmorphia between DYRK1A and Cockayne syndromes. (A) Patient 4 with DYRK1A syndrome. Image previously published in Bronicki et al., 2015 (patient #10). (B) CS patient with ERCC6 mutations at a similar age. DYRK1A syndrome is characterized by bitemporal narrowing, deep-set eyes, a thin upper vermilion and dysplastic ears. In Cockayne syndrome, facial dysmorphia changes over time and is mainly marked by microcephaly and loss of subcutaneous fat, resulting in a protruding nose and sunken eyes.
SUPPLEMENTARY TABLE S1 | Primer sequences for ATF3 and ATF3-dependent genes expression level studies.
SUPPLEMENTARY TABLE S2 | Detailed clinical features of the DYRK1A patients included in the present cohort (Y = presence or N = absence of each sign at latest known visit; NA and empty cells = not available data; SD: standard deviation).
SUPPLEMENTARY TABLE S3 | Calculation of the DYRK1A clinical score.
Ananthapadmanabhan, V., Shows, K. H., Dickinson, A. J., and Litovchick, L. (2023). Insights from the protein interaction universe of the multifunctional “goldilocks” kinase DYRK1A. Front. Cell Dev. Biol. 11:1277537. doi: 10.3389/fcell.2023.1277537
Bélanger, F., Roussel, C., Sawchyn, C., St-Hilaire, E., Gezzar-Dandashi, S., Kimenyi Ishimwe, A. B., et al. (2023). A genome-wide screen reveals that Dyrk1A kinase promotes nucleotide excision repair by preventing aberrant overexpression of cyclin D1 and p21. J. Biol. Chem. 299:104900. doi: 10.1016/j.jbc.2023.104900
Bronicki, L. M., Redin, C., Drunat, S., Piton, A., Lyons, M., Passemard, S., et al. (2015). Ten new cases further delineate the syndromic intellectual disability phenotype caused by mutations in DYRK1A. Eur. J. Hum. Genet. 23, 1482–1487. doi: 10.1038/ejhg.2015.29
Bujarrabal-Dueso, A., Sendtner, G., Meyer, D. H., Chatzinikolaou, G., Stratigi, K., Garinis, G. A., et al. (2023). The DREAM complex functions as conserved master regulator of somatic DNA-repair capacities. Nat. Struct. Mol. Biol. 30, 475–488. doi: 10.1038/s41594-023-00942-8
Calmels, N., Greff, G., Obringer, C., Kempf, N., Gasnier, C., Tarabeux, J., et al. (2016). Uncommon nucleotide excision repair phenotypes revealed by targeted high- throughput sequencing. Orphanet J. Rare Dis. 11:26. doi: 10.1186/s13023-016-0408-0
Courraud, J., Chater-Diehl, E., Durand, B., and Vincent, M. (2021). Integrative approach to interpret DYRK1A variants, leading to a frequent neurodevelopmental disorder. Genet. Med. 23, 2150–2159. doi: 10.1038/s41436-021-01263-1
Deciphering Developmental Disorders Study (2017). Prevalence and architecture of de novo mutations in developmental disorders. Nature 542, 433–438. doi: 10.1038/nature21062
Epanchintsev, A., Costanzo, F., Rauschendorf, M.-A., Caputo, M., Ye, T., Donnio, L.-M., et al. (2017). Cockayne’s syndrome a and B proteins regulate transcription arrest after genotoxic stress by promoting ATF3 degradation. Mol. Cell 68, 1054–1066.e6. doi: 10.1016/j.molcel.2017.11.009
Epanchintsev, A., Rauschendorf, M.-A., Costanzo, F., Calmels, N., Obringer, C., Sarasin, A., et al. (2020). Defective transcription of ATF3 responsive genes, a marker for Cockayne syndrome. Sci. Rep. 10:1105. doi: 10.1038/s41598-020-57999-4
Evers, J. M. G., Laskowski, R. A., Bertolli, M., Clayton-Smith, J., Deshpande, C., Eason, J., et al. (2017). Structural analysis of pathogenic mutations in the DYRK1A gene in patients with developmental disorders. Hum. Mol. Genet. 26, ddw409–ddw526. doi: 10.1093/hmg/ddw409
Ferri, D., Orioli, D., and Botta, E. (2020). Heterogeneity and overlaps in nucleotide excision repair disorders 97, 12–24. doi: 10.1111/cge.13545
Guard, S. E., Poss, Z. C., Ebmeier, C. C., Pagratis, M., Simpson, H., Taatjes, D. J., et al. (2019). The nuclear interactome of DYRK1A reveals a functional role in DNA damage repair. Sci. Rep. 9:6539. doi: 10.1038/s41598-019-42990-5
Himpel, S., Panzer, P., Eirmbter, K., Czajkowska, H., Sayed, M., Packman, L. C., et al. (2001). Identification of the autophosphorylation sites and characterization of their effects in the protein kinase DYRK1A. Biochem. J. 359, 497–505. doi: 10.1042/0264-6021:3590497
Ji, J., Lee, H., Argiropoulos, B., Dorrani, N., Mann, J., Martinez-Agosto, J. A., et al. (2015). DYRK1A haploinsufficiency causes a new recognizable syndrome with microcephaly, intellectual disability, speech impairment, and distinct facies. Eur. J. Hum. Genet. 23, 1473–1481. doi: 10.1038/ejhg.2015.71
Jia, N., Nakazawa, Y., Guo, C., Shimada, M., Sethi, M., Takahashi, Y., et al. (2015). A rapid, comprehensive system for assaying DNA repair activity and cytotoxic effects of DNA-damaging reagents. Nat. Protoc. 10, 12–24. doi: 10.1038/nprot.2014.194
Koob, M., Rousseau, F., Laugel, V., Meyer, N., Armspach, J.-P., Girard, N., et al. (2016). Cockayne syndrome: a diffusion tensor imaging and volumetric study. Br. J. Radiol. 89:20151033. doi: 10.1259/bjr.20151033
Kristensen, U., Epanchintsev, A., Rauschendorf, M.-A., Laugel, V., Stevnsner, T., Bohr, V. A., et al. (2013). Regulatory interplay of Cockayne syndrome B ATPase and stress-response gene ATF3 following genotoxic stress. Proc. Natl. Acad. Sci. USA 110, E2261–E2270. doi: 10.1073/pnas.1220071110
Laugel, V. (1993). “Cockayne Syndrome, in GeneReviews®”, (Eds.) M. P. Adam, J. Feldman, G. M. Mirzaa, R. A. Pagon, S. E. Wallace, and A. Amemiya (Seattle (WA): University of Washington, Seattle).
Mayne, L. V., and Lehmann, A. R. (1982). Failure of RNA synthesis to recover after UV irradiation: an early defect in cells from individuals with Cockayne’s syndrome and xeroderma pigmentosum. Cancer Res. 42, 1473–1478. doi: 10.1016/0027-5107(82)90047-1
Meissner, L. E., Macnamara, E. F., D’Souza, P., Yang, J., and Vezina, G. Undiagnosed Diseases Network, et al. (2020). DYRK1A pathogenic variants in two patients with syndromic intellectual disability and a review of the literature. Mol. Genet. Genomic Med. 8:e1544. doi: 10.1002/mgg3.1544
Méjécase, C., Way, C. M., Owen, N., and Moosajee, M. (2021). Ocular phenotype associated with DYRK1A variants. Genes (Basel) 12:234. doi: 10.3390/genes12020234
Menon, V. R., Ananthapadmanabhan, V., Swanson, S., Saini, S., Sesay, F., Yakovlev, V., et al. (2019). DYRK1A regulates the recruitment of 53BP1 to the sites of DNA damage in part through interaction with RNF169. Cell Cycle 18, 531–551. doi: 10.1080/15384101.2019.1577525
Nance, M. A., and Berry, S. A. (1992). Cockayne syndrome: review of 140 cases. Am J Med Genet 42, 68–84. doi: 10.1002/ajmg.1320420115
Nieto Moreno, N., Olthof, A. M., and Svejstrup, J. Q. (2023). Transcription-coupled nucleotide excision repair and the transcriptional response to UV-induced DNA damage. Annu. Rev. Biochem. 92, 81–113. doi: 10.1146/annurev-biochem-052621-091205
Roewenstrunk, J., Di Vona, C., Chen, J., Borras, E., Dong, C., Arató, K., et al. (2019). A comprehensive proteomics-based interaction screen that links DYRK1A to RNF169 and to the DNA damage response. Sci. Rep. 9:6014. doi: 10.1038/s41598-019-42445-x
Schalk, A., Greff, G., Drouot, N., Obringer, C., Dollfus, H., Laugel, V., et al. (2018). Deep intronic variation in splicing regulatory element of the ERCC8 gene associated with severe but long-term survival Cockayne syndrome. Eur. J. Hum. Genet. 26, 527–536. doi: 10.1038/s41431-017-0009-y
Senju, C., Nakazawa, Y., Shimada, M., Iwata, D., Matsuse, M., Tanaka, K., et al. (2022). Aicardi-Goutières syndrome with SAMHD1 deficiency can be diagnosed by unscheduled DNA synthesis test. Front. Pediatr. 10:1048002. doi: 10.3389/fped.2022.1048002
Spitz, M. A., Severac, F., Obringer, C., Baer, S., Le May, N., Calmels, N., et al. (2021). Diagnostic and severity scores for Cockayne syndrome. Orphanet J. Rare Dis. 16:63. doi: 10.1186/s13023-021-01686-8
Stafki, S. A., Turner, J., Littel, H. R., Bruels, C. C., Truong, D., Knirsch, U., et al. (2023). The Spectrum of MORC2-related disorders: a potential link to Cockayne syndrome. Pediatr. Neurol. 141, 79–86. doi: 10.1016/j.pediatrneurol.2023.01.011
Stefanini, M., Botta, E., Lanzafame, M., and Orioli, D. (2010). Trichothiodystrophy: from basic mechanisms to clinical implications. DNA Repair (Amst) 9, 2–10. doi: 10.1016/j.dnarep.2009.10.005
van Bon, B. W., Coe, B. P., de Vries, B. B., and Eichler, E. E. (1993). “DYRK1A Syndrome, in GeneReviews®”, (Eds.) M. P. Adam, J. Feldman, G. M. Mirzaa, R. A. Pagon, S. E. Wallace, and A. Amemiya (Seattle (WA): University of Washington, Seattle).
Van Bon, B. W. M., Coe, B. P., Bernier, R., Green, C., Gerdts, J., Witherspoon, K., et al. (2016). Disruptive de novo mutations of DYRK1A lead to a syndromic form of autism and ID. Mol. Psychiatry 21, 126–132. doi: 10.1038/mp.2015.5
Keywords: DYRK1A gene, nucleotide excision repair (NER), Cockayne syndrome, trichothiodystrophy, ERCC6/CSB gene, ERCC8/CSA gene
Citation: Le May N, Courraud J, Boujelbène I, Obringer C, Ogi T, Lehmann AR, Laffargue F, Lehalle D, Mizuno S, Mohammed S, Ormières C, Willems M, Laugel V and Calmels N (2025) Clinical and molecular overlap between nucleotide excision repair (NER) disorders and DYRK1A haploinsufficiency syndrome. Front. Neurosci. 19:1554093. doi: 10.3389/fnins.2025.1554093
Edited by:
Yusuke Takatsuru, Toyo University, JapanReviewed by:
Francesco Nicita, Bambino Gesù Children’s Hospital (IRCCS), ItalyCopyright © 2025 Le May, Courraud, Boujelbène, Obringer, Ogi, Lehmann, Laffargue, Lehalle, Mizuno, Mohammed, Ormières, Willems, Laugel and Calmels. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nadège Calmels, bmFkZWdlLmNhbG1lbHNAY2hydS1zdHJhc2JvdXJnLmZy
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.