 Anping Chai
Anping Chai
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Neurosci., 14 March 2025
Sec. Neurodevelopment
Volume 19 - 2025 | https://doi.org/10.3389/fnins.2025.1552145
This article is part of the Research TopicExploring E/I Balance in Animal Models of Neurodevelopmental Disorders: Effects on Circuits and Social DynamicsView all articles
Neuronal excitation-inhibition (E/I) balance is essential for maintaining neuronal stability and proper brain functioning. Disruptions in this balance are implicated in various neurological disorders, including autism spectrum disorder, schizophrenia and epilepsy. The E/I balance is thought to be primarily mediated by intrinsic excitability, governed by an array of voltage-gated ion channels, and extrinsic excitability, maintained through a counterbalance between excitatory synaptic transmission primarily mediated by excitatory transmitter glutamate acting on excitatory ion-tropic glutamate receptors and inhibitory synaptic transmissions chiefly mediated by GABA or glycine acting on their respective inhibitory ion-tropic receptors. However, recent studies reveal that neurotransmitters can exhibit interactions that extend beyond their traditional targets, leading to a phenomenon called neurotransmitter-receptor crosstalk. Examples of such crosstalks include earlier discovery of inhibitory glycine functioning as co-transmitter gating on the NMDA subtype of excitatory glutamate receptor, and the most recent demonstration that shows the excitatory glutamate transmitter binds to the inhibitory GABAA receptor, thereby allosterically potentiating its inhibitory function. These studies demonstrate structurally and physiologically important crosstalk between excitatory and inhibitory synaptic transmission, blurring the distinction between the concepts of classic excitatory and inhibitory synaptic transmission. In this article, evidence supporting the forms of excitatory and inhibitory crosstalks will be briefly summarized and their underlying mechanisms will be discussed. Furthermore, this review will discuss the implications of these crosstalks in maintaining the E/I balance, as well as their potential involvement in synaptic plasticity and cognition in the context of social conditions.
The brain relies on neurotransmitters as messengers to enable precise and efficient communication between neurons. These neurotransmitters are released and bind to receptors, conveying signals that regulate processes including neural development, synaptic plasticity and excitation-inhibition (E/I) of membrane potentials. Disruptions in the E/I balance are believed to be central to the pathogenesis of various neurological disorders, including schizophrenia and autism spectrum disorder (ASD) characterized by social deficits, as well as epilepsy (Fritschy, 2008; Lee et al., 2017; Liu et al., 2021). The E/I balance is primarily regulated by intrinsic excitability, which is controlled by a range of voltage-gated ion channels, and by extrinsic excitability, which is maintained through a balance between excitatory and inhibitory synaptic transmissions (Desai, 2003; Chen L. et al., 2022). In the mammalian central nervous system, excitatory synaptic transmission is primarily driven by the neurotransmitter glutamate, which acts on excitatory ionotropic glutamate receptors, while inhibitory synaptic transmission is predominantly mediated by γ-aminobutyric acid (GABA) or glycine, which act on inhibitory ionotropic GABAA receptor (GABAAR) or glycine receptor (GlyR) respectively (Hyman, 2005). Increasing evidence, however, reveals that neurotransmitter could bind to receptors outside their conventional pairings as mentioned before under certain conditions (Liu et al., 2010; Bianchi et al., 2011; Wen et al., 2022; Piot et al., 2023). This phenomenon of neurotransmitter-receptor crosstalk adds another layer of complexity to the regulation of information processing in the brain. It is plausible that neurotransmitter-receptor crosstalk disturbance may also contribute to E/I imbalance and synaptic plasticity deficit. Therefore, a deeper understanding of mechanism of neurotransmitter-receptor crosstalk would inform the development of therapeutic strategies for treating social disorders related to E/I imbalance.
Neurons communicate primarily via chemical synapses. Typically, the presynaptic neuron releases neurotransmitters, which are stored in vesicles at the axon terminal, transmitting signals to ionotropic or metabotropic receptors on the postsynaptic neuron to facilitate information transfer. This review focus on three types of ionotropic receptors, including the N-methyl D-aspartate receptor (NMDAR) which is cationic receptor, and GABAAR, GlyR which are anionic receptors (Table 1). Each ligand listed in the table could target more than one type of ionotropic receptor, and each ionotropic receptor was the target of more than two kinds of ligand. It is well-known that glycine acts both as an agonist on inhibitory GlyR and as a co-agonist of excitatory NMDAR in the spinal cord and brain. Surprisingly, recent studies have demonstrated that glutamate, a canonical excitatory transmitter, could positively modulate inhibitory GABAAR function in heterologous expression system and hippocampal slices of mice (Wen et al., 2022). Furthermore, one study reported that glutamate could directly activate GlyR as a positive allosteric modulator in spinal cord neurons (Liu et al., 2010), but the other study did not find the direct link (Aubrey et al., 2020). Additionally, other studies have reported that histamine, a canonical transmitter of histamine receptors, could also potentiate NMDAR in cultured hippocampal neurons or recombinant GABAAR in HEK cells (Burban et al., 2010; Bianchi et al., 2011). It should also be noted that neurotransmitter-receptor crosstalk could occur with non-ionotropic receptors. For example, Piot et al. revealed that GABA, a canonical inhibitory transmitter, could enhance glutamate delta-1 receptor (GluD1) function and long-term potentiation of inhibitory synaptic plasticity in mouse hippocampus (Piot et al., 2023). Overall, as shown in Table 1, this type of transmitter-receptor crosstalk is therefore not an exception but could be considered as a common phenomenon in the nervous system.
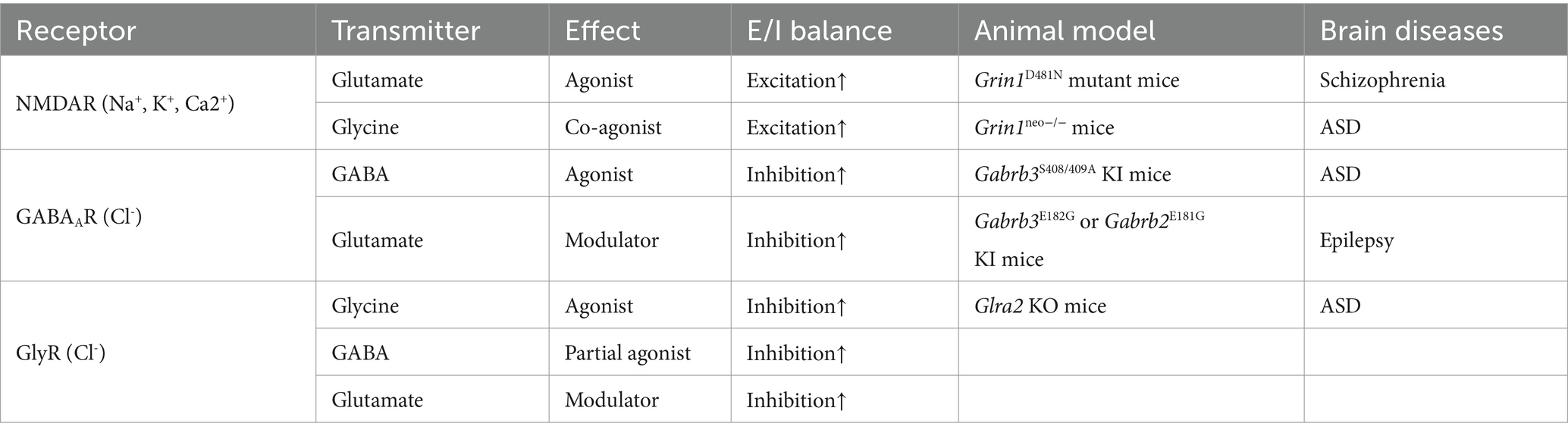
Table 1. Neurotransmitter-receptor crosstalk in animal models.
The molecular mechanisms of glycine acting as co-agonist of NMDAR have been summarized in other reviews (Hanson et al., 2024). Mutations in GluN1, such as D481N, significantly reduce glycine binding affinity in recombinant GluN1/GluN2A receptor and homozygous Grin1D481N transgenic mice (Wafford et al., 1995; Kew et al., 2000). Here, the review will focus on glutamate-GABAAR and GABA/glutamate-GlyR.
The GABAAR is a heteropentameric ionotropic receptor primarily responsible for mediating Cl− /HCO3− current in the brain. The heteropentamer is composed of a combination of 19 homologous subunits from eight classes (α1–6, β1–3, γ1–3, δ, ε, θ, π and ρ1–3). Most common configuration consisting of two α and three β aligned β-α-β-β-α, or two α, two β, and one γ subunits aligned β-α-γ-β-α counter-clockwise when viewed from the extracellular side. The GABA binding sites are located at the interface between the α and β subunits, specifically at the β+/α- interface (Sieghart and Savic, 2018).
Over 30 years ago, it was discovered that the excitatory neurotransmitter glutamate potentiated GABAAR current in acutely isolated hippocampal neurons (Stelzer and Wong, 1989). A recent study identified a novel mechanism through which glutamate can directly modulate the inhibitory GABAAR via allosteric potentiation (Wen et al., 2022). By combining electrophysiological recordings, radioligand binding assays, molecular modeling and mutational analysis, Wen et al. demonstrated that glutamate directly potentiated GABA-evoked currents in recombinant α1β2 GABAAR, with an EC50 of approximately 180 μM, comparable to nearly 1 mM concentration of glutamate in the synaptic cleft during neurotransmission (Clements et al., 1992). It is noteworthy that this potentiation was also mimicked by glutamate analogs, such as AMPA, kainate, and NMDA, as well as by the NMDA receptor antagonist AP5 in recombinant α1β2 and α1β2γ2 GABAAR, indicating some structural specificity for glutamate-like molecules. Molecular modeling and mutational analysis identified a novel glutamate binding pocket at the α+/β- subunit interface. Key residues involved in forming this pocket included K104, E137, and K155 on the α1 or α2 subunit, E181 on the β2 subunit and E182 on the β3 subunit. The mutations of these sites largely eliminated glutamate potentiation without affecting GABA binding affinity on GABAAR.
GlyR is a pentameric receptor that is also permeable to chloride ions. In mammals, there are four α subunit isoforms (α1-α4) and one β subunit. Functional GlyRs can be either homomeric α or heteromeric αβ composition. The EC50 for glycine on recombinant GlyRs generally ranges from 25 μM to 280 μM for α1 and from 46 μM to 541 μM for α2, a typical value for heteromeric receptors is around 100 μM (De Saint Jan et al., 2001; Legendre et al., 2009), which is significantly higher than the glycine concentration at NMDAR. The glycine binding sites are located at the interfaces between the N-terminal domains of neighboring subunits.
De Saint Jan et al. initially reported that GABA could potentiate recombinant human α1 and α2 homomeric glycine receptors current with an EC50 of 14.4–160 mM and 64–200 mM, respectively (De Saint Jan et al., 2001). More recently, GABA was identified as a weak partial agonist of GlyR in neurons from the medial nucleus of the trapezoid body (MNTB) of rats aged P10-P18 (Lu et al., 2008). Electrophysiological recordings, conducted in the presence of GABA receptors and glutamate receptors inhibitors or Zn2+ chelator, revealed that GABA significantly accelerated the decay kinetics of glycine-evoked currents. In MNTB patches, co-application of 10 mM GABA with 1 mM glycine (at physiological concentration) shortened the fast decay time constant and reduced the peak amplitude of glycine-evoked currents by about 25%. Although GABA may overlap with glycine binding sites, the exact binding sites on GlyR remain unclear.
Regarding the interaction between glutamate and GlyR, Liu et al. provided compelling evidence for a novel allosteric potentiation of GlyR-mediated chloride currents by glutamate [Liu et al., 2010; but also see Borghese et al. (2012); Aubrey et al. (2020)]. The glutamate binding sites on GlyR need to be determined.
All the cases of neurotransmitter-receptor crosstalk mentioned above suggested that neurotransmitters often act synergistically to enhance receptor function. This cooperative action of multiple neurotransmitters on a single receptor type indicates that neural signaling is highly cooperative. Therefore, modulating the non-canonical interactions (e.g., glutamate enhancing GABAAR) could provide new avenues for drug development that fine-tune synaptic activity without directly overstimulating canonical pathways. Investigating the non-canonical binding sites and developing compounds that target these sites will expand the toolbox for studying the physiological and pathological engagement of neurotransmitter-receptor crosstalk in the future.
Under physiological condition, excitatory and inhibitory nerve terminals are traditionally believed to be strictly segregated to ensure precise function of neural networks and E/I balance. However, accumulating evidence suggested that the E/I balance could also be modulated by mechanisms of heterosynaptic interplay and neurotransmitter-receptor crosstalk. Heterosynaptic interplay, particularly local dendritic excitatory and inhibitory synaptic crosstalk, has been documented both in vitro and in vivo (Chen et al., 2012; Hayama et al., 2013; Field et al., 2020; Ravasenga et al., 2022). The role of glutamatergic and GABAergic interplay in modulating plasticity and E/I balance has been discussed in several excellent reviews (Chapman et al., 2022; Cupolillo et al., 2024). Such synaptic interplay highlights the synaptic crosstalk occurrence between adjacent synapses within a 3–10 μm space. As for neurotransmitter-receptor crosstalk, it is unlikely that glutamate released from glutamatergic synapse would diffuse and potentiate inhibitory postsynaptic GlyR or GABAAR, given the submillimolar/millimolar concentration required for most neurotransmitter-receptor crosstalks. Here, recent findings suggested two conditions may facilitate this crosstalk.
Neurotransmitters are synthesized by biosynthetic enzymes in neurons. For example, glutamic acid decarboxylase (GAD) catalyzes the conversion of glutamate to GABA. After synthesis, neurotransmitters are transported and concentrated to reach millimolar level in synaptic vesicles (SVs; Edwards, 2007). The uptake of neurotransmitters into SVs is governed by specific vesicular transporters (VTs). These include vesicular glutamate transporters VGLUT1, VGLUT2, and VGLUT3 for glutamate, vesicular GABA transporter/vesicular inhibitory amino acid transporter VGAT/VIAAT for GABA and glycine (Blakely and Edwards, 2012). Distinct VTs in the same SVs are essential for corresponding neurotransmitter acting in neurotransmitter-receptor crosstalk. That is, the co-localization of VGLUT and VGAT in the same SV is necessary for glutamate-GABAAR crosstalk. Neurotransmitters are then released through a calcium-dependent SV membrane fusion process triggered by presynaptic depolarization. This section focuses on co-expression of VGLUT and VGAT in the same SV, a phenomenon which occurs more frequently in the brain (Upmanyu et al., 2022).
Regarding the allosteric potentiation of GABAAR by glutamate, accumulating evidence supported GABA/glutamate co-release in subsets of GABAergic synapses (Figure 1A). (1) Using electron microscopic postembedding immunogold, Stensrud et al. revealed co-expression of VGLUT3 and VGAT in the same vesicle membrane of GABAergic terminals in the hippocampus and cortex of rats and mice (Stensrud et al., 2013; Stensrud et al., 2015), indicating GABA/glutamate co-packaging in same vesicle. Following work by Pelkey et al. showed that VGLUT3/VGAT overlapping in cholecystokinin (CCK+) expressing hippocampal CA1 and CA3 interneurons was minimal in early postnatal days, but gradually increased from the juvenile stage to adulthood (Pelkey et al., 2020), suggesting GABA/glutamate could co-release to pyramidal neurons in adult mice. (2) Coexistence of VGLUT2 and VGAT has been found in excitatory mossy fiber terminals of hippocampal CA3 region, cerebellar mossy fiber terminals and GABAergic basket cells terminals in postnatal day 5–15 rat but not adult, indicating co-localization of glutamate and GABA in same vesicles at these terminals (Zander et al., 2010). Immunogold staining also showed localization of α/β subunits of GABAAR in mossy fiber synapses (Bergersen et al., 2003). Subsequent functional tests found that activation of mossy fiber boutons attached to CA3 neurons could induce GABAergic inhibitory postsynaptic currents (IPSCs) in two-week-old rats (Beltrán and Gutiérrez, 2012; but also (see Cabezas et al., 2012)). (3) Surprisingly, co-expression of GABA and glutamate in mossy fiber terminals was also discovered in hippocampi from adolescents with epilepsy (Sandler and Smith, 1991). In adult rat epilepsy model, a single pentylenetetrazol (PTZ) treatment elicited a fast inhibitory postsynaptic potential (IPSP) component in mossy fiber of CA3 (Gutierrez, 2000), indicating that glutamate and GABA co-release may occur under pathological condition. (4) Furthermore, Omiya et al. found VGLUT3-positive CCK+ cell terminals and α1-containing GABAAR at invaginating synapse in basal amygdala of adult mice (Omiya et al., 2015). (5)Additionally, one study reported GABA/glutamate co-release from individual vesicles at entopeduncular nucleus-lateral habenula (EP-LHb) synapses, while the other study provide evidence that GABA and glutamate are stored and released from distinct, non-overlapping pools of synaptic vesicles at the same synapse. A follow-up study implemented a combination of optogenetics, electrophysiology and computational modeling determined that glutamate and GABA are co-packaged in the same synaptic vesicles in EP Sst + neurons projecting to the LHb (Kim et al., 2022).
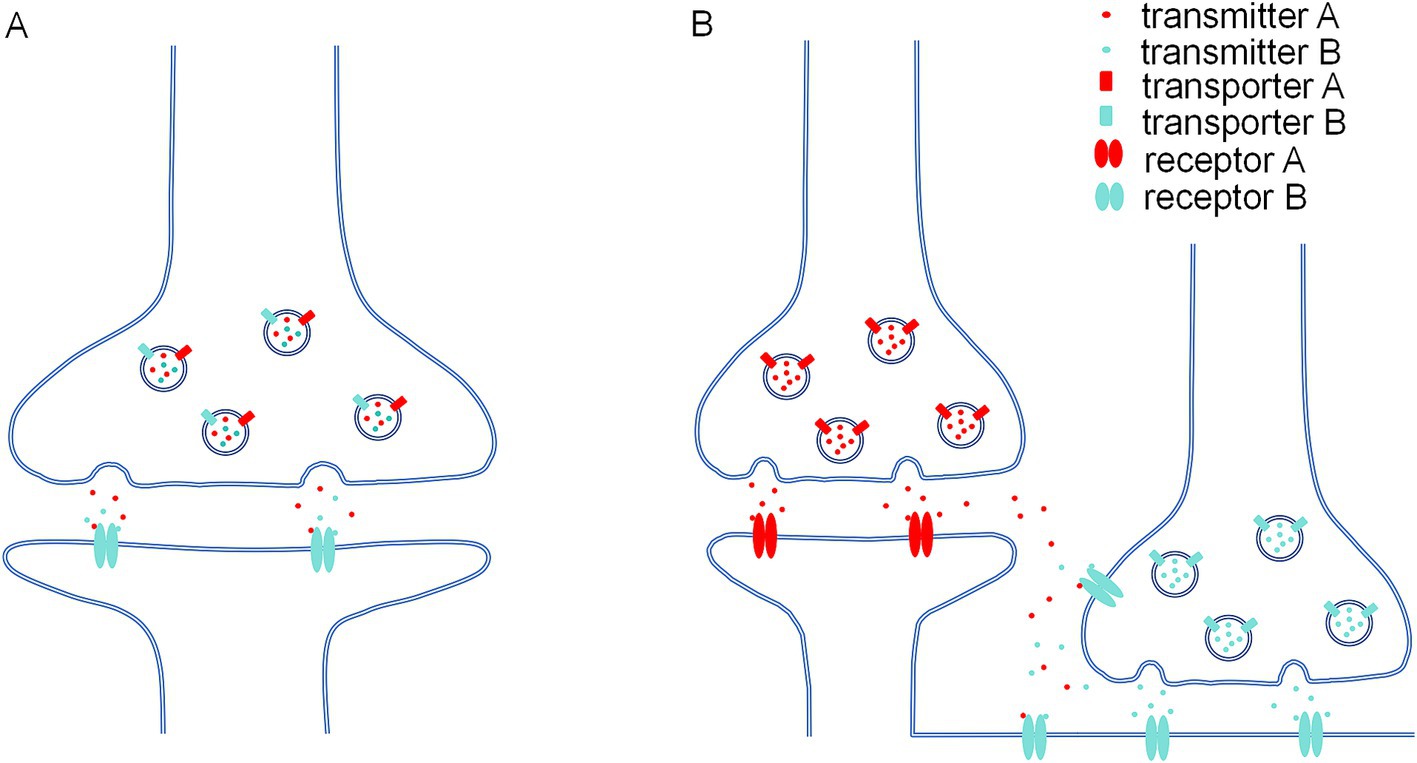
Figure 1. Two possible conditions in which neurotransmitter-receptor crosstalk occurs. (A) At the co-release synapses which two types of transmitters (e.g., glutamate and GABA) are co-packaging in the same presynaptic vesicles due to co-localization of two kinds of synaptic vesicular transporter (e.g., VGLUT and VGAT). Postsynaptic receptor binds to both transmitters (e.g., glutamate could allosterically potentiate GABA dependent GABAAR current). (B) Certain physiological or pathological conditions would cause neurotransmitter spillover, both transmitter A and transmitter B would bind to extrasynaptic or presynaptic receptor B (e.g., high frequency activity would induce glutamate and GABA spillover on extrasynaptic or presynaptic GABAAR).
In terms of GABA-GlyR crosstalk, it is facilitated by the vesicular inhibitory amino acid transporter (VIAAT, also known as VGAT), which could package both glycine and GABA into the same synaptic vesicles (Wojcik et al., 2006). The co-expression of GABA and glycine has been observed in rat spinal cord neurons (Ishibashi et al., 2013), adult mouse retina amacrine neurons (Pérez-León et al., 2022), P10-P23 rat auditory brain stem nucleus (Lu et al., 2008) and adult mouse cerebellum (Milanese et al., 2014), indicating potential crosstalk between GABA and GlyR.
Recent technological advances, such as spatiotemporal omics technology (Xu et al., 2022), have enabled brain-wide mapping of the spatial distribution of neurotransmitter transporters and synthetic enzymes in neurons, providing clearer insights into potential sites for co-releasing transmitters. However, it is important to recognize that the detection of transcripts for neurotransmitter synthetic enzymes does not necessarily imply the presence of the enzymes themselves. Therefore, functional and protein-level verification about neurotransmitter-receptor crosstalk in these neural circuits is warranted.
Neurotransmitter-receptor interaction could also been observed at non-postsynaptic locations, including extrasynaptic and presynaptic sites (Figure 1B). Neurotransmitters released at central synapses may exceed the capacity for removal by reuptake and degradation, diffuse beyond their intended synaptic cleft and activate receptors at adjacent synapses or extrasynaptic sites. The concentration of extrasynaptic neurotransmitter is at low-micromolar level under physiological condition. For instance, the concentration of extrasynaptic glycine and glutamate is around 1–2.5 μM in striatum of conscious rats (Harsing and Matyus, 2013). But under circumstances such as high synaptic activity, or pathological conditions like stroke, epilepsy and transporter dysfunctions, the concentration of extrasynaptic neurotransmitter spillover could reach to submillimolar level. For example, glutamate concentrations can increase to up to 200 μM in the plasma of ischemic stroke patients (Castillo et al., 2016). Glutamate spillover has been found at ectopic sites or induced by high frequency stimulation from parallel fiber synapses in cerebellum and the olfactory bulb of rat (Isaacson, 1999; Balakrishnan et al., 2014). Mutations in the glutamate transporter GLAST (EAAT1) lead to transporter dysfunction and seizures in human (Jen et al., 2005). Genetic knockout of the glutamate transporter GLT-1 (EAAT2) in mice leads to severe epilepsy and increased extracellular glutamate levels (Tanaka et al., 1997).
Ionotropic receptors at extrasynaptic sites exhibit relatively high affinity for neurotransmitter, given the relative low concentration of neurotransmitter spillover. In adult hippocampus and cortex, synaptic NMDAR are primarily composed of the low affinity GluN2A-containing subtype for glutamate and glycine, whereas extrasynaptic NMDAR are predominantly the high affinity GluN2B-containing subtype for both neurotransmitters (Ge and Wang, 2023). Synaptic GABAARs typically consist of α1-3, β2-3 and γ2 subunits, which exhibit low affinity to GABA and mediate fast phasic currents. In contrast, extrasynaptic GABAARs often contain α4-6, β2-3 and/or δ subunits, enabling them to respond to low ambient GABA and regulate slow tonic currents (Brickley and Mody, 2012; Liu et al., 2018). For instance, extrasynaptic α5-containing GABAARs have been detected at hippocampal pyramidal cells from P56-P84 mice (Magnin et al., 2019). In cultured hippocampal neurons, the glutamate analog AP5 potentiated both phasic inhibitory postsynaptic currents and tonic currents (Wen et al., 2022), suggesting that allosteric modulation occurs at both synaptic and extrasynaptic GABAAR. Also, the α6-containing GABAAR could be activated by GABA spillover at cerebellum of 12-day-old rats (Rossi and Hamann, 1998). Similarly, glutamate spillover has been reported in cerebellar climbing fiber to molecular layer interneurons from P12-P25 mice (Malhotra et al., 2021). These results suggest potential modulatory effect of glutamate on extrasynaptic GABAAR. Interestingly, GABA released from the histamine/GABA co-releasing neurons in hypothalamic tuberomammillary nucleus (TMN) acts on extrasynaptic GABAAR (Yu et al., 2015), though whether histamine could modulate these GABAAR needs further investigation. Functional extrasynaptic α2 or α3 homomeric GlyRs have been found in hippocampus from 3-month-old rats (Aroeira et al., 2011), and in dorsal striatum, prefrontal cortex (PFC) and hippocampus from P21–P50 mice (McCracken et al., 2017). But the glycine EC50 were reported around 500 μM in these regions, and the mechanisms by which GABA or glutamate spillover modulates extrasynaptic GlyRs remains to be elucidated.
Accumulating studies have demonstrated the presence of ionotropic receptors at presynaptic sites, acting as autoreceptors. Presynaptic GABAARs have been identified at thalamocortical glutamatergic terminals of P28-P30 rats (Wang et al., 2019). It has also been reported that α2 subunit and δ subunit containing GABAAR are positioned at hippocampal mossy fiber boutons, where they could co-release GABA and glutamate in 3–12 week-old rats as mentioned before (Ruiz et al., 2003; Alle and Geiger, 2007; Ruiz et al., 2010). These findings suggest that presynaptic GABAAR could function as autoreceptors for the co-released neurotransmitters. Similarly, NMDAR composed of GluN2A, GluN2B or GluN3A have also been identified as presynaptic autoreceptor in various regions, including rat cerebellar parallel fiber-Purkinje cell synapses, the spinal cord dorsal horn, hippocampal glutamatergic terminals, and the mice visual cortex at different ages (Liu et al., 1994; Charton et al., 1999; Bidoret et al., 2009; Larsen et al., 2011; Musante et al., 2011). The activation of presynaptic NMDAR is facilitated by the co-release of glycine from glutamatergic synapses (Cubelos et al., 2005; Raiteri et al., 2005; Muller et al., 2013; Cubelos et al., 2014).
Presynaptic homomeric GlyRs have been discovered in rat excitatory synapse, including calyx of Held in MNTB, spinal cords and ventral tegmental area (VTA) GABAergic neurons during development (Colin et al., 1998; Turecek and Trussell, 2001; Ye et al., 2004; Hruskova et al., 2012). The release of glutamate or GABA in these synapses enables the crosstalk with presynaptic GlyR. These findings suggest that presynaptic ionotropic receptors, such as GABAAR, NMDAR and GlyR, could be modulated by presynaptic neurotransmitters co-release or spillover.
Among the neurotransmitter-receptor crosstalks listed in Table 1, particularly those involving glycine-NMDAR, glutamate-GABAAR, and GABA-GlyR, a common feature is that the cross-matched ligand consistently acts in the same direction as the orthosteric neurotransmitter. For instance, glycine enhances NMDAR function together with glutamate, glutamate potentiates GABAAR with GABA, and GABA facilitates GlyR as glycine. This synergistic action observed in the crosstalk between different neurotransmitters may have important physiological and functional implications for neural circuits. E/I balance is the equilibrium between excitatory and inhibitory synaptic inputs within neural circuits. This balance is essential for maintaining optimal neuronal firing rates and overall network stability. Disruption of the E/I balance can contribute to pathogenesis of various neurological disorders characterized by hyperactivity in neural circuits, including epilepsy, ASD and schizophrenia characterized by social deficits.
Activated by the principal excitatory neurotransmitter glutamate, NMDAR is one of the major ionotropic glutamate receptors responsible for excitatory synaptic inputs. Missense mutations in the GRIN1 gene, which encodes the GluN1 subunit of NMDAR targeting by glycine, have been associated with epileptic encephalopathy or schizophrenia (Ohba et al., 2015; Ju and Cui, 2016). Transgenic Grin1neo-/- mice exhibited schizophrenic-like phenotypes, including hyperlocomotor activity, increased stereotypic movement, impaired sensorimotor gating and reduced social interactions (Gandal et al., 2012). The NMDAR hypoactivity mediated GABAergic inhibitory dysfunction was believed associating with pathogenesis of schizophrenia (Cohen et al., 2015). The D481N mutation in GluN1 lead to marked reduction in glycine co-agonist affinity at the NMDAR. Homozygous Grin1D481N mice exhibited impairment in hippocampal long-term potentiation (LTP), a synaptic plasticity mechanism underlying learning and memory (Bliss and Collingridge, 1993). Furthermore, these mice showed significant deficits in hippocampus dependent spatial learning task, such as Morris Water Maze, and reduced sensitivity to NMDA-induced seizures (Kew et al., 2000). Importantly, restoring the expression of wild type GluN1 in adult mice with congenital loss-of-function allele of Grin1 could rescue most of the behavioral phenotypes (Mielnik et al., 2021). Interestingly, a recent study reported that injection of HA-996, a blocker of the glycine-binding site on NMDAR, in periaqueductal gray (PAG) of juvenile rat could interfere the social play behavior, while blocking of astrocytic glycine transporter 1 (GlyT1) inhibitor ALX-5407 could reverse the deficits (Dvorzhak et al., 2024). It should be noted that D-serine or another partial agonist at the glycine-binding site of NMDAR, D-cycloserine, could enhance hippocampal LTP in wild-type rats and alleviate social abnormality in ASD mouse models (Yang et al., 2003; Won et al., 2012; Li et al., 2015). In conclusion, glycine-NMDAR crosstalk is crucial for maintaining E/I balance and facilitating synaptic plasticity, both of which are essential for cognitive processes like spatial learning and social interaction.
GABAAR is the main target of treating epilepsy, insomnia, anxiety and ASD characterized by E/I imbalance (Ghit et al., 2021; Vien et al., 2015). As for glutamate-GABAAR crosstalk, although there is no available inhibitor specifically blocking the binding of glutamate to GABAAR, two lines of transgenic knock-in (KI) mice carrying different mutations that impaired the glutamate allosteric potentiation on the GABAAR, have been generated in two recent studies (Wen et al., 2022; Du et al., 2023). Wen et al. generated KI mice carrying a mutation in the β2 subunit of GABAAR (β2E181G). Du et al. generated KI mice harboring another mutation in the β3 subunit (β3E182G). These two mutations effectively disrupted the allosteric potentiation by glutamate without affecting the GABA-mediated activation of GABAAR. In β2E181G KI mice, electrophysiological recording in CA1 neurons of hippocampal slice from these mice confirming the impairment of glutamate or glutamate-like AP5 potentiation of GABAAR current. Since the GABAAR current was induced by either micropressure injection of GABA or electrical theta-burst stimulation, indicating a predominant extrasynaptic GABAAR component. The β2E181G KI mice exhibited behavioral phenotypes indicative of increased neuronal excitability, including heightened sensitivity to noxious mechanical and temperature stimuli and a reduced threshold for seizure induction by kainate acid. More comprehensive tests were conducted on β3E182G KI mice. The β3E182G KI mice also had increased susceptibility to seizures induced by kainate acid, and decreased thresholds to both pressure and temperature, indicating elevated E/I imbalance. Furthermore, β3E182G KI mice exhibited enhanced hippocampus-related learning and memory, and impaired social interactions. These mice also showed reduced anxiety-like behaviors. Importantly, re-expression of wild-type α1β3-containing GABAARs in the dorsal hippocampal CA1 was sufficient to rescue the observed abnormalities in glutamate potentiation and behavioral deficits, supporting the critical role of extrasynaptic glutamate-GABAAR crosstalk in maintaining E/I balance and normal brain function.
Glycine-gated GlyR is also an important inhibitory ionotropic receptor in mammalian spinal cord and brain. Mutations in α1 subunit of GlyR have been linked to hyperekplexia characterized by exaggerated startle reflex and increased muscle tone. Mutations in the α2 subunit are associated with ASD (Pilorge et al., 2016; Chen X. et al., 2022). As for GABA-GlyR or glutamate-GlyR crosstalk, since the exact binding sites are still unknown and there are no specific antagonists targeting the binding sites, or transgenic animals with binding site mutation, the physiological role of GABA-GlyR or glutamate-GlyR crosstalk is largely elusive. In rat MNTB where GABA was co-packaging with glycine in same synaptic vesicles, Lu et al. demonstrated that co-releasing GABA could enhance the temporal precision of glycine inhibition, which might be important for sound localization (Lu et al., 2008). Therefore, the physiological significance of GABA or glutamate modulating GlyR requires further investigation.
Under pathological conditions such as ischemia or epilepsy, increased glycine and glutamate concentrations due to spillover can lead to excessive activation of extrasynaptic GluN2B-dependent NMDAR and downstream signaling, potentially contributing to excitotoxicity and neuronal damage (Lasley, 1991; Ge et al., 2020; Ge and Wang, 2023) Pathological spillover can also result from altered expression or function of neurotransmitter transporters, such as the excitatory amino acid transporters (EAATs) for glutamate. Dysfunction of these transporters can lead to increased extracellular levels of glutamate, excessively promoting receptor activation and potentially leading to neurotoxic outcomes. For example, mutations and dysfunction of EAAT2 (GLT-1) have been linked to epilepsy and schizophrenia (Rakhade and Loeb, 2008; Wang et al., 2022). Animal studies demonstrated that EAAT2 dysfunction can lead to increased seizure and cortical injury susceptibility due to impaired glutamate clearance from synapses (Tanaka et al., 1997). However, Nong et al. demonstrated that glycine treatment at 100 μM, but not 1 μM, primes the NMDAR for clathrin-dependent internalization, a process that requires subsequent activation by glutamate (Nong et al., 2003). By regulating the surface expression of NMDARs, glycine-NMDAR crosstalk may help mitigate excitotoxic damage during conditions like stroke or seizure.
As summarized in Table 1, the consequence of neurotransmitter-receptor crosstalk disruption have been always linked to pathological phenotypes, including social deficits and seizures, in transgenic knock-in mice with mutations in cross-matched ligand binding sites. A critical question that arises is how to investigate the potential causal relationship between neurotransmitter-receptor crosstalk and cognitive functions under physiological and psychological conditions during behavior? One direct approach is the development of novel antagonists that specifically target on the cross-matched ligand binding sites of receptor and preventing the potentiation without affecting orthosteric neurotransmitter activation. But such antagonists are often lacking, as their development requires the knowledge of binding sites based on co-crystallization of the ligand and receptor, which is challenging. As mentioned above and summarized in Table 1, another approach involves using transgenic knock-in mice with mutations in the cross-matched ligand binding site of the receptor, leaving the orthosteric neurotransmitter binding unaffected. However, this method has several limitations, including potential developmental compensation by other receptors or signaling pathways, as well as difficulties in manipulating the system temporally and spatially across the whole brain. Given that these neurotransmission crosstalks phenomenon occur in various neural circuits and at different cellular positions across developmental stages, a more targeted approach is necessary. For example, in order to study the role of neurotransmitter-receptor crosstalk in certain neural circuit in regulating social behaviors, one could use transgenic mice with a tamoxifen-inducible system to reduce the VGLUT gene, flanked by loxP sites, by injecting a VGAT-Cre-containing virus into specific neural circuits to inhibit glutamate-GABAAR crosstalk in a temporally and spatially controlled manner. Therefore, a combination of these methods is required to fully understand the physiological and pathological role of neurotransmitter-receptor crosstalk. It is crucial to develop potential tools for manipulating neurotransmitter-receptor crosstalk and observe the impact on network dynamics during behavior. Studying of the crosstalk at network level will be an important direction for future research, involving the modeling of specific networks and sub-networks.
The review has discussed the concept of neurotransmitter-receptor crosstalk, focusing on the molecular and cellular mechanisms in both physiological and pathological conditions. The review has also highlighted the importance of the crosstalk in maintaining the E/I balance in the nervous system. Disruption of this balance can contribute to neurological disorders such as epilepsy, ASD and schizophrenia. To further investigate the functional implications of neurotransmitter-receptor crosstalk, the review has discussed the use of transgenic mice and other techniques to study the causal link between crosstalk and cognitive functions, as well as their implications in physiological and psychological conditions.
However, there are still many challenges in studying neurotransmitter-receptor crosstalk. For example, the specific binding sites for many crosstalk interactions are still unknown, and there is a lack of specific tools for manipulating crosstalk in vivo. For the therapeutic significance, extension of the current rodent model study to non-human primates or human subject is necessary. Future research is needed to address these challenges and to further elucidate the functional roles of neurotransmitter-receptor crosstalk in health and disease.
To conclude, the study of neurotransmitter-receptor crosstalk has already yielded important insights into the complexity of neural signaling. Understanding how neurotransmitter-receptor crosstalk contributes to E/I balance is critical for developing new therapeutic strategies for social disorders in the future.
AC: Conceptualization, Writing – original draft, Writing – review & editing.
The author(s) declare that financial support was received for the research and/or publication of this article. This research has been funded by Key-Area Research and Development Program of GuangDong Province (2023B0303010001) was received for research and publication; National Natural Science Foundation of China (No. 32161133022 and 82350710223) were received for research and publication; NSFC-Guangdong Joint Fund (U20A6005) was received for research and publication; Shenzhen Key Laboratory of Translational Research for Brain Diseases (ZDSYS20200828154800001) was received for research and publication.
The author is thankful to reviewer and Prof. Yutian Wang (SUAT) for critical reading the manuscript and providing valuable advice.
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Alle, H., and Geiger, J. R. (2007). GABAergic spill-over transmission onto hippocampal mossy fiber boutons. J. Neurosci. 27, 942–950. doi: 10.1523/JNEUROSCI.4996-06.2007
Aroeira, R. I., Ribeiro, J. A., Sebastiao, A. M., and Valente, C. A. (2011). Age-related changes of glycine receptor at the rat hippocampus: from the embryo to the adult. J. Neurochem. 118, 339–353. doi: 10.1111/j.1471-4159.2011.07197.x
Aubrey, K. R., Sheipouri, D., Balle, T., Vandenberg, R. J., and Otsu, Y. (2020). Glutamate, d-(−)-2-Amino-5-Phosphonopentanoic acid, and N-methyl-d-aspartate do not directly modulate Glycine receptors. Mol. Pharmacol. 98, 719–729. doi: 10.1124/molpharm.120.000127
Balakrishnan, S., Dobson, K. L., Jackson, C., and Bellamy, T. C. (2014). Ectopic release of glutamate contributes to spillover at parallel fibre synapses in the cerebellum. J. Physiol. 592, 1493–1503. doi: 10.1113/jphysiol.2013.267039
Beltrán, J. Q., and Gutiérrez, R. (2012). Co-release of glutamate and GABA from single, identified mossy fibre giant boutons. J. Physiol. 590, 4789–4800. doi: 10.1113/jphysiol.2012.236372
Bergersen, L., Ruiz, A., Bjaalie, J. G., Kullmann, D. M., and Gundersen, V. (2003). GABA and GABAA receptors at hippocampal mossy fibre synapses. Eur. J. Neurosci. 18, 931–941. doi: 10.1046/j.1460-9568.2003.02828.x
Bianchi, M. T., Clark, A. G., and Fisher, J. L. (2011). The wake-promoting transmitter histamine preferentially enhances α-4 subunit-containing GABAA receptors. Neuropharmacology 61, 747–752. doi: 10.1016/j.neuropharm.2011.05.020
Bidoret, C., Ayon, A., Barbour, B., and Casado, M. (2009). Presynaptic NR2A-containing NMDA receptors implement a high-pass filter synaptic plasticity rule. Proc. Natl. Acad. Sci. USA 106, 14126–14131. doi: 10.1073/pnas.0904284106
Blakely, R. D., and Edwards, R. H. (2012). Vesicular and plasma membrane transporters for neurotransmitters. Cold Spring Harb. Perspect. Biol. 4:1–24. doi: 10.1101/cshperspect.a005595
Bliss, T. V., and Collingridge, G. L. (1993). A synaptic model of memory: long-term potentiation in the hippocampus. Nature 361, 31–39. doi: 10.1038/361031a0
Borghese, C. M., Blednov, Y. A., Quan, Y., Iyer, S. V., Xiong, W., Mihic, S. J., et al. (2012). Characterization of two mutations, M287L and Q266I, in the alpha1 glycine receptor subunit that modify sensitivity to alcohols. J. Pharmacol. Exp. Ther. 340, 304–316. doi: 10.1124/jpet.111.185116
Brickley, S. G., and Mody, I. (2012). Extrasynaptic GABA(a) receptors: their function in the CNS and implications for disease. Neuron 73, 23–34. doi: 10.1016/j.neuron.2011.12.012
Burban, A., Faucard, R., Armand, V., Bayard, C., Vorobjev, V., and Arrang, J. M. (2010). Histamine potentiates N-methyl-D-aspartate receptors by interacting with an allosteric site distinct from the polyamine binding site. J. Pharmacol. Exp. Ther. 332, 912–921. doi: 10.1124/jpet.109.158543
Cabezas, C., Irinopoulou, T., Gauvain, G., and Poncer, J. C. (2012). Presynaptic but not postsynaptic GABA signaling at unitary mossy fiber synapses. J. Neurosci. 32, 11835–11840. doi: 10.1523/JNEUROSCI.5543-11.2012
Castillo, J., Loza, M. I., Mirelman, D., Brea, J., Blanco, M., Sobrino, T., et al. (2016). A novel mechanism of neuroprotection: blood glutamate grabber. J. Cereb. Blood Flow Metab. 36, 292–301. doi: 10.1177/0271678X15606721
Chapman, C. A., Nuwer, J. L., and Jacob, T. C. (2022). The yin and Yang of GABAergic and glutamatergic synaptic plasticity: opposites in balance by Crosstalking mechanisms. Front Synaptic Neurosci 14:911020. doi: 10.3389/fnsyn.2022.911020
Charton, J. P., Herkert, M., Becker, C. M., and Schroder, H. (1999). Cellular and subcellular localization of the 2B-subunit of the NMDA receptor in the adult rat telencephalon. Brain Res. 816, 609–617. doi: 10.1016/s0006-8993(98)01243-8
Chen, L., Li, X., Tjia, M., and Thapliyal, S. (2022). Homeostatic plasticity and excitation-inhibition balance: the good, the bad, and the ugly. Curr. Opin. Neurobiol. 75:102553. doi: 10.1016/j.conb.2022.102553
Chen, J. L., Villa, K. L., Cha, J. W., So, P. T. C., Kubota, Y., and Nedivi, E. (2012). Clustered dynamics of inhibitory synapses and dendritic spines in the adult neocortex. Neuron 74, 361–373. doi: 10.1016/j.neuron.2012.02.030
Chen, X., Wilson, K. A., Schaefer, N., De Hayr, L., Windsor, M., Scalais, E., et al. (2022). Loss, gain and altered function of GlyR alpha2 subunit mutations in neurodevelopmental disorders. Front. Mol. Neurosci. 15:886729. doi: 10.3389/fnmol.2022.886729
Clements, J. D., Lester, R. A., Tong, G., Jahr, C. E., and Westbrook, G. L. (1992). The time course of glutamate in the synaptic cleft. Science 258, 1498–1501. doi: 10.1126/science.1359647
Cohen, S. M., Tsien, R. W., Goff, D. C., and Halassa, M. M. (2015). The impact of NMDA receptor hypofunction on GABAergic neurons in the pathophysiology of schizophrenia. Schizophr. Res. 167, 98–107. doi: 10.1016/j.schres.2014.12.026
Colin, I., Rostaing, P., Augustin, A., and Triller, A. (1998). Localization of components of glycinergic synapses during rat spinal cord development. J. Comp. Neurol. 398, 359–372.
Cubelos, B., Gimenez, C., and Zafra, F. (2005). Localization of the GLYT1 glycine transporter at glutamatergic synapses in the rat brain. Cereb. Cortex 15, 448–459. doi: 10.1093/cercor/bhh147
Cubelos, B., Leite, C., Gimenez, C., and Zafra, F. (2014). Localization of the glycine transporter GLYT1 in glutamatergic synaptic vesicles. Neurochem. Int. 73, 204–210. doi: 10.1016/j.neuint.2013.09.002
Cupolillo, D., Regio, V., and Barberis, A. (2024). Synaptic microarchitecture: the role of spatial interplay between excitatory and inhibitory inputs in shaping dendritic plasticity and neuronal output. Front. Cell. Neurosci. 18:1513602. doi: 10.3389/fncel.2024.1513602
De Saint Jan, D., David-Watine, B., Korn, H., and Bregestovski, P. (2001). Activation of human alpha1 and alpha2 homomeric glycine receptors by taurine and GABA. J. Physiol. 535, 741–755. doi: 10.1111/j.1469-7793.2001.t01-1-00741.x
Desai, N. S. (2003). Homeostatic plasticity in the CNS: synaptic and intrinsic forms. J. Physiol. Paris 97, 391–402. doi: 10.1016/j.jphysparis.2004.01.005
Du, Y., Li, J., Wang, M., Tian, Q., Pang, Y., Wen, Y., et al. (2023). Genetic inhibition of glutamate allosteric potentiation of GABA(a)Rs in mice results in hyperexcitability, leading to neurobehavioral abnormalities. MedComm 4:e235. doi: 10.1002/mco2.235
Dvorzhak, A., Brecht, M., and Schmitz, D. (2024). Social play behavior is driven by glycine-dependent mechanisms. Curr. Biol. 34, 3654–3664.e6. doi: 10.1016/j.cub.2024.06.073
Edwards, R. H. (2007). The neurotransmitter cycle and quantal size. Neuron 55, 835–858. doi: 10.1016/j.neuron.2007.09.001
Field, R. E., D'Amour, J. A., Tremblay, R., Miehl, C., Rudy, B., Gjorgjieva, J., et al. (2020). Heterosynaptic plasticity determines the set point for cortical excitatory-inhibitory balance. Neuron 106, 842–854.e4. doi: 10.1016/j.neuron.2020.03.002
Fritschy, J. M. (2008). Epilepsy, E/I balance and GABA(a) receptor plasticity. Front. Mol. Neurosci. 1:5. doi: 10.3389/neuro.02.005.2008
Gandal, M. J., Anderson, R. L., Billingslea, E. N., Carlson, G. C., Roberts, T. P., and Siegel, S. J. (2012). Mice with reduced NMDA receptor expression: more consistent with autism than schizophrenia? Genes Brain Behav. 11, 740–750. doi: 10.1111/j.1601-183X.2012.00816.x
Ge, Y., Chen, W., Axerio-Cilies, P., and Wang, Y. T. (2020). NMDARs in cell survival and death: implications in stroke pathogenesis and treatment. Trends Mol. Med. 26, 533–551. doi: 10.1016/j.molmed.2020.03.001
Ge, Y., and Wang, Y. T. (2023). GluN2B-containing NMDARs in the mammalian brain: pharmacology, physiology, and pathology. Front. Mol. Neurosci. 16:1190324. doi: 10.3389/fnmol.2023.1190324
Ghit, A., Assal, D., Al-Shami, A. S., and Hussein, D. E. E. (2021). GABA(a) receptors: structure, function, pharmacology, and related disorders. J. Genet. Eng. Biotechnol. 19:123. doi: 10.1186/s43141-021-00224-0
Gutierrez, R. (2000). Seizures induce simultaneous GABAergic and glutamatergic transmission in the dentate gyrus-CA3 system. J. Neurophysiol. 84, 3088–3090. doi: 10.1152/jn.2000.84.6.3088
Hanson, J. E., Yuan, H., Perszyk, R. E., Banke, T. G., Xing, H., Tsai, M. C., et al. (2024). Therapeutic potential of N-methyl-D-aspartate receptor modulators in psychiatry. Neuropsychopharmacology 49, 51–66. doi: 10.1038/s41386-023-01614-3
Harsing, L. G. Jr., and Matyus, P. (2013). Mechanisms of glycine release, which build up synaptic and extrasynaptic glycine levels: the role of synaptic and non-synaptic glycine transporters. Brain Res. Bull. 93, 110–119. doi: 10.1016/j.brainresbull.2012.12.002
Hayama, T., Noguchi, J., Watanabe, S., Takahashi, N., Hayashi-Takagi, A., Ellis-Davies, G. C., et al. (2013). GABA promotes the competitive selection of dendritic spines by controlling local Ca2+ signaling. Nat. Neurosci. 16, 1409–1416. doi: 10.1038/nn.3496
Hruskova, B., Trojanova, J., Kulik, A., Kralikova, M., Pysanenko, K., Bures, Z., et al. (2012). Differential distribution of glycine receptor subtypes at the rat calyx of held synapse. J. Neurosci. 32, 17012–17024. doi: 10.1523/JNEUROSCI.1547-12.2012
Isaacson, J. S. (1999). Glutamate spillover mediates excitatory transmission in the rat olfactory bulb. Neuron 23, 377–384. doi: 10.1016/s0896-6273(00)80787-4
Ishibashi, H., Yamaguchi, J., Nakahata, Y., and Nabekura, J. (2013). Dynamic regulation of glycine-GABA co-transmission at spinal inhibitory synapses by neuronal glutamate transporter. J. Physiol. 591, 3821–3832. doi: 10.1113/jphysiol.2012.250647
Jen, J. C., Wan, J., Palos, T. P., Howard, B. D., and Baloh, R. W. (2005). Mutation in the glutamate transporter EAAT1 causes episodic ataxia, hemiplegia, and seizures. Neurology 65, 529–534. doi: 10.1212/01.wnl.0000172638.58172.5a
Ju, P., and Cui, D. (2016). The involvement of N-methyl-D-aspartate receptor (NMDAR) subunit NR1 in the pathophysiology of schizophrenia. Acta Biochim. Biophys. Sin. Shanghai 48, 209–219. doi: 10.1093/abbs/gmv135
Kew, J. N., Koester, A., Moreau, J. L., Jenck, F., Ouagazzal, A. M., Mutel, V., et al. (2000). Functional consequences of reduction in NMDA receptor glycine affinity in mice carrying targeted point mutations in the glycine binding site. J. Neurosci. 20, 4037–4049. doi: 10.1523/JNEUROSCI.20-11-04037.2000
Kim, S., Wallace, M. L., El-Rifai, M., Knudsen, A. R., and Sabatini, B. L. (2022). Co-packaging of opposing neurotransmitters in individual synaptic vesicles in the central nervous system. Neuron 110, 1371–1384.e7. doi: 10.1016/j.neuron.2022.01.007
Larsen, R. S., Corlew, R. J., Henson, M. A., Roberts, A. C., Mishina, M., Watanabe, M., et al. (2011). NR3A-containing NMDARs promote neurotransmitter release and spike timing-dependent plasticity. Nat. Neurosci. 14, 338–344. doi: 10.1038/nn.2750
Lasley, S. M. (1991). Roles of neurotransmitter amino acids in seizure severity and experience in the genetically epilepsy-prone rat. Brain Res. 560, 63–70. doi: 10.1016/0006-8993(91)91215-m
Lee, E., Lee, J., and Kim, E. (2017). Excitation/inhibition imbalance in animal models of autism Spectrum disorders. Biol. Psychiatry 81, 838–847. doi: 10.1016/j.biopsych.2016.05.011
Legendre, P., Forstera, B., Juttner, R., and Meier, J. C. (2009). Glycine receptors caught between genome and proteome - functional implications of RNA editing and splicing. Front. Mol. Neurosci. 2:23. doi: 10.3389/neuro.02.023.2009
Li, J., Chai, A., Wang, L., Ma, Y., Wu, Z., Yu, H., et al. (2015). Synaptic P-Rex1 signaling regulates hippocampal long-term depression and autism-like social behavior. Proc. Natl. Acad. Sci. USA 112, E6964–E6972. doi: 10.1073/pnas.1512913112
Liu, Y., Ouyang, P., Zheng, Y., Mi, L., Zhao, J., Ning, Y., et al. (2021). A selective review of the excitatory-inhibitory imbalance in schizophrenia: underlying biology, genetics, microcircuits, and symptoms. Front. Cell Dev. Biol. 9:664535. doi: 10.3389/fcell.2021.664535
Liu, H., Wang, H., Sheng, M., Jan, L. Y., Jan, Y. N., and Basbaum, A. I. (1994). Evidence for presynaptic N-methyl-D-aspartate autoreceptors in the spinal cord dorsal horn. Proc. Natl. Acad. Sci. USA 91, 8383–8387. doi: 10.1073/pnas.91.18.8383
Liu, J., Wu, D. C., and Wang, Y. T. (2010). Allosteric potentiation of glycine receptor chloride currents by glutamate. Nat. Neurosci. 13, 1225–1232. doi: 10.1038/nn.2633
Liu, S., Xu, L., Guan, F., Liu, Y. T., Cui, Y., Zhang, Q., et al. (2018). Cryo-EM structure of the human alpha5beta3 GABA(a) receptor. Cell Res. 28, 958–961. doi: 10.1038/s41422-018-0077-8
Lu, T., Rubio, M. E., and Trussell, L. O. (2008). Glycinergic transmission shaped by the corelease of GABA in a mammalian auditory synapse. Neuron 57, 524–535. doi: 10.1016/j.neuron.2007.12.010
Magnin, E., Francavilla, R., Amalyan, S., Gervais, E., David, L. S., Luo, X., et al. (2019). Input-specific synaptic location and function of the alpha5 GABA(a) receptor subunit in the mouse CA1 hippocampal neurons. J. Neurosci. 39, 788–801. doi: 10.1523/JNEUROSCI.0567-18.2018
Malhotra, S., Banumurthy, G., Pennock, R. L., Vaden, J. H., Sugihara, I., Overstreet-Wadiche, L., et al. (2021). Climbing Fiber-mediated spillover transmission to interneurons is regulated by EAAT4. J. Neurosci. 41, 8126–8133. doi: 10.1523/JNEUROSCI.0616-21.2021
McCracken, L. M., Lowes, D. C., Salling, M. C., Carreau-Vollmer, C., Odean, N. N., Blednov, Y. A., et al. (2017). Glycine receptor alpha3 and alpha2 subunits mediate tonic and exogenous agonist-induced currents in forebrain. Proc. Natl. Acad. Sci. USA 114, E7179–E7186. doi: 10.1073/pnas.1703839114
Mielnik, C. A., Binko, M. A., Chen, Y., Funk, A. J., Johansson, E. M., Intson, K., et al. (2021). Consequences of NMDA receptor deficiency can be rescued in the adult brain. Mol. Psychiatry 26, 2929–2942. doi: 10.1038/s41380-020-00859-4
Milanese, M., Romei, C., Usai, C., Oliveri, M., and Raiteri, L. (2014). A new function for glycine GlyT2 transporters: stimulation of gamma-aminobutyric acid release from cerebellar nerve terminals through GAT1 transporter reversal and ca(2+)-dependent anion channels. J. Neurosci. Res. 92, 398–408. doi: 10.1002/jnr.23321
Muller, E., Bakkar, W., Martina, M., Sokolovski, A., Wong, A. Y., Legendre, P., et al. (2013). Vesicular storage of glycine in glutamatergic terminals in mouse hippocampus. Neuroscience 242, 110–127. doi: 10.1016/j.neuroscience.2013.03.007
Musante, V., Summa, M., Cunha, R. A., Raiteri, M., and Pittaluga, A. (2011). Pre-synaptic glycine GlyT1 transporter--NMDA receptor interaction: relevance to NMDA autoreceptor activation in the presence of Mg2+ ions. J. Neurochem. 117, 516–527. doi: 10.1111/j.1471-4159.2011.07223.x
Nong, Y., Huang, Y. Q., Ju, W., Kalia, L. V., Ahmadian, G., Wang, Y. T., et al. (2003). Glycine binding primes NMDA receptor internalization. Nature 422, 302–307. doi: 10.1038/nature01497
Ohba, C., Shiina, M., Tohyama, J., Haginoya, K., Lerman-Sagie, T., Okamoto, N., et al. (2015). GRIN1 mutations cause encephalopathy with infantile-onset epilepsy, and hyperkinetic and stereotyped movement disorders. Epilepsia 56, 841–848. doi: 10.1111/epi.12987
Omiya, Y., Uchigashima, M., Konno, K., Yamasaki, M., Miyazaki, T., Yoshida, T., et al. (2015). VGluT3-expressing CCK-positive basket cells construct invaginating synapses enriched with endocannabinoid signaling proteins in particular cortical and cortex-like amygdaloid regions of mouse brains. J. Neurosci. 35, 4215–4228. doi: 10.1523/JNEUROSCI.4681-14.2015
Pelkey, K. A., Calvigioni, D., Fang, C., Vargish, G., Ekins, T., Auville, K., et al. (2020). Paradoxical network excitation by glutamate release from VGluT3(+) GABAergic interneurons. eLife 9, 1–26. doi: 10.7554/eLife.51996
Pérez-León, J. A., Espinal-Centeno, A., Mendoza-Gonzalez, Z., Camacho, A. C., Lopez, A. B., Perez, R. A., et al. (2022). Identification of amacrine neurons with a glycinergic and GABAergic phenotype in the mouse retina. Med Res Arch 10, 1–25. doi: 10.18103/mra.v10i1.2624
Pilorge, M., Fassier, C., Le Corronc, H., Potey, A., Bai, J., De Gois, S., et al. (2016). Genetic and functional analyses demonstrate a role for abnormal glycinergic signaling in autism. Mol. Psychiatry 21, 936–945. doi: 10.1038/mp.2015.139
Piot, L., Heroven, C., Bossi, S., Zamith, J., Malinauskas, T., Johnson, C., et al. (2023). GluD1 binds GABA and controls inhibitory plasticity. Science 382, 1389–1394. doi: 10.1126/science.adf3406
Raiteri, L., Stigliani, S., Siri, A., Passalacqua, M., Melloni, E., Raiteri, M., et al. (2005). Glycine taken up through GLYT1 and GLYT2 heterotransporters into glutamatergic axon terminals of mouse spinal cord elicits release of glutamate by homotransporter reversal and through anion channels. Biochem. Pharmacol. 69, 159–168. doi: 10.1016/j.bcp.2004.08.029
Rakhade, S. N., and Loeb, J. A. (2008). Focal reduction of neuronal glutamate transporters in human neocortical epilepsy. Epilepsia 49, 226–236. doi: 10.1111/j.1528-1167.2007.01310.x
Ravasenga, T., Ruben, M., Regio, V., Polenghi, A., Petrini, E. M., and Barberis, A. (2022). Spatial regulation of coordinated excitatory and inhibitory synaptic plasticity at dendritic synapses. Cell Rep. 38:110347. doi: 10.1016/j.celrep.2022.110347
Rossi, D. J., and Hamann, M. (1998). Spillover-mediated transmission at inhibitory synapses promoted by high affinity alpha6 subunit GABA(a) receptors and glomerular geometry. Neuron 20, 783–795. doi: 10.1016/s0896-6273(00)81016-8
Ruiz, A., Campanac, E., Scott, R. S., Rusakov, D. A., and Kullmann, D. M. (2010). Presynaptic GABAA receptors enhance transmission and LTP induction at hippocampal mossy fiber synapses. Nat. Neurosci. 13, 431–438. doi: 10.1038/nn.2512
Ruiz, A., Fabian-Fine, R., Scott, R., Walker, M. C., Rusakov, D. A., and Kullmann, D. M. (2003). GABAA receptors at hippocampal mossy fibers. Neuron 39, 961–973. doi: 10.1016/s0896-6273(03)00559-2
Sandler, R., and Smith, A. D. (1991). Coexistence of GABA and glutamate in mossy fiber terminals of the primate hippocampus: an ultrastructural study. J. Comp. Neurol. 303, 177–192. doi: 10.1002/cne.903030202
Sieghart, W., and Savic, M. M. (2018). International Union of Basic and Clinical Pharmacology. CVI: GABA(a) receptor subtype- and function-selective ligands: key issues in translation to humans. Pharmacol. Rev. 70, 836–878. doi: 10.1124/pr.117.014449
Stelzer, A., and Wong, R. K. (1989). GABAA responses in hippocampal neurons are potentiated by glutamate. Nature 337, 170–173. doi: 10.1038/337170a0
Stensrud, M. J., Chaudhry, F. A., Leergaard, T. B., Bjaalie, J. G., and Gundersen, V. (2013). Vesicular glutamate transporter-3 in the rodent brain: vesicular colocalization with vesicular gamma-aminobutyric acid transporter. J. Comp. Neurol. 521, 3042–3056. doi: 10.1002/cne.23331
Stensrud, M. J., Sogn, C. J., and Gundersen, V. (2015). Immunogold characteristics of VGLUT3-positive GABAergic nerve terminals suggest corelease of glutamate. J. Comp. Neurol. 523, 2698–2713. doi: 10.1002/cne.23811
Tanaka, K., Watase, K., Manabe, T., Yamada, K., Watanabe, M., Takahashi, K., et al. (1997). Epilepsy and exacerbation of brain injury in mice lacking the glutamate transporter GLT-1. Science 276, 1699–1702. doi: 10.1126/science.276.5319.1699
Turecek, R., and Trussell, L. O. (2001). Presynaptic glycine receptors enhance transmitter release at a mammalian central synapse. Nature 411, 587–590. doi: 10.1038/35079084
Upmanyu, N., Jin, J., Emde, H. V., Ganzella, M., Bosche, L., Malviya, V. N., et al. (2022). Colocalization of different neurotransmitter transporters on synaptic vesicles is sparse except for VGLUT1 and ZnT3. Neuron 110, 1483–1497.e7. doi: 10.1016/j.neuron.2022.02.008
Vien, T. N., Modgil, A., Abramian, A. M., Jurd, R., Walker, J., Brandon, N. J., et al. (2015). Compromising the phosphodependent regulation of the GABAAR beta3 subunit reproduces the core phenotypes of autism spec trum disorders. Proc Natl Acad Sci USA, 112: 14805–14810.
Wafford, K. A., Kathoria, M., Bain, C. J., Marshall, G., Le Bourdelles, B., Kemp, J. A., et al. (1995). Identification of amino acids in the N-methyl-D-aspartate receptor NR1 subunit that contribute to the glycine binding site. Mol. Pharmacol. 47, 374–380. doi: 10.1016/S0026-895X(25)08550-5
Wang, L., Kloc, M., Maher, E., Erisir, A., and Maffei, A. (2019). Presynaptic GABAA receptors modulate Thalamocortical inputs in layer 4 of rat V1. Cereb. Cortex 29, 921–936. doi: 10.1093/cercor/bhx364
Wang, L., Ma, T., Qiao, D., Cui, K., Bi, X., Han, C., et al. (2022). Polymorphism of rs12294045 in EAAT2 gene is potentially associated with schizophrenia in Chinese Han population. BMC Psychiatry 22:171. doi: 10.1186/s12888-022-03799-1
Wen, Y., Dong, Z., Liu, J., Axerio-Cilies, P., Du, Y., Li, J., et al. (2022). Glutamate and GABA(a) receptor crosstalk mediates homeostatic regulation of neuronal excitation in the mammalian brain. Signal Transduct. Target. Ther. 7:340. doi: 10.1038/s41392-022-01148-y
Wojcik, S. M., Katsurabayashi, S., Guillemin, I., Friauf, E., Rosenmund, C., Brose, N., et al. (2006). A shared vesicular carrier allows synaptic corelease of GABA and glycine. Neuron 50, 575–587. doi: 10.1016/j.neuron.2006.04.016
Won, H., Lee, H. R., Gee, H. Y., Mah, W., Kim, J. I., Lee, J., et al. (2012). Autistic-like social behaviour in Shank2-mutant mice improved by restoring NMDA receptor function. Nature 486, 261–265. doi: 10.1038/nature11208
Xu, J., Jo, A., DeVries, R. P., Deniz, S., Cherian, S., Sunmola, I., et al. (2022). Intersectional mapping of multi-transmitter neurons and other cell types in the brain. Cell Rep. 40:111036. doi: 10.1016/j.celrep.2022.111036
Yang, Y., Ge, W., Chen, Y., Zhang, Z., Shen, W., Wu, C., et al. (2003). Contribution of astrocytes to hippocampal long-term potentiation through release of D-serine. Proc. Natl. Acad. Sci. USA 100, 15194–15199. doi: 10.1073/pnas.2431073100
Ye, J. H., Wang, F., Krnjevic, K., Wang, W., Xiong, Z. G., and Zhang, J. (2004). Presynaptic glycine receptors on GABAergic terminals facilitate discharge of dopaminergic neurons in ventral tegmental area. J. Neurosci. 24, 8961–8974. doi: 10.1523/JNEUROSCI.2016-04.2004
Yu, X., Ye, Z., Houston, C. M., Zecharia, A. Y., Ma, Y., Zhang, Z., et al. (2015). Wakefulness is governed by GABA and histamine Cotransmission. Neuron 87, 164–178. doi: 10.1016/j.neuron.2015.06.003
Keywords: neurotransmitter, synaptic transmission, excitation-inhibition balance, autism spectrum disorder, schizophrenia, epilepsy
Citation: Chai A (2025) Pleiotropic neurotransmitters: neurotransmitter-receptor crosstalk regulates excitation-inhibition balance in social brain functions and pathologies. Front. Neurosci. 19:1552145. doi: 10.3389/fnins.2025.1552145
Edited by:
Kazuhiko Sawada, Tsukuba International University, JapanReviewed by:
Andrea Barberis, Italian Institute of Technology (IIT), ItalyCopyright © 2025 Chai. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anping Chai, YXAuY2hhaUBzaWF0LmFjLmNu
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.