 María Belén Martín-Sanz
María Belén Martín-Sanz Delvis Lucas-Muñoz2
Delvis Lucas-Muñoz2- 1Research Group of Humanities and Qualitative Research in Health Science, King Juan Carlos University, Alcorcón, Spain
- 2Hospital Pediátrico Dr. Hugo Mendoza, Santo Domingo, Dominican Republic
- 3Universidad Autónoma de Santo Domingo, Santo Domingo, Dominican Republic
Spinal muscular atrophy (SMA) is a progressive genetic neuromuscular condition affecting spinal motor neurons. The underlying cause of SMA is deletions or mutations in the SMN gene. It is classified into five variants based on age and clinical manifestations of the patient. In this report, we present the case discovery of a four-month-old male patient with SMA type 1, presenting with generalized hypotonia and regression of acquired neurodevelopmental milestones. Our study aims to illustrate, through a case report, the clinical analysis, therapeutic interventions, and progression until the patient’s demise. This aims to share the challenges in managing such patients and the strategies employed in their care plan. By documenting this case, our goal is to contribute to the understanding of SMA type 1 and emphasize the ongoing need for learning effective care strategies.
1 Introduction
Spinal muscular atrophy (SMA) is a rare progressive genetic neuromuscular disorder affecting spinal motor neurons, caused by defects in both copies of the SMN1 gene, leading to degeneration of alpha motor neurons in the anterior horn of the spinal cord (Masson et al., 2021; Day et al., 2022; Verhaart et al., 2017). SMA is an autosomal recessive neurodegenerative disease, with homozygous mutation on chromosome 5q being the primary genetic cause of infant mortality in the absence of treatment (Day et al., 2022; Eugenio Mercuri et al., 2018). Its incidence is estimated between 1 in 6,000 to 1 in 10,000 newborns (Verhaart et al., 2017; Eugenio Mercuri et al., 2018; Chen et al., 2020). Clinically, hypotonia is the initial symptom in 60% of SMA cases (Pera et al., 2020). Clinical presentations include weakness and atrophy of proximal muscles, areflexia, paradoxical breathing pattern, gastrointestinal dysfunction, metabolic disturbances, orthopedic complications, cognitive impairments, and deficits in social interaction and expression (Masson et al., 2021; Day et al., 2022; Gusset et al., 2021; Aponte Ribero et al., 2023; Corsello et al., 2021; Saladini et al., 2020). Severe cases result in severe mobility limitations and ventilatory insufficiency that can lead to patient mortality (Day et al., 2022).
Four subtypes of SMA (Type 0, I, II, III, IV) have been described based on age of onset, clinical severity, and life expectancy (Giannotta et al., 2024; Schorling et al., 2020). Within the SMA subtype classification, this case corresponds to Type I, also known as Werdnig-Hoffmann disease or severe SMA. This subtype manifests within the first 6 months of life and carries a mortality rate of 50% by 1 year of age, with a survival rate of 8% at 20 months (Gregoretti et al., 2013; Lally et al., 2017). Addressing this condition requires new paradigms for multidisciplinary coordination to maximize autonomy and address multiple healthcare needs, alongside updated knowledge of healthcare interventions for these patients (Finkel et al., 2018; Perego et al., 2020; Bach et al., 2024).
Herein, we present the clinical findings, intervention, and evolution of what, to our knowledge, is the first documented case of SMA Type 1 detected in the Dominican Republic. Despite initial clinical presentation and applied treatment, the patient succumbed at 6 months of age.
2 Patient information and clinical findings
The male patient debuted at 3 months of age with generalized hypotonia, impaired mobility, and regression in previously acquired neurodevelopmental milestones. Regarding perinatal history, the child was born via cesarean section at 38 weeks to a 30-year-old mother. This was her third pregnancy and third cesarean section. The patient was admitted after a three-day history of fever accompanied by 2 days of respiratory distress, diagnosed with bronchial syndrome and community-acquired pneumonia.
Three weeks later, he returned for outpatient consultation requested by his parents at the pediatric neurology service due to concerns about symptoms like those presented by a deceased male sibling at 3 months of age 2 years earlier (loss of crying, generalized hypotonia, and loss of mobility). The parents reported that the sibling died from cardiopulmonary arrest after a month in intensive care, necessitating tracheotomy, with no confirmed differential diagnosis. In Figure 1, a photo of the patient depicts trunk and limb weakness, loss of head control, and inability to roll over. There are no known cases in other family lines.
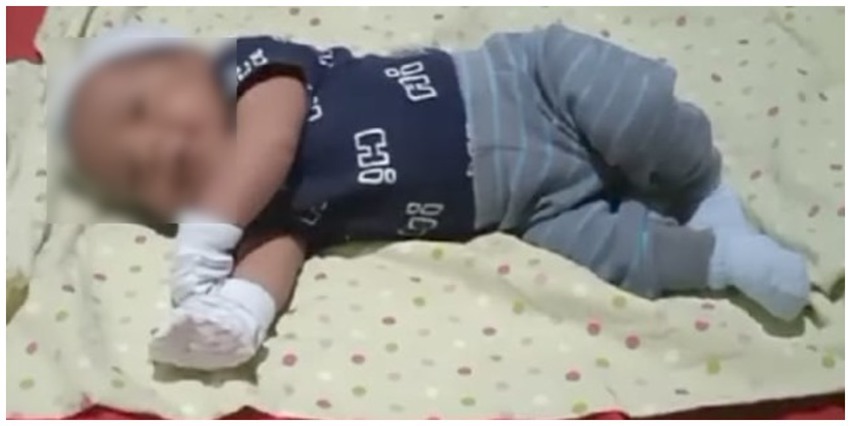
Figure 1. Generalized hypotonia observed in the patient.
In this physical examination, the patient is hypoactive, afebrile, eupneic, and well-hydrated. He presents with a symmetrical and normodynamic thorax, although subcostal retractions are noted. Oral cavity lesions, cough, and rhinorrhea were observed in this patient, findings commonly associated in such cases with bulbar muscle weakness due to impaired secretion clearance and increased risk of secondary respiratory and oral infections. Despite the presence of crackles, rhonchi, and crepitations, lung ventilation appears adequate with audible vesicular breath sounds. Cardiac rhythms are regular with no audible murmurs. The abdomen is flat, depressible, non-tender on palpation, without palpable masses or organomegaly. Hemodynamically, the patient is stable. Upper and lower extremities are symmetrical without edema, and peripheral pulses are normal. There are no allergies reported, and the patient has completed the appropriate vaccination schedule for his age. Neurologically, the patient scores 7 points on the Hammersmith Infant Neurological Examination scale and 3 points on the Children’s Hospital of Philadelphia Infant Test of Neuromuscular Disorders (CHOP INTEND) for motor function evaluation. Genetic testing has been requested due to the patient’s current clinical history and family background, confirming a diagnosis of SMA type 1.
At 4 months of age, the patient was readmitted urgently due to severe respiratory distress. Parents reported significant difficulty swallowing the previous day and a two-day history of fever. The patient exhibited pallor of the skin and mucous membranes, with centrally and peripherally strong and intense pulses and a capillary refill of 4 sec while receiving dobutamine support at 10 mcg/kg/min. Blood pressure was 115/65 mmHg, and heart rate was 113 beats per minute. Ventilation-wise, audible vesicular breath sounds were present, signs of pneumonia (See Figure 2) under ventilation mode pa/c fio2:80, pins 16, fr:32, and 98% oxygen saturation.
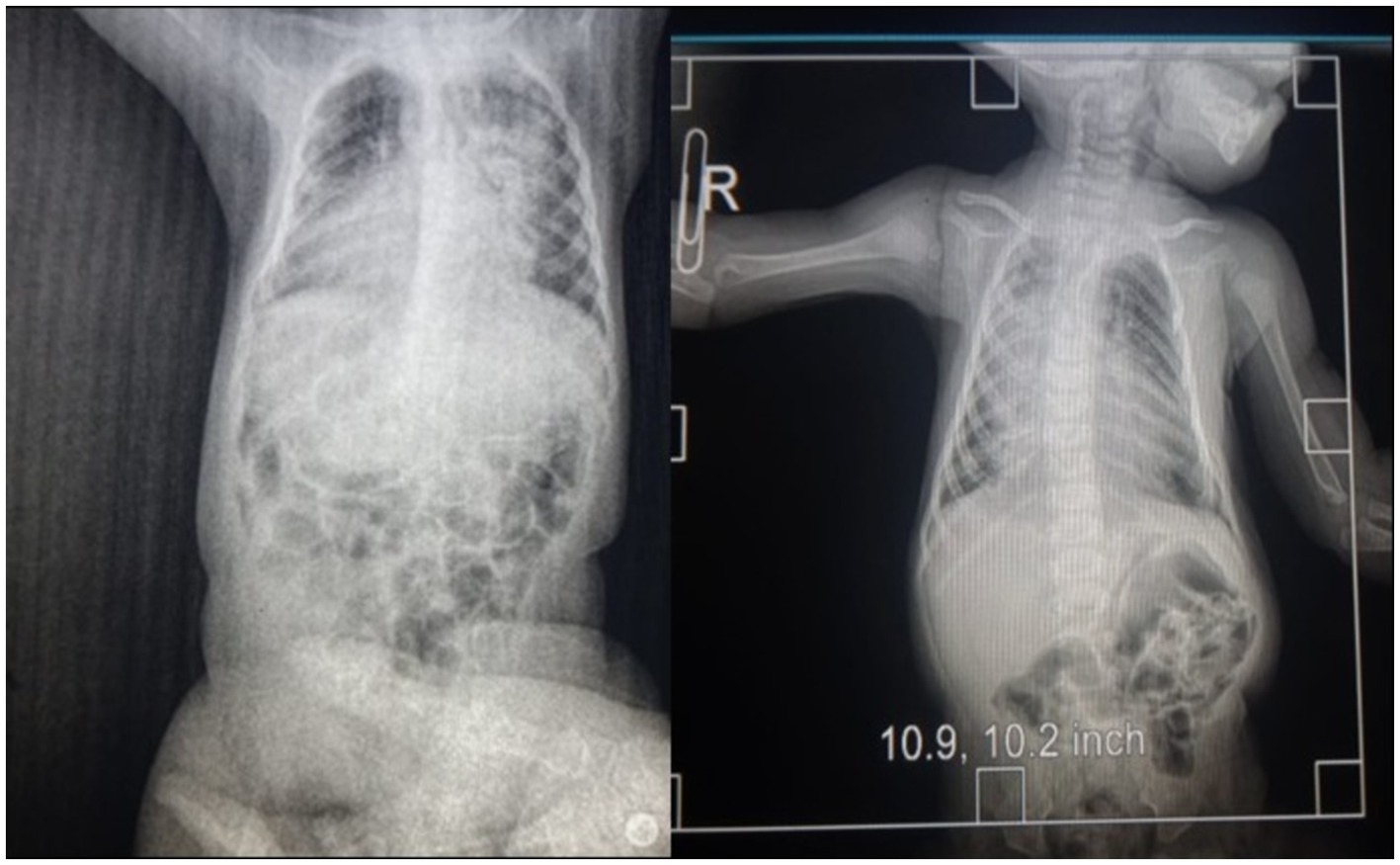
Figure 2. Radiology image.
The patient presents with a distended abdomen, visible peristalsis, and palpable hepatomegaly measuring 2 cm. Laboratory tests indicate normal findings. Neurologically, the patient has equal and reactive pupils measuring 3 mm. Cranial CT scan results show no significant abnormalities. Electromyogram could not be performed due to services covered by your insurance.
The patient received rescue therapy with salbutamol nebulization (0.15 mg/kg every 5 h, not exceeding 2.5 mg per dose), along with N-acetylcysteine (200 mg every 8 h) and methylprednisolone (1.5 mg/kg, not exceeding 60 mg/day). During the chest X-ray procedure, the patient experienced cardiac arrest, attributed to acute decompensation rather than X-ray exposure. Resuscitation required the administration of three doses of adrenaline. Subsequently, the patient was intubated and transferred to the Intensive Care Unit, where sedation with midazolam and fentanyl was administered. The patient remained in intensive care for 2 months until his death due to cardiorespiratory arrest. In Figure 3, a timeline summary of the patient’s most relevant clinical events is depicted.
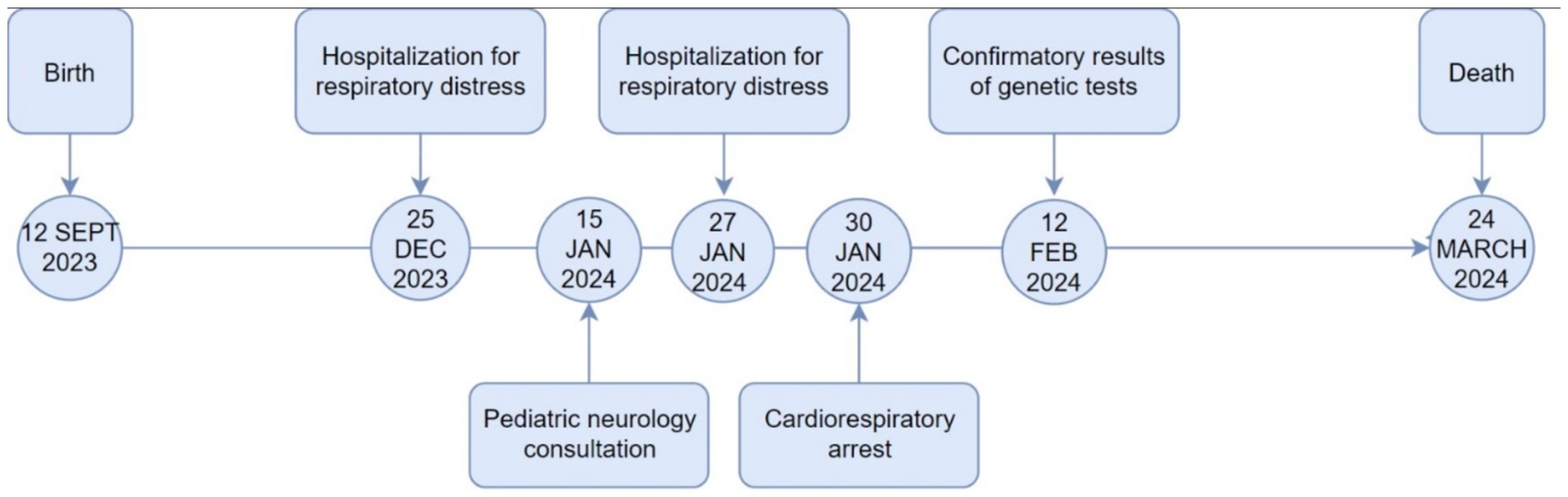
Figure 3. Timeline of key clinical events.
3 Diagnostic assessment
Based on the clinical history and findings from the physical examination, a provisional diagnosis of SMA was established. To confirm this diagnosis, genetic testing was performed using a buccal swab sample to extract DNA. The method employed was Multiplex Ligation-dependent Probe Amplification (MLPA), specifically analyzing the copy numbers of exons 7 and 8 of the SMN1 and SMN2 genes. The test had to be requested from a laboratory located outside the country to be conducted in the Dominican Republic. This testing confirmed SMA Type 1 by identifying a homozygous deletion in the SMN1 gene. Figure 4 displays the results of the genetic test conducted on this patient.

Figure 4. Genetic test results.
4 Therapeutic interventions
The treatment prescribed by the Pediatric Neurology Service was Nusinersen, administered intrathecally at a dose of 12 mg/5 mL. Nusinersen is an antisense oligonucleotide that modulates RNA splicing to increase production of the SMN (Survival Motor Neuron) protein. This protein is deficient in patients with SMA due to mutations in the SMN1 gene. The treatment regimen included loading doses on days 0, 14, 28, and 60, followed by a maintenance dose at 4 months. However, this treatment could not be administered due to bureaucratic barriers in obtaining approval for the drug within the country’s High-Cost Medication Program. Approval for administration was granted 15 days after the patient’s death.
5 Discussion
SMA presents significant challenges in clinical practice due to its rapid progression and severe complications (Nishio et al., 2023). The intervention followed recommendations from prior studies advocating for an interdisciplinary approach addressing respiratory, nutritional, gastroenterological, orthopedic, and psychosocial issues (Corsello et al., 2021; Kirschner et al., 2020). Management focused on enhancing quality of life and disease progression rather than employing a palliative approach, aligning with previous research findings, which was a key strength in this case (Eugenio Mercuri et al., 2018; Corsello et al., 2021; Finkel et al., 2018; Kirschner et al., 2020). In other patients with SMA, physical training programs lasting at least 12 weeks have been shown to improve muscular and cardiorespiratory function. However, this approach was not implemented in this case due to limited access to specialized physical rehabilitation services (Bartels et al., 2019).
Prescribed treatment included new clinical therapies such as Nusinersen, which has demonstrated improvements in psychomotor function and patient survival rates (Day et al., 2022; Erdos and Wild, 2022; Qiao et al., 2023). Recently, the FDA approved Onasemnogene abeparvovec for SMA type 1, a gene therapy utilizing viral vectors to deliver a functional SMN1 gene copy to motor neurons through a single intravenous infusion (Ogbonmide et al., 2023). Risdiplam has also shown increased overall survival rates in treated patients (Ribero et al., 2022; Ratni et al., 2018). A significant limitation encountered was the accessibility barriers to these treatments in the Dominican Republic. Other proposed interventions suggest the use of gene therapy through a single intravenous injection of an AAV9 vector, which has demonstrated a significant improvement in both life expectancy and motor function in patients. In our case, this clinical approach differed from what was feasible for our patient due to access barriers within the healthcare services of our country (Barkats, 2020). A genotype–phenotype correlation suggests that SMN2 is a potent disease modifier in spinal muscular atrophy (SMA). Recently, two innovative treatments targeting SMN have been approved: antisense oligonucleotides (ASOs) and virus-mediated gene therapy. However, these treatments are not available within the Dominican Republic’s healthcare system, limiting therapeutic options for SMA patients in the country (Chen, 2020).
Another challenge was the inability to initiate presymptomatic treatment, despite literature supporting early screening techniques for timely intervention (Corsello et al., 2021; Institute for Quality and Efficiency in Health Care (IQWiG), 2020; Strauss et al., 2022). Early screening has shown to improve clinical prognosis by addressing muscle weakness in affected patients (Vill et al., 2019), yet difficulties persist in achieving early diagnosis due to limited access to diagnostic evaluations and resource constraints in the region (Pera et al., 2020). Notably, neonatal screening services are not covered by the Dominican Social Security System, thereby creating barriers to accessing high-impact healthcare services necessary for rare disease treatments in our country. High economic costs associated with specific healthcare interventions further strained clinical decision-making and medication administration in this case (Dangouloff et al., 2021). This aligns with previous studies highlighting the economic burden of SMA on healthcare services, negatively influencing decision-making and access to clinical resources (Toro et al., 2023; Yang et al., 2022). Disparities in healthcare access and resources across different geographic areas have been identified, resulting in unmet patient care needs (Paracha et al., 2022; Landfeldt et al., 2021). Such reports help identify gaps in healthcare delivery based on varying healthcare systems, policies, and challenges in implementing internationally established protocols for managing this condition across countries (Farrar et al., 2018).
Moreover, addressing SMA goes beyond clinical treatment to understanding its broader social and familial impacts, as previous studies have shown that having a rare disease significantly affects economic, structural, emotional, and social aspects within families (Dias et al., 2023; Sandilands et al., 2022). This underscores the need for comprehensive biopsychosocial support and assistance for all family members from healthcare resource managers in managing these conditions, emphasizing the importance of increased research in this area (Dias et al., 2023; Sandilands et al., 2022).
In conclusion, documenting unique clinical experiences is crucial for continuous learning and ongoing reevaluation of clinical practice, thereby enhancing understanding and patient care for these conditions.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics statement
Written informed consent was obtained from the minor(s)’ legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
Author contributions
MBM-S: Writing – original draft, Writing – review & editing, Conceptualization, Investigation, Methodology, Supervision, Validation, Visualization. DL-M: Conceptualization, Investigation, Methodology, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. MC-H: Conceptualization, Investigation, Methodology, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Aponte Ribero, V., Martí, Y., Batson, S., Mitchell, S., Gorni, K., Gusset, N., et al. (2023). Systematic literature review of the natural history of spinal muscular atrophy: motor function, scoliosis, and contractures. Neurology 101, e2103–e2113. doi: 10.1212/WNL.0000000000207878
Bach, J. R., Saporito, L., and Weiss, W. (2024). Spinal muscular atrophy type 1 survival without new pharmacotherapies: two treatment paradigms. Am. J. Phys. Med. Rehabil. 103, 233–237. doi: 10.1097/PHM.0000000000002354
Barkats, M. (2020). SMA: from gene discovery to gene therapy. Med. Sci. 36, 137–140. doi: 10.1051/medsci/2020010
Bartels, B., Montes, J., van der Pol, W. L., and de Groot, J. F. (2019). Physical exercise training for type 3 spinal muscular atrophy. Cochrane Database Syst. 2019:CD012120. doi: 10.1002/14651858.CD012120.pub2
Chen, T. H. (2020). New and developing therapies in spinal muscular atrophy: from genotype to phenotype to treatment and where do we stand? Int. J. Mol. Sci. 21:3297. doi: 10.3390/ijms21093297
Chen, X., Sanchis-Juan, A., French, C. E., Connell, A. J., Delon, I., Kingsbury, Z., et al. (2020). Spinal muscular atrophy diagnosis and carrier screening from genome sequencing data. Genet. Med. 22, 945–953. doi: 10.1038/s41436-020-0754-0
Corsello, A., Scatigno, L., Pascuzzi, M. C., Calcaterra, V., Dilillo, D., Vizzuso, S., et al. (2021). Nutritional, gastrointestinal and Endo-metabolic challenges in the Management of Children with spinal muscular atrophy type 1. Nutrients 13:2400. doi: 10.3390/nu13072400
Dangouloff, T., Botty, C., Beaudart, C., Servais, L., and Hiligsmann, M. (2021). Systematic literature review of the economic burden of spinal muscular atrophy and economic evaluations of treatments. Orphanet J. Rare Dis. 16:47. doi: 10.1186/s13023-021-01695-7
Day, J. W., Howell, K., Place, A., Long, K., Rossello, J., Kertesz, N., et al. (2022). Advances and limitations for the treatment of spinal muscular atrophy. BMC Pediatr. 22:632. doi: 10.1186/s12887-022-03671-x
Dias, A. G., Daher, A., Barrera Ortiz, L., Carreño-Moreno, S., Hafez, H. S. R., Jansen, A. M., et al. (2023). Rarecare: a policy perspective on the burden of rare diseases on caregivers in Latin America. Front. Public Health 11:1127713. doi: 10.3389/fpubh.2023.1127713
Erdos, J., and Wild, C. (2022). Mid- and long-term (at least 12 months) follow-up of patients with spinal muscular atrophy (SMA) treated with nusinersen, onasemnogene abeparvovec, risdiplam or combination therapies: a systematic review of real-world study data. Eur. J. Paediatr. Neurol. 39, 1–10. doi: 10.1016/j.ejpn.2022.04.006
Eugenio MercuriFinkel, R., Muntoni, F., Wirth, B., Montes, J., Main, M., et al. (2018). Diagnosis and management of spinal muscular atrophy: part 1: recommendations for diagnosis, rehabilitation, orthopedic and nutritional care. Neuromuscul. Disord. 28, 103–115. doi: 10.1016/j.nmd.2017.11.005
Farrar, M. A., Carey, K. A., Paguinto, S. G., Chambers, G., and Kasparian, N. A. (2018). Financial, opportunity and psychosocial costs of spinal muscular atrophy: an exploratory qualitative analysis of Australian carer perspectives. BMJ Open 8:e020907. doi: 10.1136/bmjopen-2017-020907
Finkel, R. S., Mercuri, E., Meyer, O. H., Simonds, A. K., Schroth, M. K., Graham, R. J., et al. (2018). Diagnosis and management of spinal muscular atrophy: part 2: pulmonary and acute care; medications, supplements and immunizations; other organ systems; and ethics. Neuromuscul. Disord. 28, 197–207. doi: 10.1016/j.nmd.2017.11.004
Giannotta, G., Ruggiero, M., De Rinaldis, M., and Trabacca, A. (2024). Exploring variability in cognitive functioning in patients with spinal muscular atrophy: a scoping review. Neurol. Sci. 45, 3699–3710. doi: 10.1007/s10072-024-07503-x
Gregoretti, C., Ottonello, G., Chiarini Testa, M. B., Mastella, C., Ravà, L., Bignamini, E., et al. (2013). Survival of patients with spinal muscular atrophy type 1. Pediatrics 131, e1509–e1514. doi: 10.1542/peds.2012-2278
Gusset, N., Stalens, C., Stumpe, E., Klouvi, L., Mejat, A., Ouillade, M. C., et al. (2021). Understanding European patient expectations towards current therapeutic development in spinal muscular atrophy. Neuromuscul. Disord. 31, 419–430. doi: 10.1016/j.nmd.2021.01.012
Institute for Quality and Efficiency in Health Care (2020). Newborn screening for 5q-linked spinal muscular atrophy: IQWiG reports – Commission no. S18-02. Cologne, Germany: Institute for Quality and Efficiency in Health Care IQWiG.
Kirschner, J., Butoianu, N., Goemans, N., Haberlova, J., Kostera-Pruszczyk, A., Mercuri, E., et al. (2020). European ad-hoc consensus statement on gene replacement therapy for spinal muscular atrophy. Eur. J. Paediatr. Neurol. 28, 38–43. doi: 10.1016/j.ejpn.2020.07.001
Lally, C., Jones, C., Farwell, W., Reyna, S. P., Cook, S. F., and Flanders, W. D. (2017). Indirect estimation of the prevalence of spinal muscular atrophy type I, II, and III in the United States. Orphanet J. Rare Dis. 12:175. doi: 10.1186/s13023-017-0724-z
Landfeldt, E., Pechmann, A., McMillan, H. J., Lochmüller, H., and Sejersen, T. (2021). Costs of illness of spinal muscular atrophy: a systematic review. Appl. Health Econ. Health Policy 19, 501–520. doi: 10.1007/s40258-020-00624-2
Masson, R., Brusa, C., Scoto, M., and Baranello, G. (2021). Brain, cognition, and language development in spinal muscular atrophy type 1: a scoping review. Dev. Med. Child Neurol. 63, 527–536. doi: 10.1111/dmcn.14798
Nishio, H., Niba, E. T. E., Saito, T., Okamoto, K., Takeshima, Y., and Awano, H. (2023). Spinal muscular atrophy: the past, present, and future of diagnosis and treatment. Int. J. Mol. Sci. 24:11939. doi: 10.3390/ijms241511939
Ogbonmide, T., Rathore, R., Rangrej, S. B., Hutchinson, S., Lewis, M., Ojilere, S., et al. (2023). Gene therapy for spinal muscular atrophy (SMA): a review of current challenges and safety considerations for Onasemnogene Abeparvovec (Zolgensma). Cureus 15:e36197. doi: 10.7759/cureus.36197
Paracha, N., Hudson, P., Mitchell, S., and Sutherland, C. S. (2022). Systematic literature review to assess the cost and resource use associated with spinal muscular atrophy management. PharmacoEconomics 40, 11–38. doi: 10.1007/s40273-021-01105-7
Pera, M. C., Coratti, G., Berti, B., D'Amico, A., Sframeli, M., Albamonte, E., et al. (2020). Diagnostic journey in spinal muscular atrophy: is it still an odyssey? PLoS One 15:e0230677. doi: 10.1371/journal.pone.0230677
Perego, M. G. L., Galli, N., Nizzardo, M., Govoni, A., Taiana, M., Bresolin, N., et al. (2020). Current understanding of and emerging treatment options for spinal muscular atrophy with respiratory distress type 1 (SMARD1). Cell. Mol. Life Sci. 77, 3351–3367. doi: 10.1007/s00018-020-03492-0
Qiao, Y., Chi, Y., Gu, J., and Ma, Y. (2023). Safety and efficacy of Nusinersen and Risdiplam for spinal muscular atrophy: a systematic review and Meta-analysis of randomized controlled trials. Brain Sci. 13:1419. doi: 10.3390/brainsci13101419
Ratni, H., Ebeling, M., Baird, J., Bendels, S., Bylund, J., Chen, K. S., et al. (2018). Discovery of Risdiplam, a selective survival of motor Neuron-2 (SMN2) gene splicing modifier for the treatment of spinal muscular atrophy (SMA). J. Med. Chem. 61, 6501–6517. doi: 10.1021/acs.jmedchem.8b00741
Ribero, V. A., Daigl, M., Martí, Y., Gorni, K., Evans, R., Scott, D. A., et al. (2022). How does risdiplam compare with other treatments for types 1-3 spinal muscular atrophy: a systematic literature review and indirect treatment comparison. J. Comp. Eff. Res. 11, 347–370. doi: 10.2217/cer-2021-0216
Saladini, M., Nizzardo, M., Govoni, A., Taiana, M., Bresolin, N., Comi, G. P., et al. (2020). Spinal muscular atrophy with respiratory distress type 1: clinical phenotypes, molecular pathogenesis and therapeutic insights. J. Cell. Mol. Med. 24, 1169–1178. doi: 10.1111/jcmm.14874
Sandilands, K., Williams, A., and Rylands, A. J. (2022). Carer burden in rare inherited diseases: a literature review and conceptual model. Orphanet J. Rare Dis. 17:428. doi: 10.1186/s13023-022-02561-w.36494728
Schorling, D. C., Pechmann, A., and Kirschner, J. (2020). Advances in treatment of spinal muscular atrophy - new phenotypes, new challenges, new implications for Care. J. Neuromuscul. Dis. 7, 1–13. doi: 10.3233/JND-190424
Strauss, K. A., Farrar, M. A., Muntoni, F., Saito, K., Mendell, J. R., Servais, L., et al. (2022). Onasemnogene abeparvovec for presymptomatic infants with two copies of SMN2 at risk for spinal muscular atrophy type 1: the phase III SPR1NT trial. Nat. Med. 28, 1381–1389. doi: 10.1038/s41591-022-01866-4
Toro, W., Yang, M., Georgieva, M., Song, W., Patel, A., Jiang, A. X., et al. (2023). Health Care resource utilization and costs for patients with spinal muscular atrophy: findings from a retrospective US claims database analysis. Adv. Ther. 40, 4589–4605. doi: 10.1007/s12325-023-02621-y
Verhaart, I. E. C., Robertson, A., Wilson, I. J., Aartsma-Rus, A., Cameron, S., Jones, C. C., et al. (2017). Prevalence, incidence and carrier frequency of 5q-linked spinal muscular atrophy - a literature review. Orphanet J. Rare Dis. 12:124. doi: 10.1186/s13023-017-0671-8
Vill, K., Kölbel, H., Schwartz, O., Blaschek, A., Olgemöller, B., Harms, E., et al. (2019). One year of newborn screening for SMA - results of a German pilot project. J. Neuromuscul. Dis. 6, 503–515. doi: 10.3233/JND-190428
Keywords: spinal muscular atrophy (SMA), type I, SMN1, Werdnig Hoffmann disease, motor neuron disease, progressive muscular atrophies
Citation: Martín-Sanz MB, Lucas-Muñoz D and Colomé-Hidalgo M (2025) Spinal muscular atrophy type 1 in the Caribbean: the first case report from the Dominican Republic. Front. Neurosci. 18:1476977. doi: 10.3389/fnins.2024.1476977
Edited by:
Edoardo Gioele Spinelli, Vita-Salute San Raffaele University, ItalyReviewed by:
Partha Sarathi Sarkar, University of Texas Medical Branch at Galveston, United StatesArsen Hunanyan, Duke University, United States
Copyright © 2025 Martín-Sanz, Lucas-Muñoz and Colomé-Hidalgo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: María Belén Martín-Sanz, YmVsZW4ubWFydGluQHVyamMuZXM=
†ORCID: María Belén Martín-Sanz, orcid.org/0000-0001-8628-0979
Manuel Colomé-Hidalgo, orcid.org/0000-0002-4562-6491