 Grace A. Rosen1,2,3
Grace A. Rosen1,2,3 Daniel Kirsch4
Daniel Kirsch4 Raymond Nicks5
Raymond Nicks5 Hunter Kelley1,3,5Rebecca Mathias1,2
Hunter Kelley1,3,5Rebecca Mathias1,2 Kerry A. Cormier1,2,5,6Caroline A. Kubilus2,5,6Bryan Dec5
Kerry A. Cormier1,2,5,6Caroline A. Kubilus2,5,6Bryan Dec5 Thor D. Stein1,2,4,5Victor E. Alvarez1,2,3,5,6
Thor D. Stein1,2,4,5Victor E. Alvarez1,2,3,5,6 Michael L. Alosco2,5,7,8
Michael L. Alosco2,5,7,8 Ann C. McKee1,2,4,5,6
Ann C. McKee1,2,4,5,6 Bertrand R. Huber1,2,3,5*
Bertrand R. Huber1,2,3,5*- 1VA Boston Healthcare System, US Department of Veterans Affairs, Boston, MA, United States
- 2Department of Neurology, Boston University Chobanian and Avedisian School of Medicine, Boston, MA, United States
- 3National Center for PTSD, US Department of Veterans Affairs, Boston, MA, United States
- 4Department of Pathology and Laboratory Medicine, Boston University Chobanian and Avedisian School of Medicine, Boston, MA, United States
- 5Boston University Alzheimer's Disease Research Center and Boston University CTE Center, Boston, MA, United States
- 6VA Bedford Healthcare System, US Department of Veterans Affairs, Bedford, MA, United States
- 7Department of Neurology, Boston Medical Center, Boston, MA, United States
- 8Department of Anatomy and Neurobiology, Boston University Chobanian and Avedisian School of Medicine, Boston, MA, United States
Postmortem human brain tissue is a critical resource for studying neurodegenerative disease, providing critical insights into cellular morphology, pathology, and network connectivity. To improve standard microscopy and enable high-resolution, three-dimensional (3D) images of tissues at the subcellular level, tissue-clearing methods have been developed. These 3D images allow for the analysis of large regions of interest and can be used to study structural and spatial changes that occur during neurodegeneration. Additionally, 3D imaging facilitates the visualization of whole-cell morphology, especially in cells with long processes that would otherwise be truncated in single-plane images. Human brain tissue is especially challenging for tissue clearing due to the abundance of lipids in myelin and the need for optimal fixation and low postmortem intervals. Formaldehyde-based fixatives, commonly used in preserving tissue, hinder antibody binding by crosslinking important antibody epitopes, and fluorescent microscopy requires the incorporation of fluorescent labels through passive diffusion or electrophoresis. Recent studies have focused on optimally fixed human brain tissue with short postmortem intervals, limiting the general applicability of these methods. To address these challenges, we developed SHARD (SHIELD, antigen retrieval, and delipidation), a simple and widely applicable method for clearing and labeling human brain tissue, which can be applied to long-term banked human brain tissue preserved in formaldehyde. SHARD is a novel addition to the SHIELD tissue clarification method, combining antigen retrieval, tissue clearing, and staining of 200-μm sections from long-term banked human brain tissue. The SHARD method is effective for postmortem intervals (PMIs) ranging from 10 to 72 h in multiple neurodegenerative diseases and control samples. In this study, we demonstrate that the SHARD method significantly enhances the immunostaining of glial fibrillary acidic protein (GFAP), an astrocytic cytoskeletal marker. Overall, the combination of antigen retrieval and tissue delipidation holds great potential for achieving detailed 3D immunostaining in long-term formaldehyde-fixed postmortem human brain tissue, opening new avenues for research and discovery.
1 Introduction
Postmortem human brain tissue is an invaluable resource for studying microscopic structural changes associated with neurodegenerative and psychiatric disorders. Three-dimensional tissue imaging offers advantages over standard microscopy by enabling improved visualization of cellular morphology and spatial relationships between cells. However, this technique requires optically transparent tissue and the accessibility of target epitopes to antibodies, both of which present technical challenges.
The three major approaches for volumetric imaging are hydrophobic, hydrophilic, and hydrogel-based clearing methods. Hydrophobic methods such as 3DISCO lead to well-preserved cleared organs within a few days, with downsides of tissue shrinkage, reagents that include toxic organic solvents, and the possibility of fluorescent signal degradation over time due to protein instability in clearing solutions (Ertürk et al., 2012; Ueda, 2020). Hydrophilic clearing methods such as CUBIC have the advantage of preserving protein structure and function due to their use of water-soluble reagents (Matsumoto et al., 2019). Hydrogel-based methods offer additional structural stabilization through crosslinking proteins, nucleic acids, and hydrogel monomers that are in place in preparation for lipid removal (Park et al., 2019). Compared to formalin fixation alone, hydrogel-embedding of brain tissue also results in minimal protein loss during clearing (Chung et al., 2013).
Due to the advantages of hydrogel-based methods, we opted to use the SHIELD protocol (stabilization under harsh conditions via intramolecular epoxide linkages to prevent degradation). After the SHIELD procedure, lipids are washed from the tissue using sodium dodecyl sulfate (SDS). If actively clearing, SDS is driven into the tissue with an electric current (Kim et al., 2015).
The fully cleared tissue can be fluorescently labeled and incubated in an index-matching solution so that its refractive index matches that of a glass cover slip. Previous SHIELD-based studies have successfully characterized the morphology and spatial distribution of cells in tissue sections up to 8 mm thick (Chung and Deisseroth, 2013; Ueda, 2020); tissue thickness is limited by the index of refraction inhomogeneity, which blurs images, and by the optical limitations of the microscope. Another challenge to 3D imaging is laser absorption and scattering by pigmented tissue features such as melanocytes.
Tissue clearing and fluorescent labeling have been successfully employed to image whole mouse brains, approximately 13.2 mm × 11.4 mm × 8 mm (Chung and Deisseroth, 2013; Kim et al., 2015; Yun et al., 2019; Wang et al., 2020) and many human organs, including kidney (approximately 12 × 8 × 5 cm), pancreas (approximately 11.5 × 4 × 2 cm), heart (approximately 12 × 8 × 6 cm), lung (approximately 13 × 8 × 0.5 cm), spleen (approximately 13 × 8 × 4 cm), and 2 mm thick brain 6 cm × 8 cm coronals (Ku et al., 2020; Mai et al., 2022; Shi et al., 2022). Previous studies clearing postmortem human brain sections have required minimal fixation several months of clearing (Chung et al., 2013; Liebmann et al., 2016; Liu et al., 2016; Phillips et al., 2016; Lai et al., 2018; Morawski et al., 2018), or the use of 100 μm sections, cleared over a few weeks, that resulted in warping of the tissue surface (Murray et al., 2015). The utility of tissue clearing has been demonstrated through the qualitative visualization of Alzheimer’s disease (AD) amyloid plaques in humans (Ando et al., 2014). Moreover, 2–5 mm human brain sections have been successfully immunostained and volumetrically visualized with a hydrophobic clearing method (Hildebrand et al., 2019).
One barrier to successful immunofluorescent staining in postmortem human brain tissue is signal specificity. Postmortem human tissue is autofluorescent due to sources such as lipids, lipofuscin, hemoglobin, hemosiderin, collagen, and myelin. Recent protocols have added a photobleaching step to reduce background signal from autofluorescence (Duong and Han, 2013; Ku et al., 2020). Furthermore, antigen sites for stains may be masked by formaldehyde crosslinking. Formaldehyde fixation results in inter- and intramolecular crosslinking that results in redistribution of electrostatic charges and physical hindrance of the antigen site availability (Rait et al., 2004; Boenisch, 2006). Antigen retrieval is a process in which the chemical crosslinking that happens between formaldehyde and amino acids is reversed, making epitopes available for immunochemistry (D’Amico et al., 2009). Protocols for antigen retrieval involve incubating tissue in acidic or basic solutions at near boiling heat, resulting in increased staining quality. The utility of antigen retrieval has been demonstrated in pre-fixed frozen mouse tissue (Ino, 2003), frozen human tissue (Alelú-Paz et al., 2008), as well as thin formaldehyde-fixed paraffin-embedded (FFPE) 3 μm to tens of μm sections of human tissue (Namimatsu et al., 2005; Shi et al., 2007). There is a growing body of literature on optimizing antigen retrieval in thicker sections than light scattering in FFPE allows (Evers and Uylings, 1994; Jiao et al., 1999). Alkaline antigen retrieval has been successfully implemented in 500 μm sections of postmortem human brain tissue to visualize neuronal, glial, and vasculature markers (Pesce et al., 2022). Sodium dodecyl sulfate (SDS) has also been used as a medium for antigen retrieval for tissue-clearing organoids and human organs (Messal et al., 2021).
In this study, we introduced SHARD (SHIELD, antigen retrieval, and delipidation), a method by which SHIELD-treated tissue undergoes citrate-based antigen retrieval, delipidation with SDS, and stained for three-dimensional rendering of targets of interest via fluorescent antibodies and small-molecule dyes. SHARD works well across a range of postmortem intervals and disease states and can also be used in a multiplex staining with up to seven colors. We tested it alongside a SHIELD-only protocol and with an additional photobleaching step. To the best of our knowledge, this is the first instance of quantitative analysis of different tissue-clearing treatments across various neurodegenerative disease states using long-term formaldehyde-fixed human tissue3W. The protocol we developed provides a method for tissue clearing and antigen unmasking that is compatible with long-term formaldehyde-fixed human brain tissue and works with the majority of autopsy tissue currently available from brain banks. SHARD offers key benefits of scalability, simplicity, and the ability to stain and image up to six targets, and it can be widely applied to render markers of interest in three dimensions in a few weeks’ time.
2 Materials and equipment
2.1 Reagents
• Paraformaldehyde (PFA, Sigma-Aldrich # S812315 325).
• Sodium Phosphate, dibasic (Sigma-Aldrich # S9763).
• Sodium phosphate, monobasic (Sigma-Aldrich # S8282).
• Sodium-m-periodate (Sigma-Aldrich # 311448).
• L Lysine (Sigma-Aldrich # L8662).
• Sodium Hydroxide (Fisher Scientific # S318).
• Potassium phosphate, dibasic (Sigma-Aldrich # P3786).
• SHIELD Kit (LifeCanvas # SH-250, includes SHIELD-Buffer Solution, SHIELD-Epoxy Solution, and SHIELD ON).
• Triton X-100 (Fisher Scientific # BP151).
• Normal Donkey Serum (Jackson ImmunoResearch # NC9624464).
• Sodium chloride (Sigma-Aldrich # S5886).
• Agarose, low gelling (Sigma-Aldrich # A9414).
• Citric acid (Sigma-Aldrich # C7129).
• Sodium citrate tribasic dihydrate (Sigma-Aldrich # S4641).
• Sodium dodecyl sulfate (Fisher Scientific # BP166).
• Boric acid (Sigma-Aldrich # B7901).
• Sodium sulfite anhydrous (Fisher Scientific # S430).
• Tris Base (Fisher Scientific # BP154).
• EDTA 0.5 M, pH 8 (Invitrogen # 15575–038).
• Tween (Thermo Scientific # J20605).
• Hydrogen peroxide (Fisher Scientific # H325).
• Sucrose (Sigma-Aldrich # S0389).
• 10% buffered formalin phosphate (Fisher Scientific # SF100).
• Sodium azide (Sigma-Aldrich # S2002).
• Tomato lectin DyLight 488 conjugate (Vector Laboratories # DL-1174).
• Primary antibodies: see Table 1.
• Alexa Fluor® 750-Anti-Mouse (abcam # ab175738).
• Alexa Fluor® 647-Anti-Mouse (Jackson ImmunoResearch # 715–605-150).
• Alexa Fluor® 647-Anti-Rabbit (Jackson ImmunoResearch # 711–605-152).
• Alexa Fluor® 594-Anti-Chicken (Jackson ImmunoResearch # 703–585-155).
• Alexa Fluor® 594-Anti-Goat (Jackson ImmunoResearch # 705–585-003).
• Cy3-Anti-Goat (Jackson ImmunoResearch # 705–165-147).
• Cy3-Anti-Rabbit (Jackson ImmunoResearch # 711–167-003).
• Cy3-Anti-Chicken (Jackson ImmunoResearch # 703–166-155).
• Alexa Fluor® 488-Anti-Mouse (Jackson ImmunoResearch # 715–545-150).
• Alexa Fluor® 488-Anti-Rabbit (Jackson ImmunoResearch # 711–545-152).
• EasyIndex (LifeCanvas # EI-500-1.52).
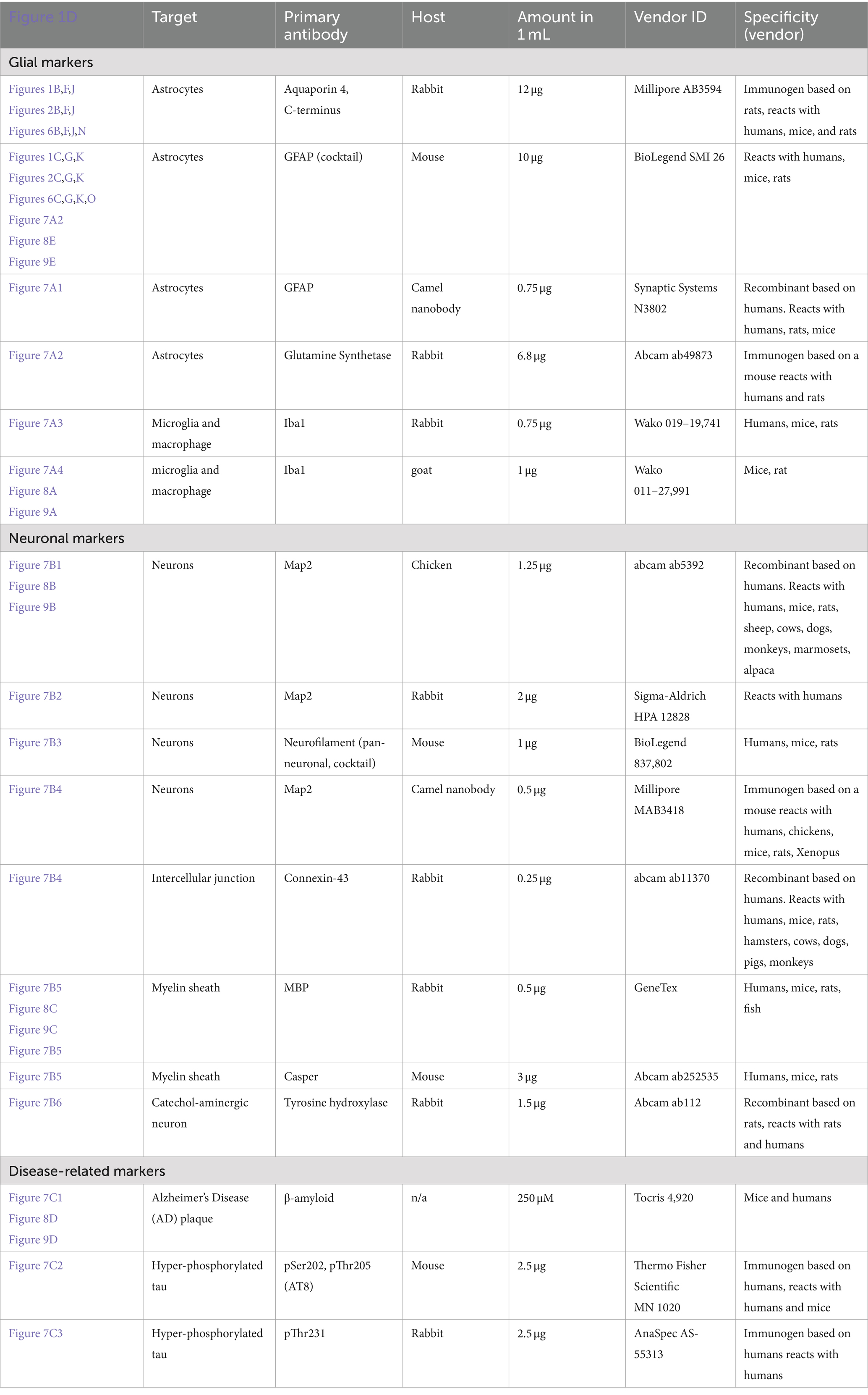
Table 1. Detailed information on the primary antibodies used.
2.2 Materials/equipment
• Vibratome (Precisionary Instruments, Inc. # VF-700-0Z).
• Microscope Slides, Precleaned (Fisher Scientific # 12–550-15).
• No. 1.5 Coverglass, 22 × 30 mm (Corning # 2980–223).
• Zeiss 880 LSM Confocal Microscope with Airyscan and 20x PLAN APO Lens, 0.8 numerical aperture (NA), (Zeiss).
• Nikon Ti2 Eclipse Crest V2 spinning disc confocal microscope with 60x PLAN APO λD, 1.42 NA oil, and 100x CFI PLAN APO D 1.45 NA oil objective lenses, as well as a 20x CFI PLAN APO D, 0.8 NA lens.
• Nikon AX-R confocal microscope with 40x CFI PLAN APO silicone oil lens, 1.25 NA with cover glass thickness correction collar set to 0.17 mm, 60x CFI PLAN APO λD, 1.42 NA oil lens, 20x CFI PLAN APO λD, 0.8 NA lens, or 4x CFI PLAN APO λD 0.20 NA lens, equipped with one multi-alkaloid (MA) detector, two tunable GaAsP detectors, one non-tunable GaAsP detector, and seven laser lines: 405, 488, 514, 561, 594, 633, and 730 nm.
2.3 Solution formulations
• Periodate-Lysine-Paraformaldehyde (PLP): Heat 1 L of deionized water to 60°C. Add 80 g of paraformaldehyde and 44 g of sodium phosphate dibasic. Cool the solution to room temperature using a water bath. Add 12 g of sodium phosphate monobasic and 8.6 g L of L-lysine Filter the solution and add deionized (DI) water to reach a total volume of 4 L. Adjust the pH to 7.4 using sodium hydroxide.
• SHIELD OFF solution (per 25 mm × 25 mm × 3 mm sample): Mix 5 mL of DI water, 5 mL of SHIELD-buffer solution, and 10 mL of SHIELD-epoxy solution.
• 25x phosphate-buffered saline (PBS): Dissolve 188 g of potassium phosphate dibasic in approximately 800 mL of deionized water, heating as needed. Add sodium phosphate monobasic and sodium chloride and stir until fully dissolved. Cool the solution and dilute it to a final volume of 1 L.
• PBS: Mix 40 mL of 25x stock solution with 960 mL of deionized water.
• 10 mM citrate: Mix 14 mL of 0.1 M citric acid with 86 mL of 0.1 M sodium citrate. Add 900 mL of deionized water to achieve a total volume of 1 L.
• 30% sucrose: Dissolve 300 g of sucrose into 1 L of PBS.
• Passive clearing buffer: Prepare a buffer containing 300 mM sodium dodecyl sulfate (SDS), 10 mM boric acid, and 100 mM sodium sulfite. Adjust the pH to 9.
• PBST: Add 4 mL of Triton X-100 to 996 mL of PBS to make 1 L of PBST.
• Alkaline antigen retrieval solution: Prepare a solution with 10 mM Tris base, 1 mM EDTA, and 0.05% Tween. Adjust the pH to 9.
• 5% donkey serum (DKS): Dilute 50 μL of donkey serum in 950 μL of PBST.
• Primary antibody solution: Add the specified amounts of primary antibodies from Table 1, along with 10 μL of DKS, and adjust the volume to 1 mL with PBST.
• Secondary antibody solution: Prepare the secondary antibody of choice at a 2:1 molar ratio of secondary to the primary antibody, add 10 μL of DKS, and adjust the volume to 1 mL with PBST.
• 4% PFA 1 Prepare a 1:2.5 dilution of 10% PFA stock in PBS to make 4% PFA.
• 0.02% sodium azide tissue storage solution: Dissolve 0.2 g of sodium azide in 1 L of PBS.
2.4 Software and computer
• Zen (Black Edition) with Airyscan Deconvolution (Zeiss).
• Nikon Elements Advanced Research 6.02.01 (Nikon).
• Imaris 10.0 (Oxford Instruments) running on a Windows 10 system with an Intel® Xeon® Gold 5,218 CPU @ 2.30 GHz and 2.29 GHz processors, 192 GB of RAM, and an NVIDIA RTX A6000 Graphics Card.
• Arivis Pro, running on a Windows 11 system with an AMD Ryzen Threadripper PRO 5995WX with 64 cores @ 2.70 GHz, 512 GB of RAM, and an NVIDIA RTX A6000 Graphics Card.
3 Methods
3.1 Study design and brain donors
Autopsy participants included 21 brain donors from three brain banks housed at VA Boston Healthcare System with harmonized neuropathological processing protocols and diagnostic procedures: Understanding Neurological Injury and Traumatic Encephalopathy (UNITE, n = 13), Boston University Alzheimer’s Disease Research Center (ADRC n = 3), and the National Posttraumatic Stress Disorder Brain Bank (NPBB, n = 5). UNITE’s objective is to characterize the neuropathology and clinical-pathological correlates of CTE and repetitive head impacts (Mez et al., 2015; Alosco et al., 2021). The ADRC longitudinally follows its participants to collaborate with the National Alzheimer’s Coordinating Center to study Alzheimer’s Disease (AD) and related dementia (Gavett et al., 2012). The NPBB facilitates research on the cause, progression, and treatment of PTSD (Friedman et al., 2017). Consents for brain donation and research participation were provided by the donors’ next of kin. Institutional review boards from the Boston University Medical Center and VA Boston Healthcare System approved brain donation, postmortem clinical record review, neuropathological evaluation, and clinical interviews with donor family members. Cases were coded as “u,” “p,” or “a,” depending on whether they came from the UNITE, NPBB, or ADRC brain banks (Table 2).
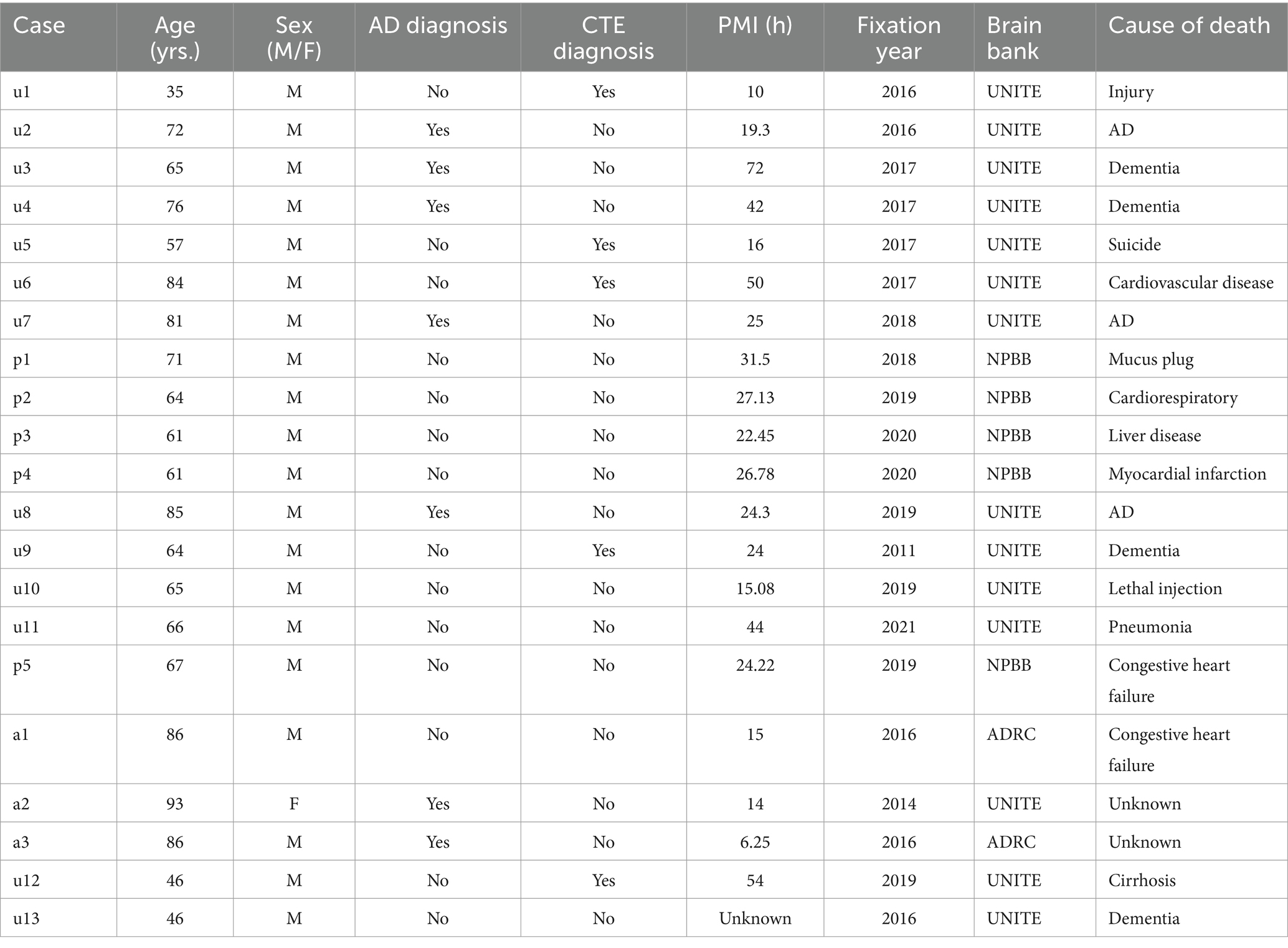
Table 2. Demographics and neuropathological diagnoses of the cases studied.
We examined 11 cases for quantification of the effectiveness of the three different treatment conditions and 10 further cases for staining with additional antibodies. Cases u1-7 and p1-4 were used for the quantification of tomato lectin, aquaporin-4, and GFAP staining. The postmortem interval (PMI) of these cases ranged from 10 to 72 h. The PMI here is defined by the difference between the time of death and brain removal. The cases had been stored at 4° C in Periodate-Lysine-Paraformaldehyde (PLP) for up to five years at the time of staining. There was a mix of healthy controls (n = 4), CTE cases (n = 3), and AD cases (n = 4). The 10 cases stained with additional antibodies had PMI values ranging from 6.25 to 54 h and included cases that had been stored in PLP for 1–10 years at the time of staining.
3.2 Clinical assessment and diagnosis
Demographic information, medical history, and other antemortem clinical variables were obtained during retrospective clinical evaluation with informants for all brain donors and included a detailed assessment of repetitive head impact exposure and traumatic brain injury history for the UNITE brain bank (Mez et al., 2015; Alosco et al., 2021). Tissue acquisition, processing, diagnostic assessment protocols, ethical considerations, governance, and oversight of the VA National PTSD Brain Bank were previously described (Friedman et al., 2017). Details on the procedures of the BU ADRC brain bank are available in prior publications (Gavett et al., 2012; Alosco et al., 2021). Diagnoses for Alzheimer’s disease were assigned according to the National Institute on Aging-Alzheimer’s Association guidelines (Montine et al., 2012).
3.3 Tissue processing and pathological assessment
Postmortem brain tissue was fixed in periodate-lysine-paraformaldehyde (PLP) for at least 3 months at 4°C. Neuropathological assessment was performed using procedures previously established (Vonsattel et al., 2008; Mez et al., 2015). Neuropathological evaluations were made by board-certified neuropathologists (ACM, TDS, BRH) according to published diagnostic criteria and were kept blinded to antemortem clinical information (Mez et al., 2015).
3.4 SHIELD post-fixation
PLP-fixed dorsolateral prefrontal cortex (DLF, Brodmann area 46) tissue blocks were harvested with a 16 × 16 mm2 leather punch with a thickness of 3 mm. Tissue harvesting was based on a standardized blocking scheme that is used in all cases. The 16 × 16 × 3 mm tissue sections were incubated in SHIELD OFF solution (LifeCanvas SHIELD-buffer solution, LifeCanvas SHIELD-Epoxy solution, and deionized water) for 3 days with shaking at 4°C (LifeCanvas, Cambridge, MA). Sections were transferred to LifeCanvas SHIELD ON buffer and incubated for 24 h. with shaking at 37°C and stored in phosphate-buffered saline (PBS) with 0.02% sodium azide for up to several weeks. SHIELD-preserved sections were embedded in 2% agarose gel and cut into 200 μm slices using a vibratome (VF-700-0Z Microtome, Precisionary Instruments, Inc., Natick, MA). Slices were stored in PBS with 0.02% sodium azide for up to a few months.
3.5 Tissue clearing and antigen retrieval
A summary of the three tissue treatment conditions quantified can be found in Table 3.
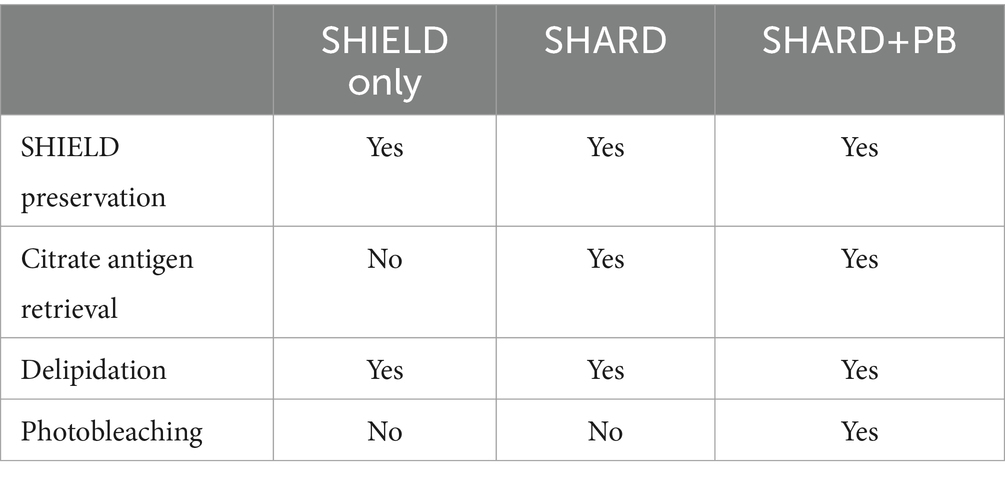
Table 3. Summary of tissue treatment conditions.
3.5.1 SHIELD only condition
For tissue subjected to the “SHIELD only” treatment, the delipidation step was started immediately after the SHIELD treatment. Delipidation consists of overnight incubation at 37°C with light shaking in a passive clearing buffer. Slices were subsequently washed twice with PBST at room temperature (RT) with light shaking to wash out the SDS.
3.5.2 SHARD condition
Tissue slices that underwent antigen retrieval were incubated with 10 mM citrate buffer overnight at 4°C. The next day, slices were incubated in 10 mM citrate buffer at 95°C for 15 min before transferring to 30% sucrose in PBS at 4°C, where they remained for a few hours or overnight until they sank in the solution. Slices were then delipidated overnight as described above. The “SHARD” condition refers to tissue slices that were stained immediately after this point.
3.5.3 SHARD+PB condition
After antigen retrieval with citrate and delipidation, sections were photobleached with white light LEDs. Sections that were photobleached were done so for 5 days in PBST, as previously described (Ku et al., 2020). This treatment is referred to as “SHARD+PB” throughout the paper.
In summary, these three conditions will be called “SHIELD only” (delipidation without antigen retrieval or photobleaching), “SHARD” (SHIELD and antigen retrieval), and “SHARD+PB” (SHIELD, antigen retrieval, and photobleaching). These conditions are compared qualitatively and quantitatively in Figures 1–5.
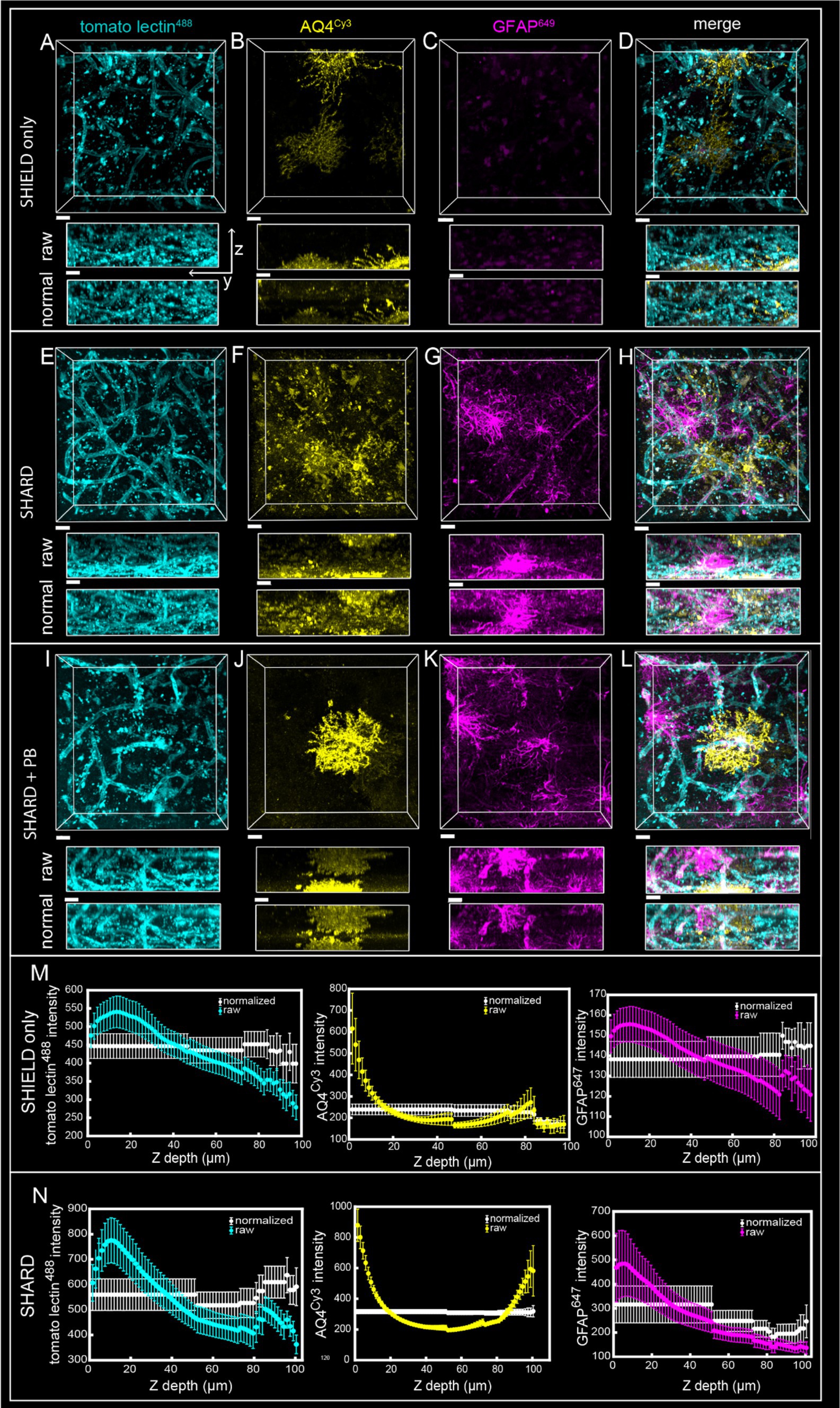
Figure 1. Comparison of SHIELD only, SHARD, and SHARD+PB conditions in the gray matter. 3D image stacks of immunofluorescent stains in the DLF gray matter resulting from the three experimental conditions: (A–D) SHIELD only, (E–H) SHARD, (I–L) SHARD+PB. The three stains shown are tomato lectin488 (A,E,I), anti-AQ4Cy3 (B,F,J), and anti-GFAP647 (C,G,K). The overlay of all three is shown in (D,H,L). All images are from the same 64-year-old case with no neurodegenerative disease. Scale bar = 20 μm, 72 μm Z-stacks. Display parameters for the dynamic range were held constant for each stain between conditions. (M,N) Intensity vs. imaging depth plots averaged over the 11 quantified cases for tomato lectin488 (left), AQ4Cy3 (middle), and GFAP647 (right). Raw pixel intensity (colored) and normalized pixel intensities (white) are both plotted for SHIELD-only tissue treatment (M) and SHARD tissue treatment (N).
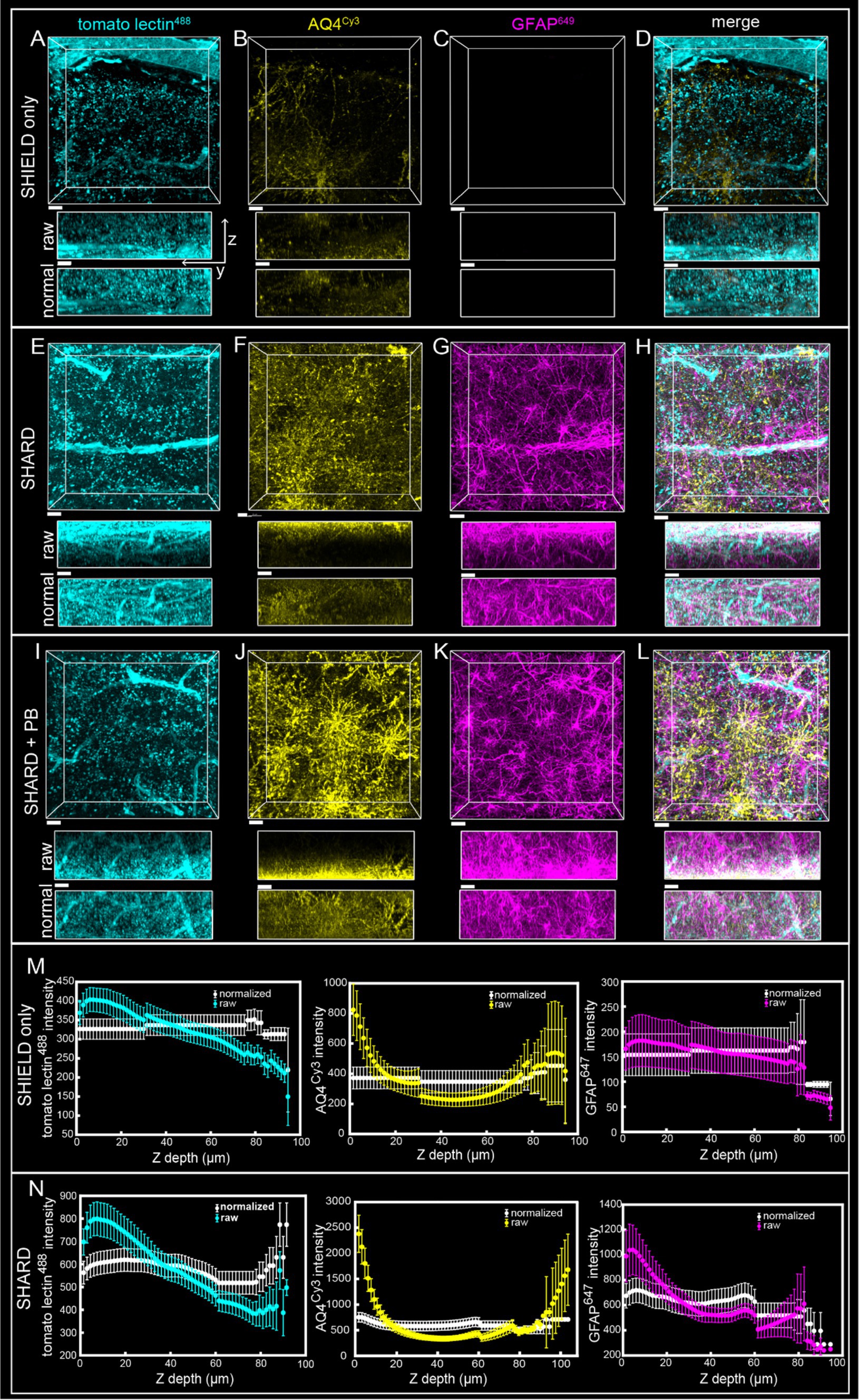
Figure 2. Comparison of SHIELD only, SHARD, and SHARD+PB conditions in the white matter. 3D image stacks of immunofluorescent stains in the DLF white matter resulting from the three experimental conditions: (A–D) SHIELD only, (E–H) SHARD, and (I–L) SHARD+PB. For each image, the views displayed are xy (top) and yz (middle and bottom). The middle yz view shows raw pixel intensity, whereas the bottom yz view shows pixel intensity after running normalized layers preprocessing in Imaris. The three stains shown are tomato lectin488 (A,E,I), anti-AQ4Cy3 (B,F,J), and anti-GFAP647 (C,G,K). The overlay of all three is shown in (D,H,L). Images are taken using tissue donated by the same individual as Figure 1. Scale bar = 20 μm, 72 μm Z-stacks. Display parameters for the dynamic range were held constant for each stain between conditions. (M,N) Intensity vs. imaging depth plots averaged over the 11 quantified cases for tomato lectin488 (left), AQ4Cy3 (middle), and GFAP647 (right). Raw pixel intensity (colored) and normalized pixel intensities (white) are both plotted for SHIELD-only tissue treatment (M) and SHARD tissue treatment (N).
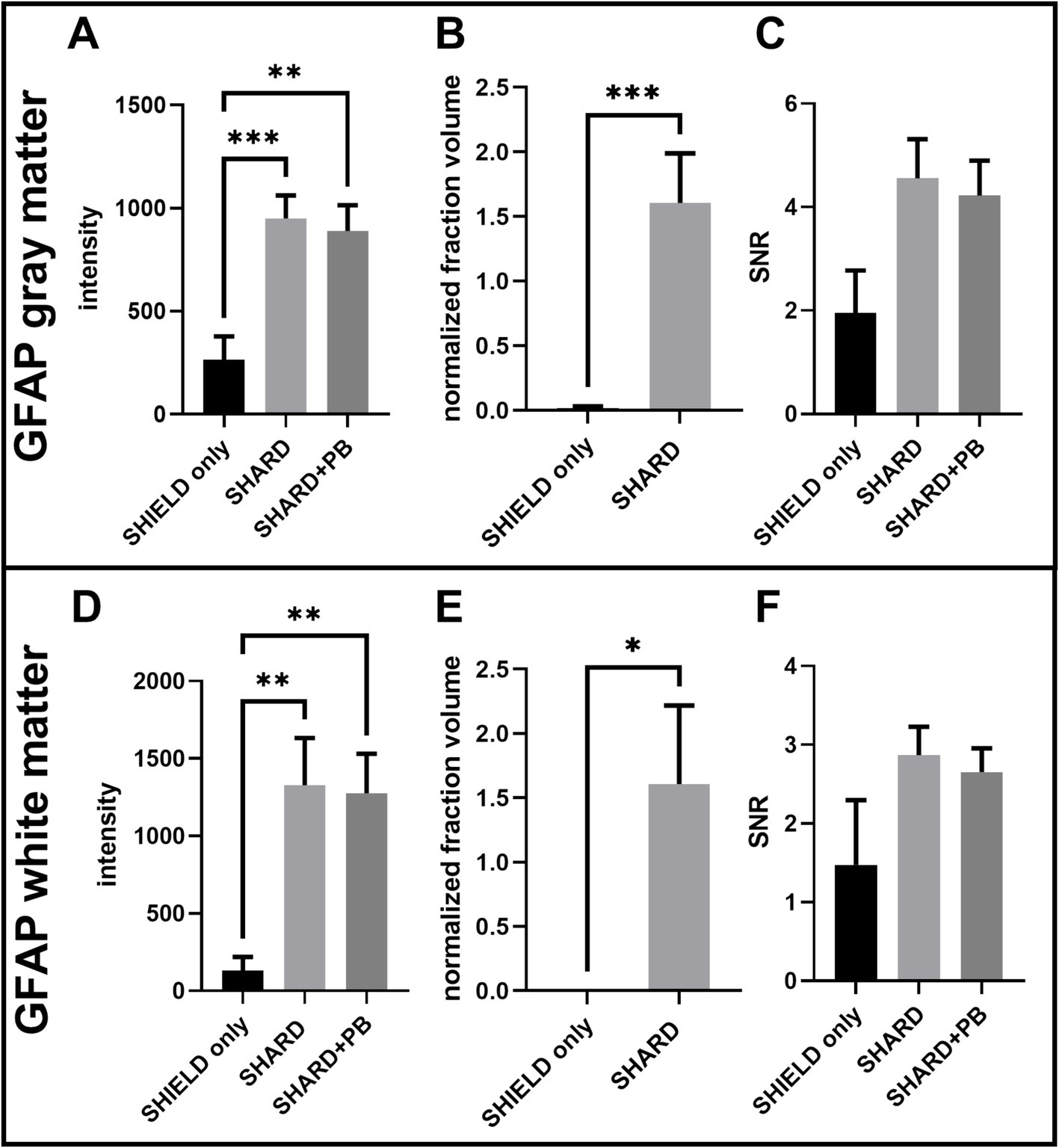
Figure 3. Quantification of GFAP647 staining across the three experimental conditions in DLF tissue. (A) GFAP647 signal intensity in the gray matter, (B) GFAP647 normalized detected fraction volume in the gray matter, (C) GFAP647 signal-to-noise ratio in the gray matter, (D) GFAP647 signal intensity in the white matter, (E) GFAP647 normalized detected fraction volume in the white matter, and (F) GFAP647 signal-to-noise ratio in the white matter. *p < 0.05, **p < 0.01, ***p < 0.001.
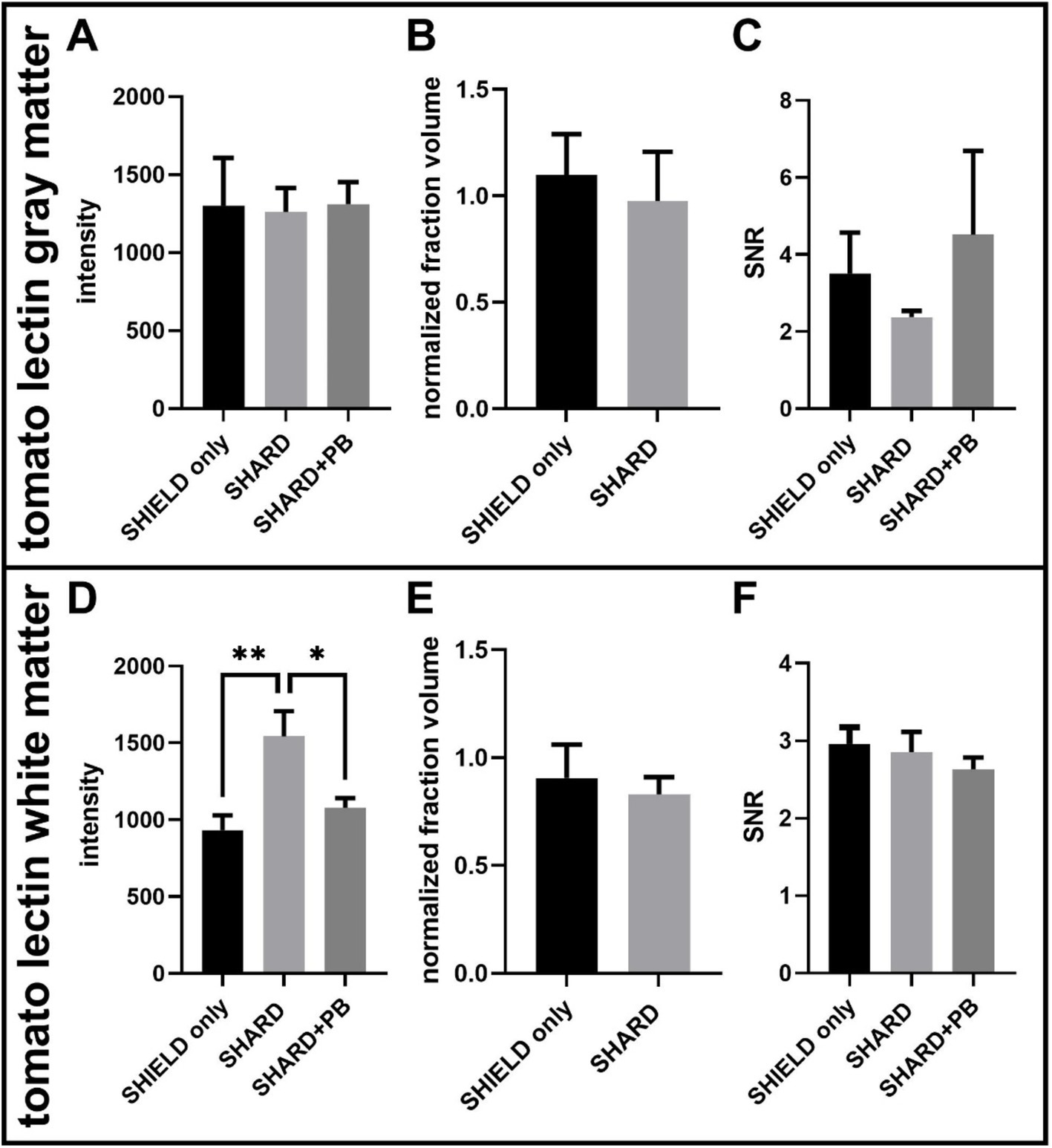
Figure 4. Quantification of tomato lectin488 staining across the three experimental conditions in DLF tissue. (A) Tomato lectin488 signal intensity in the gray matter, (B) tomato lectin488 normalized detected fraction volume in the gray matter, (C) tomato lectin488 signal to noise ratio in the gray matter, (D) tomato lectin488 signal intensity in the white matter, (E) tomato lectin488 normalized detected fraction volume in the white matter, and (F) tomato lectin488 signal to noise ratio in the white matter. *p < 0.05, **p < 0.01. For each condition, the number of cases was 11 cases.
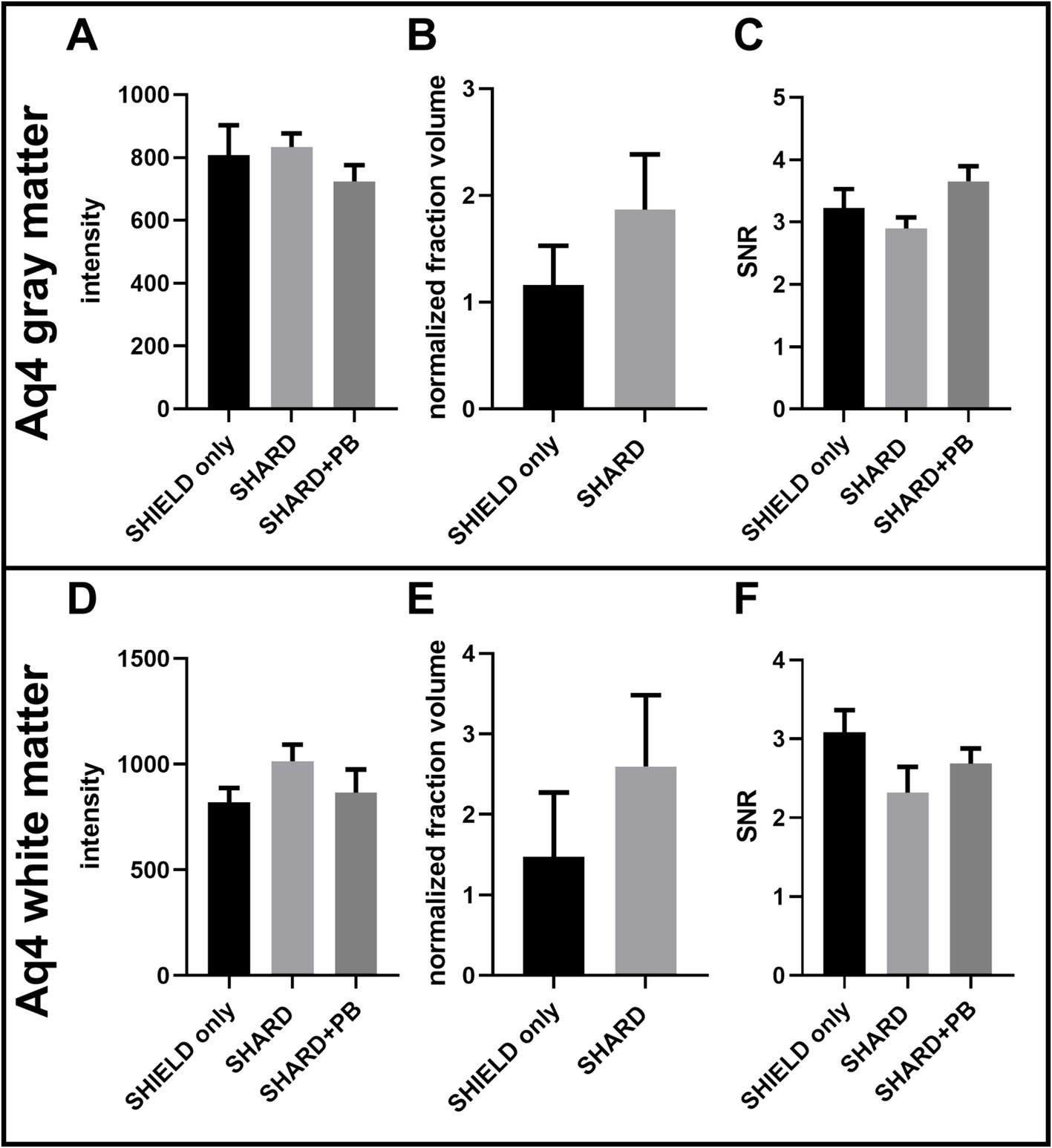
Figure 5. Quantification of Aq4Cy3 staining across the three experimental conditions in DLF tissue. (A) AQ4 Cy3 signal intensity in the gray matter, (B) AQ4 Cy3 normalized detected fraction volume in the gray matter, (C) AQ4 Cy3 signal to noise ratio in the gray matter, (D) AQ4 Cy3 signal intensity in the white matter, (E) AQ4 Cy3 normalized detected fraction volume in the white matter, and (F) AQ4Cy3 signal to noise ratio in the white matter. For each condition, n = 11 cases.
3.6 Alkaline decolorization treatment
A side-by-side comparison was completed on case u11 DLF tissue using the SHARD+PB condition and a previously published alkaline decolorization treatment (Scardigli et al., 2021). Briefly, before SHIELD processing, the section was treated with 30% (vol/vol) H2O2 diluted in deionized (DI) water for 1 h at RT and then washed three times in PBS for 10 min each. The sample was incubated in an alkaline antigen retrieval solution for 10 min at 95°C. The specimen was then cooled at RT for 30 min and washed in DI water for 5 min before incubating for 1 h in PBS. The section was immediately stained. This comparison is shown in Figure 6.

Figure 6. Qualitative Comparison of SHARD+PB vs. an alkaline antigen retrieval treatment. 3D renderings of immunofluorescent stains in the DLF of case u11 following treatment with the SHARD+PB condition (A–D,I–L) and the alkaline AR protocol (E–H,M–P) in the gray matter (A–H, 97.5 μm Z-stacks) and the white matter (I–P, 79.5 μm Z-stacks). The three stains shown are tomato lectin488 (A,E,I,M), anti-AQ4Cy3 (B,F,J,N), and anti-GFAP647 (C,G,K,O). The overlay of all three is shown in (D,H,L, P). All stains are shown as xy views (top) and yz views (bottom), both of which are raw data. Scale bar = 20 μm. (Q) Whole sample scans at 4x of u11 treated with the alkaline AR protocol (Q) and the SHARD+PB condition (R), with a scale bar of 1 mm for both. (S) Plots of stain intensity vs. tissue Z depth in the gray matter (top row) and white matter (bottom row). Stain intensities in the tissue treated with alkaline antigen retrieval are plotted in white, and intensities in tissue treated with SHARD are plotted in the same colors in which they are displayed (A–P).
3.7 Staining
Sections were incubated for 2 h in a blocking solution of PBST with 5% normal donkey serum immediately before staining. Staining was conducted over two 24-h RT incubations with gentle shaking. For the sections used to compare different experimental conditions, each slice was incubated with a 10 μg anti-GFAP antibody cocktail (BioLegend, cat# 837602, San Diego, CA) and 12 μg anti-Aquaporin-4 (Abcam Ab 3,594, Boston, MA) in 1% normal donkey serum in PBST for a total volume of 1 mL. On the 2nd day, each slice was incubated with 40 μg tomato lectin488 (Vector Laboratories, Newark, CA), as previously described (Rosen et al., 2023), 6.7 μg Cy3-conjugated donkey anti-Rabbit secondary antibody, and 10 μg Alexa Fluor®488 (AF488)-conjugated donkey anti-mouse secondary (Jackson ImmunoResearch, 711–165-152 and 715–605-150, respectively, West Grove, PA) in PBST for a total volume of 1 mL. These immunofluorescence stains will be abbreviated as AQ4Cy3 and GFAP647.
Samples shown in Figure 7 were prepared with SHARD and stained with primary antibodies listed in Table 1. After 24 h of incubation with the primary antibody, sections were washed three times in PBST. Unless the stain was a small-molecule dye or conjugated primary, sections were incubated in the appropriate secondary antibody from Jackson ImmunoResearch such that the ratio of secondary to primary antibody was 2:1.
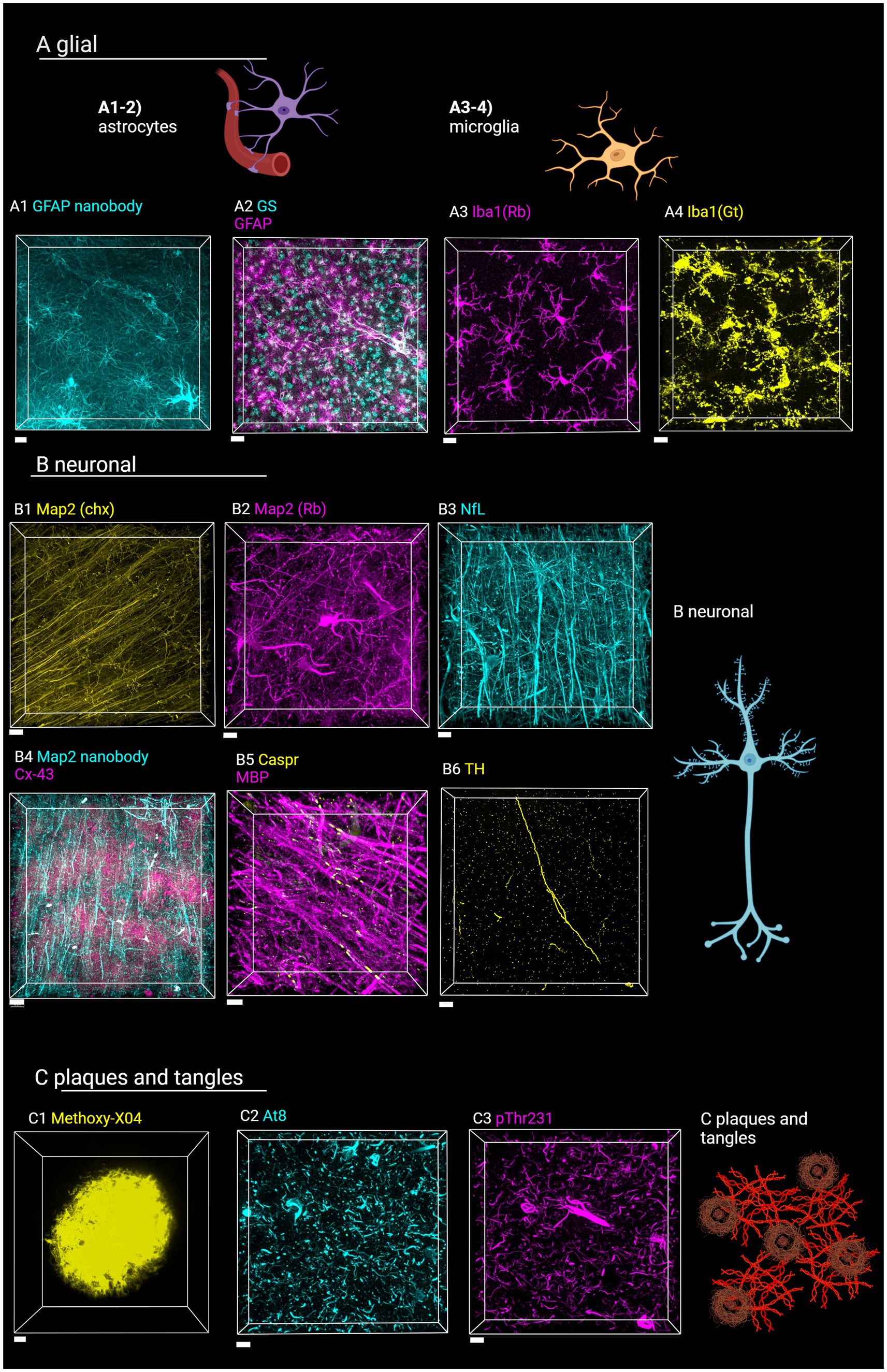
Figure 7. Staining of various targets of interest in the human brain with SHARD. Antibodies and stains are defined in Table 1. Images are taken in the DLF unless otherwise indicated. (A) Astrocytes and microglia. (A1) AF647-conjugated camelid anti-GFAP fluorescence in case u8, taken at 60x, 31.5 μm Z-stack, scale bar 10 μm. (A2) anti-GSCy3 and anti-GFAP647 fluorescence in case u9 taken at 20x, 90 μm Z-stack, scale bar 30 μm. (A3) anti-Iba1647 (rabbit host species) fluorescence in case u10 taken at 60x, 45 μm Z-stack, scale bar 15 μm. (A4) Anti-Iba1Cy3 (goat host species) fluorescence in case u11 taken at 40x, 45 μm Z-stack, scale bar 15 μm. (B) Neuronal stains (B1) Anti-Map2Cy3 (chicken host species) fluorescence in case a3 parietal lobe taken at 40x, 10 μm scale bar, 94 μm Z-stack. (B2) Anti-Map2 (rabbit host species) fluorescence in case u13 taken at 60x, 68 μm Z-stack, 15 μm scale bar. (B3) Anti-neurofilament647 fluorescence in case u12 taken at 40x, 103 μm Z-stack, 20 μm scale bar (B4) anti-Map2555 nanobody and anti-Cx43647 fluorescence in case p4 taken at 40x, 103 μm Z-stack, 30 μm scale bar. (B5) Anti-MBP647 and anti-caspr488 fluorescence in case p5 entorhinal cortex taken at 60x, 48 μm Z-stack, 10 μm scale bar. (B6) Anti-TH647 fluorescence in case a1 taken at 40x, 53 μm Z-stack, 20 μm scale bar. (C1) Methoxy-X04 fluorescence in case u3, taken at 100x, 36 μm Z-stack, 5 μm scale bar. (C2) Anti-AT8647 fluorescence in case u9 taken at 20x, 34 μm Z-stack, 15 μm sale bar. (C3) Anti-pThr231488 fluorescence in case u9 taken at 20x, 46 μm Z-stack, 15 μm scale bar. Created in BioRender (Rosen, 2024), BioRender.com/f02b703.
After the staining procedure, the samples were washed three times with PBST for 30 min each wash and incubated overnight in 4% PFA to post-fix the dye and antibodies to the tissue. The next day, the samples were washed three times, 2 h per wash, in PBS. Samples were incubated overnight in EasyIndex (LifeCanvas) before mounting onto a 25 × 75 × 1.0 mm glass slide (Fisher, Pittsburgh, PA) and covered with a No. 1.5, 22 × 30 mm glass coverslip (Corning GlassWorks, Corning, NY). EasyIndex (LifeCanvas, Cambridge, MA) was used for mounting media.
3.8 Imaging of cleared tissue
To quantify the three conditions, tissue slides were imaged on a Zeiss 880 confocal using a PLAN APO 20x/0.8 NA objective with Zen Black software (Zeiss, White Plains, NY). DyLight488, Cy3, and Alexa 647 were excited with 488 nm, 561 nm, and 633 nm wavelengths, respectively. Z-stacks of the available working distance were taken with optical sections spaced 1.5 μm apart, with a zoom of 1.8 and 4 line averaging. Pixel size was 0.115 × 0.115 × 1.5 μm and 16-bit. Pixel dwell was 2.05 μs. Emitted light was sent to the Airyscan detector, and 3D Airyscan processing was completed in Zen Black prior to image analysis. The pinhole radius was 266 μm for the tomato lectin488 channel, 449 μm for the Aq4Cy3 channel, and 598 μm for the GFAP647 channel. All image stacks were taken in the cortical gyri of the DLF, defined as the top third of the distance between the gyral crest and sulcus.
Images in Figure 7 were taken using either a Nikon Ti2 Eclipse Crest V2 spinning disc microscope or a Nikon AX-R microscope (Nikon, Melville, NY). The pinhole was set to 1 AU with a zoom size of 1.0 and pixel dwell time of 0.24 μs for all images acquired with the AX-R. The spinning disc has 70 μm pinholes. Nikon AX-R images were all taken with unidirectional scanning. Figures 7A1,A3,B2,B5 were taken with a CFI PLAN APO λ 60x/1.42 NA oil immersion objective with the Nikon spinning disc microscope. Figures 7A2,C2,C3 were taken with a CFI PLAN APO λ 20x/0.8 NA air objective on the Nikon spinning disc microscope. Figure 7C1 was taken with a CFI PLAN APO λ 100X/1.45 NA oil immersion objective with the Nikon spinning disc microscope. Figures 7A4,B1,B3,B6 were taken with a CFI PLAN APO 40x/1.25 NA silicone oil lens on the Nikon AX-R microscope using unidirectional scanning. Figures 7B4–5 was taken with a CFI PLAN APO λ 60x/1.42 NA oil immersion objective with the Nikon AX-R microscope.
Images in Figures 8, 9 were acquired with a Nikon AX-R confocal microscope with two tunable gallium arsenide phosphide (GaAsP) detectors, a multi-alkaloid detector with emission filters, and a fixed GaAsP detector with emission filters. The pinhole was set to 1 AU for all channels with a zoom size of 1.0, unidirectional scanning, 4 line averaging, and pixel size of Parameters for the optical configurations used for the 7-color multiplex shown in Figures 8, 9 are displayed in Table 4. Imaging was done with a CFI PLAN APO 40x/1.25 NA silicone oil lens and unidirectional scanning. Data from the fluorophores was divided into two optical configurations: one with methoxy-X04, AlexaFluor488, autofluorescence, and AlexaFluor594, and the other with Cy3, AlexaFluor647, and AlexaFluor750. In this way, both optical configurations had two dyes that used both the tunable GaAsP detectors. Z-resolution was 0.4 μm. Images on a single plane were taken with both optical configurations before moving in Z onto the next plane.

Figure 8. Imaging of a seven-color multiplex in the gray matter of the parietal lobe in case of a2, all in the same field of view. The scale bar is 30 μm for all. (A) Iba1594, (B) Map2Cy3, (C) MBP647, (D) Methoxy-X04, (E) GFAP750, (F) autofluorescence, (G) tomato lectin488, (H) tomato lectin488 after autofluorescence subtraction and image segmentation, (I) Overlay of stains shown in (A–E,H). The z-stack is 148 μm, with a Z-step of 0.5 μm. yz view of image stacks is shown with both raw pixel intensities and pixel intensities after layer normalization preprocessing.

Figure 9. Imaging of a seven-color multiplex in the white matter of the parietal lobe in case of a2, all in the same field of view. The scale bar is 30 μm for all. (A) Iba1594, (B) Map2Cy3, (C) MBP647, (D) Methoxy-X04, (E) GFAP750, (F) autofluorescence, (G) tomato lectin488, (H) tomato lectin488 after autofluorescence subtraction and image segmentation, (I) Overlay of stains shown in (A–E,H). The z-stack is 116 μm with a Z-step of 0.5 μm. yz view of image stacks is shown with both raw pixel intensities and pixel intensities after layer normalization preprocessing.
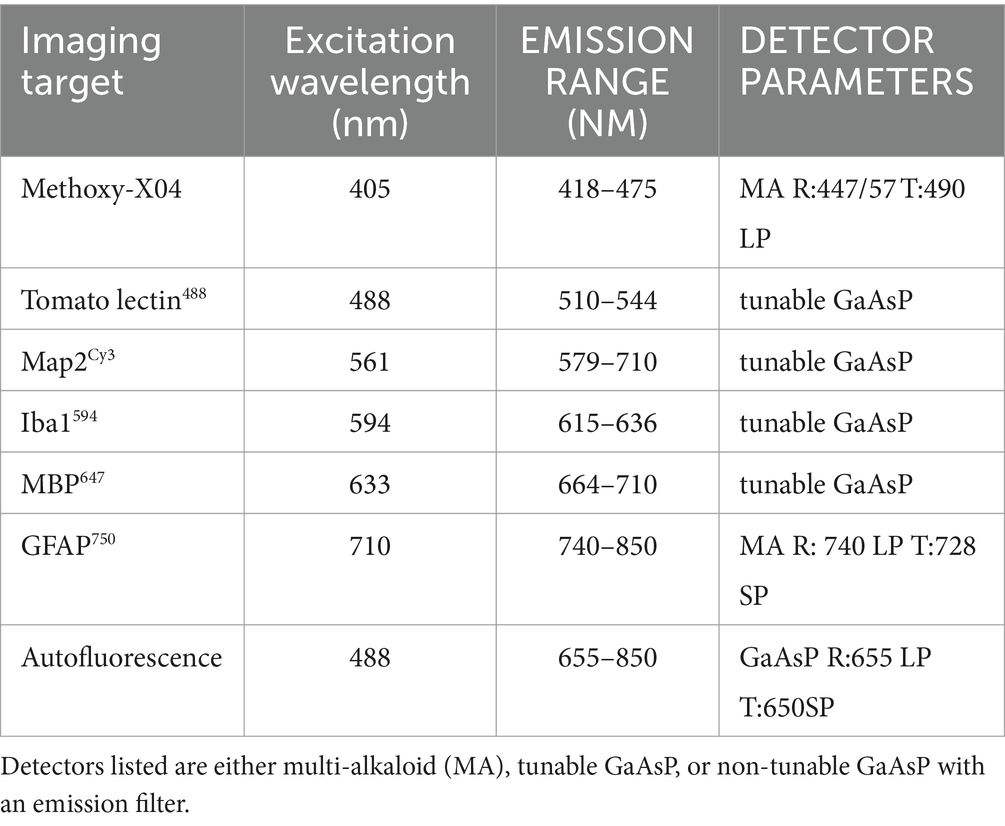
Table 4. Nikon AX-R optical configuration parameters for immunofluorescent 7-plex in cleared postmortem human brain tissue.
A surface for the autofluorescence was created in Imaris 10 with segmentation based on pixel intensity and then thresholded to a volume of segmentation greater than 100 pixels. The intensity threshold was manually set to encompass all of the signals, as the purpose of this channel was to capture the low-intensity signals that appear in all channels. To subtract autofluorescence from the tomato lectin488 channel, pixels inside of the autofluorescent surface in the tomato lectin488 channel were set to zero, and the tomato lectin488 staining was also segmented based on pixel intensity and volume. Pixels outside of the resulting segmentation were set to zero, resulting in the background-subtracted tomato lectin488 images. The rationale for removing autofluorescence before segmentation is that in the fluorophore channels, there is no way to distinguish between autofluorescent pixel intensity and pixel intensity coming from fluorophores. It is easier to create an additional background subtracted channel and segment than to manually edit segmented surfaces individually to remove autofluorescence, which is a time-consuming process in Imaris. All images displayed are after a layer-normalizing preprocessing step that accounts for the effects of photobleaching.
3.9 Quantification
Segmentation was completed using Imaris (v10.0, Bitplane, Belfast, United Kingdom). Surface creation for each channel (GFAP647, AQ4Cy3, and tomato lectin488) was based on background-subtracted pixel intensity and segment volume. Only these two parameters were used for the segmentation of GRAP647 and AQ4 Cy3. For the tomato lectin488 channel, an additional filter was used for sphericity, removing sphere-shaped objects as blood vessels have tubular morphology. For each case, parameters for segmentation were created based on the SHARD+PB condition white matter and gray matter images, giving a total of six parameter sets (one gray matter, one white matter, for each channel). These parameters were then applied to the image sets from conditions 1 and 2 for all six channels, gray matter and white matter. The average pixel intensity in the created surface was the “signal” measurement used in the signal-to-noise ratio calculation. The average intensity of pixels outside of the surface was the “noise” intensity measurement. The signal-to-noise ratio (SNR) was calculated as the mean signal intensity divided by the mean noise intensity.
The fraction volume for each stain in each image stack was calculated as the total volume of the segmented surface divided by the total volume of the image stack. To account for any biological difference in fraction volume on a case-by-case basis, we normalized the fraction volume of the SHIELD only and SHARD conditions to the fraction volume of the SHARD+PB condition. Therefore, the normalized fraction volume of the SHARD+PB condition was always 1.
To quantify stain penetration depth and assess the utility of the normalized layer function in Imaris, we imported raw and normalized data into Arivis Pro for quantification. The mean pixel intensity was calculated for each plane in each of the raw and normalized channels. This was done for each of the 22 Z-stacks of each tissue treatment condition from 11 cases, as each case had two Z-stacks per condition, one in the white matter and one in the gray matter. For each condition and stain, standard errors were calculated for each Z position using the number of cases that had corresponding images at each Z position.
3.10 Statistical analyses
The fluorescence microscopy data were analyzed using SPSS (v. 27, IBM, Inc., Armonk, NY) and GraphPad Prism (v. 9.0.0, GraphPad Software, La Jolla, CA). An analysis of variance (ANOVA) was used to obtain means of signal intensity and SNR for tomato lectin, aquaporin, and GFAP stains in each experimental group. Because fraction volume was normalized based on the SHARD+PB condition, normalized fraction volume in the SHARD+PB condition had a standard error of zero and was not included in statistical comparisons. An unpaired t-test was used to compare the normalized fraction volume of the SHIELD-only condition versus the SHARD condition. Associations among measured image quality quantifiers were measured with linear regression models with age of death or PMI as the independent variable and signal intensity, noise intensity, normalized fraction volume, and SNR as the dependent variables. The significance level was set a priori to 5%.
4 Results
4.1 Antigen retrieval results in robust fluorescent microscopy images on cleared postmortem human brain tissue
SHIELD tissue treatment, passive delipidation, and staining were successful in visualizing blood vessels and astrocytes in three dimensions. Cases u1-7 and p1-4 were each subjected to SHIELD only, SHARD, and SHARD+PB conditions. Figure 1 shows tomato lectin488, AQ4Cy3, and GFAP647 fluorescence in the gray matter, and Figure 2 shows tomato lectin488, AQ4Cy3, and GFAP647 fluorescence in the white matter. Fluorescence was distinctive in the gray and white matter for all three stains. Tomato lectin488 fluorescence shows many branching curving vessels in the gray matter and more linear vessels in the white matter with fewer branches (Figures 1A,E,I compared to Figures 2A,E,I). Astrocyte density was generally greater in the white matter compared to the gray matter, as white matter showed more AQ4Cy3 and GFAP647 fluorescence density (Figures 2B,C,F,G,J,K) than the gray matter (Figures 2B,C,F,G,J,K). Staining quality appeared comparable among the three experimental conditions for tomato lectin488 (Figures 1, 2A vs. Figure 2E vs. Figure 2I) and AQ4Cy3 (Figures 1, 2B vs. Figure 2F vs. Figure 2J). However, the SHIELD-only condition produced minimal, if any, staining for GFAP647 (Figures 1, 2C) compared to the SHARD or SHARD+PB conditions (Figures 1, 2G,K).
Both photobleaching during confocal imaging and stain penetration into the tissue can be quantified by stain intensity vs. depth plots. Plots of raw data intensity vs. depth showed an initial decrease in staining intensity with increasing depth for all three stains (Figures 1, 2M,N, colored plots). The normalized layer function in Arivis, which standardizes the mean intensity of each channel it is applied to in each Z-slice, can be used to effectively eliminate the decrease of intensity through Z (Figures 1, 2M,N, white plots). Qualitatively, the normalized layer function creates uniformity of intensity in each Z slice, as seen in the yz views in Figures 1, 2A–L. Intensity vs. depth curves were U-shaped for AQ4Cy3, with planes at the ends of the Z-stack containing more intense pixels than the middle (Figures 1, 2M,N, middle yellow plots).
4.2 Antigen retrieval dramatically improved GFAP647 stain
Antigen retrieval significantly increased both mean signal brightness and mean detected volume of GFAP647 fluorescence in the DLF gray matter. In the gray matter, the mean segmented signal intensity was greater in the SHARD (1,000 ± 100) and SHARD+PB (900 ± 100) conditions than they were for the SHIELD only condition (300 ± 100), p = 0.0007 and 0.0019, respectively (Figure 3A). The mean normalized fraction volume was similarly higher in the SHARD condition (1.6 ± 0.4) compared to the SHIELD-only condition (0.02 ± 0.01), p < 0.0001 (Figure 3B). Only three cases had any GFAP647 detected by the same surface-building parameters applied to the SHARD+PB condition in the gray matter. The ratio of average pixel intensity inside the segmented surface vs. that outside the segmented surface (SNR) was 2.0 ± 0.8 in the SHIELD-only condition, 4.5 ± 0.8 in the SHARD condition, and 4.2 ± 0.7 in the SHARD+PB condition, none of which were significantly different (Figure 3C).
The same increases in signal brightness and normalized fraction volume of GFAP647 fluorescence were measured in the GFAP647 white matter. GFAP647 white matter signal intensity was significantly greater in the SHARD+PB condition (1,300 ± 200) compared to the SHIELD-only condition (130 ± 90, p = 0.0034) (Figure 3D). The GFAP647 white matter signal intensity in the SHARD condition (1,300 ± 300) was also significantly greater than that in the SHIELD-only condition (p = 0.0077). The normalized fraction volume of GFAP647 in the white matter was 1.7 ± 0.6, significantly greater than the equivalent in the SHIELD-only condition, 0.0004 ± 0.0009, p = 0.003 (Figure 3E). None of the cases showed any detected GFAP647 volume in the white matter in the SHIELD-only condition. Again, the mean SNR values of the three conditions were not significantly different, with values of 1.4 ± 0.8 for SHIELD only, 2.9 ± 0.4 for the SHARD condition, and 2.6 ± 0.3 for the SHARD+PB condition (Figure 3F).
4.3 Tomato lectin488 fluorescence was consistent across experimental conditions
There were no statistically significant differences in the gray matter for tomato lectin488 signal fluorescence intensity, normalized fraction volume, or SNR (Figures 4A–C). The white matter tomato lectin488 signal intensity in the SHARD condition (1,500 ± 200) was significantly greater than the signal intensity in the SHIELD-only condition (900 ± 100, p = 0.0021) and in the SHARD+PB condition (1,080 ± 60, p = 0.0207, Figure 4D). There were also no statistically significant differences in the white matter for tomato lectin488 normalized fraction volume, or SNR (Figures 4E,F).
4.4 AQ4Cy3 fluorescence was consistent across experimental conditions
There were no statistically significant differences in AQ4 Cy3 signal intensity, normalized fraction volume, or SNR between the three conditions in the white matter or the gray matter (Figure 5). The gray and white matter AQ4 normalized fraction volume in the SHARD condition appears to be trending higher than in the SHIELD-only condition in both the gray and white matter, but this difference is not statistically significant (Figures 5B,E).
4.5 Alkaline antigen retrieval produced comparable image quality to SHARD but fragile tissue
High-quality immunofluorescence images were also produced using antigen retrieval in a pH nine solution and bleaching with hydrogen peroxide (Figures 6E–H,M). For direct comparison, a section from the same case was treated with the pH 6 SHARD+PB condition (Figures 6A–D,I). While most of the metrics of image quality were comparable, the alkaline antigen retrieval and peroxide bleaching were more effective at reducing background in the tomato lectin488 channel. The tomato lectin488 SNR of the SHARD+PB condition was 1.5 in the gray matter and 1.6 in the white matter. The tomato lectin488 SNR in the alkaline AR + peroxide bleach condition was higher at 3.2 in the gray matter and 2.4 in the white matter. The SHARD+PB condition detected about twice as much GFAP647 in the gray matter, with a fraction volume of 0.02 compared to 0.008 in the alkaline AR + peroxide bleach condition. While the alkaline AR + peroxide bleach condition did produce high-quality images, a major drawback to this technique was that the tissue was torn and brittle at the end of treatment (Figures 6Q,R). Penetration of all three stains was comparable regardless of SHARD or alkaline antigen retrieval treatment (Figure 6S).
4.6 3D visualization of additional markers with SHARD
We also tested 14 additional antibodies and one small-molecule dye for staining different cell types and pathology-associated markers with the antigen retrieval and tissue-clearing protocol (Table 1). The hope is to provide a reference by which future studies on human tissue may be completed. Images of these stains are shown in Figure 7, along with cartoons of the cell type or marker detected. Through these stains, astrocytes, microglia, neurons, plaques, and hyperphosphorylated tau tangles were successfully visualized.
Glial cells are a major target group we sought to detect. As described in sections 3.2 and 3.4, the GFAP and AQ4 antibodies detect astrocytes, with GFAP especially detecting the vast network of processes. As an alternative to the GFAP from BioLegend, a camelid GFAP nanobody conjugated to AlexaFluor647 dye also detects activated astrocytes (Figure 7A1). Nanobodies are engineered antibodies that are advantageous due to their small (~15 kDa) size (Sun et al., 2021). We further detected astrocytes with a glutamine synthetase antibody (Figure 7A2). Detecting microglia was successful with a well-established ionized calcium-binding adapter molecule I (Iba1) antibody from Wako (Figure 7A3). As another alternative, we also tested Wako’s Iba1 produced in goats (Figure 7A4).
Secondly, different types of neurons and neuronal components could be detected with the SHARD method. Microtubule-associated protein 2 (Map2) proved to be a successful target for visualizing neuronal cell bodies and processes, with a primary antibody produced in both chickens (Figure 7B1) and rabbits (Figure 7B2). Pan-neuronal staining is visualized with an anti-neurofilament antibody (Figure 7B3). As a direct staining alternative, an AlexaFluor555-conjugated Map2 primary nanobody also detected neuronal processes (Figure 7B4). A GeneTex myelin basic protein antibody was effective for visualizing the myelin sheath, and an Abcam antibody against the contactin-associated protein (caspr) highlights the ends of the axonal internodes (Figure 7B5). Intercellular gap junctions were visualized with an anti-connexin43 (Cx43) antibody (Figure 7B4). The anti-tyrosine hydroxylase (TH) antibody detects catecholaminergic principal neurons and processes (Figure 7B6).
Finally, we detected a few targets for neuropathological disease markers. Small-molecule dye Methoxy-X04 from Tocris Bioscience detected Aβ plaques, a hallmark of AD (Figure 7C1). Tangles of hyperphosphorylated tau (ptau) are also present in AD and in the pathognomonic lesion of CTE. We detected two variants of ptau with a widely used AT8 antibody (Figure 7C2) and an anti-pThr231 antibody (Figure 7C3).
4.7 Highly multiplexed immunofluorescence in cleared tissue
The toolbox of various stains described, plus the SHARD method, allowed for the visualization of six fluorescently stained targets in addition to autofluorescence. Provided that each primary antibody is from a different host species, multiple targets can be stained simultaneously without the cross-reactivity of fluorescent secondary antibodies. A seven-color multiplex was imaged in both gray matter (Figure 8) and white matter (Figure 9). With the Nikon AX-R, customizable detectors, and six different laser lines, we imaged a multiplex constituting of Iba1594 (goat host species, Figures 8A, 9B), Map2Cy3 (chicken host species, Figures 8B, 9B), MBP647 (rabbit host species, Figures 8C, 9C), Methoxy-X04 (small-molecule dye, Figures 8D, 9D), GFAP750 (mouse host species, Figures 8E, 9E), autofluorescence (Figures 8F, 9F), and tomato lectin488 (small-molecule dye, Figures 8G,H, 9G,H). Figures 8G, 9G show the tomato lectin488 raw data. Background-subtracted tomato lectin488 images are shown in Figures 8H, 9H.
Fluorophores were strategically paired with their targets. Because of the multitude of dyes in this multiplex, the Cy3 channel has the smallest range of detection without risking cross-detection from the AlexaFluor594 channel. For this reason, a robust antibody such as Map2 best suits this dye in the multiplex. From our experience, myelin is autofluorescent in red, and thus, we chose the AlexaFluor647 dye for labeling MBP. Similarly, autofluorescence of blood vessels is excited with a 488 nm light source, so tomato lectin488 was chosen to stain blood vessels. With these two strategic dye placements, nontarget autofluorescence was minimized across channels. Finally, autofluorescence in postmortem human tissue is usually strongest when excited by the 488 laser. Another reason that the DyLight 488 fluorophore was chosen to stain blood vessels is that blood vessels have obvious morphology that can be easily segmented.
A separate channel was also designated for autofluorescence. In our experience, autofluorescence can have a large Stokes shift. Because of this, we were able to excite autofluorescence with the 488 laser and use the same detector range as the AlexaFluor647 channel. After segmentation, pixels designated as autofluorescence were subtracted from the tomato lectin488 channel (see Figure 8G vs. Figure 8H and Figure 9G vs. Figure 9H). This autofluorescence can be segmented and subtracted off of any imaged channel to remove background.
4.8 Effectiveness of antigen retrieval persists across brain banks and AD status
There were no differences in the means of variables measured in cases without neurodegenerative disease compared to cases with neurodegenerative disease. Of the 11 cases quantified in the three conditions, four cases had AD. The means of signal intensities, signal-to-noise ratios, noise intensities, and normalized fraction volumes did not differ for tomato lectin, AQ4, or GFAP647 staining in the gray or white matter (Supplementary Table S1). There were also no significant differences when cases were grouped by Brain Bank of donation (NPBB vs. UNITE, Supplementary Table S2).
4.9 Effects of age and postmortem interval
Fluorescent noise intensity decreased with increasing age with tomato lectin, GFAP647, and AQ4 Cy3 stains. This negative correlation was present in all three experimental conditions for all stains that had signal (see Table 5 for details).
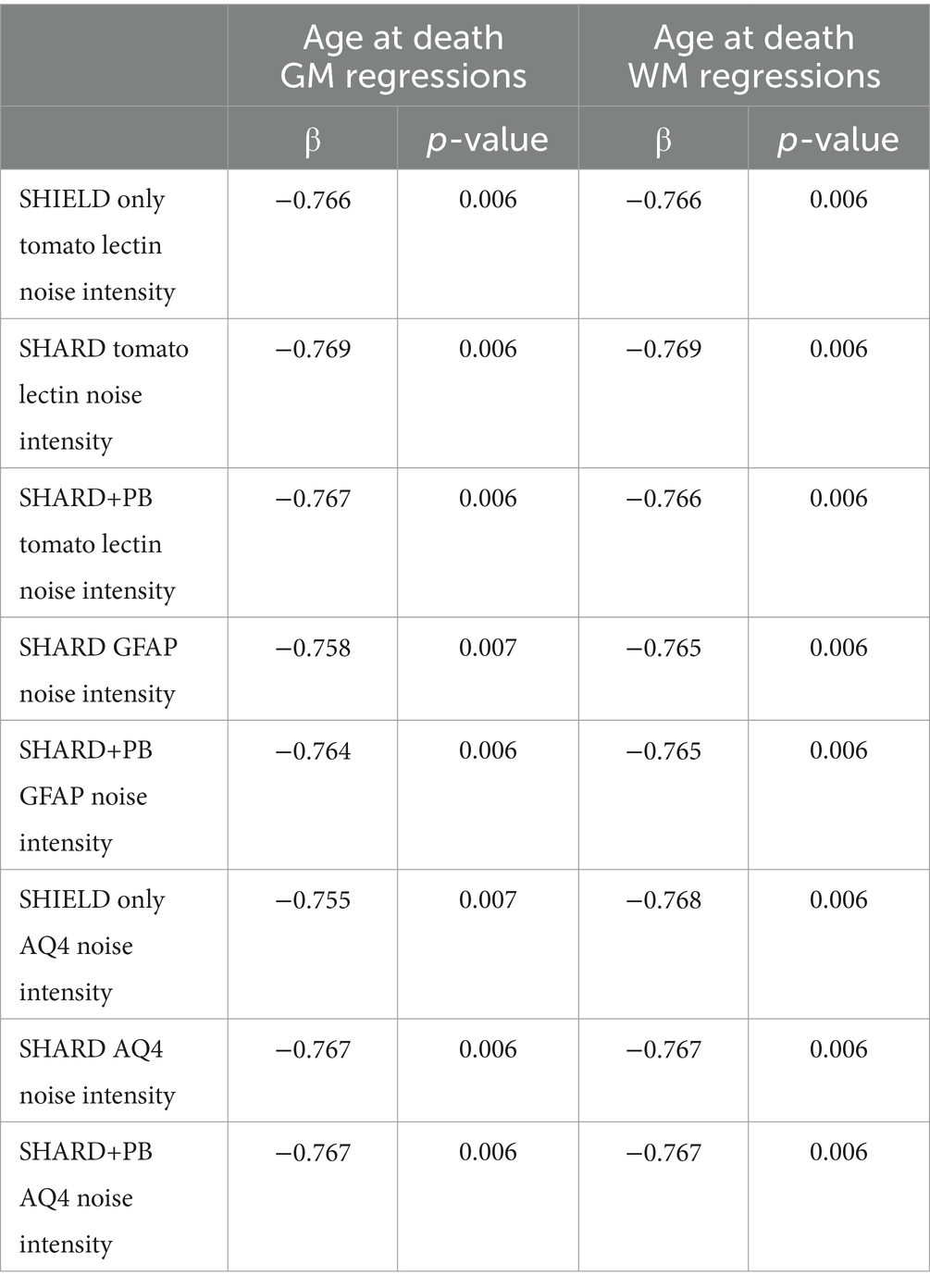
Table 5. Linear regressions of the age of death vs. measured variables in the gray matter (first two columns) and the white matter (last two columns).
There were a few increases in astrocytic staining measurements that correlated with increased PMI (Table 6). The white matter AQ4Cy3 normalized fraction volume showed a significant correlation with PMI. The SNR of tomato lectin488 fluorescence in the white matter increased significantly with PMI in the SHARD+PB condition (standardized β = 0.793, p = 0.004). The normalized white matter AQ4Cy3 fraction volume increased with PMI in the SHIELD-only condition (standardized β = 0.787, p = 0.004) and the SHARD condition (standardized β = 0.783, p = 0.004). There were no correlations of any measured quantification of image quality with the year the sample was received.
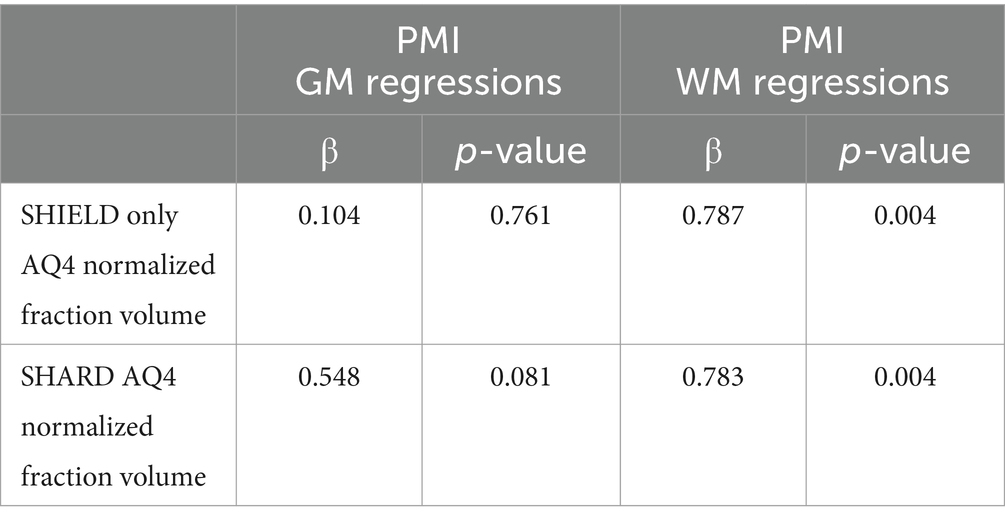
Table 6. Linear regressions of PMI vs. measured variables in the gray matter (first two columns and the white matter, last two columns).
5 Discussion
We quantified the effectiveness of the SHARD method for robust immunostaining of cleared, long-term formaldehyde-fixed human postmortem brain tissue using tomato lectin488, AQ4Cy3, and GFAP647 stains. We compared immunostaining across delipidated tissues treated with SHIELD, SHARD, and SHARD followed by photobleaching with white light (SHARD+PB).
Acidic antigen retrieval significantly enhanced staining with the cytoskeletal GFAP antibody, as indicated by higher mean segmented signal intensity and a greater mean fraction of the image stack exhibiting the detected signal. Alkaline antigen retrieval produced comparable images, though acidic treatment had the advantage of preserving tissue integrity.
Both antigen retrieval and tissue clearing were effective across the three quantified markers and the three brain banks from which the samples were sourced, including those with AD. Additionally, we visualized a broader panel of antibodies compatible with the citrate antigen retrieval method. Notably, increased age at death correlated negatively with image noise across all channels.
Antigen retrieval with citrate dramatically increased the staining effectiveness of an intracellular marker. Without antigen retrieval, the GFAP647 signal was virtually absent. Heating in acidic conditions breaks formaldehyde crosslinking, which is crucial for unmasking binding sites, particularly cellular components like GFAP. In contrast, AQ4Cy3 and tomato lectin488 stains were effective not only in the SHARD conditions but also in the SHIELD-only condition. Tomato lectin likely does not require antigen retrieval due to its small-molecule structure, which experiences less steric hindrance when binding to its target, even in the presence of formaldehyde crosslinking. On the other hand, AQ4 is a cell membrane-bound target. Since SDS-based clearing dissolves cellular membranes, our findings suggest that SDS treatment alone is sufficient to expose the AQ4 antigen.
SHARD is equal to or more widely applicable than the few previously published studies combining antigen retrieval and tissue clearing. We found that a previously published method using alkaline antigen retrieval and hydrogen peroxide bleach (Scardigli et al., 2021) worked with our samples for microscopic regions but resulted in torn tissue sections. SHARD uses 10 mM citrate buffer, an order of magnitude lower than the citrate buffer used in more recent work in the field (Woelfle et al., 2023) but in line with methods documented a few decades ago (Jiao et al., 1999; Brorson, 2002; Ino, 2003; Namimatsu et al., 2005; Shi et al., 2007). The advantage of the method described here is that it is compatible with long-term, immersion-fixed human brain tissue stored in formaldehyde, and the treatment results are intact tissue sections. Our analyses show that this method works well despite any unavoidable variability of using human tissue, such as neurodegenerative disease status, varying age of death, and postmortem interval.
Intensity vs. depth analysis revealed uneven tissue penetration with SHARD. Because of confocal imaging, it is impossible to conclude that inhomogeneity in average pixel intensity throughout Z is entirely due to differential stain diffusion throughout the tissue. However, the U-shaped curves seen with AQ4Cy3 staining intensity would only be seen if there was a higher concentration of the stain at the edges of the sample. GFAP647 also sometimes displayed a U-shaped curve (Figure 6S), indicating that the edges of the sample were exposed to a higher concentration of the stain. The relatively uniform yz views of tomato lectin488 staining raw data would indicate that most of the decrease in staining intensity vs. imaging depth is due to photobleaching. A small-molecule dye would likely achieve more homogenous diffusion throughout samples than immunofluorescent antibodies. Even though samples were 200-μm thick, the acquired Z-stacks were closer to 100 μm due to both the sample not being flat and sample compression upon mounting. Further optimization is needed to achieve immunofluorescent staining homogeneity, but in the meantime, a computational correction such as the normalized layers function in Imaris results in uniformity of pixel intensities throughout a Z-stack (see Figures 1, 2M,N and yz views in Figures 1, 2A–L, 6A–P, 7, 8). As the normalized layer function leads to higher noise at deeper stacks, staining homogeneity through Z would be most advantageous. The use of expansion microscopy, nanobodies, fluorescently conjugated primary antibodies, and/or small-molecule dyes to stain tissue would also lead to more uniform staining.
Acidic antigen retrieval worked well across various conditions, including PMI, duration of storage in fixative, and age. We show data and images from samples with PMIs from 10 to 72 h and with ages of death ranging from 35 to 93 years. PMI correlated positively with normalized AQ4Cy3 fraction volume in the white matter. This is likely due to the breakdown of the membranes prior to clearing, causing AQ4 to spread beyond localized astrocytic cell bodies in fresher tissue. Interestingly, age at death had a negative correlation with noise intensity in all three quantified stains in both the gray and white matter. This was the only correlation that was universally true regardless of tissue treatment and stain and is likely due to the decrease in total brain lipids after the first 40–50 years of life (Naudí et al., 2015).
SHARD is highly suited for high multiplexing, allowing up to seven stains to be visualized simultaneously, provided that attention is given to the primary antibodies’ host species and the emission spectra of the detected stains. For optimal noise reduction, aligning the optical parameters of a target’s autofluorescence with the fluorophore used for immunofluorescence visualization is highly advantageous. For example, based on our experience, myelin exhibits autofluorescent when excited by a 633 nm laser, making myelin basic protein an ideal candidate for the AlexaFluor647 dye. In this way, the myelin autofluorescence from the 633 nm laser will overlap with the AlexaFluor647 fluorescence intensity at the target when bound to a secondary antibody for MBP. However, if AlexaFluor647 is used to stain a different target, there may still be autofluorescence corresponding to myelin morphology.
Non-specific background can also be significantly reduced by imaging an additional channel configured specifically for autofluorescence and subtracting that channel’s segmentation from the immunofluorescent channels during image analysis. In our 7-plex experiment, autofluorescence was subtracted from the tomato lectin488 channel. The distinct morphology of blood vessels makes them an ideal target for the AlexaFluor488 channel, which exhibits the highest spectral autofluorescence.
Photobleaching as a background-reducing step had less of an effect than expected, based on previous studies (Duong and Han, 2013; Ku et al., 2020). The photobleaching protocol was believed to eliminate autofluorescence so that the SHARD+PB signals would consist purely of immunofluorescence without autofluorescence.
However, signal and noise measurements were not statistically different between the SHARD and SHARD+PB conditions for GFAP647, AQ4Cy3, and gray matter tomato lectin488. Interestingly, white matter tomato lectin488 signal intensity was significantly lower in the SHARD+PB condition compared to the SHARD condition. This decrease suggests that significant noise may have contributed to the tomato lectin488 signal in the SHARD condition. Despite this, the SNR of white matter tomato lectin488 was not significantly affected by photobleaching, indicating that photobleaching likely did not impact overall image quality. Instead, creating a separate channel for autofluorescence proved more effective than photobleaching for removing non-specific fluorescent signals.
The limitations of this study include the small sample size and the fact that only male donors were used for comparison between tissue treatment conditions. The diversity of the study population was constrained by the availability of tissue from the UNITE, NPBB, and ADRC brain banks. Future studies aimed at refining tissue treatment methods should include donors of different genders to increase generalizability. Despite the small sample size, to the best of our knowledge, this is the largest systematically quantified comparison of different tissue treatment techniques for 3D microscopy. This study is not intended to serve as an endpoint in SHIELD tissue clearing optimization; rather, it represents a waypoint for further advancements. Further development could explore the use of thicker tissue sections, employing electrophoresis-driven clearing and staining, multi-photon or light sheet microscopy, and potentially higher multiplexing through fluorescence lifetime imaging (FLIM).
Overall, the SHARD method paves the way for new areas of research using optically cleared postmortem human brain tissue that has been preserved long-term. The combination of antigen retrieval and tissue clearing provides a powerful approach for visualizing targets that might otherwise be undetectable. The markers highlighted in this study can be utilized to investigate human disease through microscopy, with potential applications in a variety of research contexts.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics statement
The studies involving humans were approved by VA Boston Healthcare Institutional Review Board affiliated with the National Center for PTSD Brain Bank. The studies were conducted in accordance with the local legislation and institutional requirements. The human samples used in this study were acquired from primarily isolated as part of your previous study for which ethical approval was obtained. The donors' next of kin provided written informed consent for brain donation and research participation.
Author contributions
GR: Conceptualization, Data curation, Formal analysis, Methodology, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. DK: Formal analysis, Writing – review & editing. RN: Resources, Writing – review & editing. HK: Resources, Writing – review & editing. RM: Resources, Writing – review & editing. KC: Resources, Writing – review & editing. CK: Resources, Writing – review & editing. BD: Writing – review & editing. TS: Writing – review & editing. VA: Resources, Writing – review & editing. MA: Resources, Writing – review & editing. AM: Resources, Writing – review & editing. BH: Conceptualization, Funding acquisition, Investigation, Methodology, Project administration, Resources, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was supported by the NINDS F31NS127449, NINDS/NIA RF1NS122854, United States Department of Veterans Affairs, Veterans Health Administration, BLRD Merit Award (I01BX005933), National Institute of Neurological Disorders and Stroke (U54NS115266), and National Institute of Aging Boston University AD Research Center (P30AG072978).
Acknowledgments
The authors gratefully acknowledge the use of resources and facilities at the VA Boston Healthcare System and the National Center for PTSD Brain Bank. We also thank Ambar Hart González for providing feedback on the manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Correction note
This article has been corrected with minor changes. These changes do not impact the scientific content of the article.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnins.2024.1474617/full#supplementary-material
References
Alelú-Paz, R., Iturrieta-Zuazo, I., Byne, W., Haroutunian, V., García-Villanueva, M., Rábano, A., et al. (2008). A new antigen retrieval technique for human brain tissue. PLoS One 3:e3378. doi: 10.1371/journal.pone.0003378
Alosco, M. L., Mian, A. Z., Buch, K., Farris, C. W., Uretsky, M., Tripodis, Y., et al. (2021). Structural MRI profiles and tau correlates of atrophy in autopsy-confirmed CTE. Alzheimers Res. Ther. 13:193. doi: 10.1186/s13195-021-00928-y
Ando, K., Laborde, Q., Lazar, A., Godefroy, D., Youssef, I., Amar, M., et al. (2014). Inside Alzheimer brain with CLARITY: senile plaques, neurofibrillary tangles and axons in 3-D. Acta Neuropathol. 128, 457–459. doi: 10.1007/s00401-014-1322-y
Boenisch, T. (2006). Heat-induced antigen retrieval: what are we retrieving? J Histochem Cytochem 54, 961–964. doi: 10.1369/jhc.6P6945.2006
Brorson, S.-H. (2002). pH-dependent effect of heat-induced antigen retrieval of epoxy section for electron microscopy. Micron 33, 481–482. doi: 10.1016/S0968-4328(01)00040-3
Chung, K., and Deisseroth, K. (2013). CLARITY for mapping the nervous system. Nat. Methods 10, 508–513. doi: 10.1038/nmeth.2481
Chung, K., Wallace, J., Kim, S. Y., Kalyanasundaram, S., Andalman, A. S., Davidson, T. J., et al. (2013). Structural and molecular interrogation of intact biological systems. Nature 497, 332–337. doi: 10.1038/nature12107
D’Amico, F., Skarmoutsou, E., and Stivala, F. (2009). State of the art in antigen retrieval for immunohistochemistry. J. Immunol. Methods 341, 1–18. doi: 10.1016/j.jim.2008.11.007
Duong, H., and Han, M. (2013). A multispectral LED array for the reduction of background autofluorescence in brain tissue. J. Neurosci. Methods 220, 46–54. doi: 10.1016/j.jneumeth.2013.08.018
Ertürk, A., Becker, K., Jährling, N., Mauch, C. P., Hojer, C. D., Egen, J. G., et al. (2012). Three-dimensional imaging of solvent-cleared organs using 3DISCO. Nat. Protoc. 7, 1983–1995. doi: 10.1038/nprot.2012.119
Evers, P., and Uylings, H. B. M. (1994). Effects of microwave pretreatment on immunocytochemical staining of vibratome sections and tissue blocks of human cerebral cortex stored in formaldehyde fixative for long periods. J. Neurosci. Methods 55, 163–172. doi: 10.1016/0165-0270(94)90208-9
Friedman, M. J., Huber, B. R., Brady, C. B., Ursano, R. J., Benedek, D. M., Kowall, N. W., et al. (2017). VA’s national PTSD brain Bank: a National Resource for research. Curr. Psychiatry Rep. 19:73. doi: 10.1007/s11920-017-0822-6
Gavett, B. E., Lou, K. R., Daneshvar, D. H., Green, R. C., Jefferson, A. L., and Stern, R. A. (2012). Diagnostic accuracy statistics for seven neuropsychological assessment battery (NAB) test variables in the diagnosis of Alzheimer’s disease. Appl. Neuropsychol. Adult 19, 108–115. doi: 10.1080/09084282.2011.643947
Hildebrand, S., Schueth, A., Herrler, A., Galuske, R., and Roebroeck, A. (2019). Scalable labeling for Cytoarchitectonic characterization of large optically cleared human neocortex samples. Sci. Rep. 9:10880. doi: 10.1038/s41598-019-47336-9
Ino, H. (2003). Antigen retrieval by heating En bloc for pre-fixed frozen material. J Histochem Cytochem 51, 995–1003. doi: 10.1177/002215540305100803
Jiao, Y., Sun, Z., Lee, T., Fusco, F. R., Kimble, T. D., Meade, C. A., et al. (1999). A simple and sensitive antigen retrieval method for free-floating and slide-mounted tissue sections. J. Neurosci. Methods 93, 149–162. doi: 10.1016/S0165-0270(99)00142-9
Kim, S.-Y., Cho, J. H., Murray, E., Bakh, N., Choi, H., Ohn, K., et al. (2015). Stochastic electrotransport selectively enhances the transport of highly electromobile molecules. Proc. Natl. Acad. Sci. U. S. A. 112, E6274–E6283. doi: 10.1073/pnas.1510133112
Ku, T., Guan, W., Evans, N. B., Sohn, C. H., Albanese, A., Kim, J. G., et al. (2020). Elasticizing tissues for reversible shape transformation and accelerated molecular labeling. Nat. Methods 17, 609–613. doi: 10.1038/s41592-020-0823-y
Lai, H. M., Liu, A. K. L., Ng, H. H. M., Goldfinger, M. H., Chau, T. W., DeFelice, J., et al. (2018). Next generation histology methods for three-dimensional imaging of fresh and archival human brain tissues. Nat. Commun. 9:1066. doi: 10.1038/s41467-018-03359-w
Liebmann, T., Renier, N., Bettayeb, K., Greengard, P., Tessier-Lavigne, M., and Flajolet, M. (2016). Three-dimensional study of Alzheimer’s disease hallmarks using the iDISCO clearing method. Cell Rep. 16, 1138–1152. doi: 10.1016/j.celrep.2016.06.060
Liu, A. K. L., Hurry, M. E. D., Ng, O. T. W., DeFelice, J., Lai, H. M., Pearce, R. K. B., et al. (2016). Bringing CLARITY to the human brain: visualization of Lewy pathology in three dimensions. Neuropathol. Appl. Neurobiol. 42, 573–587. doi: 10.1111/nan.12293
Mai, H., Rong, Z., Zhao, S., Cai, R., Steinke, H., Bechmann, I., et al. (2022). Scalable tissue labeling and clearing of intact human organs. Nat. Protoc. 17, 2188–2215. doi: 10.1038/s41596-022-00712-8
Matsumoto, K., Mitani, T. T., Horiguchi, S. A., Kaneshiro, J., Murakami, T. C., Mano, T., et al. (2019). Advanced CUBIC tissue clearing for whole-organ cell profiling. Nat. Protoc. 14, 3506–3537. doi: 10.1038/s41596-019-0240-9
Messal, H. A., Almagro, J., Zaw Thin, M., Tedeschi, A., Ciccarelli, A., Blackie, L., et al. (2021). Antigen retrieval and clearing for whole-organ immunofluorescence by FLASH. Nat. Protoc. 16, 239–262. doi: 10.1038/s41596-020-00414-z
Mez, J., Solomon, T. M., Daneshvar, D. H., Murphy, L., Kiernan, P. T., Montenigro, P. H., et al. (2015). Assessing clinicopathological correlation in chronic traumatic encephalopathy: rationale and methods for the UNITE study. Alzheimers Res. Ther. 7:62. doi: 10.1186/s13195-015-0148-8
Montine, T. J., Phelps, C. H., Beach, T. G., Bigio, E. H., Cairns, N. J., Dickson, D. W., et al. (2012). National Institute on Aging–Alzheimer’s association guidelines for the neuropathologic assessment of Alzheimer’s disease: a practical approach. Acta Neuropathol. 123, 1–11. doi: 10.1007/s00401-011-0910-3
Morawski, M., Kirilina, E., Scherf, N., Jäger, C., Reimann, K., Trampel, R., et al. (2018). Developing 3D microscopy with CLARITY on human brain tissue: towards a tool for informing and validating MRI-based histology. NeuroImage 182, 417–428. doi: 10.1016/j.neuroimage.2017.11.060
Murray, E., Cho, J. H., Goodwin, D., Ku, T., Swaney, J., Kim, S. Y., et al. (2015). Simple, scalable proteomic imaging for high-dimensional profiling of intact systems. Cell 163, 1500–1514. doi: 10.1016/j.cell.2015.11.025
Namimatsu, S., Ghazizadeh, M., and Sugisaki, Y. (2005). Reversing the effects of formalin fixation with Citraconic anhydride and heat: a universal antigen retrieval method. J. Histochem. Cytochem. 53, 3–11. doi: 10.1177/002215540505300102
Naudí, A., Cabré, R., Jové, M., Ayala, V., Gonzalo, H., Portero-Otín, M., et al. (2015). Lipidomics of human brain aging and Alzheimer’s disease pathology. Int. Rev. Neurobiol. 122, 133–189. doi: 10.1016/bs.irn.2015.05.008
Park, Y.-G., Sohn, C. H., Chen, R., McCue, M., Yun, D. H., Drummond, G. T., et al. (2019). Protection of tissue physicochemical properties using polyfunctional crosslinkers. Nat. Biotechnol. 37, 73–83. doi: 10.1038/nbt.4281
Pesce, L., Scardigli, M., Gavryusev, V., Laurino, A., Mazzamuto, G., Brady, N., et al. (2022). 3D molecular phenotyping of cleared human brain tissues with light-sheet fluorescence microscopy. Commun. Biol. 5:447. doi: 10.1038/s42003-022-03390-0
Phillips, J., Laude, A., Lightowlers, R., Morris, C. M., Turnbull, D. M., and Lax, N. Z. (2016). Development of passive CLARITY and immunofluorescent labelling of multiple proteins in human cerebellum: understanding mechanisms of neurodegeneration in mitochondrial disease. Sci. Rep. 6:26013. doi: 10.1038/srep26013
Rait, V. K., Xu, L., O'Leary, T. J., and Mason, J. T. (2004). Modeling formalin fixation and antigen retrieval with bovine pancreatic RNase a II. Interrelationship of crosslinking, immunoreactivity, and heat treatment. Lab. Investig. 84, 300–306. doi: 10.1038/labinvest.3700041
Rosen, G., Kirsch, D., Horowitz, S., Cherry, J. D., Nicks, R., Kelley, H., et al. (2023). Three dimensional evaluation of cerebrovascular density and branching in chronic traumatic encephalopathy. Acta Neuropathol. Commun. 11:123. doi: 10.1186/s40478-023-01612-y
Scardigli, M., Pesce, L., Brady, N., Mazzamuto, G., Gavryusev, V., Silvestri, L., et al. (2021). Comparison of different tissue clearing methods for three-dimensional reconstruction of human brain cellular anatomy using advanced imaging techniques. Front. Neuroanat. 15:752234. doi: 10.3389/fnana.2021.752234
Shi, S.-R., Liu, C., and Taylor, C. R. (2007). Standardization of immunohistochemistry for formalin-fixed, paraffin-embedded tissue sections based on the antigen-retrieval technique: from experiments to hypothesis. J. Histochem. Cytochem. 55, 105–109. doi: 10.1369/jhc.6P7080.2006
Shi, L., Wei, M., Miao, Y., Qian, N., Shi, L., Singer, R. A., et al. (2022). Highly-multiplexed volumetric mapping with Raman dye imaging and tissue clearing. Nat. Biotechnol. 40, 364–373. doi: 10.1038/s41587-021-01041-z
Sun, S., Ding, Z., Yang, X., Zhao, X., Zhao, M., Gao, L., et al. (2021). Nanobody: a small antibody with big implications for tumor therapeutic strategy. Int. J. Nanomedicine 16, 2337–2356. doi: 10.2147/IJN.S297631
Ueda, H. R. (2020). Tissue clearing and its applications in neuroscience. Nat. Rev. Neurosci. :19. doi: 10.1038/s41583-019-0250-1
Vonsattel, J. P. G., del Amaya, M. P., and Keller, C. E. (2008). Twenty-first century brain banking. Processing brains for research: the Columbia University methods. Acta Neuropathol. 115, 509–532. doi: 10.1007/s00401-007-0311-9
Wang, Q., Ding, S. L., Li, Y., Royall, J., Feng, D., Lesnar, P., et al. (2020). The Allen mouse brain common coordinate framework: a 3D reference atlas. Cell 181, 936–953.e20. doi: 10.1016/j.cell.2020.04.007
Woelfle, S., Deshpande, D., Feldengut, S., Braak, H., del Tredici, K., Roselli, F., et al. (2023). CLARITY increases sensitivity and specificity of fluorescence immunostaining in long-term archived human brain tissue. BMC Biol. 21:113. doi: 10.1186/s12915-023-01582-6
Keywords: optical clearing, antigen retrieval, volumetric imaging, 3D confocal imaging, 3D confocal laser scanning microscopy, human brain banking, immunofluorescence
Citation: Rosen GA, Kirsch D, Nicks R, Kelley H, Mathias R, Cormier KA, Kubilus CA, Dec B, Stein TD, Alvarez VE, Alosco ML, McKee AC and Huber BR (2024) SHARD: an improved method for staining and visualizing multiplex immunofluorescence in optically cleared postmortem human brain tissue. Front. Neurosci. 18:1474617. doi: 10.3389/fnins.2024.1474617
Edited by:
Ana-Maria Oros-Peusquens, Helmholtz Association of German Research Centres (HZ), GermanyReviewed by:
Ludovico Silvestri, University of Florence, ItalyPraju Vikas Anekal, Harvard University, United States
Copyright © 2024 Rosen, Kirsch, Nicks, Kelley, Mathias, Cormier, Kubilus, Dec, Stein, Alvarez, Alosco, McKee and Huber. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bertrand R. Huber, aHViZXIuYmVydHJhbmRAZ21haWwuY29t