
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Neurosci. , 06 September 2024
Sec. Neurodevelopment
Volume 18 - 2024 | https://doi.org/10.3389/fnins.2024.1462893
This article is part of the Research Topic Advances in DYRK1A Syndrome: Underlying Mechanisms, Disease Models, and Novel Therapeutic Approaches View all 4 articles
 Elysabeth D. Otte
Elysabeth D. Otte Randall J. Roper*
Randall J. Roper*DYRK1A syndrome results from a reduction in copy number of the DYRK1A gene, which resides on human chromosome 21 (Hsa21). DYRK1A has been implicated in the development of cognitive phenotypes associated with many genetic disorders, including Down syndrome (DS) and Alzheimer’s disease (AD). Additionally, overexpression of DYRK1A in DS has been implicated in the development of abnormal skeletal phenotypes in these individuals. Analyses of mouse models with Dyrk1a dosage imbalance (overexpression and underexpression) show skeletal deficits and abnormalities. Normalization of Dyrk1a copy number in an otherwise trisomic animal rescues some skeletal health parameters, and reduction of Dyrk1a copy number in an otherwise euploid (control) animal results in altered skeletal health measurements, including reduced bone mineral density (BMD) in the femur, mandible, and skull. However, little research has been conducted thus far on the implications of DYRK1A reduction on human skeletal health, specifically in individuals with DYRK1A syndrome. This review highlights the skeletal phenotypes of individuals with DYRK1A syndrome, as well as in murine models with reduced Dyrk1a copy number, and provides potential pathways altered by a reduction of DYRK1A copy number, which may impact skeletal health and phenotypes in these individuals. Understanding how decreased expression of DYRK1A in individuals with DYRK1A syndrome impacts bone health may increase awareness of skeletal traits and assist in the development of therapies to improve quality of life for these individuals.
Dual-specificity tyrosine phosphorylation-regulated kinase 1A (DYRK1A) is a highly conserved gene located on human chromosome 21 (Hsa21) with known roles in neuronal and skeletal development. Variations in DYRK1A copy number lead to phenotypes associated with several developmental disorders, suggesting that a critical gene-dosage balance of DYRK1A is required for proper early development. DYRK1A syndrome, also known as DYRK1A haploinsufficiency syndrome and Intellectual developmental disorder, autosomal dominant 7 (MRD7) (OMIM:614104) is generally caused by the presence of a pathogenic variant of DYRK1A, that reduces the functional copy number of DYRK1A from two to one, and likely decreases DYRK1A protein levels (Arranz et al., 2019). This disorder is presently characterized by intellectual disability, the development of autism spectrum disorder (ASD), and behavioral problems. Individuals with DYRK1A syndrome also display non-cognitive phenotypes including altered facies, intrauterine growth restriction, short stature, and reduced skeletal growth. The potential skeletal abnormalities associated with DYRK1A syndrome have not been well characterized, but insights from the consequences of dosage imbalance of DYRK1A and information from mouse models with altered Dyrk1a copy number may provide a basis for further examining the skeletal health of individuals with DYRK1A syndrome.
The purpose of this review is to examine existing case reports and studies detailing skeletal abnormalities in individuals with DYRK1A syndrome, as well as to highlight skeletal parameters in Dyrk1a haploinsufficient mouse models. Our objective is to call attention to potential skeletal phenotypes affecting individuals with DYRK1A syndrome, and provide potential molecular pathways leading to these abnormalities, so that therapies may be found to improve quality of life for these individuals.
DYRK1A gene dosage imbalance has been associated with multiple disorders affecting both cognitive ability and skeletal health, including Down syndrome (DS), Alzheimer’s disease (AD), Parkinson’s disease (PD), and Huntington disease (HD) (Deboever et al., 2022). DYRK1A is a member of the DYRK family of kinases. DYRK protein kinases are activated by autophosphorylation on a tyrosine residue in the activation loop, which catalyzes the phosphorylation of exogenous serine and threonine residues. DYRK proteins are evolutionally conserved, and orthologs of DYRK1A are found in animal models including mouse (Mus musculus--Dyrk1a), fruit fly (Drosophila melanogaster--Mnb), and nematode (Caenorhabditis elegans--mbk-1) (Figure 1) (Alvarez et al., 2007; Rammohan et al., 2022). Null mutations in or removal of both copies of the DYRK1A gene are embryonic lethal in humans and mammalian models, but non-mammalian knockout models, including C. elegans strains without mbk-1, are viable. In other experimental systems, models containing either one or three copies of the gene are utilized to understand more about the effects of DYRK1A dosage imbalance. Together, data examining the phenotypes associated with both increased and decreased Dyrk1a expression indicate a dosage-dependent role of Dyrk1a on neural and skeletal system development and function.
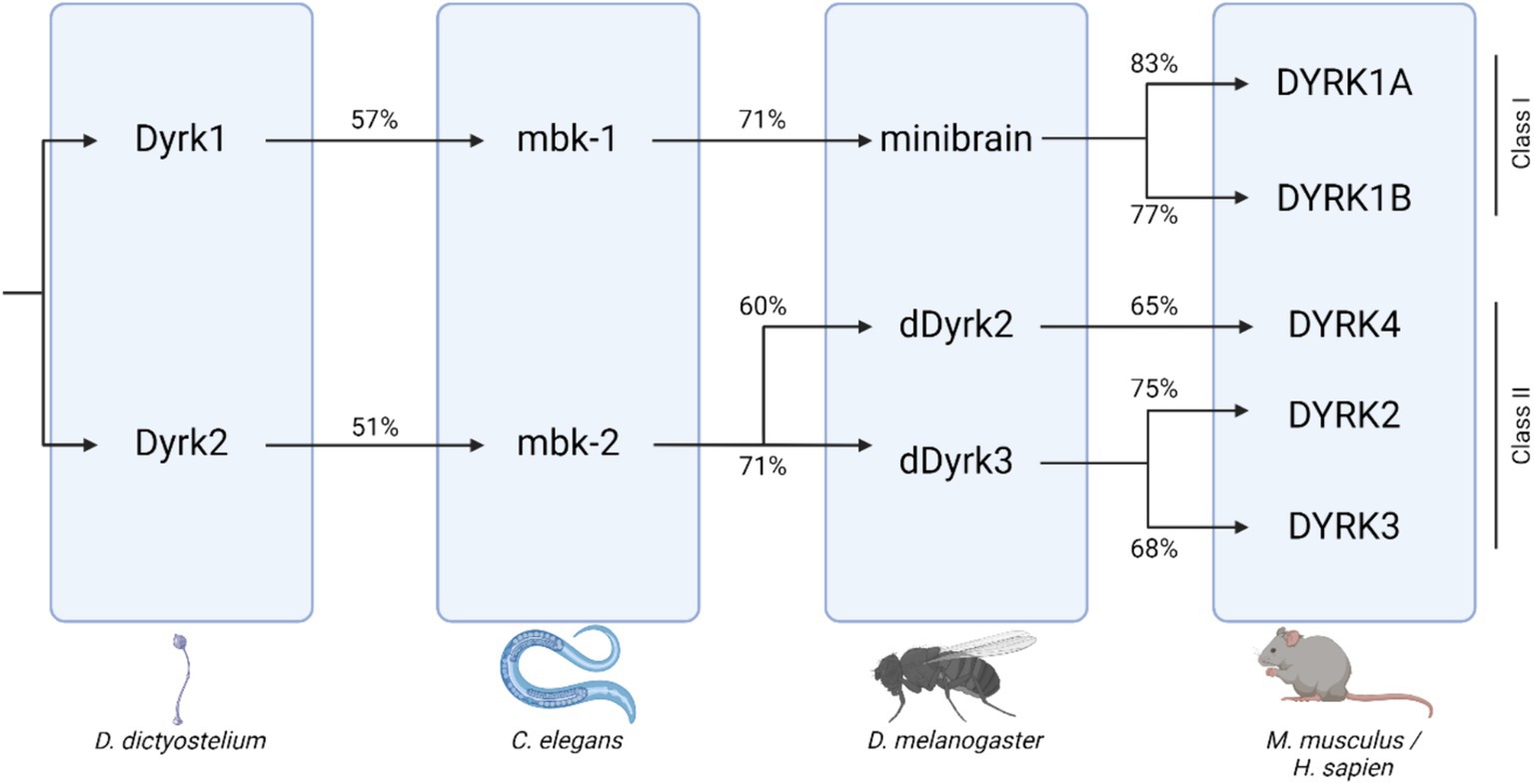
Figure 1. DYRK protein kinase family tree across species, including D. Dictyostelium, C. elegans, D. melanogaster, and mammals. Percent orthology between species shown beneath connecting arrows [adapted from Deboever et al. (2022); created with BioRender].
DYRK1A is ubiquitously expressed in all human tissues and is essential for neurogenesis, cell cycle regulation, and synaptic plasticity. DYRK1A/MNB is involved in the phosphorylation of microtubule dynamics, which help to regulate dendritic patterning and neuronal function (Ori-McKenney et al., 2016; Deboever et al., 2022). The effects of mammalian Dyrk1a are found throughout embryonic development and continue into neonatal neurogenesis, and high levels of DYRK1A have been detected in the adult cerebellum (Deboever et al., 2022). Dyrk1a has also been implicated in the development of craniofacial features. Alterations in face size and neural crest cells in animal models with Dyrk1a deficiency and overexpression may be explained by increased cell death and decreased cellular proliferation in cellular precursors to the face (McElyea et al., 2016; Johnson et al., 2024).
Illustrating the dosage-dependent properties of DYRK1A, DYRK1A syndrome is an autosomal dominant disorder often caused by a loss-of-function mutation within the DYRK1A gene. Findings in individuals with DYRK1A syndrome suggest a potential heterozygous de novo pathogenic variant in DYRK1A, leading to the development of this diverse disorder (van Bon et al., 2015). Over 120 disease-related variants have been reported in DYRK1A, the majority of which are small deletion mutations, missense mutations, nonsense mutations, and small insertion mutations. The majority of missense pathogenic variants of the DYRK1A gene are located in the kinase domain, leading to a disruption of the catalytic domain and kinase activity of the protein (van Bon et al., 2015). In addition, some studies suggest that phenotypes associated with DYRK1A syndrome may be compounded by mutations in nearby genes, including potassium inwardly rectifying channel subfamily J member 6 (KCNJ6), also located nearby on Hsa21. Mutations in KCNJ6 have been associated with the development of Keppen-Lubinsky syndrome, leading to severe developmental delay, facial dysmorphism, intellectual disability, and seizures (Valetto et al., 2012).
Individuals with DYRK1A syndrome display cognitive and behavioral phenotypes including the development of ASD, anxiety, epilepsy, and sleep disturbances (Ji et al., 2015; van Bon et al., 2015; van Bon et al., 2016). DYRK1A syndrome is believed to account for up to 0.5% of individuals with intellectual disability, and studies have found that DYRK1A is one of the most frequent de novo mutated genes in ASD (Arbones et al., 2019). The cognitive phenotypes associated with this disorder have been the most widely characterized, as intellectual disability and developmental delay are some of the most common symptoms present in these individuals (van Bon et al., 2015).
Other phenotypes commonly associated with individuals with DYRK1A syndrome include intrauterine growth restriction (IUGR), microcephaly, hypertonia, urogenital abnormalities, characteristic facial features, micrognathia, feeding difficulties, short stature, and musculoskeletal anomalies (Earl et al., 2017; Meissner et al., 2020; Huang et al., 2023). These phenotypes are often noted perinatally, and many persist after birth. Slow head growth rates into adulthood result in an increasingly severe microcephaly phenotype. Altered skull development and formation may be a result of not only neuronal impacts of reduced DYRK1A expression but may also be a product of altered growth of the skull in these individuals. Facial dysmorphisms have been reported in approximately 98% of published cases of DYRK1A syndrome, supporting the hypothesis of the dosage-dependent role of DYRK1A in craniofacial development (Earl et al., 2017). Dental anomalies, including widely spaced teeth, delayed primary dentition, and supernumerary teeth have also been reported in individuals with DYRK1A syndrome, along with typical facial gestalt (Desai, 1997; van Bon et al., 2016; Huang et al., 2023).
In comparison to the cognitive and behavioral phenotypes associated with the disorder, skeletal and growth abnormalities are largely unexplored and reflect limited research examining the potential impacts of DYRK1A syndrome on skeletal health. One study evaluating the stature and growth of individuals with DYRK1A syndrome found that over half of individuals surveyed had a short stature and low birth weight (Lanvin et al., 2024). Short stature may be due to prenatal and/or early childhood development, while low birth weight often persists later into life. Skeletal abnormalities amongst these individuals commonly impact the hands and feet and associated bones. Several case studies of individuals with DYRK1A syndrome have described contractures of the ankles, feet, and hands, as well as cavovarus foot deformities, tibial osteochondrosis, high arched feet, long hallux, and arachnodactyly (Earl et al., 2017; Huang et al., 2023). Additionally, approximately 10% of individuals with DYRK1A haploinsufficiency develop musculoskeletal abnormalities, including scoliosis, kyphosis, and pectus excavatum. These musculoskeletal phenotypes target the spine, sternum, and ribs, which may be indicative of the effects of DYRK1A haploinsufficiency on skeletal development and growth. Further understanding the dosage-dependent role of DYRK1A in skeletal development, and skull formation and growth may help to elucidate how these phenotypes may be closely associated.
Individuals with DS may have up to 300 protein-coding genes triplicated, including DYRK1A. This dosage imbalance of DYRK1A is present in almost all individuals with Trisomy 21, leading to DS (OMIM:190685). This imbalance, in combination with other triplicated genes, has been implicated in the development of many phenotypes associated with DS (Blazek et al., 2015; Deboever et al., 2022; Redhead et al., 2023; Lana-Elola et al., 2024). Individuals with DS often present with alterations in multiple organ systems, including congenital heart defects, musculoskeletal abnormalities, cognitive deficits, respiratory issues, and intestinal problems (Antonarakis et al., 2020). Mouse models of DS have been used to verify the role of DYRK1A in the development of these phenotypes. For example, in the Ts65Dn DS mouse model, with about half of the genes homologous to Hsa21 in three copies, increased DYRK1A protein levels in neural stem cells of the cerebellar cortex lengthens the G1 phase and reduce neuronal production, supporting the role of DYRK1A in the development of cognitive phenotypes associated with DS (Najas et al., 2015). Like those with DYRK1A haploinsufficiency, individuals with DS often present with intellectual disability, along with craniofacial abnormalities, and musculoskeletal deficits, potentially due to the roles of DYRK1A in bone formation, osteoclastogenesis, neurogenesis, and synaptogenesis (Lee et al., 2009; Park and Chung, 2013).
Skeletal abnormalities, including small body size, reduced bone mineral density (BMD) and weaker bone strength are often characteristic phenotypes of individuals with Trisomy 21. Deficits in skeletal microarchitecture often lead to the development of osteopenia or osteoporosis in individuals with DS (Angelopoulou et al., 1999). Additionally, these individuals often show delayed skeletal age in relation to chronological age, and reach skeletal maturation earlier than individuals without DS, due to a pronounced period of growth during a shorter period of skeletal maturation (de Moraes et al., 2008). Many of these characteristics have been, at least in part, attributed the gene-dose imbalance of DYRK1A in these individuals (Arron et al., 2006; Thomas and Roper, 2021; Sloan et al., 2023).
A reduction of DYRK1A to one copy (DYRK1A haploinsufficiency) or an increase to three copies (Trisomy 21), may lead to similar tissues that are affected and comparable phenotypes (Figure 2 and Table 1), thus illustrating the effect of DYRK1A dosage imbalance (underexpression or overexpression) in humans. The impact of DYRK1A dosage imbalance may be better understood in experimental models where Dyrk1a dosage can be manipulated and studied. From our studies of DS mouse models (Dyrk1a overexpression) and Dyrk1a haploinsufficiency mouse models (Dyrk1a underexpression), DYRK1A dosage imbalance may lead to skeletal alterations that need to be better characterized in individuals with DYRK1A haploinsufficiency syndrome.
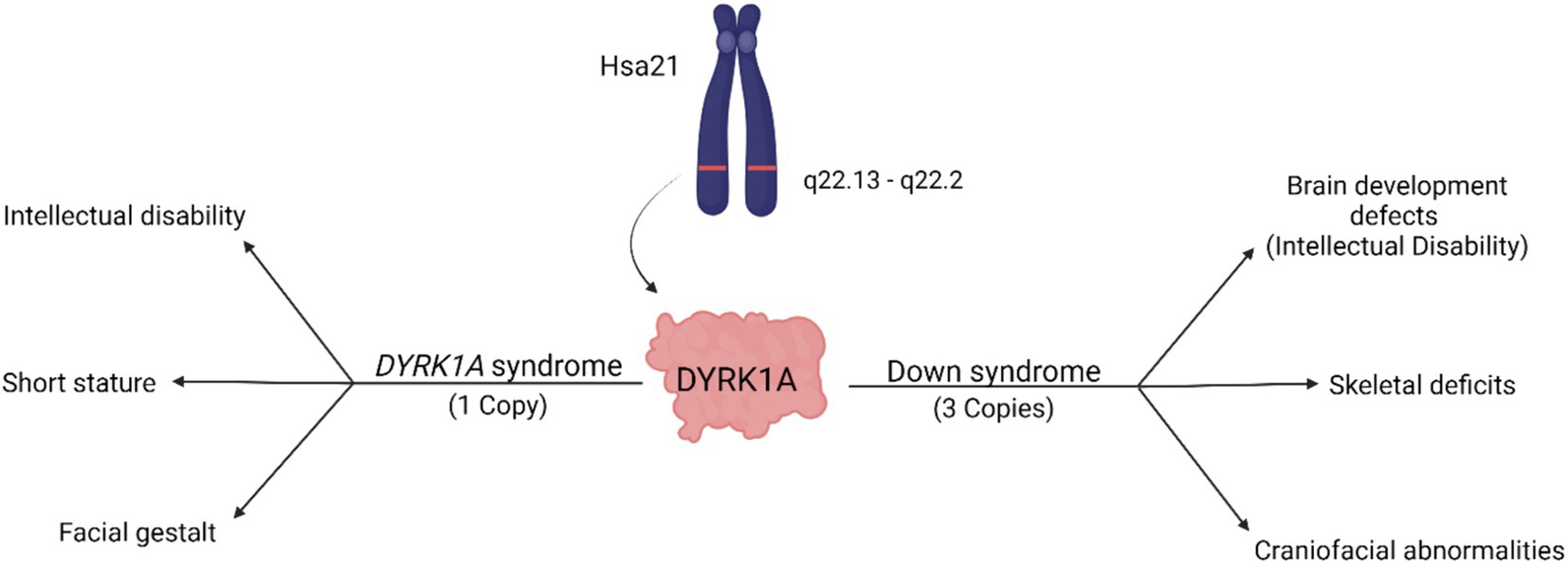
Figure 2. Dosage-dependent effects of the DYRK1A gene. Overexpression of DYRK1A is associated with Down syndrome phenotypes, while underexpression of DYRK1A results in the development of DYRK1A syndrome. Both disorders present with phenotypes affecting cognition, craniofacial development, and skeletal health [adapted from Laham et al. (2021); Created with BioRender].
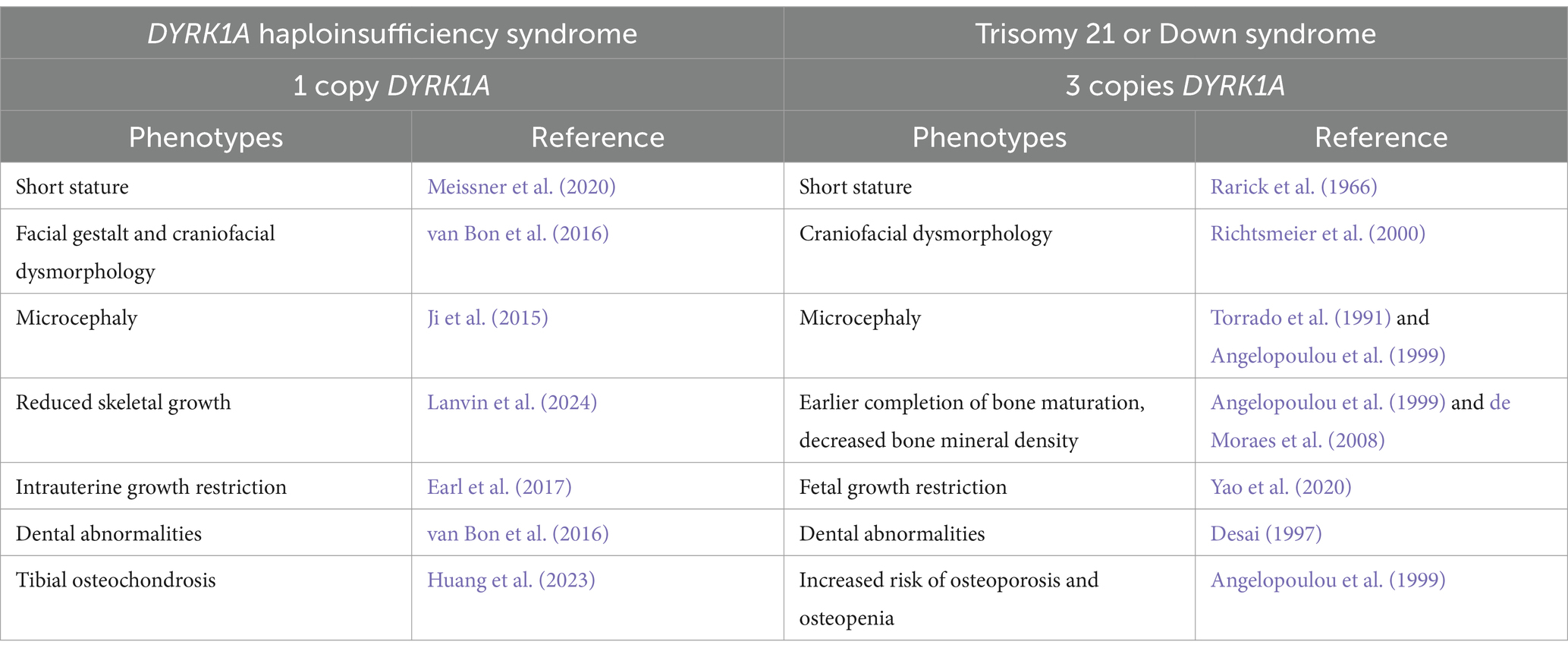
Table 1. Skeletal phenotypes associated with haploinsufficiency (1 copy – DYRK1A syndrome) or trisomy (3 copies – Down syndrome) of DYRK1A.
Observations in mouse models with dosage imbalance of Dyrk1a lead to a hypothesis that a reduction of Dyrk1a, as in DYRK1A syndrome, and an increase of Dyrk1a, as in Trisomy 21, both cause skeletal deficits, providing a foundation for the study of these phenotypes in humans. Male and female mice transgenic for extra human DYRK1A exhibited reduced trabecular bone measures, but did not show changes in cortical thickness (Lee et al., 2009). Additionally, these mice had reduced osteoclastogenesis and defective osteoblastogenesis. Mouse models of DYRK1A haploinsufficiency have been created by mutating exons 3, 5, or 6 of the Dyrk1a gene. These mice have either been bred to wild-type mice, and approximately half of the progeny will carry a single copy of Dyrk1a, or these mice have been bred to trisomic mice where approximately a quarter of the mice may carry a single copy of Dyrk1a (Figure 3).
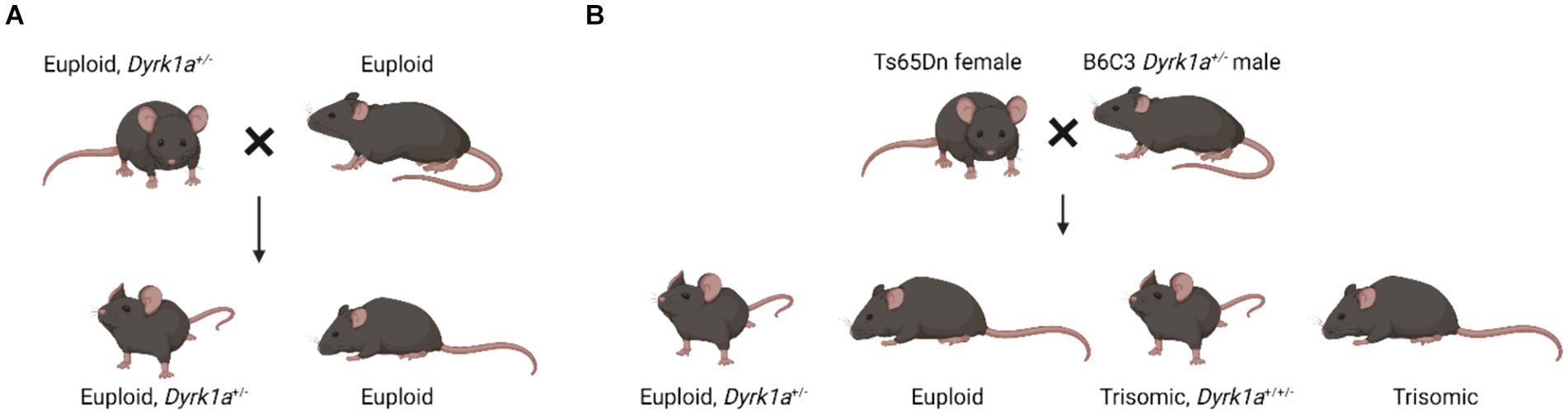
Figure 3. Breeding schematics used to generate euploid, Dyrk1a+/− mice (Fotaki et al., 2002; LaCombe et al., 2024; Created with BioRender).
Mouse models with only one functional copy of Dyrk1a show phenotypes similar to those identified in humans with DYRK1A syndrome. Dyrk1a heterozygous knockout mice containing a frameshift mutation in exon 3 of Dyrk1a have been used to examine the cognitive and behavioral phenotypes of the disorder, and have shown that haploinsufficient mice display cognitive flexibility deficits, as well as sociability and communication impairments (Raveau et al., 2018). Mice containing targeted disruptions of Dyrk1a gene in exons 5 and 6 also present with neurological traits including hyperthermia-induced seizures and defective social interactions, closely mimicking the phenotypes observed in humans with DYRK1A haploinsufficiency (Arranz et al., 2019). These Dyrk1a haploinsufficient mice also display reductions in hippocampally-dependent memory tasks and impairments in novel object recognition tasks when compared to euploid controls (Arque et al., 2008).
Many of these cognitive phenotypes are believed to be tied to glial cell abnormalities and impaired neocortical circuit development caused by Dyrk1a dosage imbalance. Dyrk1a heterozygous mice compared to control mice of the same background have a decreased brain size and have been found to present with increased astrogliogenesis in the neocortex and delays in cortical oligodendrocyte progenitor cell production (Fotaki et al., 2002). Additionally, axonal conductivity impairment is believed to be altered in these animals, caused by decreased myelination and thinner axons in the corpus callosum (Pijuan et al., 2022). These haploinsufficient mice displayed abnormal proportions of excitatory and inhibitory neocortical neurons, caused by altered excitatory neuron production during early and mid-corticogenesis, and present with smaller, less branched pyramidal cells in the neocortex (Benavides-Piccione et al., 2005; Arranz et al., 2019).
Neurological traits associated with DYRK1A imbalance may additionally be contributed to dopaminergic neuron imbalance caused by abnormal activity of the caspase9-dependent apoptotic pathway (Barallobre et al., 2014). Mice haploinsufficient for Dyrk1a display hypoactivity and altered gait, suggesting a role of Dyrk1a in the development of the neuromotor system, and which may be directly correlated with altered dopaminergic activity (Fotaki et al., 2004; Martinez de Lagran et al., 2007). Administration of dopaminergic antagonists resulted in increased seizure activity in wild-type animals that was not seen in Dyrk1a haploinsufficient animals, suggesting that Dyrk1a haploinsufficient mice may have altered self-regulatory mechanisms of dopamine neurons (Martinez de Lagran et al., 2007). Additional studies from our lab examining DS have yielded mice with Dyrk1a haploinsufficiency and have begun to examine the skeletal phenotypes of these animals.
Compared to the effects of triplication of Dyrk1a on skeletal health, there are far fewer studies examining the effects of Dyrk1a haploinsufficiency on skeletal health and development using mammalian models. Similar to individuals with DYRK1A haploinsufficiency, mouse models with one copy of Dyrk1a have been used to characterize more than cognitive and behavioral deficits; yet many growth and skeletal deficits have been observed in these mice. Our laboratory has utilized a Dyrk1a+/− mouse with a mutation spanning exons 5 and 6 that shows a number of skeletal abnormalities (Fotaki et al., 2002; Arranz et al., 2019).
Male and female mouse models with only one copy of Dyrk1a exhibit smaller body mass as compared to control mice at postnatal day (P)30 and P36, which may be indicative of lower bone mass (Figure 4) (Fotaki et al., 2002; LaCombe et al., 2024). Some changes in cranial face and mandible but not cranial base or vault were observed in 6-week-old Dyrk1a haploinsufficient as compared to control mice (McElyea et al., 2016). Reduction of Dyrk1a copy number from conception in otherwise euploid 6-week-old male mice led to a significant decrease in mandible and skull BMD, along with reductions in femur BMD (Blazek et al., 2015). Otherwise normal male mice with Dyrk1a haploinsufficiency displayed a decrease in trabecular number, thickness, area, and perimeter in the 6-week-old (a time of bone formation roughly equivalent to the skeletal age of humans under the age of 20) femur when compared with euploid littermates (Blazek et al., 2015). Measurement of mechanical properties of the same animals showed decreases in ultimate force and stiffness in femurs from Dyrk1a haploinsufficient animals when compared to euploid mice. The skeletal measurements in Dyrk1a haploinsufficient mice were similar to those found in trisomic or DS model mice, potentially due to alterations in osteoclastogenesis caused by Dyrk1a gene dosage imbalance, and these skeletal phenotypes were normalized in trisomic animals with a normalized copy number of Dyrk1a (Blazek et al., 2015). The skeletal abnormalities found in Dyrk1a haploinsufficient mice may portend similar skeletal deficiencies in humans with DYRK1A haploinsufficiency.
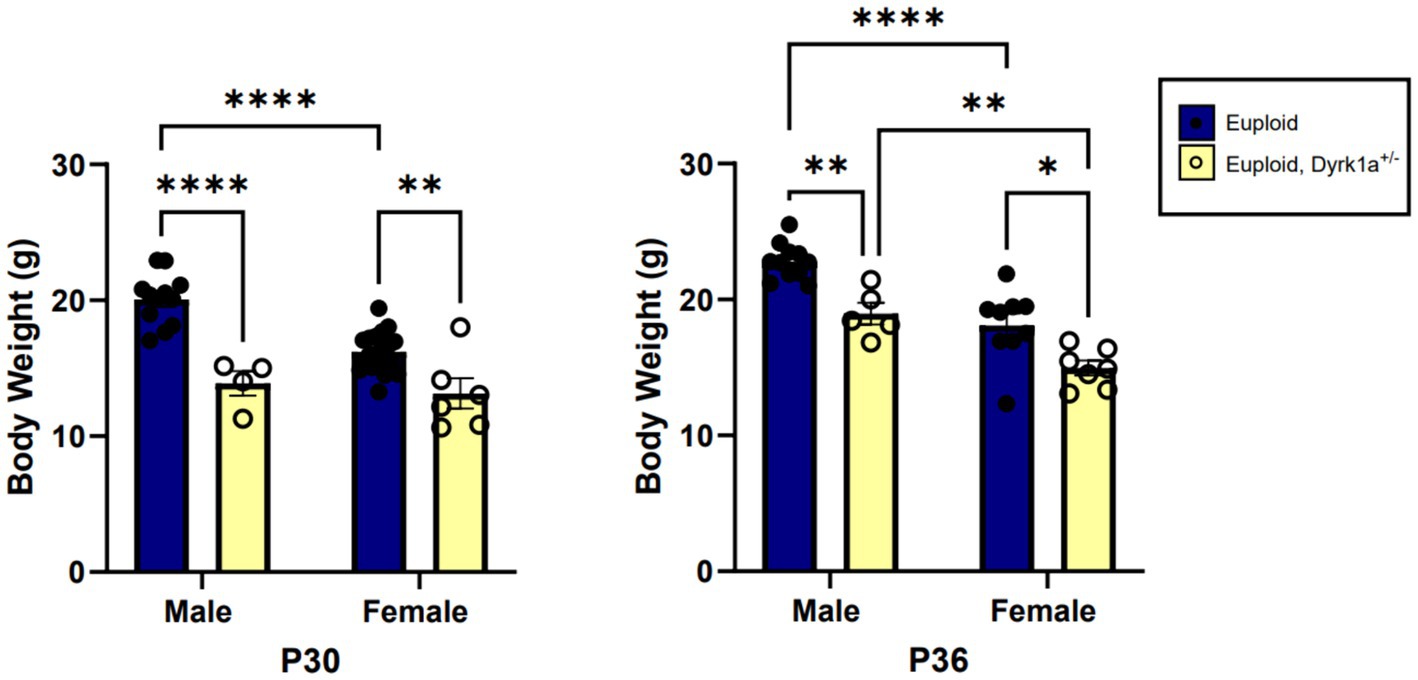
Figure 4. Body weight comparisons between euploid and euploid, Dyrk1a+/− male and female animals at P30 and P36, highlighting reduced weight in both male and female euploid, Dyrk1a+/− animals. Data analyzed using two-way ANOVA. Male: euploid (n = 12), euploid, Dyrk1a+/− (n = 4); female: euploid (n = 19), euploid, Dyrk1a+/− (n = 6). *p < 0.05, **p < 0.01, ****p < 0.0001. [Data from LaCombe et al. (2024)].
Additionally, we have shown that mice with a single copy of Dyrk1a may result in sexually dimorphic skeletal phenotypes, with skeletal deficits occurring earlier in male mice. In the femur of Dyrk1a haploinsufficient mice at P30 and P36, both trabecular and cortical bone are more severely affected in male than in female mice (Figure 5) (Thomas et al., 2021; LaCombe et al., 2024). In Dyrk1a haploinsufficient mice at P30 (an early time of bone formation), male animals haploinsufficient for Dyrk1a displayed reductions in trabecular BMD, bone volume fraction, and thickness in the femur when compared to euploid mice (Figure 6). Dyrk1a haploinsufficent male mice also displayed reductions in cortical cross-sectional area, thickness, periosteal perimeter, and section modulus (Figure 7). Female Dyrk1a haploinsufficient animals did not exhibit significant trabecular or cortical deficits at P30. At P36, femurs from male mice with only one copy of Dyrk1a displayed reductions in trabecular thickness and section modulus. Once again, female mice with one copy of Dyrk1a did not display significant trabecular or cortical deficits at P36 (LaCombe et al., 2024). These data emphasize both potential confounding effects of sex and age on skeletal deficits in Dyrk1a haploinsufficent animals, and similar sex-and temporally-specific skeletal abnormalities may be seen in humans with DYRK1A syndrome. For example, female individuals with DYRK1A syndrome may exhibit bone deficits later than males. Additionally, there may be periodic normalizations of some skeletal phenotypes throughout development (LaCombe et al., 2024; Lamantia et al., 2024). Knowing these sex-and temporally-specific details may provide a timeline for identifying skeletal phenotypes associated with DYRK1A syndrome and a “target” timeframe for possible therapeutic interventions.
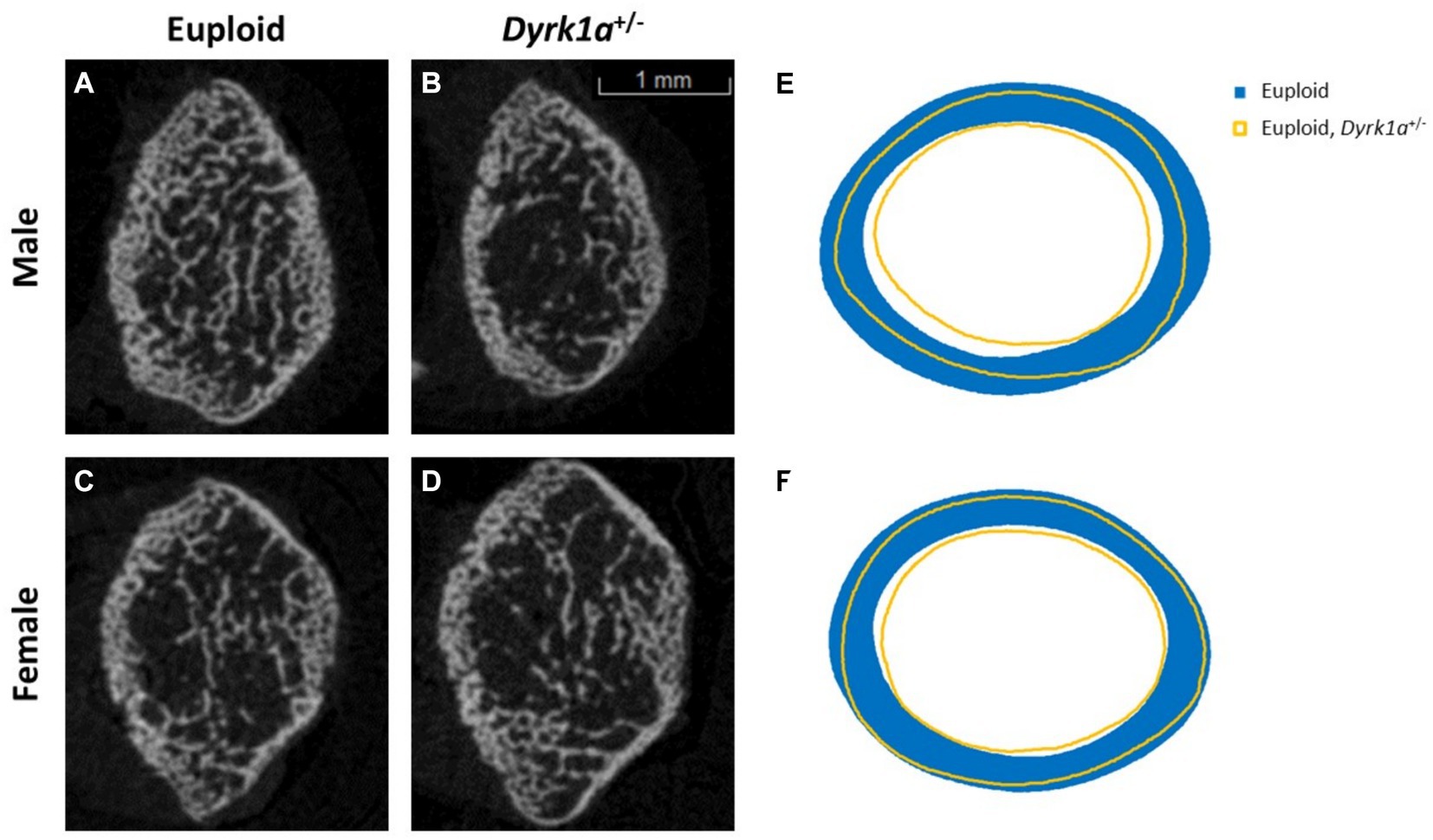
Figure 5. Trabecular and cortical bone are affected by Dyrk1a copy number at P30. Representative images of trabecular structure taken at P30 of euploid males (A), euploid, Dyrk1a+/− males (B), euploid females (C), and euploid, Dyrk1a+/− females (D) as revealed by μCT analysis. Radar graphs of average periosteal and endocortical perimeter measurements of male (E) and female (F) euploid and euploid, Dyrk1a+/− animals [Data from LaCombe et al. (2024)].
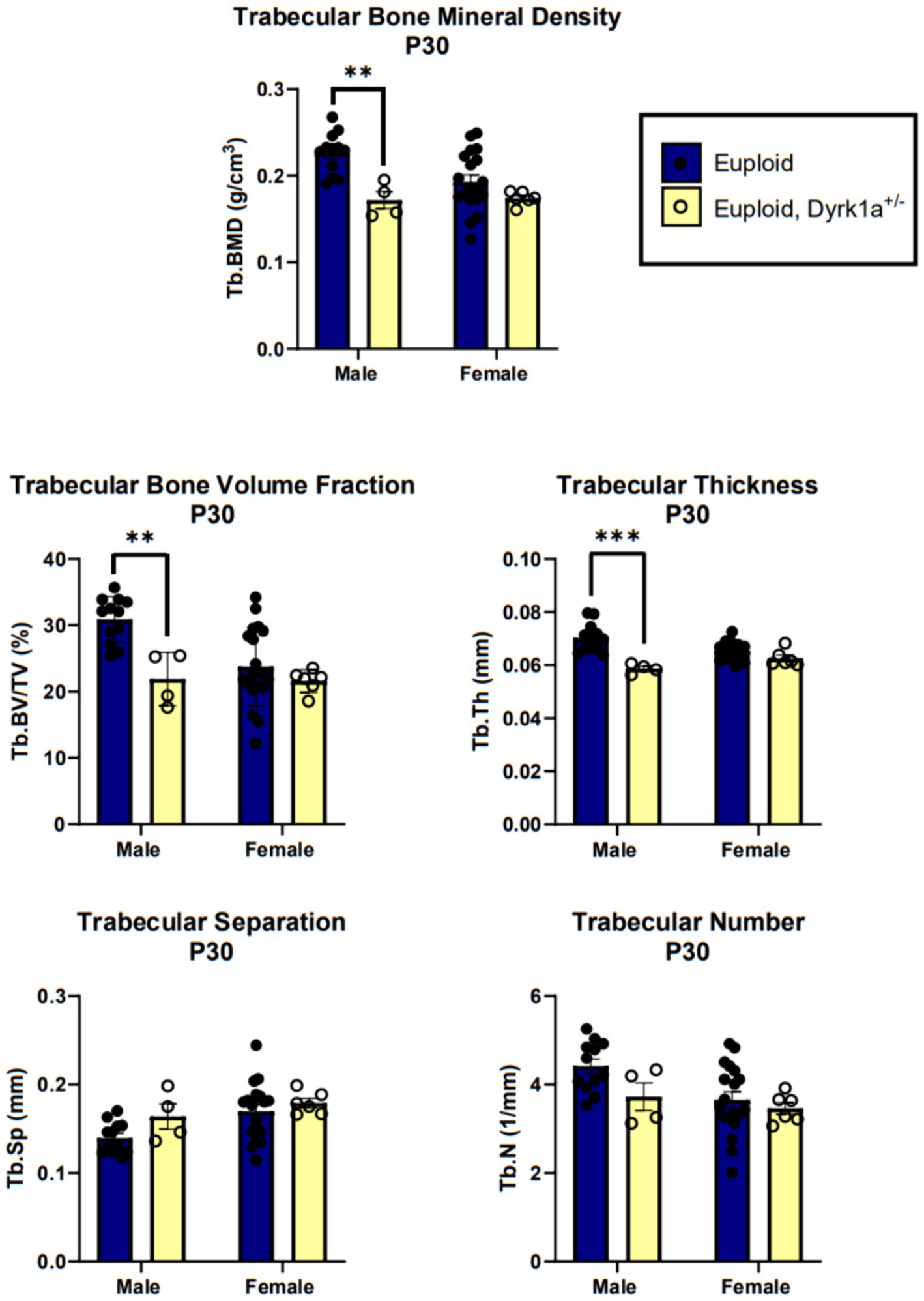
Figure 6. Trabecular bone measurements of male and female euploid and euploid, Dyrk1a+/− animals at P30. Data analyzed using one-way ANOVA and Tukey/Games-Howell post hoc analysis. Male: euploid (n = 12), euploid, Dyrk1a+/− (n = 4); female: euploid (n = 19), euploid, Dyrk1a+/− (n = 6). **p < 0.01, ***p < 0.001. [Data from LaCombe et al. (2024)].
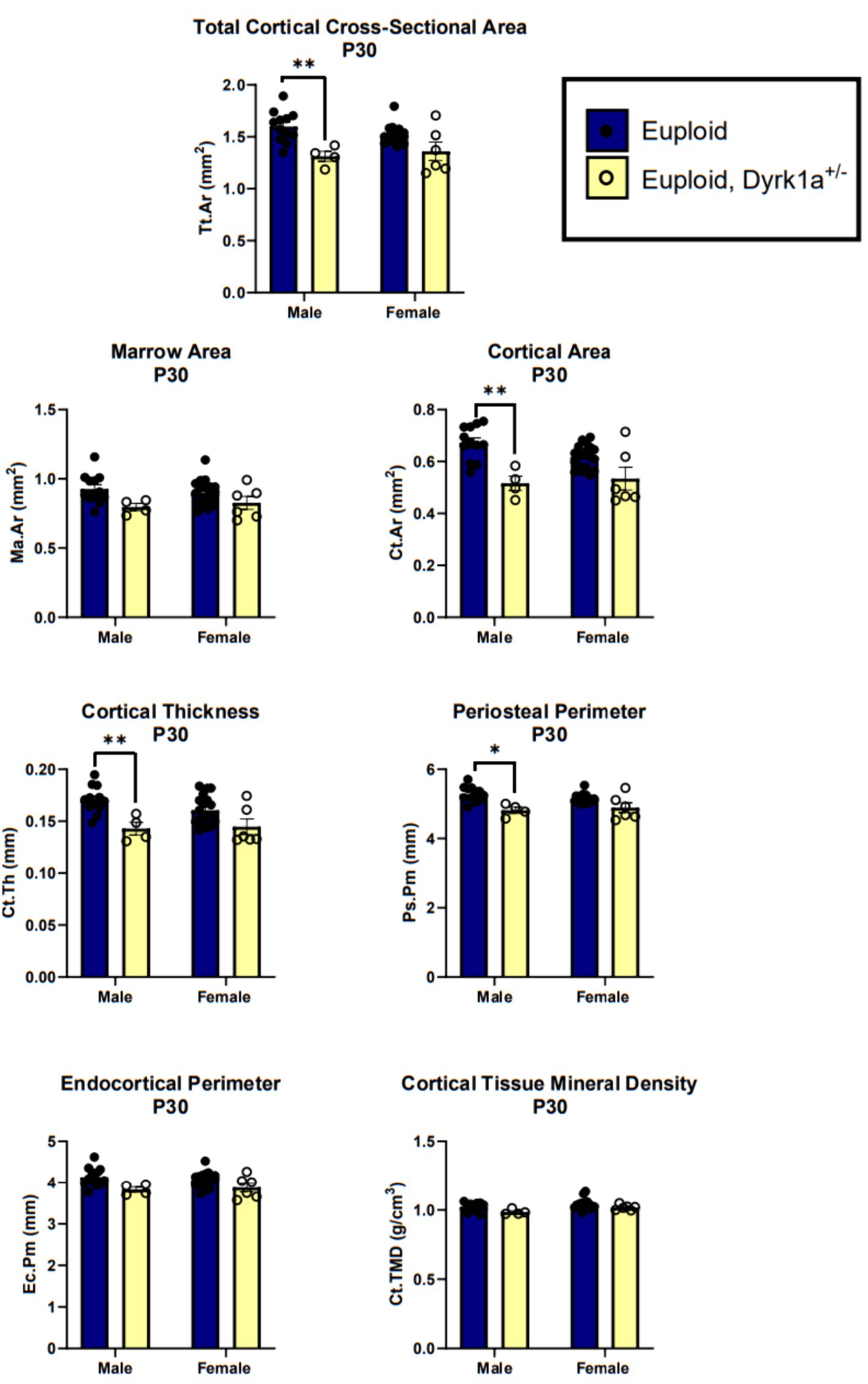
Figure 7. Cortical bone measurements of male and female euploid and euploid, Dyrk1a+/− animals at P30. Data analyzed using one-way ANOVA and Tukey/Games-Howell post hoc analysis. Male: euploid (n = 12), euploid, Dyrk1a+/− (n = 4); female: euploid (n = 19), euploid, Dyrk1a+/− (n = 6). *p < 0.05, **p < 0.01. [Data from LaCombe et al. (2024)].
The use of DYRK1A inhibitors has been a topic of research for several years as a potential pharmacotherapeutic treatment for DS phenotypes. The use of these inhibitors in DS mouse models has also shown the possible effects of these DYRK1A inhibitors in euploid (normal) littermate controls. Treatment of 3-week-old euploid animals with a purported DYRK1A inhibitor, epigallocatechin-3-gallate (EGCG), resulted in a significant increase in femoral bone volume as well as trabecular number and thickness when compared to trisomic Ts65Dn animals. Treatment of euploid animals with 9 mg/kg/day EGCG resulted in significantly increased percent bone volume, but did not alter the mechanical properties or BMD of the femur (Blazek et al., 2015). As treatment with EGCG increased, male euploid animals exhibited decreased cortical thickness (Jamal et al., 2022). Treatment with 50 mg/kg/day EGCG of male euploid animals showed slight reductions in cortical bone phenotypes. These data further support the dosage-dependent hypothesis regarding Dyrk1a and suggest that pharmaceutical reduction of DYRK1A activity may alter skeletal formation and health. The effects of EGCG in DS animal models and control mice suggested that this inhibitor may affect products of Dyrk1a or other trisomic genes in the Ts65Dn mouse model and negatively impact bone structure and function. Additional studies examining how EGCG may impact skeletal health have found that different supplements exhibit different effects on skeletal measures, with some resulting in improvement, and others being detrimental (Abeysekera et al., 2016). Treatments using DYRK1A inhibitors must carefully be administered at proper levels so that characteristics of DYRK1A syndrome, especially in bone, are not induced (Stringer et al., 2017).
The effects of reduced DYRK1A likely involve many molecular pathways to produce skeletal and other deficits. DYRK1A acts on the PI3K/AKT/mTOR pathway via modulation of brain-derived neurotrophic factor (BDNF) (Rammohan et al., 2022). BDNF has been suggested to play a role in fracture healing and bone remodeling (Xue et al., 2021). Additionally, the PI3K/AKT/mTOR and NFATc pathways play a role in skeletal development and homeostasis (Arron et al., 2006; Chen, 2018).
The skeletal system contains a widely distributed neurovascular system that undergoes active remodeling during bone regeneration and repair. Nearly all the sensory nerves in bone express neurotrophic receptor tyrosine kinase type 1 (TrkA), which is a receptor for nerve growth factor (NGF). NGF activation of TrkA plays a role in the activation of osteogenic cues leading to bone formation through the Wnt/β-catenin pathway. DYRK1A overexpression has negative effects on NGF signal transduction in vitro, suggesting another potential pathway through which DYRK1A dosage may influence skeletal growth and health (Stefos et al., 2013; Tomlinson et al., 2017).
DYRK1A also interacts with the RE1-silencing transcription factor (REST), also known as the neuron restrictive silencing factor (NRSF), which acts as an inhibitor of cell proliferation and differentiation and has been suggested to be a regulator of early osteoblast differentiation. REST is traditionally known as a master neuronal negative regulator transcription factor that functions to maintain transcriptional silence of neuronal genes, though recent research has begun to examine its effects outside of neuronal maintenance. Studies have found that REST regulates early osteoblast differentiation via an unknown pathway. Alterations in Dyrk1a expression have been found to modify Rest/Nrsf gene expression, which may in turn change osteoblast differentiation and lead to skeletal abnormalities in individuals with altered levels of DYRK1A expression (Lepagnol-Bestel et al., 2009; Liu et al., 2015).
Additionally, DYRK1A has been found to be a promoter of fibroblast-like synoviocytes (FLS), which play a role in cartilage/bone destruction in rheumatoid arthritis (RA) via activation of the ERK/MAPK pathway (Guo et al., 2018). More recent data has shown that conditional reduction of Dyrk1a in murine chondrocytes results in increased progression of osteoarthritis (OA), suggesting a protective effect of Dyrk1a in disease progression, potentially via modulation of the EGFR-ERK signaling in articular chondrocytes (Liu et al., 2023). These influences on the ERK signaling pathway may alter bone growth and development through modulation of osteoblast differentiation (Kim et al., 2022). We hypothesize that by critically examining Dyrk1a haploinsufficent mice, we will gain a better understanding of how reduced Dyrk1a copy number leads to skeletal deficits at the molecular level, including specific Dyrk1a mutations that may lead to particular skeletal deficits.
Though no studies have concentrated on skeletal health parameters in individuals with DYRK1A syndrome, reports of individuals with DYRK1A haploinsufficiency have shown the development of several phenotypes that may indicate skeletal deficits, including intrauterine growth abnormalities, short stature, craniofacial and dental abnormalities, and developmental abnormalities in hand, foot, and ankle. Skeletal abnormalities are a common phenotype seen in individuals with intellectual disabilities, potentially partially due to decreased activity levels in these individuals. However, examination of the molecular connections that may cause both cognitive and skeletal phenotypes is worth considering. Mouse model studies with overexpression and underexpression of Dyrk1a provide foundational evidence that DYRK1A copy number dosage imbalance alters skeletal development and health, showing that both increased and decreased gene dosage results in skeletal health deficits. Additional studies designed to examine skeletal development in mouse models of DYRK1A syndrome should be conducted to provide evidence of the impacts of underexpressed DYRK1A on bone health, especially concentrating on molecular mechanisms. These data would provide additional avenues for the development of therapies targeting DYRK1A syndrome, to increase the quality of life of these individuals. Current early intervention treatments recommended for individuals diagnosed with DYRK1A haploinsufficiency include physical therapy to increase mobility and modulate risk for scoliosis and other musculoskeletal symptoms (van Bon et al., 2015). These early treatments may additionally modulate skeletal phenotypes that have yet to be uncovered, as chronic exercise is a known inducer of bone growth and remodeling. Current evidence supports the role of DYRK1A in skeletal formation and health and suggests that skeletal phenotypes should be examined in individuals with DYRK1A syndrome. From the data presented, these skeletal deficits may appear in males earlier than females with DYRK1A haploinsufficiency. Molecular pathways influenced by DYRK1A that may lead to both cognitive and skeletal phenotypes must also be studied, and the effects of a gene-dosage imbalance of DYRK1A need to be fully characterized. Further understanding the roles of this dosage sensitive gene in the development of multiple organ systems may aid in the potential development of pharmacotherapies to treat the symptoms associated with both DS and DYRK1A syndrome.
EO: Conceptualization, Visualization, Writing – original draft, Writing – review & editing. RR: Funding acquisition, Supervision, Writing – review & editing.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This research in this manuscript was supported by funds from National Institutes of Health Eunice Kennedy Shriver National Institute for Child Health and Human Development (HD090603) and the National Institute of Arthritis and Musculoskeletal and Skin Diseases (AR078663).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abeysekera, I., Thomas, J., Georgiadis, T. M., Berman, A. G., Hammond, M. A., Dria, K. J., et al. (2016). Differential effects of Epigallocatechin-3-gallate containing supplements on correcting skeletal defects in a down syndrome mouse model. Mol. Nutr. Food Res. 60, 717–726. doi: 10.1002/mnfr.201500781
Alvarez, M., Altafaj, X., Aranda, S., and de la Luna, S. (2007). DYRK1A autophosphorylation on serine residue 520 modulates its kinase activity via 14-3-3 binding. Mol. Biol. Cell 18, 1167–1178. doi: 10.1091/mbc.e06-08-0668
Angelopoulou, N., Souftas, V., Sakadamis, A., and Mandroukas, K. (1999). Bone mineral density in adults with Down's syndrome. Eur. Radiol. 9, 648–651. doi: 10.1007/s003300050726
Antonarakis, S. E., Skotko, B. G., Rafii, M. S., Strydom, A., Pape, S. E., Bianchi, D. W., et al. (2020). Down syndrome. Nat Rev Dis Primers 6:9. doi: 10.1038/s41572-019-0143-7
Arbones, M. L., Thomazeau, A., Nakano-Kobayashi, A., Hagiwara, M., and Delabar, J. M. (2019). DYRK1A and cognition: a lifelong relationship. Pharmacol. Ther. 194, 199–221. doi: 10.1016/j.pharmthera.2018.09.010
Arque, G., Fotaki, V., Fernandez, D., Martinez de Lagran, M., Arbones, M. L., and Dierssen, M. (2008). Impaired spatial learning strategies and novel object recognition in mice haploinsufficient for the dual specificity tyrosine-regulated kinase-1A (Dyrk1A). PLoS One 3:e2575. doi: 10.1371/journal.pone.0002575
Arranz, J., Balducci, E., Arato, K., Sanchez-Elexpuru, G., Najas, S., Parras, A., et al. (2019). Impaired development of neocortical circuits contributes to the neurological alterations in DYRK1A haploinsufficiency syndrome. Neurobiol. Dis. 127, 210–222. doi: 10.1016/j.nbd.2019.02.022
Arron, J. R., Winslow, M. M., Polleri, A., Chang, C. P., Wu, H., Gao, X., et al. (2006). NFAT dysregulation by increased dosage of DSCR1 and DYRK1A on chromosome 21. Nature 441, 595–600. doi: 10.1038/nature04678
Barallobre, M. J., Perier, C., Bove, J., Laguna, A., Delabar, J. M., Vila, M., et al. (2014). DYRK1A promotes dopaminergic neuron survival in the developing brain and in a mouse model of Parkinson's disease. Cell Death Dis. 5:e1289. doi: 10.1038/cddis.2014.253
Benavides-Piccione, R., Dierssen, M., Ballesteros-Yanez, I., Martinez de Lagran, M., Arbones, M. L., Fotaki, V., et al. (2005). Alterations in the phenotype of neocortical pyramidal cells in the Dyrk1A+/− mouse. Neurobiol. Dis. 20, 115–122. doi: 10.1016/j.nbd.2005.02.004
Blazek, J. D., Abeysekera, I., Li, J., and Roper, R. J. (2015). Rescue of the abnormal skeletal phenotype in Ts65Dn down syndrome mice using genetic and therapeutic modulation of trisomic Dyrk1a. Hum. Mol. Genet. 24, 5687–5696. doi: 10.1093/hmg/ddv284
Chen, J. L. F. (2018). mTOR signaling in skeletal development and disease. Bone Res. 6:1. doi: 10.1038/s41413-017-0004-5
de Moraes, M. E., Tanaka, J. L., de Moraes, L. C., Filho, E. M., and de Melo Castilho, J. C. (2008). Skeletal age of individuals with down syndrome. Spec. Care Dentist. 28, 101–106. doi: 10.1111/j.1754-4505.2008.00020.x
Deboever, E., Fistrovich, A., Hulme, C., and Dunckley, T. (2022). The omnipresence of DYRK1A in human diseases. Int. J. Mol. Sci. 23:9355. doi: 10.3390/ijms23169355
Desai, S. S. (1997). Down syndrome: a review of the literature. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. Endod. 84, 279–285. doi: 10.1016/S1079-2104(97)90343-7
Earl, R. K., Turner, T. N., Mefford, H. C., Hudac, C. M., Gerdts, J., Eichler, E. E., et al. (2017). Clinical phenotype of ASD-associated DYRK1A haploinsufficiency. Mol. Autism. 8:54. doi: 10.1186/s13229-017-0173-5
Fotaki, V., Dierssen, M., Alcantara, S., Martinez, S., Marti, E., Casas, C., et al. (2002). Dyrk1A haploinsufficiency affects viability and causes developmental delay and abnormal brain morphology in mice. Mol. Cell. Biol. 22, 6636–6647. doi: 10.1128/MCB.22.18.6636-6647.2002
Fotaki, V., Martinez De Lagran, M., Estivill, X., Arbones, M., and Dierssen, M. (2004). Haploinsufficiency of Dyrk1A in mice leads to specific alterations in the development and regulation of motor activity. Behav. Neurosci. 118, 815–821. doi: 10.1037/0735-7044.118.4.815
Guo, X., Zhang, D., Zhang, X., Jiang, J., Xue, P., Wu, C., et al. (2018). Dyrk1A promotes the proliferation, migration and invasion of fibroblast-like synoviocytes in rheumatoid arthritis via down-regulating Spry2 and activating the ERK MAPK pathway. Tissue Cell 55, 63–70. doi: 10.1016/j.tice.2018.10.002
Huang, C., Luo, H., Zeng, B., Feng, C., Chen, J., Yuan, H., et al. (2023). Identification of two novel and one rare mutation in DYRK1A and prenatal diagnoses in three Chinese families with intellectual Disability-7. Front. Genet. 14:1290949. doi: 10.3389/fgene.2023.1290949
Jamal, R., LaCombe, J., Patel, R., Blackwell, M., Thomas, J. R., Sloan, K., et al. (2022). Increased dosage and treatment time of Epigallocatechin-3-gallate (EGCG) negatively affects skeletal parameters in normal mice and down syndrome mouse models. PLoS One 17:e0264254. doi: 10.1371/journal.pone.0264254
Ji, J., Lee, H., Argiropoulos, B., Dorrani, N., Mann, J., Martinez-Agosto, J. A., et al. (2015). DYRK1A haploinsufficiency causes a new recognizable syndrome with microcephaly, intellectual disability, speech impairment, and distinct facies. Eur. J. Hum. Genet. 23, 1473–1481. doi: 10.1038/ejhg.2015.71
Johnson, H. K., Wahl, S. E., Sesay, F., Litovchick, L., and Dickinson, A. J. (2024) Dyrk1a is required for craniofacial development in Xenopus laevis. Dev. Biol. 511:63–75. doi: 10.1016/j.ydbio.2024.04.004
Kim, J. M., Yang, Y. S., Hong, J., Chaugule, S., Chun, H., van der Meulen, M. C. H., et al. (2022). Biphasic regulation of osteoblast development via the ERK MAPK-mTOR pathway. eLife 17:11:e78069. doi: 10.7554/eLife.78069
LaCombe, J. M., Sloan, K., Thomas, J. R., Blackwell, M. P., Crawford, I., Bishop, F., et al. (2024) Sex specific emergence of trisomic Dyrk1a-related skeletal phenotypes in the development of a down syndrome mouse model. doi: 10.1242/dmm.050914
Laham, A. J., Saber-Ayad, M., and El-Awady, R. (2021). DYRK1A: a down syndrome-related dual protein kinase with a versatile role in tumorigenesis. Cell. Mol. Life Sci. 78, 603–619. doi: 10.1007/s00018-020-03626-4
Lamantia, J., Sloan, K., Wallace, J. M., and Roper, R. J. (2024). Compromised femoral and lumbovertebral bone in the Dp(16)1Yey down syndrome mouse model. Bone 181:117046. doi: 10.1016/j.bone.2024.117046
Lana-Elola, E., Aoidi, R., Llorian, M., Gibbins, D., Buechsenschuetz, C., Bussi, C., et al. (2024). Increased dosage of DYRK1A leads to congenital heart defects in a mouse model of down syndrome. Sci. Transl. Med. 16:eadd6883. doi: 10.1126/scitranslmed.add6883
Lanvin, P. L., Goronflot, T., Isidor, B., Nizon, M., Durand, B., el Chehadeh, S., et al. (2024). Growth charts in DYRK1A syndrome. Am. J. Med. Genet. A 194, 9–16. doi: 10.1002/ajmg.a.63412
Lee, Y., Ha, J., Kim, H. J., Kim, Y. S., Chang, E. J., Song, W. J., et al. (2009). Negative feedback inhibition of NFATc1 by DYRK1A regulates bone homeostasis. J. Biol. Chem. 284, 33343–33351. doi: 10.1074/jbc.M109.042234
Lepagnol-Bestel, A. M., Zvara, A., Maussion, G., Quignon, F., Ngimbous, B., Ramoz, N., et al. (2009). DYRK1A interacts with the REST/NRSF-SWI/SNF chromatin remodelling complex to deregulate gene clusters involved in the neuronal phenotypic traits of down syndrome. Hum. Mol. Genet. 18, 1405–1414. doi: 10.1093/hmg/ddp047
Liu, B., Cheng, S., Xing, W., Pourteymoor, S., and Mohan, S. (2015). RE1-silencing transcription factor (Rest) is a novel regulator of osteoblast differentiation. J. Cell. Biochem. 116, 1932–1938. doi: 10.1002/jcb.25148
Liu, Z., Hu, S., Wu, J., Quan, X., Shen, C., Li, Z., et al. (2023). Deletion of DYRK1A accelerates osteoarthritis progression through suppression of EGFR-ERK signaling. Inflammation 46, 1353–1364. doi: 10.1007/s10753-023-01813-6
Martinez de Lagran, M., Bortolozzi, A., Millan, O., Gispert, J. D., Gonzalez, J. R., Arbones, M. L., et al. (2007). Dopaminergic deficiency in mice with reduced levels of the dual-specificity tyrosine-phosphorylated and regulated kinase 1A, Dyrk1A(+/−). Genes Brain Behav. 6, 569–578. doi: 10.1111/j.1601-183X.2006.00285.x
McElyea, S. D., Starbuck, J. M., Tumbleson-Brink, D. M., Harrington, E., Blazek, J. D., Ghoneima, A., et al. (2016). Influence of prenatal EGCG treatment and Dyrk1a dosage reduction on craniofacial features associated with down syndrome. Hum. Mol. Genet. 25, 4856–4869. doi: 10.1093/hmg/ddw309
Meissner, L. E., Macnamara, E. F., D'Souza, P., Yang, J., and Vezina, G.Undiagnosed Diseases Network, et al. (2020). DYRK1A pathogenic variants in two patients with syndromic intellectual disability and a review of the literature. Mol. Genet. Genomic Med. 8:e1544. doi: 10.1002/mgg3.1544
Najas, S., Arranz, J., Lochhead, P. A., Ashford, A. L., Oxley, D., Delabar, J. M., et al. (2015). DYRK1A-mediated cyclin D1 degradation in neural stem cells contributes to the neurogenic cortical defects in down syndrome. EBioMedicine 2, 120–134. doi: 10.1016/j.ebiom.2015.01.010
Ori-McKenney, K. M., McKenney, R. J., Huang, H. H., Li, T., Meltzer, S., Jan, L. Y., et al. (2016). Phosphorylation of beta-tubulin by the down syndrome kinase, Minibrain/DYRK1a, regulates microtubule dynamics and dendrite morphogenesis. Neuron 90, 551–563. doi: 10.1016/j.neuron.2016.03.027
Park, J., and Chung, K. C. (2013). New perspectives of Dyrk1A role in neurogenesis and Neuropathologic features of down syndrome. Exp. Neurobiol. 22, 244–248. doi: 10.5607/en.2013.22.4.244
Pijuan, I., Balducci, E., Soto-Sanchez, C., Fernandez, E., Barallobre, M. J., and Arbones, M. L. (2022). Impaired macroglial development and axonal conductivity contributes to the neuropathology of DYRK1A-related intellectual disability syndrome. Sci. Rep. 12:19912. doi: 10.1038/s41598-022-24284-5
Rammohan, M., Harris, E., Bhansali, R. S., Zhao, E., Li, L. S., and Crispino, J. D. (2022). The chromosome 21 kinase DYRK1A: emerging roles in cancer biology and potential as a therapeutic target. Oncogene 41, 2003–2011. doi: 10.1038/s41388-022-02245-6
Rarick, G. L., Rapaport, I. F., and Seefeldt, V. (1966). Long bone growth in Down's syndrome. Am. J. Dis. Child. 112, 566–571. doi: 10.1001/archpedi.1966.02090150110012
Raveau, M., Shimohata, A., Amano, K., Miyamoto, H., and Yamakawa, K. (2018). DYRK1A-haploinsufficiency in mice causes autistic-like features and febrile seizures. Neurobiol. Dis. 110, 180–191. doi: 10.1016/j.nbd.2017.12.003
Redhead, Y., Gibbins, D., Lana-Elola, E., Watson-Scales, S., Dobson, L., Krause, M., et al. (2023). Craniofacial dysmorphology in down syndrome is caused by increased dosage of Dyrk1a and at least three other genes. Development 150:dev201077. doi: 10.1242/dev.201077
Richtsmeier, J. T., Baxter, L. L., and Reeves, R. H. (2000). Parallels of craniofacial maldevelopment in down syndrome and Ts65Dn mice. Dev. Dyn. 217, 137–145. doi: 10.1002/(SICI)1097-0177(200002)217:2<137::AID-DVDY1>3.0.CO;2-N
Sloan, K., Thomas, J., Blackwell, M., Voisard, D., Lana-Elola, E., Watson-Scales, S., et al. (2023). Genetic dissection of triplicated chromosome 21 orthologs yields varying skeletal traits in down syndrome model mice. Dis. Model. Mech. 16:dmm049927. doi: 10.1242/dmm.049927
Stefos, G. C., Soppa, U., Dierssen, M., and Becker, W. (2013). NGF upregulates the plasminogen activation inhibitor-1 in neurons via the calcineurin/NFAT pathway and the down syndrome-related proteins DYRK1A and RCAN1 attenuate this effect. PLoS One 8:e67470. doi: 10.1371/journal.pone.0067470
Stringer, M., Goodlett, C. R., and Roper, R. J. (2017). Targeting trisomic treatments: optimizing Dyrk1a inhibition to improve down syndrome deficits. Mol. Genet. Genomic Med. 5, 451–465. doi: 10.1002/mgg3.334
Thomas, J. R., and Roper, R. J. (2021). Current analysis of skeletal phenotypes in down syndrome. Curr. Osteoporos. Rep. 19, 338–346. doi: 10.1007/s11914-021-00674-y
Thomas, J. R., Sloan, K., Cave, K., Wallace, J. M., and Roper, R. J. (2021). Skeletal deficits in male and female down syndrome model mice Arise independent of normalized Dyrk1a expression in osteoblasts. Genes (Basel) 12:1729. doi: 10.3390/genes12111729
Tomlinson, R. E., Li, Z., Li, Z., Minichiello, L., Riddle, R. C., Venkatesan, A., et al. (2017). NGF-TrkA signaling in sensory nerves is required for skeletal adaptation to mechanical loads in mice. Proc. Natl. Acad. Sci. USA 114, E3632–E3641. doi: 10.1073/pnas.1701054114
Torrado, C., Bastian, W., Wisniewski, K. E., and Castells, S. (1991). Treatment of children with down syndrome and growth retardation with recombinant human growth hormone. J. Pediatr. 119, 478–483. doi: 10.1016/S0022-3476(05)82068-2
Valetto, A., Orsini, A., Bertini, V., Toschi, B., Bonuccelli, A., Simi, F., et al. (2012). Molecular cytogenetic characterization of an interstitial deletion of chromosome 21 (21q22.13q22.3) in a patient with dysmorphic features, intellectual disability and severe generalized epilepsy. Eur. J. Med. Genet. 55, 362–366. doi: 10.1016/j.ejmg.2012.03.011
van Bon, B. W., Coe, B. P., Bernier, R., Green, C., Gerdts, J., Witherspoon, K., et al. (2016). Disruptive de novo mutations of DYRK1A lead to a syndromic form of autism and ID. Mol. Psychiatry 21, 126–132. doi: 10.1038/mp.2015.5
van Bon, B. W. M., Coe, B. P., de Vries, B. B. A., and Eichler, E. E. (2015). DYRK1A syndrome in GeneReviews® [Internet]. Seattle, WA: University of Washington.
Xue, F., Zhao, Z., Gu, Y., Han, J., Ye, K., and Zhang, Y. (2021). 7,8-Dihydroxyflavone modulates bone formation and resorption and ameliorates ovariectomy-induced osteoporosis. eLife 10:e64872. doi: 10.7554/eLife.64872
Keywords: haploinsufficiency, gene dosage, mouse models, Down syndrome, Hsa21
Citation: Otte ED and Roper RJ (2024) Skeletal health in DYRK1A syndrome. Front. Neurosci. 18:1462893. doi: 10.3389/fnins.2024.1462893
Edited by:
Oliver Glass, Duke University, United StatesReviewed by:
Nathalie Janel, Université Paris Cité, FranceCopyright © 2024 Otte and Roper. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Randall J. Roper, cmpyb3BlckBpdS5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.