 Chu-Min Ou1†
Chu-Min Ou1† Ke Ning
Ke Ning- 1Guangdong Celconta Biotechnology Co., Ltd., Dongguan, Guangdong, China
- 2School of Biological Engineering, Dalian Polytechnic University, Dalian, China
- 3Sheffield Institute of Translational Neuroscience (SITraN), University of Sheffield, Sheffield, United Kingdom
An incurable neurogenerative illness, Alzheimer’s disease, is the cause of most global health, medical, and social disasters. The two main symptoms are cognitive impairment and neuronal loss. Current medications that target tau protein tangles and Aβ plaques are not very effective because they only slow the symptoms of AD and do not repair damaged cells. Stem cell-based treatments, however, present an alternative strategy in the treatment of AD. They have the capacity to divide into specialized adult cells, have self-renewal abilities, and multiplication. Stem cells can now be employed as a donor source for cell therapy due to developments in stem cell technology. This review covers preclinical and clinical updates on studies based on targeting the tau protein tangles and Aβ plaque, as well as four types of stem cells employed in AD treatment. The review also outlines the two basic pathologic aspects, tau protein tangles and Aβ plaques, of AD.
1 Introduction
In 2021, WHO posted the seventh most common cause of death globally is Alzheimer’s disease (after Ischaemic heart disease, COVID-19, Stroke, Chronic obstructive pulmonary disease, Lower respiratory infections, Trachea, bronchus, lung cancer), which is a neurological illness linked to advanced progressive dementia. Symptoms of the condition include mood swings, sleep disturbances, behavioral changes, and cognitive impairment. Severe symptoms include malnutrition, neuronal necrosis that results in multiple organ failure, and even brain death as AD progresses to advanced stages (Ma et al., 2022).
Age 65 can be used to distinguish between two types of AD, early-onset EOAD (before age 65) and late-onset LOAD (after age 65), both of which share similar clinical characteristics (Penke et al., 2023). According to estimates, EOAD has a heritability of 90–100% (Wingo et al., 2012), whereas LOAD heritability, which ranges from 60% to 80%, is marginally lower than that of other diseases (Gatz et al., 1997). An important contributing factor to autosomal dominant early-onset Alzheimer’s disease (EOAD) is a mutation in the presenilin-1 (PS-1) gene (Hanisch and Kölmel, 2004). Several genetic loci or polygenic effects work together to produce the genetic composition of LOAD, which makes this disease far more complex than EOAD. According to genetic research, there are multiple components of Alzheimer’s disease, and the most significant risk factor for LOAD (cholesterol synthesis and transport) is ApoE4 (Sims et al., 2020). And we can see the different between FAD and SAD in Table 1.
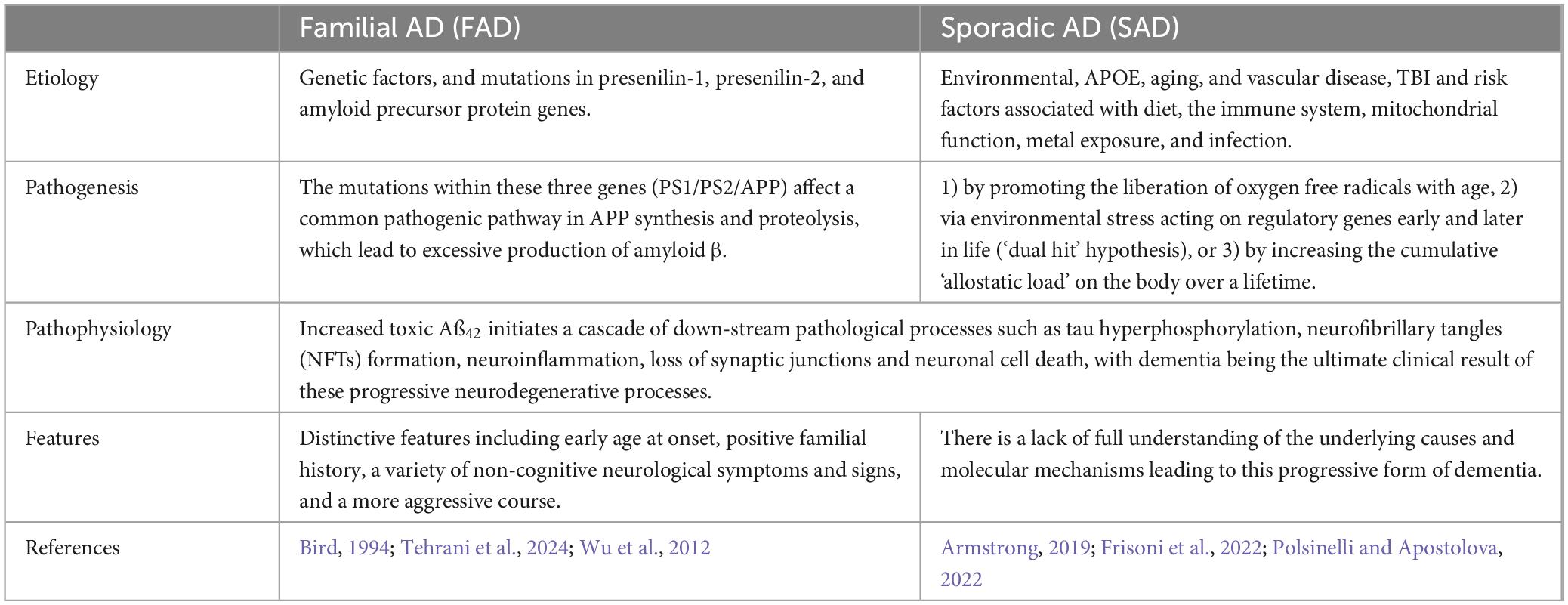
Table 1. Sporadic and familial forms of AD.
Owing to the disease’s prevalence, the rising use of memory consults, and advancements in treatment, it is becoming increasingly obvious that Alzheimer’s disease (AD) must be diagnosed early. In the early stages of AD, a variety of strategies have been developed to identify patients who are at risk. It has been suggested that biological markers have been detected in CSF or blood. Considerable research has been done on other anatomical markers, such as the use of nuclear magnetic resonance imaging (MRI) to detect hippocampal atrophy. However, the clinical approach continues to be the most commonly employed in the early diagnosis field. However, it has only been able to identify the earliest cognitive disorders and explain the various phases of memory system alteration associated with the progression of lesions within the hippocampus. Non-cognitive or behavioral deficits are still considered late and are associated with the spread of the lesion to the neocortex, especially to the frontal lobes (Andreasen et al., 2001; Bature et al., 2017; Michel et al., 2010). Involvement of the cerebral cortex involved in higher levels of processing occurs at later stages and eventually much of the brain becomes affected leading to profound consequences in terms of the ability of the individual to lead an independent life.
A novel therapeutic option for AD has been developed based on the remarkable outcomes of stem cell therapies, which have recently been shown in preclinical and clinical trials. In this review, we address treatment approaches for AD-related diseases and discuss new developments in stem cell therapy for AD.
2 Characterization of AD pathology
Based on the examination of autopsy records, scientists have concluded that extracellular Aβ deposits (plaques) and intracellular neurofibrillary tangles (NFTs) are the two key pathological features associated with AD.
2.1 Aβ deposits
The brain contains a variety of Aβ deposits, also known as plaques. These include diffuse plaques, which are a mature form of Aβ deposits that appear in brain regions in the early stages of β-amyloidosis and have dense amyloid cores and neuroinflammatory pathology; cored neurologic plaques, which appear in the later stages of Aβ deposition and are a mature form of Aβ deposits that usually have dense Aβ cores; and neurologic plaques that have been associated with dementia (Ikonomovic et al., 2020).
2.1.1 The formation of Aβ deposits
The APP protein is linked to Aβ deposits. Amyloid precursor protein (APP) is a transmembrane protein that has both cytoplasmic C-tails and soluble extracellular structural domains. There are 18 exons in the human APP gene, which is found on chromosome 21. The APP isoforms APP770, APP751, and APP695, as well as APLP1 and APLP2, are members of the human APP family (Cho et al., 2022; Galvão et al., 2019; O’Brien and Wong, 2011). APP affects a number of physiological processes, including blood coagulation regulation, cell adhesion, neuronal synapse formation, proliferation of neural stem or progenitor cells (NSPCs), and synaptogenesis (Dawkins and Small, 2014). Subsequently, two hydrolases, extracellular β-secretase and intracellular γ-secretase, cleave APP to generate Aβ (Esch et al., 1990; Galvão et al., 2019). When a mutation happens in APP, it results in elevated production of Aβ, which causes degenerative alterations in peripheral neurons and the development of age spots.
According to the amyloid cascade hypothesis, amyloid-β peptide (Aβ) is a key element in the pathophysiology of AD and is considered the primary cause of AD development (Hardy and Selkoe, 2002; Karran et al., 2011). Monomeric Aβ, a neuropeptide with 40–42 amino acids, is a physiological neuroprotectant whose in vivo concentrations are strongly linked to its physiological functions: (1) at picomolar concentrations, it regulates synaptic function, neural circuits, organelle transport, neurogenesis, neuroinflammation, and cognitive processes; (2) the Aβ monomer blocks microbial infections and stops the blood-brain barrier (BBB) from leaking at low concentrations (“vascular plug”); and (3) when present at physiological concentrations, Aβ safeguards the BBB, encourages brain damage healing, and suppresses tumor growth. In addition, Aβ monomers start to group together to produce unique Aβ oligomers, which subsequently form neuritic plaques (Devkota et al., 2021; Jeong et al., 2022; Kent et al., 2020; Mohamed Asik et al., 2021; Nichols et al., 2022; Penke et al., 2020).
2.1.2 Predisposing factors for AD amyloidosis
The cause of AD amyloidosis is multifactorial. First, there is a disruption in cellular autophagy. In addition, autophagy is a comparable pathway for processing APP and APLP1 in neuronal cells under stress, as shown by Fangfang Zhou et al. Given the strong correlation between Aβ generation and APP processing, autophagy within neuronal cells could account for the accumulation of Aβ in AD (Zhou et al., 2011). ER stress and calcium dysregulation are the second most common causes. Since the ER is a crucial organelle for the control of calcium homeostasis, ER stress is typically linked to disruption of calcium homeostasis (Görlach et al., 2006). According to the findings of Eun Sun Jung et al., acute ER stress causes the development of chaperones associated with ERAD, which quickly breaks down APP via the ubiquitin-dependent proteasome pathway to eliminate APP that is misfolded or unfolded and stop the synthesis of Aβ. Conversely, APP buildup in the ER and increased Aβ synthesis are caused by proteasome malfunction induced by persistent ER stress (Jung et al., 2015). Third, inadequate cerebral blood flow can result in a lack of oxygen and energy in the brain, which can impact the synthesis of Aβ and its clearance mechanisms. Reduced cerebral blood flow is linked to the early identification of AD (Fazlollahi et al., 2020). Fourth, patients have elevated cholesterol levels. A protein found in the central nervous system called low-density lipoprotein receptor-related protein 1 (LRP1) aids in the removal of Aβ amyloidosis (Sagare et al., 2007). On the other hand, high cholesterol exacerbates apoptosis in the brain and causes cognitive impairment in AD mice by decreasing LRP1 expression, increasing the expression of the late glycosylation end product receptor RAGE, and increasing the expression of Aβ40 in brain microvascular endothelial cells. This results in deficient Aβ transport at the BBB (Zhou et al., 2021). Fifth, the acetylcholine dose is decreased. The key neurotransmitter that sustains consciousness is acetylcholine (Ach), and patients with AD exhibit a considerable decrease in Ach, which results in symptoms such as dementia. There are numerous possible explanations for the decreased acetylcholine concentration. For instance, Aβ1–42 directly induces a persistent increase in presynaptic Ca2+ levels and disturbs neuronal communication by blocking nicotine-mediated activation of the presynaptic nAChR, as discovered by John J. Dougherty’s team (Dougherty et al., 2003). However, one of the current therapeutic objectives for AD is the prevention of acetylcholinesterase oversynthesis, which successfully mitigates acetylcholine degradation (Rees and Brimijoin, 2003).
2.2 Neurofibrillary degeneration (NFT)
In neuroinflammatory (senile) plaques, neurofibrillary degeneration manifests as neurofibrillary tangles, nerve fiber bundles and dystrophic neuronal protrusions surrounding an amyloid core. Among the vast number of isolated tangles in the AD brain, the primary protein subunit of neuroprogenitor fiber tangles/paired helical filaments was identified by Western blot analysis as the microtubule-associated protein Tau (Grundke-Iqbal et al., 1986).
2.2.1 Tau
Tau, a microtubule-associated protein (MAPT), is essential for the maintenance of complex neuronal cell microstructures, including microtubule assembly and stabilization, particularly in axons. It polymerizes microtubule proteins into microtubules (Avila et al., 2004). Microtubules and nerve cell plasma membranes are also susceptible to tau. Tau appears to influence synaptic function and encourage neuronal maturation through its presence in synapses and dendrites. Neuronal function can be impaired, and even AD can result from altered synaptic dispersion and disruption of the interaction of neurons with synaptic proteins. Among these pathways, one of the most illustrative pathogenetic pathways causing AD is tau hyperphosphorylation. In large filamentous tangles, Alonso AC et al. demonstrated that, in solution, normal tau binds nonsaturatingly to AD hyperphosphorylated tau (AD P-tau) to form large fine particles with a diameter of 3.3 ± 0.7 nm. In contrast, the ability of AD P-tau to aggregate with normal tau was eliminated by dephosphorylation with alkaline phosphatase, which also prevented the formation of tangles (Alonso et al., 1996). Bin Li and colleagues showed that separating normal brain tau and MAP2, an aberrantly hyperphosphorylated tau protein, from the cytoplasmic lysate of Alzheimer’s disease brains (AD P-tau) inhibits microtubule network construction and causes disruptions (Li et al., 2007). This implies that the hyperphosphorylation of Tau causes self-assembly of aggregates, segregation of normal MAPs, and disruption of microtubules, resulting in tangles of nerve fibres that impede normal nerve function and cause dementia symptoms.
2.2.2 Tau hyperphosphorylation
Furthermore, the following are the variables that lead to Tau hyperphosphorylation: (1) An increasing number of studies validate the link between inflammation and Tau pathology in AD. According to Johanna Ojala et al., people with AD have higher amounts of IL-18 in their brains and CSF. IL-18 is found in neurons as well as blood vessels and neuroglia linked to Aβ plaques. IL-18 could play a significant role in the neuroinflammatory response vicious cycle that AD sufferers’ brains experience (Ojala et al., 2009). Christina Ising et al. also showed that loss of NLRP3 inflammatory vesicle function reduces tau hyperphosphorylation and aggregation by regulating tau kinase and phosphatase, that Tau activates NLRP3 inflammatory vesicles and that intracerebral injection of fibrillar β-amyloid-containing brain homogenate induces tau pathology in an NLRP3-dependent manner (Ising et al., 2019). (2) Another significant factor in Tau hyperphosphorylation is the gene. The most potent genetic risk factor for AD is APOE4, which has been shown to increase brain amyloid-β plaques more than other ApoE isoforms. However, Yang Shi et al. showed that the brains of P301S/E4 mice had significantly greater levels of tau protein and that P301S/E4 P301S/E4 mice had significantly greater brain atrophy and neuroinflammation than did P301S/E2 and P301S/E3 mice. In contrast, P301S/EKO mice were largely protected from these changes, indicating that ApoE influences tau pathogenesis, neuroinflammation, and tau-mediated neurodegeneration independently of Aβ pathology. While an absence of ApoE is protective against tau-mediated neurodegeneration, ApoE4 plays a “toxic” role in the process of tau accumulation (Shi et al., 2017). (3) Additionally, through dysregulation or in certain ways, protein kinases and phosphatases can hyperphosphorylate Tau. Among these pathways, the PI3K/AKT/GSK-3β pathway is a crucial pathway that induces Tau hyperphosphorylation in AD; in addition, aberrant Tau hyperphosphorylation is linked to glycogen synthase kinase-3β (GSK-3β), cell cycle protein-dependent kinase 5 (CDK5), and p38 mitogen-activated protein kinase (p38-MAPK)(Hu et al., 2013; Wu et al., 2008). (4) Tau phosphorylation may result from specific medical conditions, therapeutic regimens, or prescribed drugs. It is increasingly recognized that cognitive dysfunction is a significant comorbidity of diabetes. Over the past ten years, there has been a convergence of evidence from brain autopsy studies suggesting that persons with type 2 diabetes mellitus (T2DM) and those without this disease do not exhibit any greater prevalence of the basic neuropathologic characteristics of AD (Arvanitakis et al., 2006). The main characteristic of insulin resistance is interference with insulin signaling, which results in insulin resistance. Insulin resistance impacts systemic metabolism and the brain directly by interfering with the brain’s insulin pathway (Arnold et al., 2018).
3 Directions for AD treatment and the current status of clinical trials
Several variables are involved in the pathogenesis of Alzheimer’s disease, and these targets have been extensively studied by researchers worldwide. Currently, donepezil and memantine are FDA-approved medications for the treatment of AD. However, these medications are limited to treating mild and moderate AD symptoms, and to have any therapeutic benefit, they must be given as soon as AD symptoms start. Currently, there are no FDA-approved medications that target core pathologies or provide long-term treatment options for advanced AD patients. Numerous medications have been tested in clinical trials, but they have all failed at different points and have shown a number of negative side effects. The clinical studies that have been conducted in recent years to treat Aβ amyloid deposition plaques and tau-related neurofibrillary tangles are included in Tables 2, 3.
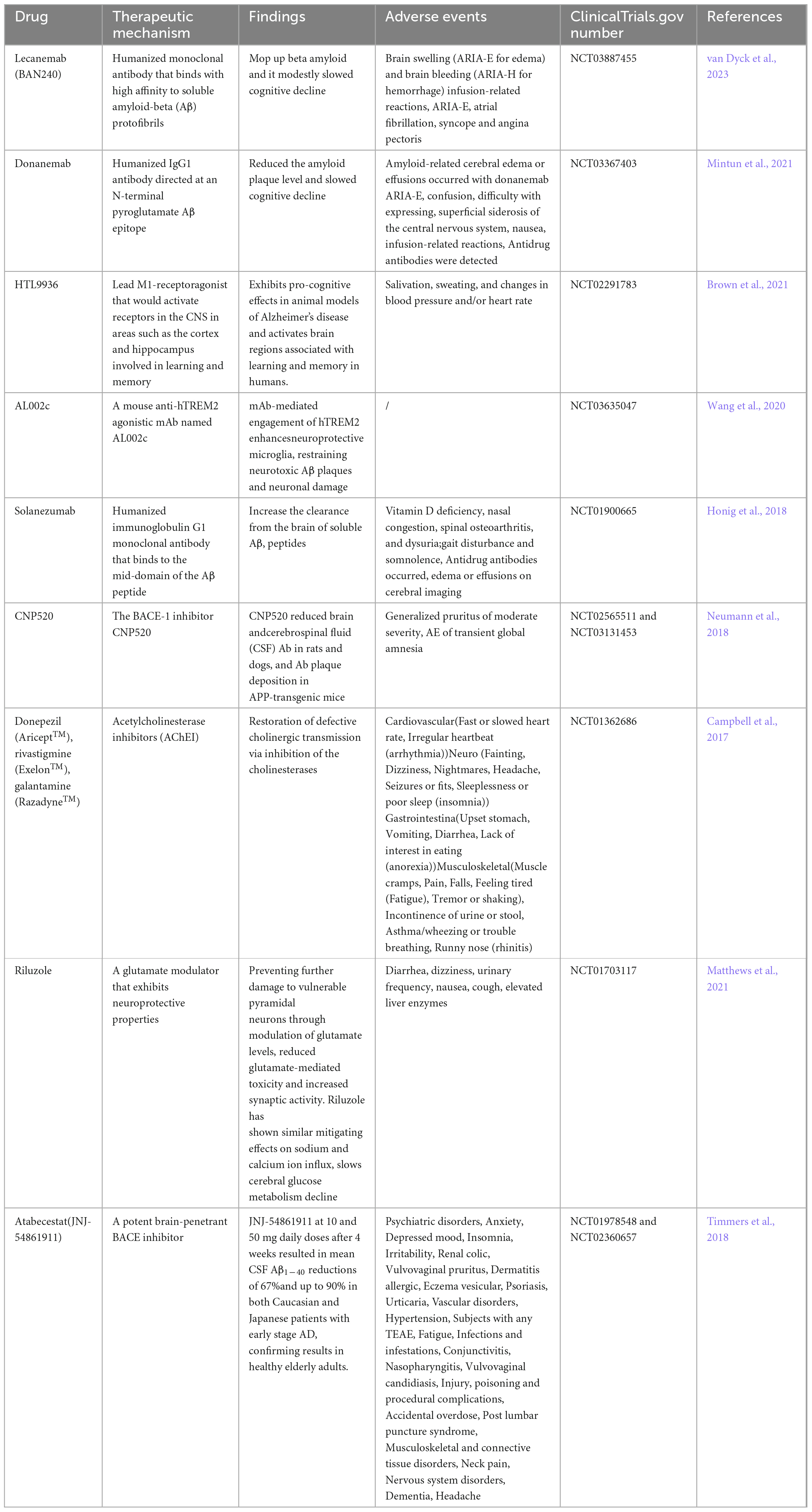
Table 2. Treatments based on Aβ plaques.
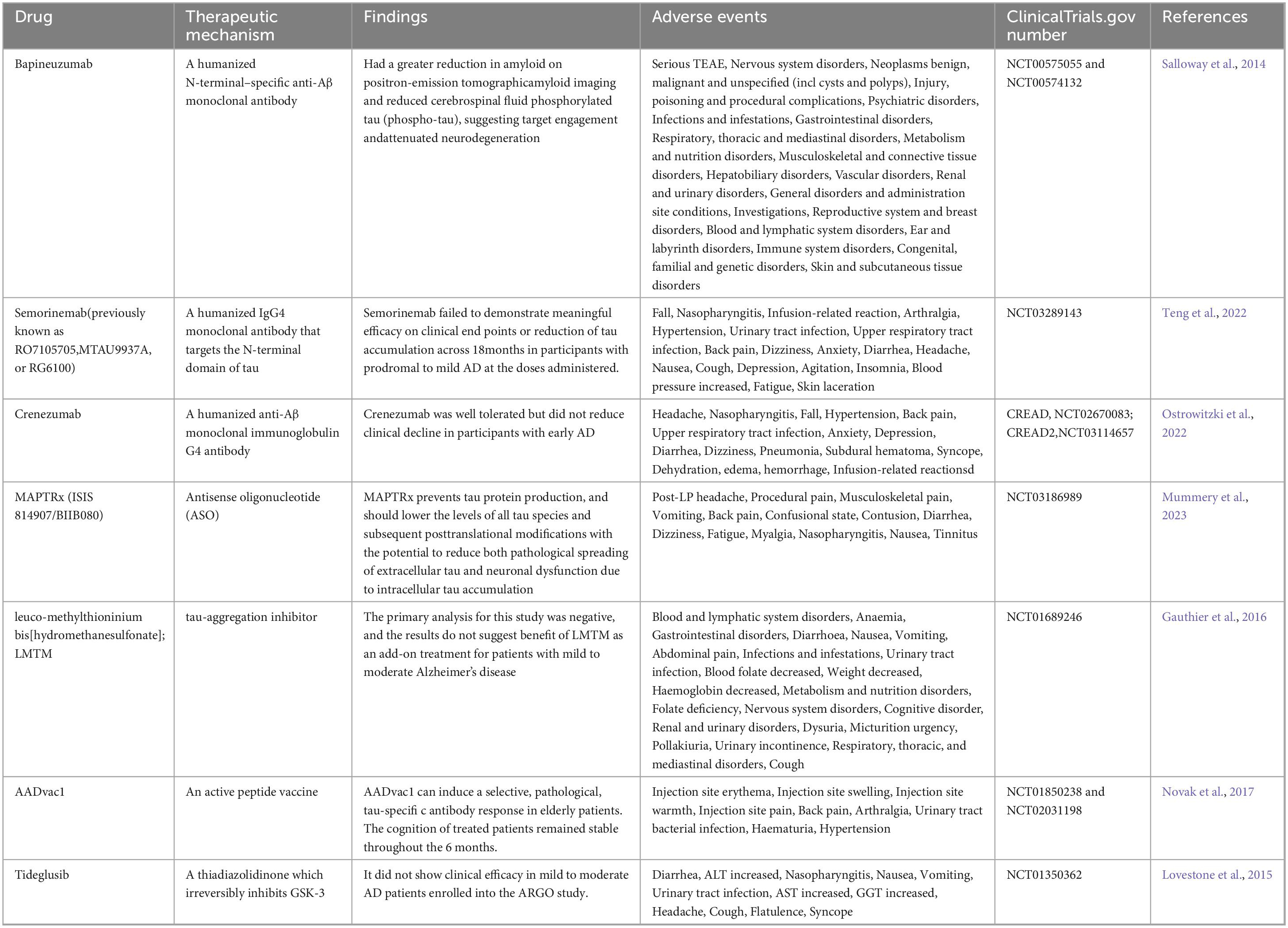
Table 3. Clinical studies involving Tau and NFTs.
The two main treatments being tested in clinical trials for Aβ amyloid deposition plaques are small molecule inhibitors and immunotherapy. Among these, small molecule inhibitors primarily focus on inhibiting β- and γ-secretases. A recent announcement regarding the drug’s withdrawal from the phase III trial revealed that atabecestat, also referred to as JNJ-54861911, a BACE inhibitor, decreased CSF Aβ1–40 in patients with early AD after 4 weeks of dosing. Additionally, the drug has been shown to cause a variety of adverse effects, primarily elevated liver enzymes (Timmers et al., 2018). And the newer generation of “γ-secretase allosteric stabilizers” (GSAS) are being tested. The prototype, introduced in 2006 by Eisai (Yu et al., 2014), targets an allosteric site in the PSEN subunit (Cai et al., 2017; Yang et al., 2021), which are stabilizers of the enzyme that enhances APP processing in affected neurons (De Strooper and Karran, 2024). Additionally, acetylcholinesterase inhibitors (AChEIs) are utilized to extend cholinergic neurotransmission and signaling in patients with symptomatic AD, but the study population has also experienced adverse effects in the CNS and musculoskeletal system (Bond et al., 2012; Campbell et al., 2017). Among the most promising immunotherapies, however, are humanized monoclonal antibodies such as Lecanemab (BAN2401), which bind highly affinity to soluble amyloid-β (Aβ), eliminating β-amyloid and only slightly slowing cognitive decline in the early stages of the disease. Despite being approved, however, research on potentially severe brain hemorrhage and swelling is still lacking (Couzin-Frankel, 2023; Honig et al., 2018; Mintun et al., 2021; van Dyck et al., 2023).
Analogous to Aβ immunotherapy, the humanized IgG4 monoclonal antibody semorinemab has been developed to block Tau aggregation, but it has not been found to decrease or clinically slow the rate of cerebral tau accumulation in prodromal patients with mild Alzheimer’s disease (Teng et al., 2022). In contrast, AADvac1, an active peptide vaccine, generates IgG antibodies that can target tau microtubule site epitopes, decrease neuroprogenitor fibre tangles and ameliorate deleterious phenotypes in Tau patients, and in older patients, it causes a pathologic, selective, tau-specific antibody response (Novak et al., 2017). The microtubule-associated protein tau (MAPT) gene encodes the tau protein. MAPTRx (ISIS 814907/BIIB080) is an antisense oligodeoxynucleotide (ASO) intended to lower the mRNA concentration of MAPT and consequently the protein level of tau. However, 94% of subjects treated with MAPT experienced adverse events, the most common of which was headache following lumbar puncture (LP)(Mummery et al., 2023). An analog of thiadiazolidinone, tideglusib, inhibits GSK-3 irreversiblely. In multiple laboratory models, tideglusib has been demonstrated to decrease tau hyperphosphorylation, Aβ deposition, neuronal loss, and inflammatory and neurotrophic deficiency markers; however, in ARGO research, tideglusib did not demonstrate any therapeutic efficacy in patients with mild to moderate AD (Lovestone et al., 2015).
The most common therapeutic approach to stop, slow, or even reverse the progression of AD pathology has been to decrease tau or Aβ levels. In clinical trials, however, they are experiencing trouble because the effects do not seem to be correlated with improved symptoms. It is believed that the problem with anti-Aβ amyloid deposition monoclonal antibody-based treatments is that they are being used when the disease is already very advanced. That is why they are currently being tested in young people carrying familial variants in PSEN1, PSEN2 and APP to study if they can delay or prevent the development of the disease (Liu et al., 2024; Wagemann et al., 2024). At the same time, early markers of the disease are being sought that allow patients to be diagnosed before cognitive deterioration, which could improve the effectiveness of treatments. Ubiquitin-Proteasome System in the Different Stages of Dominantly In review Inherited Alzheimer’s Disease (DIAD) have been noticed by Eric McDade, and a positive association was observed with imaging markers (PiB PET, tau PET) and a negative correlation with markers of neurodegeneration (FDG PET, MRI), highlighting a significant link between UPS dysregulation and neurodegenerative processes, implicating the UPS as a potential therapeutic target in AD pathogenesis (McDade et al., 2024). There are more markers for the early diagnosis of Alzheimer’s Disease, such as Osteopontin, apathy and anxiety, and the crosstalk Between Amyloid-β, retina, and sleep, contributing to the treatment of Alzheimer’s disease (De Guia et al., 2024; Johansson et al., 2020; Quesnel et al., 2024).
4 Stem cells in animal models of AD
Currently, FDA-approved medications have substantial adverse effects, can only be used to treat symptoms, and cannot fully cure AD. Approaches to treating AD have been revolutionized by stem cell therapy, which aims to restore damaged neurons. With stem cell therapy emerging as a promising treatment option for AD, stem cell technology is becoming increasingly sophisticated and has already shown promise in preclinical tests. Embryonic stem cells (ESCs), mesenchymal stem cells (MSCs), brain-derived neural stem cells (NSCs), and induced pluripotent stem cells (IPSCs) are the cells most frequently employed in AD research, and we also conclude in Table 4.
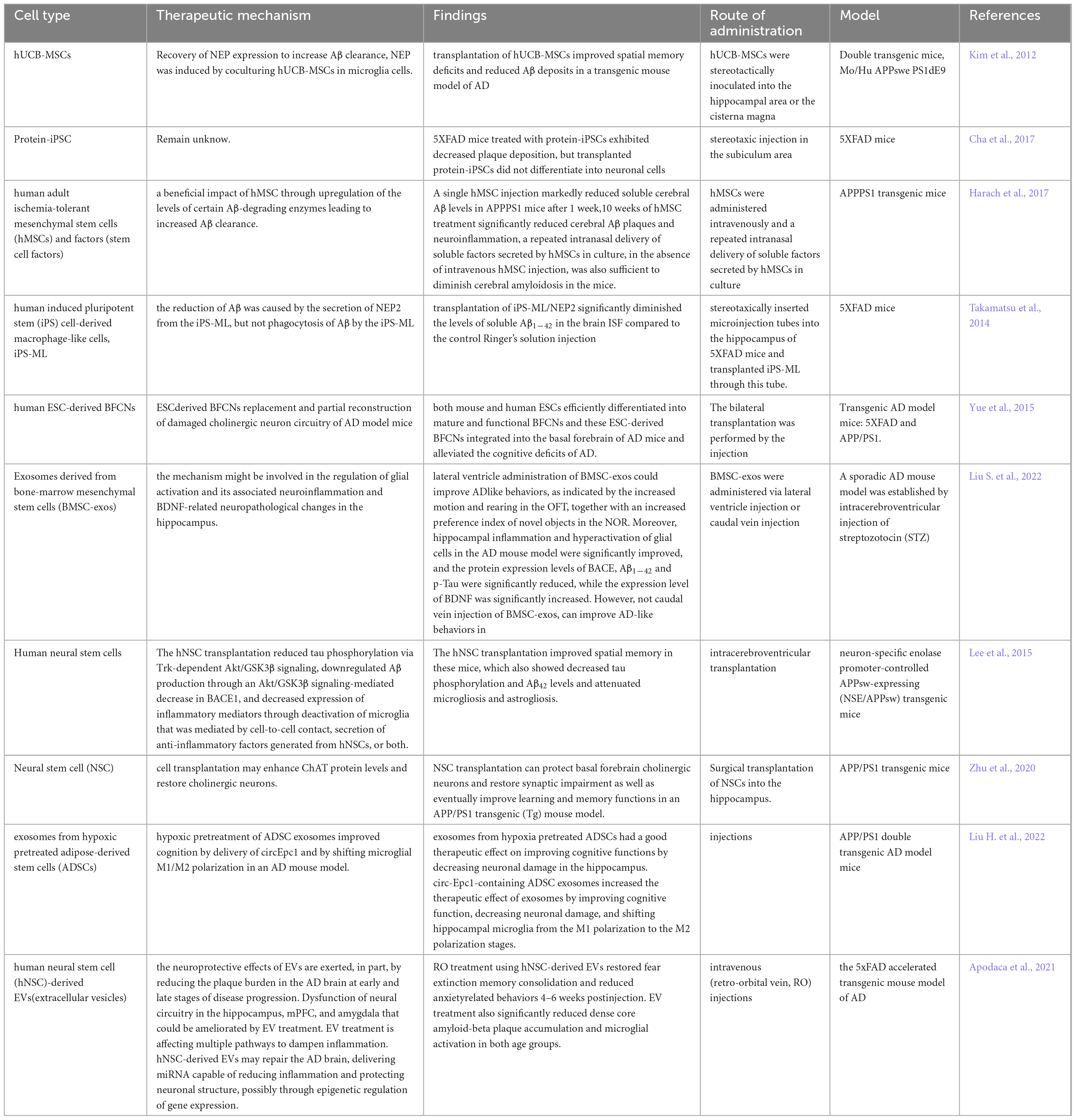
Table 4. Models of stem cell therapy for AD.
4.1 ESCs
With the exception of the placenta, pluripotent stem cells called embryonic stem cells (ESCs) can be grown in vitro and still differentiate into each of the three embryonic germ layers (Conley et al., 2004). Embryonic stem cells can differentiate into basal forebrain cholinergic neurons or BFCNs. Following the transplantation of BFCNs generated from mouse and human ESCs into the basal forebrain of AD model mice, Wei Yue et al. reported that animals with AD exhibited enhanced learning and memory performance when BFCN progenitors mostly developed into mature cholinergic neurons. These neurons then functionally integrate into the endogenous basal forebrain cholinergic projection system, alleviating cognitive impairments in AD animals (Yue et al., 2015). As above, Yan Liu et al. differentiated human embryonic stem cells (hESCs) into a nearly homogenous population of NKX2.1 MGE-like progenitor cells. These progenitor cells give rise to GABA interneurons and BFCNs, which are connected with host hippocampal neurons to rectify learning and memory deficits. The mice with medial septal lesions were then transplanted with these MGE-like progenitor cells (Liu et al., 2013). Although there are many data supporting the pluripotency of ESCs, there are inherent therapeutic risks involved, in which their clinical application their is limited by immunogenic rejection, unregulated cell proliferation, and cancer-causing potential (Lee et al., 2013). At present, one of the main areas of research is the use of ESCs to create animal disease models (Honda et al., 2016; Kim et al., 2020; Mancuso et al., 2019).
4.2 MSCs
MSCs have the capacity to differentiate into mesodermal cells, which include adipocytes, chondrocytes, and osteoblasts. MSCs and neural cells may interact, primarily through bystander processes. MSCs can produce a neuroprotective milieu by exerting anti-inflammatory and antiproliferative effects on microglia and astrocytes and rescue neurons and oligodendrocytes from apoptosis by releasing trophic and antiapoptotic chemicals. Furthermore, MSCs can stimulate the growth and development of localized neural precursor cells, allowing them to differentiate into adult neurons and oligodendrocytes (Uccelli et al., 2008). Several studies have even demonstrated that MSCs may have immunomodulatory effects and certain suppressive effects on T-cell proliferation. These properties could aid in preventing immunological rejection following MSC transplantation (Ankrum et al., 2014; Bartholomew et al., 2002; Di Nicola et al., 2002).
MSC-based treatments for AD have also demonstrated some success in preclinical research. APPPS1 transgenic mice were given intravenous injections of hMSCs by Taoufiq Harach et al. These findings indicate that hMSCs have amyloid-degrading and anti-inflammatory properties. A single injection of hMSCs significantly reduced soluble brain Aβ levels in APPPS1 mice after one week, and a 10-week treatment with hMSCs significantly reduced the number of brain Aβ plaques and neuroinflammation in APPPS1 mice without increasing cerebral amyloid angiopathy or microhemorrhages (Harach et al., 2017). Furthermore, the significance of stem cell-derived exosomes has been steadily highlighted in recent research. BMSC-exosomes (produced from bone marrow mesenchymal stem cells) carry out the functions of BMSCs. Sen Liu et colleagues. found that injecting BMSC-exos via the lateral ventricle significantly reduced hippocampal inflammation and neuroglial overactivation in an AD animal model and improved AD-like behavioral performance in mice. Significant decreases in the protein expression levels of BACE, Aβ1–42, and p-Tau were observed, while significant increases in BDNF expression were observed (Liu S. et al., 2022).
4.3 NSCs
Neural stem cells are also multipotent stem cells that are mostly present in the developing and central nervous systems of mammals. They can differentiate into neurons, astrocytes, and oligodendrocytes (Gincberg et al., 2012). In AD therapy, neural stem cells (NSCs) function primarily through two mechanisms. First, damaged neurons are repaired through direct transplantation. The other group controls brain synaptic activity by secreting a range of neurotrophic factors, which are mediated via paracrine actions.
An AD amyloid precursor protein (APP)/progerin 1 (PS1) transgenic (Tg) mouse model was generated by seeding human brain-derived neural stem cells (hNSCs) by Xueyuan Li et al. In AD mice, hNSC transplantation corrected impairments in identification, learning, and memory but not in anxiety tests. Aβ plaques did not significantly decrease, but the densities of neurons, synapses, and neurofibrils in the frontal cortex and hippocampus of hNSC-treated AD mice significantly increased (Li et al., 2016). Similarly, Qing Zhu et al. reported increased expression of the protein choline acetyltransferase (ChAT) and restoration of cholinergic neurons in the basal forebrain following NSC transplantation. Furthermore, the expression of synaptophysin (SYP), postsynaptic density protein 95 (PSD-95), and microtubule-associated protein 2 (MAP-2) was considerably upregulated in the hippocampal regions of AD mice treated with NSCs, and spatial learning and memory were enhanced (Zhu et al., 2020). With the guidance of the rat neuron-specific enolase (NSE) promoter, Il-Shin Lee et al. investigated the potential therapeutic benefits of human fetal brain-derived NSCs in NSE/APPsw transgenic mice harboring a Swedish mutant of APP. They discovered that the implanted cells migrated and engrafted extensively and that some of the cells differentiated into neurons and glial cells, which improved the spatial memory of the mice. Trk-dependent Akt/GSK3β signaling decreased tau phosphorylation, downregulated Aβ production through the Akt/GSK3β signaling-mediated reduction in BACE1, and decreased the expression of inflammatory mediators through microglia-mediated inactivation or secretion of anti-inflammatory factors generated by hNSCs or both. By providing nutrients and acting as an antiapoptotic agent, hNSCs also enhance synaptic plasticity (Lee et al., 2015).
4.4 iPSCs
In 2006, Yamanaka published a groundbreaking study on the reprogramming of cells to become pluripotent stem cells from mouse embryonic or adult fibroblasts by introducing four factors: Oct3/4, Sox2, c-Myc, and Klf4. This process is termed “induced pluripotent stem” (iPS) and was carried out under ESC culture conditions (Takahashi and Yamanaka, 2006). This work was awarded the 2012 Nobel Prize in Medicine together with Sir John Gurdon’s previous study on reprogramming utilizing somatic cell nuclear transplantation (Abbott, 2012). The practical differentiation capability of iPS cells is believed to be equivalent to that of ESC, and iPSC acquisition minimizes some immunogenic and ethical issues.
Researchers Moon-Yong Cha et al. discovered that transplanting Protein-iPSCs into a 5XFAD transgenic AD mouse model resulted in glial cell differentiation, decreased plaque formation, and decreased cognitive impairment (Cha et al., 2017). On the other hand, Edsel M. Abud’s group discovered that microglia-like cells (iMGLs) that were differentiated from iPSCs grow in vitro similarly to microglia in vivo. Their functional evaluation revealed that these cells migrate through calcium transients, strongly phagocytose CNS substrates, and secrete cytokines in response to inflammatory stimuli. When iMGLs are implanted into human brain organoids and transgenic mice in vivo, the iMGLs resemble microglia (Abud et al., 2017). Enkephalinase-2 (NEP2), a protease with Aβ-degrading activity, was expressed by Koutaro Takamatsu et al. in iPS-ML, a macrophage-like myeloid cell of iPS cell origin. In vitro, NEP2 expression enhanced the reduction in the level of soluble Aβ oligomers in the culture medium and attenuated Aβ neurotoxicity. Since intracerebral delivery of iPS-ML/NEP2 to 5XFAD mice resulted in a considerable decrease in Aβ levels in the brain interstitial fluid, these findings imply that iPS-ML/NEP2 may be a viable therapeutic agent for the treatment of AD (Takamatsu et al., 2014).
5 Clinical trials of stem cells for AD treatment
When Alzheimer’s disease and stem cells were entered into ClinicalTrials.gov, a search of seven completed clinical studies was performed, as shown in Table 5.
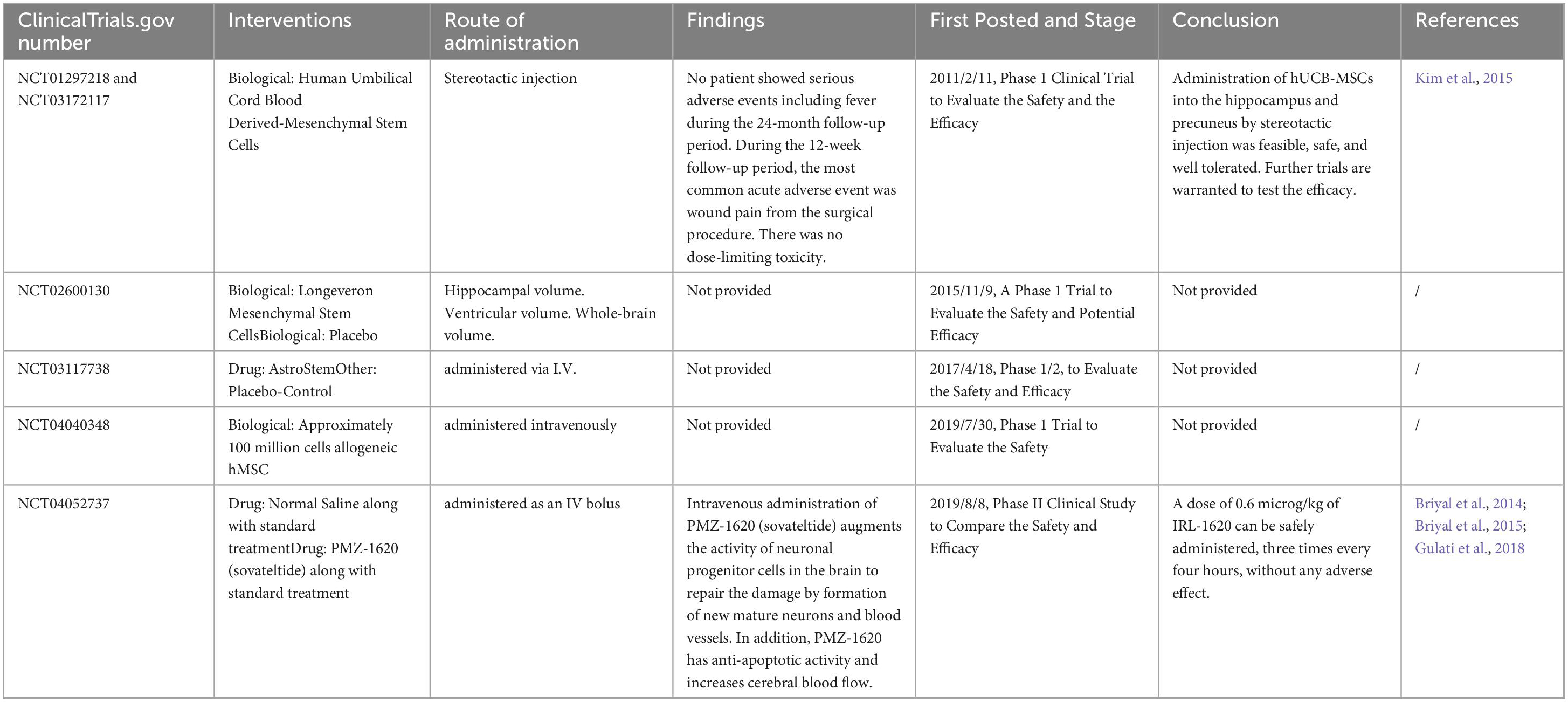
Table 5. Stem cells in clinical trials for AD.
The majority of their research was in Phase 1 or Phase 2, with the main objective being to assess the effectiveness and safety of stem cell preparations. For instance, Hee Jin Kim et al. conducted a phase 1 clinical study (NCT01297218) in Korea to assess the safety and dose-limiting toxicity of stereotactic intracerebral injection of mesenchymal stem cells from human umbilical cord blood (hUCB-MSCs) (Kim et al., 2015). Additionally, none of the patients experienced serious adverse events, such as fever, during the 24-month follow-up period, and during the 12-week follow-up period, the most common acute adverse event was only wound pain from surgery (n = 59), with no toxicity that exceeded a certain dose. These results demonstrated that stereotactic injection of hUCB-MSCs into the hippocampus and precuneus is safe, feasible, and well tolerated (Kim et al., 2015). Hee Jin Kim et al. subsequently assessed the safety and dose-limiting toxicity of three ventricular injections of human umbilical cord blood mesenchymal stem cells (hUCB-MSCs) in the context of NCT03172117 (Kim et al., 2021). Four weeks before the patients received MSC treatment, Ommaya reservoirs were inserted into their right ventricles. Three MSC injections were administered to each patient, which were separated by four weeks. Following their initial hUCB-MSC injection, these patients were monitored for up to 12 weeks and then for an additional 36 months as part of an extended observational study. Three repeated injections of hUCB-MSCs into the lateral ventricle via the Ommaya reservoir appear to be feasible, relatively safe, and well tolerated. Fever (n = 9) was the most common adverse reaction following hUCB-MSC injection, and five participants completed the 36-month extended observational study without experiencing any additional serious adverse events (Kim et al., 2021). A phase II clinical trial by Briyal et al. also compared the safety and effectiveness of treating mild to moderate Alzheimer’s disease patients with PMZ-1620 (Briyal et al., 2014). As an agonist of the endothelin B (ETB) receptor, PMZ-1620 promotes neurovascular remodeling and repair as well as nerve regeneration when it binds to the ETB receptor. After brain injury, dormant stem cells in the brain become activated. When PMZ-1620 (sauvatide) is injected intravenously, it activates the brain’s neural progenitor cells, which create new, mature neurons and blood vessels to heal the damage. PMZ-1620 also has antiapoptotic effects and enhances blood flow to the brain. The findings in NCT04052737 demonstrated that IRL-1620 can be taken three times every four hours at a dose of 0.6 μg/kg without the risk of side effects (Briyal et al., 2014; Briyal et al., 2015; Gulati et al., 2018).
In addition, other clinical trials involving stem cell therapy for Alzheimer’s disease are currently being conducted. The first is a phase IIa study led by Stemedica Cell Technologies, which aimed to assess the safety and tolerability of ischemia-tolerant allogeneic human mesenchymal stem cells (hMSCs) administered intravenously to individuals with mild-to-moderate dementia induced by the disease. The participants were divided into two groups of 20, with each group randomly assigned a 1:1 ratio to receive either the active study drug or a placebo, and the participants were scheduled to conclude on December 31, 2024. The second trial is a phase 1 open-label safety study in which Regenerative BioMedicine, Inc., started to investigate the impact of increasing the volume of intracerebral injections of this investigational product on the brain volume of in vitro-expanded autologous adipose-derived stem cells (ADSCs) in individuals with mild-to-moderate Alzheimer’s disease (ADS). The experimental product, called RB-ADSC, is made of stem cells that were extracted through liposuction from the adipose tissue of participants. Following their extraction, the stem cells were grown and cultivated in a laboratory setting before being reinfused into the original patient. Under the scalp, an Ommaya reservoir—a soft plastic reservoir attached to the ventricle of the brain—was inserted. When determining the recommended Phase 2 clinical trial dose, the main goal will be to assess major adverse events, and dose-limiting toxicities. This trial is anticipated to be completed by 2025–03.
Besides, the Development of iPS From Donated Somatic Cells of Patients With Neurological Diseases (NCT00874783) is active, but not recruiting. The major goal of the project is to develop human iPS cells from cell cultures from skin biopsies or the patient’s hair. The iPS cells will be developed primarily for modeling diseases and drug discovery as well as basic research, and for developing the technology that may eventually allow the use of iPS cells for future transplantation therapy. And the Rajavtihi Neuronal Adult Stem Cells Project (RNASc)(NCT00927108) was withdrawn, with the reason remains unknown.
6 Discussion and perspective
Additional studies are needed to demonstrate the safety and effectiveness of stem cell therapy in humans, despite the large number of pertinent preclinical trials that have been conducted. Why is there still poor translation between animal research and human trials in stem cell therapy for AD? What challenges does the development of stem cell treatment currently face, and how may these challenges be overcome? Currently, experts are interested in how they may be successfully used for diseases that are difficult to cure, such as AD.
6.1 Safety and immaturity of stem cell technology
Pharmaceutical drugs derived from stem cells have potential unforeseen effects, even if they have clinical potential. In response, the FDA issued a warning regarding the safety of unproven stem cell treatments; highlighted a number of reports of major adverse outcomes, including the development of spine tumors and blindness; and announced tighter regulation and oversight of stem cell clinics (Marks et al., 2017).
One of the primary safety concerns associated with stem cell therapy for AD is the immunological response resulting from allografting. MSCs have been demonstrated to assist in lowering immunological rejection (Di Nicola et al., 2002); however, this is still a cause for worry. Systemic immunosuppression remains the standard approach in clinical practice for lowering the chance of graft rejection, although this medication typically has a number of negative side effects in addition to possibly decreasing stem cell activity. Tobias Deuse’s team demonstrated that when major histocompatibility complex (MHC) class I and II genes are inactivated and CD47 is overexpressed, both mouse and human iPSCs lose their immunogenicity but retain their pluripotent stem cell potential and differentiation capacity, which is derived from low immunogenicity. However, genetic manipulation of stem cells to reduce or actively counteract immune rejection has also recently been performed. Treatment with mouse or human iPSCs, endothelial cells, smooth muscle cells, or cardiomyocytes consistently prevents immune rejection in fully MHC-mismatched allogeneic recipients, and patients live for extended periods of time without the need for immunosuppressive measures (Deuse et al., 2019). Secondly, threat tumor development and inadvertent differentiation. The tumorigenicity of stem cells is one of the main barriers to employing them as a foundation for regenerative medicine therapies because of their unique capacity to self-renew and specialize into multiple cell types, which is also mechanistically tied to their tumorigenic activity (Tung and Knoepfler, 2015). The case of a boy with ataxia telangiectasia who underwent neural stem cell therapy and developed a brain tumor was first reported by Gideon Rechavi and colleagues. Molecular and cytogenetic studies revealed that the tumor was not of host origin, indicating that it may have originated from transplanted neural stem cells (Amariglio et al., 2009). This anecdotal case report demonstrates that there is real danger of stem cell tumor growth and that this topic needs to be carefully evaluated. Furthermore, stem cells injected directly into the body may unintentionally differentiate into other tissues. As a result, methods such as initial differentiation into certain cells in vitro and assessment of their cellular purity to weed out undifferentiated cells should be used, as well as optimization of the stem cell differentiation technique Thirdly, additional concerns include uncontrolled in vivo environmental conditions, displacement of cells from the site of implantation, responses at the site of administration, and contaminated cells prior to injection.
6.2 There are no well-established SAD animal models
Sporadic AD (SAD), which affects most AD patients, is caused mostly by a combination of environmental risk factors and genetic variables. These include insulin resistance (Gupta et al., 2019; Kamat et al., 2016), inflammation (Heneka et al., 2015), hypercholesterolemia (Wu et al., 2022; Xu et al., 2020), natural aging, and brain damage (Depp et al., 2023). There is yet no animal model that can accurately depict sporadic AD disease; however, since familial AD (FAD) accounts for a tiny percentage of cases and has a well-established family history of genetic or chromosomal abnormalities, it is comparatively simple to create an animal disease model of FAD. This is among the causes of the numerous studies that have demonstrated superior outcomes in animal trials but not in human trials. Nevertheless, there are currently few animal models of SAD available for stem cell therapy (Table 3). Additionally, because most animals do not develop AD and because rodents and primates differ significantly in terms of neuronal function and anatomy, the effects of mouse models are not representative of most AD pathologies and may replicate only a limited subset of human AD pathology, not pathological features such as neuronal loss. In other words, research utilizing a variety of animal models must be performed if stem cell treatment is to effectively cure AD.
7 Conclusion
Since the development of stem cell technology and the capacity to differentiate stem cells into various CNS neurons and glial cells, stem cell therapy has advanced to human clinical trials and shown promise in animal models of AD. However, a full solution for AD has not been developed as yet. To properly transform stem cell research into useful applications for patients with various disorders, scientists, physicians, regulators, and ethicists must collaborate.
Author contributions
C-MO: Writing – original draft. W-WX: Writing – original draft. DL: Writing – original draft. LM: Writing – original draft. H-TX: Writing – original draft. KN: Data curation, Supervision, Visualization, Writing – review and editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Conflict of interest
C-MO, W-WX, DL, LM, H-TX, and KN were employed by Guangdong Celconta Biotechnology Co., Ltd.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
AD, Alzheimer’s disease; A β, Amyloid beta; ApoE4, apolipoprotein-E4; APP, Amyloid precursor protein; AChEIs, acetylcholinesterase inhibitors; ASO, antisense oligodeoxynucleotide; ADSCs, adipose-derived stem cells; BACE-1: β -site APP-cleaving enzyme 1 BFCNs, basal forebrain cholinergic neurons; CDK5, cycle protein-dependent kinase 5; ChAT, choline acetyltransferase; EOAD, Early-onset Alzheimer’s disease; ER, endoplasmic reticulum; ERAD, Endoplasmic Reticulum-Associated Degradation; ESCs, embryonic stem cells; ETB, endothelin B; GSK-3 β, glycogen synthase kinase-3 β ; iPSCs, induced pluripotent stem cells; iMGLs, microglia-like cells; LOAD, Late-onset Alzheimer’s disease; LRP1, low-density lipoprotein receptor-related protein 1; LP, lumbar puncture; MAPT, microtubule-associated protein tau; MSCs, mesenchymal stem cells; MHC, major histocompatibility complex; NFTs, Intracellular Neurofibrillary Tangles; NLRP3, Nucleotide-binding oligomerization domain, leucine-rich repeat and pyrin domain-containing 3; NSPCs, Neural stem or progenitor cells; NSCs, brain-derived neural stem cells; NSE, neuron-specific enolase; NEP2, enkephalinase-2; PS-1, progerin-1; tau, hyperphosphorylated tau; PSD-95, postsynaptic density protein 95; p38-MAPK, p38 mitogen-activated protein kinase; RAGE, Receptor of Advanced Glycation Endproducts; SYP, synaptophysin; T2DM, type 2 diabetes mellitus.
References
Abud, E., Ramirez, R., Martinez, E., Healy, L., Nguyen, C., and Newman, S. (2017). iPSC-derived human microglia-like cells to study neurological diseases. Neuron 94, 278–293.e9
Alonso, A., Grundke-Iqbal, I., and Iqbal, K. (1996). Alzheimer’s disease hyperphosphorylated tau sequesters normal tau into tangles of filaments and disassembles microtubules. Nat. Med. 2, 783–787. doi: 10.1038/nm0796-783
Amariglio, N., Hirshberg, A., Scheithauer, B., Cohen, Y., Loewenthal, R., Trakhtenbrot, L., et al. (2009). Donor-derived brain tumor following neural stem cell transplantation in an ataxia telangiectasia patient. PLoS Med. 6:e1000029. doi: 10.1371/journal.pmed.1000029
Andreasen, N., Minthon, L., Davidsson, P., Vanmechelen, E., Vanderstichele, H., Winblad, B., et al. (2001). Evaluation of CSF-tau and CSF-Abeta42 as diagnostic markers for Alzheimer disease in clinical practice. Arch Neurol. 58, 373–379. doi: 10.1001/archneur.58.3.373
Ankrum, J., Ong, J., and Karp, J. (2014). Mesenchymal stem cells: Immune evasive, not immune privileged. Nat. Biotechnol. 32, 252–260. doi: 10.1038/nbt.2816
Apodaca, L., Baddour, A., Garcia, C. Jr., Alikhani, L., Giedzinski, E., Ru, N., et al. (2021). Human neural stem cell-derived extracellular vesicles mitigate hallmarks of Alzheimer’s disease. Alzheimers Res Ther. 13:57. doi: 10.1186/s13195-021-00791-x
Armstrong, R. (2019). Risk factors for Alzheimer’s disease. Folia Neuropathol. 57, 87–105. doi: 10.5114/fn.2019.85929
Arnold, S., Arvanitakis, Z., Macauley-Rambach, S., Koenig, A., Wang, H., Ahima, R., et al. (2018). Brain insulin resistance in type 2 diabetes and Alzheimer disease: Concepts and conundrums. Nat. Rev. Neurol. 14, 168–181. doi: 10.1038/nrneurol.2017.185
Arvanitakis, Z., Schneider, J., Wilson, R., Li, Y., Arnold, S., Wang, Z., et al. (2006). Diabetes is related to cerebral infarction but not to AD pathology in older persons. Neurology 67, 1960–1965. doi: 10.1212/01.wnl.0000247053
Avila, J., Lucas, J., Perez, M., and Hernandez, F. (2004). Role of tau protein in both physiological and pathological conditions. Physiol. Rev. 84, 361–384. doi: 10.1152/physrev.00024.2003
Bartholomew, A., Sturgeon, C., Siatskas, M., Ferrer, K., McIntosh, K., Patil, S., et al. (2002). Mesenchymal stem cells suppress lymphocyte proliferation in vitro and prolong skin graft survival in vivo. Exp. Hematol. 30, 42–48. doi: 10.1016/s0301-472x(01)00769-x
Bature, F., Guinn, B., Pang, D., and Pappas, Y. (2017). Signs and symptoms preceding the diagnosis of Alzheimer’s disease: A systematic scoping review of literature from 1937 to 2016. BMJ Open 7:e015746. doi: 10.1136/bmjopen-2016-015746
Bond, M., Rogers, G., Peters, J., Anderson, R., Hoyle, M., and Miners, A. (2012). The effectiveness and cost-effectiveness of donepezil, galantamine, rivastigmine and memantine for the treatment of Alzheimer’s disease (review of Technology Appraisal No. 111): A systematic review and economic model. Health Technol. Assess. 16, 1–470. doi: 10.3310/hta16210
Briyal, S., Nguyen, C., Leonard, M., and Gulati, A. (2015). Stimulation of endothelin B receptors by IRL-1620 decreases the progression of Alzheimer’s disease. Neuroscience 20, 1–11. doi: 10.1016/j.neuroscience.2015.05.044
Briyal, S., Shepard, C., and Gulati, A. (2014). Endothelin receptor type B agonist, IRL-1620, prevents beta amyloid (Aβ) induced oxidative stress and cognitive impairment in normal and diabetic rats. Pharmacol. Biochem. Behav. 120, 65–72. doi: 10.1016/j.pbb.2014.02.008
Brown, A., Bradley, S., Marshall, F., Brown, G., Bennett, K., and Brown, J. (2021). From structure to clinic: Design of a muscarinic M1 receptor agonist with potential to treatment of Alzheimer’s disease. Cell 184, 5886–5901.e22. doi: 10.1016/j.cell.2021.11.001
Cai, T., Yonaga, M., and Tomita, T. (2017). Activation of γ-secretase trimming activity by topological changes of transmembrane domain 1 of Presenilin 1. J. Neurosci. 37, 12272–12280. doi: 10.1523/JNEUROSCI.1628-17.2017
Campbell, N., Perkins, A., Gao, S., Skaar, T., Li, L., Hendrie, H., et al. (2017). Adherence and tolerability of Alzheimer’s disease medications: A pragmatic randomized trial. J. Am. Geriatr. Soc. 65, 1497–1504. doi: 10.1111/jgs.14827
Cha, M., Kwon, Y., Ahn, H., Jeong, H., Lee, Y., and Moon, M. (2017). Protein-induced pluripotent stem cells ameliorate cognitive dysfunction and reduce Aβ deposition in a mouse model of Alzheimer’s disease. Stem Cells Transl. Med. 6, 293–305. doi: 10.5966/sctm.2016-0081
Cho, Y., Bae, H., Okun, E., Arumugam, T., and Jo, D. (2022). Physiology and pharmacology of amyloid precursor protein. Pharmacol. Ther. 235:108122. doi: 10.1016/j.pharmthera.2022.108122
Conley, B., Young, J., Trounson, A., and Mollard, R. (2004). Derivation, propagation and differentiation of human embryonic stem cells. Int. J. Biochem. Cell Biol. 36, 555–567. doi: 10.1016/j.biocel.2003.07.003
Couzin-Frankel, J. (2023). Side effects loom over Alzheimer’s drugs. Science 381, 466–467. doi: 10.1126/science.adk0830
Dawkins, E., and Small, D. (2014). Insights into the physiological function of the β-amyloid precursor protein: Beyond Alzheimer’s disease. J. Neurochem. 129, 756–769. doi: 10.1111/jnc.12675
De Guia, I., Eslick, S., Naismith, S., Kanduri, S., Shah, T., and Martins, R. (2024). The crosstalk between Amyloid-β, retina, and sleep for the early diagnosis of Alzheimer’s disease: A narrative review. J. Alzheimers Dis. Rep. 8, 1009–1021. doi: 10.3233/ADR-230150
De Strooper, B., and Karran, E. (2024). New precision medicine avenues to the prevention of Alzheimer’s disease from insights into the structure and function of γ-secretases. EMBO J. 43, 887–903. doi: 10.1038/s44318-024-00057-w
Depp, C., Sun, T., Sasmita, A., Spieth, L., Berghoff, S., and Nazarenko, T. (2023). Myelin dysfunction drives amyloid-β deposition in models of Alzheimer’s disease. Nature 618, 349–357. doi: 10.1038/s41586-023-06120-6
Deuse, T., Hu, X., Gravina, A., Wang, D., Tediashvili, G., De, C., et al. (2019). Hypoimmunogenic derivatives of induced pluripotent stem cells evade immune rejection in fully immunocompetent allogeneic recipients. Nat. Biotechnol. 37, 252–258. doi: 10.1038/s41587-019-0016-3
Devkota, S., Williams, T., and Wolfe, M. (2021). Familial Alzheimer’s disease mutations in amyloid protein precursor alter proteolysis by γ-secretase to increase amyloid β-peptides of =45 residues. J. Biol. Chem. 296:100281. doi: 10.1016/j.jbc.2021.100281
Di Nicola, M., Carlo-Stella, C., Magni, M., Milanesi, M., Longoni, P., Matteucci, P., et al. (2002). Human bone marrow stromal cells suppress T-lymphocyte proliferation induced by cellular or nonspecific mitogenic stimuli. Blood 99, 3838–3843. doi: 10.1182/blood.v99.10.3838
Dougherty, J., Wu, J., and Nichols, R. (2003). Beta-amyloid regulation of presynaptic nicotinic receptors in rat hippocampus and neocortex. J. Neurosci. 23, 6740–6747. doi: 10.1523/JNEUROSCI.23-17-06740.2003
Esch, F., Keim, P., Beattie, E., Blacher, R., Culwell, A., Oltersdorf, T., et al. (1990). Cleavage of amyloid beta peptide during constitutive processing of its precursor. Science 248, 1122–1124. doi: 10.1126/science.2111583
Fazlollahi, A., Calamante, F., Liang, X., Bourgeat, P., Raniga, P., Dore, V., et al. (2020). Australian imaging biomarkers and lifestyle (AIBL) research group. increased cerebral blood flow with increased amyloid burden in the preclinical phase of Alzheimer’s disease. J. Magn. Reson. Imaging 51, 505–513. doi: 10.1002/jmri.26810
Frisoni, G., Altomare, D., Thal, D., Ribaldi, F., van der Kant, R., Ossenkoppele, R., et al. (2022). The probabilistic model of Alzheimer disease: The amyloid hypothesis revised. Nat. Rev. Neurosci. 23, 53–66. doi: 10.1038/s41583-021-00533-w
Galvão, F., Grokoski, K., da Silva, B., Lamers, M., and Siqueira, I. (2019). The amyloid precursor protein (APP) processing as a biological link between Alzheimer’s disease and cancer. Ageing Res. Rev. 49, 83–91. doi: 10.1016/j.arr.2018.11.007
Gatz, M., Pedersen, N., Berg, S., Johansson, B., Johansson, K., Mortimer, J., et al. (1997). Heritability for Alzheimer’s disease: The study of dementia in Swedish twins. J. Gerontol. A Biol. Sci. Med. Sci. 52, M117–M125. doi: 10.1093/gerona/52a.2.m117
Gauthier, S., Feldman, H., Schneider, L., Wilcock, G., Frisoni, G., and Hardlund, J. (2016). Efficacy and safety of tau-aggregation inhibitor therapy in patients with mild or moderate Alzheimer’s disease: A randomized, controlled, double-blind, parallel-arm, phase 3 trial. Lancet 388, 2873–2884. doi: 10.1016/S0140-6736(16)31275-2
Gincberg, G., Arien-Zakay, H., Lazarovici, P., and Lelkes, P. (2012). Neural stem cells: Therapeutic potential for neurodegenerative diseases. Br. Med. Bull. 104, 7–19. doi: 10.1093/bmb/lds024
Görlach, A., Klappa, P., and Kietzmann, T. (2006). The endoplasmic reticulum: Folding, calcium homeostasis, signaling, and redox control. Antioxid. Redox Signal. 8, 1391–1418. doi: 10.1089/ars.2006.8.1391
Grundke-Iqbal, I., Iqbal, K., Quinlan, M., Tung, Y., Zaidi, M., and Wisniewski, H. (1986). Microtubule-associated protein tau. A component of Alzheimer paired helical filaments. J. Biol. Chem. 261, 6084–6089.
Gulati, A., Hornick, M., Briyal, S., and Lavhale, M. S. (2018). A novel neuroregenerative approach using ET(B) receptor agonist, IRL-1620, to treat CNS disorders. Physiol. Res. 67, (Suppl. 1), S95–S113. doi: 10.33549/physiolres.933859
Gupta, S., Singhal, N., Ganesh, S., and Sandhir, R. (2019). Extending arms of insulin resistance from diabetes to Alzheimer’s disease: Identification of potential therapeutic targets. CNS Neurol Disord. Drug Targets 18, 172–184. doi: 10.2174/1871527317666181114163515
Hanisch, F., and Kölmel, H. (2004). Genotype-phenotype analysis in early-onset Alzheimer’s disease due to presenilin-1 mutations at codon 139. Eur. J. Med. Res. 9, 361–364.
Harach, T., Jammes, F., Muller, C., Duthilleul, N., Cheatham, V., Zufferey, V., et al. (2017). Administrations of human adult ischemia-tolerant mesenchymal stem cells and factors reduce amyloid beta pathology in a mouse model of Alzheimer’s disease. Neurobiol. Aging 51, 83–96. doi: 10.1016/j.neurobiolaging.2016.11.009
Hardy, J., and Selkoe, D. (2002). The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 297, 353–356. doi: 10.1126/science.1072994
Heneka, M., Carson, M., El Khoury, J., Landreth, G., Brosseron, F., and Feinstein, D. (2015). Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 14, 388–405. doi: 10.1016/S1474-4422(15)70016-5
Honda, M., Minami, I., Tooi, N., Morone, N., Nishioka, H., Uemura, K., et al. (2016). The modelling of Alzheimer’s disease by the overexpression of mutant Presenilin 1 in human embryonic stem cells. Biochem. Biophys. Res. Commun. 469, 587–592. doi: 10.1016/j.bbrc.2015.12.025
Honig, L., Vellas, B., Woodward, M., Boada, M., Bullock, R., and Borrie, M. (2018). Trial of solanezumab for mild dementia due to Alzheimer’s disease. N. Engl. J. Med. 378, 321–330. doi: 10.1056/NEJMoa1705971
Hu, Y., Long, N., Pigino, G., Brady, S., and Lazarov, O. (2013). Molecular mechanisms of environmental enrichment: Impairments in Akt/GSK3β, neurotrophin-3 and CREB signaling. PLoS One 8:e64460. doi: 10.1371/journal.pone.0064460
Ikonomovic, M., Buckley, C., Abrahamson, E., Kofler, J., Mathis, C., Klunk, W., et al. (2020). Postmortem analyses of PiB and flutemetamol in diffuse and cored amyloid-β plaques in Alzheimer’s disease. Acta Neuropathol. 140, 463–476. doi: 10.1007/s00401-020-02175-1
Ising, C., Venegas, C., Zhang, S., Scheiblich, H., Schmidt, S., and Vieira-Saecker, A. (2019). NLRP3 inflammasome activation drives tau pathology. Nature 575, 669–673. doi: 10.1038/s41586-019-1769-z
Jeong, H., Shin, H., Hong, S., and Kim, Y. (2022). Physiological roles of monomeric amyloid-β and implications for Alzheimer’s disease therapeutics. Exp. Neurobiol. 31, 65–88. doi: 10.5607/en22004IF
Johansson, M., Stomrud, E., Lindberg, O., Westman, E., Johansson, P., van Westen, D., et al. (2020). Apathy and anxiety are early markers of Alzheimer’s disease. Neurobiol. Aging 85, 74–82. doi: 10.1016/j.neurobiolaging.2019.10.008
Jung, E., Hong, H., Kim, C., and Mook-Jung, I. (2015). Acute ER stress regulates amyloid precursor protein processing through ubiquitin-dependent degradation. Sci. Rep. 5:8805. doi: 10.1038/srep08805
Kamat, P., Kalani, A., Rai, S., Tota, S., Kumar, A., and Ahmad, A. (2016). Streptozotocin intracerebroventricular-induced neurotoxicity and brain insulin resistance: A therapeutic intervention for treatment of sporadic Alzheimer’s disease (sad)-like pathology. Mol. Neurobiol. 53, 4548–4562. doi: 10.1007/s12035-015-9384-y
Karran, E., Mercken, M., and De Strooper, B. (2011). The amyloid cascade hypothesis for Alzheimer’s disease: An appraisal for the development of therapeutics. Nat. Rev. Drug Discov. 10, 698–712. doi: 10.1038/nrd3505
Kent, S., Spires-Jones, T., and Durrant, C. (2020). The physiological roles of tau and Aβ: Implications for Alzheimer’s disease pathology and therapeutics. Acta Neuropathol. 140, 417–447. doi: 10.1007/s00401-020-02196-w
Kim, D., Choi, S., Lee, J., Kim, H., Kim, H., Lee, J., et al. (2020). Feasibility and efficacy of intra-arterial administration of embryonic stem cell derived-mesenchymal stem cells in animal model of Alzheimer’s disease. J. Alzheimers Dis. 76, 1281–1296. doi: 10.3233/JAD-200026
Kim, H., Cho, K., Jang, H., Lee, N., Jung, Y., Kim, J., et al. (2021). Intracerebroventricular injection of human umbilical cord blood mesenchymal stem cells in patients with Alzheimer’s disease dementia: A phase I clinical trial. Alzheimers Res. Ther. 13:154. doi: 10.1186/s13195-021-00897-2
Kim, H., Seo, S., Chang, J., Lee, J., Kim, C., Chin, J., et al. (2015). Stereotactic brain injection of human umbilical cord blood mesenchymal stem cells in patients with Alzheimer’s disease dementia: A phase 1 clinical trial. Alzheimers Dement. 1, 95–102. doi: 10.1016/j.trci.2015.06.007
Kim, J., Kim, D., Kim, J., Lee, D., Jeon, H., Kwon, S., et al. (2012). Soluble intracellular adhesion molecule-1 secreted by human umbilical cord blood-derived mesenchymal stem cell reduces amyloid-β plaques. Cell Death Differ. 19, 680–691. doi: 10.1038/cdd.2011.140
Lee, A., Tang, C., Rao, M., Weissman, I., and Wu, J. (2013). Tumorigenicity as a clinical hurdle for pluripotent stem cell therapies. Nat. Med. 19, 998–1004. doi: 10.1038/nm.3267
Lee, I., Jung, K., Kim, I., Lee, H., Kim, M., Yun, S., et al. (2015). Human neural stem cells alleviate Alzheimer-like pathology in a mouse model. Mol. Neurodegener. 10:38. doi: 10.1186/s13024-015-0035-6
Li, B., Chohan, M., Grundke-Iqbal, I., and Iqbal, K. (2007). Disruption of microtubule network by Alzheimer abnormally hyperphosphorylated tau. Acta Neuropathol. 113, 501–511. doi: 10.1007/s00401-007-0207-8
Li, X., Zhu, H., Sun, X., Zuo, F., Lei, J., Wang, Z., et al. (2016). Human neural stem cell transplantation rescues cognitive defects in APP/PS1 model of Alzheimer’s disease by enhancing neuronal connectivity and metabolic activity. Front. Aging Neurosci. 23:282. doi: 10.3389/fnagi.2016.00282
Liu, H., Jin, M., Ji, M., Zhang, W., Liu, A., and Wang, T. (2022). Hypoxic pretreatment of adipose-derived stem cell exosomes improved cognition by delivery of circ-Epc1 and shifting microglial M1/M2 polarization in an Alzheimer’s disease mice model. Aging (Albany NY) 14, 3070–3083. doi: 10.18632/aging.203989
Liu, H., Li, J., Ziegemeier, E., Adams, S., McDade, E., and Clifford, D. (2024). Dominantly inherited alzheimer network trials unit (DIAN-TU): Trial satisfaction and attitudes towards future clinical trials. J. Prev. Alzheimers Dis. 11, 558–566. doi: 10.14283/jpad.2024.61
Liu, S., Fan, M., Xu, J., Yang, L., Qi, C., Xia, Q., et al. (2022). Exosomes derived from bone-marrow mesenchymal stem cells alleviate cognitive decline in AD-like mice by improving BDNF-related neuropathology. J. Neuroinflammation 19:35. doi: 10.1186/s12974-022-02393-2
Liu, Y., Weick, J., Liu, H., Krencik, R., Zhang, X., Ma, L., et al. (2013). Medial ganglionic eminence-like cells derived from human embryonic stem cells correct learning and memory deficits. Nat. Biotechnol. 31, 440–447. doi: 10.1038/nbt.2565
Lovestone, S., Boada, M., Dubois, B., Hüll, M., Rinne, J., Huppertz, H., et al. (2015). ARGO investigators. A phase II trial of tideglusib in Alzheimer’s disease. J. Alzheimers Dis. 45, 75–88. doi: 10.3233/JAD-141959
Ma, C., Hong, F., and Yang, S. (2022). Amyloidosis in Alzheimer’s disease: Pathogeny, etiology, and related therapeutic directions. Molecules 27:1210. doi: 10.3390/molecules27041210IF
Mancuso, R., Van Den Daele, J., Fattorelli, N., Wolfs, L., Balusu, S., and Burton, O. (2019). Stem-cell-derived human microglia transplanted in mouse brain to study human disease. Nat. Neurosci. 22, 2111–2116. doi: 10.1038/s41593-019-0525-x
Marks, P., Witten, C., and Califf, R. (2017). Clarifying Stem-cell therapy’s benefits and risks. N. Engl. J. Med. 376, 1007–1009. doi: 10.1056/NEJMp1613723
Matthews, D., Mao, X., Dowd, K., Tsakanikas, D., Jiang, C., and Meuser, C. (2021). Riluzole, a glutamate modulator, slows cerebral glucose metabolism decline in patients with Alzheimer’s disease. Brain 144, 3742–3755. doi: 10.1093/brain/awab222
McDade, E., Liu, H., Bui, Q., Hassenstab, J., Gordon, B., and Benzinger, T. (2024). Ubiquitin-proteasome system in the different stages of dominantly inherited Alzheimer’s disease. Res Sq [Preprint] doi: 10.21203/rs.3.rs-4202125/v1
Michel, B., Luciani, V., Geda, Y., Sambuchi, N., Paban, V., and Azorin, J. (2010). Dans la maladie d’Alzheimer, l’expression des troubles psychologiques et comportementaux est précoce et spécifique des stades lésionnels [In Alzheimer’s disease, the clinical expression of behavioral and psychological signs and symptoms is early and specific of neuropathological stages]. Encephale 36, 314–325. doi: 10.1016/j.encep.2009.10.012
Mintun, M., Lo, A., Duggan Evans, C., Wessels, A., Ardayfio, P., and Andersen, S. (2021). Donanemab in early Alzheimer’s disease. N. Engl. J. Med. 384, 1691–1704. doi: 10.1056/NEJMoa2100708
Mohamed Asik, R., Suganthy, N., Aarifa, M., Kumar, A., Szigeti, K., Mathe, D., et al. (2021). Alzheimer’s disease: A molecular view of β-amyloid induced morbific events. Biomedicines 9:1126. doi: 10.3390/biomedicines9091126
Mummery, C., Börjesson-Hanson, A., Blackburn, D., Vijverberg, E., De Deyn, P., and Ducharme, S. (2023). Tau-targeting antisense oligonucleotide MAPTRx in mild Alzheimer’s disease: A phase 1b, randomized, placebo-controlled trial. Nat. Med. 29, 1437–1447. doi: 10.1038/s41591-023-02326-3
Neumann, U., Ufer, M., Jacobson, L., Rouzade-Dominguez, M., Huledal, G., and Kolly, C. (2018). The BACE-1 inhibitor CNP520 for prevention trials in Alzheimer’s disease. EMBO Mol. Med. 10:e9316. doi: 10.15252/emmm.201809316
Nichols, R., Gulisano, W., and Puzzo, D. (2022). Editorial: Beta amyloid: From physiology to pathogenesis. Front. Mol. Neurosci. 15:876224. doi: 10.3389/fnmol.2022.876224
Novak, P., Schmidt, R., Kontsekova, E., Zilka, N., Kovacech, B., and Skrabana, R. (2017). Safety and immunogenicity of the tau vaccine AADvac1 in patients with Alzheimer’s disease: A randomized, double-blind, placebo-controlled, phase 1 trial. Lancet Neurol. 16, 123–134. doi: 10.1016/S1474-4422(16)30331
O’Brien, R., and Wong, P. (2011). Amyloid precursor protein processing and Alzheimer’s disease. Annu. Rev. Neurosci. 34, 185–204. doi: 10.1146/annurev-neuro-061010-113613
Ojala, J., Alafuzoff, I., Herukka, S., van Groen, T., Tanila, H., and Pirttilä, T. (2009). Expression of interleukin-18 is increased in the brains of Alzheimer’s disease patients. Neurobiol. Aging 30, 198–209. doi: 10.1016/j.neurobiolaging.2007.06.006
Ostrowitzki, S., Bittner, T., Sink, K., Mackey, H., Rabe, C., and Honig, L. (2022). Evaluating the safety and efficacy of crenezumab vs placebo in adults with early Alzheimer disease: Two phase 3 randomized placebo-controlled trials. JAMA Neurol. 79, 1113–1121. doi: 10.1001/jamaneurol.2022.2909
Penke, B., Szűcs, M., and Bogár, F. (2020). Oligomerization and conformational change turn monomeric β-amyloid and tau proteins toxic: Their role in Alzheimer’s pathogenesis. Molecules 25:1659. doi: 10.3390/molecules25071659
Penke, B., Szűcs, M., and Bogár, F. (2023). New pathways identify novel drug targets for the prevention and treatment of Alzheimer’s disease. Int. J. Mol. Sci. 24:5383. doi: 10.3390/ijms24065383
Polsinelli, A., and Apostolova, L. (2022). Atypical Alzheimer disease variants. Continuum (Minneap Minn) 28, 676–701. doi: 10.1212/CON.0000000000001082
Quesnel, M., Labonté, A., Picard, C., Bowie, D., Zetterberg, H., Blennow, K., et al. (2024). Disease neuroimaging initiative; PREVENT-AD Research group. osteopontin: A novel marker of pre-symptomatic sporadic Alzheimer’s disease. Alzheimers Dement. doi: 10.1002/alz.14065 Online ahead of print.
Rees, T., and Brimijoin, S. (2003). The role of acetylcholinesterase in the pathogenesis of Alzheimer’s disease. Drugs Today (Barc) 39, 75–83. doi: 10.1358/dot.2003.39.1.740206
Sagare, A., Deane, R., Bell, R., Johnson, B., Hamm, K., Pendu, R., et al. (2007). Clearance of amyloid-beta by circulating lipoprotein receptors. Nat. Med. 13, 1029–1031. doi: 10.1038/nm1635
Salloway, S., Sperling, R., Fox, N., Blennow, K., Klunk, W., and Raskind, M. (2014). Bapineuzumab 301 and 302 clinical trial investigators. two phase 3 trials of bapineuzumab in mild-to-moderate Alzheimer’s disease. N. Engl. J. Med. 370, 322–333. doi: 10.1056/NEJMoa1304839
Shi, Y., Yamada, K., Liddelow, S., Smith, S., Zhao, L., and Luo, W. (2017). ApoE4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy. Nature 549, 523–527. doi: 10.1038/nature24016
Sims, R., Hill, M., and Williams, J. (2020). The multiplex model of the genetics of Alzheimer’s disease. Nat. Neurosci. 23, 311–322. doi: 10.1038/s41593-020-0599-5
Takahashi, K., and Yamanaka, S. (2006). Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126, 663–676. doi: 10.1016/j.cell.2006.07.024
Takamatsu, K., Ikeda, T., Haruta, M., Matsumura, K., Ogi, Y., Nakagata, N., et al. (2014). Degradation of amyloid beta by human induced pluripotent stem cell-derived macrophages expressing Neprilysin-2. Stem Cell Res. 13(3 Pt A), 442–453. doi: 10.1016/j.scr.2014.10.001
Tehrani, M., Matsuda, I., Yamagata, A., Kodama, Y., Matsunaga, T., Sato, M., et al. (2024). E22G Aβ40 fibril structure and kinetics illuminate how Aβ40 rather than Aβ42 triggers familial Alzheimer’s. Nat. Commun. 15:7045. doi: 10.1038/s41467-024-51294-w
Teng, E., Manser, P., Pickthorn, K., Brunstein, F., Blendstrup, M., and Sanabria Bohorquez, S. (2022). Safety and efficacy of semorinemab in individuals with prodromal to mild Alzheimer disease: A randomized clinical trial. JAMA Neurol. 79, 758–767. doi: 10.1001/jamaneurol.2022.1375
Timmers, M., Streffer, J., Russu, A., Tominaga, Y., Shimizu, H., and Shiraishi, A. (2018). Pharmacodynamics of atabecestat (JNJ-54861911), an oral BACE1 inhibitor in patients with early Alzheimer’s disease: Randomized, double-blind, placebo-controlled study. Alzheimers Res. Ther. 10:85. doi: 10.1186/s13195-018-0415-6
Tung, P., and Knoepfler, P. (2015). Epigenetic mechanisms of tumorigenicity manifesting in stem cells. Oncogene 34, 2288–2296. doi: 10.1038/onc.2014.172
Uccelli, A., Moretta, L., and Pistoia, V. (2008). Mesenchymal stem cells in health and disease. Nat. Rev. Immunol. 8, 726–736. doi: 10.1038/nri2395
van Dyck, C., Swanson, C., Aisen, P., Bateman, R., Chen, C., and Gee, M. (2023). Lecanemab in early Alzheimer’s disease. N. Engl. J. Med. 388, 9–21. doi: 10.1056/NEJMoa2212948
Wagemann, O., Liu, H., Wang, G., Shi, X., Bittner, T., and Scelsi, M. (2024). Dominantly inherited Alzheimer network–trials unit. downstream biomarker effects of gantenerumab or solanezumab in dominantly inherited Alzheimer disease: The DIAN-TU-001 randomized clinical trial. JAMA Neurol. 81, 582–593. doi: 10.1001/jamaneurol.2024.0991
Wang, S., Mustafa, M., Yuede, C., Salazar, S., Kong, P., and Long, H. (2020). Anti-human TREM2 induces microglia proliferation and reduces pathology in an Alzheimer’s disease model. J. Exp. Med. 217:e20200785. doi: 10.1084/jem.20200785
Wingo, T., Lah, J., Levey, A., and Cutler, D. (2012). Autosomal recessive causes likely in early-onset Alzheimer disease. Arch Neurol. 69, 59–64. doi: 10.1001/archneurol.2011.221
Wu, H., Lu, D., Jiang, H., Xiong, Y., Qu, C., Li, B., et al. (2008). Simvastatin-mediated upregulation of VEGF and BDNF, activation of the PI3K/Akt pathway, and increase of neurogenesis are associated with therapeutic improvement after traumatic brain injury. J. Neurotrauma 25, 130–139. doi: 10.1089/neu.2007.0369
Wu, L., Rosa-Neto, P., Hsiung, G., Sadovnick, A., Masellis, M., Black, S., et al. (2012). Early-onset familial Alzheimer’s disease (EOFAD). Can. J. Neurol. Sci. 39, 436–445. doi: 10.1017/s0317167100013949
Wu, M., Zhai, Y., Liang, X., Chen, W., Lin, R., and Ma, L. (2022). Connecting the dots between hypercholesterolemia and Alzheimer’s disease: A potential mechanism based on 27-Hydroxycholesterol. Front. Neurosci. 16:842814. doi: 10.3389/fnins.2022.842814
Xu, C., Apostolova, L., Oblak, A., and Gao, S. (2020). Association of Hypercholesterolemia with Alzheimer’s disease pathology and cerebral amyloid angiopathy. J. Alzheimers Dis. 73, 1305–1311. doi: 10.3233/JAD-191023
Yang, G., Zhou, R., Guo, X., Yan, C., Lei, J., and Shi, Y. (2021). Structural basis of γ-secretase inhibition and modulation by small molecule drugs. Cell 184, 521–533.e14. doi: 10.1016/j.cell.2020.11.049
Yu, Y., Logovinsky, V., Schuck, E., Kaplow, J., Chang, M., Miyagawa, T., et al. (2014). Safety, tolerability, pharmacokinetics, and pharmacodynamics of the novel γ-secretase modulator, E2212, in healthy human subjects. J. Clin. Pharmacol. 54, 528–536. doi: 10.1002/jcph.249
Yue, W., Li, Y., Zhang, T., Jiang, M., Qian, Y., Zhang, M., et al. (2015). ESC-derived basal forebrain cholinergic neurons ameliorate the cognitive symptoms associated with Alzheimer’s disease in mouse models. Stem Cell Rep. 5, 776–790. doi: 10.1016/j.stemcr.2015.09.010
Zhou, F., van Laar, T., Huang, H., and Zhang, L. (2011). APP and APLP1 are degraded through autophagy in response to proteasome inhibition in neuronal cells. Protein Cell. 2, 377–383. doi: 10.1007/s13238-011-1047-9
Zhou, R., Chen, L., Yang, H., Li, L., Liu, J., Chen, L., et al. (2021). Effect of high cholesterol regulation of LRP1 and RAGE on Aβ transport across the blood-brain barrier in Alzheimer’s disease. Curr. Alzheimer Res. 18, 428–442. doi: 10.2174/1567205018666210906092940
Zhu, Q., Zhang, N., Hu, N., Jiang, R., Lu, H., Xuan, A., et al. (2020). Neural stem cell transplantation improves learning and memory by protecting cholinergic neurons and restoring synaptic impairment in an amyloid precursor protein/presenilin 1 transgenic mouse model of Alzheimer’s disease. Mol. Med. Rep. 21, 1172–1180. doi: 10.3892/mmr.2020.10918
Keywords: Alzheimer’s disease, stem cell therapy, Aβ deposits, nFTS, clinical trials
Citation: Ou C-M, Xue W-W, Liu D, Ma L, Xie H-T and Ning K (2024) Stem cell therapy in Alzheimer’s disease: current status and perspectives. Front. Neurosci. 18:1440334. doi: 10.3389/fnins.2024.1440334
Received: 29 May 2024; Accepted: 09 October 2024;
Published: 21 November 2024.
Edited by:
Suren A. Tatulian, University of Central Florida, United StatesReviewed by:
Leonardo Romorini, Fundación Para la Lucha Contra las Enfermedades Neurológicas de la Infancia (FLENI), ArgentinaAlan Hazell, University of Montreal, Canada
Copyright © 2024 Ou, Xue, Liu, Ma, Xie and Ning. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ke Ning, a2UubmluZ0B4a2RiaW8uY29t; orcid.org/0000-0002-0771-1134; Hai-Tao Xie, NDA1NTE1MTc2QHFxLmNvbQ==; orcid.org/0009-0000-2849-4012
†These authors have contributed equally to this work