 Eric W. Ottesen
Eric W. Ottesen Natalia N. Singh
Natalia N. Singh Joonbae Seo
Joonbae Seo Ravindra N. Singh
Ravindra N. Singh
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Neurosci., 12 July 2024
Sec. Neurogenomics
Volume 18 - 2024 | https://doi.org/10.3389/fnins.2024.1412893
This article is part of the Research TopicRNA Targeting in CNS TherapeuticsView all 4 articles
Introduction: The U1 small nuclear RNA (snRNA) forms ribonucleoprotein particles (RNPs) such as U1 snRNP and U1-TAF15 snRNP. U1 snRNP is one of the most studied RNPs due to its critical role in pre-mRNA splicing in defining the 5′ splice site (5′ss) of every exon through direct interactions with sequences at exon/intron junctions. Recent reports support the role of U1 snRNP in all steps of transcription, namely initiation, elongation, and termination. Functions of U1-TAF15 snRNP are less understood, though it associates with the transcription machinery and may modulate pre-mRNA splicing by interacting with the 5′ss and/or 5′ss-like sequences within the pre-mRNA. An anti-U1 antisense oligonucleotide (ASO) that sequesters the 5′ end of U1 snRNA inhibits the functions of U1 snRNP, including transcription and splicing. However, it is not known if the inhibition of U1 snRNP influences post-transcriptional regulation of pre-mRNA splicing through deep intronic sequences.
Methods: We examined the effect of an anti-U1 ASO that sequesters the 5′ end of U1 snRNA on transcription and splicing of all internal exons of the spinal muscular atrophy (SMA) genes, SMN1 and SMN2. Our study was enabled by the employment of a multi-exon-skipping detection assay (MESDA) that discriminates against prematurely terminated transcripts. We employed an SMN2 super minigene to determine if anti-U1 ASO differently affects splicing in the context of truncated introns.
Results: We observed substantial skipping of multiple internal exons of SMN1 and SMN2 triggered by anti-U1 treatment. Suggesting a role for U1 snRNP in interacting with deep intronic sequences, early exons of the SMN2 super minigene with truncated introns were resistant to anti-U1 induced skipping. Consistently, overexpression of engineered U1 snRNAs targeting the 5′ss of early SMN1 and SMN2 exons did not prevent exon skipping caused by anti-U1 treatment.
Discussion: Our results uncover a unique role of the U1 snRNA-associated RNPs in splicing regulation executed through deep intronic sequences. Findings are significant for developing novel therapies for SMA based on deep intronic targets.
Alternative splicing is an essential process by which a single gene generates multiple unique transcripts in a cell-specific manner by differential usage of splice sites (Baralle and Giudice, 2017). Alternative splicing of a given exon is subject to dynamic combinatorial control by numerous proteins and cis-elements, including RNA structure (Singh and Singh, 2018; Baralle et al., 2019; Shenasa and Hertel, 2019; Singh et al., 2022). Overexpression of splicing factors and/or mutations of cis-elements are associated with pathogenic conditions, including neurodegenerative diseases and cancer (Tazi et al., 2009; Lee and Rio, 2015; Schilling et al., 2019; Wei et al., 2024). U1 snRNP, an RNA-protein particle (RNP) comprised of U1 snRNA, seven Sm proteins (B, D1, D2, D3, E, F and G) and three U1-specific proteins (U1-70K, U1-A and U1-C), is a critical component of the splicing machinery that regulates both constitutive and alternative splicing (Buratti and Baralle, 2010). U1 snRNP plays an essential role in spliceosome assembly and recognition of the 5′ splice site (5′ss) through both RNA-protein interactions and base-pairing of U1 snRNA with the sequences at the 5′ss defined by the last three nucleotides of the exon and the first six nucleotides of an intron (Du and Rosbash, 2002; Lund and Kjems, 2002; Ptok et al., 2019). Interaction of U1 snRNA with the 5′ss can occur in different registers in the vicinity of the 5′ss with no apparent effect on splicing outcomes (Tan et al., 2016). Growing evidence supports that U1 snRNP can influence the selection of the 5′ss through sequences away from the 5′ss in the absence of the usual recruitment of U1 snRNP at the 5′ss (Singh R. N. and Singh N. N. 2019). Independent of 5′ss selection, U1 snRNP can affect spliceosome assembly by interacting with factors that define the 3′ss (Barabino et al., 1990). Other than spliceosome assembly, U1 snRNP determines the directionality of transcription and transcript length through interactions with both the transcription machinery and the newly synthesized RNA (Venters et al., 2019). There also exists a unique RNP, U1-TAF15 snRNP, encompassing entirely different protein components but containing exactly the same U1 snRNA present in the U1 snRNP (Jobert et al., 2009). U1-TAF15 snRNP and its components have been implicated in transcription and/or splicing regulation (Jobert et al., 2009; Ibrahim et al., 2013). For the sake of simplicity, here we refer to any RNP assembled on U1 snRNA as U1 RNP, including U1 snRNP and U1-TAF15 snRNP. As per the exon definition model of splicing, cross-exon interactions facilitated by recruitment of a single U1 snRNP at the 5′ss play an important role in defining exonic sequences (De Conti et al., 2013). However, limited attention has been paid towards the intron-definition model in which cross-intron interactions could be facilitated by potential recruitment of multiple U1 RNPs at 5′ss-like sequences within intronic sequences. In other words, it is not known if the interaction of U1 RNPs with 5′ss-like sequences within the intron serves as an important initial step towards defining the boundaries of an exon.
Spinal muscular atrophy (SMA) is a leading genetic disease of infants and children (Awano et al., 2014; Ahmad et al., 2016; Wirth et al., 2020). SMA is caused by homozygous deletion of or mutations in the Survival Motor Neuron 1 (SMN1) gene which codes for the essential SMN protein (Singh R. N. et al., 2017). Humans carry a second copy of the SMN gene, SMN2, which fails to compensate for the loss of SMN1 due to predominant skipping of exon 7 (Lorson et al., 1999; Monani et al., 1999a). Transcripts lacking exon 7 of SMN2 produce a truncated protein, SMNΔ7, which is less stable and is only partial functional (Vitte et al., 2007; Burnett et al., 2009; Cho and Dreyfuss, 2010; Gray et al., 2018). Strategies aimed at the restoration of SMN2 exon 7 inclusion are approved therapies for SMA (Singh et al., 2020). For the sake of simplicity, we use SMN1/2 to refer to both SMN1 and SMN2 as these two genes are expressed at the same level and are spliced similarly except for exon 7 (Echaniz-Laguna et al., 1999; Monani et al., 1999b). Multiple factors and cis-elements have been implicated in regulation of SMN1/2 exon 7 splicing (Singh and Singh, 2018). It has been also established that the 5′ss of SMN2 exon 7 is presented within an inhibitory context encompassing several negative cis-elements, including intronic splicing silencer N1 (ISS-N1), a terminal stem-loop structure and an intronic structure facilitated by a long-distance interaction (Singh et al., 2004b, 2006, 2007, 2013; Hua et al., 2008; Beusch et al., 2017; Frederiksen et al., 2021). Interaction of TIA1, an SMA modifying factor, with sequences immediately downstream of the 5′ss of exon 7 of SMN2 has been proposed to enhance the recruitment of U1 snRNP and promote exon 7 inclusion (Singh et al., 2011; Howell et al., 2017a). Consistently, engineered U1 snRNAs (eU1s) with extended base pairing at the 5′ss of exon 7 restore SMN2 exon 7 inclusion (Singh et al., 2007). Also, eU1s targeting intronic sequences located downstream of the 5′ss of exon 7 restore SMN2 exon 7 inclusion, supporting the role of U1 RNP in splice site selection from a distance (Rogalska et al., 2016; Singh et al., 2017a).
In addition to exon 7, exons 3 and 5 of SMN1/2 undergo skipping alone or in combination with other exons, as captured by a multi-exon skipping detection assay (MESDA) (Singh et al., 2012; Seo et al., 2016b). We previously reported that the inhibition of U1 RNP in HeLa cells using an antisense oligonucleotide (ASO) triggers skipping of multiple internal exons of SMN1/2 (Singh et al., 2017a). However, it is not known if this skipping might be tied to intronic sequences located away from the 5′ss. Recently we reported an SMN2 super minigene encompassing SMN2 promoter, all exons and their flanking intronic sequences and the entire 3′ untranslated region (3′UTR) (Ottesen et al., 2024). Despite introns in the super minigene being truncated, SMN2 super minigene showed a nearly identical splicing pattern to that of the endogenous SMN2. Also, inhibition of transcription elongation produced a similar effect on splicing of exons 3 and 7 in the SMN2 super minigene and endogenous SMN1/2, suggesting that kinetic coupling of SMN2 transcription and splicing of exons 3 and 7 is not impacted by intron truncations or the chromatin context specific to the large intronic sequences (Ottesen et al., 2024). At the same time, depletion of DHX9 promoted skipping of exons 3, 4 and 5 in the context of endogenous SMN1/2 but not in the context of the super minigene, underscoring the role of the intronic sequences missing in the context of the super minigene. These results supported that the SMN2 super minigene could be exploited to examine if the intronic sequences missing from the super minigene have significance under specific situations such as the depletion of splicing factors.
Here we compare the effect of U1 RNP inhibition on removal of each intron of SMN1/2 in HeLa, neuronal SH-SY5Y and SMA patient cells. Our results reveal susceptibility of most internal exons to skipping under conditions of U1 RNP inhibition with the effect being more pronounced on the alternatively spliced exons 3, 5 and 7. We employed the SMN2 super minigene to determine if deletion of internal sequences within introns would have any bearing on the ability of U1 RNP inhibition to change splicing of SMN1/2 exons. Surprisingly, we observed the loss of the inhibitory effect of U1 RNP inhibition on splicing of exons flanked by the truncated introns. Our findings support a hypothesis that U1 RNPs promote definition of exons in the context of endogenous SMN1/2 through intronic sequences outside the canonical 5′ss present at the exon/intron junction. Supporting this argument, inclusion of none of the internal exons affected by U1 RNP inhibition were fully restored by eU1s targeting their respective 5′ss. Our results reveal a novel post-transcriptional role of U1 RNPs in general splicing regulation through an intron definition model. Our findings are significant for developing novel SMA therapies through elevating U1 RNP levels so that introns of SMN2 are efficiently excised.
All cell culture reagents and media were obtained from Life Technologies unless otherwise specified. Human cervical adenocarcinoma (HeLa) cells were obtained from the American Type Culture Collection (ATCC) and were grown in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS). SH-SY5Y neuroblastoma cells were obtained from ATCC and grown in a 1:1 mix of minimum essential medium (MEM) and F12 growth medium and supplemented with 10% FBS. GM03813 SMA patient cells were obtained from Coriell Institute of Medical Research and were grown in MEM supplemented with 1X GlutaMAX and 15% FBS. ASOs were obtained from Dharmacon and contained 2′-O-methyl modifications at each base and phosphorothioate backbones. The sequences of oligonucleotides are as follows: 10mer control ASO (non-targeting ASO): UUGCCUUUCU, 11mer anti-U1 ASO: CAGGUAAGUAU. To transfect cells: HeLa and SH-SY5Y cells were counted and seeded at a density of 1 × 105 or 3.5 × 105 cells per well of 24-well plates, respectively. GM03813 cells were counted and seeded at a density of 2.8 × 105 cells per well of 6-well plates. Sixteen hours later, cells were transfected with the indicated plasmids and/or ASOs using Lipofectamine 2000 following the manufacturer’s instructions. All ASO transfections were adjusted to a total concentration of 200 nM using non-targeting 10mer ASO. For co-transfection of the SMN2 super minigene (SMN2Sup) and ASO, we used 200 nM of ASO and 0.2 μg of SMN2Sup. For co-transfections of eU1s and SMN2Sup, we used 0.25 μg eU1 expression construct or pCI-Neo empty vector and 0.25 μg of SMN2Sup. For co-transfections of ASO, eU1, and SMN2Sup, we used 200 nM of ASO, 0.25 μg of eU1 expression construct, and 0.1 μg of SMN2Sup. For co-transfections using SMN2∆I6 minigene and eU1s, we used 0.3 μg of SMN2∆I6 and 0.5 μg of an eU1 expression construct or pCI-Neo empty vector. Six hours after transfection, media containing transfection complexes was replaced with fresh media. Twenty-four hours after transfection, cells were lysed directly in TRIzol reagent (Life Technologies) for RNA isolation.
All primers used in this study were previously described (Singh et al., 2004a; Ottesen et al., 2024). RNA was isolated using TRIzol reagent (Life Technologies) following the manufacturer’s instructions. After isolation, RNA was treated with RQ1 RNase-free DNase (Promega) to remove contaminating DNA, following the manufacturer’s instructions. RNA was then re-purified by phenol:chloroform extraction and ethanol precipitation. To generate cDNA, reverse transcription (RT) was carried out in 5 μL reactions containing 0.5–1.0 μg RNA using Superscript III reverse transcriptase (Life Technologies) following the manufacturer’s instructions. To generate cDNA for qPCR and MESDA, RT reactions were primed with random primers (Promega) and gene-specific primers, respectively.
MESDA was carried out as previously described (Singh et al., 2012) using 5′ end-32P-labeled reverse primer. For SMN2Sup MESDA, the forward primer annealed within the FLAG tag region and reverse primer annealed to the first 25 bases of exon 8, while for endogenous MESDA, the forward primer annealed to the exon 1 region across the ATG start codon, which is interrupted by the FLAG tag in SMN2Sup (Ottesen et al., 2024). Splice isoforms were quantified by densitometric quantification using ImageJ software. For each isoform, percentage inclusion was calculated by dividing the intensity of its band by the total signal in each lane.
Quantitative PCR (qPCR) was carried out using PowerUp SYBR green master mix (Life technologies). Each 20 μL reaction contained 3 μL of 1:40 diluted cDNA (equivalent to 7.5 ng RNA) and 0.6 μM of each primer. Templates for standard curves were generated as previously described (Ottesen et al., 2024). Copies/cell were estimated using the assumption that each HeLa cell contains approximately 30 pg. of RNA. For each experiment, SMN2Sup and endogenous SMN1/2 transcripts were normalized against HPRT by calculating the relative expression of HPRT in each individual sample relative to the average across a given experiment, then dividing each SMN2Sup or SMN1/2 measurement by that value.
5′ss scores were calculated using MaxEntScan (Yeo and Burge, 2004). Putative intronic 5′ss-like sequences were identified using ESEFinder 3.0 and mapped to the SMN1/2 locus similar to Cartegni et al. (2003) and Kaida et al. (2010).
Excel (Microsoft, Version 16.62) was used for all calculations and generation of plots. Data was expressed as mean ± SEM. p values were calculated using an unpaired Student’s t-test. Unless otherwise mentioned, experiments were performed in triplicate, and p values were two-tailed and the level of statistical significance was set as p < 0.05.
In order to determine the effect of inhibition of U1 RNP on transcription and splicing of SMN1/2, we transfected cells with 50 or 200 nM of anti-U1 ASO that sequesters the first 11 nucleotides at the 5′ end of U1 snRNA (Figure 1A). As a control, we used a 10 nucleotide-long non-targeting ASO. We performed this experiment in three cell types: HeLa cells, SH-SY5Y neuroblastoma cells, and GM03813 SMA patient fibroblasts. Of note, the 5′ss of an exon forms an RNA:RNA duplex with the U1 snRNA during the early stages of spliceosome assembly. A strong 5′ss:U1 snRNA duplex is expected to be favorable for defining the 5′ss, particularly at low concentrations of functional U1 snRNP. Each exon of SMN1/2 has a different sequence at the 5′ss that forms a unique 5′ss:U1 snRNA duplex (Figure 1A). Our approach is likely to capture any correlation between the size of the 5′ss:U1 snRNA duplex and exon inclusion under conditions of U1 snRNP inhibition. We employed MESDA to analyze the splicing of all internal exons of SMN1/2. In HeLa cells, both concentrations of anti-U1 ASO decreased the proportion of full-length transcript of SMN1/2, while skipping of multiple exons was increased, especially at 200 nM (Figure 1B). The strongest effect was observed on the alternatively spliced exons 3, 5 and 7 as we captured increased levels of co-skipped transcripts such as ∆5,7, ∆3,7, and ∆3,5,7. We also captured enhanced skipping of constitutive exons 2A, 2B and 4 in different combinations along with skipping of exons 3, 5 and 7. The most prominent short transcript was ∆2A,2B,3,4,5,7 in which only exons 1, 6, and 8 were included. These results supported that the inhibition of U1 snRNP by an anti-U1 ASO was not absolute and still allowed recognition of strong 5′ss. Of note, exons 1 and 6 happen to have the strongest 5′ss:U1 snRNA duplex (8 bp) among all exons of SMN1/2. When we adjusted conditions to capture extremely short transcripts of SMN1/2, we detected a very faint band corresponding to the skipping of all internal exons of SMN1/2 (Supplementary Figure S1). However, this band was observed only when HeLa cells were transfected with the highest concentration of anti-U1 ASO. As compared to HeLa cells, the effect of anti-U1 ASO on splicing of SMN1/2 exons was less pronounced in case of SH-SY5Y and GM03813 cells (Figure 1B). We attribute this to the decreased transfectability of neuronal SH-SY5Y and GM03813 cells compared to HeLa cells, although we cannot rule out that these cell types might have relatively high tolerances for decreased levels of functional U1 RNPs. To measure the impact of U1 RNP inhibition on total SMN1/2 mRNA levels, we performed qPCR using primers annealing to exon 1 and the junction between exons 1 and 2A (Figure 1C). Use of these primers allows capture of a vast majority of spliced isoforms of SMN1/2 as well as any prematurely terminated transcripts that contain at least the first two exons, although variants lacking exon 2A will be missed. Transfection alone caused a slight increase in SMN1/2 levels in HeLa cells, while, surprisingly, there was a concentration-dependent increase in total SMN1/2 levels upon transfection with anti-U1 ASO (Figure 1C, top panel). This trend was also captured in case of SH-SY5Y and GM03813 cells, although the effect was significant only at 200 nM of anti-U1 ASO (Figure 1C, lower two panels). Overall, our results support a model in which skipping of multiple exons was caused by pairing of splice sites separated by large distances, such as between the 5′ss of exon 2B and the 3′ss of exon 6 (Figure 1D).
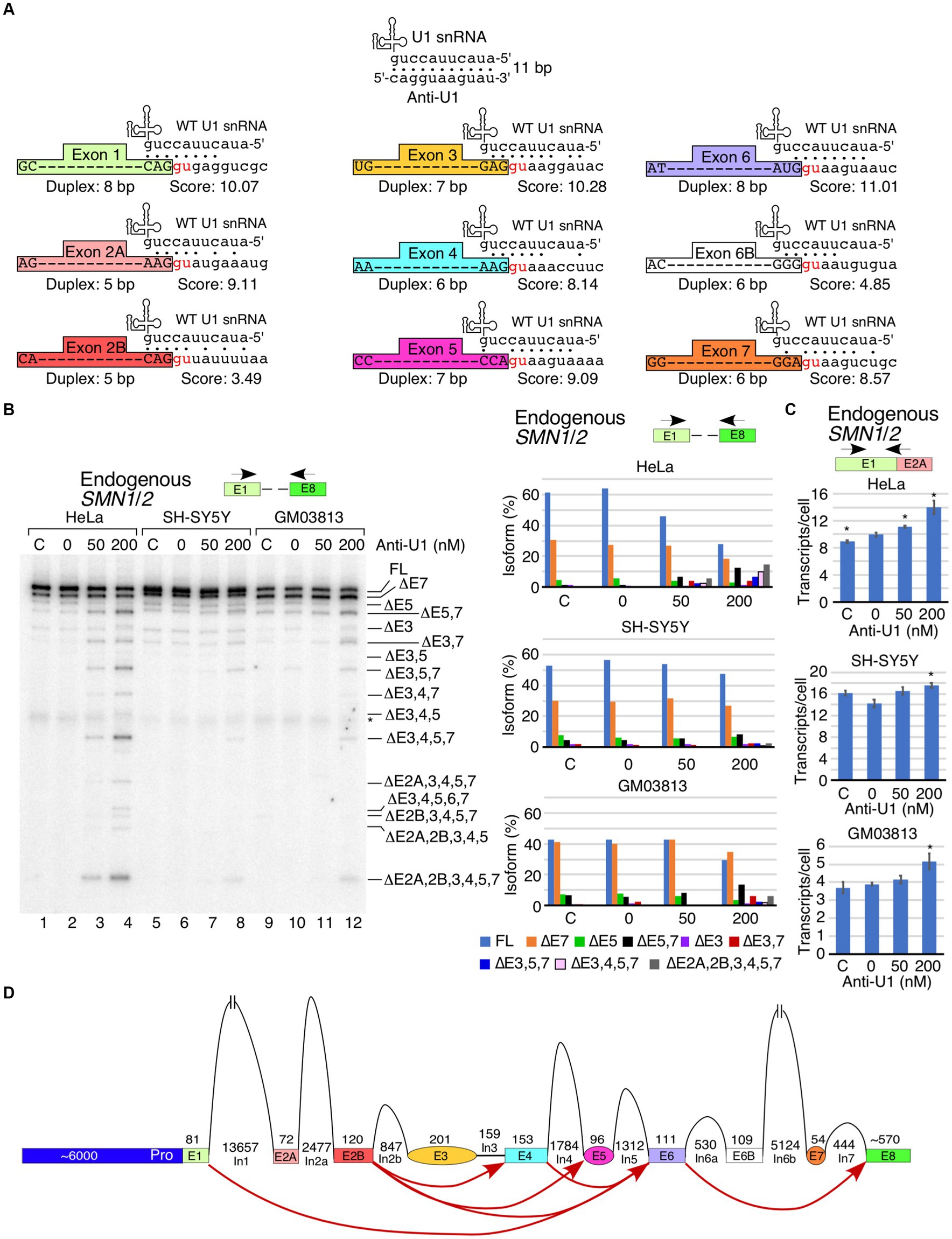
Figure 1. Cell type- and exon-specific effect of U1 RNP inhibition on SMN1/2 transcripts. (A) Diagrammatic overview of WT U1 snRNA and its interactions with anti-U1 ASO or each of the 5′ss of SMN1/2 exons. Exons are shown as different colored boxes. Dots indicate base pairing between U1 snRNA and anti-U1 or 5′ss. GU dinucleotides that make up the first two intronic positions of each 5′ss are shown in red. The lengths of the longest contiguous stretch of base pairs (bp) in each interaction and splice site scores are indicated. “Anti-U1” indicates anti-U1 ASO. (B) Left panel: Splicing pattern of endogenous SMN1/2 transcripts isolated from HeLa, SH-SY5Y, or GM03813 cells transfected with anti-U1 ASO as determined by MESDA. Locations of primers used are indicated above the gel. Cell types and treatments are labeled at the top. “C” indicates untransfected control cells and “0” indicates transfection of nontargeting control ASO. Splice isoforms are labeled at the right side of the gel. “*” indicates a nonspecific background signal. Right panels: Quantification of relative isoform abundance depicted in the left panel. Isoform color coding is indicated at the bottom. Cell type is indicated above each graph. (C) Total transcript level of endogenous SMN1/2 in cells transfected with anti-U1 ASO as determined by qPCR. Locations of primers used are indicated at the top. Cell type is indicated above each graph. Error bars represent the standard error of the mean (SEM), n = 3. *p < 0.05. (D) Diagrammatic overview of the SMN1/2 genes and exon skipping events triggered by anti-U1 ASO. Exons are shown as colored shapes, introns as black lines/broken lines. Red curved arrows denote splicing events triggered by anti-U1 ASO.
We have recently reported an SMN2 super minigene (SMN2Sup) encompassing all internal exons flanked by their intronic sequences but carrying large internal deletions within introns (Figure 2A) (Ottesen et al., 2024). In order to examine the potential role of intronic sequences away from splice sites in U1 RNP-dependent splicing of SMN1/2 exons, we transfected HeLa cells with SMN2Sup and different concentrations of anti-U1 ASO. We monitored splicing using MESDA with primers designed to specifically amplify transcripts generated from either SMN2Sup or endogenous SMN1/2. As expected, increasing concentrations of anti-U1 ASO promoted skipping of the alternatively spliced exons 3, 5 and 7 as well as constitutive exons 2A, 2B and 4 in different combinations in case of endogenous SMN1/2 (Figure 2B). Surprisingly, in case of SMN2Sup, we only observed an increase in skipping of exon 7 and co-skipping of exons 5 and 7. Even the highest concentration of anti-U1 ASO had no effect on splicing of any of the early exons, including the alternatively spliced exon 3, in transcripts generated from SMN2Sup (Figure 2C). We observed a reduction in intensity of MESDA bands for SMN2Sup but not for endogenous SMN1/2, suggesting that the inhibition of U1 RNP had a negative effect on levels of transcripts generated from SMN2Sup which contains shortened introns. This observation was confirmed by the results of qPCR that showed a clear decrease in SMN2Sup transcript levels with the increasing concentrations of anti-U1 ASO (Figure 2C, right panel). In contrast, expression of endogenous SMN1/2 moderately increased at all anti-U1 ASO concentrations, although the effect was statistically significant only at 100 nM of anti-U1 ASO (Figure 2B, lower right panel). Notice that earlier described increase in the levels of endogenous SMN1/2 transcripts upon U1 RNP inhibition was observed in HeLa cells that were not simultaneously transfected with SMN2Sup (Figure 1C). Hence, less impressive rise in the levels of the endogenous SMN1/2 presented in Figure 2B could be attributed to the presence of SMN2Sup transcripts in the same cells.
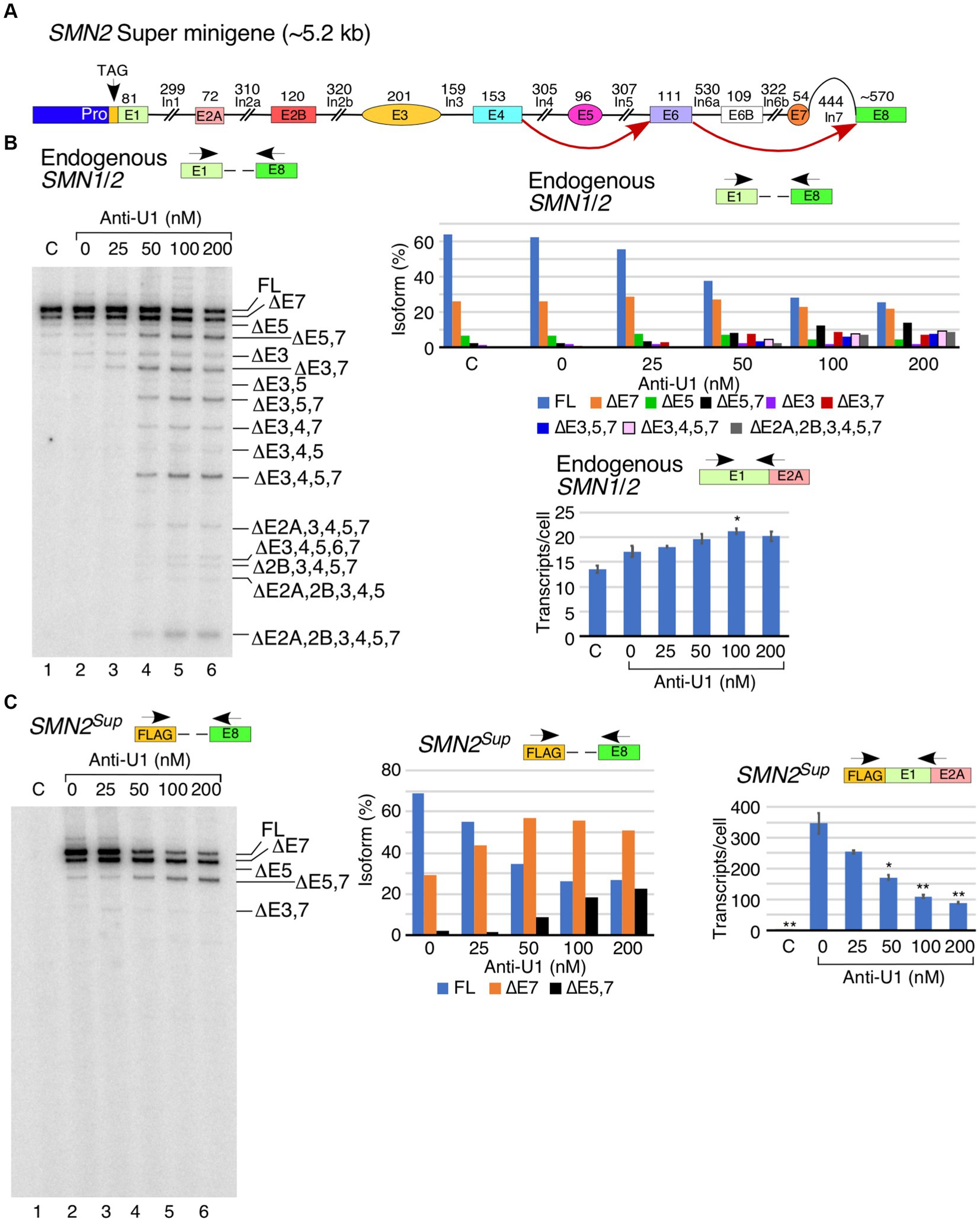
Figure 2. Comparison of the effect of anti-U1 ASO on endogenous SMN1/2 and SMN2 super minigene. (A) Diagrammatic overview of the SMN2 super minigene (SMN2Sup) and exon skipping events triggered by anti-U1 ASO. Exons are shown as colored shapes, introns as black lines/broken lines. Red curved arrows denote splicing events triggered by anti-U1 ASO. (B) Left panel: Splicing pattern of endogenous SMN1/2 transcripts isolated from HeLa cells co-transfected with anti-U1 ASO and SMN2Sup as determined by MESDA. Locations of primers used are indicated above the gel. Anti-U1 treatments are labeled at the top. “C” indicates untransfected control cells and “0” indicates transfection of non-targeting control ASO. Splice isoforms are labeled at the right side of the gel. Upper right panel: Quantification of relative isoform abundance depicted in the left panel. Isoform color coding is indicated at the bottom. Lower right panel: Total transcript level of endogenous SMN1/2 transcripts isolated from HeLa cells co-transfected with anti-U1 ASO and SMN2Sup as determined by qPCR. Locations of primers used are indicated at the top. Error bars represent SEM, n = 3. *p < 0.05. (C) Left panel: Splicing pattern of SMN2Sup transcripts isolated from HeLa cells co-transfected with anti-U1 ASO and SMN2Sup as determined by MESDA. Labeling is the same as in (B). Middle panel: Quantification of relative isoform abundance depicted in the left panel. Isoform color coding is indicated at the bottom. Right panel: Total transcript level of SMN2Sup transcripts isolated from HeLa cells co-transfected with anti-U1 ASO and SMN2Sup as determined by qPCR. Locations of primers used are indicated at the top. Error bars represent SEM, n = 3. *p < 0.05, **p < 0.01.
Having examined the effect of U1 RNP inhibition on splicing of SMN1/2 exons, we next tested the effect of overexpression of eU1s on splicing of each exon of SMN1/2. Prior studies support that increasing the size of the 5′ss:U1 snRNA duplex has an stimulatory effect on inclusion of exons (Singh, 2007; Zychlinski et al., 2009; Hicks et al., 2010; Singh et al., 2017a). We designed eU1s such that the size of the 5′ss:U1 snRNA duplex of the target exon is increased to 11 bp (Figure 3A). We transfected HeLa cells with eU1 snRNA-expressing plasmids along with SMN2Sup and monitored splicing by MESDA (Figure 3B). Compared to control, overexpression of eU1s targeting early exons of SMN1/2, namely eU1-E1, eU1-E2A, eU1-E2B, and eU1-E3, triggered an increase in skipping of exon 7, and to a lesser extent, co-skipping of exons 5 and 7 in SMN2Sup transcripts (Figure 3B, left panel). eU1-E1, eU1-E2B, and eU1-E3 also increased co-skipping of exons 3 and 7 in SMN2Sup transcripts. The fact that an eU1 targeting the 5′ss of exon 3 was somewhat deleterious for its splicing indicates that the nature of the 5′ss of exon 3 does not contribute significantly to its skipping. eU1-E4 and eU1-E6 did not significantly alter splicing of any of the exons of SMN2Sup transcripts, supporting that the recruitment of U1 snRNP at the 5′ss of the constitutively spliced exons 4 and 6 is not a limiting factor for splicing of the neighboring alternatively spliced exons 3, 5 and 7 (Figure 3B, left panel). Surprisingly, although eU1-E5 did not change splicing of exon 5 by itself, it significantly reduced skipping of exon 7 and co-skipping of exons 5 and 7 in SMN2Sup transcripts (Figure 3B, left panel). We have previously shown that a cryptic exon (exon 6B) derived from an Alu element in intron 6 is included in some transcripts of SMN1 and SMN2 (Seo et al., 2016a; Ottesen et al., 2017). Co-transfection of eU1-E6B with SMN2Sup resulted in complete inclusion of exon 6B in full-length transcripts as well as exon 7-skipped and exon 5- and 7-co-skipped splice isoforms generated from SMN2Sup, with no effect on splicing of other exons (Figure 3B, left panel). Overexpression of eU1-E7 triggered almost complete inclusion of exon 7 in SMN2Sup transcripts as demonstrated by the disappearance of the ∆7 band and drastic increase of the full-length (FL) band (Figure 3B, left panel). Consistently, co-skipping of exons 5 and 7 was decreased while skipping of exon 5 alone was increased. Unexpectedly, there was an increase in co-skipping of exons 5, 6, and 7 in SMN2Sup transcripts in the presence of overexpressed eU1-E7.
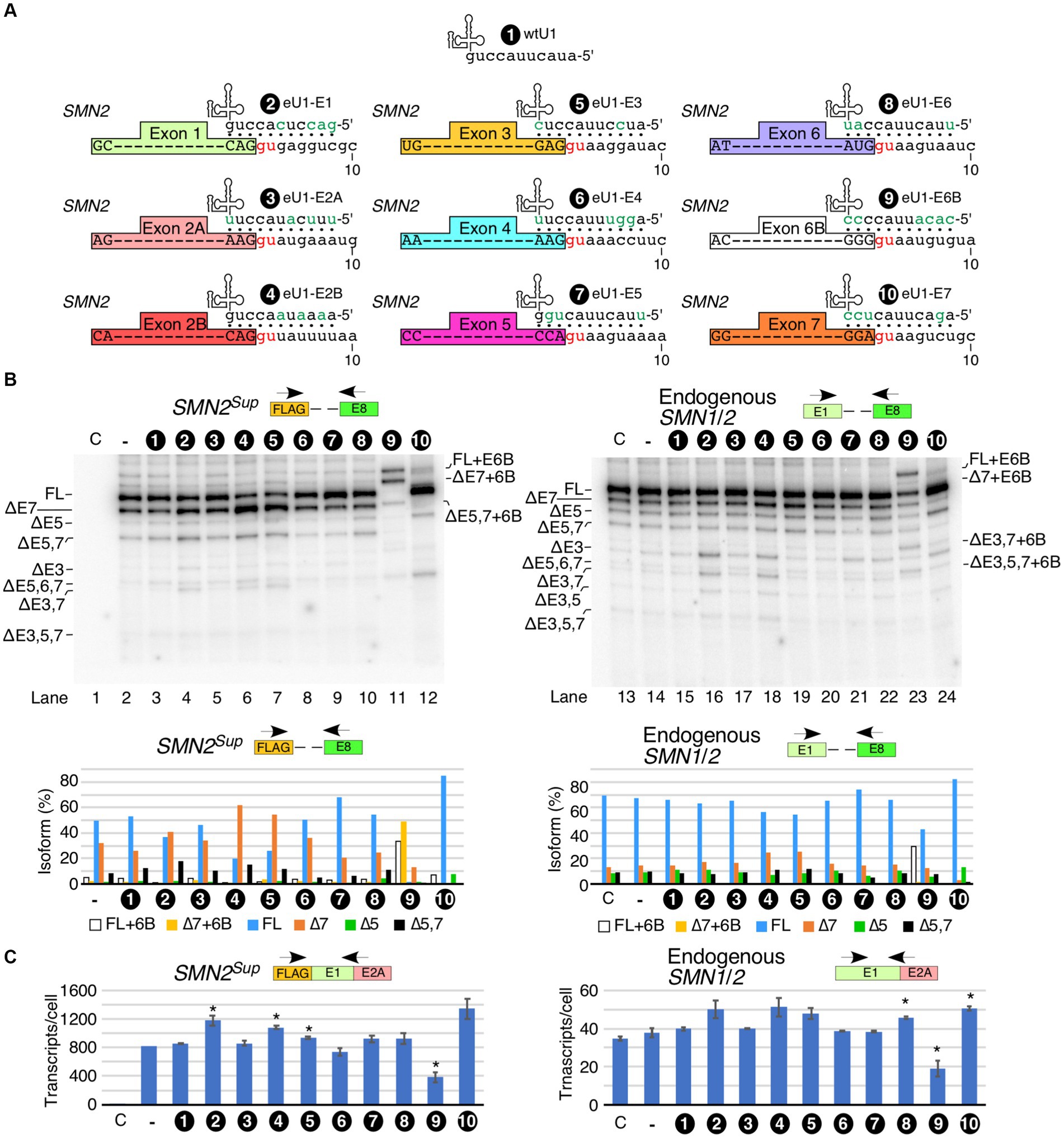
Figure 3. Effect of engineered U1 snRNA overexpression on splicing of SMN1/2 exons. (A) Diagrammatic representation of base pairing formed between eU1s and the 5′ss of SMN1/2 exons. Three nucleotides upstream and eight nucleotides downstream of each GU are shown. The GU dinucleotides are indicated by red letters. Base pairing is marked by black dots; mutated nucleotides within U1 snRNAs are indicated in green. (B) Splicing pattern of SMN2Sup (left panel) and endogenous SMN1/2 (right panel) transcripts isolated from HeLa cells in the presence of the overexpressed eU1s shown in (A) as determined by MESDA. The identity of eU1 constructs is marked at the top of the gel. Splice isoforms are indicated at the left and right sides of each gel. “C” indicates untransfected control cells and “-“indicates transfection of SMN2Sup only. Quantification of the relative amount of the indicated splice products is given in the lower panel as a bar diagram. (C) Total transcript level of SMN2Sup (left panel) and endogenous SMN1/2 (right panel) transcripts isolated from HeLa cells in the presence of eU1 overexpression. Locations of primers used are indicated at the top. Error bars represent SEM, n = 3. *p < 0.05 compared to wtU1 control.
We examined the effects of overexpression of eU1s on splicing of endogenous SMN1/2 in the same samples transfected with SMN2Sup. Overall, the effects of eU1s were less prominent on endogenous SMN1/2 transcripts than on SMN2Sup transcripts, likely due to the presence of a high background of untransfected cells (Figure 3B, right panel). However, we noted increased skipping of exon 3 and co-skipping of exons 3 and 7 caused by overexpression of eU1-E1 and eU1-E2B, and increased skipping of exon 7 triggered by overexpression of eU1s targeting exons 2B and 3 (Figure 3B, right panel). Overexpression of eU1-6B promoted inclusion of exon 6B in full-length transcripts as well as transcripts with skipped exons 3, 5, and 7 in different combinations (Figure 3B, right panel). To capture the non-specific effect of eU1s on splicing of exon 7, we employed the SMN2∆I6 minigene that lacks sequences upstream of exon 6 and carries a large internal deletion within intron 6 (Singh et al., 2004a). As expected, overexpression of eU1-E7 almost completely restored inclusion of exon 7 in transcripts generated from SMN2∆I6 (Supplementary Figure S2). Consistent with the results observed for SMN2Sup, overexpression of eU1-E1 and eU1-E3 increased skipping of exon 7, whereas eU1-E5 promoted inclusion of exon 7 in transcripts generated from SMN2∆I6 (Supplementary Figure S2).
We tested how overexpression of eU1s affected levels of transcripts derived from SMN2Sup. Overexpression of eU1s targeting the 5′ss of exons 1, 2B and 3 showed statistically significant increases in transcript levels (Figure 3C). eU1s targeting the 5′ss of exons 5, 6 and 7 showed increase in transcripts generated from SMN2Sup as well, although the effects were not statistically significant. In case of eU1 targeting the 5′ss of exons 6B, we observed a noticeable decrease in levels of transcripts generated from SMN2Sup. This is likely due to susceptibility of the exon 6B-containing transcripts to NMD as previously reported (Seo et al., 2016a). Effects of eU1s on endogenous SMN1/2 transcript levels were largely similar to those we observed for transcripts produced from SMN2Sup, although the effects were smaller probably due to the high background of untransfected cells (Figure 3C).
We asked whether U1 RNP inhibition by anti-U1 ASO could be counteracted by the overexpression of eU1s targeting specific exons. We based our experiment on the premise that the 11-bp-long duplex formed between eU1 and its target 5′ss would be thermodynamically more stable than the 8-bp or shorter duplex formed between anti-U1 ASO and an eU1. Therefore, we hoped to capture SMN1/2 exons that are vulnerable to skipping due to small size of the duplex formed between the 5′ss and U1 snRNA. Among eU1s that targeted the first five exons, eU1-E3 and eU1-E4 partially neutralized the negative effect of anti-U1 ASO (Figure 4A). These results confirmed that the small size of the duplex formed between the 5′ss and U1 snRNA contributes at least in part towards the skipping of exon 3 and 4 under conditions of inhibition of U1 snRNP. However, our results do not rule out the possibility that the stimulatory effect of eU1s could be due to interaction with the same deep intronic motifs that engage with the wild type U1 RNP. Our results supported that the inhibitory effect of the anti-U1 ASO is not associated with the poor recruitment of the U1 snRNP to the 5′ss of exons 2A and 2B and the effect of inhibition of U1 RNP is likely exerted by intronic sequences away from the splice sites. Of note, considering that the effect of U1 snRNP inhibition on splicing of exons 2A and 2B was not captured in the context of super minigene, we rule out that poor recruitment of eU1s at the 5′ss of exons 2A and 2B is the reason behind their skipping under conditions of inhibition of U1 snRNP (Figure 2). Overexpression of eU1-E3 and eU1-E4 had the most prominent “rescue” effects on ∆3,4,5,7, which is one of the major isoforms produced by the inhibition of U1 RNP (Figure 4A). We also observed a clear reduction in the levels of several other small isoforms, including ∆2A,3,4,5,7, ∆3,4,5,6,7, ∆2B,3,4,5,7, ∆2A,2B,3,4,5, and ∆2A,2B,3,4,5,7 upon overexpression of eU1-E3 and eU1-E4 in the presence of anti-U1 ASO (Figure 4A). Overexpression of eU1-E5 was unable to counteract the inhibitory effect of anti-U1 ASO on splicing of exon 5 of SMN1/2, suggesting that the recruitment of U1 snRNP at the 5′ss of exons 5 is not the limiting factor for splicing and the effect of inhibition of U1 RNP is likely exerted by intronic sequences away from the splice sites (Figure 4B). Splicing of exon 6 is not impacted by the inhibition of U1 RNP. Consistently, overexpression of eU1-E6 had no effect on splicing of exon 6 with or without anti-U1 ASO (Figure 4B). Surprisingly, a strong RNA:RNA duplex formed between the 5′ss of exon 6B and eU1-6B lost most of its ability to enhance exon 6B inclusion in the presence of anti-U1 ASO. These results indicate that the small RNA:RNA duplex formed between the 5′ss and U1 snRNA is one of the major limiting factors for the inclusion of the exon 6B in full-length transcripts of SMN1/2. We observed a partial rescue of exon 7 inclusion by eU1-E7 in the presence of anti-U1 ASO (Figure 4B).
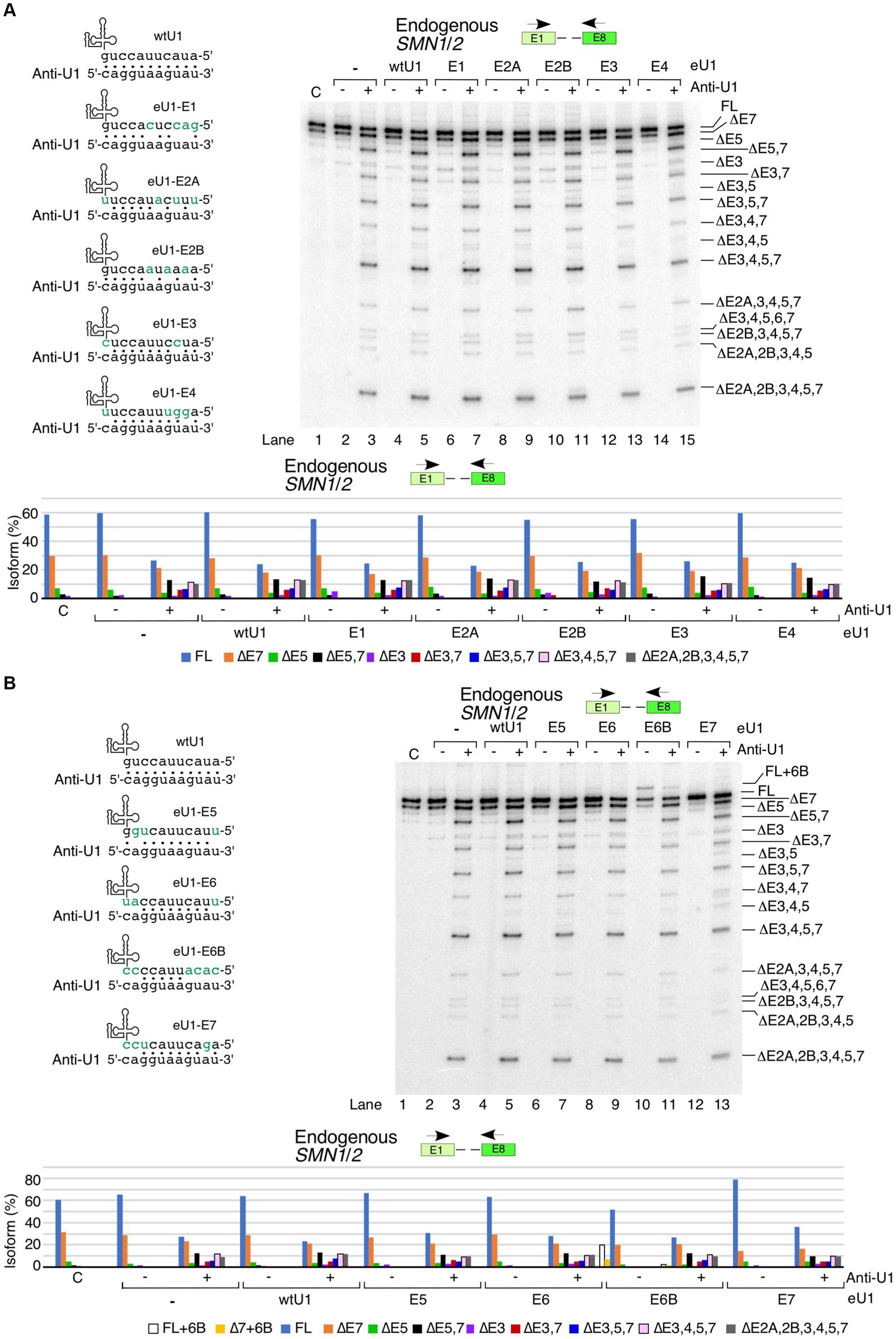
Figure 4. Functional interactions between engineered U1 snRNAs and anti-U1 ASO affecting splicing of endogenous SMN1/2 transcripts. (A) Effect of co-transfection of anti-U1 ASO and eU1s targeting early exons of SMN1/2. Upper left panel: Base pairing between eU1s targeting early exons of SMN1/2 and anti-U1. Coloring and labeling are the same as in Figure 3A. Upper right panel: Splicing pattern of endogenous SMN1/2 isolated from HeLa cells co-transfected with 200 nM anti-U1 or control ASO and plasmids expressing the indicated eU1s, as determined by MESDA. Primer locations are indicated. The target exon of each eU1 and absence (−) or presence (+) of anti-U1 are indicated at the top. “C” – untransfected control. Splice isoforms are labeled at the right side of the gel. Lower panel: Quantification of relative isoform abundance depicted in the top right panel. Isoform color coding is indicated at the bottom. (B) Effect of co-transfection of anti-U1 ASO and eU1s targeting late exons of SMN1/2. All labeling and coloring are the same as in (A).
We performed similar rescue experiments in the context of SMN2Sup by co-transfecting it with anti-U1 ASO and eU1s (Figure 5). As in case of the endogenous SMN1/2 transcripts, eU1s targeting early exons did not prevent the negative effects of anti-U1 ASO on splicing of exons 5 and 7 in SMN2Sup transcripts (Figure 5A). In fact, eU1-E3 had a noticeable additive inhibitory effect on splicing of exons 5 and 7 in the presence of anti-U1 ASO. Interestingly, eU1-E2A and eU1-E2B triggered enhanced skipping of exons 3 and 5 in the presence of anti-U1 ASO, as shown by a slight increase in the levels of ∆3,5 and ∆3,4,5,7 isoforms (Figure 5A). Unlike the effect of eU1-E5 on splicing of the endogenous SMN1/2 transcripts, this eU1 was able to fully counteract the inhibitory effect of anti-U1 ASO on splicing of exon 5 in transcripts derived from SMN2Sup transcripts (Figure 5B). In addition, eU1-E5 reduced the impact of anti-U1 ASO on skipping of exon 7. Likewise, eU1-E6B retained its ability to promote exon 6B inclusion in the context of SMN2Sup transcripts even in the presence of anti-U1 ASO, although it was not able to prevent skipping of exon 7 or co-skipping of exons 5 and 7 (Figure 5B). Of note, due to their similar sizes, exon 7-skipped transcripts and transcripts in which exons 5 and 7 are co-skipped along with exon 6B inclusion (∆5,7 + 6B) are indistinguishable by electrophoresis. Therefore, we confirmed the identity of the product in question by cloning and sequencing (Figure 5B, lane 11). eU1-E7 completely prevented skipping of exon 7 in SMN2Sup transcripts in the presence of anti-U1 ASO, but did not have a strong positive impact on splicing of exon 5 (Figure 5B). However, considering we and others have previously shown that the recruitment of eU1s at cryptic 5′ss downstream of the canonical 5′ss of SMN2 exon 7 promote inclusion of exon 7, we cannot rule out that the stimulatory effect of eU1-E7 on exon 7 splicing is not due to the interaction of eU1-E7 with intronic motifs downstream of the 5′ss of exon 7.
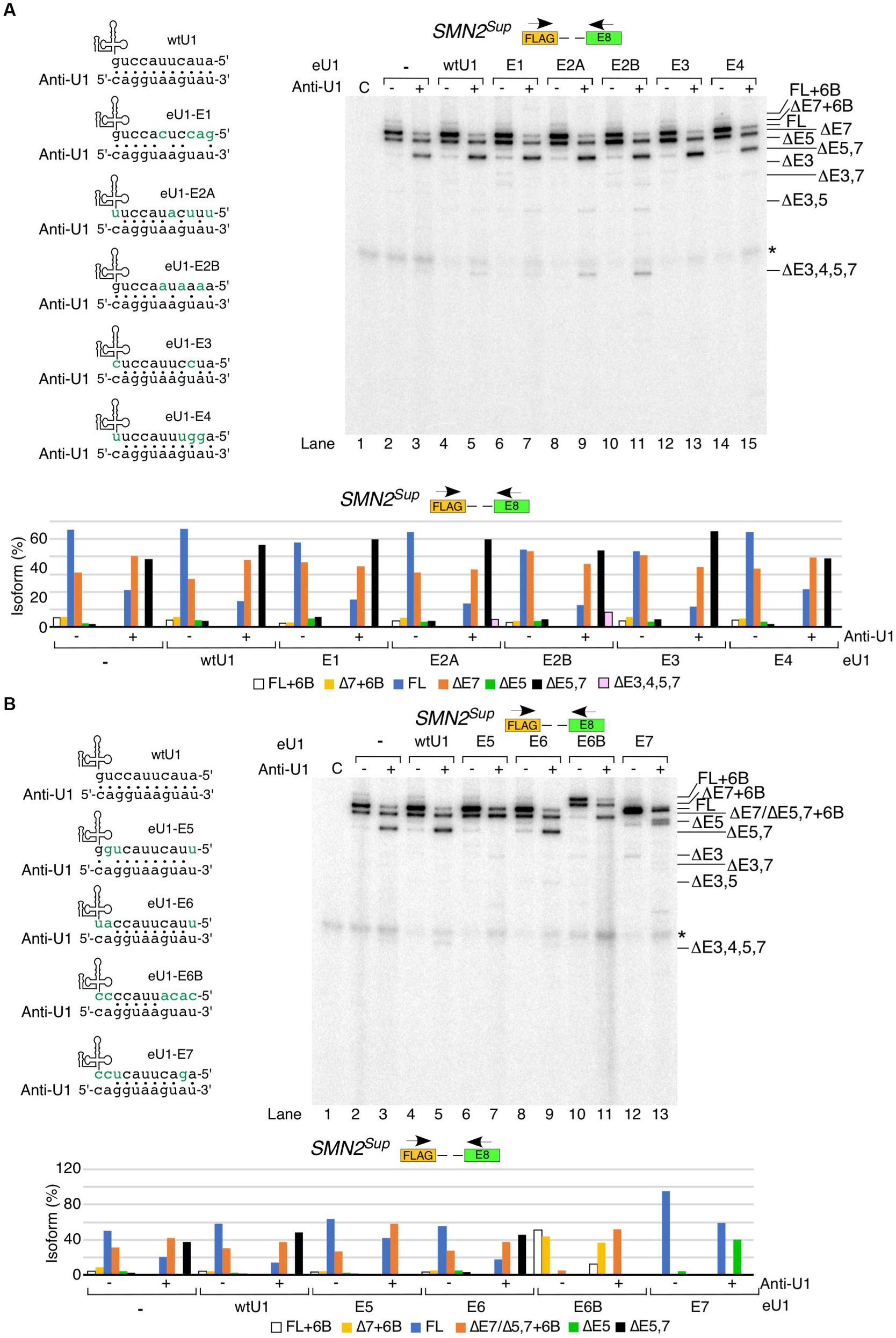
Figure 5. Functional interactions between engineered U1 snRNAs and anti-U1 ASO affecting splicing of SMN2Sup transcripts. (A) Effect of co-transfection of anti-U1 ASO and eU1s targeting early exons of SMN2Sup. Upper left panel: Base pairing between eU1s targeting early exons of SMN1/2 and anti-U1. Coloring and labeling are the same as in Figure 3A. Upper right panel: Splicing pattern of SMN2Sup transcripts isolated from HeLa cells co-transfected with 200 nM anti-U1 or control ASO, plasmids expressing the indicated eU1s, and SMN2Sup, as determined by MESDA. Primer locations are indicated. The target exon of each eU1 and absence (−) or presence (+) of anti-U1 are indicated at the top. “C” – untransfected control. Splice isoforms are labeled at the right side of the gel. “*” indicates a nonspecific background signal. Lower panel: Quantification of relative isoform abundance depicted in the top left panel. Isoform color coding is indicated at the bottom. (B) Effect of co-transfection of anti-U1 ASO and eU1s targeting late exons of SMN2Sup. All labeling and coloring are the same as in (A).
Here we report a novel role of U1 RNP in transcription and splicing regulation through intronic sequences located away from the 5′ss. Unlike previous studies that captured the effect of strong U1 RNP inhibition using a 25-mer ASO on transcription and splicing during the process of transcription elongation (Kaida et al., 2010; Berg et al., 2012), we have focused on the effect of U1 RNP inhibition on splicing events that likely occur post-transcriptionally. We used a shorter 11-mer ASO to specifically block the critical 5′-end of U1 snRNA while not perturbing the assembly of the U1 snRNP. Use of shorter ASO was also aimed at reducing the off-target effects. Of note, longer ASOs are known to produce more off-target effects than shorter ASOs (Scharner et al., 2020; Ottesen et al., 2021). Our study was enabled by the employment of MESDA that specifically captured the relative abundance of all spliced transcripts containing the first and the last annotated exons. Our approach eliminated the complex analysis of short transcripts generated upon U1 snRNP inhibition due to random pre-mature cleavage and polyadenylation. Inhibition of U1 snRNP led to the enhanced skipping of almost all exons in different combinations. We observed similar effects in all cell types examined, although the effect was more pronounced in HeLa cells (Figure 1). This is likely due to relatively high sensitivity of HeLa cells to U1 RNP inhibition and/or an efficient transfection of ASO used for the U1 snRNP inhibition. Notably, inhibition of U1 RNP caused a slight increase in the levels of endogenous SMN1/2 transcripts as measured by qPCR using primers that annealed to early exons (Figure 1C). Inhibition of U1 RNP is associated with premature transcription termination (Di et al., 2019). Therefore, it is likely that U1 RNP inhibition leads to an increase in levels of the only truncated transcripts generated from endogenous SMN1/2. Suppression of transcription has been linked to increased association of U1-TAF15 snRNP with RNA polymerase II (Jobert et al., 2009). Hence, it is likely that anti-U1 ASO decreases the association of U1-TAF15 snRNP with RNA polymerase II, leading to the enhanced transcription of SMN1/2 we captured in this study.
Based on the profile of endogenous SMN1/2 transcripts affected by U1 RNP inhibition, skipping of the alternative exons 3, 5 and 7 appeared to have occurred largely independently of each other (Figure 1B). Consistently, we detected different combinations of co-skipping that involved exons 3, 5, and 7. We detected skipping of exon 4 along with skipping of exons 3 and 5, suggesting that the co-skipping of exons 3, 4 and 5 is a single event enabled by pairing of the 5′ss of exon 2B with the 3′ss of exon 6 (Figures 1D, 6). We also observed a prominent transcript lacking the first five internal exons. We attribute generation of this transcript to pairing of the 5′ss of exon 1 with the 3′ss of exon 6 (Figures 1D, 6). It appears that the co-skipping of exons 2A, 2B, 3, 4 and 5 competes with co-skipping of exons 3, 4 and 5 since the 3′ss of exon 6 is used for both of these events. We observed very small but detectable levels of transcripts lacking all internal exons, suggesting that pairing of the 5′ss of exon 1 with the 3′ss of exon 8 of SMN1/2 pre-mRNA is a rare event with or without U1 RNP inhibition (Supplementary Figure S1). The most abundant short transcript produced upon U1 RNP inhibition harbored exon 6, suggesting that the 3′ss and/or 5′ss of exon 6 are the strongest splice sites among all internal exons of SMN1/2. Consistently, the 5′ss of exon 6 forms the longest (8 bp) RNA:RNA duplex with U1 snRNA as compared to other 5′ss of internal exons of SMN1/2 (Figure 1A). Considering inclusion of exon 6 was not affected under the conditions of inhibition of U1 snRNP supports that the concentration of the residual functional U1 snRNP was sufficient to promote exon 6 inclusion through the relatively strong RNA:RNA duplex formed between U1 snRNA and the 5′ss of exon 6. Of note, U1 snRNP is not absolutely required for splicing of certain exons (Fukumura et al., 2009). Hence, it remains a possibility that the splicing of exon 6 of SMN1/2 belongs to the category of exons that are not regulated by U1 snRNP.
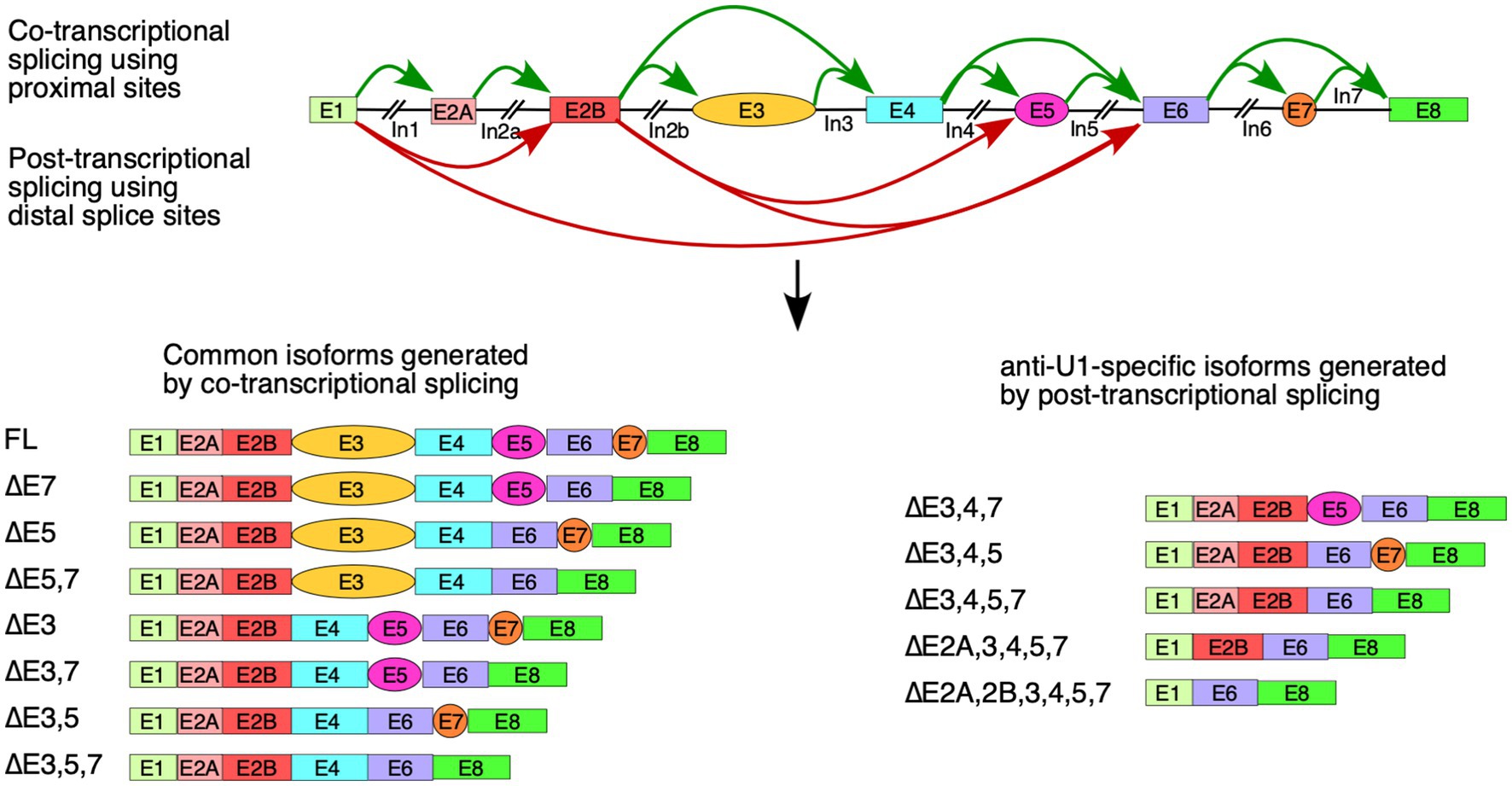
Figure 6. Model of co-transcriptional and delayed splicing of SMN1/2 exons. Diagrammatic overview of the SMN1/2 genes. Exons are shown as colored boxes and introns as black lines/broken lines. Green curved arrows indicate splice site pairings between nearby exons supported by co-transcriptional splicing model. Red curved arrows indicate splice site pairings using distal splice sites that are triggered by anti-U1 ASO. Graphical representations of major splicing products are shown below.
U1 snRNP interacts directly with the elongating polymerase II and can remain tethered throughout transcription of the downstream introns (Leader et al., 2021; Zhang et al., 2021). This allows for efficient linear splicing as the proximal 3′ss to a given 5′ss is brought in very close proximity during transcription, allowing for immediate formation of the spliceosomal complexes. Anti-U1 ASO appears to prevent or interfere with such co-transcriptional splicing, particularly that of introns 1 and 2B, allowing for co-skipping of exons 2A through 5 or exons 3 through 5 (Figure 6). This may be due to a delayed interaction of U1 snRNP with its target 5′ss, preventing it from forming a functional complex with elongating polymerase II, or it may be due to disruption of the U1 snRNP-polymerase II complex during transcription of long introns, discouraging co-transcriptional splicing. Future studies will show whether treatment with anti-U1 ASO affects the levels of SMN1/2 circular RNAs (Ottesen et al., 2019), which also require interactions between introns separated by multiple exons.
Employment of the SMN2 super minigene allowed evaluation of the effect of U1 RNP inhibition on splicing of all internal exons in the context of large deletions in the middle of introns. Surprisingly, U1 RNP inhibition did not impact the splicing of any of the first five exons (Figure 2C), supporting that the limited supply of U1 RNP does not influence the definition of these early exons when flanked by short introns. However, splicing of the alternatively spliced exon 5 that was also flanked by the relatively short introns was adversely impacted by U1 RNP inhibition. Splicing of the alternatively spliced exon 7 that was flanked by the relatively large introns was adversely impacted as well. Our findings may support a novel role of U1 RNP in splicing regulation through an intron definition model, in which U1 RNP is recruited at multiple sites within introns allowing cross-intron communication that favors pairing of the 5′ss with the adjacent 3′ss. Consistently, all of the introns of SMN1/2 except for the relatively short introns 2B, 3 and 7 contain at least one putative GU-containing-5′ss-like sequence that may serve as a binding site for U1 RNP (Supplementary Figure S3). Interaction of U1 snRNP with intronic sequences that may not resemble putative GU-containing-5′ss but still capable of forming RNA:RNA duplex is possible (Parada et al., 2014; Erkelenz et al., 2018). However, distribution of such sequences is not accurately predictable. Intronic sequences of SMN1/2 harbor inverted Alu repeats capable of forming inhibitory secondary structures that are unwound by DHX9, an RNA helicase (Ottesen et al., 2019; Singh N. N. and Singh N. R. 2019). Consistently, depletion of DHX9 promotes skipping of internal exons in the context of the endogenous SMN1/2 transcripts, but not in transcripts derived from the SMN2 super minigene carrying large intronic deletions and, hence, lacks Alu elements (Ottesen et al., 2024). Therefore, it is likely that the recruitment of U1 RNPs within intronic sequences abrogates secondary structures formed by the inverted Alu repeats.
Interactions of U1 RNP with chromatin structure impacts all steps of transcription, including initiation, elongation and termination (Chiu et al., 2018; Fiszbein et al., 2019; Zhang et al., 2021; Mimoso and Adelman, 2023). Chromatin is also formed on plasmid DNA, although its nature is dictated by the plasmid size (Shao et al., 2019). Unlike in the case of the endogenous SMN1/2 transcripts, inhibition of U1 RNP significantly decreased the levels of the SMN2 transcripts generated from the super minigene (Figure 2C). This could be due to adverse effects on one or more steps associated with transcription, including initiation, elongation and termination. Of note, our super minigene contains a short promoter encompassing only the most highly utilized transcription start sites. It is possible that these transcription start sites are differentially susceptible to inhibition of U1 RNPs. It is also possible that inhibition of U1 RNP leads to an increase in the rate of premature termination of transcription during initiation and/or promoter proximal pause (Kamieniarz-Gdula and Proudfoot, 2019). Future studies employing gene-editing based approaches would reveal any correlation between the transcription start site and rate of transcription termination during promoter proximal pause of SMN1/2. Such studies may also uncover the effect of the 5′UTR on selection of the first exon and vice versa as recently reported (Fiszbein et al., 2019). We have recently shown that inhibition of transcription elongation triggers skipping of exon 3 in the context of both the super minigene and endogenous SMN1/2 (Ottesen et al., 2024). Considering we did not observe skipping of exon 3 in the context of SMN2Sup-derived transcripts under the conditions of U1 RNP inhibition, we rule out the role of transcription elongation inhibition as a potential mechanism behind reduced levels of SMN2 transcripts generated from the super minigene. Poor transcription initiation and/or premature termination of transcription appears to be the cause of reduction in the levels of transcripts derived from SMN2Sup under the conditions of U1 RNP inhibition.
For more than two decades, researchers have focused on SMN2 exon 7 inclusion as one of the primary avenues for the treatment of SMA (Singh et al., 2017b, 2020). New ideas of combined therapies with multiple compounds affecting splicing and transcription of SMN2 are being advanced to improve the efficacy of existing therapies (Oh et al., 2020; Marasco et al., 2022; Ottesen et al., 2023; Ottesen and Singh, 2024). Our finding that U1 RNPs could affect splicing outcomes through intronic sequences located away from the splice sites opens up yet additional avenues for SMA therapy. Considering the SMN complex is involved in snRNP biogenesis, even a slight increase in SMN production would elevate the levels of U1 RNP, which in turn would further increase the levels of SMN (Jodelka et al., 2010). Of note, the SMN complex is also proposed to be involved in disassembly of snRNPs and assembly of U1-TAF15 snRNP (Chari et al., 2008). However, the role of U1-TAF15 snRNP biogenesis in SMA and/or other pathological conditions has not yet been investigated. An inhibitory sequence located in the middle of intron 7 is known to modulate SMN2 exon 7 splicing (Singh et al., 2010, 2013; Howell et al., 2017b). Future studies will reveal how U1 RNP-responsive deep intronic sequences within SMN2 could be exploited to enhance SMN levels. To a broader significance, our findings uncover a novel role of U1 RNP in splicing regulation that is independent of spliceosome assembly and intimately tied to deep intronic sequences. Mutations of U1 snRNA and/or misregulation of U1 RNP is associated with many pathological conditions, including amyotrophic lateral sclerosis, Alzheimer’s disease, and cancer (Yu et al., 2015; Bai, 2018; Shuai et al., 2019; Suzuki et al., 2019; Oh et al., 2020). Our findings may provide a mechanistic basis as to how low levels of U1 RNPs may impact splicing of specific genes due to the presence of their responsive cis-elements within deep intronic sequences. Many variants of U1 snRNAs are expressed in humans (O'Reilly et al., 2013). Future experiments will determine if some of the effects of inhibition of U1 snRNP is due to inhibition of the less expressed variants of U1 snRNAs. Overexpression of U1 snRNP is also associated with pathological conditions (Cheng et al., 2017, 2018; Kumari et al., 2019). It would be of interest to see if intronic sequences away from the 5′ss engage in aberrant splicing under conditions of the overexpression of U1 snRNP. An intronic mutation within the ATM gene leading to the abrogation of a putative U1 snRNA binding site away from the 5′ss of the upstream exon is associated with ataxia-telangiectasia (Pagani et al., 2002). Therefore, we envision many scenarios in which deep intronic mutations associated with the interaction with U1 snRNP could lead to pathological conditions.
The original contributions presented in the study are included in the article/Supplementary material, further inquiries can be directed to the corresponding author.
EO: Conceptualization, Formal analysis, Investigation, Validation, Writing – original draft, Writing – review & editing. NS: Writing – original draft, Writing – review & editing, Formal analysis, Investigation, Validation. JS: Investigation, Writing – original draft, Writing – review & editing. RS: Conceptualization, Funding acquisition, Supervision, Writing – original draft, Writing – review & editing, Project administration, Resources.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was supported by grants from the National Institutes of Health R01 NS055925 to RS.
Authors acknowledge Dr. Diou Luo for critical reading of the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnins.2024.1412893/full#supplementary-material
Ahmad, S., Bhatia, K., Kannan, A., and Gangwani, L. (2016). Molecular mechanisms of neurodegeneration in spinal muscular atrophy. J. Exp. Neurosci. 10, JEN.S33122–JEN.S33149. doi: 10.4137/jen.s33122
Awano, T., Kim, J. K., and Monani, U. (2014). Spinal muscular atrophy: journeying from bench to bedside. Neurotherapeutics 11, 786–795. doi: 10.1007/s13311-014-0293-y
Bai, B. (2018). U1 snRNP alteration and neuronal cell cycle reentry in Alzheimer disease. Front. Aging Neurosci. 10:75. doi: 10.3389/fnagi.2018.00075
Barabino, S. M., Blencowe, B. J., Ryder, U., Sproat, B. S., and Lamond, A. I. (1990). Targeted snRNP depletion reveals an additional role for mammalian U1 snRNP in spliceosome assembly. Cell 63, 293–302. doi: 10.1016/0092-8674(90)90162-8
Baralle, F. E., and Giudice, J. (2017). Alternative splicing as a regulator of development and tissue identity. Nat. Rev. Mol. Cell Biol. 18, 437–451. doi: 10.1038/nrm.2017.27
Baralle, F. E., Singh, R. N., and Stamm, S. (2019). RNA structure and splicing regulation. Biochim. Biophys. Acta Gene Regul. Mech. 1862:194448. doi: 10.1016/j.bbagrm.2019.194448
Berg, M. G., Singh, L. N., Younis, I., Liu, Q., Pinto, A. M., Kaida, D., et al. (2012). U1 snRNP determines mRNA length and regulates isoform expression. Cell 150, 53–64. doi: 10.1016/j.cell.2012.05.029
Beusch, I., Barraud, P., Moursy, A., Cléry, A., and Allain, F. H. T. (2017). Tandem hnRNP A1 RNA recognition motifs act in concert to repress the splicing of survival motor neuron exon 7. eLife 6:35. doi: 10.7554/eLife.25736
Buratti, E., and Baralle, D. (2010). Novel roles of U1 snRNP in alternative splicing regulation. RNA Biol. 7, 412–419. doi: 10.4161/rna.7.4.12153
Burnett, B. G., Muñoz, E., Tandon, A., Kwon, D. Y., Sumner, C. J., and Fischbeck, K. H. (2009). Regulation of SMN protein stability. Mol. Cell. Biol. 29, 1107–1115. doi: 10.1128/mcb.01262-08
Cartegni, L., Wang, J. H., Zhu, Z. W., Zhang, M. Q., and Krainer, A. R. (2003). ESEfinder: a web resource to identify exonic splicing enhancers. Nucleic Acids Res. 31, 3568–3571. doi: 10.1093/nar/gkg616
Chari, A., Golas, M. M., Klingenhäger, M., Neuenkirchen, N., Sander, B., Englbrecht, C., et al. (2008). An assembly chaperone collaborates with the SMN complex to generate spliceosomal SnRNPs. Cell 135, 497–509. doi: 10.1016/j.cell.2008.09.020
Cheng, Z., Du, Z. Q., Zhai, B. H., Yang, Z., and Zhang, T. (2018). U1 small nuclear RNA overexpression implicates autophagic-lysosomal system associated with AD. Neurosci. Res. 136, 48–55. doi: 10.1016/j.neures.2018.01.006
Cheng, Z., Shang, Y. C., Gao, S., and Zhang, T. (2017). Overexpression of U1 snRNA induces decrease of U1 spliceosome function associated with Alzheimer's disease. J. Neurogenet. 31, 337–343. doi: 10.1080/01677063.2017.1395425
Chiu, A. C., Suzuki, H. I., Wu, X., Mahat, D. B., Kriz, A. J., and Sharp, P. A. (2018). Transcriptional pause sites delineate stable nucleosome-associated premature polyadenylation suppressed by U1 snRNP. Mol. Cell 69, 648–663.e7. doi: 10.1016/j.molcel.2018.01.006
Cho, S. C., and Dreyfuss, G. (2010). A degron created by SMN2 exon 7 skipping is a principal contributor to spinal muscular atrophy severity. Genes Dev. 24, 438–442. doi: 10.1101/gad.1884910
De Conti, L., Baralle, M., and Buratti, E. (2013). Exon and intron definition in pre-mRNA splicing. Wiley Interdiscip. Rev. RNA 4, 49–60. doi: 10.1002/wrna.1140
Di, C., So, B. R., Cai, Z., Arai, C., Duan, J., and Dreyfuss, G. (2019). U1 snRNP Telescripting roles in transcription and its mechanism. Cold Spring Harb. Symp. Quant. Biol. 84, 115–122. doi: 10.1101/sqb.2019.84.040451
Du, H., and Rosbash, M. (2002). The U1 snRNP protein U1C recognizes the 5′ splice site in the absence of base pairing. Nature 419, 86–90. doi: 10.1038/nature00947
Echaniz-Laguna, A., Miniou, P., Bartholdi, D., and Melki, J. (1999). The promoters of the survival motor neuron gene (SMN) and its copy (SMNc) share common regulatory elements. Am. J. Hum. Genet. 64, 1365–1370. doi: 10.1086/302372
Erkelenz, S., Theiss, S., Kaisers, W., Ptok, J., Walotka, L., Müller, L., et al. (2018). Ranking noncanonical 5′ splice site usage by genome-wide RNA-seq analysis and splicing reporter assays. Genome Res. 28, 1826–1840. doi: 10.1101/gr.235861.118
Fiszbein, A., Krick, K. S., Begg, B. E., and Burge, C. B. (2019). Exon-mediated activation of transcription starts. Cell 179, 1551–1565.e17. doi: 10.1016/j.cell.2019.11.002
Frederiksen, S. B., Holm, L. L., Larsen, M. R., Doktor, T. K., Andersen, H. S., Hastings, M. L., et al. (2021). Identification of SRSF10 as a regulator of SMN2 ISS-N1. Hum. Mutat. 42, 246–260. doi: 10.1002/humu.24149
Fukumura, K., Taniguchi, I., Sakamoto, H., Ohno, M., and Inoue, K. (2009). U1-independent pre-mRNA splicing contributes to the regulation of alternative splicing. Nucleic Acids Res. 37, 1907–1914. doi: 10.1093/nar/gkp050
Gray, K. M., Kaifer, K. A., Baillat, D., Wen, Y., Bonacci, T. R., Ebert, A. D., et al. (2018). Self-oligomerization regulates stability of survival motor neuron protein isoforms by sequestering an SCFSlmb degron. Mol. Biol. Cell 29, 96–110. doi: 10.1091/mbc.E17-11-0627
Hicks, M. J., Mueller, W. F., Shepard, P. J., and Hertel, K. J. (2010). Competing upstream 5′ splice sites enhance the rate of proximal splicing. Mol. Cell. Biol. 30, 1878–1886. doi: 10.1128/mcb.01071-09
Howell, M. D., Ottesen, E. W., Singh, N. N., Anderson, R. L., Seo, J., Sivanesan, S., et al. (2017a). TIA1 is a gender-specific disease modifier of a mild mouse model of spinal muscular atrophy. Sci. Rep. 7:7183. doi: 10.1038/s41598-017-07468-2
Howell, M. D., Ottesen, E. W., Singh, N. N., Anderson, R. L., and Singh, R. N. (2017b). Gender-specific amelioration of SMA phenotype upon disruption of a deep Intronic structure by an oligonucleotide. Mol. Ther. 25, 1328–1341. doi: 10.1016/j.ymthe.2017.03.036
Hua, Y., Vickers, T. A., Okunola, H. L., Bennett, C. F., and Krainer, A. R. (2008). Antisense masking of an hnRNP A1/A2 intronic splicing silencer corrects SMN2 splicing in transgenic mice. Am. J. Hum. Genet. 82, 834–848. doi: 10.1016/j.ajhg.2008.01.014
Ibrahim, F., Maragkakis, M., Alexiou, P., Maronski, M. A., Dichter, M. A., and Mourelatos, Z. (2013). Identification of in vivo, conserved, TAF15 RNA binding sites reveals the impact of TAF15 on the neuronal transcriptome. Cell Rep. 3, 301–308. doi: 10.1016/j.celrep.2013.01.021
Jobert, L., Pinzón, N., Van Herreweghe, E., Jády, B. E., Guialis, A., Kiss, T., et al. (2009). Human U1 snRNA forms a new chromatin-associated snRNP with TAF15. EMBO Rep. 10, 494–500. doi: 10.1038/embor.2009.24
Jodelka, F. M., Ebert, A. D., Duelli, D. M., and Hastings, M. L. (2010). A feedback loop regulates splicing of the spinal muscular atrophy-modifying gene, SMN2. Hum. Mol. Genet. 19, 4906–4917. doi: 10.1093/hmg/ddq425
Kaida, D., Berg, M. G., Younis, I., Kasim, M., Singh, L. N., Wan, L., et al. (2010). U1 snRNP protects pre-mRNAs from premature cleavage and polyadenylation. Nature 468, 664–668. doi: 10.1038/nature09479
Kamieniarz-Gdula, K., and Proudfoot, N. J. (2019). Transcriptional control by premature termination: a forgotten mechanism. Trends Genet. 35, 553–564. doi: 10.1016/j.tig.2019.05.005
Kumari, E., Shang, Y. C., Cheng, Z., and Zhang, T. (2019). U1 snRNA over-expression affects neural oscillations and short-term memory deficits in mice. Cogn. Neurodyn. 13, 313–323. doi: 10.1007/s11571-019-09528-x
Leader, Y., Maor, G. L., Sorek, M., Shayevitch, R., Hussein, M., Hameiri, O., et al. (2021). The upstream 5′ splice site remains associated to the transcription machinery during intron synthesis. Nat. Commun. 12:4545. doi: 10.1038/s41467-021-24774-6
Lee, Y., and Rio, D. C. (2015). “Mechanisms and regulation of alternative pre-mRNA splicing” in Annual review of biochemistry. ed. R. D. Kornberg (Palo Alto, CA: Palo Alto Annual Reviews), 291–323.
Lorson, C. L., Hahnen, E., Androphy, E. J., and Wirth, B. (1999). A single nucleotide in the SMN gene regulates splicing and is responsible for spinal muscular atrophy. Proc. Natl. Acad. Sci. USA 96, 6307–6311. doi: 10.1073/pnas.96.11.6307
Lund, M., and Kjems, J. (2002). Defining a 5′ splice site by functional selection in the presence and absence of U1 snRNA 5′ end. RNA 8, 166–179. doi: 10.1017/s1355838202010786
Marasco, L. E., Dujardin, G., Sousa-Luís, R., Liu, Y. H., Stigliano, J. N., Nomakuchi, T., et al. (2022). Counteracting chromatin effects of a splicing-correcting antisense oligonucleotide improves its therapeutic efficacy in spinal muscular atrophy. Cell 185, 2057–2070.e15. doi: 10.1016/j.cell.2022.04.031
Mimoso, C. A., and Adelman, K. (2023). U1 snRNP increases RNA pol II elongation rate to enable synthesis of long genes. Mol. Cell 83, 1264–1279.e10. doi: 10.1016/j.molcel.2023.03.002
Monani, U. R., Lorson, C. L., Parsons, D. W., Prior, T. W., Androphy, E. J., Burghes, A. H. M., et al. (1999a). A single nucleotide difference that alters splicing patterns distinguishes the SMA gene SMN1 from the copy gene SMN2. Hum. Mol. Genet. 8, 1177–1183. doi: 10.1093/hmg/8.7.1177
Monani, U. R., McPherson, J. D., and Burghes, A. H. (1999b). Promoter analysis of the human centromeric and telomeric survival motor neuron genes (SMNC and SMNT). Biochim. Biophys. Acta 1445, 330–336. doi: 10.1016/s0167-4781(99)00060-3
Oh, J. M., Venters, C. C., Di, C., Pinto, A. M., Wan, L., Younis, I., et al. (2020). U1 snRNP regulates cancer cell migration and invasion in vitro. Nat. Commun. 11:1. doi: 10.1038/s41467-019-13993-7
O'Reilly, D., Dienstbier, M., Cowley, S. A., Vazquez, P., Drozdz, M., Taylor, S., et al. (2013). Differentially expressed, variant U1 snRNAs regulate gene expression in human cells. Genome Res. 23, 281–291. doi: 10.1101/gr.142968.112
Ottesen, E. W., Luo, D., Seo, J., Singh, N. N., and Singh, R. N. (2019). Human survival motor neuron genes generate a vast repertoire of circular RNAs. Nucleic Acids Res. 47, 2884–2905. doi: 10.1093/nar/gkz034
Ottesen, E. W., Luo, D., Singh, N. N., and Singh, R. N. (2021). High concentration of an ISS-N1-targeting antisense oligonucleotide causes massive perturbation of the transcriptome. Int. J. Mol. Sci. 22:33. doi: 10.3390/ijms22168378
Ottesen, E. W., Seo, J., Luo, D., Singh, N. N., and Singh, R. N. (2024). A super minigene with a short promoter and truncated introns recapitulates essential features of transcription and splicing regulation of the SMN1 and SMN2 genes. Nucleic Acids Res. 52, 3547–3571. doi: 10.1093/nar/gkad1259
Ottesen, E. W., Seo, J., Singh, N. N., and Singh, R. N. (2017). A multilayered control of the human of the survival motor neuron gene expression by Alu elements. Front. Microbiol. 8:2252. doi: 10.3389/fmicb.2017.02252
Ottesen, E. W., and Singh, R. N. (2024). Synergistic effect of an antisense oligonucleotide and small molecule on splicing correction of the spinal muscular atrophy gene. Neurosci. Insights 19:26331055241233596. doi: 10.1177/26331055241233596
Ottesen, E. W., Singh, N. N., Luo, D., Kaas, B., Gillette, B. J., Seo, J., et al. (2023). Diverse targets of SMN2-directed splicing-modulating small molecule therapeutics for spinal muscular atrophy. Nucleic Acids Res. 51, 5948–5980. doi: 10.1093/nar/gkad259
Pagani, F., Buratti, E., Stuani, C., Bendix, R., Dörk, T., and Baralle, F. E. (2002). A new type of mutation causes a splicing defect in ATM. Nat. Genet. 30, 426–429. doi: 10.1038/ng858
Parada, G. E., Munita, R., Cerda, C. A., and Gysling, K. (2014). A comprehensive survey of non-canonical splice sites in the human transcriptome. Nucleic Acids Res. 42, 10564–10578. doi: 10.1093/nar/gku744
Ptok, J., Müller, L., Theiss, S., and Schaal, H. (2019). Context matters: regulation of splice donor usage. Biochim. Biophys. Acta Gene Regul. Mech. 1862:194391. doi: 10.1016/j.bbagrm.2019.06.002
Rogalska, M. E., Tajnik, M., Licastro, D., Bussani, E., Camparini, L., Mattioli, C., et al. (2016). Therapeutic activity of modified U1 core spliceosomal particles. Nat. Commun. 7:11168. doi: 10.1038/ncomms11168
Scharner, J., Ma, W. K., Zhang, Q., Lin, K. T., Rigo, F., Bennett, C. F., et al. (2020). Hybridization-mediated off-target effects of splice-switching antisense oligonucleotides. Nucleic Acids Res. 48, 802–816. doi: 10.1093/nar/gkz1132
Schilling, J., Broemer, M., Atanassov, I., Duernberger, Y., Vorberg, I., Dieterich, C., et al. (2019). Deregulated splicing is a major mechanism of RNA-induced toxicity in Huntington's disease. J. Mol. Biol. 431, 1869–1877. doi: 10.1016/j.jmb.2019.01.034
Seo, J., Singh, N. N., Ottesen, E. W., Lee, B. M., and Singh, R. N. (2016a). A novel human-specific splice isoform alters the critical C-terminus of survival motor neuron protein. Sci. Rep. 6:30778. doi: 10.1038/srep30778
Seo, J., Singh, N. N., Ottesen, E. W., Sivanesan, S., Shishimorova, M., and Singh, R. N. (2016b). Oxidative stress triggers body-wide skipping of multiple exons of the spinal muscular atrophy gene. PLoS One 11:e0154390. doi: 10.1371/journal.pone.0154390
Shao, W., Alcantara, S. G., and Zeitlinger, J. (2019). Reporter-ChIP-nexus reveals strong contribution of the Drosophila initiator sequence to RNA polymerase pausing. eLife 8:e41461. doi: 10.7554/eLife.41461
Shenasa, H., and Hertel, K. J. (2019). Combinatorial regulation of alternative splicing. Biochim. Biophys. Acta Gene Regul. Mech. 1862:194392. doi: 10.1016/j.bbagrm.2019.06.003
Shuai, S. M., Suzuki, H., Diaz-Navarro, A., Nadeu, F., Kumar, S. A., Gutierrez-Fernandez, A., et al. (2019). The U1 spliceosomal RNA is recurrently mutated in multiple cancers. Nature 574:712-+. doi: 10.1038/s41586-019-1651-z
Singh, R. N. (2007). Evolving concepts on human SMN pre-mRNA splicing. RNA Biol. 4, 7–10. doi: 10.4161/rna.4.1.4535
Singh, N. N., Androphy, E. J., and Singh, R. N. (2004a). An extended inhibitory context causes skipping of exon 7 of SMN2 in spinal muscular atrophy. Biochem. Biophys. Res. Commun. 315, 381–388. doi: 10.1016/j.bbrc.2004.01.067
Singh, N. N., Androphy, E. J., and Singh, R. N. (2004b). In vivo selection reveals combinatorial controls that define a critical exon in the spinal muscular atrophy genes. RNA 10, 1291–1305. doi: 10.1261/rna.7580704
Singh, N. N., Del Rio-Malewski, J. B., Luo, D., Ottesen, E. W., Howell, M. D., and Singh, R. N. (2017a). Activation of a cryptic 5′ splice site reverses the impact of pathogenic splice site mutations in the spinal muscular atrophy gene. Nucleic Acids Res. 45, 12214–12240. doi: 10.1093/nar/gkx824
Singh, N. N., Hollinger, K., Bhattacharya, D., and Singh, R. N. (2010). An antisense microwalk reveals critical role of an intronic position linked to a unique long-distance interaction in pre-mRNA splicing. RNA 16, 1167–1181. doi: 10.1261/rna.2154310
Singh, N. N., Howell, M. D., Androphy, E. J., and Singh, R. N. (2017b). How the discovery of ISS-N1 led to the first medical therapy for spinal muscular atrophy. Gene Ther. 24, 520–526. doi: 10.1038/gt.2017.34
Singh, R. N., Howell, M. D., Ottesen, E. W., and Singh, N. N. (2017). Diverse role of survival motor neuron protein. Biochim. Biophys. Acta Gene Regul. Mech. 1860, 299–315. doi: 10.1016/j.bbagrm.2016.12.008
Singh, N. N., Lawler, M. N., Ottesen, E. W., Upreti, D., Kaczynski, J. R., and Singh, R. N. (2013). An intronic structure enabled by a long-distance interaction serves as a novel target for splicing correction in spinal muscular atrophy. Nucleic Acids Res. 41, 8144–8165. doi: 10.1093/nar/gkt609
Singh, N. N., O'Leary, C. A., Eich, T., Moss, W. N., and Singh, R. N. (2022). Structural context of a critical exon of spinal muscular atrophy gene. Front. Mol. Biosci. 9:928581. doi: 10.3389/fmolb.2022.928581
Singh, N. N., Seo, J., Ottesen, E. W., Shishimorova, M., Bhattacharya, D., and Singh, R. N. (2011). TIA1 prevents skipping of a critical exon associated with spinal muscular atrophy. Mol. Cell. Biol. 31, 935–954. doi: 10.1128/MCB.00945-10
Singh, N. N., Seo, J., Rahn, S. J., and Singh, R. N. (2012). A multi-exon-skipping detection assay reveals surprising diversity of splice isoforms of spinal muscular atrophy genes. PLoS One 7:e49595. doi: 10.1371/journal.pone.0049595
Singh, R. N., Seo, J., and Singh, N. N. (2020). RNA in spinal muscular atrophy: therapeutic implications of targeting. Expert Opin. Ther. Targets 24, 731–743. doi: 10.1080/14728222.2020.1783241
Singh, R. N., and Singh, N. N. (2018). Mechanism of splicing regulation of spinal muscular atrophy genes. Adv. Neurobiol. 20, 31–61. doi: 10.1007/978-3-319-89689-2_2
Singh, N. N., and Singh, R. N. (2019). How RNA structure dictates the usage of a critical exon of spinal muscular atrophy gene. Biochim. Biophys. Acta Gene Regul. Mech. 1862:194403. doi: 10.1016/j.bbagrm.2019.07.004
Singh, R. N., and Singh, N. N. (2019). A novel role of U1 snRNP: splice site selection from a distance. Biochim. Biophys. Acta Gene Regul. Mech. 1862, 634–642. doi: 10.1016/j.bbagrm.2019.04.004
Singh, N. N., Singh, R. N., and Androphy, E. J. (2007). Modulating role of RNA structure in alternative splicing of a critical exon in the spinal muscular atrophy genes. Nucleic Acids Res. 35, 371–389. doi: 10.1093/nar/gkl1050
Singh, N. K., Singh, N. N., Androphy, E. J., and Singh, R. N. (2006). Splicing of a critical exon of human survival motor neuron is regulated by a unique silencer element located in the last intron. Mol. Cell. Biol. 26, 1333–1346. doi: 10.1128/MCB.26.4.1333-1346.2006
Suzuki, H., Kumar, S. A., Shuai, S. M., Diaz-Navarro, A., Gutierrez-Fernandez, A., De Antonellis, P., et al. (2019). Recurrent noncoding U1 snRNA mutations drive cryptic splicing in SHH medulloblastoma. Nature 574:707. doi: 10.1038/s41586-019-1650-0
Tan, J., Ho, J. X., Zhong, Z., Luo, S., Chen, G., and Roca, X. (2016). Noncanonical registers and base pairs in human 5′ splice-site selection. Nucleic Acids Res. 44, 3908–3921. doi: 10.1093/nar/gkw163
Tazi, J., Bakkour, N., and Stamm, S. (2009). Alternative splicing and disease. BBA Mol. Basis Dis. 1792, 14–26. doi: 10.1016/j.bbadis.2008.09.017
Venters, C. C., Oh, J. M., Di, C., So, B. R., and Dreyfuss, G. (2019). U1 snRNP Telescripting: suppression of premature transcription termination in introns as a new layer of gene regulation. Cold Spring Harb. Perspect. Biol. 11:15. doi: 10.1101/cshperspect.a032235
Vitte, J., Fassier, C., Tiziano, F. D., Dalard, C., Soave, S., Roblot, N., et al. (2007). Refined characterization of the expression and stability of the SMN gene products. Am. J. Pathol. 171, 1269–1280. doi: 10.2353/ajpath.2007.070399
Wei, Y., Chen, Z. S., Li, Y. W., and Song, K. (2024). The splicing factor WBP11 mediates MCM7 intron retention to promote the malignant progression of ovarian cancer. Oncogene 43, 1565–1578. doi: 10.1038/s41388-024-03015-2
Wirth, B., Karakaya, M., Kye, M. J., and Mendoza-Ferreira, N. (2020). Twenty-five years of spinal muscular atrophy research: from phenotype to genotype to therapy, and what comes next. Annu. Rev. Genomics Hum. Genet. 21, 231–261. doi: 10.1146/annurev-genom-102319-103602
Yeo, G., and Burge, C. B. (2004). Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J. Comput. Biol. 11, 377–394. doi: 10.1089/1066527041410418
Yu, Y., Chi, B., Xia, W., Gangopadhyay, J., Yamazaki, T., Winkelbauer-Hurt, M. E., et al. (2015). U1 snRNP is mislocalized in ALS patient fibroblasts bearing NLS mutations in FUS and is required for motor neuron outgrowth in zebrafish. Nucleic Acids Res. 43, 3208–3218. doi: 10.1093/nar/gkv157
Zhang, S., Aibara, S., Vos, S. M., Agafonov, D. E., Lührmann, R., and Cramer, P. (2021). Structure of a transcribing RNA polymerase II-U1 snRNP complex. Science 371, 305–309. doi: 10.1126/science.abf1870
Keywords: pre-mRNA splicing, U1 snRNP, U1 snRNA, spinal muscular atrophy (SMA), survival motor neuron (SMN), super minigene
Citation: Ottesen EW, Singh NN, Seo J and Singh RN (2024) U1 snRNA interactions with deep intronic sequences regulate splicing of multiple exons of spinal muscular atrophy genes. Front. Neurosci. 18:1412893. doi: 10.3389/fnins.2024.1412893
Edited by:
Jiuyong Xie, University of Manitoba, CanadaReviewed by:
Sue Fletcher, PYC Therapeutics, AustraliaCopyright © 2024 Ottesen, Singh, Seo and Singh. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ravindra N. Singh, c2luZ2hyQGlhc3RhdGUuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.