 Daniela Formicola1†
Daniela Formicola1† Irina Podda2
Irina Podda2 Elia Dirupo3
Elia Dirupo3 Elena Andreucci3
Elena Andreucci3 Sabrina Giglio4
Sabrina Giglio4 Paola Cipriani5
Paola Cipriani5 Clara Bombonato5
Clara Bombonato5 Filippo Maria Santorelli1,5*
Filippo Maria Santorelli1,5* Anna Chilosi5‡
Anna Chilosi5‡- 1Department of Neurobiology and Molecular Medicine, IRCCS Fondazione Stella Maris, Pisa, Italy
- 2Parole al Centro Studio di Logopedia, Genoa, Italy
- 3Medical Genetics Unit, Meyer Children’s University Hospital IRCCS, Florence, Italy
- 4Medical Genetics Unit, Department of Medical Sciences and Public Health, University of Cagliari, Cagliari, Italy
- 5Department of Developmental Neuroscience, IRCCS Fondazione Stella Maris Scientific Institute, Pisa, Italy
Background: Childhood apraxia of speech (CAS) is a genetically heterogeneous pediatric motor speech disorder. The advent of whole exome sequencing (WES) and whole genome sequencing techniques has led to increased identification of pathogenic variants in CAS genes. In an as yet uncharacterized Italian cohort, we aimed both to identify new pathogenic gene variants associated with CAS, and to confirm the disease-related role of genes already reported by others. We also set out to refine the clinical and neurodevelopmental characterization of affected children, with the aim of identifying specific, gene-related phenotypes.
Methods: In a single-center study aiming to explore the genetic etiology of CAS in a cohort of 69 Italian children, WES was performed in the families of the 34 children found to have no copy number variants. Each of these families had only one child affected by CAS.
Results: High-confidence (HC) gene variants were identified in 7/34 probands, in two of whom they affected KAT6A and CREBBP, thus confirming the involvement of these genes in speech impairment. The other probands carried variants in low-confidence (LC) genes, and 20 of these variants occurred in genes not previously reported as associated with CAS. UBA6, ZFHX4, and KAT6A genes were found to be more enriched in the CAS cohort compared to control individuals. Our results also showed that most HC genes are involved in epigenetic mechanisms and are expressed in brain regions linked to language acquisition processes.
Conclusion: Our findings confirm a relatively high diagnostic yield in Italian patients.
1 Introduction
Childhood Apraxia of Speech (CAS) is a pediatric neurological motor speech disorder whose core features are lengthened and disrupted coarticulatory transitions, inconsistent errors on consonants and vowels, and abnormal word and phrase prosody due to impaired planning and programming of speech movements (American Speech-Language-Hearing Association, 2007). In CAS the precision and consistency of movements underlying speech are impaired in the absence of central or peripheral neuromuscular deficits and of structural oro-facial abnormalities. Clinically, CAS is diagnosed based on a qualitative assessment of speech samples gathered across different tasks, at the syllable, word, phrase and sentence level (see Supplementary Figure 1 for the operationalization of CAS signs). A set of speech features must be detected in the speech samples, such as difficulties in achieving the initial articulatory condition and in transitioning between different speech movements, increasing difficulties in multisyllabic sequences, syllable segregation, erroneous stress assignment, inconsistent devoicing, inconsistent errors with consonants and vowels, slow speech and diadochokinetic rate, groping, vocalic epenthesis and altered nasal resonance (Shriberg et al., 2011; Chenausky et al., 2020). These features result in poor intelligibility and effortful speech production. Typically, speech disruptions in CAS persist over time and necessitate protracted and intensive therapy. Ultimately, CAS can affect children’s expressive skills, academic performance (Miller and Lewis, 2022; Lewis et al., 2023) and participation in age-appropriate activities, as well as self-esteem and psychological well-being (Lewis et al., 2021; Cassar et al., 2023).
Although CAS presents in isolation in a relatively small proportion of cases, more often it co-occurs with prevalently expressive language impairment (Murray et al., 2019; Chilosi et al., 2022; Lewis et al., 2023), and with other complex neurodevelopmental disorders, such as Intellectual Disability (ID), Autism Spectrum Disorder (ASD), Attention Deficit Hyperactivity Disorder (ADHD) and Developmental Coordination Disorder (DCD) (Shriberg et al., 2019a; Stein et al., 2020; Chilosi et al., 2022; Chenausky et al., 2023; Morgan et al., 2024). CAS was estimated to occur in 1 child per 1,000 in the general pediatric population (Shriberg et al., 1997) and in 2.4% in a clinical population of children with speech sound disorders (Shriberg et al., 2019b). The prevalence was estimated to be higher in males than in females, with 2–3:1 male/female ratio (Hall et al., 1993; Lewis et al., 2004).
The etiology of CAS is still largely unknown. Unlike acquired apraxia of speech in adults, CAS is not linked to specific brain lesions, even though cerebral abnormalities are occasionally found (Chilosi et al., 2022; Chenausky et al., 2023; Chilosi et al., 2015). Nonetheless, advanced brain imaging techniques reveal volumetric and connectivity alterations in cortical areas and in subcortical networks underlying speech production (Kadis et al., 2014; Fiori et al., 2016; Conti et al., 2020).
A recent paper by Morgan et al. (2024) highlights that FOXP2 dysfunction remains relatively specific to speech disorder, as compared with other recently identified monogenic conditions associated with CAS (Morison et al., 2023). In addition to FOXP2 variants, single variants and small indels in other genes (such as FOXP1 and CNTNAP2) and pathogenic copy number variants (CNVs) (e.g., 16p11.2 deletion) have been described in Chilosi et al. (2022). These variants often occur de novo and may involve genes already associated with other neurodevelopmental disorders, possibly determining phenotypic overlapping, with important implications for case management. The phenotypic spectrum can, therefore, be expanded to include “milder presentations,” defined by primary speech disorder in children with average cognitive performance (Morgan et al., 2024).
The advent of whole exome sequencing (WES) and whole genome sequencing techniques has led to increased identification of monogenic variants in CAS-associated genes. The list of these genes is provided in Supplementary Table 1. Despite the genetic heterogeneity of CAS, many implicated proteins functionally converge on pathways involved in chromatin modification or transcriptional regulation, opening the door to precision diagnosis and therapies (Morgan et al., 2024).
In a single-center study, we conducted WES analysis and deep phenotyping of speech and language profiles in a cohort of children with isolated CAS and no co-occurring neurodevelopmental disorders, except for language impairment. Our aim was to identify new pathogenic gene variants associated with CAS and to confirm the disease-related role of genes already reported by others. We also set out to refine the clinical and neurodevelopmental characterization of the affected children in our sample.
2 Materials and methods
2.1 Population study
Sixty-nine children (53 boys and 16 girls) were diagnosed with idiopathic CAS during evaluation for speech disorders at the Neurolinguistic and Neuropsychology Unit of IRCCS Stella Maris, a tertiary care hospital for children with neurological, neurodevelopmental, and psychiatric disorders. The CAS diagnosis was based on the presence of the three American Speech-Language-Hearing Association core speech characteristics1 and of at least five of the ten speech signs included in the Mayo checklist (Shriberg et al., 2011).
All the children underwent a comprehensive clinical and instrumental assessment that included structural brain imaging at 1.5 T and the standardized phenotypic assessment protocol that we adopt in complex neuropsychological and neurodevelopmental disorders (Chilosi et al., 2023).
Children were eligible for inclusion in the study if they were aged ≥4 years at clinical evaluation, if Italian was the only or primary language spoken at home, and if they were able to complete a full neurological and speech and language assessment. After an extensive neuropsychiatric evaluation by a specialized team, using specific procedures and carefully applying the DSM-5 clinical criteria, any children diagnosed with concomitant neurodevelopmental disorders such as ID, ASD and ADHD were excluded from the study. Other exclusion criteria were orofacial structural abnormalities liable to affect speech, hearing impairment, and the presence of “dysarthria only” speech features (Iuzzini-Seigel et al., 2022).
2.2 Deep speech and language phenotyping
Deep speech and language phenotyping was conducted through face-to-face evaluations by speech-language pathologists experienced in the assessment of pediatric motor speech disorders.
Speech evaluation included the following measures: (a) consonantal phonetic repertoire; (b) percentage of inaccurate productions of the items on a list of 46 probe words characterized by increasingly complex motor speech and syllable patterns; (c) percentage of inconsistent errors on the number of incorrectly produced probe words; (d) percentage of words with omitted syllables in the same probe word task; and (e) diadochokinetic (DDK) rate, as the number of fast repetitions of a nonsense three-syllable sequence (i.e., /pataka/). Overall intelligibility was rated by two independent scorers, who reviewed videos of speech samples and provided, independently, an intelligibility score ranging from 0 (never intelligible) to 5 (fully intelligible).
Language skills were assessed through the administration of standardized tests of receptive and expressive vocabulary and grammar. The level of expressive grammar complexity was evaluated according to the Grid for the Analysis of Spontaneous Speech (Chilosi et al., 2019). To estimate the overall level of speech and language proficiency, two composite severity scores were calculated based, respectively, on five speech and four language measures, as previously reported (Chilosi et al., 2022). Each measure was assigned a score of 0 when normal or borderline, and 1 when deficient. The maximum severity score was, therefore, 5 for speech and 4 for language. The speech composite severity score took into account consonantal phonetic repertoire, word inaccuracy and inconsistency of errors, syllable omissions, and DDK rate.
2.3 Cognitive assessment
Depending on the child’s age, non-verbal intelligence quotient (IQ) was assessed using the Wechsler Preschool and Primary Scale of Intelligence 3rd Edition (WPPSI-III) or the Wechsler Intelligence Scale for Children 3rd Edition (WISC-III) or 4th Edition (WISC-IV). Children were enrolled if their non-verbal IQ was >70.
2.4 Molecular studies
Prior to this study, all 69 patients had been investigated for pathogenic CNVs as part of their routine clinical assessment. The investigation was based on chromosome microarray analysis (CMA) and it revealed CNVs in 26 children (see Supplementary Table 2). Five of these children presented chromosome 16p11.2 deletion syndrome. Clinical data were insufficient in nine “negative” cases. In the remaining 34 cases, who tested negative for CNVs on CMA, we performed WES on the family trio. We prioritized cases that were completely negative for CNVs for exome sequencing. In 22 families the index case was the only affected individual, whereas in 10 kindred one of the two parents presented a history of speech language disorder. In a further family the father and brother of the index case also had a history of language delay. In the last family the father did not consent to molecular investigations (Supplementary Figure 2).
Total genomic DNA was extracted from peripheral blood using the QIAamp Mini Kit (QIAGEN®, Hilden, Germany) and samples were anonymized by means of numeric codes. Construction of enzymatic fragmentation libraries was followed by end repair, A-tailing, adapter ligation, and library amplification. Libraries were hybridized to a commercially available WES capture array (SeqCap EZ Exome v3, Nimblegen, Roche, Basel, Switzerland) and sequenced with NextSeq500/550 (Illumina Inc., San Diego, CA). The reads were aligned with the human reference hg19 genome using the Burrows-Wheeler Aligner (10.1093/bioinformatics/btp324), and mapped and analyzed with the IGV software package (Thorvaldsdottir et al., 2013). Downstream alignment processing was performed using the Genome Analysis Toolkit Unified Genotyper Module (Menke et al., 2016), SAMtool (Li et al., 2009), and Picard toolkit.2 Variants were annotated using Annovar (Wang et al., 2010) to obtain information such as variant frequency in different populations, and the predictions of the variant effect were obtained using a variety of software (CADD, SIFT, Polyphen2, MutationTaster, MutationAssessor, FATHMM and FATHMM MKL). Quality control of sequencing showed that 96% of the reads were mapped to the reference genome (hg19), and 97% of the targeted regions were covered by ≥30X reads with an average depth of 100X.
Clinically relevant germline variants were filtered step by step to identify the potentially interesting candidates; we retained only the single nucleotide variants and indel gene variants with a minor allele frequency (MAF) < 1% in the gnomAD v.2.1 database. Non-coding variants were excluded. In addition, we removed variants with a read depth < 20 and an alternative allele frequency < 35% of the normal allele (proportion of variant reads). Trio analyses were performed on the remaining gene variants by assuming a de novo, autosomal recessive, and X-linked mode of inheritance. We evaluated genes panel in-silico using the SFARI database and the Human Phenotype Ontology (HPO) of speech apraxia: HP: 0011098 and HP:0001249 to consider potential inherited variants from parents in light of genetic phenomena such as incomplete penetrance and variable expressivity. All reported variants were also inspected with IGV v2.7.
We analyzed exome data for loss-of-function (LoF) variants and predicted damaging missense variants. In particular, we focused on nonsense, missense, frameshift, and splice-region variant types, including synonymous variants, with a MAF < 0.03% in the gnomAD v2.1 database, in consideration of potentially causal variants for CAS.
The genes were further screened to assess biological significance for neural development, and known neurological disorders and functions, using the Online Mendelian Inheritance in Man (OMIM) database and consulting the PubMed website.
To verify whether detected variants were located in genes already known to be associated with ASD or neurodevelopmental disorders, we scrutinized the SFARI database.3
Classification of variants complied with the American College of Medical Genetics (ACMG) guidelines (Richards et al., 2015), and variants were therefore split into two categories (Hildebrand et al., 2020) according to whether they occurred in high-confidence (HC) or low-confidence (LC) genes.
The HC variants met the following criteria: (i) LoF variants (nonsense, frameshift and splicing variant); for splicing variants we retained those with SpliceAI scores >0.2; for damaging missense variants we retained those satisfying at least one of the following conditions: CADD Phred score ≥ 20 or a Revel score ≥ 0.644; (ii) variant located in genes associated with CAS phenotype, and in which segregation analysis was supportive.
Variants were defined LC if they involved (i)OMIM genes not associated with disease, (ii) class 3 variants in the ACMG classification, (iii) or class 4 variants located in genes not fully consistent with the proband’s phenotype, (iiii) or when family segregation was not supportive.
2.4.1 Gene ontology analysis
For the HC gene list in our analyses, we queried the TOPPGENE database to perform a Gene Ontology (GO) analysis, focusing on identifying enriched biological processes, cellular components, and molecular functions associated with the genes. We also conducted a comprehensive set enrichment analysis based on the Human Phenotype Ontology (HPO), cellular components, and biological processes. This approach allowed us to identify relevant biological pathways and cellular components associated with our gene set, following methodologies similar to those in the recent studies by Zhang et al. (2023) and Maleki et al. (2020).
2.4.2 Gene expression data analysis
The Gene-level RNA-seq RPKM expression data were retrieved from the Developmental.
Transcriptome section4 of the BrainSpan Atlas of the Developing Human Brain database (Miller et al., 2014). Two custom selections were generated, for the HC and LC gene lists, respectively. Data were sorted by structure, and the resulting expression matrices were downloaded. The downloaded files were subsequently imported into R, and heatmaps were generated for visualization employing the ComplexHeatmap package.5
3 Results
The 34 probands who underwent WES analysis had a mean age of 71.8 ± 26.2 months (range: 47–159 months). WES studies did not identify any HC or LC variants in one child (proband #60). Instead, HC variants were identified in 7 children, 3 males and 4 females (Figure 1A). The mean age of this HC group was 67 ± 19.1 months (range: 50–94 months). The variants affected 7 different genes (Table 1). LC variants (Figure 1B; Table 2), on the other hand, were detected in 26 children, 24 males and 2 females, aged 73.1 ± 28.4 months (range: 47–159 months). Table 3 synthesizes the comparisons between the clinical characteristics of the children with HC and LC variants. Whilst speech severity did not differ significantly between the two groups, language impairment was more frequent and more severe in the HC than in the LC group. Moreover, in the HC group language impairment involved more frequently also receptive language skills. Tables 4, 5 report the phenotypic characteristics of the children belonging to the two groups.
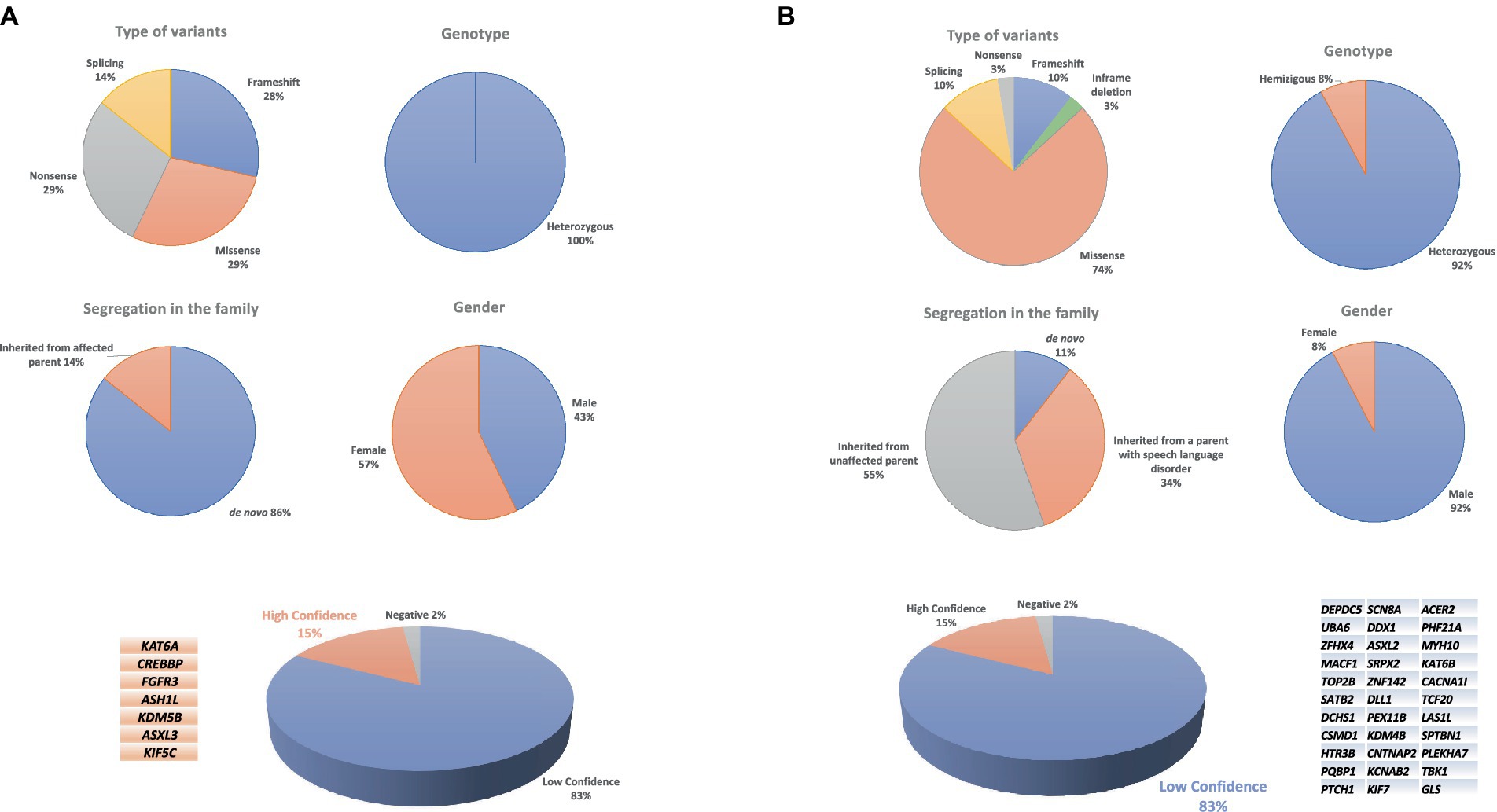
Figure 1. Chart of high-confidence (A) and low-confidence variants (B) in a CAS cohort.
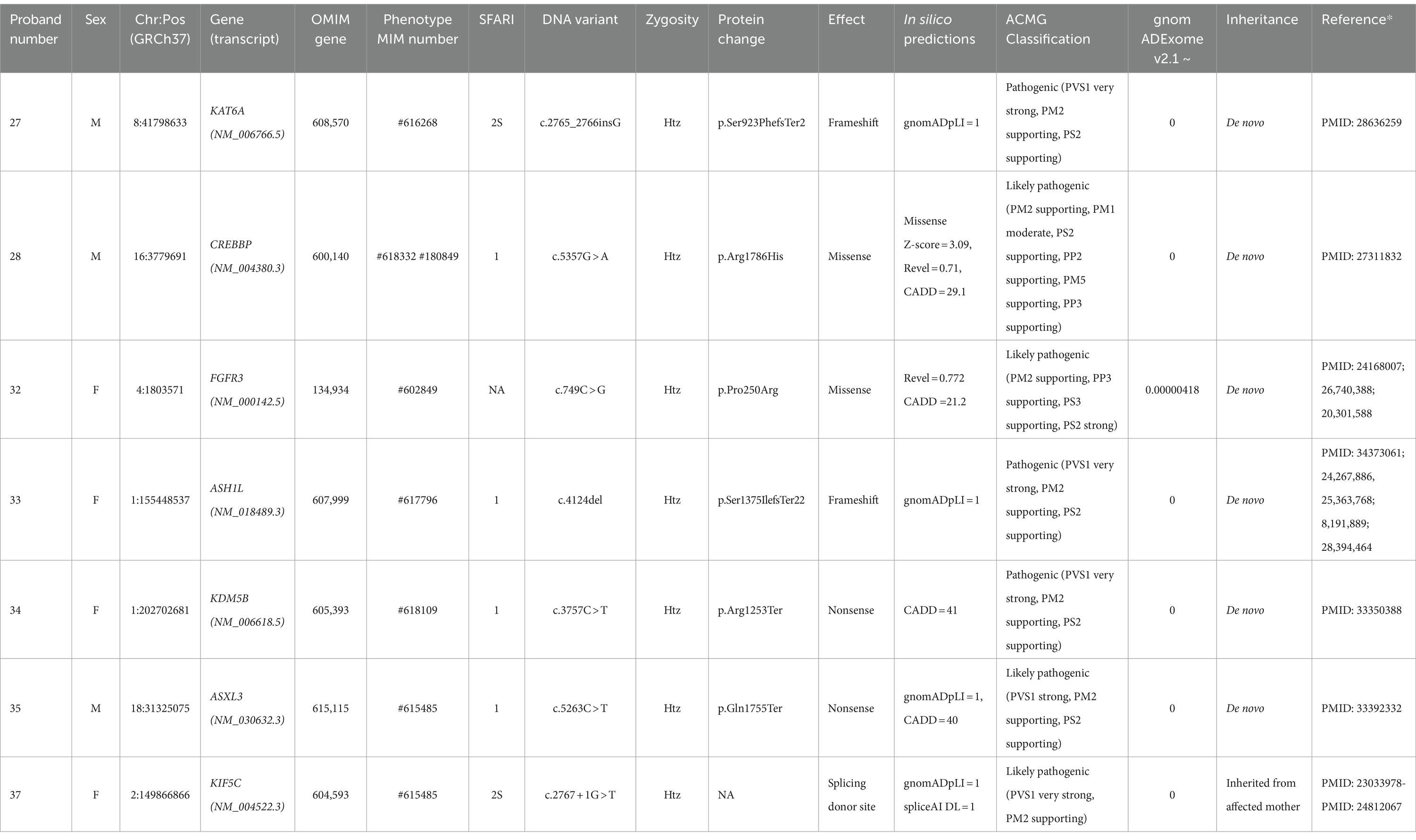
Table 1. High-confidence gene variants in individuals with CAS.
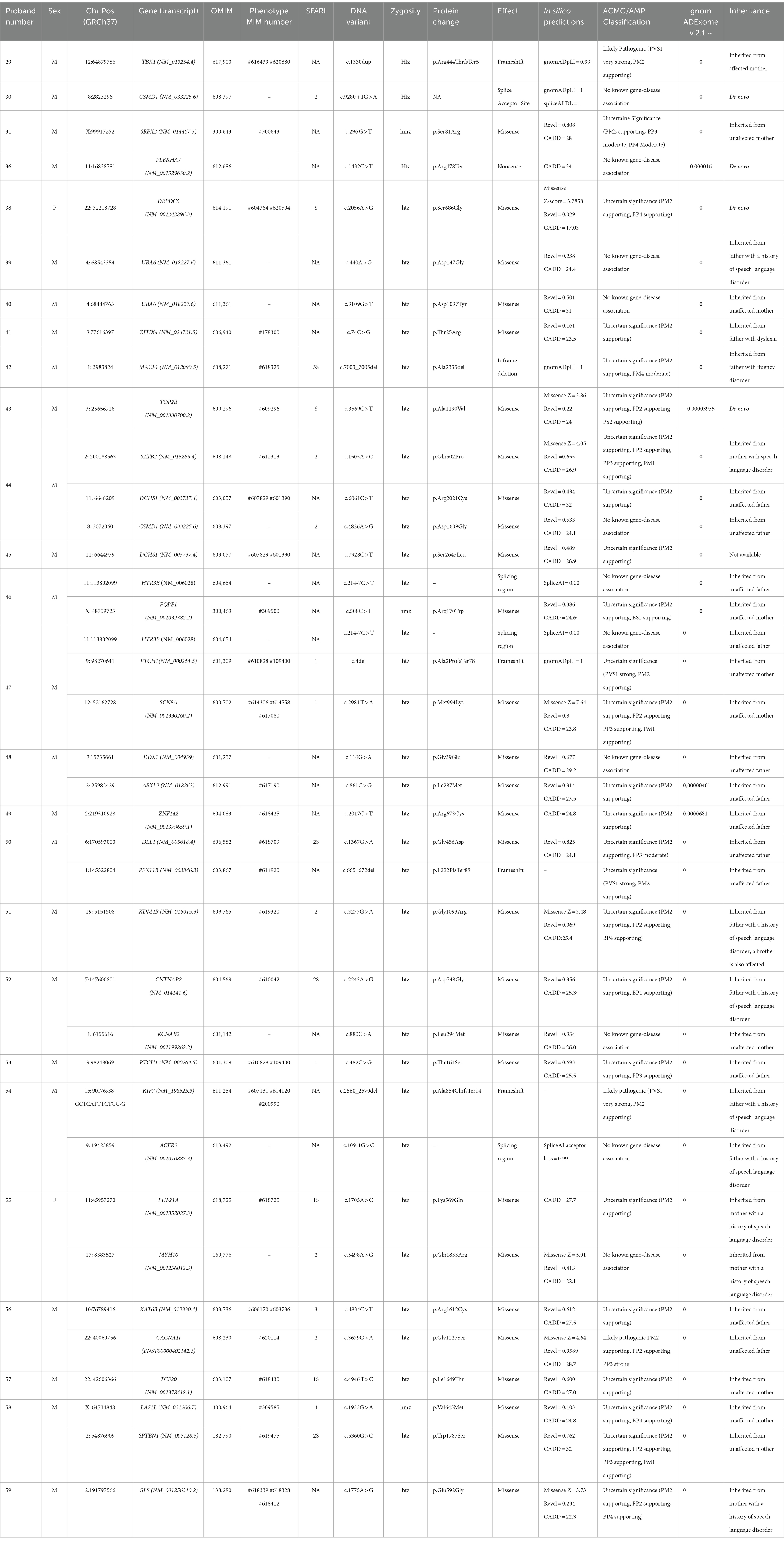
Table 2. Low-confidence variants in individuals with CAS.
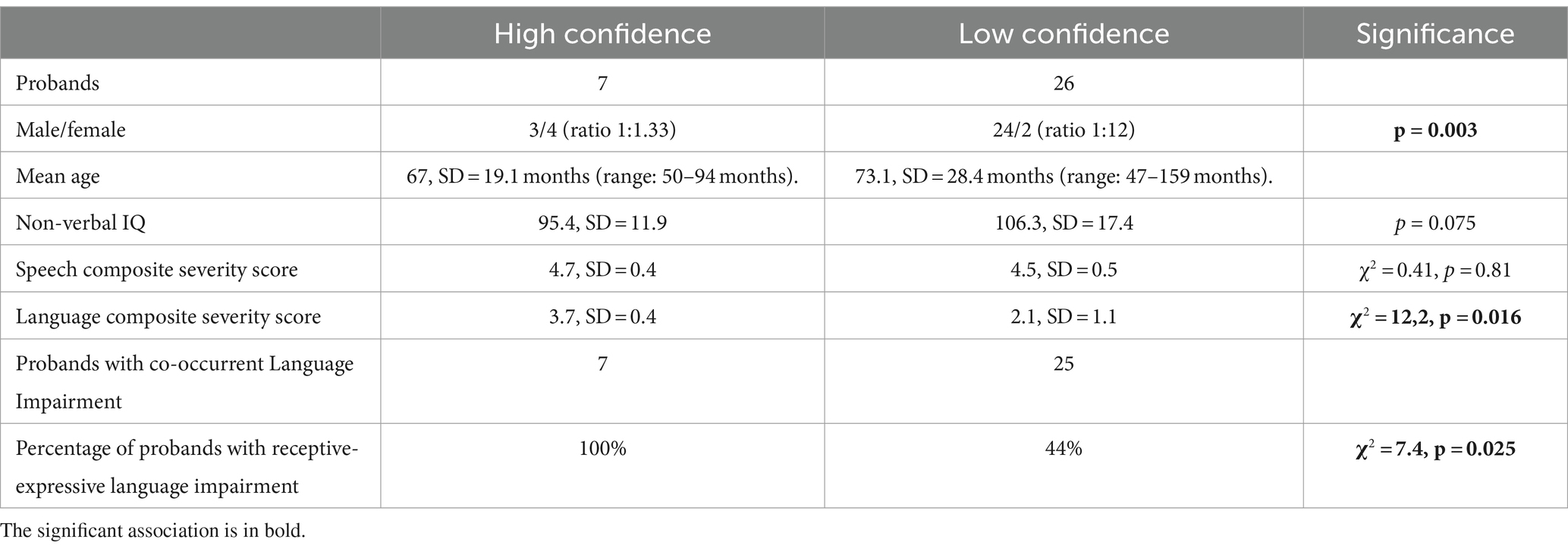
Table 3. Comparison between clinical characteristics of children with HC and LC gene alterations.
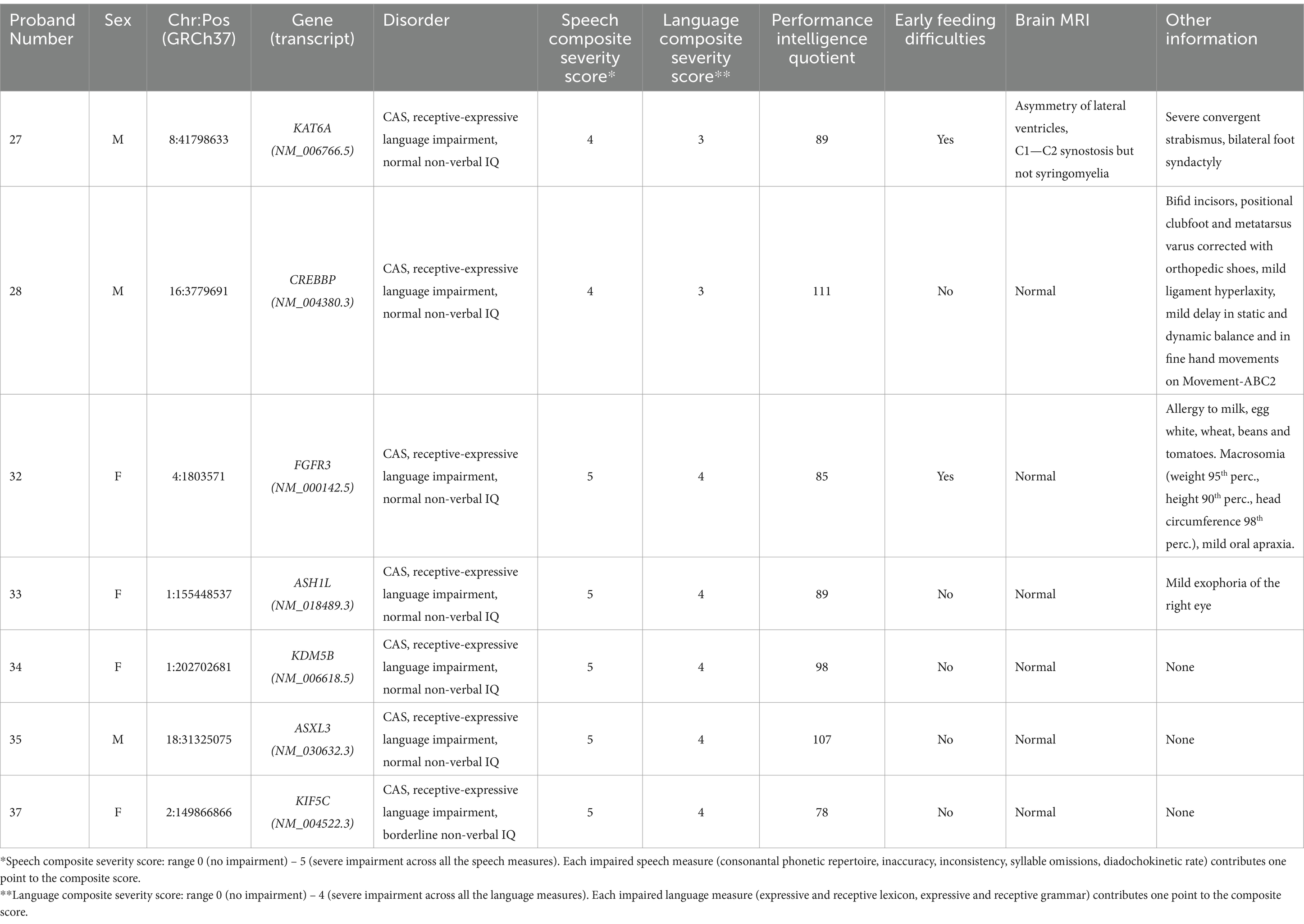
Table 4. Clinical features of individuals with CAS and high-confidence variants.
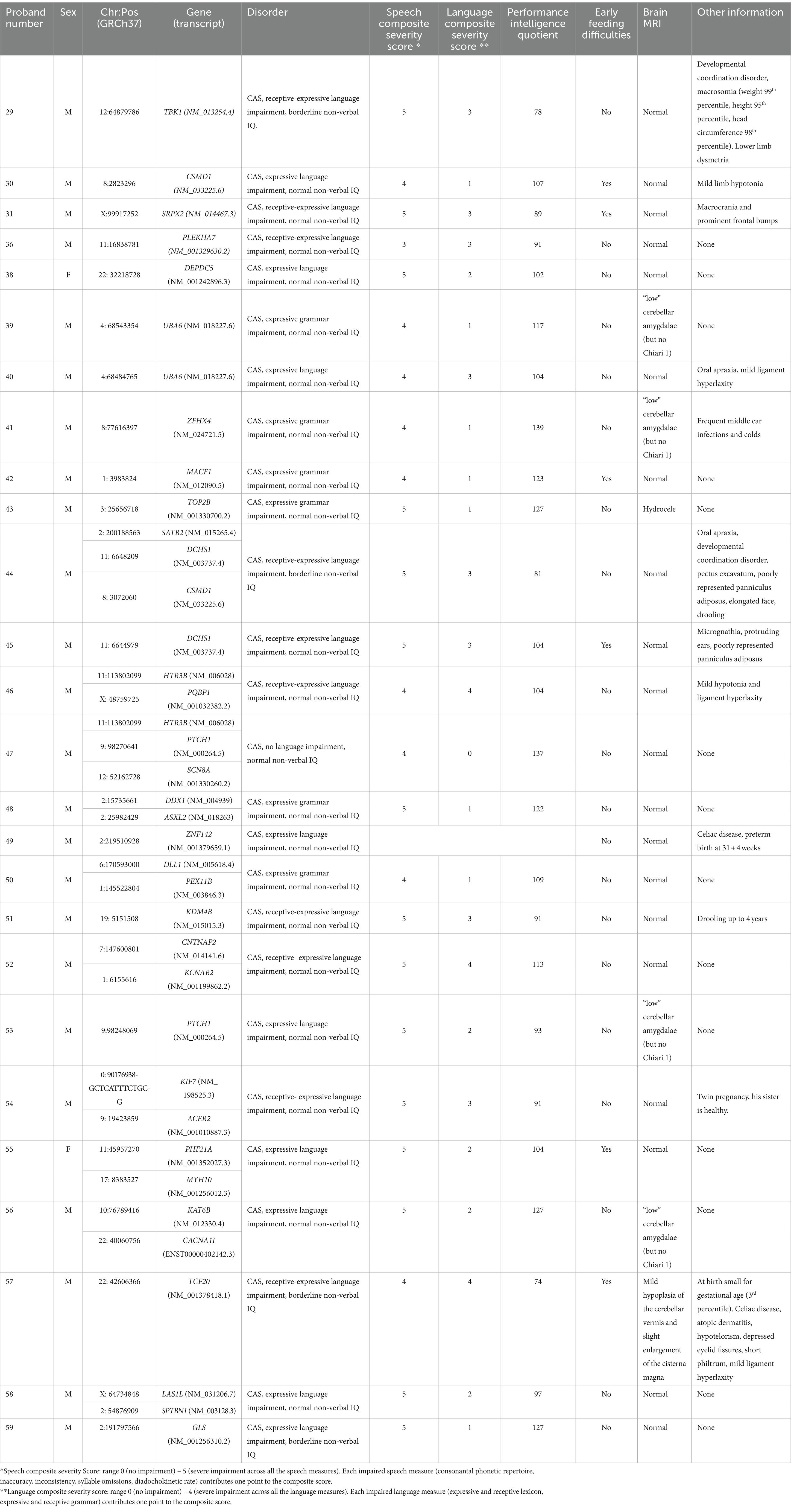
Table 5. Clinical features of individuals with CAS and low-confidence variants.
3.1 HC: distribution of variants and associated genes
Figure 1A shows the distribution of the variant types, levels of zygosity, and family segregation patterns, including the percentage of de novo variants (6/7, 86%), found in the patients with HC variants. Supplementary Table 3A provides the CAS speech features of HC genes.
HC variants occurred in genes already associated with language disorders. In particular, variants were detected in the following genes: KAT6A (Zwaveling-Soonawala et al., 2017), CREBBP (Menke et al., 2016) and FGFR3 (Aravidis et al., 2014; Kruszka et al., 2016; Kruszka et al., 1993; Robin et al., 1998). Interestingly, in proband #32 we identified the reported c.749C > G (p.Pro250Arg) variant in FGFR3 that has been associated, in multiple cases, with Muenke syndrome (MIM: 602849), a condition characterized by considerable phenotypic variability (Kruszka et al., 1993). About 40% of individuals with Muenke syndrome present ID, and 66.3% show developmental delay; some patients, however, as in the case of our proband #32, lack the typical clinical or radiographic features of Muenke syndrome (Kruszka et al., 2016; Kruszka et al., 1993; Robin et al., 1998; Moko and Blandin de Chalain, 2001).
Seven of our CAS probands harbored HC variants — specifically, LoF variants (two frameshift, two non-sense, and one splicing) and two missense variants in putatively novel CAS genes, not yet associated with impaired language phenotypes.
In proband #33, we identified a c.4124del de novo frameshift in ASH1L regarded as “likely pathogenic.” This gene plays a critical role in development by activating homeobox (HOX) genes and it is associated with autosomal dominant intellectual disability (MIM: 617796) (Okamoto et al., 2017; Wang et al., 2016; Stessman et al., 2017). Table 1 shows the remaining HC variants detected in novel genes.
3.2 LC variants: distribution of variants and associated genes
LC variants were identified in 26 children who underwent WES analysis (76% of the sample) (Table 2). These variants are currently classified as class 3 or 4 (see Figure 1B for variant types and zygosity). For variants in genes that do not have a recognized gene-disease association according to OMIM, it was not possible to apply the ACMG classification criteria. As a result, we have annotated these cases in the table as ‘no known gene-disease association’ rather than assigning an ACMG classification. Ten children had more than one variant involving different genes. The Supplementary Table 3B provides the CAS speech features of LC genes.
In the LC subgroup, we identified variants in 33 genes associated with syndromic disorders or other complex neurodevelopmental disorders whose features include speech delay or LI. Fourteen of these genes are listed in the SFARI gene database (Table 2).
Most of the variants were inherited from an unaffected parent (21/34), whereas 13 variants were inherited from a parent reporting early developmental language and speech delay, not otherwise characterized. In 4/38 (11%) cases, variants occurred de novo (Figure 1B). Other variants of note included a de novo missense variant in DEPDC5 found in proband #38. LoF variants in DEPDC5 have been reported in patients with dominantly inherited familial focal epilepsy with variable foci (Samanta, 2022). We also found a de novo LC variant in TOP2B in case #43. TOP2B codes for topoisomerase II isoenzyme beta, which is abundant in both the developing and the adult brain. Defects of topoisomerase can cause developmental delay, ID, hypotonia, microcephaly, and autistic features (Lam et al., 2017; Hiraide et al., 2020). In proband #44 we identified three variants in SATB2, CSMD1, and DCHS1. Both SATB2 and CSMD1 are SFARI genes with a score of 2, and in our study were transmitted by an affected mother and an unaffected father, respectively. Alongside variants in CSMD1 and DCHS1 recurred in different CAS probands; indeed, it is interesting to note that in another CAS proband (#30), we identified a splicing de-novo variant in the CSMD1 gene, highlighting a potential role of this gene in the CAS phenotype. An ultrarare c.214-7C>T variant in HTR3B was identified in two different CAS cases (#46 and #47). Although its effects on splicing are uncertain (see Supplementary Table 4), we evaluated the recurrence of the variant c.214-7C > T in the HTR3B gene in our CAS cohort, compared to control individuals. Additional LC genes are listed in Table 2. Also interesting is the involvement of the TBK1 gene, though there is no evidence of a child neurodevelopmental phenotype associated with this variant. Likely pathogenic c.1330dup (p. Arg444ThrfsTer5) variant in TBK1 was detected in proband #29, in whom CAS was associated with receptive-expressive LI, and borderline non-verbal IQ. Pathogenic variants in TBK1 have previously been associated with a combined frontotemporal dementia-amyotrophic lateral sclerosis syndrome, a neurodegenerative condition characterized by adult-onset cognitive decline, behavioral abnormalities, dysarthria, and upper and lower motor neuron involvement. Similarly, a nonsense variant in TBK1 has already been described in a patient with dysarthria and an asymmetric akinetic-rigid syndrome (Lamb et al., 2019). Although pathogenic variants in TBK1 have previously been associated with frontotemporal dementia and amyotrophic lateral sclerosis, the phenotype is highly variable, with some cases reported to show primary progressive aphasia and impaired speech production without any other cognitive symptoms (Swift et al., 2021; Price, 2012).
Among the numerous genes considered in this study, some have previously been suggested to be related to CAS. In our study, we identified various variants in genes associated with CAS in the literature, both for HC and LC genes. For example, in two probands (cases #39 and #40), c.440A > G (p.Asp147Gly) and c.3109G > T (p.Asp1037Tyr) were identified in UBA6, a known CAS gene (Hildebrand et al., 2020). In case #41, c.74C > G (p. Thr25Arg) was found in ZFHX4, which others have already shown to be involved (Kaspi et al., 2023; Eising et al., 2019). Also, HC variant identified in KAT6A has already been described as CAS-associated variants (Eising et al., 2019; Kaspi et al., 2023). A gene enrichment analysis (Supplementary Figure 3; Supplementary Table 5), performed by sequencing data drawn from the “1,000 Genomes” database and referring to 107 healthy Italians (Tuscany) plus 395 healthy Europeans of multiple genetic backgrounds (CEU, FIN, GBR, IBS), revealed that rare variants (frequency < 1%) in recurrent genes UBA6, ZFHX4, and KAT6A are more common in children with CAS.
3.3 Gene ontology and expression profile of CAS genes
In an effort to provide a biological interpretation of our data, we performed Gene Ontology (GO) analysis of all the HC genes, using the TOPPGENE database. “Absent speech,” “Intellectual disability, severe” and “Intellectual disability, moderate” (Figure 2A) were found to be the prevalent HPO terms associated with candidate HC genes. GO analysis of the molecular functions, in which the HC genes are involved, suggested the presence of epigenetic mechanisms (Figure 2A). Indeed, these genes encode for DNA-binding proteins and may act as transcriptional activators or suppressors by modulating the expression of target genes. Most HC genes also participate in biological processes involved in chromatin remodeling (Figure 2A), a mechanism closely related to neurodevelopment. Finally, using the BrainSpan database,6 we were able to derive the spatial and temporal expressions of both the HC and the LC genes found in this study and relate them to different brain areas. Preliminary observation, based on the visual analysis of color intensities of the gene enrichment analysis heatmap (Figure 2B). (Figure 2B) showed that KIF5C is highly expressed (red) in many brain regions, indicating strong expression in these tissues; three HC genes (CREBBP, ASHL1 and KDM5B), in addition to four LC ones (DDX1, PQBP1, MYH10, PEX11B) (Supplementary Figure 4), were highly co-expressed throughout the brain, although the cerebellum and dorsal thalamus were the most involved areas. Both of these brain areas contribute to speech and language processing in normal and pathological conditions (Silveri, 2021; Price, 2012; Klostermann et al., 2013).
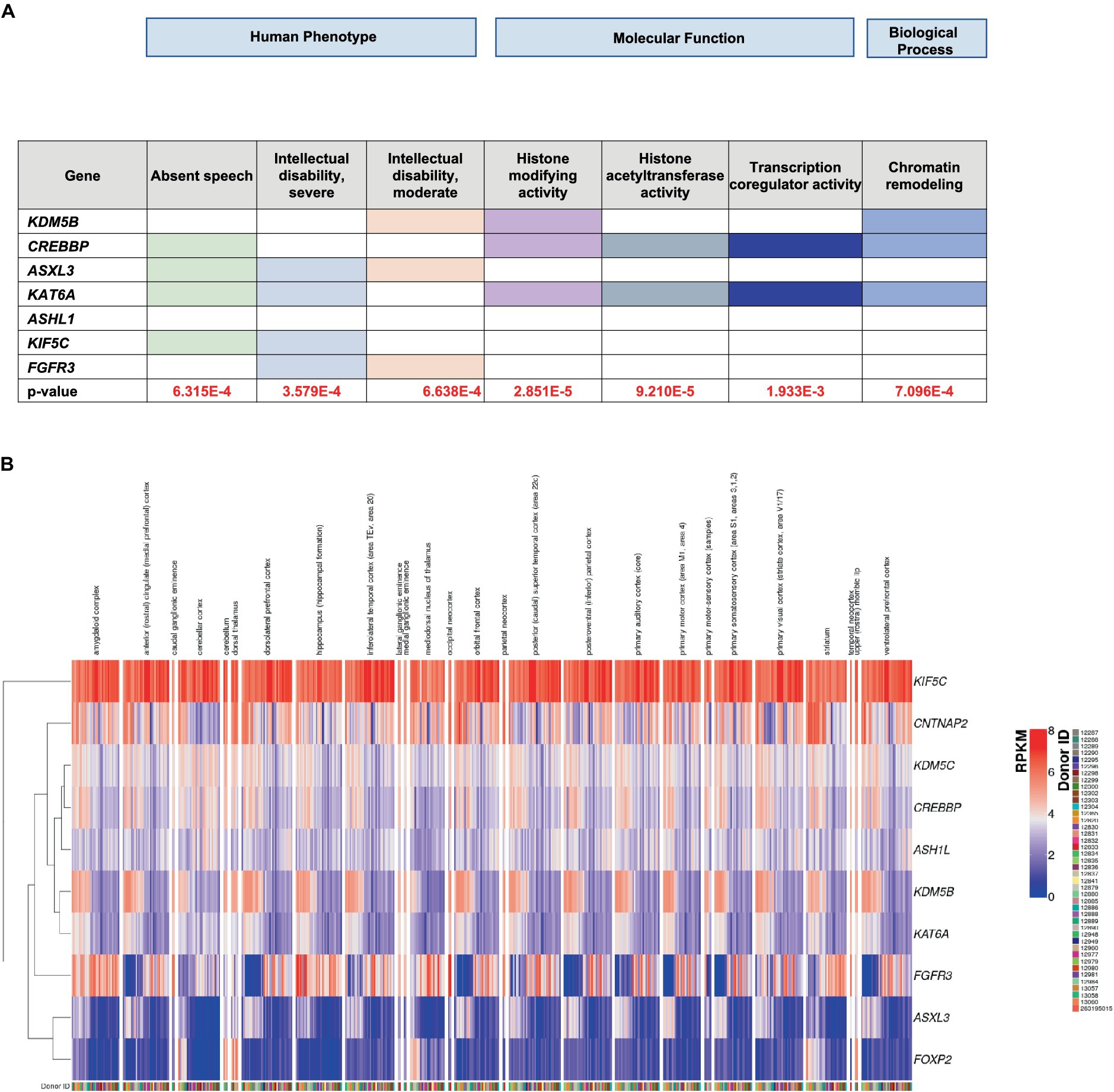
Figure 2. (A) ToppGene enrichment analysis of the GO human phenotype ontology, molecular function and biological process. (B) RNA-seq RPKM values obtained from the BrainSpan database for the set of high-confidence (HC) genes. For each donor available in the BrainSpan Atlas of the Developing Human Brain database, the RNA-seq expression values of the HC genes are plotted using the ComplexHeatmap 2 R package. The heatmap is divided into submodules, each representing the genes expression values for a particular brain area, described at the top of the plot. Note that each donor is associated with one or more columns, since the RPKM values are available for multiple brain areas. Genes are also clustered based on their RPKM values, as represented by the dendogram on the left, in order to better show expression similarity between genes.
4 Discussion
This single-center study performed in Italian children to describe the expanding molecular landscape of CAS yielded potentially relevant findings.
4.1 Molecular characterization and findings
First, molecular characterization was achieved in 21.35% of the cohort, a diagnostic yield that is lower than those found by others using WES. Hildebrand and coworkers obtained a diagnostic rate of 32% (Hildebrand et al., 2020), Eising and collaborators (Eising et al., 2019) a diagnostic rate of 42% and Kaspi and colleagues (Eising et al., 2019) recorded a 26% rate. These findings, however, show that this group of Italian CAS children, like other cohorts, is well characterized, with the identification of some causal genes confirming the remarkable genetic heterogeneity of this disorder. Our findings are consistent with those of previous studies, including a recent review by Morgan et al. (2024), which identified significant genetic heterogeneity in CAS with many implicated genes converging on pathways involved in chromatin modification and transcriptional regulation (Morgan et al., 2024). This reinforces the importance of these pathways in the etiology of CAS and supports the potential for precision diagnosis and targeted therapies.
Second, we described a number of new pathogenic/likely pathogenic variants and novel potential CAS genes, and defined a network that includes both new and previously reported CAS genes. Moreover, we aimed to enhance our understanding of genotype–phenotype correlations. The integration of new findings allows genetic characterization and more precise phenotype identification in most children.
Unlike our earlier study (Chilosi et al., 2022), the present research focused on children with CAS and no co-occurrent ID, ASD and ADHD. Using WES, we found both HC (7 children) and LC (26 children) variants.
Third, all the HC genes found in this study are involved in processes underlying speech and language acquisition, and their alterations yield phenotypes characterized by delayed expressive language, poor or absent speech, LI and ID.
4.2 HC genes, haploinsufficiency, and clinical relevance
Our findings highlight a role for pathogenic/likely pathogenic variants in genes that have either already been related to speech and language impairment (namely, KAT6A, CREBBP and FGFR3), or can be recognized as new possible players, closely linked to neurodevelopmental disorders. It is also of note that our CAS cohort was enriched in variants occurring in genes that encode proteins involved in epigenetic mechanisms and are expressed in brain regions closely linked to language acquisition processes (Silveri, 2021; Price, 2012; Klostermann et al., 2013).
Although HC genes were selected based on their known involvement in language and neurodevelopment, the identification of specific variants provides new insights into their role in CAS. Indeed, HC variants in ASH1L and KDM5B were first reported in relation to CAS phenotypes. In line with what is known in the literature for ASH1L, where loss-of-function variants predominate in intellectual disability, we also identified variants causing premature termination codons in our probands. This is consistent with previous studies that have shown a high prevalence of PTVs in dominant developmental disorders (Faundes et al., 2018).
ASH1L and KDM5B are genes that encode components of the epigenetic apparatus (Miyazaki et al., 2013; Shao et al., 2014), and the superimposed cognitive features such as speech/language deficits that have been reported in chromatin-related disorders (Ng et al., 2023), seem to indicate molecular effects on neural systems dedicated to language and oromotor functioning. Heterozygous variants in ASH1L have been identified in patients with autosomal dominant ID, absent speech, and behavioral abnormalities (OMIM: 617796) (Okamoto et al., 2017; Wang et al., 2016; Stessman et al., 2017). This gene plays a critical role in neurodevelopment by activating HOX genes. It catalyzes mono- and demethylation of H3K36, promoting gene expression (Miyazaki et al., 2013), and it is highly expressed in both embryonic and adult brains (Okamoto et al., 2017). KDM5B was first related to autosomal recessive ID in 2018 (Faundes et al., 2018), however scientific evidence suggested that, unlike homozygous LoF variants, which are likely fully penetrant, heterozygous LoF variants of the same gene have incomplete penetrance and are implicated in autosomal dominant ID and ASD (Borroto et al., 2024.).
As in other neurodevelopmental conditions, the detection of pathogenic or likely pathogenic variants is crucial to allow early identification and prioritization of cases needing further in-depth clinical assessment. Our proband #27, for example, harbored a de novo frameshift alteration in KAT6A, a relatively rare etiology in CAS, ASD, ID, and severe language and communication disorders (Kennedy et al., 2019; St John et al., 2022). Alongside complex and severe presentations, some alterations in KAT6A can also show milder profiles and minor behavioral symptoms (St John et al., 2022). Our findings are in line with those of Kennedy et al., (Kennedy et al., 2019) who observed greater severity of developmental delay and an increased frequency associated with truncating pathogenic variants in the last two exons, suggesting that nonsense-mediated decay may play a role in mitigating the phenotype. Nevertheless, the presence of other clinical features — our case, for example, showed strabismus, early feeding difficulties, and hypotonia, associated with markedly delayed and effortful acquisition of speech and the absence of microcephaly, cardiac abnormalities, and gastrointestinal complications — can facilitate earlier molecular identification. Moreover, detailed neuropsychological phenotyping, performed in a longitudinal perspective, may provide precious information on the long-term outcome of CAS, making it possible to hypothesize a grading of clinical severity based on the deleteriousness of the gene variant. As an example, proband #41 harbored a missense variant in ZFHX4, a gene in which LoF variants usually result in a complex neuropsychological phenotype characterized by developmental delay, ID, and motor speech and language impairment (Fontana et al., 2021). Long-term follow up of this case allowed us to rule out cognitive difficulties, to track changes in language acquisition up to normal functioning, but also to detect the persistence of the motor speech disorder and the emergence of academic learning difficulties at school age.
In our work, the use of dataset enrichment to interpret clinical phenotype is further corroborated by the evidence concerning the LC genes, of which 18/38 (namely, SATB2, SRPX2, LAS1L, SPTBN1, GLS, ZNF142, TCF20, SCN8A, CNTNAP2, KCNAB2, MACF1, CACNA1I, ASXL2, PTCH1, DLL1, KAT6B, PHF21A, KDM4B) are involved in delayed speech and language development (HP:0000750, p = 6.433E-8), language impairment (HP:0002463, p = 9.713E-9), and feeding difficulties (HP:0011968. p = 1.531E-6). Additionally, 11/38 LC genes (ZNF142, LASL1, TCF20, SCN8A, MACF1, PEX11B, DLL1, PQBP1, PHF21A, KIF7, SCN8A) are significantly associated with ID (DisGeNET database, p = 3.933E-11). It is tempting to speculate that minor genetic hits, such as those resulting from LC variants, have some effects during development and influence variable clinical expressivity linked to other factors, including non-genetic ones.
4.3 Gene expression, enrichment analysis, and brain structures involvement
Finally, our attempt to correlate novel etiologies in CAS with the relative genes’ molecular function and spatio-temporal expression in the brain seemed to suggest a role in CAS for two brain structures (cerebellum and dorsal thalamus), both highly involved in neurodevelopment and speech acquisition. A theoretical model of speech motor control acquisition, the DIVA model, maintains that the cerebellum plays a crucial role in the generation of feed-forward motor commands for speech, thus allowing the efficient production of pre-programmed and pre-planned movement sequences. Children with CAS are hypothesized to be unable to generate accurate anticipatory motor commands, and to therefore retain an immature control strategy that relies on slow, retroactive mechanisms (Terband et al., 2009). We ourselves (Fiori et al., 2016), using a connectome approach to study the brains of children with CAS, found reduced connectivity in three subnetworks, one of which (subnetwork 2) also includes the right cerebellum. Finally, a body of literature reports a wide range of symptoms related to speech, language, executive functioning, visuo-spatial processing, academic learning, and behavioral disturbances in children with cerebellar dysfunctions, either acquired or congenital (Riva and Giorgi, 2000; Koziol et al., 2014; Salman and Tsai, 2016; Panagopoulos et al., 2022). The thalamus and the higher-order dorsal thalamic nuclei in particular are richly connected to the prefrontal motor cortices, whose role in speech generation and motor control is well known. Interestingly, the dorsal thalamus appears to play a role in informing several cortical areas of an upcoming motor command and in providing an anticipatory model of the expected sensory consequences of a motor response (Sherman, 2007). Beyond the suggested implications of the dorsal thalamus in motor control, some authors also highlight its functional contributions to distinct cognitive and executive skills, such as working memory, new learning, adaptive decision making, attention and language (Ouhaz et al., 2018). Relationships between speech, language and thalamic function have also been studied after thalamotomy in children (Nass et al., 2000). The role of the cerebellum in generating motor commands for speech and the thalamus’s function in motor and cognitive control further highlight their importance in CAS.
Deep speech and language phenotyping revealed a different profile between the HC and LC groups. With regard to the severity of the speech disorder, our comparison of these two groups did not reveal any statistically significant difference, but the number of children with receptive-expressive language impairment was higher among those with HC variants. This finding is of clinical relevance. If confirmed in other study cohorts, and for speakers of other languages, it should inform therapeutic choices and allow a better approach to the comorbid manifestations that can be present in children with CAS (Miller et al., 2019). Although additional studies are necessary to consolidate this information, our expansion of the genetic landscape in CAS allowed us to focus on molecular pathways and regional and anatomical brain expression. This approach could potentially help to disentangle the complex mechanisms leading to impaired language acquisition processes, and favor the development of new tools for early detection and better care. Given the genetic heterogeneity and pleiotropy, the assessment of children with CAS should be multidisciplinary, as some genetic variants could be associated not only to other neurodevelopmental disorders, but also to multiple organs alterations, such as eye defects (i.e., in KAT6A). Moreover, the frequent association of CAS with language disorder, the increased risk of developing academic learning difficulties and atypical implicit learning (Bombonato et al., 2022), mandates intensive therapy and educational management aimed to provide specific and dynamic support to the children and to their families across the different needs emerging in development. Alternative strategies for communications should be implemented to support children with the most severe, persistent and complex presentations of the disorder. Deep speech, language and cognitive phenotyping is crucial for future genetic research and for clinical decision making.
Data availability statement
The data presented in the study are deposited in the European Variation Archive (EVA) at EMBL-EBI, accession number PRJEB74924 (https://www.ebi.ac.uk/eva/?eva-study=PRJEB74924).
Ethics statement
The studies involving humans were approved by the CEPR Comitato Etico Pediatrico Regione Toscana. The studies were conducted in accordance with local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin.
Author contributions
DF: Conceptualization, Data curation, Supervision, Writing – original draft, Writing – review & editing, Formal analysis, Investigation, Methodology, Resources, Software, Validation, Visualization. IP: Formal analysis, Investigation, Methodology, Writing – original draft, Writing – review & editing. ED: Software, Writing – review & editing. EA: Writing – review & editing. SG: Writing – review & editing. PC: Funding acquisition, Writing – original draft, Writing – review & editing. CB: Cognitive assessments, Writing – review & editing. FMS: Data curation, Writing – original draft, Writing – review & editing, Conceptualization, Funding acquisition, Project administration, Supervision. AC: Conceptualization, Data curation, Funding acquisition, Investigation, Methodology, Project administration, Supervision, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. The research activity was supported by Italian Ministry of Health grants and by Regional Pediatric Ethics Committee (CEP) 19-03-2018/RF2016-0236156 to AC and Ricerca Corrente 2023 to FMS. The publication of this paper was fully supported by FMS through the “RC 5x1000” funding.
Acknowledgments
The authors are deeply grateful to the children and families who participated in this work. We would also like to thank the speech therapists B. Franchi, L. Diridoni, A. Ridulfo, and I. Baroni who helped with the speech and language clinical tests.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnins.2024.1396240/full#supplementary-material
Footnotes
1. ^https://www.asha.org/policy/tr2007-00278/
2. ^https://broadinstitute.github.io/picard/ accessed on 14 December 2023
3. ^https://www.sfari.org/ last accessed on December 31, 2022
4. ^https://www.brainspan.org/rnaseq/searches
5. ^http://bioconductor.org/packages/release/bioc/html/ComplexHeatmap.html
References
American Speech-Language-Hearing Association. (2007). Childhood apraxia of speech [technical report]. Available at: https://www.asha.org/policy/tr2007-00278
Aravidis, C., Konialis, C. P., Pangalos, C. G., and Kosmaidou, Z. (2014). A familial case of Muenke syndrome. Diverse expressivity of the FGFR3 Pro252Arg mutation--case report and review of the literature. J. Matern. Fetal Neonatal Med. 27, 1502–1506. doi: 10.3109/14767058.2013.860520
Bombonato, C., Casalini, C., Pecini, C., Angelucci, G., Vicari, S., Podda, I., et al. (2022). Implicit learning in children with childhood apraxia of speech. Res. Dev. Disabil. 122:104170. doi: 10.1016/j.ridd.2021.104170
Borroto, M. C., Michaud, C., Hudon, C., Agrawal, P. B., Agre, K., Applegate, C. D., et al. (2024). A genotype/phenotype study of KDM5B-associated disorders suggests a pathogenic effect of dominantly inherited missense variants. Genes 15:1033. doi: 10.3390/genes15081033
Cassar, C., McCabe, P., and Cumming, S. (2023). “I still have issues with pronunciation of words”: a mixed methods investigation of the psychosocial and speech effects of childhood apraxia of speech in adults. Int. J. Speech Lang. Pathol. 25, 193–205. doi: 10.1080/17549507.2021.2018496
Chenausky, K. V., Baas, B., Stoeckel, R., Brown, T., Green, J. R., Runke, C., et al. (2023). Comorbidity and severity in childhood apraxia of speech: a retrospective chart review. J. Speech Lang. Hear. Res. 66, 1–13. doi: 10.1044/2022_JSLHR-22-00436
Chenausky, K. V., Brignell, A., Morgan, A., Gagné, D., Norton, A., Tager-Flusberg, H., et al. (2020). Factor analysis of signs of childhood apraxia of speech. J. Commun. Disord. 87:106033. doi: 10.1016/j.jcomdis.2020.106033
Chilosi, A. M., Brovedani, P., Cipriani, P., and Casalini, C. (2023). Sex differences in early language delay and in developmental language disorder. J. Neurosci. Res. 101, 654–667. doi: 10.1002/jnr.24976
Chilosi, A. M., Lorenzini, I., Fiori, S., Graziosi, V., Rossi, G., Pasquariello, R., et al. (2015). Behavioral and neurobiological correlates of childhood apraxia of speech in Italian children. Brain Lang. 150, 177–185. doi: 10.1016/j.bandl.2015.10.002
Chilosi, A. M., Pfanner, L., Pecini, C., Salvadorini, R., Casalini, C., Brizzolara, D., et al. (2019). Which linguistic measures distinguish transient from persistent language problems in late talkers from 2 to 4 years? A study on Italian speaking children. Res. Dev. Disabil. 89, 59–68. doi: 10.1016/j.ridd.2019.03.005
Chilosi, A. M., Podda, I., Ricca, I., Comparini, A., Franchi, B., Fiori, S., et al. (2022). Differences and commonalities in children with childhood apraxia of speech and comorbid neurodevelopmental disorders: a multidimensional perspective. J Pers Med 12:313. doi: 10.3390/jpm12020313
Conti, E., Retico, A., Palumbo, L., Spera, G., Bosco, P., Biagi, L., et al. (2020). Autism Spectrum disorder and childhood apraxia of speech: early language-related hallmarks across structural MRI study. J Pers Med 10:275. doi: 10.3390/jpm10040275
Eising, E., Carrion-Castillo, A., Vino, A., Strand, E. A., Jakielski, K. J., Scerri, T. S., et al. (2019). A set of regulatory genes co-expressed in embryonic human brain is implicated in disrupted speech development. Mol. Psychiatry 24, 1065–1078. doi: 10.1038/s41380-018-0020-x
Faundes, V., Newman, W. G., Bernardini, L., Canham, N., Clayton-Smith, J., Dallapiccola, B., et al. (2018). Histone lysine methylases and demethylases in the landscape of human developmental disorders. Am. J. Hum. Genet. 102, 175–187. doi: 10.1016/j.ajhg.2017.11.013
Fiori, S., Guzzetta, A., Mitra, J., Pannek, K., Pasquariello, R., Cipriani, P., et al. (2016). Neuroanatomical correlates of childhood apraxia of speech: a connectomic approach. Neuroimage Clin 12, 894–901. doi: 10.1016/j.nicl.2016.11.003
Fontana, P., Ginevrino, M., Bejo, K., Cantalupo, G., Ciavarella, M., Lombardi, C., et al. (2021). A ZFHX4 mutation associated with a recognizable neuropsychological and facial phenotype. Eur. J. Med. Genet. 64:104321. doi: 10.1016/j.ejmg.2021.104321
Hall, P. K., Jordan, L. S., and Robin, D. A. (1993). Developmental apraxia of speech: Theory and clinical practice. Virginia: Pro-Ed, Università della.
Hildebrand, M. S., Jackson, V. E., Scerri, T. S., Van Reyk, O., Coleman, M., Braden, R. O., et al. (2020). Severe childhood speech disorder: gene discovery highlights transcriptional dysregulation. Neurology 94, e2148–e2167. doi: 10.1212/WNL.0000000000009441
Hiraide, T., Watanabe, S., Matsubayashi, T., Yanagi, K., Nakashima, M., Ogata, T., et al. (2020). A de novo TOP2B variant associated with global developmental delay and autism spectrum disorder. Mol. Genet. Genomic Med. 8:e1145. doi: 10.1002/mgg3.1145
Iuzzini-Seigel, J., Allison, K. M., and Stoeckel, R. (2022). A tool for differential diagnosis of childhood apraxia of speech and dysarthria in children: a tutorial. Lang. Speech Hear. Serv. Sch. 53, 926–946. doi: 10.1044/2022_LSHSS-21-00164
Kadis, D. S., Goshulak, D., Namasivayam, A., Pukonen, M., Kroll, R., De Nil, L. F., et al. (2014). Cortical thickness in children receiving intensive therapy for idiopathic apraxia of speech. Brain Topogr. 27, 240–247. doi: 10.1007/s10548-013-0308-8
Kaspi, A., Hildebrand, M. S., Jackson, V. E., Braden, R., van Reyk, O., Howell, T., et al. (2023). Genetic aetiologies for childhood speech disorder: novel pathways co-expressed during brain development. Mol. Psychiatry 28, 1647–1663. doi: 10.1038/s41380-022-01764-8
Kennedy, J., Goudie, D., Blair, E., Chandler, K., Joss, S., McKay, V., et al. (2019). KAT6A syndrome: genotype-phenotype correlation in 76 patients with pathogenic KAT6A variants. Genet. Med. 21, 850–860. doi: 10.1038/s41436-018-0259-2
Klostermann, F., Krugel, L. K., and Ehlen, F. (2013). Functional roles of the thalamus for language capacities. Front. Syst. Neurosci. 7:32. doi: 10.3389/fnsys.2013.00032
Koziol, L. F., Budding, D., Andreasen, N., D’Arrigo, S., Bulgheroni, S., Imamizu, H., et al. (2014). Consensus paper: the cerebellum’s role in movement and cognition. Cerebellum 13, 151–177. doi: 10.1007/s12311-013-0511-x
Kruszka, P., Addissie, Y. A., Yarnell, C. M., Hadley, D. W., Guillen Sacoto, M. J., Platte, P., et al. (2016). Muenke syndrome: an international multicenter natural history study. Am. J. Med. Genet. A 170, 918–929. doi: 10.1002/ajmg.a.37528
Kruszka, P., Rolle, M., Kahle, K. T., and Muenke, M. (1993). “Muenke Syndrome” in GeneReviews((R)). eds. M. P. Adam, J. Feldman, G. M. Mirzaa, R. A. Pagon, S. E. Wallace, and L. J. H. Bean, et al. (Seattle (WA): University of Washington).
Lam, C. W., Yeung, W. L., and Law, C. Y. (2017). Global developmental delay and intellectual disability associated with a de novo TOP2B mutation. Clin. Chim. Acta 469, 63–68. doi: 10.1016/j.cca.2017.03.022
Lamb, R., Rohrer, J. D., Real, R., Lubbe, S. J., Waite, A. J., Blake, D. J., et al. (2019). A novel TBK1 mutation in a family with diverse frontotemporal dementia spectrum disorders. Cold Spring Harb Mol Case Stud 5:a003913. doi: 10.1101/mcs.a003913
Lewis, B. A., Benchek, P., Tag, J., Miller, G., Freebairn, L., Taylor, H. G., et al. (2021). Psychosocial comorbidities in adolescents with histories of childhood apraxia of speech. Am. J. Speech Lang. Pathol. 30, 2572–2588. doi: 10.1044/2021_AJSLP-21-00035
Lewis, B. A., Freebairn, L. A., Hansen, A. J., Iyengar, S. K., and Taylor, H. G. (2004). School-age follow-up of children with childhood apraxia of speech. Lang. Speech Hear. Serv. Sch. 35, 122–140. doi: 10.1044/0161-1461(2004/014)
Lewis, B. A., Miller, G. J., Iyengar, S. K., Stein, C., and Benchek, P. (2023). Long-term outcomes for individuals with childhood apraxia of speech. J. Speech Lang. Hear. Res. 1–17. doi: 10.1044/2023_JSLHR-22-00647
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079. doi: 10.1093/bioinformatics/btp352
Maleki, F., Ovens, K., Hogan, D. J., and Kusalik, A. J. (2020). Gene Set Analysis: Challenges, Opportunities, and Future Research. Front. Genet. 11:654. doi: 10.3389/fgene.2020.00654
Menke, L. A., van Belzen, M. J., Alders, M., Cristofoli, F., Ehmke, N., Fergelot, P., et al. (2016). CREBBP mutations in individuals without Rubinstein-Taybi syndrome phenotype. Am. J. Med. Genet. A 170, 2681–2693. doi: 10.1002/ajmg.a.37800
Miller, G. J., Lewis, B., Benchek, P., Freebairn, L., Tag, J., Budge, K., et al. (2019). Reading outcomes for individuals with histories of suspected childhood apraxia of speech. Am. J. Speech Lang. Pathol. 28, 1432–1447. doi: 10.1044/2019_AJSLP-18-0132
Miller, G. J., and Lewis, B. A. (2022). Reading skills in children with suspected childhood apraxia of speech and children with Reading disorders: same or different? Lang. Speech Hear. Serv. Sch. 53, 985–1005. doi: 10.1044/2022_LSHSS-21-00149
Miller, J. A., Ding, S. L., Sunkin, S. M., Smith, K. A., Ng, L., Szafer, A., et al. (2014). Transcriptional landscape of the prenatal human brain. Nature 508, 199–206. doi: 10.1038/nature13185
Miyazaki, H., Higashimoto, K., Yada, Y., Endo, T. A., Sharif, J., Komori, T., et al. (2013). Ash1l methylates Lys36 of histone H3 independently of transcriptional elongation to counteract polycomb silencing. PLoS Genet. 9:e1003897. doi: 10.1371/journal.pgen.1003897
Moko, S. B., and Blandin de Chalain, T. M. (2001). New Zealand Maori family with the pro250arg fibroblast growth factor receptor 3 mutation associated with craniosynostosis. J. Craniomaxillofac. Surg. 29, 22–24.
Morgan, A., Amor, J. D., John, M. D., Sheffer, I. E., and Hildebrand, M. S. (2024). Genetic architecture of childhood speech disorder: a review. Mol. Psychiatry 29, 1281–1292. doi: 10.1038/s41380-024-02409-8
Morison, L. D., Meffert, E., Stampfer, M., Wilke, I. S., Vollmer, B. V., Schulze, K., et al. (2023). In-depth characterisation of a cohort of individuals with missense and loss-of-function variants disrupting FOXP2. J. Med. Genet. 60, 597–607. doi: 10.1136/jmg-2022-108734
Murray, E., Thomas, D., and McKechnie, J. (2019). Comorbid morphological disorder apparent in some children aged 4-5 years with childhood apraxia of speech: findings from standardised testing. Clin. Linguist. Phon. 33, 42–59. doi: 10.1080/02699206.2018.1513565
Nass, R., Boyce, L., Leventhal, F., Levine, B., Allen, J., Maxfield, C., et al. (2000). Acquired aphasia in children after surgical resection of left-thalamic tumors. Dev. Med. Child Neurol. 42, 580–590. doi: 10.1017/s0012162200001109
Ng, R., Harris, J., Kleefstra, T., Morgan, A. T., and Simpson, B. (2023). Editorial: characterizing the neurobehavioral phenotype of mendelian disorders of epigenetic machinery. Front. Genet. 14:1338078. doi: 10.3389/fgene.2023.1338078
Okamoto, N., Miya, F., Tsunoda, T., Kato, M., Saitoh, S., Yamasaki, M., et al. (2017). Novel MCA/ID syndrome with ASH1L mutation. Am. J. Med. Genet. A 173, 1644–1648. doi: 10.1002/ajmg.a.38193
Ouhaz, Z., Fleming, H., and Mitchell, A. S. (2018). Cognitive functions and neurodevelopmental disorders involving the prefrontal cortex and mediodorsal thalamus. Front. Neurosci. 12:33. doi: 10.3389/fnins.2018.00033
Panagopoulos, D., Stranjalis, G., Gavra, M., Boviatsis, E., Korfias, S., Karydakis, P., et al. (2022). The entity of cerebellar mutism syndrome: a narrative review centered on the etiology, diagnostics, prevention, and therapeutic options. Children 10:83. doi: 10.3390/children10010083
Price, C. J. (2012). A review and synthesis of the first 20 years of PET and fMRI studies of heard speech, spoken language and reading. NeuroImage 62, 816–847. doi: 10.1016/j.neuroimage.2012.04.062
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424. doi: 10.1038/gim.2015.30
Riva, D., and Giorgi, C. (2000). The cerebellum contributes to higher functions during development: evidence from a series of children surgically treated for posterior fossa tumours. Brain 123, 1051–1061. doi: 10.1093/brain/123.5.1051
Robin, N. H., Scott, J. A., Cohen, A. R., and Goldstein, J. A. (1998). Nonpenetrance in FGFR3-associated coronal synostosis syndrome. Am. J. Med. Genet. 80, 296–297. doi: 10.1002/(SICI)1096-8628(19981116)80:3<296::AID-AJMG25>3.0.CO;2-6
Salman, M. S., and Tsai, P. (2016). The role of the pediatric cerebellum in motor functions, cognition, and behavior: a clinical perspective. Neuroimaging Clin. N. Am. 26, 317–329. doi: 10.1016/j.nic.2016.03.003
Samanta, D. (2022). DEPDC5-related epilepsy: a comprehensive review. Epilepsy Behav. 130:108678. doi: 10.1016/j.yebeh.2022.108678
Shao, G. B., Chen, J. C., Zhang, L. P., Huang, P., Lu, H. Y., Jin, J., et al. (2014). Dynamic patterns of histone H3 lysine 4 methyltransferases and demethylases during mouse preimplantation development. In Vitro Cell. Dev. Biol. Anim. 50, 603–613. doi: 10.1007/s11626-014-9741-6
Sherman, S. M. (2007). The thalamus is more than just a relay. Curr. Opin. Neurobiol. 17, 417–422. doi: 10.1016/j.conb.2007.07.003
Shriberg, L. D., Aram, D. M., and Kwiatkowski, J. (1997). Developmental apraxia of speech: I. Descriptive and theoretical perspectives. J. Speech Lang. Hear. Res. 40, 273–285. doi: 10.1044/jslhr.4002.273
Shriberg, L. D., Kwiatkowski, J., and Mabie, H. L. (2019a). Estimates of the prevalence of motor speech disorders in children with idiopathic speech delay. Clin. Linguist. Phon. 33, 679–706. doi: 10.1080/02699206.2019.1595731
Shriberg, L. D., Potter, N. L., and Strand, E. A. (2011). Prevalence and phenotype of childhood apraxia of speech in youth with galactosemia. J. Speech Lang. Hear. Res. 54, 487–519. doi: 10.1044/1092-4388(2010/10-0068)
Shriberg, L. D., Strand, E. A., Jakielski, K. J., and Mabie, H. L. (2019b). Estimates of the prevalence of speech and motor speech disorders in persons with complex neurodevelopmental disorders. Clin. Linguist. Phon. 33, 707–736. doi: 10.1080/02699206.2019.1595732
Silveri, M. C. (2021). Contribution of the cerebellum and the basal ganglia to language production: speech, word fluency, and sentence construction-evidence from pathology. Cerebellum 20, 282–294. doi: 10.1007/s12311-020-01207-6
St John, M., Amor, D. J., and Morgan, A. T. (2022). Speech and language development and genotype-phenotype correlation in 49 individuals with KAT6A syndrome. Am. J. Med. Genet. A 188, 3389–3400. doi: 10.1002/ajmg.a.62899
Stein, C. M., Benchek, P., Miller, G., Hall, N. B., Menon, D., Freebairn, L., et al. (2020). Feature-driven classification reveals potential comorbid subtypes within childhood apraxia of speech. BMC Pediatr. 20:519. doi: 10.1186/s12887-020-02421-1
Stessman, H. A., Xiong, B., Coe, B. P., Wang, T., Hoekzema, K., Fenckova, M., et al. (2017). Targeted sequencing identifies 91 neurodevelopmental-disorder risk genes with autism and developmental-disability biases. Nat. Genet. 49, 515–526. doi: 10.1038/ng.3792
Swift, I. J., Bocchetta, M., Benotmane, H., Woollacott, I. O., Shafei, R., and Rohrer, J. D. (2021). Variable clinical phenotype in TBK1 mutations: case report of a novel mutation causing primary progressive aphasia and review of the literature. Neurobiol. Aging 99, 100 e109–100 e115. doi: 10.1016/j.neurobiolaging.2020.08.014
Terband, H., Maassen, B., Guenther, F. H., and Brumberg, J. (2009). Computational neural modeling of speech motor control in childhood apraxia of speech (CAS). J. Speech Lang. Hear. Res. 52, 1595–1609. doi: 10.1044/1092-4388(2009/07-0283)
Thorvaldsdottir, H., Robinson, J. T., and Mesirov, J. P. (2013). Integrative genomics viewer (IGV): high-performance genomics data visualization and exploration. Brief. Bioinform. 14, 178–192. doi: 10.1093/bib/bbs017
Wang, K., Li, M., and Hakonarson, H. (2010). ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 38:e164. doi: 10.1093/nar/gkq603
Wang, T., Guo, H., Xiong, B., Stessman, H. A., Wu, H., Coe, B. P., et al. (2016). De novo genic mutations among a Chinese autism spectrum disorder cohort. Nat. Commun. 7:13316. doi: 10.1038/ncomms13316
Zhang, Y., Wang, Q., Zhang, Y., Chen, Y., Liu, T., Zhao, J., et al. (2023). Functional enrichment analysis of mutated genes in neuropsychiatric disorders using TOPPGENE. Neuropsychiatric Genetics 19, 215–230. doi: 10.1093/neuogen/gny123
Keywords: childhood apraxia of speech, exome sequencing, high confidence genes, low confidence genes, gene ontology and expression profile of CAS genes
Citation: Formicola D, Podda I, Dirupo E, Andreucci E, Giglio S, Cipriani P, Bombonato C, Santorelli FM and Chilosi A (2024) Expanding the molecular landscape of childhood apraxia of speech: evidence from a single-center experience. Front. Neurosci. 18:1396240. doi: 10.3389/fnins.2024.1396240
Edited by:
Silvia Pellegrini, University of Pisa, ItalyReviewed by:
Benedetto Vitiello, University of Turin, ItalyDavid Amor, The University of Melbourne, Australia
Else Eising, Max Planck Institute for Psycholinguistics, Netherlands
Copyright © 2024 Formicola, Podda, Dirupo, Andreucci, Giglio, Cipriani, Bombonato, Santorelli and Chilosi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Filippo Maria Santorelli, ZmlsaXBwbzMzNjRAZ21haWwuY29t
†Present address: Daniela Formicola, U.O.C. Medical Genetics Laboratory, Department of Experimental Medicine, San Camillo Forlanini Hospital - Sapienza University, Rome, Italy
‡ORCID: Anna Chilosi, http://orcid.org/0000-0002-0005-7512