 Esther García-García1,2,3
Esther García-García1,2,3 Maria Carreras-Caballé1,2Albert Coll-Manzano1Alba Ramón-Lainez1,2,3Gisela Besa-Selva1,2,3
Maria Carreras-Caballé1,2Albert Coll-Manzano1Alba Ramón-Lainez1,2,3Gisela Besa-Selva1,2,3 Esther Pérez-Navarro1,2,3
Esther Pérez-Navarro1,2,3 Cristina Malagelada1,2,3
Cristina Malagelada1,2,3 Jordi Alberch1,2,3,4
Jordi Alberch1,2,3,4 Mercè Masana1,2,3*†
Mercè Masana1,2,3*† Manuel J. Rodríguez1,2,3*†
Manuel J. Rodríguez1,2,3*†- 1Department of Biomedical Sciences, School of Medicine and Health Sciences, Institute of Neurosciences, Universitat de Barcelona, Barcelona, Spain
- 2August Pi i Sunyer Biomedical Research Institute (IDIBAPS), Barcelona, Spain
- 3Centro de Investigación Biomédica en Red de Enfermedades Neurodegenerativas (CIBERNED), Instituto de Salud Carlos III, Barcelona, Spain
- 4Production and Validation Center of Advanced Therapies (Creatio), Faculty of Medicine and Health Science, University of Barcelona, Barcelona, Spain
VPS13A disease and Huntington’s disease (HD) are two basal ganglia disorders that may be difficult to distinguish clinically because they have similar symptoms, neuropathological features, and cellular dysfunctions with selective degeneration of the medium spiny neurons of the striatum. However, their etiology is different. VPS13A disease is caused by a mutation in the VPS13A gene leading to a lack of protein in the cells, while HD is due to an expansion of CAG repeat in the huntingtin (Htt) gene, leading to aberrant accumulation of mutant Htt. Considering the similarities of both diseases regarding the selective degeneration of striatal medium spiny neurons, the involvement of VPS13A in the molecular mechanisms of HD pathophysiology cannot be discarded. We analyzed the VPS13A distribution in the striatum, cortex, hippocampus, and cerebellum of a transgenic mouse model of HD. We also quantified the VPS13A levels in the human cortex and putamen nucleus; and compared data on mutant Htt-induced changes in VPS13A expression from differential expression datasets. We found that VPS13A brain distribution or expression was unaltered in most situations with a decrease in the putamen of HD patients and small mRNA changes in the striatum and cerebellum of HD mice. We concluded that the selective susceptibility of the striatum in VPS13A disease and HD may be a consequence of disturbances in different cellular processes with convergent molecular mechanisms already to be elucidated.
1 Introduction
Basal ganglia disorders are a heterogeneous group of neurodegenerative diseases that affect selectively a group of neuronal populations in cortical and subcortical circuitries. VPS13A disease (chorea-acanthocytosis, ChAc, OMIM: 200150; Jung et al., 2011; Peikert et al., 2023) and Huntington’s disease (HD, OMIM: 143100; Ross et al., 2014) are two basal ganglia disorders that are often clinically difficult to distinguish because they have similar symptoms, magnetic resonance imaging findings (Suzuki et al., 2020) and neuropathological features with selective degeneration of the medium spiny neurons (MSNs) in the striatum (Liu et al., 2019). However, the etiology of both diseases is different. ChAc is a rare autosomal recessive neurodegenerative disease caused by a mutation in the VPS13A gene located on chromosome 9, which encodes the protein VPS13A (Ueno et al., 2001). Meanwhile, HD is an autosomal dominant disease that is due to an expansion of CAG repeat in the huntingtin (Htt) gene located on chromosome 4 (Ross et al., 2014). Thus, MSNs have a selective vulnerability to the lack of VPS13A (Henkel et al., 2008; Walterfang et al., 2011) and to the accumulation of mutant Htt (mHtt; Vonsattel and DiFiglia, 1998; Kassubek et al., 2004), while other neuronal populations in the striatum, such as interneurons, are more resistant. Therefore, the understanding of the mechanisms that make MSNs more vulnerable can be useful in developing therapeutical strategies to protect these neurons and their circuitries.
The low frequency of ChAc patients determines the little knowledge so far about the localization and function of VPS13A. This is a large ubiquitous protein highly expressed in the brain with a distinct VPS13A distribution that contributes to explaining the ChAc neuropathology (García-García et al., 2021). However, although the main neuropathological feature in ChAc patients is the selective degeneration of the caudate nucleus and putamen, the concentration of VPS13A in basal ganglia nuclei is weak (Kurano et al., 2007; García-García et al., 2021). Thus, the vulnerability of striatal neurons to VPS13A disease seems not to be related to the amount of protein present in the cell, but to specific striatal functional properties, circuitry, and MSN cell processes specifically affected by the lack of VPS13A.
VPS13A has a large variety of cellular functions that can affect the selective vulnerability of distinct neuronal populations. At the cellular level, VPS13A is a lipid transport protein localized in the contact sites between organelles (Kumar et al., 2018). Its lack of function has been associated with endocytic trafficking and lysosomal degradation impairment (Muñoz-Braceras et al., 2019), impaired autophagic degradation (Muñoz-Braceras et al., 2015), and abnormal calcium homeostasis (Pelzl et al., 2017). Furthermore, although VPS13A is not enriched in the synaptic compartment (Kurano et al., 2007; García-García et al., 2021), it is important for maintaining the neuronal optimal synaptic activity. Indeed, synaptic plasticity impairment and deficient glutamatergic and BDNF transmissions have been related to mouse corticostriatal VPS13A knockdown (García-García et al., 2023). In this line, enhanced neurite outgrowth and ramifications have been described in MSNs differentiated from hiPSC derived from fibroblast of ChAc (Stanslowsky et al., 2016), reinforcing the role of VPS13A in shaping neuronal dendritic morphology.
Interestingly, in HD, mHtt expression is also ubiquitous (Marques Sousa and Humbert, 2013). It accumulates in neurons as insoluble and hardly removable aggregates leading to synaptic abnormalities. Impairment of corticostriatal synaptic plasticity has been widely documented in HD mouse models and patients, with important glutamatergic and BDNF transmission disturbances in the molecular basis of these impairments (Canals et al., 2004; Del Toro et al., 2006). Furthermore, the accumulation of mHtt impairs a plethora of cell functions, including disruption of the ERK1/2 and the PKA signaling pathways (Saavedra et al., 2011; Tyebji et al., 2015). More interestingly mHtt also alters autophagy (Ravikumar et al., 2004), RNA splicing processes (Fernández-Nogales et al., 2016), and the rate of synthesis of selective proteins in the striatum (Creus-Muncunill et al., 2019), being these alterations at the basis of the molecular mechanisms of MSN dysfunction.
Considering all that, the involvement of VPS13A in the molecular mechanisms of HD pathophysiology cannot be discarded. The first step to approach this hypothesis is to depict the distribution of VPS13A in the HD brain, which should help to further understand its role in both basal ganglia synaptic plasticity and connectivity, and the ChAc neurodegenerative mechanisms. Thus, this study is focused on the analysis of the putative changes in VPS13A brain distribution induced by mHtt, especially in the striatum, cortex, hippocampus, and cerebellum.
2 Materials and methods
2.1 Human post-mortem nervous tissue
Human post-mortem tissue samples of the motor cortex and putamen were used to assess VPS13A concentration. Samples were collected at autopsy from individuals who had suffered a clinical history of HD (n = 3 female + 4 male, age: 54.4 (28–72) years; postmortem intervals of 4–18 h), and from non-HD controls (n = 4 female +2 male, age: 62.7 (39–81) years; postmortem intervals of 4–17 h; Table 1).

Table 1. Human post-mortem Huntington’s disease samples used in this study.
2.2 HD mouse model
Male and female R6/1 transgenic mice expressing the human exon-1 of mHtt containing 145 CAG repeats, and their corresponding wildtype (WT) littermates were obtained from Jackson Laboratory (Bar Harbor, ME, United States) and maintained in a B6CBA background. Animals were housed together in groups of mixed genotypes and kept under a 12:12 h light/dark cycle in a room at 19–22 °C and 40–60% humidity, with free access to food and water.
2.3 Mouse brain tissue sampling
Mice aged 20 weeks were used in the experiments. At this age, R6/1 mice show motor disturbances and synaptic plasticity impairment (Fernández-García et al., 2020; Kim et al., 2020). For immunohistochemical analysis, mice were anesthetized with a mixture of ketamine plus xylazine (100 + 10 mg/kg, i.p.) and transcardially perfused with ice-cooled 0.1 M PBS, followed by 4% paraformaldehyde (PFA). Then, brains were removed and fixed by immersion in 4% PFA at 4°C overnight. All PFA-fixed brains were cryoprotected with 30% sucrose in 0.1 M PBS and 0.02% sodium azide and frozen in dry ice-cooled isopentane. Specimens were stored at −80°C until sectioning. Sagittal serial sections were collected at 14 μm with a cryotome. For biochemical analysis, mice underwent euthanasia by cervical dislocation, and the brains were removed, dissected, and kept at −80°C.
2.4 Fluorescence in situ hybridization
A FISH procedure was performed using the RNAscope® 2.5 High Definition–Red Assay kit (Advanced Cell Diagnostics, Newark, CA, United States), following the instructions of the manufacturer and as previously reported in García-García et al. (2021). The target probe for the mouse Vps13a gene (Probe-Mm-Vps13a-E61-E71-C2, Advanced Cell Diagnostics, Newark, CA, United States) was hybridized for 2 h at 40°C, followed by a series of signal amplification and washing steps. Hybridizations were performed in a HybEZTM Hybridization System (Advanced Cell Diagnostics, Newark, CA, United States). Negative controls were performed with a negative control probe (targeting the DapB gene from the Bacillus subtilis strain SMY) provided by the kit. Specific hybridization signals were detected by fluorescence, and RNA staining was identified as red dots.
Images of the Vps13a expression staining were obtained with an inverted microscope (Leica DMI6000 B, Thermo Fisher Scientific, Waltham, MA, United States). For qualitative visual analysis of the intensity of Vps13a mRNA labeling in the slices, digital images were processed using an 8-bit 16-color lookup table with ImageJ 1.51a (National Institutes of Health, Bethesda, MD, United States).
2.5 Quantitative real-time PCR
The aqueous phase containing total RNA was isolated from the different mouse brain regions using QIAzol (Qiagen, Hilden, Germany), following the protocol of the manufacturer. Total RNA isolation, reverse transcription of RNA and qRT-PCR were performed as already described (García-García et al., 2021). PrimeTime qPCR assays were used as recommended by the provider (assay code Mm.PT.56a.8500899, sequence NM_173028(1) for Vps13a, and assay code Mm.PT.39a.1 sequence NM_008084 for GAPDH; IDT technologies, United States). The expression level was determined using a standard curve and normalized to housekeeper Gapdh gene mRNA levels. The ΔΔCt method was used to analyze the data.
2.6 Western blot
Tissues were homogenized in lysis buffer and protein samples were resolved in SDS-PAGE and NuPAGE gels as already published (García-García et al., 2023). Immunoblots were probed with anti- VPS13A (1:1500, Cat: HPA021662, Sigma-Aldrich, St. Louis, MI, United States). Immunoreactive bands were visualized using the Western blotting Luminol Reagent (Santa Cruz Biotechnology, Dallas, TX, United States). Images were acquired using Chemidoc™ (Bio-Rad, Hercules, CA, United States) and quantified by a computer-assisted densitometer (ImageLab™, Bio-Rad, Hercules, CA, United States).
2.7 Statistical analysis
All experiments were blinded and randomized. All results are reported as mean ± SEM. Normal distribution of data was assumed when the Shapiro–Wilk test was positive for normality. Statistical analyses were performed using either a two-tailed Student’s t-test or one-way ANOVA followed by the Bonferroni test. Lineal regression models were generated to assess the relationship between the VPS13A levels in human brain samples and the HD Vonsattel’s grade or the number of CAG repeats. All statistic tests were performed on GraphPad Prism 9.0 (GraphPad Software, San Diego). Differential expression datasets from Microarray and RNA-seq analysis were identified in the Gene Expression Omnibus (GEO) repository from the National Center for Biotechnology Information (NCBI). The comparisons between control and HD conditions were performed with the GEO2R tool of the repository. In all analyses, values of p < 0.05 were considered statistically significant.
3 Results
To study the distribution of vps13a mRNA, we performed FISH on sagittal brain sections from WT and R6/1 mice at the age of 20 weeks. First, we focused on the brain regions of interest (Figure 1A) and we found different staining intensity profiles as previously published (García-García et al., 2021). We observed a staining enrichment in the cerebellum, notably in Purkinje cells and the granular layer; the pyramidal layers of the hippocampus and granular layer of dentate gyrus also presented high levels of staining. We found moderate staining in the somatosensory cortex and with wide expression along all cortical layers with a marked signal in layer V. Staining in the striatum was weak. We found no apparent differences in staining intensity between WT and R6/1 mice (Figure 1A).
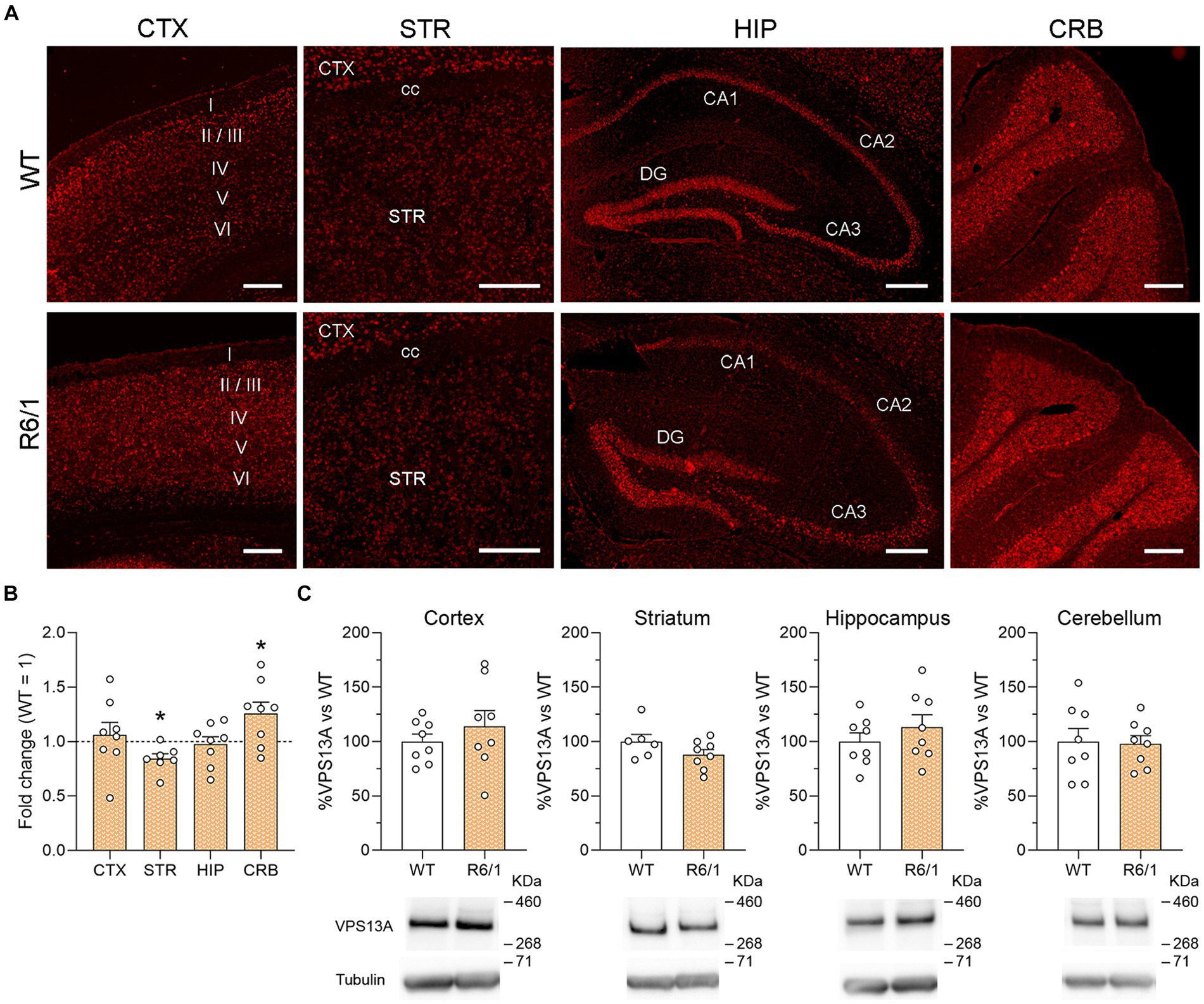
Figure 1. VPS13A mRNA and protein levels in representative brain regions of R6/1 mice. (A) Specific labeling of Vps13a mRNA in illustrative sagittal sections of striatum (STR), somatosensorial cortex (CTX), hippocampal formation (HIP) and cerebellum (CRB); cc, corpus callosum; STR, striatum; I, II, III, IV, V and VI are cortical layers; n = 3 WT and 3 R6/1 mice. Scale bar 250 μm. (B) Vps13a mRNA levels were analyzed by qRT-PCR in the striatum somatosensory cortex, striatum, hippocampus, and cerebellum. Values are expressed as mean ± SEM. Differences were analyzed by One-way ANOVA followed by the Bonferroni post-hoc test. *p < 0.05. Each point represents data from an individual mouse. n = 8 WT and 8 R6/1. (C) VPS13A protein levels were analyzed by western blot. Values are expressed as mean ± SEM. Differences were analyzed by Student’s t-test. Each point represents data from an individual mouse. n = 8 Ctrl and 8 R6/1 (CTX, HIP and CRB) and 6 WT and 8 R6/1 (STR).
We then analyzed the effects of mHtt on the VPS13A expression in the cortex, striatum, hippocampus, and cerebellum of 20-week-old R6/1 mice. After mRNA quantification by qRT-PCR, we found a significant 19% VPS13A expression decrease in the striatum and a 23% increase in the cerebellum of R6/1 mice when compared with WT animals (Figure 1B). However, these changes were not corroborated by western blot since we found no mHtt-induced changes in the VPS13A protein concentration in any of the four studied areas (Figure 1C).
We next analyzed whether this lack of significant VPS13A changes in symptomatic R6/1 mice was also present in the cerebral cortex and the putamen of HD patients. We found no significant changes in VPS13A levels in the motor cortex. However, we detected a significant 34% decrease in the putamen of HD patients compared with non-affected individuals by western blot (Figure 2A). The regression models showed no relationship between the levels of VPS13A protein with the HD Vonsattel’s grade in the motor cortex or the putamen (r2 = 0.005, p = 0.893 for the motor cortex and r2 = 0.098, p = 0.493 for the putamen; Figure 2B). Finally, we found no significant correlation between the levels of VPS13A and the number of CAG repeats in the motor cortex (r2 = 0.023, p = 0.772) or the putamen (r2 < 0.001, p = 0.994; Figure 2C).
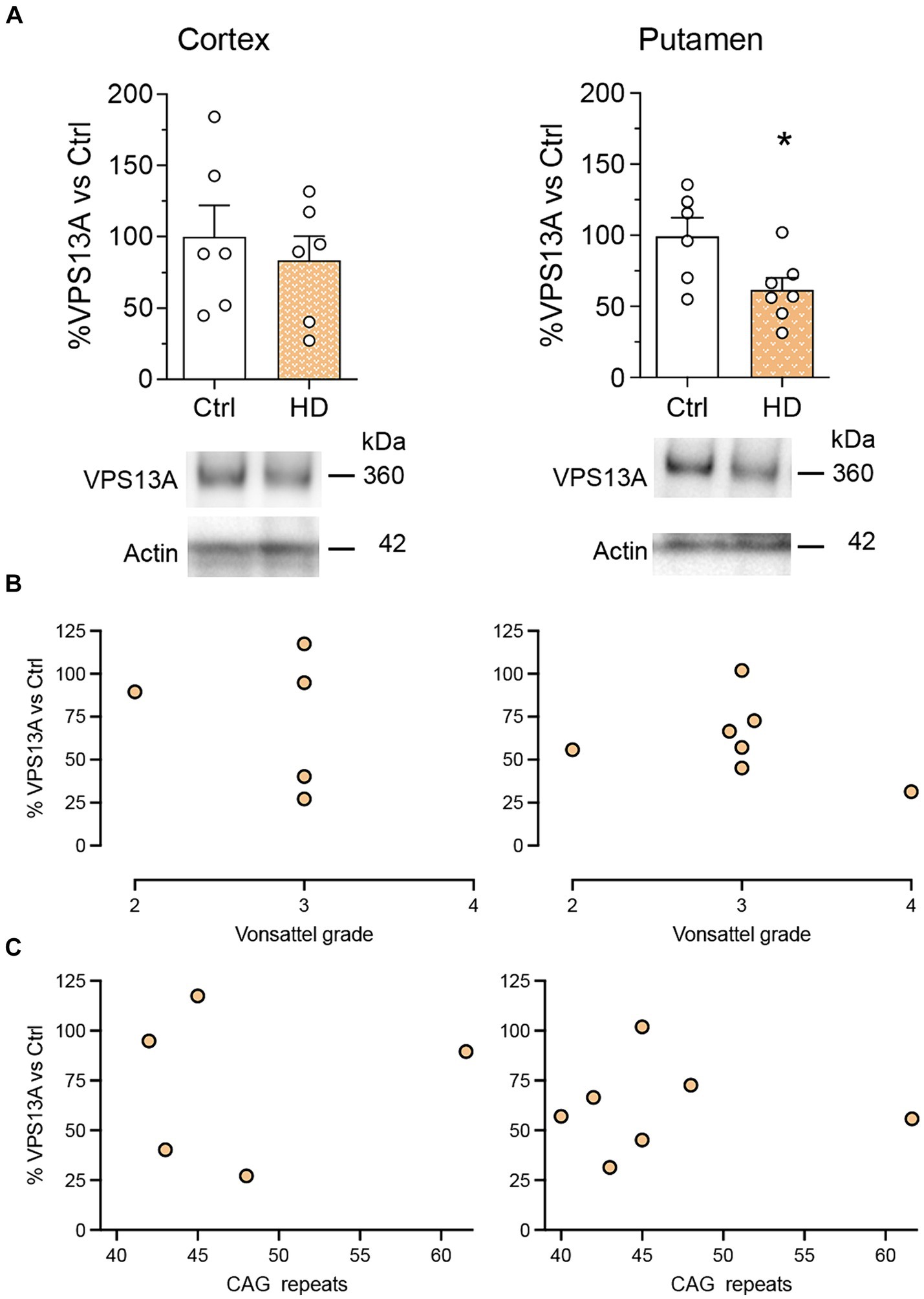
Figure 2. The levels of the VPS13A protein do not correlate with the HD condition. VPS13A protein levels were analyzed by western blot. (A) Representative immunoblots are shown. Values are expressed as mean ± SEM. Differences were analyzed by un-paired Student’s t-test. *p < 0.05. Dot plots show the lack of correlation of the VPS13A levels in the motor cortex and the putamen with either the neuropathological stage (Vonsalttel grade) (B) or with the number of CAG repeats in the Htt gene (C). Each point corresponds to the value from an individual sample. n = 6 Ctrl and 6 HD (cortex) and 6 Ctrl and 7 HD (putamen).
To validate these results, we finally assessed the involvement of the VPS13A gene expression changes in HD in a series of differential-expression datasets published in the GEO. We compared the VPS13A expression in either, the motor cortex of HD-patients vs. controls (Lin et al., 2016), the whole blood of HD patients vs. controls (Hu et al., 2011), the iPSC-derived MSNs of HD vs. control patients (Chiu et al., 2015), the striatum of R6/2 mice vs. WT mice (Labbadia et al., 2011), and the conditionally immortalized HD STHdhQ111/Q111 striatal neuronal progenitor cell line vs. conditionally immortalized WT STHdhQ7/Q7 striatal neuronal progenitor cell line (Sadri-Vakili et al., 2007). According to the fold-change parameter of the VPS13A gene expression, with the p-value adjusted by the false discovery rate (Table 2), we found no significant HD effect in the VPS13A expression in any of the datasets analyzed.
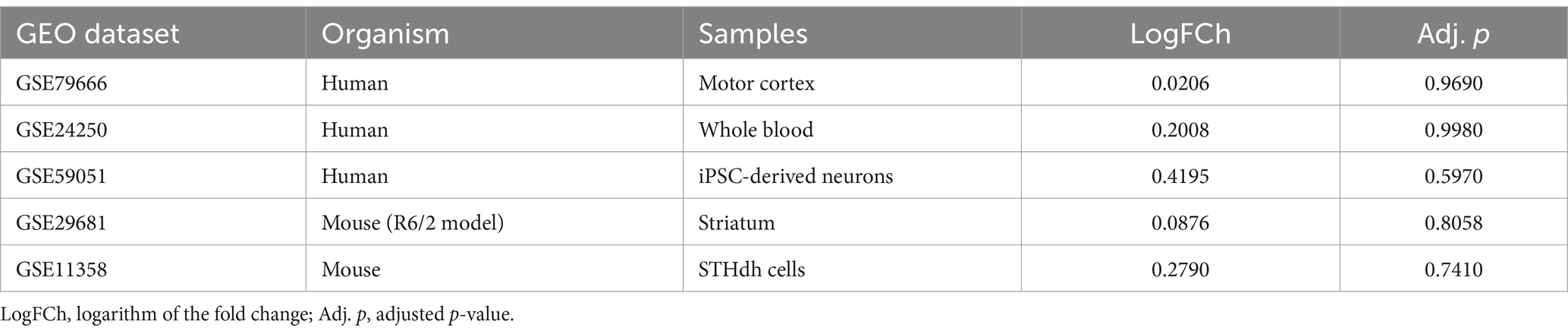
Table 2. Differential expression analyses of changes in VPS13A expression in the human and mouse HD datasets.
4 Discussion
With the evidence from the results obtained, we found that mHtt accumulation only subtly alters VPS13A, with some significant changes in the mRNA and protein concentration in the striatum but does not influence the tissue distribution. This indicates that VPS13A is a very stable protein with crucial functions in neuronal functioning. Previous results found that VPS13A neuronal content is stable over time and that its concentration is not modulated by the overactivation of cholinergic, dopaminergic, or glutamatergic systems (García-García et al., 2021). Altogether, these data suggest that VPS13A has a stable presence and role in neurons with a fine regulation of protein levels that maintains its steady role.
Our results validate previous observations of the heterogeneous VPS13A brain distribution (Kurano et al., 2007; García-García et al., 2021). Of the four analyzed regions, the striatum showed the lowest expression, also in the HD brain, although it is the most affected area in ChAc patients, while the cerebellum showed the highest expression, also in the R6/1 mouse model of HD. The fact that mHtt does not modify the brain distribution of VPS13A indicates a lack of interdependence of both proteins. Consequently, it suggests that their respective mutations might trigger convergent pathological mechanisms affecting the weak cellular properties of the MSNs. Although the protein tissue distribution is unaffected, the decrease in protein levels in the putamen of HD patients suggests that a direct interaction of VPS13A with mHtt cannot be discarded. As proposed for many other proteins (Wanker et al., 2019), a pathological interaction of VPS13A with mHtt may limit its functionality and biological functions, leading to VPS13A dysfunction, subsequent deficient phospholipid homeostasis (Miltenberger-Miltenyi et al., 2023), and mitochondrial function impairment.
In this regard, both VPS13A and mHtt have been involved in some common cellular processes. For example, data suggest the VPS13A involvement in the protein degradation machinery and autophagy in ChAc pathophysiology (Muñoz-Braceras et al., 2015; Lupo et al., 2016; Vonk et al., 2017), a process that is also altered in HD (Ravikumar et al., 2004). However, among all these common pathophysiological processes, mitochondrial dysfunction stands out. Recent studies showed that VPS13A is localized at sites where the endoplasmic reticulum (ER) and mitochondria are in close contact to enable lipid transfer required for mitochondria and lipid-droplet-related processes in cell lines (Yeshaw et al., 2019). Interestingly, structural and functional changes in the ER-mitochondria contact sites leading to mitochondrial dysfunction (Shirendeb et al., 2011; Cherubini et al., 2020), and aberrant lipid homeostasis or calcium signaling (Aditi et al., 2016; Pchitskaya et al., 2018; Tshilenge et al., 2023) have been reported to contribute to the specific degeneration of the striatum in HD (Browne, 2008).
Additionally, despite the low expression of VPS13A and mHtt in the striatum, both VPS13A reduction and mHtt accumulation have been associated with striatal synaptic plasticity impairment (Parievsky et al., 2017; García-García et al., 2023) and a reduction of signaling molecules important for synaptic functioning such as BDNF and CX3CL1 (Canals et al., 2004; Giralt et al., 2009; Kim et al., 2020; Azman and Zakaria, 2022; García-García et al., 2023). Strong evidence links these two proteins with altered neuronal communication and deficient long-term depression induction in the corticostriatal circuitry of HD (Baydyuk et al., 2011; Besusso et al., 2013; Kim et al., 2020) Thus, defining the role of VPS13A in striatal synaptic plasticity and MSN communication may constitute a key point to understanding the specific striatal vulnerability not only in ChAc, but also in HD.
Finally, the low number of HD-patient samples and transgenic animals herein analyzed constitutes a limitation of the study, as it may cause a loss of statistical power to detect differences in VPS3A expression and protein. Keeping this in mind, we conclude that VPS13A brain distribution is not substantially affected by mHtt. Therefore, the selective susceptibility of MSN in both ChAc and HD might be a consequence of disturbances in convergent cellular processes and circuitry alterations with divergent molecular mechanisms. Further experiments are necessary to evaluate the role of the VPS13A function in mediating these convergent mechanisms that determine MSN-specific vulnerability in basal ganglia disorders.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics statement
Human tissue samples were obtained from the Neurological Tissue Bank of the Biobanc- Hospital Clínic-Institut d’Investigacions Biomèdiques August Pi i Sunyer (IDIBAPS, Barcelona, Spain). The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study. All animal procedures were conducted in accordance with the Spanish RD 53/2013 and European 2010/63/UE regulations for the care and use of laboratory animals and approved by the animal experimentation Ethics Committee of the Universitat de Barcelona (368/19) and Generalitat de Catalunya (11193).
Author contributions
EG-G: Writing – original draft, Methodology, Investigation, Formal analysis. MC-C: Writing – review & editing, Investigation, Formal analysis. AC-M: Writing – review & editing, Investigation, Formal analysis. AR-L: Writing – review & editing, Investigation, Formal analysis. GB-S: Writing – review & editing, Formal analysis. EP-N: Writing – review & editing, Resources, Investigation. CM: Writing – review & editing, Resources, Funding acquisition. JA: Writing – review & editing, Validation, Resources, Project administration, Funding acquisition. MM: Writing – review & editing, Validation, Project administration, Funding acquisition, Conceptualization. MR: Writing – original draft, Supervision, Funding acquisition, Data curation, Conceptualization.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This study was supported by grants from the Ministerio de Ciencia e Innovación (Spain), under projects no. PID2020-119386RB-I00 (JA and MR), no. PID2021-124896OA-I00 (MM), and no. SAF2017-88812-R and PID2020-119236RB-I00 /AEI/10.13039/501100011033/ (CM); Instituto de Salud Carlos III, Ministerio de Ciencia, Innovación y Universidades and European Regional Development Fund (ERDF) [CIBERNED, to JA], Spain; and ChAc Foundation (JA, MM and MR) Spain. Also, the project has been supported by María de Maeztu Unit of Excellence (CEX2021-001159), Institute of Neurosciences of the University of Barcelona, Ministry of Science, Innovation, and Universities. This research is part of NEUROPA. The NEUROPA Project has received funding from the European Union’s Horizon 2020 Research and Innovation Program under Grant Agreement No. 863214 (MM).
Acknowledgments
We are very grateful to Carmen Andrade (Dept. Biomedical Sciences, UB) for her technical support on histological procedures, to Ana Maria Lopez (María de Maeztu Unit of Excellence, Institute of Neurosciences of the University of Barcelona, CEX2021-001159, Ministry of Science, Innovation and Universities) and Silvia Artigas (CIBERNED) for excellent mouse colony management.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Aditi, K., Shakarad, M. N., and Agrawal, N. (2016). Altered lipid metabolism in Drosophila model of Huntington’s disease. Sci. Rep. 6:31411. doi: 10.1038/SREP31411
Azman, K. F., and Zakaria, R. (2022). Recent advances on the role of brain-derived neurotrophic factor (BDNF) in neurodegenerative diseases. Int. J. Mol. Sci. 23:6827. doi: 10.3390/IJMS23126827
Baydyuk, M., Russell, T., Liao, G. Y., Zang, K., An, J. J., Reichardt, L. F., et al. (2011). TrkB receptor controls striatal formation by regulating the number of newborn striatal neurons. Proc. Natl. Acad. Sci. USA 108, 1669–1674. doi: 10.1073/PNAS.1004744108
Besusso, D., Geibel, M., Kramer, D., Schneider, T., Pendolino, V., Picconi, B., et al. (2013). BDNF-TrkB signaling in striatopallidal neurons controls inhibition of locomotor behavior. Nat. Commun. 4:2031. doi: 10.1038/NCOMMS3031
Browne, S. E. (2008). Mitochondria and Huntington’s disease pathogenesis: insight from genetic and chemical models. Ann. N. Y. Acad. Sci. 1147, 358–382. doi: 10.1196/ANNALS.1427.018
Canals, J. M., Pineda, J. R., Torres-Peraza, J. F., Bosch, M., Martín-Ibañez, R., Muñoz, M. T., et al. (2004). Brain-derived neurotrophic factor regulates the onset and severity of motor dysfunction associated with enkephalinergic neuronal degeneration in Huntington’s disease. J. Neurosci. 24, 7727–7739. doi: 10.1523/JNEUROSCI.1197-04.2004
Cherubini, M., Lopez-Molina, L., and Gines, S. (2020). Mitochondrial fission in Huntington’s disease mouse striatum disrupts ER-mitochondria contacts leading to disturbances in Ca2+ efflux and reactive oxygen species (ROS) homeostasis. Neurobiol. Dis. 136:104741. doi: 10.1016/J.NBD.2020.104741
Chiu, F. L., Lin, J. T., Chuang, C. Y., Chien, T., Chen, C. M., Chen, K. H., et al. (2015). Elucidating the role of the A2A adenosine receptor in neurodegeneration using neurons derived from Huntington’s disease iPSCs. Hum. Mol. Genet. 24, 6066–6079. doi: 10.1093/HMG/DDV318
Creus-Muncunill, J., Badillos-Rodríguez, R., Garcia-Forn, M., Masana, M., Garcia-Díaz Barriga, G., Guisado-Corcoll, A., et al. (2019). Increased translation as a novel pathogenic mechanism in Huntington’s disease. Brain 142, 3158–3175. doi: 10.1093/brain/awz230
Del Toro, D., Canals, J. M., Ginés, S., Kojima, M., Egea, G., and Alberch, J. (2006). Mutant huntingtin impairs the post-Golgi trafficking of brain-derived neurotrophic factor but not its Val66Met polymorphism. J. Neurosci. 26, 12748–12757. doi: 10.1523/JNEUROSCI.3873-06.2006
Fernández-García, S., Conde-Berriozabal, S., García-García, E., Gort-Paniello, C., Bernal-Casas, D., García-Díaz Barriga, G., et al. (2020). M2 cortex-dorsolateral striatum stimulation reverses motor symptoms and synaptic deficits in Huntington’s disease. eLife 9, 1–24. doi: 10.7554/eLife.57017
Fernández-Nogales, M., Santos-Galindo, M., Hernández, I. H., Cabrera, J. R., and Lucas, J. J. (2016). Faulty splicing and cytoskeleton abnormalities in Huntington’s disease. Brain Pathol. 26, 772–778. doi: 10.1111/BPA.12430
García-García, E., Chaparro-Cabanillas, N., Coll-Manzano, A., Carreras-Caballé, M., Giralt, A., Del Toro, D., et al. (2021). Unraveling the spatiotemporal distribution of VPS13A in the mouse brain. Int. J. Mol. Sci. 22:13018. doi: 10.3390/IJMS222313018
García-García, E., Ramón-Lainez, A., Conde-Berriozabal, S., del Toro, D., Escaramis, G., Giralt, A., et al. (2023). VPS13A knockdown impairs corticostriatal synaptic plasticity and locomotor behavior in a new mouse model of chorea-acanthocytosis. Neurobiol. Dis. 187:106292. doi: 10.1016/J.NBD.2023.106292
Giralt, A., Rodrigo, T., Martín, E. D., Gonzalez, J. R., Milà, M., Ceña, V., et al. (2009). Brain-derived neurotrophic factor modulates the severity of cognitive alterations induced by mutant huntingtin: involvement of phospholipaseCgamma activity and glutamate receptor expression. Neuroscience 158, 1234–1250. doi: 10.1016/J.NEUROSCIENCE.2008.11.024
Henkel, K., Walterfang, M., Velakoulis, D., Danek, A., and Kassubek, J. (2008). “Volumetric neuroimaging in Neuroacanthocytosis” in Neuroacanthocytosis syndromes II. eds. R. H. Walker, S. Saiki, and A. Danek (Berlin Heidelberg: Springer), 175–185.
Hu, Y., Chopra, V., Chopra, R., Locascio, J. J., Liao, Z., Ding, H., et al. (2011). Transcriptional modulator H2A histone family, member Y (H2AFY) marks Huntington disease activity in man and mouse. Proc. Natl. Acad. Sci. USA 108, 17141–17146. doi: 10.1073/pnas.1104409108
Jung, H. H., Danek, A., and Walker, R. H. (2011). Neuroacanthocytosis syndromes. Orphanet J. Rare Dis. 6:68. doi: 10.1186/1750-1172-6-68
Kassubek, J., Landwehrmeyer, G. B., Ecker, D., Juengling, F. D., Muche, R., Schuller, S., et al. (2004). Global cerebral atrophy in early stages of Huntington’s disease: quantitative MRI study. Neuroreport 15, 363–365. doi: 10.1097/00001756-200402090-00030
Kim, A., García-García, E., Straccia, M., Comella-Bolla, A., Miguez, A., Masana, M., et al. (2020). Reduced Fractalkine levels Lead to striatal synaptic plasticity deficits in Huntington’s disease. Front. Cell. Neurosci. 14:163. doi: 10.3389/FNCEL.2020.00163
Kumar, N., Leonzino, M., Hancock-Cerutti, W., Horenkamp, F. A., Li, P. Q., Lees, J. A., et al. (2018). VPS13A and VPS13C are lipid transport proteins differentially localized at ER contact sites. J. Cell Biol. 217, 3625–3639. doi: 10.1083/JCB.201807019
Kurano, Y., Nakamura, M., Ichiba, M., Matsuda, M., Mizuno, E., Kato, M., et al. (2007). In vivo distribution and localization of chorein. Biochem. Biophys. Res. Commun. 353, 431–435. doi: 10.1016/J.BBRC.2006.12.059
Labbadia, J., Cunliffe, H., Weiss, A., Katsyuba, E., Sathasivam, K., Seredenina, T., et al. (2011). Altered chromatin architecture underlies progressive impairment of the heat shock response in mouse models of Huntington disease. J. Clin. Invest. 121, 3306–3319. doi: 10.1172/JCI57413
Lin, L., Park, J. W., Ramachandran, S., Zhang, Y., Tseng, Y. T., Shen, S., et al. (2016). Transcriptome sequencing reveals aberrant alternative splicing in Huntington’s disease. Hum. Mol. Genet. 25, 3454–3466. doi: 10.1093/HMG/DDW187
Liu, J., Heinsen, H., Grinberg, L. T., Alho, E., Amaro, E., Pasqualucci, C. A., et al. (2019). Pathoarchitectonics of the cerebral cortex in chorea-acanthocytosis and Huntington’s disease. Neuropathol. Appl. Neurobiol. 45, 230–243. doi: 10.1111/NAN.12495
Lupo, F., Tibaldi, E., Matte, A., Sharma, A. K., Brunati, A. M., Alper, S. L., et al. (2016). A new molecular link between defective autophagy and erythroid abnormalities in chorea-acanthocytosis. Blood 128, 2976–2987. doi: 10.1182/BLOOD-2016-07-727321
Marques Sousa, C., and Humbert, S. (2013). Huntingtin: here, there, everywhere! J Huntington’s Dis 2, 395–403. doi: 10.3233/JHD-130082
Miltenberger-Miltenyi, G., Jones, A., Tetlow, A. M., Conceição, V. A., Crary, J. F., Ditzel, R. M., et al. (2023). Sphingolipid and phospholipid levels are altered in human brain in chorea-Acanthocytosis. Mov. Disord. 38, 1535–1541. doi: 10.1002/MDS.29445
Muñoz-Braceras, S., Calvo, R., and Escalante, R. (2015). TipC and the chorea-acanthocytosis protein VPS13A regulate autophagy in Dictyostelium and human HeLa cells. Autophagy 11, 918–927. doi: 10.1080/15548627.2015.1034413
Muñoz-Braceras, S., Tornero-Écija, A. R., Vincent, O., and Escalante, R. (2019). VPS13A is closely associated with mitochondria and is required for efficient lysosomal degradation. Dis. Model. Mech. 12:dmm036681. doi: 10.1242/DMM.036681
Parievsky, A., Moore, C., Kamdjou, T., Cepeda, C., Meshul, C. K., and Levine, M. S. (2017). Differential electrophysiological and morphological alterations of thalamostriatal and corticostriatal projections in the R6/2 mouse model of Huntington’s disease. Neurobiol. Dis. 108, 29–44. doi: 10.1016/J.NBD.2017.07.020
Pchitskaya, E., Popugaeva, E., and Bezprozvanny, I. (2018). Calcium signaling and molecular mechanisms underlying neurodegenerative diseases. Cell Calcium 70, 87–94. doi: 10.1016/J.CECA.2017.06.008
Peikert, K., Dobson-Stone, C., Rampoldi, L., Miltenberger-Miltenyi, G., Neiman, A., De Camilli, P., et al. (2023). VPS13A disease. Curated Reference Collection in Neurosci. Biobehav. Psychol. 217–219. doi: 10.1016/B978-0-12-809324-5.00521-6
Pelzl, L., Elsir, B., Sahu, I., Bissinger, R., Singh, Y., Sukkar, B., et al. (2017). Lithium sensitivity of store operated Ca2+ entry and survival of fibroblasts isolated from chorea-Acanthocytosis patients. Cell. Physiol. Biochem. 42, 2066–2077. doi: 10.1159/000479901
Ravikumar, B., Vacher, C., Berger, Z., Davies, J. E., Luo, S., Oroz, L. G., et al. (2004). Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat. Genet. 36, 585–595. doi: 10.1038/NG1362
Ross, C. A., Pantelyat, A., Kogan, J., and Brandt, J. (2014). Determinants of functional disability in Huntington’s disease: role of cognitive and motor dysfunction. Mov. Disord. 29, 1351–1358. doi: 10.1002/MDS.26012
Saavedra, A., Giralt, A., Rué, L., Xifró, X., Xu, J., Ortega, Z., et al. (2011). Striatal-enriched protein tyrosine phosphatase expression and activity in Huntington’s disease: a STEP in the resistance to excitotoxicity. J. Neurosci. 31, 8150–8162. doi: 10.1523/JNEUROSCI.3446-10.2011
Sadri-Vakili, G., Bouzou, B., Benn, C. L., Kim, M. O., Chawla, P., Overland, R. P., et al. (2007). Histones associated with downregulated genes are hypo-acetylated in Huntington’s disease models. Hum. Mol. Genet. 16, 1293–1306. doi: 10.1093/HMG/DDM078
Shirendeb, U., Reddy, A. P., Manczak, M., Calkins, M. J., Mao, P., Tagle, D. A., et al. (2011). Abnormal mitochondrial dynamics, mitochondrial loss and mutant huntingtin oligomers in Huntington’s disease: implications for selective neuronal damage. Hum. Mol. Genet. 20, 1438–1455. doi: 10.1093/HMG/DDR024
Stanslowsky, N., Reinhardt, P., Glass, H., Kalmbach, N., Naujock, M., Hensel, N., et al. (2016). Neuronal dysfunction in iPSC-derived medium spiny neurons from chorea-Acanthocytosis patients is reversed by Src kinase inhibition and F-actin stabilization. J. Neurosci. 36, 12027–12043. doi: 10.1523/JNEUROSCI.0456-16.2016
Suzuki, F., Sato, N., Ota, M., Sugiyama, A., Shigemoto, Y., Morimoto, E., et al. (2020). Discriminating chorea-acanthocytosis from Huntington’s disease with single-case voxel-based morphometry analysis. J. Neurol. Sci. 408:116545. doi: 10.1016/j.jns.2019.116545
Tshilenge, K. T., Aguirre, C. G., Bons, J., Gerencser, A. A., Basisty, N., Song, S., et al. (2023). Proteomic analysis of Huntington’s disease medium spiny neurons identifies alterations in lipid droplets. Molecular & Cellular Proteomics: MCP 22:100534. doi: 10.1016/J.MCPRO.2023.100534
Tyebji, S., Saavedra, A., Canas, P. M., Pliassova, A., Delgado-García, J. M., Alberch, J., et al. (2015). Hyperactivation of D1 and A2A receptors contributes to cognitive dysfunction in Huntington’s disease. Neurobiol. Dis. 74, 41–57. doi: 10.1016/J.NBD.2014.11.004
Ueno, S. I., Maruki, Y., Nakamura, M., Tomemori, Y., Kamae, K., Tanabe, H., et al. (2001). The gene encoding a newly discovered protein, chorein, is mutated in chorea-acanthocytosis. Nat. Genet. 28, 121–122. doi: 10.1038/88825
Vonk, J. J., Yeshaw, W. M., Pinto, F., Faber, A. I. E., Lahaye, L. L., Kanon, B., et al. (2017). Drosophila Vps13 is required for protein homeostasis in the brain. PLoS One 12:e0170106. doi: 10.1371/JOURNAL.PONE.0170106
Vonsattel, J. P. G., and DiFiglia, M. (1998). Huntington disease. J. Neuropathol. Exp. Neurol. 57, 369–384. doi: 10.1097/00005072-199805000-00001
Walterfang, M., Looi, J. C. L., Styner, M., Walker, R. H., Danek, A., Niethammer, M., et al. (2011). Shape alterations in the striatum in chorea-acanthocytosis. Psychiatry Res. 192, 29–36. doi: 10.1016/J.PSCYCHRESNS.2010.10.006
Wanker, E. E., Ast, A., Schindler, F., Trepte, P., and Schnoegl, S. (2019). The pathobiology of perturbed mutant huntingtin protein-protein interactions in Huntington’s disease. J. Neurochem. 151, 507–519. doi: 10.1111/JNC.14853
Keywords: chorea-acanthocytosis, VPS13A, huntingtin, Huntington’s disease, movement disorders, basal ganglia, neurodegeneration
Citation: García-García E, Carreras-Caballé M, Coll-Manzano A, Ramón-Lainez A, Besa-Selva G, Pérez-Navarro E, Malagelada C, Alberch J, Masana M and Rodríguez MJ (2024) Preserved VPS13A distribution and expression in Huntington’s disease: divergent mechanisms of action for similar movement disorders? Front. Neurosci. 18:1394478. doi: 10.3389/fnins.2024.1394478
Edited by:
Anuska V. Andjelkovic, University of Michigan, United StatesReviewed by:
Nutan Sharma, Harvard Medical School, United StatesRoberto Rodríguez-Labrada, Cuban Neuroscience Center, Cuba
Copyright © 2024 García-García, Carreras-Caballé, Coll-Manzano, Ramón-Lainez, Besa-Selva, Pérez-Navarro, Malagelada, Alberch, Masana and Rodríguez. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Manuel J. Rodríguez, marodriguez@ub.edu; Mercè Masanammasana@ub.edu
†These authors have contributed equally to this work