
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
PERSPECTIVE article
Front. Neurosci., 10 November 2023
Sec. Translational Neuroscience
Volume 17 - 2023 | https://doi.org/10.3389/fnins.2023.1282267
This article is part of the Research TopicDeveloping Personalized Treatment in Neurodegenerative Disorders: Role of Genomics and Novel Technologies in Identifying Actionable Targets and Developing Interventions in Rare-DiseasesView all 5 articles
 Valentino Rački1,2*†
Valentino Rački1,2*† Mario Hero1,2†
Mario Hero1,2† Eliša Papić1,2
Eliša Papić1,2 Gloria Rožmarić1
Gloria Rožmarić1 Nada Starčević Čizmarević3
Nada Starčević Čizmarević3 Darko Chudy4
Darko Chudy4 Borut Peterlin5
Borut Peterlin5 Vladimira Vuletić1,2
Vladimira Vuletić1,2In this perspective article, we highlight the possible applicability of genetic testing in Parkinson’s disease and dystonia patients treated with deep brain stimulation (DBS). DBS, a neuromodulatory technique employing electrical stimulation, has historically targeted motor symptoms in advanced PD and dystonia, yet its precise mechanisms remain elusive. Genetic insights have emerged as potential determinants of DBS efficacy. Known PD genes such as GBA, SNCA, LRRK2, and PRKN are most studied, even though further studies are required to make firm conclusions. Variable outcomes depending on genotype is present in genetic dystonia, as DYT-TOR1A, NBIA/DYTPANK2, DYT-SCGE and X-linked dystonia-parkinsonism have demonstrated promising outcomes following GPi-DBS, while varying outcomes have been documented in DYT-THAP1. We present two clinical vignettes that illustrate the applicability of genetics in clinical practice, with one PD patient with compound GBA mutations and one GNAL dystonia patient. Integrating genetic testing into clinical practice is pivotal, particularly with advancements in next-generation sequencing. However, there is a clear need for further research, especially in rarer monogenic forms. Our perspective is that applying genetics in PD and dystonia is possible today, and despite challenges, it has the potential to refine patient selection and enhance treatment outcomes.
Deep brain stimulation (DBS) is a functional neuromodulatory procedure that entails the utilization of a neurostimulator to administer electrical impulses to the brain (Pycroft et al., 2018). For several decades, it has been utilized predominantly in the context of advanced Parkinson’s disease (PD) and dystonia to mitigate the motor symptoms associated with the conditions (Hickey and Stacy, 2016). The precise mechanisms underlying the therapeutic effectiveness of DBS have not been fully elucidated. Nevertheless, DBS has demonstrated benefits in a wide range of neurological and psychiatric disorders, including dystonia, essential tremor, Tourette syndrome, intractable pain, epilepsy, treatment-resistant depression, and obsessive-compulsive disorder (Ashkan et al., 2017). The technique is commonly employed to selectively focus on particular regions inside the brain, such as the subthalamic nucleus (STN), globus pallidus internus (GPi) or the thalamic ventral intermedial nucleus (VIM; Reese and Volkmann, 2017; Lozano et al., 2019). However, it is worth noting that the impact of DBS can be seen on whole neural networks, as it modulates activity in both upstream and downstream fashion (Ashkan et al., 2017). In this context, it has the potential to impact a wide range of symptoms, encompassing nonmotor symptoms observed in neurodegenerative disorders (Kurtis et al., 2017). There is a noted variability in patient outcomes with both mentioned conditions that is dependent on several factors. The main factors for outcomes are the target, accuracy of lead placement and stimulation parameters, which are more technical and can be mitigated by proper planning and procedure (Bari et al., 2015; Hartmann et al., 2019; Koeglsperger et al., 2019; Vitek et al., 2022). Other factors that influence outcomes are the disease subtypes and patient characteristics, specifically genetic background, which has increased in importance in recent years (Rizzone et al., 2019; Tisch, 2022).
Genetic basis of diseases cannot be discovered without testing, which is becoming an integral part of the clinical workup in neurological patients, especially in movement disorders or neuromuscular disease patients (Lefroy et al., 2020). Novel next generation sequencing techniques have made an enormous impact in genetic neurological disorders, and greatly expanded the diagnostic yields, while also leading to new gene discoveries in the field (Johnson, 2015). Still, even with these expanded capabilities, experts are still hesitant to routinely seek genetic testing for their patients with PD, with a recent study showing that 41 percent of movement disorder experts did not refer patients to genetic testing and more than 80 percent referred less than 11 patients in 1 year (Alcalay et al., 2020). Use of genetic testing in dystonia is more common than in PD, especially in younger patients, albeit still underutilized (Pozojevic et al., 2021). Cost–benefit of genetic testing is different depending on the medical field and specialty (Córdoba et al., 2018), and has not been established yet in movement disorders (Pal et al., 2023). However, it is important to note that the price for next generation sequencing has been decreasing with each new innovation in the field (Satam et al., 2023), and in some cases constituted a lesser cost compared to aggregated cost of various diagnostic procedures (Córdoba et al., 2018). Clinical algorithms have been put forth to guide clinical decisions on whether to test patients, but they have been focused on diagnostic yields, not therapeutic outcomes (Zech et al., 2021). Our team is comprised of neurologists, neurosurgeons and geneticists in three separate clinical centers. In this perspective, we offer our view of the applicability of genetic testing in dystonia and Parkinson’s disease patients planned to be, or treated with, deep brain stimulation, based on our experience combining the two fields in recent years, as well as two clinical vignettes that best exemplify our perspective.
DBS has been used for advanced Parkinson’s disease treatment since the 1980’s (Frey et al., 2022), and it has a proven long-term benefit for patients properly selected for treatment (Limousin and Foltynie, 2019). The question of patient selection is an important one, and the current determinants include disease subtype, age, time to surgery, psychiatric comorbidities and cognitive issues (Rački et al., 2022). Genetic background of patients is another important determinant for disease progression, quality of life and outcome (Iwaki et al., 2019; Rizzone et al., 2019). Current evidence mainly encompasses the more common causative genes of PD, such as GBA, SNCA, LRRK2, and PRKN.
The most common genetic risk factor for PD are mutations in the GBA gene (Kovanda et al., 2022), and is the gene with the clearest evidence thus far, indicating that both STN and GPi DBS can have a solid impact on motor symptoms, albeit with an increased risk of cognitive decline than in noncarriers (Weiss et al., 2012; Lythe et al., 2017). This was confirmed in a recent composite analysis study with 366 subjects, where GBA mutation carriers that were treated with STN-DBS showed faster cognitive decline than GBA carriers with no DBS (Mattis Dementia rating Scale decline 1.71 points/year more), and even more so than noncarriers treated with DBS (Mattis Dementia rating Scale decline 1.49 points/year more; Pal et al., 2022). The patient groups were not evenly distributed but had enough participants (58 GBA + DBS+, 82 GBA + DBS-, 98 GBA-DBS+, 128 GBA-DBS-subjects), and the severity of the variant played a role in the speed of the decline. Even though there was a disparity in the number of patients in groups between the mild (non-neuronopathic) and severe (neuronopathic) variants with DBS (24 risk variant, 23 mild variant, 11 severe variant), it is important to keep in mind that there are differences between variants in clinical phenotype and therapeutic outcome (Pal et al., 2022). Regardless of risk, the benefit on motor outcomes and fluctuations is becoming clearer, as beneficial effects up to a 40% reduction have been seen on the Unified Parkinson’s Disease Rating Scale Part III (UPDRS III) and almost complete reduction (97%) in the Unified Parkinson’s Disease Rating Scale Part IV (UPDRS IV) scores (Angeli et al., 2013; Mangone et al., 2020). Finally, long-term follow-ups are important, as it has been shown that GBA-DBS patients can have axial deterioration and postural instability, despite positive earlier improvements (Straccia et al., 2022). Milder variants, like N370S, were shown to have beneficial effects with reduced incidence of cognitive or axial side effects compared to more severe variants like L444P (Artusi and Lopiano, 2023), highlighting that variant-to-variant differences will influence therapeutical decisions in the future. Furthermore, PD can be seen during the clinical presentation of other genetic diseases such as Gaucher’s disease type 1 (GD1), which is caused by homozygous or compound heterozygous GBA variants, with an age of onset after 60 years old and common cognitive impairment (Chetrit et al., 2013).
Various outcomes have been described for SNCA mutation carriers, with duplication patients initially displaying good to excellent motor outcomes (Antonini et al., 2012), but faster cognitive decline than noncarriers, especially in case of missense mutations (Youn et al., 2022). On the other hand, reports of LRRK2 mutation carriers, especially G2019S, show that the response can be even better than in noncarriers (Sayad et al., 2016). Worse outcomes are found in R114G mutation carriers compared to other missense mutations in LRRK2 (de Oliveira et al., 2019), indicating that we must consider both gene and mutation location when considering DBS in these patients. PRKN mutation carriers exhibit excellent long-term benefits of DBS in both STN and GPi, especially in patients with good responses to levodopa (Rizzone et al., 2019; Covolo et al., 2023), and can be considered good candidates for DBS. Patients that can show benefit from DBS are those with motor complications associated with levodopa treatment, with marked Unified Parkinsons’s Disease Rating Scale IV reduction (Aasly, 2020). For other, rarer mutations, such as PINK1, DJ1, VPS35 and CHCHD2, the evidence is mostly based on case reports and is thus too early to give firm recommendations in routine practice. In general, we must keep in mind the benefits and potential side-effects of DBS therapy, such as postural instability (Ahrweiller et al., 2019) or selective cognitive decline (Rački et al., 2022), as these can be potentially exacerbated depending on certain monogenic causes like DJ1, PRKN or SNCA (Marsili et al., 2021). Taken together, we can see the value that genetic testing is beginning to have in PD, and we must keep in mind that patients with different variants in one gene can have different phenotypes and outcomes to advanced therapy. It is important to highlight that all evidence in this field is not based on randomized clinical trials, and further research is clearly needed, especially in rarer monogenic causes (Aasly, 2020; Kamo et al., 2022).
The subject of this case vignette is a male patient, aged 43, who has been diagnosed with GD1, as a result of compound heterozygous mutations in the GBA gene (p.H294Q, p.N409S, p.D448H), which was previously published in Movement Disorders Clinical Practice (Racki et al., 2021). The patient had progressive parkinsonian symptoms characterized by marked tremor and rigidity, which responded to levodopa, but with early motor fluctuations and dyskinesias 3 years after initiating treatment. The utilization of DBS was suggested as a potential intervention to mitigate the progressive decline in the individual’s health status. Due to previous knowledge of possible limitations of treating GBA associated PD with DBS, we performed extensive neuropsychological testing. The results of cognitive examinations indicated the presence of mild depression, but no evidence of cognitive loss was observed. Consequently, based on these findings, the patient was deemed appropriate for STN-DBS. There was a notable and significant reduction in motor symptoms and dyskinesias after the procedure, resulting in a rapid favorable outcome. The overall levodopa dose was significantly reduced by 68%, with continuous improvements in the patient’s overall quality of life and a restoration of his independence. We decided to follow the cognitive functioning in this patient yearly through neuropsychological testing (Wechsler Memory Scale, Animal Naming Test, Controlled Oral World Association Test, various personality tests depending on age) and alternating different versions (8.1, 8.2, 8.3) of the Montreal Cognitive Assessment (MoCA), and testing throughout the last 5 years show an absence of any cognitive decline (Table 1). The improvements to quality of life and motor symptoms are still present as the patient is being able to care independently for his underage son. There are no issues with axial symptoms or other non-motor symptoms yet. This vignette highlights the potential advantages of initiating DBS at an early stage for individuals with GD1-PD who experience variable responses to levodopa. It emphasizes the importance of doing comprehensive cognitive evaluations prior to the treatment and regularly assessing cognitive functioning after DBS in patients with GBA mutations, and a way we can individualize treatment plans depending on the present genotype. Finally, to the best of our knowledge, this is the longest follow-up reported for a GD1-PD patient treated with DBS, as the previous reports mention great symptomatic improvement but no time course (Chetrit et al., 2013).
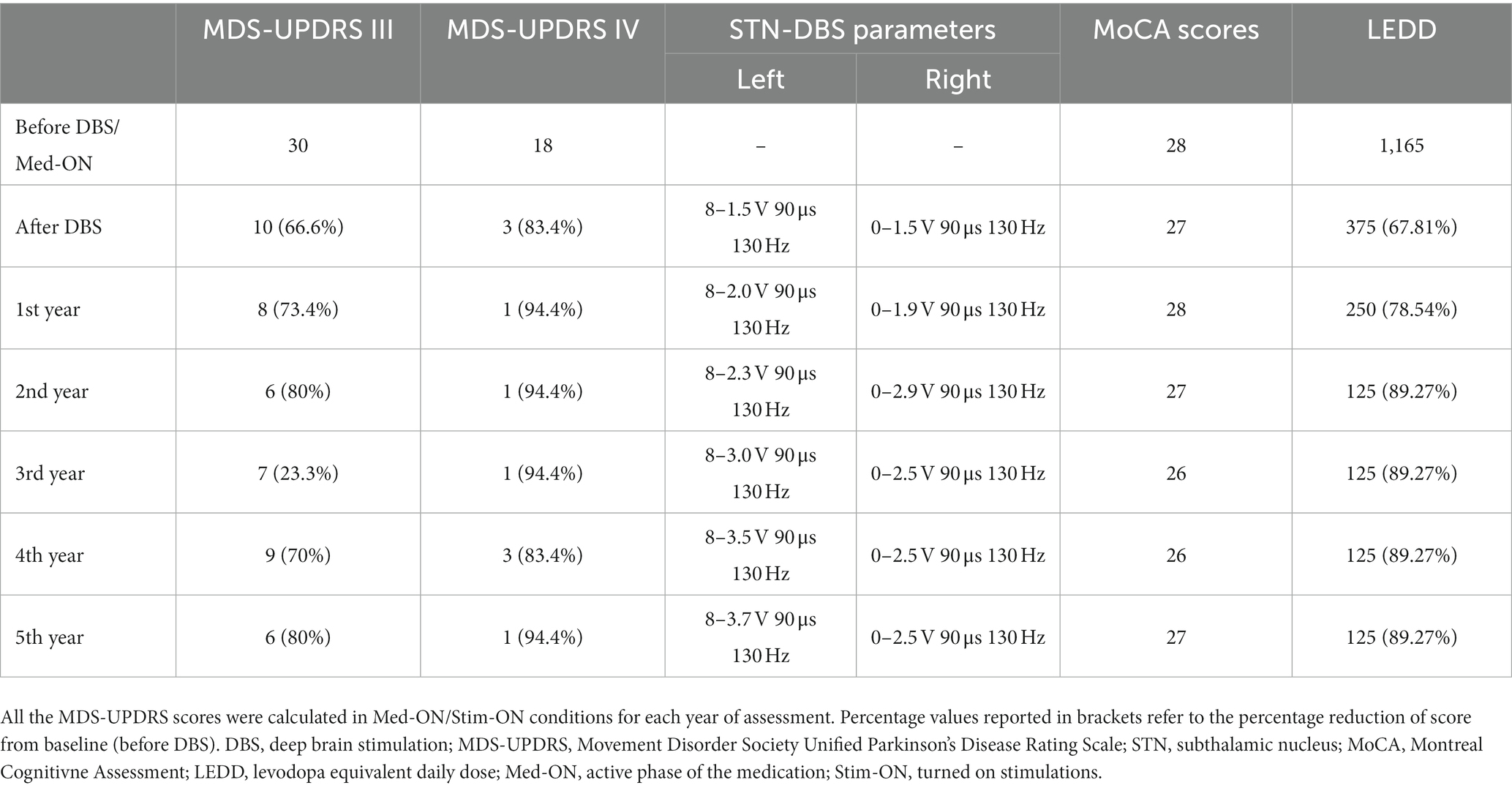
Table 1. Clinical characteristics of the GD1-PD patient through 5 years.
Dystonia is defined as a movement disorder marked by sustained or intermittent muscle contractions that result in unusual, frequently repetitive movements, postures, or a combination of both (Albanese et al., 2013). Genetic forms of dystonia are defined as those wherein an underlying gene has been found as the source of the disease. As more and more dystonia-related genes are uncovered, our understanding of the genetic basis of the disorder has grown (Pozojevic et al., 2021). The most effective method for treating segmental and generalized dystonia that is refractory to medical treatment is globus pallidus internus deep brain stimulation (GPi DBS), which is suitable in children and adults (Tisch and Kumar, 2021). Shorter duration of the disease, younger age at onset and the specific subtype of dystonia are all predictors of the degree of improvement after GPi DBS surgery, where most cases of idiopathic isolated dystonia have better results than those with combined dystonia (Eltahawy et al., 2004; Andrews et al., 2010; Reese and Volkmann, 2014; Artusi et al., 2020). However, there are exceptions, such as acquired tardive dystonia caused on by exposure to neuroleptic drugs and dystonia noticed in the setting of neurodegeneration by brain iron accumulation (NBIA/DYT-PANK2; Tisch and Kumar, 2021). Also, idiopathic isolated craniofacial and laryngeal dystonia have a variable response to DBS, highlighting the importance the underlying cause and the body distribution when predicting the results of DBS for dystonia (Limotai et al., 2011; Finger et al., 2020; Tisch and Kumar, 2021). The response to DBS seems to differ among different monogenic forms of dystonia, showing varying degrees of clinical improvement for each. GPi-DBS has a positive effect on both short- and long-term motor and functional results in individuals with DYT-TOR1A, DYT-THAP1, and NBIA/DYTPANK2, in contrast to the other monogenic forms of dystonia, with DYT-TOR1A having the most significant clinical improvement (Artusi et al., 2020). Although DYT1 dystonia typically displays positive responsiveness to GPi-DBS, a study conducted by Cif et al. documented cases of secondary deterioration occurring after a few years among a subset of DYT1 dystonia patients who initially responded well at the 12-month mark (Cif et al., 2010). Similar was found in a study by Tsuboi et al., where results revealed that 11 out of 132 patients with DYT1 dystonia experienced secondary worsening within 6 months to 3 years following DBS. This decline was linked to an earlier age of onset, more rapid disease progression, and involvement of cranial regions (Tsuboi et al., 2020).
In contrast to Artusi et al., Tisch et al. described a worse and more variable effect of DBS in THAP1 dystonia (Tisch and Kumar, 2021). As per literature findings, the reported average improvement in Burke-Fahn-Marsden Dystonia Rating Scale (BFMDRS) is approximately 35%, and this can vary from 16 to 72% (Groen et al., 2010; Zittel et al., 2010; Panov et al., 2012; Krause et al., 2015). Additionally, there is a tendency for limited or negligible improvements in speech and bulbar function (Groen et al., 2010; Panov et al., 2012; Tisch and Kumar, 2021). Due to uncertainties regarding the efficacy of GPi DBS in THAP1 dystonia, alternative targets for deep brain stimulation are being investigated, such as the ventral lateral anterior thalamic nucleus (Mure et al., 2014). Several studies have shown that GPi DBS generally yields positive outcomes in the management of myoclonus dystonia arising from DYT-SCGE (DYT11) (Gruber et al., 2010; Kurtis et al., 2010; Kim et al., 2014). A study conducted by Kosutzka et al. presented 9 patients with genetically confirmed DYT-SCGE dystonia in whom GPi DBS achieved improvement in myoclonus by 94%, dystonia by 71 and 88% improvement in disability scores (Kosutzka et al., 2019). Favorable results due to GPi DBS have also been recorded in X-linked dystonia-parkinsonism (DYT3), with post-operative improvements in dystonia, alongside with improvements in the UPDRS III (Brüggemann et al., 2019). The effects of GPi DBS in other monogenic forms of both isolated and combined dystonia such as DYT-GNAL (DYT25), DYT-KMT2B (DYT28), DYT-ATP1A3 (DYT12), and DYT-ANO3 (DYT24) have been documented in smaller patient cohorts and showed different outcomes (Tisch and Kumar, 2021; Rajan et al., 2022). More specifically, patients with GNAL gene variants had faster responses than DYT6 patients, although the long term effects were similar and favorable in both reported groups (Ahn et al., 2019; Sarva et al., 2019). Even though GPi DBS is the most used and reported target, VIM DBS has also been reported as beneficial for dystonic tremor and myoclonus in DYT-SCGE, and can be used along with pallidal stimulation (Trottenberg et al., 2001; Wang et al., 2017). Due to the rare nature of genetic dystonia, most of the current findings are based on case reports and meta-analysis, which limit the interpretation of findings with a clear necessity for further investigations, like in PD.
We present a 39-year-old woman with a novel likely pathogenic variant in the GNAL gene (NM_182978.4: c.394A > G: p.Lys132Glu). Pathogenicity of the variant was assessed using the ACMG/ACGS (American College of Medical Genetics and Genomics/Association for Clinical Genomic Science) criteria, based on the findings that the variant was not present in the control population, is located within the functional domain, has multiple in-silico predictors of pathogenicity and clinical picture that corresponds with GNAL patients (Fuchs et al., 2013). First symptoms began at the age of 7 years as the involuntary movements of the right hand and difficulties with fine motor skills, along with a mild cervical dystonia. Ten years later, during high school, the facial spasms started. The patient was initially treated at Clinical Hospital Center Zagreb, where she received botulinum toxin injections due to torticollis. The first control at our center in Rijeka occurred in 2016, when blepharospasm and cervical dystonia with writer’s cramp, action tremor and dystonic body posture were described. The patient was initially treated with 200 IU of botulinum toxin injections, but with consistent worsening. Due to the generalized dystonia and young age, DBS was proposed to the patient, which she refused. Several years after whole-exome sequencing was performed, and the results showed a heterogeneous variant in the GNAL gene. DBS was proposed due to the findings in the literature that this might be effective for her genotype (Ahn et al., 2019; Sarva et al., 2019; Tisch and Kumar, 2021). After through discussion with the patient, she agreed to the procedure and underwent bilateral directional GPi-DBS. She has a positive early response that improved even further in the first 6 months of treatment and now has a significant reduction in the BFMDRS from 40 preoperative (31 movement scale, 9 disability scale) to 10 (8 movement scale, 2 disability scale) 6 months after the procedure. Most beneficial were improvements in blepharospasm, cervical dystonia and posture. This vignette highlights the usefulness of genetic testing in routine clinical practice, which enabled the patient to make a better-informed decision, which was based on the currently available evidence.
When initiating any diagnostic procedure, all physicians ask themselves what information can be gained and how it can be used to help our patients. The question of genetic testing is multifaceted and cannot be only approached through outcome, applicability or cost–benefit, as every patient has a basic right to the most accurate diagnosis if they choose to pursue it, which can in turn impact whole families. However, in this perspective, we wanted to highlight how genetics can be applied today to improve our decision making and treatment plans for PD and dystonia patients, and we believe it has a use in routine clinical practice.
DBS in PD has established long-term benefits for well-selected patients. Previously, patient selection involved factors like disease subtype, age, surgery timing, and cognitive issues, while recent studies suggest genetic influences on post-DBS outcomes. Genes like GBA, SNCA, LRRK2, and PRKN are the most common and studied. We can use the knowledge we have today to tailor our approaches to patients depending on their genotype, and it is part of our clinical practice. While various genetic factors impact DBS outcomes, evidence remains non-randomized, requiring further research, particularly for rarer genetic causes of the disease.
Genetic testing should be a standard in the assessment and management of dystonia patients due to the significant impact of underlying genetic mutations on treatment outcomes. This is especially apparent in cases where specific genetic forms of dystonia respond well to GPi DBS. According to the literature listed above, GPi DBS has shown varying degrees of success depending on the specific genetic subtype, some with significant clinical improvement. Genetic mutations of dystonia such as DYT-TOR1A, NBIA/DYTPANK2, DYT-SCGE and X-linked dystonia-parkinsonism have demonstrated promising outcomes following GPi-DBS, while varying outcomes have been documented in DYT-THAP1. For other genetic forms of dystonia, we still lack clinical data and studies with a larger number of subjects to determine the effectiveness of DBS therapy.
The choice of genetic test is still unclear in this rapidly changing field. Our pipeline generally involves whole-exome sequencing with multiplex-ligation dependent probe amplification to detect chromosomal abnormalities, which is a common approach (Olgiati et al., 2016). Even though we can use software defined panels in analysis for more accurate diagnostic procedures, performing whole-exome sequencing enables easier reassessment and adding novel genes as they are discovered. Whole-genome sequencing is currently used on a case-by-case basis depending on the complexity of phenotypes and the decision to find novel genes, which is what can be said for optical genome mapping as well. Targeted gene panels can be of use in larger scale testing, but defining and designing them can be challenging (Bean et al., 2020). While all procedures have a place in current testing pipelines, we must stress the importance of reassessment, and that patients should be informed that a current negative test can be changed in the future, especially in the case of variants of unknown significance (Salinas et al., 2020).
It must be said that the current evidence is still insufficient to make firm claims and many challenges remain, especially in cases of rarer monogenic causes, but we must be aware that the only way to improve is to continue to combine and try to apply new genetic findings for advanced treatment of movement disorders. This has to be highlighted in light of the findings that many specialists do not view this as applicable and consider it a purely scientific exercise, which we do not think is the case.
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author.
The studies involving humans were approved by Ethics Committee of the Clinical Hospital Center Rijeka. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
VR: Conceptualization, Data curation, Investigation, Resources, Writing – original draft, Writing – review & editing. MH: Conceptualization, Data curation, Investigation, Resources, Writing – original draft, Writing – review & editing. EP: Data curation, Investigation, Methodology, Writing – review & editing. GR: Investigation, Methodology, Writing – original draft, Writing – review & editing. NČ: Investigation, Supervision, Writing – review & editing. DC: Investigation, Supervision, Writing – review & editing. BP: Conceptualization, Investigation, Supervision, Writing – review & editing. VV: Conceptualization, Funding acquisition, Investigation, Methodology, Resources, Supervision, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was funded by the University of Rijeka grant (uniri-biomed-18-1981353) and the Croatian Science Foundation grant (IP-2019-04-7276).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Aasly, J. O. (2020). Long-term outcomes of genetic Parkinson’s disease. J Mov Disord 13, 81–96. doi: 10.14802/jmd.19080
Ahn, J. H., Kim, A. R., Kim, N. K. D., Park, W.-Y., Kim, J. S., Kim, M., et al. (2019). The effect of Globus pallidus Interna deep brain stimulation on a dystonia patient with the GNAL mutation compared to patients with DYT1 and DYT6. J Mov Disord 12, 120–124. doi: 10.14802/jmd.19006
Ahrweiller, K., Houvenaghel, J. F., Riou, A., Drapier, S., Sauleau, P., Haegelen, C., et al. (2019). Postural instability and gait disorders after subthalamic nucleus deep brain stimulation in Parkinson’s disease: a PET study. J. Neurol. 266, 2764–2771. doi: 10.1007/s00415-019-09482-y
Albanese, A., Bhatia, K., Bressman, S. B., DeLong, M. R., Fahn, S., Fung, V. S. C., et al. (2013). Phenomenology and classification of dystonia: a consensus update: dystonia: phenomenology and classification. Mov. Disord. 28, 863–873. doi: 10.1002/mds.25475
Alcalay, R. N., Kehoe, C., Shorr, E., Battista, R., Hall, A., Simuni, T., et al. (2020). Genetic testing for Parkinson disease: current practice, knowledge, and attitudes among US and Canadian movement disorders specialists. Genet. Med. 22, 574–580. doi: 10.1038/s41436-019-0684-x
Andrews, C., Aviles-Olmos, I., Hariz, M., and Foltynie, T. (2010). Which patients with dystonia benefit from deep brain stimulation? A metaregression of individual patient outcomes. J. Neurol. Neurosurg. Psychiatry 81, 1383–1389. doi: 10.1136/jnnp.2010.207993
Angeli, A., Mencacci, N. E., Duran, R., Aviles-Olmos, I., Kefalopoulou, Z., Candelario, J., et al. (2013). Genotype and phenotype in Parkinson’s disease: lessons in heterogeneity from deep brain stimulation. Mov. Disord. 28, 1370–1375. doi: 10.1002/mds.25535
Antonini, A., Pilleri, M., Padoan, A., Landi, A., Ferla, S., Biundo, R., et al. (2012). Successful subthalamic stimulation in genetic Parkinson’s disease caused by duplication of the α-synuclein gene. J. Neurol. 259, 165–167. doi: 10.1007/s00415-011-6162-2
Artusi, C. A., Dwivedi, A., Romagnolo, A., Bortolani, S., Marsili, L., Imbalzano, G., et al. (2020). Differential response to pallidal deep brain stimulation among monogenic dystonias: systematic review and meta-analysis. J. Neurol. Neurosurg. Psychiatry 91, 426–433. doi: 10.1136/jnnp-2019-322169
Artusi, C. A., and Lopiano, L. (2023). Should we offer deep brain stimulation to Parkinson’s disease patients with GBA mutations? Front. Neurol. 14:1158977. doi: 10.3389/fneur.2023.1158977
Ashkan, K., Rogers, P., Bergman, H., and Ughratdar, I. (2017). Insights into the mechanisms of deep brain stimulation. Nat. Rev. Neurol. 13, 548–554. doi: 10.1038/nrneurol.2017.105
Bari, A. A., Fasano, A., Munhoz, R. P., and Lozano, A. M. (2015). Improving outcomes of subthalamic nucleus deep brain stimulation in Parkinson’s disease. Expert. Rev. Neurother. 15, 1151–1160. doi: 10.1586/14737175.2015.1081815
Bean, L., Funke, B., Carlston, C. M., Gannon, J. L., Kantarci, S., Krock, B. L., et al. (2020). Diagnostic gene sequencing panels: from design to report—a technical standard of the American College of Medical Genetics and Genomics (ACMG). Genet. Med. 22, 453–461. doi: 10.1038/s41436-019-0666-z
Brüggemann, N., Domingo, A., Rasche, D., Moll, C. K. E., Rosales, R. L., Jamora, R. D. G., et al. (2019). Association of Pallidal Neurostimulation and Outcome Predictors with X-linked dystonia parkinsonism. JAMA Neurol. 76:211. doi: 10.1001/jamaneurol.2018.3777
Chetrit, E. B., Alcalay, R. N., Steiner-Birmanns, B., Altarescu, G., Phillips, M., Elstein, D., et al. (2013). Phenotype in patients with Gaucher disease and Parkinson disease. Blood Cells Mol. Dis. 50, 218–221. doi: 10.1016/j.bcmd.2012.11.011
Cif, L., Vasques, X., Gonzalez, V., Ravel, P., Biolsi, B., Collod-Beroud, G., et al. (2010). Long-term follow-up of DYT1 dystonia patients treated by deep brain stimulation: an open-label study: clinical course of DYT1 dystonia with DBS. Mov. Disord. 25, 289–299. doi: 10.1002/mds.22802
Córdoba, M., Rodriguez-Quiroga, S. A., Vega, P. A., Salinas, V., Perez-Maturo, J., Amartino, H., et al. (2018). Whole exome sequencing in neurogenetic odysseys: an effective, cost- and time-saving diagnostic approach. PLoS One 13:e0191228. doi: 10.1371/journal.pone.0191228
Covolo, A., Imbalzano, G., Artusi, C. A., Montanaro, E., Ledda, C., Bozzali, M., et al. (2023). 15-year subthalamic deep brain stimulation outcome in a Parkinson’s disease patient with Parkin gene mutation: a case report. Neurol. Sci. 44, 2939–2942. doi: 10.1007/s10072-023-06789-7
de Oliveira, L. M., Barbosa, E. R., Aquino, C. C., Munhoz, R. P., Fasano, A., and Cury, R. G. (2019). Deep brain stimulation in patients with mutations in Parkinson’s disease-related genes: a systematic review. Mov Disord Clin Pract 6, 359–368. doi: 10.1002/mdc3.12795
Eltahawy, H. A., Saint-Cyr, J., Giladi, N., Lang, A. E., and Lozano, A. M. (2004). Primary dystonia is more responsive than secondary dystonia to Pallidal interventions: outcome after Pallidotomy or Pallidal deep brain stimulation. Neurosurgery 54, 613–621. doi: 10.1227/01.NEU.0000108643.94730.21
Finger, M. E., Siddiqui, M. S., Morris, A. K., Ruckart, K. W., Wright, S. C., Haq, I. U., et al. (2020). Auditory-perceptual evaluation of deep brain stimulation on voice and speech in patients with dystonia. J. Voice 34, 636–644. doi: 10.1016/j.jvoice.2019.02.010
Frey, J., Cagle, J., Johnson, K. A., Wong, J. K., Hilliard, J. D., Butson, C. R., et al. (2022). Past, present, and future of deep brain stimulation: hardware, software, imaging, physiology and novel approaches. Front. Neurol. 13:825178. doi: 10.3389/fneur.2022.825178
Fuchs, T., Saunders-Pullman, R., Masuho, I., Luciano, M. S., Raymond, D., Factor, S., et al. (2013). Mutations in GNAL cause primary torsion dystonia. Nat. Genet. 45, 88–92. doi: 10.1038/ng.2496
Groen, J. L., Ritz, K., Contarino, M. F., van de Warrenburg, B. P., Aramideh, M., Foncke, E. M., et al. (2010). DYT6 dystonia: mutation screening, phenotype, and response to deep brain stimulation. Mov. Disord. 25, 2420–2427. doi: 10.1002/mds.23285
Gruber, D., Kühn, A. A., Schoenecker, T., Kivi, A., Trottenberg, T., Hoffmann, K.-T., et al. (2010). Pallidal and thalamic deep brain stimulation in myoclonus-dystonia. Mov. Disord. 25, 1733–1743. doi: 10.1002/mds.23312
Hartmann, C. J., Fliegen, S., Groiss, S. J., Wojtecki, L., and Schnitzler, A. (2019). An update on best practice of deep brain stimulation in Parkinson’s disease. Ther. Adv. Neurol. Disord. 12:1756286419838096. doi: 10.1177/1756286419838096
Hickey, P., and Stacy, M. (2016). Deep brain stimulation: a paradigm shifting approach to treat Parkinson’s disease. Front. Neurosci. 10:173. doi: 10.3389/fnins.2016.00173
Iwaki, H., Blauwendraat, C., Leonard, H. L., Liu, G., Maple-Grødem, J., Corvol, J.-C., et al. (2019). Genetic risk of Parkinson disease and progression. Neurol Genet 5:e348. doi: 10.1212/NXG.0000000000000348
Johnson, N. E. (2015). Whole-exome sequencing in neurologic practice: reducing the diagnostic odyssey. Neurol. Genet. 1:e37. doi: 10.1212/NXG.0000000000000037
Kamo, H., Oyama, G., Nishioka, K., Funayama, M., and Hattori, N. (2022). Deep brain stimulation for a patient with familial Parkinson’s disease harboring CHCHD2 p.T61I. Mov Disord Clin Pract 9, 407–409. doi: 10.1002/mdc3.13428
Kim, J. H., Na, Y. C., Lee, W. H., Chang, W. S., Jung, H. H., and Chang, J. W. (2014). Bilateral Globus pallidus Interna deep-brain stimulation in a patient with myoclonus-dystonia: a case report. Neuromodulation 17, 724–728. doi: 10.1111/ner.12162
Koeglsperger, T., Palleis, C., Hell, F., Mehrkens, J. H., and Bötzel, K. (2019). Deep brain stimulation programming for movement disorders: current concepts and evidence-based strategies. Front. Neurol. 10:410. doi: 10.3389/fneur.2019.00410
Kosutzka, Z., Tisch, S., Bonnet, C., Ruiz, M., Hainque, E., Welter, M., et al. (2019). Long-term GPi-DBS improves motor features in myoclonus-dystonia and enhances social adjustment. Mov. Disord. 34, 87–94. doi: 10.1002/mds.27474
Kovanda, A., Rački, V., Bergant, G., Georgiev, D., Flisar, D., Papić, E., et al. (2022). A multicenter study of genetic testing for Parkinson’s disease in the clinical setting. NPJ Parkinsons Dis. 8, 149–144. doi: 10.1038/s41531-022-00408-6
Krause, P., Brüggemann, N., Völzmann, S., Horn, A., Kupsch, A., Schneider, G.-H., et al. (2015). Long-term effect on dystonia after pallidal deep brain stimulation (DBS) in three members of a family with a THAP1 mutation. J. Neurol. 262, 2739–2744. doi: 10.1007/s00415-015-7908-z
Kurtis, M. M., Rajah, T., Delgado, L. F., and Dafsari, H. S. (2017). The effect of deep brain stimulation on the non-motor symptoms of Parkinson’s disease: a critical review of the current evidence. NPJ Parkinsons Dis 3:16024. doi: 10.1038/npjparkd.2016.24
Kurtis, M. M., San Luciano, M., Yu, Q., Goodman, R. R., Ford, B., Raymond, D., et al. (2010). Clinical and neurophysiological improvement of SGCE myoclonus–dystonia with GPi deep brain stimulation. Clin. Neurol. Neurosurg. 112, 149–152. doi: 10.1016/j.clineuro.2009.10.001
Lefroy, H., Harrison, V., and Németh, A. H. (2020). Genetic testing in neurology. Medicine 48, 545–549. doi: 10.1016/j.mpmed.2020.05.009
Limotai, N., Go, C., Oyama, G., Hwynn, N., Zesiewicz, T., Foote, K., et al. (2011). Mixed results for GPi-DBS in the treatment of cranio-facial and cranio-cervical dystonia symptoms. J. Neurol. 258, 2069–2074. doi: 10.1007/s00415-011-6075-0
Limousin, P., and Foltynie, T. (2019). Long-term outcomes of deep brain stimulation in Parkinson disease. Nat. Rev. Neurol. 15, 234–242. doi: 10.1038/s41582-019-0145-9
Lozano, A. M., Lipsman, N., Bergman, H., Brown, P., Chabardes, S., Chang, J. W., et al. (2019). Deep brain stimulation: current challenges and future directions. Nat. Rev. Neurol. 15, 148–160. doi: 10.1038/s41582-018-0128-2
Lythe, V., Athauda, D., Foley, J., Mencacci, N. E., Jahanshahi, M., Cipolotti, L., et al. (2017). GBA-associated Parkinson’s disease: progression in a deep brain stimulation cohort. J. Parkinsons Dis. 7, 635–644. doi: 10.3233/JPD-171172
Mangone, G., Bekadar, S., Cormier-Dequaire, F., Tahiri, K., Welaratne, A., Czernecki, V., et al. (2020). Early cognitive decline after bilateral subthalamic deep brain stimulation in Parkinson’s disease patients with GBA mutations. Parkinsonism Relat. Disord. 76, 56–62. doi: 10.1016/j.parkreldis.2020.04.002
Marsili, L., Vizcarra, J. A., Sturchio, A., Dwivedi, A. K., Keeling, E. G., Patel, D., et al. (2021). When does postural instability appear in monogenic parkinsonisms? An individual-patient meta-analysis. J. Neurol. 268, 3203–3211. doi: 10.1007/s00415-020-09892-3
Mure, H., Morigaki, R., Koizumi, H., Okita, S., Kawarai, T., Miyamoto, R., et al. (2014). Deep brain stimulation of the thalamic ventral lateral anterior nucleus for DYT6 dystonia. Stereotact. Funct. Neurosurg. 92, 393–396. doi: 10.1159/000365577
Olgiati, S., Quadri, M., and Bonifati, V. (2016). Genetics of movement disorders in the next-generation sequencing era. Mov. Disord. 31, 458–470. doi: 10.1002/mds.26521
Pal, G., Cook, L., Schulze, J., Verbrugge, J., Alcalay, R. N., Merello, M., et al. (2023). Genetic testing in Parkinson’s disease. Mov. Disord. 38, 1384–1396. doi: 10.1002/mds.29500
Pal, G., Mangone, G., Hill, E. J., Ouyang, B., Liu, Y., Lythe, V., et al. (2022). Parkinson disease and subthalamic nucleus deep brain stimulation: cognitive effects in GBA mutation carriers. Ann. Neurol. 91, 424–435. doi: 10.1002/ana.26302
Panov, F., Tagliati, M., Ozelius, L. J., Fuchs, T., Gologorsky, Y., Cheung, T., et al. (2012). Pallidal deep brain stimulation for DYT6 dystonia. J. Neurol. Neurosurg. Psychiatry 83, 182–187. doi: 10.1136/jnnp-2011-300979
Pozojevic, J., Beetz, C., and Westenberger, A. (2021). The importance of genetic testing for dystonia patients and translational research. J. Neural Transm. 128, 473–481. doi: 10.1007/s00702-021-02329-9
Pycroft, L., Stein, J., and Aziz, T. (2018). Deep brain stimulation: an overview of history, methods, and future developments. Brain Neurosci Adv 2:2398212818816017. doi: 10.1177/2398212818816017
Rački, V., Hero, M., Rožmarić, G., Papić, E., Raguž, M., Chudy, D., et al. (2022). Cognitive impact of deep brain stimulation in Parkinson’s disease patients: a systematic review. Front. Hum. Neurosci. 16:867055. doi: 10.3389/fnhum.2022.867055
Racki, V., Papic, E., Almahariq, F., Chudy, D., and Vuletic, V. (2021). The successful three-year outcome of deep brain stimulation in Gaucher disease type 1 associated Parkinson’s disease: a case report. Mov Disord Clin Pract 8, 604–606. doi: 10.1002/mdc3.13185
Rajan, R., Garg, K., Saini, A., Radhakrishnan, D. M., Carecchio, M., BK, B., et al. (2022). GPi-DBS for KMT2B-associated dystonia: systematic review and meta-analysis. Mov. Disord. Clin. Pract. 9, 31–37. doi: 10.1002/mdc3.13374
Reese, R., and Volkmann, J. (2014). “Deep brain stimulation for dystonia” in Deep Brain Stimulation: Technology and Applications, vol. 2 (Unitec House, 2 Albert Place, London N3 1QB, UK: Future Medicine Ltd), 18–31.
Reese, R., and Volkmann, J. (2017). Deep brain stimulation for the Dystonias: evidence, knowledge gaps, and practical considerations. Mov. Disord. Clin. Pract. 4, 486–494. doi: 10.1002/mdc3.12519
Rizzone, M. G., Martone, T., Balestrino, R., and Lopiano, L. (2019). Genetic background and outcome of deep brain stimulation in Parkinson’s disease. Parkinsonism Relat. Disord. 64, 8–19. doi: 10.1016/j.parkreldis.2018.08.006
Salinas, V., Vega, P., Marsili, L., Pérez-Maturo, J., Martínez, N., Zavala, L., et al. (2020). The odyssey of complex neurogenetic disorders: from undetermined to positive. Am. J. Med. Genet. C Semin. Med. Genet. 184, 876–884. doi: 10.1002/ajmg.c.31848
Sarva, H., Trosch, R., Kiss, Z. H. T., Furtado, S., Luciano, M. S., Glickman, A., et al. (2019). Deep brain stimulation in isolated dystonia with a GNAL mutation. Mov. Disord. 34, 301–303. doi: 10.1002/mds.27585
Satam, H., Joshi, K., Mangrolia, U., Waghoo, S., Zaidi, G., Rawool, S., et al. (2023). Next-generation sequencing technology: current trends and advancements. Biology (Basel) 12:997. doi: 10.3390/biology12070997
Sayad, M., Zouambia, M., Chaouch, M., Ferrat, F., Nebbal, M., Bendini, M., et al. (2016). Greater improvement in LRRK2 G2019S patients undergoing subthalamic nucleus deep brain stimulation compared to non-mutation carriers. BMC Neurosci. 17:6. doi: 10.1186/s12868-016-0240-4
Straccia, G., Colucci, F., Eleopra, R., and Cilia, R. (2022). Precision medicine in Parkinson’s disease: from genetic risk signals to personalized therapy. Brain Sci. 12:1308. doi: 10.3390/brainsci12101308
Tisch, S. (2022). Deep brain stimulation in dystonia: factors contributing to variability in outcome in short and long term follow-up. Curr. Opin. Neurol. 35, 510–517. doi: 10.1097/WCO.0000000000001072
Tisch, S., and Kumar, K. R. (2021). Pallidal deep brain stimulation for monogenic dystonia: the effect of gene on outcome. Front. Neurol. 11:630391. doi: 10.3389/fneur.2020.630391
Trottenberg, T., Meissner, W., Arnold, G., Einhaupl, K. M., Kupsch, A., Kabus, C., et al. (2001). Neurostimulation of the ventral intermediate thalamic nucleus in inherited myoclonus-dystonia syndrome. Mov. Disord. 16, 769–771. doi: 10.1002/mds.1119
Tsuboi, T., Cif, L., Coubes, P., Ostrem, J. L., Romero, D. A., Miyagi, Y., et al. (2020). Secondary worsening following DYT1 dystonia deep brain stimulation: a multi-country cohort. Front. Hum. Neurosci. 14:242. doi: 10.3389/fnhum.2020.00242
Vitek, J. L., Patriat, R., Ingham, L., Reich, M. M., Volkmann, J., and Harel, N. (2022). Lead location as a determinant of motor benefit in subthalamic nucleus deep brain stimulation for Parkinson’s disease. Front. Neurosci. 16:1010253. doi: 10.3389/fnins.2022.1010253
Wang, J.-W., Li, J.-P., Wang, Y.-P., Zhang, X.-H., and Zhang, Y.-Q. (2017). Deep brain stimulation for myoclonus-dystonia syndrome with double mutations in DYT1 and DYT11. Sci. Rep. 7:41042. doi: 10.1038/srep41042
Weiss, D., Brockmann, K., Srulijes, K., Meisner, C., Klotz, R., Reinbold, S., et al. (2012). Long-term follow-up of subthalamic nucleus stimulation in glucocerebrosidase-associated Parkinson’s disease. J. Neurol. 259, 1970–1972. doi: 10.1007/s00415-012-6469-7
Youn, J., Oyama, G., Hattori, N., Shimo, Y., Kuusimäki, T., Kaasinen, V., et al. (2022). Subthalamic deep brain stimulation in Parkinson’s disease with SNCA mutations: based on the follow-up to 10 years. Brain Behav. 12:e2503. doi: 10.1002/brb3.2503
Zech, M., Jech, R., Boesch, S., Škorvánek, M., Necpál, J., Švantnerová, J., et al. (2021). Scoring algorithm-based genomic testing in dystonia: a prospective validation study. Mov. Disord. 36, 1959–1964. doi: 10.1002/mds.28614
Keywords: deep brain stimulation, dystonia, Parkinson’s disease, whole-exome sequencing, genetic testing
Citation: Rački V, Hero M, Papić E, Rožmarić G, Čizmarević NS, Chudy D, Peterlin B and Vuletić V (2023) Applicability of clinical genetic testing for deep brain stimulation treatment in monogenic Parkinson’s disease and monogenic dystonia: a multidisciplinary team perspective. Front. Neurosci. 17:1282267. doi: 10.3389/fnins.2023.1282267
Edited by:
Vitor Engracia Valenti, São Paulo State University, BrazilReviewed by:
Giulia Straccia, C.T.O. Hospital, A.O.R.N. Dei Colli, ItalyCopyright © 2023 Rački, Hero, Papić, Rožmarić, Čizmarević, Chudy, Peterlin and Vuletić. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Valentino Rački, dmFsZW50aW5vLnJhY2tpQHVuaXJpLmhy
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.