 Sophie F. Hill
Sophie F. Hill Paymaan Jafar-Nejad3
Paymaan Jafar-Nejad3 Miriam H. Meisler
Miriam H. Meisler- 1Neuroscience Graduate Program, University of Michigan, Ann Arbor, MI, United States
- 2Department of Human Genetics, University of Michigan, Ann Arbor, MI, United States
- 3Ionis Pharmaceuticals, Carlsbad, CA, United States
- 4Department of Neurology, University of Michigan, Ann Arbor, MI, United States
Developmental and epileptic encephalopathies (DEEs) are severe seizure disorders with inadequate treatment options. Gain- or loss-of-function mutations of neuronal ion channel genes, including potassium channels and voltage-gated sodium channels, are common causes of DEE. We previously demonstrated that reduced expression of the sodium channel gene Scn8a is therapeutic in mouse models of sodium and potassium channel mutations. In the current study, we tested whether reducing expression of the potassium channel gene Kcnt1 would be therapeutic in mice with mutation of the sodium channel genes Scn1a or Scn8a. A Kcnt1 antisense oligonucleotide (ASO) prolonged survival of both Scn1a and Scn8a mutant mice, suggesting a modulatory effect for KCNT1 on the balance between excitation and inhibition. The cation channel blocker quinidine was not effective in prolonging survival of the Scn8a mutant. Our results implicate KCNT1 as a therapeutic target for treatment of SCN1A and SCN8A epilepsy.
1. Introduction
Developmental and epileptic encephalopathies (DEEs) are among the most severe epileptic disorders. The typical disease course begins with onset of seizures during the first year of life, followed by developmental delay, movement disorders, intellectual disability, sleep disturbances, and feeding difficulties (Scheffer and Nabbout, 2019; Meisler et al., 2021; Johannesen et al., 2022). Seizures are often resistant to treatment with current antiepileptic drugs (Scheffer and Nabbout, 2019; Meisler et al., 2021; Johannesen et al., 2022).
Many DEEs result from mutations in sodium and potassium channel genes (Lindy et al., 2018; Symonds et al., 2019). Based on their roles in the neuronal action potential, excessive sodium current or insufficient potassium current would be predicted to cause hyperexcitability and epilepsy. In agreement with expectation, many missense mutations in the voltage-gated sodium channel gene SCN8A result in excessive sodium current (“gain-of-function”, or GOF, mutations) and SCN8A-DEE (Veeramah et al., 2012; Meisler et al., 2021; Johannesen et al., 2022). Experimental expression of an SCN8A GOF mutation in excitatory neurons is sufficient to cause seizures and premature death, while expression limited to inhibitory neurons does not (Bunton-Stasyshyn et al., 2019).
Loss-of-function (LOF) mutations of the sodium channel gene SCN1A and GOF mutations of the potassium channel gene KCNT1 can also cause epilepsy (Barcia et al., 2012; Scheffer and Nabbout, 2019; Gribkoff and Winquist, 2023). SCN1A haploinsufficiency reduces the excitability of inhibitory neurons, altering excitation/inhibition balance (Cheah et al., 2012; Tai et al., 2014; Favero et al., 2018). The epileptogenic mechanism of KCNT1 GOF mutations is not well established, but a similar disinhibitory mechanism may be involved (Shore et al., 2020; Gertler et al., 2022; Wu et al., 2023).
KCNT1 is a sodium-activated potassium channel (also known as Slo2.2, KNa1.1, or Slack) with widespread expression in the central nervous system (Rizzi et al., 2016). KCNT1 regulates afterhyperpolarization amplitude and action potential threshold (Martinez-Espinosa et al., 2015; Quraishi et al., 2019; Shore et al., 2020; Gertler et al., 2022; Wu et al., 2023). KCNT1 GOF mutations enhance bursting behavior in excitatory neurons and reduce action potential firing in inhibitory neurons (Quraishi et al., 2019; Shore et al., 2020; Gertler et al., 2022; Wu et al., 2023).
In the mouse, homozygous knock-in of KCNT1 GOF mutations results in spontaneous seizures, reduced threshold for seizure induction, behavioral abnormalities, and premature lethality (Quraishi et al., 2020; Shore et al., 2020; Burbano et al., 2022; Gertler et al., 2022). Burbano et al. (2022) described an antisense oligonucleotide (ASO) that reduces expression of Kcnt1. Administration of the ASO to a homozygous Kcnt1 GOF mouse prolonged survival, reduced seizure frequency, and corrected behavioral abnormalities. Conversely, homozygous loss of Kcnt1 also improves survival after electrically induced seizrues (Quraishi et al., 2020).
Reducing expression of Scn8a prolongs survival of epileptic mice with mutations in the potassium channel genes Kcna1 and Kcnq2 (Hill et al., 2022). Here, we asked whether modulating expression of a potassium channel can improve the phenotype of sodium channel mutants. Administration of the Kcnt1 ASO (Burbano et al., 2022) on postnatal day 2 doubled the lifespan of Scn8a mutant mice and extended survival of Scn1a haploinsufficient mice. Our results suggest a new therapeutic intervention for DEEs caused by mutations of SCN1A and SCN8A.
2. Methods
2.1. Mice
The Scn8acond allele, abbreviated W, contains two tandem copies of exon 26, the final coding exon of Scn8a (Bunton-Stasyshyn et al., 2019). The upstream copy, designated 26a, is a floxed exon that encodes the wildtype channel. Deletion of exon 26a by Cre results in expression of exon 26b encoding the variant p.R1872W. This variant has been identified in multiple individuals with SCN8A epilepsy (Bunton-Stasyshyn et al., 2019; Johannesen et al., 2022). Scn8acond/cond male mice were crossed with EIIa-Cre/+ female mice (JAX 003724) to generate Scn8acond/+,EIIa-Cre double heterozygous mice expressing the R1872W variant (designated W/+ mice). Both the Scn8acond allele and the EIIa-Cre transgene were maintained on a C57Bl/6 J genetic background.
Scn1a+/− mice with deletion of exon 1 were maintained on the protective 129S6/SvEvTac strain background and activated in (C57BL/6 J X 129S6/SvEvTac) F1 mice (Miller et al., 2014). Both male and female mice were used for all experiments. Experiments were approved by the Committee on the Use and Care of Animals at the University of Michigan.
2.2. ASOs
ASOs were synthesized by Ionis Pharmaceuticals as described (Swayze et al., 2007). Both the non-targeting control and Kcnt1 ASOs are 20-bp gap-mers with 5 2’-O-methoxyethyl modifications on the first and last 5 bases and phosphorothioate modifications on all 20 bases. The Kcnt1 ASO (5’ GCT TCA TGC CAC TTT CCA GA 3′) is complementary to the 3’ UTR of mouse Kcnt1 and was previously described (Burbano et al., 2022). The non-targeting control ASO (5’ CCT ATA GGA CTA TTC AGG AA 3′) is well-tolerated and is not complementary to any transcript encoded by the mouse genome (Swayze et al., 2007). Animals treated with control ASO received a 30 μg dose.
2.3. Intracerebroventricular (ICV) injections
At postnatal day 2 (P2), mice were cryo-anesthetized for 3 min. ASO was diluted in PBS (2 μL injection volume) and manually injected into the left ventricle as described (Lenk et al., 2020). Animals were allowed to recover for 10 min at 37°C before being returned to the home cage.
2.4. qRT-PCR
Brain and spinal cord from 3-week-old mice treated with control or Kcnt1 ASO were homogenized in TRIzol (Invitrogen Cat. #15596026, Waltham, MA). RNA was extracted using the Direct-zol RNA Mini Prep kit from Zymo Research (Irvina, CA). cDNA was synthesized with the LunaScript kit from New England Biolabs (Ipwsich, MA). Scn8a (Mm00488110_m1), Kcnt1 (Mm01330661_g1), and Tbp (Mm01277042_m1) transcripts were quantified using TaqMan gene expression assays (Applied Biosystems, Foster City, CA).
2.5. Quinidine administration
Quinidine (Sigma Aldrich, St. Louis, MO) was diluted in phosphate-buffered saline (50 or 100 mg/kg) and administered by daily intraperitoneal injection beginning at P10, the youngest age at which daily intraperitoneal injections were feasible.
3. Results
3.1. Characterization of the Kcnt1 ASO
We used an ASO to reduce expression of mouse Kcnt1. The 20 base-pair “gap-mer” ASO targets the 3’ UTR of the mouse Kcnt1 gene (Figure 1A) and recruits RNaseH1 to degrade the transcript (Burbano et al., 2022). We first administered the ASO to wild-type animals by ICV injection at P2. Three weeks later, we measured gene expression in brain and spinal cord by qRT-PCR (Figure 1B; Supplementary Table S1). Kcnt1 expression was reduced in both brain and spinal cord (two-way ANOVA, p < 0.0001). For example, adminsitration of 45 μg Kcnt1 ASO reduced Kcnt1 expression in brain to 0.25 ± 0.04 of control (mean ± SD, n = 3) (Supplementary Table S1). Reduction of Kcnt1 transcript reduces KCNT1 protein expression (Burbano et al., 2022). Expression of Scn8a was unaffected by the Kcnt1 ASO (Supplementary Figure S1). No changes in Kcnt1 expression were detected in previous studies of Scn1a and Scn8a mutant mice (Sprissler et al., 2017; Hawkins et al., 2019; Valassina et al., 2022).
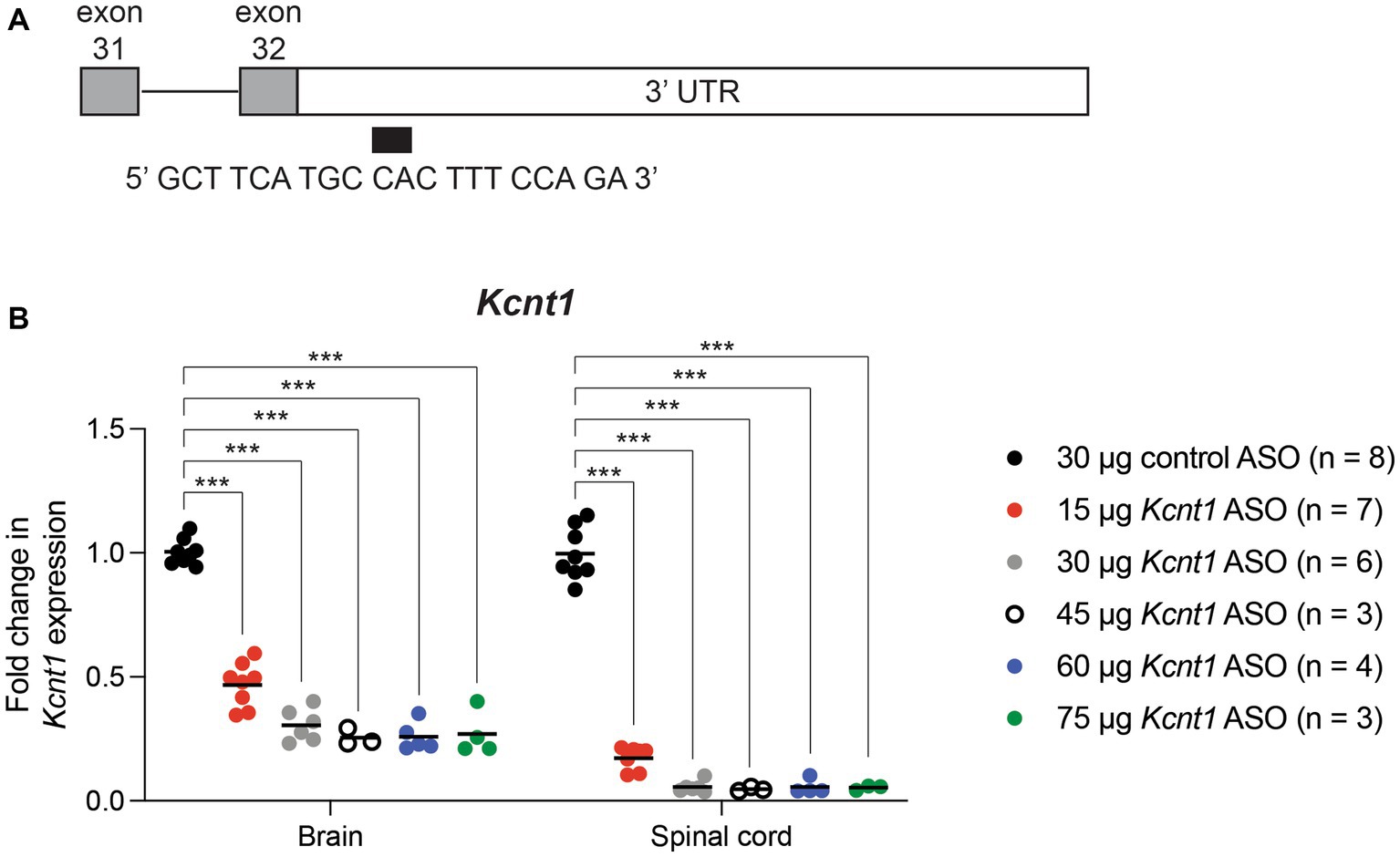
Figure 1. Kcnt1 ASO reduces Kcnt1 transcript abundance. (A) The Kcnt1 ASO targets the proximal 3’UTR of the Kcnt1 transcript. (B) Expression of Kcnt1 in brain and spinal cord from P21 wildtype mice treated with Kcnt1 ASO on P2, measured by qRT-PCR (*** = p < 0.0001, Sidak’s multiple comparisons test).
3.2. ASO-mediated reduction of Kcnt1 extends the lifespan of an SCN8A DEE mouse
We previously generated a mouse with Cre-dependent expression of the patient mutation p.R1872W (Bunton-Stasyshyn et al., 2019). Expression of this mutation by crossing with the ubiquitously expressed EIIa-Cre results in a single, lethal seizure at P14 (Bunton-Stasyshyn et al., 2019). We treated Scn8acond/+,EIIa-Cre (W/+) animals with 15–75 μg Kcnt1 ASO by ICV injection at P2. Mice treated with the control ASO exhibited median survival of 16 days (Figure 2). Mice treated with 15 μg Kcnt1 ASO lived three days longer (median survival = 19 days, p = 0.0493, Mantel-Cox log-rank test). Treatment with 30 μg Kcnt1 ASO extended median survival to 27 days (p < 0.0001, Mantel- Cox log-rank test). Mice treated with 45 μg, the optimal dose, exhibited median survival of 36 days, more than double the lifespan of control ASO-treated mice (p < 0.0001, Mantel-Cox log-rank test, Figure 2). Treatment with 60 or 75 μg Kcnt1 ASO did not further reduce Kcnt1 expression (Figure 1B) or further extend survival (Supplementary Figure S2).
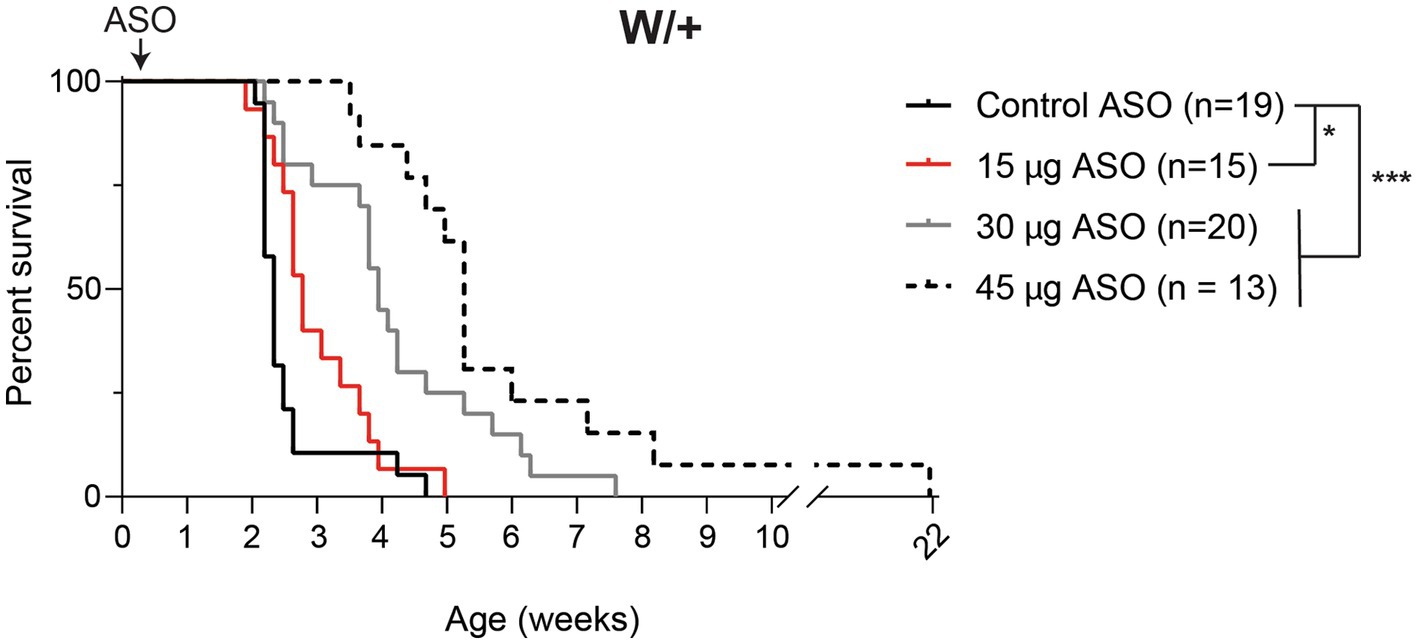
Figure 2. Kcnt1 ASO prolongs survival of Scn8a mutant mice. Survival of Scn8acond/+,EIIa-Cre (W/+) mice treated with 15–45 μg Kcnt1 ASO at P2 compared with a cohort of contemporaneous control mice treated with control ASO [Control data was previously published in Lenk et al. (2020)]. Asterisks indicate significance of Mantel-Cox log-rank tests: * = p < 0.05, *** = p < 0.0001.
3.3. Quinidine does not extend survival in the SCN8A DEE mouse
Quinidine is a nonspecific cation channel blocker used to treat cardiac arrhythmia. In vitro, quinidine blocks KCNT1 channel activity, suggesting that it could be a precision therapy for patients with gain-of-function KCNT1 mutations (Mori et al., 1998; Milligan et al., 2014). In vivo, quinidine has mixed efficacy in KCNT1 epilepsy patients (Mikati et al., 2015; Numis et al., 2018; Fitzgerald et al., 2019; Cole et al., 2021).
To determine whether inhibition of KCNT1 channels by quinidine would be therapeutic in Scn8a mutant mice, we administered 50 or 100 mg/kg quinidine by daily intraperitoneal injection beginning at P10 (Figure 3). Untreated mice exhibited median survival of 15 days (n = 47). Treatment with 50 or 100 mg/kg quinidine did not extend the lifespan of the SCN8A-DEE mice (median survival = 14 days; n = 7 & 9, respectively; Figure 3).
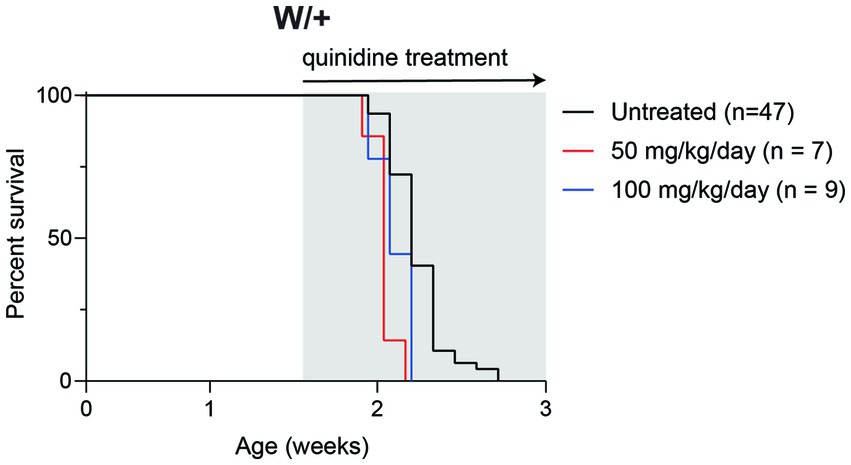
Figure 3. Quinidine does not prolong survival of Scn8a mutant mice. Survival of Scn8acond/+,EIIa-Cre (W/+) mice daily treated with 50 or 100 mg/kg quinidine compared to untreated mice. Grey shading indicates treatment period.
3.4. ASO-mediated reduction of Kcnt1 extends the lifespan of a mouse model of SCN1A haploinsufficiency
We also tested the effect of the Kcnt1 ASO in Scn1a+/− mice, a model of Dravet Syndrome. Consistent with previous studies (Miller et al., 2014; Favero et al., 2018), approximately 1/3 of untreated Scn1a+/− mice died between 3 and 4 weeks of age, and during the remaining 6-month monitoring period, there were several sporadic deaths (Figure 4). We administered 45 μg Kcnt1 ASO to Scn1a+/− mice at P2. None of the treated mice died in the first 4 weeks, indicating that reduced Kcnt1 expression during this critical period is sufficient to prevent death (p = 0.0513, Mantel-Cox log-rank test, Figure 4). There were four deaths during the 6-month monitoring period, all after 9 weeks of age (Figure 4). Since quinidine was not effective in the Scn8a mutant mice, we did not treat the Scn1a mutant mice.
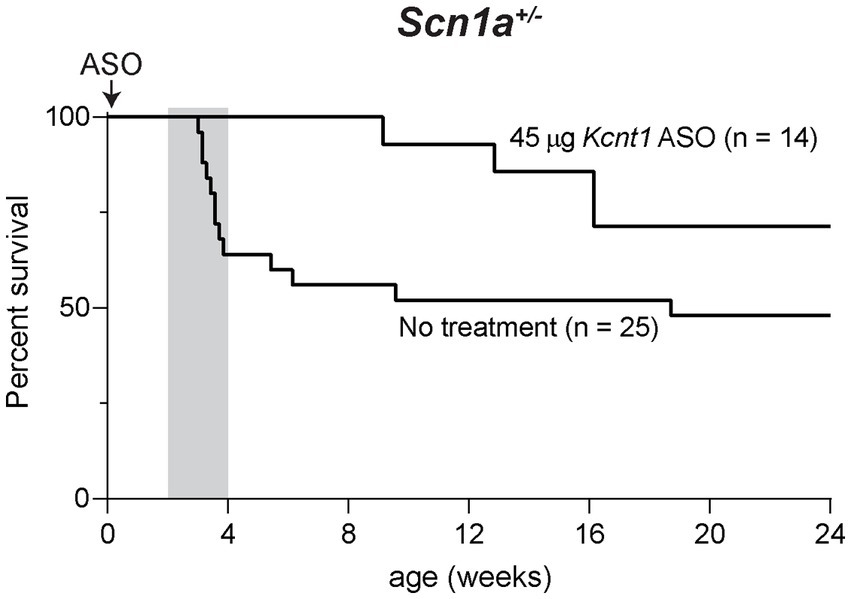
Figure 4. Kcnt1 ASO prolongs survival of Scn1a+/− mice. Survival of Scn1a+/− mice treated with 45 μg Kcnt1 ASO at P2 compared to untreated mice (p = 0.0513, Mantel-Cox log-rank test). Grey shading indicates the critical period in the development of Dravet Syndrome.
4. Discussion
Developmental and epileptic encephalopathies are frequently caused by pathological variants of ion channel genes. Here, we showed that reduction of Kcnt1 expression is protective in mouse models of Scn1a and Scn8a epilepsy. Our findings suggest that patients with mutations of SCN1A and SCN8A could benefit from treatment with a KCNT1 ASO or KCNT1-specific channel blocker.
A previous study demonstrated that the Kcnt1 ASO improved the survival, seizure, and behavioral phenotypes of Kcnt1 GOF mice (Burbano et al., 2022). Interestingly, the Kcnt1 ASO was therapeutic at lower doses in Kcnt1 mutant mice than in the Scn8a mutant studied here. In neonatal Kcnt1 mutant mice, 3.4 μg extended the median survival by more than 100 days (Burbano et al., 2022). In contrast, doses of 15–45 μg added only 20 days to survival of the Scn8a mutant mice. These observations suggest that the effect in the Scn8a mutant may be indirect. For example, gain-of-function of Kcnt1 reduces excitability of parvalbumin interneurons (Gertler et al., 2022). Reduced expression of Kcnt1 may enhance excitability of parvalbumin interneurons and thereby reduce seizure susceptibility. Consistent with this hypothesis, homozygous knockout of Kcnt1 reduces the lethality of electrically-induced seizures by more than half (Quraishi et al., 2020). Further investigation may identify other types of epilepsy that respond to reduction of KCNT1.
We previously demonstrated that reducing Scn8a expression is therapeutic in Scn1a+/− mice (Lenk et al., 2020) and in mice with epilepsy caused by loss of the potassium channel genes Kcna1 and Kcnq2 (Hill et al., 2022). P2 administration of the Scn8a ASO completely rescued the Scn1a+/− mice (Lenk et al., 2020). In contrast, 4/14 of the Scn1a+/− mice treated with the Kcnt1 ASO died between two and six months of age. The deaths after 2 months may result from turnover of the Kcnt1 ASO; alternatively, reduced Kcnt1 may be effective only in the interval between 3–4 weeks. The long-term effectiveness of the Scn8a ASO in Scn1a mutant mice is interesting, since the effect on Scn8a expression persists for only 6 weeks (Lenk et al., 2020). Viral overexpression of the Kcna1 channel is protective against seizures induced by tetanus neurotoxin or pentylenetetrazole (Snowball et al., 2019; Qiu et al., 2022). Taken together, these observations suggest that modulation of ion channel expression to compensate for epileptogenic mutations is a promising therapeutic strategy.
Among the ion channel genes that could be targeted to treat channelopathies, KCNT1 is an attractive choice because reduced expression is well tolerated. Heterozygous loss-of-function mutations of KCNT1 are present in the general population and not associated with disease (Lek et al., 2016; Karczewski et al., 2020). Kcnt1−/− mice are healthy and fertile, with minor abnormalities such as impaired reversal learning and slightly elevated pain sensitivity (Bausch et al., 2015; Lu et al., 2015; Martinez-Espinosa et al., 2015; Quraishi et al., 2020). In contrast, heterozygous loss of Scn8a is not present in the healthy population (probability of loss-of-function intolerance, pLI = 1) (Lek et al., 2016; Karczewski et al., 2020) and homozygous loss is lethal in the mouse (Burgess et al., 1995).
Quinidine has been proposed as a therapy for patients with KCNT1 epilepsy because of the effectiveness of high doses for correction of GOF mutations in vitro (Milligan et al., 2014; Numis et al., 2018). The effects of quinidine are not specific to KCNT1 (Roden, 2014). Clinical application of quinidine in KCNT1 epilepsy has mixed success. Some individuals achieved seizure freedom (Mikati et al., 2015; Fitzgerald et al., 2019), but most patients report no benefit or worsening seizures (Mikati et al., 2015; Numis et al., 2018; Cole et al., 2020). Quinidine concentration sufficiently high to block KCNT1 may be difficult to achieve in vivo without deleterious effects on other ion channels (Liu et al., 2022). We found that quinidine was not protective in Scn8a mutant mice. More specific KCNT1 channel blockers (Cole et al., 2020; Griffin et al., 2021) may be more effective for treatment of KCNT1, SCN8A, and SCN1A epilepsy.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary material, further inquiries can be directed to the corresponding author.
Ethics statement
The animal study was approved by University of Michigan Institutional Animal Care & Use Committee. The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
SH: Conceptualization, Data curation, Formal Analysis, Investigation, Methodology, Visualization, Writing – original draft, Writing – review & editing. PJ-N: Conceptualization, Resources, Writing – review & editing. FR: Conceptualization, Resources, Writing – review & editing. MM: Conceptualization, Funding acquisition, Supervision, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. Supported by NINDS grant R01 NS34509 (MM) and a Rackham Predoctoral Fellowship from the Rackham School of Graduate Studies, University of Michigan (SH).
Conflict of interest
PJ-N and FR are paid employees of Ionis Pharmaceuticals.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnins.2023.1282201/full#supplementary-material
References
Barcia, G., Fleming, M. R., Deligniere, A., Gazula, V. R., Brown, M. R., Langouet, M., et al. (2012). De novo gain-of-function KCNT1 channel mutations cause malignant migrating partial seizures of infancy. Nat. Gen. 44, 1255–1259. doi: 10.1038/ng.2441
Bausch, A. E., Dieter, R., Nann, Y., Hausmann, M., Meyerdierks, N., Kaczmarek, L. K., et al. (2015). The sodium-activated potassium channel slack is required for optimal cognitive flexibility in mice. Learn. Mem. 22, 323–335. doi: 10.1101/lm.037820.114
Bunton-Stasyshyn, R. K. A., Wagnon, J. L., Wengert, E. R., Barker, B. S., Faulkner, A., Wagley, P. K., et al. (2019). Prominent role of forebrain excitatory neurons in SCN8A encephalopathy. Brain 142, 362–375. doi: 10.1093/brain/awy324
Burbano, L. E., Li, M., Jancovski, N., Jafar-Nejad, P., Richards, K., Sedo, A., et al. (2022). Antisense oligonucleotide therapy for KCNT1 encephalopathy. JCI Insight 7:e146090. doi: 10.1172/JCI.INSIGHT.146090
Burgess, D. L., Kohrman, D. C., Galt, J., Plummer, N. W., Jones, J. M., Spear, B., et al. (1995). Mutation of a new sodium channel gene, Scn8a, in the mouse mutant “motor endplate disease”. Nat. Genet. 10, 461–465. doi: 10.1038/ng0895-461
Cheah, C. S., Yu, F. H., Westenbroek, R. E., Kalume, F. K., Oakley, J. C., Potter, G. B., et al. (2012). Specific deletion of NaV1.1 sodium channels in inhibitory interneurons causes seizures and premature death in a mouse model of Dravet syndrome. Proc. Natl. Acad. Sci. U. S. A. 109, 14646–14651. doi: 10.1073/pnas.1211591109
Cole, B. A., Clapcote, S. J., Muench, S. P., and Lippiat, J. D. (2021). Targeting KNa1.1 channels in KCNT1-associated epilepsy. Trends Pharmacol. Sci. 42, 700–713. doi: 10.1016/J.TIPS.2021.05.003
Cole, B. A., Johnson, R. M., Dejakaisaya, H., Pilati, N., Fishwick, C. W. G., Muench, S. P., et al. (2020). Structure-based identification and characterization of inhibitors of the epilepsy-associated KNa1.1 (KCNT1) Potassium Channel. iScience 23:101100. doi: 10.1016/J.ISCI.2020.101100
Favero, M., Sotuyo, N. P., Lopez, E., Kearney, J. A., and Goldberg, E. M. (2018). A transient developmental window of fast-spiking interneuron dysfunction in a mouse model of dravet syndrome. J. Neurosci. 38, 7912–7927. doi: 10.1523/JNEUROSCI.0193-18.2018
Fitzgerald, M. P., Fiannacca, M., Smith, D. M., Gertler, T. S., Gunning, B., Syrbe, S., et al. (2019). Treatment responsiveness in KCNT1-related epilepsy. Neurotherapeutics 16, 848–857. doi: 10.1007/s13311-019-00739-y
Gertler, T. S., Cherian, S., DeKeyser, J. M., Kearney, J. A., and George, A. L. Jr. (2022). KNa1.1 gain-of-function preferentially dampens excitability of murine parvalbumin-positive interneurons. Neurobiol. Dis. 168:105713. doi: 10.1016/j.nbd.2022.105713
Gribkoff, V. K., and Winquist, R. J. (2023). Potassium channelopathies associated with epilepsy-related syndromes and directions for therapeutic intervention. Biochem. Pharmacol. 208:115413. doi: 10.1016/J.BCP.2023.115413
Griffin, A. M., Kahlig, K. M., Hatch, R. J., Hughes, Z. A., Chapman, M. L., Antonio, B., et al. (2021). Discovery of the first orally available, selective K(Na)1.1 inhibitor: in vitro and in vivo activity of an Oxadiazole series. ACS Med. Chem. Lett. 12, 593–602. doi: 10.1021/acsmedchemlett.0c00675
Hawkins, N. A., Calhoun, J. D., Huffman, A. M., and Kearney, J. A. (2019). Gene expression profiling in a mouse model of Dravet syndrome. Exp. Neurol. 311, 247–256. doi: 10.1016/J.EXPNEUROL.2018.10.010
Hill, S. F., Ziobro, J. M., Jafar-Nejad, P., Rigo, F., and Meisler, M. H. (2022). Genetic interaction between Scn8a and potassium channel genes Kcna1 and Kcnq2. Epilepsia 63, e125–e131. doi: 10.1111/EPI.17374
Johannesen, K. M., Liu, Y., Koko, M., Gjerulfsen, C. E., Sonnenberg, L., Schubert, J., et al. (2022). Genotype-phenotype correlations in SCN8A-related disorders reveal prognostic and therapeutic implications. Brain 145, 2991–3009. doi: 10.1093/BRAIN/AWAB321
Karczewski, K. J., Francioli, L. C., Tiao, G., Cummings, B. B., Alföldi, J., Wang, Q., et al. (2020). The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581, 434–443. doi: 10.1038/s41586-020-2308-7
Lek, M., Karczewski, K. J., Minikel, E. V., Samocha, K. E., Banks, E., Fennell, T., et al. (2016). Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291. doi: 10.1038/nature19057
Lenk, G. M., Jafar-Nejad, P., Hill, S. F., Huffman, L. D., Smolen, C. E., Wagnon, J. L., et al. (2020). Scn8a antisense oligonucleotide is protective in mouse models of SCN8A encephalopathy and Dravet syndrome. Ann. Neurol. 87, 339–346. doi: 10.1002/ana.25676
Lindy, A. S., Stosser, M. B., Butler, E., Downtain-Pickersgill, C., Shanmugham, A., Retterer, K., et al. (2018). Diagnostic outcomes for genetic testing of 70 genes in 8565 patients with epilepsy and neurodevelopmental disorders. Epilepsia 59, 1062–1071. doi: 10.1111/epi.14074
Liu, R., Sun, L., Wang, Y., Wang, Q., and Wu, J. (2022). New use for an old drug: quinidine in KCNT1-related epilepsy therapy. Neurol. Sci. 44, 1201–1206. doi: 10.1007/S10072-022-06521-X/FIGURES/1
Lu, R., Bausch, A. E., Kallenborn-Gerhardt, W., Stoetzer, C., Debruin, N., Ruth, P., et al. (2015). Slack channels expressed in sensory neurons control neuropathic pain in mice. J. Neurosci. Off. J. Soc. Neurosci. 35, 1125–1135. doi: 10.1523/JNEUROSCI.2423-14.2015
Martinez-Espinosa, P. L., Wu, J., Yang, C., Gonzalez-Perez, V., Zhou, H., Liang, H., et al. (2015). Knockout of Slo2.2 enhances itch, abolishes KNa current, and increases action potential firing frequency in DRG neurons. elife 4:e10013. doi: 10.7554/eLife.10013
Meisler, M. H., Hill, S. F., and Yu, W. (2021). Sodium channelopathies in neurodevelopmental disorders. Nat. Rev. Neurosci. 22, 152–166. doi: 10.1038/s41583-020-00418-4
Mikati, M. A., Jiang, Y. H., Carboni, M., Shashi, V., Petrovski, S., Spillmann, R., et al. (2015). Quinidine in the treatment of KCNT1-positive epilepsies. Ann. Neurol. 78, 995–999. doi: 10.1002/ana.24520
Miller, A. R., Hawkins, N. A., McCollom, C. E., and Kearney, J. A. (2014). Mapping genetic modifiers of survival in a mouse model of Dravet syndrome. Genes Brain Behav. 13, 163–172. doi: 10.1111/gbb.12099
Milligan, C. J., Li, M., Gazina, E. V., Heron, S. E., Nair, U., Trager, C., et al. (2014). KCNT1 gain of function in 2 epilepsy phenotypes is reversed by quinidine. Ann. Neurol. 75, 581–590. doi: 10.1002/ana.24128
Mori, K., Kobayashi, S., Saito, T., Masuda, Y., and Nakaya, H. (1998). Inhibitory effects of class I and IV antiarrhythmic drugs on the Na+−activated K+ channel current in guinea pig ventricular cells. Naunyn Schmiedeberg's Arch. Pharmacol. 358, 641–648. doi: 10.1007/pl00005306
Numis, A. L., Nair, U., Datta, A. N., Sands, T. T., Oldham, M. S., Patel, A., et al. (2018). Lack of response to quinidine in KCNT1-related neonatal epilepsy. Epilepsia 59, 1889–1898. doi: 10.1111/epi.14551
Qiu, Y., O'Neill, N., Maffei, B., Zourray, C., Almacellas-Barbanoj, A., Carpenter, J. C., et al. (2022). On-demand cell-autonomous gene therapy for brain circuit disorders. Science 378, 523–532. doi: 10.1126/SCIENCE.ABQ6656
Quraishi, I. H., Mercier, M. R., McClure, H., Couture, R. L., Schwartz, M. L., Lukowski, R., et al. (2020). Impaired motor skill learning and altered seizure susceptibility in mice with loss or gain of function of the Kcnt1 gene encoding slack (KNa1.1) Na+−activated K+ channels. Sci. Rep. 10:3213. doi: 10.1038/S41598-020-60028-Z
Quraishi, I. H., Stern, S., Mangan, K. P., Zhang, Y., Ali, S. R., Mercier, M. R., et al. (2019). An epilepsy-associated KCNT1 mutation enhances excitability of human iPSC-derived neurons by increasing slack K Na currents. J. Neurosci. 39, 7438–7449. doi: 10.1523/JNEUROSCI.1628-18.2019
Rizzi, S., Knaus, H.-G., and Schwarzer, C. (2016). Differential distribution of the sodium-activated potassium channels slick and slack in mouse brain. J. Comp. Neurol. 524, 2093–2116. doi: 10.1002/cne.23934
Roden, D. M. (2014). Pharmacology and toxicology of Nav1.5-class 1 anti-arrhythmic drugs. Card. Electrophysiol. Clin. 6, 695–704. doi: 10.1016/j.ccep.2014.07.003
Scheffer, I. E., and Nabbout, R. (2019). SCN1A-related phenotypes: epilepsy and beyond. Epilepsia 60, S17–S24. doi: 10.1111/epi.16386
Shore, A. N., Colombo, S., Tobin, W. F., Petri, S., Cullen, E. R., Dominguez, S., et al. (2020). Reduced GABAergic neuron excitability, altered synaptic connectivity, and seizures in a KCNT1 gain-of-function mouse model of childhood epilepsy. Cell Rep. 33:108303. doi: 10.1016/j.celrep.2020.108303
Snowball, A., Chabrol, E., Wykes, R. C., Shekh-Ahmad, T., Cornford, J. H., Lieb, A., et al. (2019). Epilepsy gene therapy using an engineered potassium channel. J. Neurosci. 39, 3159–3169. doi: 10.1523/JNEUROSCI.1143-18.2019
Sprissler, R. S., Wagnon, J. L., Bunton-Stasyshyn, R. K., Meisler, M. H., and Hammer, M. F. (2017). Altered gene expression profile in a mouse model of SCN8A encephalopathy. Exp. Neurol. 288, 134–141. doi: 10.1016/J.EXPNEUROL.2016.11.002
Swayze, E., Siwkowski, A. M., Wancewicz, E. V., Migawa, M. T., Wyrzykiewicz, T. K., Hung, G., et al. (2007). Antisense oligonucleotides containing locked nucleic acid improve potency but cause significant hepatotoxicity in animals. Nucleic Acids Res. 35, 687–700. doi: 10.1093/NAR/GKL1071
Symonds, J. D., Zuberi, S. M., Stewart, K., McLellan, A., O‘Regan, M., MacLeod, S., et al. (2019). Incidence and phenotypes of childhood-onset genetic epilepsies: a prospective population-based national cohort. Brain 142, 2303–2318. doi: 10.1093/BRAIN/AWZ195
Tai, C., Abe, Y., Westenbroek, R. E., Scheuer, T., and Catterall, W. A. (2014). Impaired excitability of somatostatin- and parvalbumin-expressing cortical interneurons in a mouse model of Dravet syndrome. Proc. Natl. Acad. Sci. U. S. A. 111, E3139–E3148. doi: 10.1073/pnas.1411131111
Valassina, N., Brusco, S., Salamone, A., Serra, L., Luoni, M., Giannelli, S., et al. (2022). Scn1a gene reactivation after symptom onset rescues pathological phenotypes in a mouse model of Dravet syndrome. Nat. Commun. 13:161. doi: 10.1038/S41467-021-27837-W
Veeramah, K. R., O'Brien, J. E., Meisler, M. H., Cheng, X., Dib-Hajj, S. D., Waxman, S. G., et al. (2012). De novo pathogenic SCN8A mutation identified by whole-genome sequencing of a family quartet affected by infantile epileptic encephalopathy and SUDEP. Am. J. Hum. Genet. 90, 502–510. doi: 10.1016/j.ajhg.2012.01.006
Keywords: Scn8a, Scn1a, Kcnt1, epilepsy, ASO, sodium channel, potassium channel
Citation: Hill SF, Jafar-Nejad P, Rigo F and Meisler MH (2023) Reduction of Kcnt1 is therapeutic in mouse models of SCN1A and SCN8A epilepsy. Front. Neurosci. 17:1282201. doi: 10.3389/fnins.2023.1282201
Edited by:
Evgenia Sitnikova, Institute of Higher Nervous Activity and Neurophysiology (RAS), RussiaReviewed by:
Severine Deforges, UMR5297 Institut Interdisciplinaire de Neurosciences (IINS), FranceAlexander Bryson, University of Melbourne, Australia
Copyright © 2023 Hill, Jafar-Nejad, Rigo and Meisler. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sophie F. Hill, c2ZoaWxsQHVtaWNoLmVkdQ==