 Tania Quintela-López
Tania Quintela-López Jonathan Lezmy*
Jonathan Lezmy*- Department of Neuroscience, Physiology & Pharmacology, University College London, London, United Kingdom
Introduction
The brain constantly adapts to received inputs by reshaping its neural circuits. These circuits hold the ability to strengthen or depress their activity in mechanisms underlying learning and memory, but also to keep their activity within a normal homeostatic range. Homeostatic adaptations are achieved by maintaining a relatively stable activity set-point in response to aberrant stimuli, for example by scaling the synaptic strength, stabilizing the ratio of excitatory-inhibitory inputs, and modifying the excitable properties of neurons (Turrigiano, 2011). Homeostatic plasticity in somatodendritic compartments is mediated by structural modifications underlying changes in information transmission: changes in the number or size of excitatory and inhibitory synapses, of AMPA, NMDA and GABA receptors, and of voltage-gated Nav and Kv channel-mediated currents (Desai, 2003). In addition, homeostatic adaptations were shown to occur at excitable sites along axons, where action potentials (APs) are generated: the axon initial segment (AIS) and the nodes of Ranvier of myelinated axons. These adaptations allow to adjust the output signals by modulating APs along axons. At the AIS, anchoring proteins and ion channels re-localize, and the number of axo-axonic GABAergic synapses is adjusted to tune neurons' excitability (Grubb and Burrone, 2010; Kuba et al., 2010; Pan-Vazquez et al., 2020). Nodes, which have a similar molecular composition to that of the AIS, also share the ability to modify their length in response to stimuli and may regulate the information flow along myelinated axons in a homeostatic manner (Cullen et al., 2021).
Alzheimer's disease (AD), the commonest cause of dementia, is linked to lapses in memory from its early stages due to synapse loss, followed by a cognitive decline due to ongoing neurodegeneration. It has been proposed that impairments in synaptic homeostatic plasticity mechanisms may underlie early symptoms of the disease (Frere and Slutsky, 2018; Taylor and Jeans, 2021). Interestingly, AIS homeostatic adaptations which regulate neurons' excitability may occur before homeostatic scaling of synaptic activity (Lezmy et al., 2020), raising the question whether axonal homeostatic plasticity mechanisms are involved from the earliest stages of the disease. Here, we will review recent studies showing that axonal excitable sites are targets in AD, focussing on amyloid and tau pathologies (Figure 1). Finally, we will discuss how these disruptions may interfere with axonal homeostatic adaptations and contribute to AD progression.
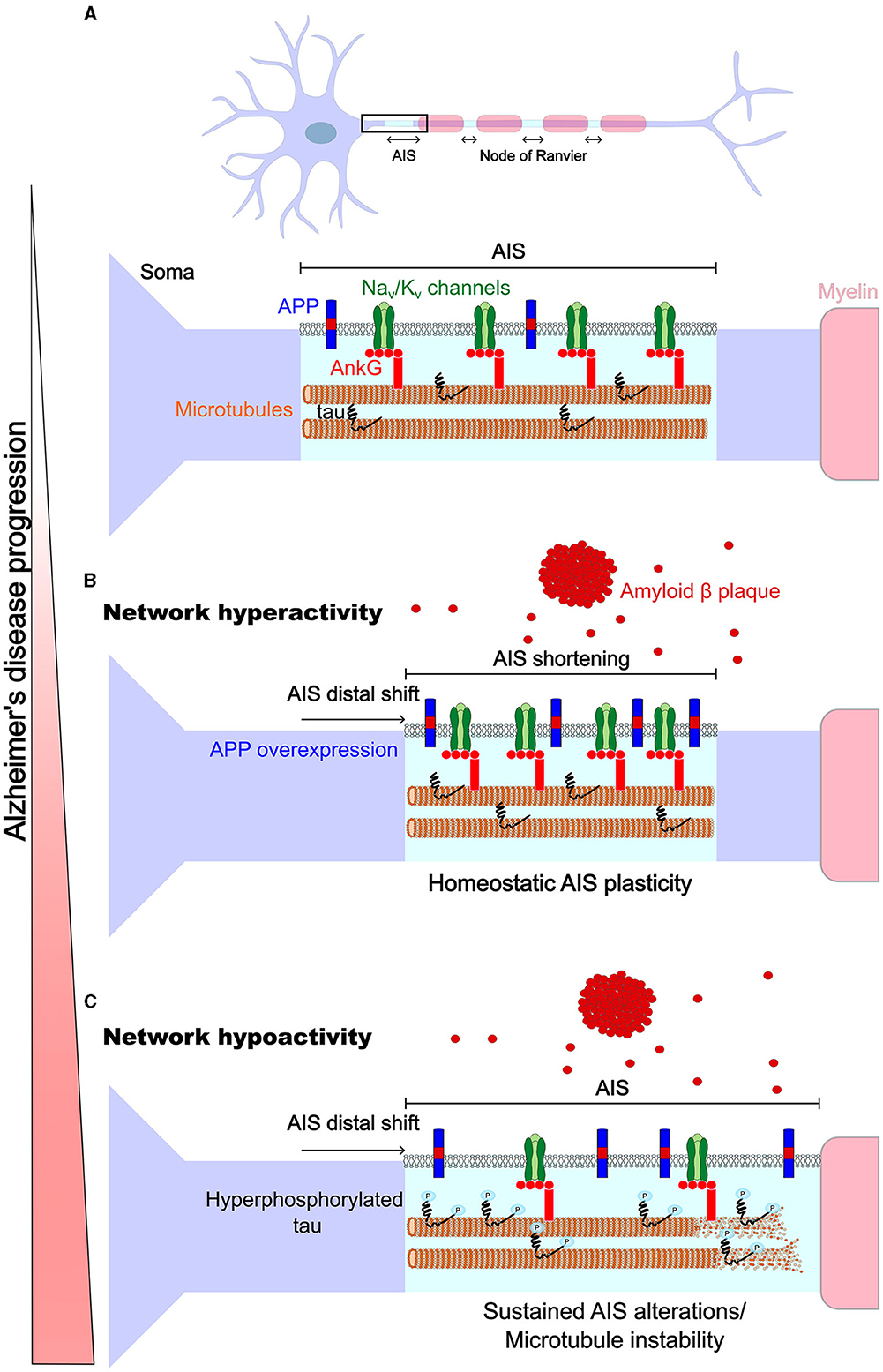
Figure 1. Homeostatic plasticity of axonal excitable sites in Alzheimer's disease. (A) Top, The AIS and nodes of Ranvier share a similar molecular composition and may be subject to similar homeostatic adaptation mechanisms, but nodes have been less studied (thus nodes are not represented in detail in the diagram). Bottom, the AIS, like the nodes, highly expresses voltage-gated Nav and Kv channels (green) which are anchored by the structural proteins βIV-spectrin (not shown) and ankyrin G (light red), and ankyrin G binds to microtubules (orange and beige). Endogenous transmembrane protein APP (blue and red) and intracellular tau (black) are expressed at axonal excitable sites. (B) Network hyperactivity early in AD is associated with amyloid pathology, where APP is overexpressed, high levels of Aβ are secreted extracellularly and Aβ plaques are formed (red). At this stage, an AIS shift away from the soma and a shortening of the AIS were reported. Thus, modifications of the AIS could be driven by mechanisms of homeostatic plasticity that counter network hyperactivity. (C) Network hypoactivity occurs at a later stage and is associated with tau pathology which follows amyloid pathology. Here, a shift of the AIS away from the soma was reported, which could reinforce the neuronal hypoactivity. This may be due to sustained AIS modifications, aberrantly mimicking the homeostatic adaptations triggered by the earlier amyloid pathology, and/or due to AIS structural modifications mediated by tau hyperphosphorylation causing the loss of microtubule stability (which may precede AIS disassembly).
Disruption of the AIS structure in amyloid pathology
Early in AD, extracellular amyloid β (Aβ) accumulates as amyloid-beta precursor protein (APP) is cleaved, and aggregates of Aβ molecules form amyloid plaques. Studies suggest that AIS structural alterations may be driven by their interaction with Aβ plaques. In an Aβ-depositing mouse model (APP/TTA), AIS length did not change in the layers 2–4 of the cortex of 6 months old mice when Aβ plaque burden is moderate, but AISs contacting plaques were shorter in 11 months old mice when Aβ plaque burden is severe (Marin et al., 2016). However, a change in AIS length was not observed in cortical pyramidal neurons of one-year old APP/PS1 mice, another Aβ-depositing model (León-Espinosa et al., 2012). Instead, this model showed a reduction in the number of GABAergic axo-axonic synapses forming on the AISs that contact Aβ plaques. Both models (APP/PS1 and APP/TTA) increase Aβ plaque load by overexpressing APP, up to 10 times more in APP/TTA mice (Marin et al., 2016). Aβ plaques accumulate as early as 6–8 weeks of age in APP/TTA mice (Jankowsky et al., 2005), but only from 6 months old in the APP/PS1 mice used (Jankowsky et al., 2004). In the hippocampus of 18 months old APPNL − F mice, a knock-in model expressing a mutant APP (thus avoiding artifacts resulting from APP overexpression) where plaques start accumulating at 6 months old (Saito et al., 2014), GABAergic synapses at the AIS were enlarged, while the total number of synapses at the AIS was unchanged (changes in AIS length were not reported here; Sos et al., 2020). Thus, although the underlying mechanism is unclear, Aβ plaque load may promote AIS structural alterations as AD progresses, by modifying the AIS length and shaping the inhibitory inputs that the AIS receives.
Alterations in the AIS structure may be mediated by APP overexpression or soluble Aβ rather than, or in addition to, Aβ plaque toxicity. In cortical and hippocampal APP/PS1 primary neurons (where APP and soluble Aβ levels, but not plaques, is expected to be high), AISs were shorter than in wild-type (WT) mice and were not responsive to homeostatic changes in AIS length usually triggered by increasing (with bicuculline) or decreasing (with TTX) neuronal activity (Martinsson et al., 2022). Alternatively, applying soluble Aβ onto cultured hippocampal neurons impaired microtubule plus-end-binding protein 3 (EB3) stability at the AIS, thereby disturbing microtubule integrity and lengthening the AIS, and was accompanied by a depolarising shift in the AP voltage threshold (Tsushima et al., 2015). This effect was mediated by Aβ inhibition of histone deacetylase 6 (HDAC6), as applying tubacin (a HDAC6 inhibitor) increased EB3 dynamics at the AIS and lengthened the AIS, similar to Aβ application. A recent study suggests that APP overexpression induced by glutamate excitotoxicity, or by overexpression via plasmid transfection carrying the APPSwe mutation or using transgenic (R1.40 model) primary neurons, drives the modifications in AIS structure (Ma et al., 2023). Elevated concentration of APP, which contacts the AIS scaffolding proteins ankyrin G and βIV-spectrin, promoted an AIS shift away from the soma and a shortening of the AIS. In this study, AIS structure was not altered by the interaction with soluble Aβ nor with plaques. Overall, divergent evidence indicates that AIS structural modification may be promoted by either APP, soluble Aβ or Aβ plaques, depending on the mouse model of AD in use, on the age of the mice (and thus the stage of amyloid pathology) and/or on the experimental procedures chosen. We discuss below how some of these findings may be linked to AIS homeostatic adaptation mechanisms.
Disruption of the AIS structure in tau pathology
Aggregation of tau protein inside neurons occurs after the onset of amyloid pathology in AD (Harris et al., 2020), as tau becomes hyperphosphorylated and dissociates from the microtubule. Hyperphosphorylated tau destabilizes axonal microtubule, and promotes an AIS shift away from the soma and a reduction in the excitability of cultured hippocampal neurons. This AIS distal shift could be prevented by the microtubule stabilizer taxol (Hatch et al., 2017). Furthermore, in CA1 pyramidal neurons of rTg4510 mice (where mutant tau is overexpressed, and thus have more hyperphosphorylated tau), the AIS was shifted away from the soma and the neuronal excitability was reduced, months before the onset of neurodegeneration in this tauopathy model (Hatch et al., 2017). In contrast, a recent study shows that, in brains from AD patients post-mortem, AISs of cortical pyramidal neurons with hyperphosphorylated tau (immunolabeled with AT8) were commonly found to be closer to the soma and longer (Antón-Fernández et al., 2022). Thus, as for amyloid pathology, disease stage may be crucial in defining the influence of tau on the AIS structure. To assess tau effects on AIS alterations, Chang et al. (2021) used a mouse model where tau is ablated (MAPT knockout). In the layer 5 of the somatosensory cortex, they observed a shortening of the AIS in excitatory pyramidal cells, and a lengthening of the AIS together with a shift closer to the soma in inhibitory parvalbumin (PV+) neurons. Furthermore, in tau knockout mice, the AIS of PV+ interneurons, but not of excitatory neurons, was irresponsive to chronic depolarisation that usually triggers an AIS shift away from the soma in wild-type mice. Thus, endogenous tau, which interacts with EB3 to stabilize ankyrin G in the AIS, may be involved in AIS homeostatic adaptation mechanisms in a cell type-dependent manner.
Disruption of the node of Ranvier structure in AD
Homeostatic adaptations at nodes of Ranvier have been much less studied than their counterparts at the AIS. Since nodes and AISs have a similar molecular composition, where Nav and Kv7 channels are highly expressed and anchored by ankyrin G and βIV-spectrin, it is reasonable to assume that at least some of the mechanisms involved in Aβ and tau pathologies discussed above apply to nodes, although experimental studies verifying this assumption will be needed in the future. Interestingly, APP was shown to be highly expressed in nodes of the optic nerve, cerebellum and spinal cord (but not in nodes of the peripheral nervous system). APP may be involved in regulating node length, as APP KO mice had longer nodes than wild-type mice (Xu et al., 2014). Furthermore, the conduction speed of spinal cord axons in APP KO mice was reduced, as well as the Na+ current density of CA1 pyramidal neurons. Nav1.6-mediated currents were elevated in HEK293 cell lines overexpressing APP (Li et al., 2016), as APP phosphorylation increased Nav1.6 surface expression (Liu et al., 2015). In addition, the C99 fragment of APP, which accumulates intracellularly in AD, binds to Kv7.2/3 channels at nodes and inhibits their activity (Manville and Abbott, 2021). Thus, high APP levels may not only induce alterations in the structure of the AIS and nodes but also directly affect the activity of ion channels shaping APs at axonal excitable sites. In this article, we do not discuss impairments in glia-axon interaction nor in myelination in AD (reviewed recently in Nasrabady et al., 2018; Hirschfeld et al., 2022; Uddin and Lim, 2022; Maitre et al., 2023), but it is important to mention that changes in glial responses and in myelin pattern may also modify the structure of the AIS and nodes. However, it is unclear whether glia-axon and myelin-mediated alterations of excitable sites would occur as a result of a disbalance in homeostatic plasticity mechanisms applying on the myelin and glia, or as a consequence of myelin damage and glia aberrant activity linked to AD.
Link between homeostatic axonal plasticity and Alzheimer's disease
Evidence described above suggests that, at least in some instances, alteration in the structure of axonal excitable sites may reflect disruption of mechanisms underlying homeostatic axonal plasticity. To speculate on this, we aligned the occurrence of the AIS alterations described above with the known changes in neuronal activity at different stages of AD (Figure 1). Although AD is a complex disease that alter many cellular mechanisms with divergent effects on neural circuits, initial Aβ rise can be broadly associated with network hyperactivity, while the later accumulation of tau can be associated with network hypoactivity (Harris et al., 2020). The initial hyperactivity may explain the finding of short AISs when Aβ plaque load is severe (Marin et al., 2016), when soluble Aβ accumulates (Martinsson et al., 2022) and when APP is overexpressed (Martinsson et al., 2022; Ma et al., 2023). An AIS shortening is expected to reduce neuronal excitability to compensate for prolonged hyperactivity in healthy neurons (although it is not always the case; see Adachi et al., 2015). The AIS distal shift found in association with APP overexpression is also expected to lower the neuronal excitability (Ma et al., 2023). In addition, larger GABAergic synapses at the AIS in the APPNL − F model reinforce the suggestion that the AIS reshapes to counter network hyperexcitability (Sos et al., 2020). However, less GABAergic synapses were found in the APP/PS1 (León-Espinosa et al., 2012), which would presumably increase neuronal excitability, opposing a homeostatic effect, although regulation of synapse number at the AIS crucially depends on whether the synapses are depolarising or hyperpolarising (Pan-Vazquez et al., 2020). Prolonged application of Aβ onto neurons also reduced neuronal excitability (Tsushima et al., 2015), but this could also reflect a loss of integrity of the AIS, as the AIS lengthens because microtubule disassembles when tau acetylates.
Generally, pathogenic tau accumulation would result in a shift of the AIS away from the soma in excitatory neurons (Hatch et al., 2017; Chang et al., 2021), which corresponds with an expected decrease in neuronal excitability (Adachi et al., 2015) that should strengthen, rather than compensate for the tau-linked hypoactivity. Tau-mediated effects at the AIS may be mostly driven by microtubule disassembly via tau phosphorylation or acetylation, as part of neurodegeneration processes. Remaining functional axons may attempt to induce homeostatic mechanisms, as the remaining AISs were usually closer to the soma in brains of AD patients post-mortem (Antón-Fernández et al., 2022). Although AIS disassembly may explain hyperphosphorylated tau-mediated effects, a possible role for tau in mediating homeostatic changes should not be neglected. Tau mutation (tauV337M) or tau knock-out prevented activity-dependent homeostatic AIS plasticity, in mechanisms involving the alteration of microtubule dynamics (Lezmy et al., 2017; Sohn et al., 2019; Chang et al., 2021). From this perspective, endogenous tau-linked homeostatic adaptations could in fact be triggered by the initial Aβ-mediated hyperactivity in AD but, when sustained, could become detrimental in the hypoactive phase linked to pathogenic tau. Finally, disruptions of homeostatic adaptations altering the expression of axonal voltage-gated channels should also be taken into consideration as, although experimental data are limited, changes in their distribution is expected to regulate axonal excitability and conduction speed in health and disease.
Conclusions
Although the knowledge in the field is still sparse, recent literature suggests that homeostatic plasticity of axonal excitable sites may play a crucial role in the pathophysiology of AD. Axonal homeostatic adaptations that are triggered at the onset of the disease may be beneficial to counter network hyperexcitability. However, sustained or aberrant activation of such mechanisms may become detrimental when AD switches to hypoactivation, and they may then contribute to memory loss and cognitive decline.
Author contributions
TQ-L: Writing—review and editing. JL: Writing—review and editing, Conceptualization, Writing—original draft.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by an EMBO fellowship (ALTF 430-2019) to JL and a Wellcome Trust Investigator Award (219366/Z/19/Z) to David Attwell.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Adachi, R., Yamada, R., and Kuba, H. (2015). Plasticity of the axonal trigger zone. Neuroscientist 21, 255–265. doi: 10.1177/1073858414535986
Antón-Fernández, A., León-Espinosa, G., DeFelipe, J., and Muñoz, A. (2022). Pyramidal cell axon initial segment in Alzheimer's disease. Scient. Rep. 12, 8722. doi: 10.1038/s41598-022-12700-9
Chang, C. W., Evans, M. D., Yu, X., Yu, G. Q., and Mucke, L. (2021). Tau reduction affects excitatory and inhibitory neurons differently, reduces excitation/inhibition ratios, and counteracts network hypersynchrony. Cell Rep. 37, 109855. doi: 10.1016/j.celrep.2021.109855
Cullen, C. L., Pepper, R. E., Clutterbuck, M. T., Pitman, K. A., Oorschot, V., Auderset, L., et al. (2021). Periaxonal and nodal plasticities modulate action potential conduction in the adult mouse brain. Cell Rep. 34, 108641. doi: 10.1016/j.celrep.2020.108641
Desai, N. S. (2003). Homeostatic plasticity in the CNS: synaptic and intrinsic forms. J. Physiol. Paris 97, 391–402. doi: 10.1016/j.jphysparis.2004.01.005
Frere, S., and Slutsky, I. (2018). Alzheimer's disease: from firing instability to homeostasis network collapse. Neuron 97, 32–58. doi: 10.1016/j.neuron.2017.11.028
Grubb, M. S., and Burrone, J. (2010). Activity-dependent relocation of the axon initial segment fine-tunes neuronal excitability. Nature 465, 1070–1074. doi: 10.1038/nature09160
Harris, S. S., Wolf, F., De Strooper, B., and Busche, M. A. (2020). Tipping the scales: peptide-dependent dysregulation of neural circuit dynamics in Alzheimer's disease. Neuron 107, 417–435. doi: 10.1016/j.neuron.2020.06.005
Hatch, R. J., Wei, Y., Xia, D., and Götz, J. (2017). Hyperphosphorylated tau causes reduced hippocampal CA1 excitability by relocating the axon initial segment. Acta Neuropathol. 133, 717–730. doi: 10.1007/s00401-017-1674-1
Hirschfeld, L. R., Risacher, S. L., Nho, K., and Saykin, A. J. (2022). Myelin repair in Alzheimer's disease: a review of biological pathways and potential therapeutics. Transl. Neurodegener. 11, 47. doi: 10.1186/s40035-022-00321-1
Jankowsky, J. L., Fadale, D. J., Anderson, J., Xu, G. M., Gonzales, V., Jenkins, N. A., et al. (2004). Mutant presenilins specifically elevate the levels of the 42 residue beta-amyloid peptide in vivo: evidence for augmentation of a 42-specific gamma secretase. Hum. Molec. Genet. 13,159–170. doi: 10.1093/hmg/ddh019
Jankowsky, J. L., Slunt, H. H., Gonzales, V., Savonenko, A. V., Wen, J. C., Jenkins, N. A., et al. (2005). Persistent amyloidosis following suppression of Aβ production in a transgenic model of Alzheimer disease. PLoS Med. 2, e355. doi: 10.1371/journal.pmed.0020355
Kuba, H., Oichi, Y., and Ohmori, H. (2010). Presynaptic activity regulates Na+ channel distribution at the axon initial segment. Nature 465, 1075–1078. doi: 10.1038/nature09087
León-Espinosa, G., Defelipe, J., and Muñoz, A. (2012). Effects of amyloid-plaque proximity on the axon initial segment of pyramidal cells. J. Alzheimer's Dis. 29, 841–852. doi: 10.3233/JAD-2012-112036
Lezmy, J., Gelman, H., Katsenelson, M., Styr, B., Tikochinsky, E., Lipinsky, M., et al. (2020). M-current inhibition in hippocampal excitatory neurons triggers intrinsic and synaptic homeostatic responses at different temporal scales. J. Neurosci. 40, 3694–3706. doi: 10.1523/JNEUROSCI.1914-19.2020
Lezmy, J., Lipinsky, M., Khrapunsky, Y., Patrich, E., Shalom, L., Peretz, A., et al. (2017). M-current inhibition rapidly induces a unique CK2-dependent plasticity of the axon initial segment. Proc. Natl. Acad. Sci. U S A. 114, E10234–E10243. doi: 10.1073/pnas.1708700114
Li, S., Wang, X., Ma, Q. H., Yang, W. L., Zhang, X. G., Dawe, G. S., et al. (2016). Amyloid precursor protein modulates Nav1.6 sodium channel currents through a Go-coupled JNK pathway. Sci. Rep. 6, 1–16. doi: 10.1038/srep39320
Liu, C., Tan, F. C. K., Xiao, Z. C., and Dawe, G. S. (2015). Amyloid precursor protein enhances Nav1.6 sodium channel cell surface expression. J. Biol. Chem. 290, 12048–12057. doi: 10.1074/jbc.M114.617092
Ma, F., Akolkar, H., Xu, J., Liu, Y., Popova, D., Xie, J., et al. (2023). The amyloid precursor protein modulates the position and length of the axon initial segment. J. Neurosci. 43, 1830–1844. doi: 10.1523/JNEUROSCI.0172-22.2023
Maitre, M., Jeltsch-David, H., Okechukwu, N. G., Klein, C., Patte-Mensah, C., and Mensah-Nyagan, A. G. (2023). Myelin in Alzheimer's disease: culprit or bystander? Acta Neuropathol. Commun. 11, 1–18. doi: 10.1186/s40478-023-01554-5
Manville, R. W., and Abbott, G. W. (2021). The amyloid precursor protein C99 fragment modulates voltage-gated potassium channels. Cell. Physiol. Biochem. 55, 157–170. doi: 10.33594/000000397
Marin, M. A., Ziburkus, J., Jankowsky, J., and Rasband, M. N. (2016). Amyloid-β plaques disrupt axon initial segments. Exp. Neurol. 281, 93–98. doi: 10.1016/j.expneurol.2016.04.018
Martinsson, I., Quintino, L., Garcia, M. G., Konings, S. C., Torres-Garcia, L., Svanbergsson, A., et al. (2022). Aβ/Amyloid precursor protein-induced hyperexcitability and dysregulation of homeostatic synaptic plasticity in neuron models of Alzheimer's disease. Front. Aging Neurosci. 14, 946297. doi: 10.3389/fnagi.2022.946297
Nasrabady, S. E., Rizvi, B., Goldman, J. E., and Brickman, A. M. (2018). White matter changes in Alzheimer's disease: a focus on myelin and oligodendrocytes. Acta Neuropathol. Commun. 6, 22. doi: 10.1186/s40478-018-0515-3
Pan-Vazquez, A., Wefelmeyer, W., Gonzalez Sabater, V., Neves, G., and Burrone, J. (2020). Activity-dependent plasticity of axo-axonic synapses at the axon initial segment. Neuron 106, 265–276.e6. doi: 10.1016/j.neuron.2020.01.037
Saito, T., Matsuba, Y., Mihira, N., Takano, J., Nilsson, P., Itohara, S., et al. (2014). Single App knock-in mouse models of Alzheimer's disease. Nat. Neurosci. 17, 661–663. doi: 10.1038/nn.3697
Sohn, P. D., Huang, C. T. L., Yan, R., Fan, L., Tracy, T. E., Camargo, C. M., et al. (2019). Pathogenic tau impairs axon initial segment plasticity and excitability homeostasis. Neuron 104, 458–470.e5. doi: 10.1016/j.neuron.2019.08.008
Sos, K. E., Mayer, M. I., Takács, V. T., Major, A., Bardóczi, Z., Beres, B. M., et al. (2020). Amyloid β induces interneuron-specific changes in the hippocampus of APPNL − F mice. PLoS ONE 15, e0233700. doi: 10.1371/journal.pone.0233700
Taylor, H. B. C., and Jeans, A. F. (2021). Friend or foe? The varied faces of homeostatic synaptic plasticity in neurodegenerative disease. Front. Cell Neurosci. 15, 782768. doi: 10.3389/fncel.2021.782768
Tsushima, H., Emanuele, M., Polenghi, A., Esposito, A., Vassalli, M., Barberis, A., et al. (2015). HDAC6 and RhoA are novel players in Abeta-driven disruption of neuronal polarity. Nat. Commun. 6, 1–14. doi: 10.1038/ncomms8781
Turrigiano, G. (2011). Too many cooks? Intrinsic and synaptic homeostatic mechanisms in cortical circuit refinement. Annu. Rev. Neurosci. 34, 89–103. doi: 10.1146/annurev-neuro-060909-153238
Uddin, M. S., and Lim, L. W. (2022). Glial cells in Alzheimer's disease: from neuropathological changes to therapeutic implications. Age. Res. Rev. 78, 101622. doi: 10.1016/j.arr.2022.101622
Keywords: axon initial segment (AIS), node of Ranvier, Alzheimer's disease, homeostatic plasticity, amyloid pathology, tau pathology
Citation: Quintela-López T and Lezmy J (2023) Homeostatic plasticity of axonal excitable sites in Alzheimer's disease. Front. Neurosci. 17:1277251. doi: 10.3389/fnins.2023.1277251
Received: 16 August 2023; Accepted: 06 October 2023;
Published: 23 October 2023.
Edited by:
Isak Martinsson, Lund University, SwedenReviewed by:
Ze-Ka Chen, University of North Carolina at Chapel Hill, United StatesCopyright © 2023 Quintela-López and Lezmy. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jonathan Lezmy, ai5sZXpteUB1Y2wuYWMudWs=