 Lena H. Nguyen
Lena H. Nguyen Manas Sharma2
Manas Sharma2 Angelique Bordey
Angelique Bordey- 1Department of Neuroscience, School of Behavioral and Brain Sciences, University of Texas at Dallas, Richardson, TX, United States
- 2Departments of Neurosurgery and Cellular & Molecular Physiology, Wu Tsai Institute, Yale University School of Medicine, New Haven, CT, United States
Hyperactivation of the mechanistic target of rapamycin complex 1 (mTORC1) pathway during neurodevelopment leads to focal cortical malformations associated with intractable seizures. Recent evidence suggests that dysregulated cap-dependent translation downstream of mTORC1 contributes to cytoarchitectural abnormalities and seizure activity. Here, we examined whether reducing cap-dependent translation by expressing a constitutively active form of the translational repressor, 4E-BP1, downstream of mTORC1 would prevent the development of cortical malformations and seizures. 4E-BP1CA was expressed embryonically either in radial glia (neural progenitor cells) that generate cortical layer 2/3 pyramidal neurons or in migrating neurons destined to layer 2/3 using a conditional expression system. In both conditions, 4E-BP1CA expression reduced mTORC1-induced neuronal hypertrophy and alleviated cortical mislamination, but a subset of ectopic neurons persisted in the deep layers and the white matter. Despite the above improvements, 4E-BP1CA expression in radial glia had no effects on seizure frequency and further exacerbated behavioral seizure severity associated with mTORC1 hyperactivation. In contrast, conditional 4E-BP1CA expression in migratory neurons mitigated the severity of behavioral seizures but the seizure frequency remained unchanged. These findings advise against targeting 4E-BPs by 4E-BP1CA expression during embryonic development for seizure prevention and suggest the presence of a development-dependent role for 4E-BPs in mTORC1-induced epilepsy.
1. Introduction
Hyperactivation of the mechanistic target of rapamycin complex 1 (mTORC1) signaling pathway during embryonic development leads to focal malformation of cortical development (FMCD) and epilepsy (Crino, 2015). The mTORC1 pathway is a key regulator of cell growth and controls a wide range of cellular processes, including protein synthesis, autophagy, and metabolism (Laplante and Sabatini, 2012). The best-characterized function of mTORC1 is the control of cap-dependent translation via eukaryotic translation initiation factor 4E (eIF4E)-binding proteins (4E-BPs) (Richter and Sonenberg, 2005; Ma and Blenis, 2009). mTORC1 promotes cap-dependent translation by directly phosphorylating and inactivating the translational repressors, 4E-BP1 and 4E-BP2. Phosphorylated 4E-BPs dissociate from eIF4E, allowing eIF4E to form the eIF4F complex and initiate translation. Accumulating evidence has supported a critical role for 4E-BP-eIF4E-mediated translation in the mechanism of mTORC1-induced FMCD pathology and epilepsy. Several studies have shown increased levels of phosphorylated 4E-BPs in human and mouse FMCD brain tissue (Baybis et al., 2004; Orlova and Crino, 2010; Hanai et al., 2017; Kim et al., 2019; Nguyen et al., 2022). Furthermore, genetic and pharmacological inhibition of eIF4E in mice expressing mTORC1-activating gene variants rescued FMCD pathologies and seizures (Kim et al., 2019). Consistent with a role for 4E-BP-eIF4E signaling in the disease pathogenesis, embryonic expression of a constitutive active 4E-BP1 mutant (4E-BP1CA) that resists mTORC1 phosphorylation (Choi et al., 2003; Schalm et al., 2003) rescued several FMCD pathologies, including cortical mislamination and neuronal overgrowth, in mice expressing constitutive active Rheb (RhebCA; the direct activator of mTORC1) (Gong et al., 2015; Lin et al., 2016). Additionally, expression of 4E-BP1CA in adolescent RhebCA mice, after the onset of seizures, alleviated the seizure frequency and associated FMCD pathologies (Nguyen et al., 2022). Considering that FCMDs are formed during cortical neurogenesis and can be detected during fetal development in humans (Glenn et al., 2012; Lerman-Sagie et al., 2021), earlier therapeutic interventions could potentially limit FCMD formation and seizure development. However, the effects of embryonic 4E-BP1CA expression on seizure development remain unknown.
In this study, we examined whether embryonic expression of 4E-BP1CA in cortical pyramidal neurons prevents mTORC1-induced seizures using the RhebCA mouse model. We co-expressed RhebCA with 4E-BP1CA, which was previously shown to reduce cap-dependent translation (Lin et al., 2016; Nguyen et al., 2022), in developing cortices via in utero electroporation (IUE). We used two experimental paradigms. In the first condition, 4E-BP1CA was expressed in radial glia (neural progenitor cells) that generate cortical layer (L) 2/3 pyramidal neurons. In the second condition, we bypassed radial glia and selectively expressed 4E-BP1CA in postmitotic migrating neurons destined to L2/3 through a conditional expression system. In both conditions, 4E-BP1CA expression reduced mTORC1-induced neuronal hypertrophy and alleviated cortical mislamination by increasing the proportion of correctly positioned neurons in the cortex. Despite these histological improvements, 4E-BP1CA expression in radial glia had no effects on seizure frequency and further exacerbated behavioral seizure severity in RhebCA mice. In contrast, selective 4E-BP1CA expression in postmitotic migratory neurons mitigated the severity of behavioral seizures but the seizure frequency remained unchanged. These findings caution against targeting 4E-BPs by 4E-BP1CA expression during embryonic development for seizure prevention and suggest the presence of a developmentally dependent role of 4E-BPs in mTORC1-induced epilepsy.
2. Materials and methods
2.1. Animals
All animal procedures were performed in accordance with Yale University Institutional Animal Care and Use Committee’s regulations. All experiments were performed on male and female CD-1 mice (Charles River Laboratories).
2.2. In utero electroporation (IUE)
IUE was performed in timed-pregnant embryonic day (E) 15 mice as previously described (Nguyen et al., 2022). Briefly, a solution of the appropriate DNA plasmid mixture was injected into the right lateral ventricle of each embryo using a glass pipette. All solutions contained 0.03% Fast Green dye to visualize the injection. For each litter, half of the embryos were injected with the experimental plasmids and the other half received the respective control plasmids. Following each injection, a 5 mm tweezer electrode was positioned on the embryo’s head and six 42 V, 50 ms pulses at 950 ms intervals were applied using a pulse generator (ECM830, BTX). The electrodes were positioned to target expression in the medial prefrontal cortex (mPFC). Pups were screened at postnatal day (P) 0 to verify successful electroporation to the targeted region by detecting fluorescent reporter expression (tdTomato or GFP) on a fluorescence-enabled stereomicroscope.
In the first experimental paradigm (RhebCA + 4E-BP1CA), mice were electroporated with a plasmid mixture consisting of pCAG-RhebCA + pCAG-4E-BP1CA (or pCAG-GFP as control) + pCAG-tdTomato. In the second condition (RhebCA + conditional [c]4E-BP1CA), mice were electroporated with a plasmid mixture consisting of pCAG-RhebCA+ pCALNL-4E-BP1CA (or pCALNL-DsRed as control) + pCIG2-DCX-Cre-IRES-GFP + pCALNL-GFP. The plasmid concentrations and sources are listed in Table 1.
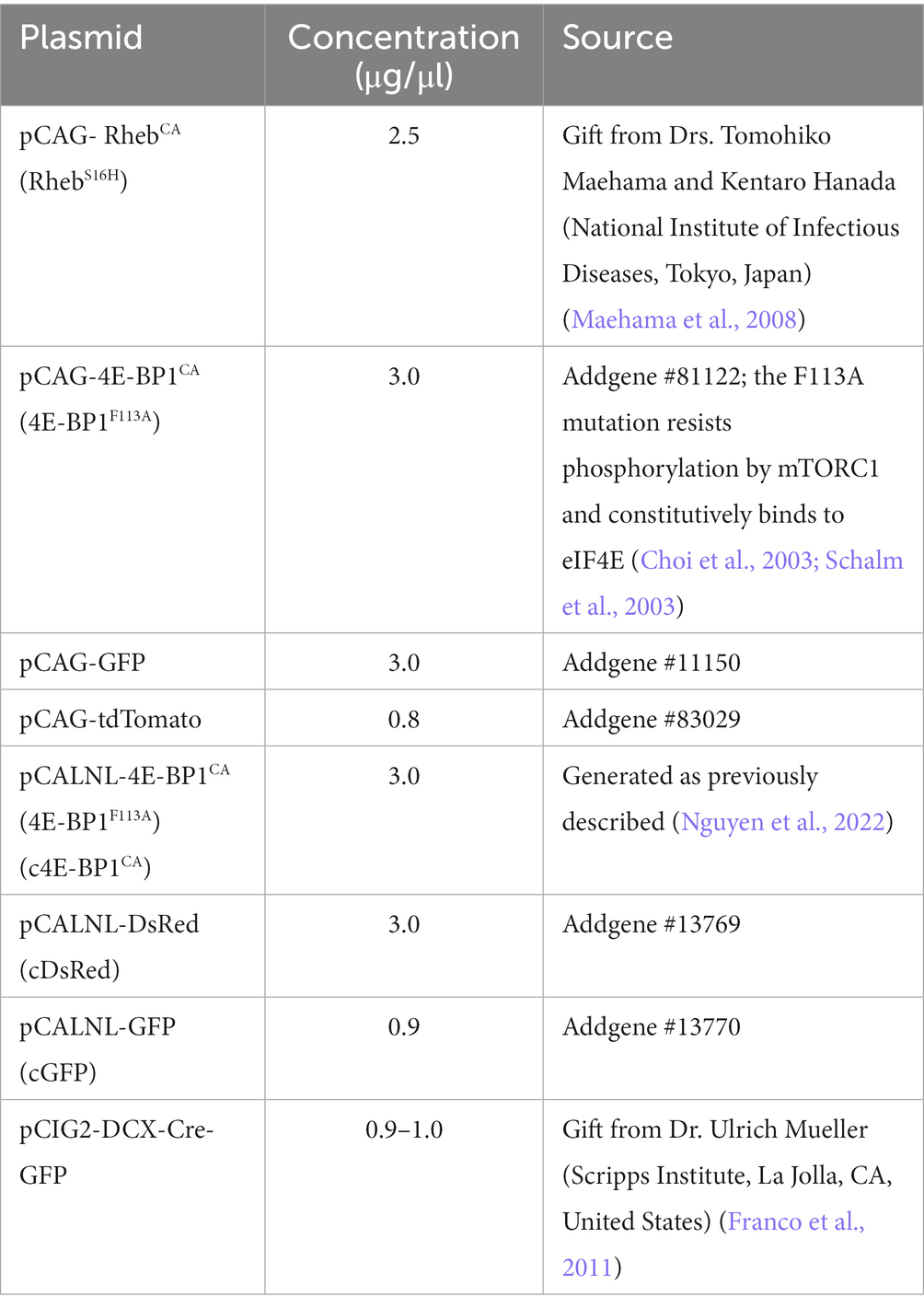
Table 1. DNA plasmid information.
2.3. Video-EEG recording and analysis
Mice were implanted at 9–10 (RhebCA + 4E-BP1CA) or 6–8 (RhebCA + c4E-BP1CA) weeks of age with prefabricated EEG headmounts (Pinnacle Technology, Inc.) for subdural cortical EEG recording as previously described (Nguyen et al., 2019, 2022). Mice were video-EEG recorded starting at 10–12 (RhebCA + 4E-BP1CA) or 7–8 (RhebCA + c4E-BP1CA) weeks of age, during which they were housed in individual recording chambers in a light-, temperature-, and humidity-controlled room with ad libitum access to food and water. Synchronous video-EEG recording was acquired using a three-channel EEG tethered system and Sirenia Acquisition software (Pinnacle Technology, Inc.). Mice were recorded 24 h/day for at least 7 consecutive days. The average recording hours per animal was 173.1 ± 18.9 SD (RhebCA + 4E-BP1CA) and 170.7 ± 13.4 SD (RhebCA + c4E-BP1CA) hours.
Seizure frequency and severity were analyzed using Sirenia Seizure Basic software (Pinnacle Technology, Inc.). All analyses were performed blinded to experimental groups. The entire EEG traces were manually reviewed for the occurrence of electrographic seizures, defined as a sudden onset of high amplitude activity with a characteristic pattern of progressive frequency and amplitude increases over the course of the event lasting ≥10 s (Nguyen et al., 2015, 2019). The mean number of seizures per day (seizures/day) was obtained by the equation: (total number of seizures/total recording hours) × 24 for each animal. For each identified seizure, synchronous video recordings were visually inspected, and the severity of the behavioral seizure correlates was scored using a modified Racine scale (Table 2).
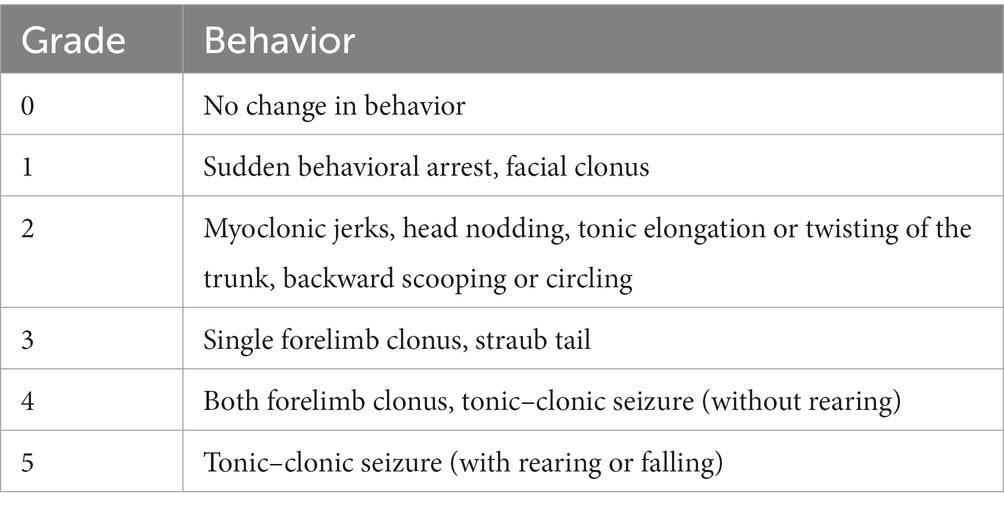
Table 2. Modified Racine scale.
2.4. Histology
Brain tissue was collected for histological analysis at the end of EEG recording. Mice were deeply anesthetized with sodium pentobarbital (85 mg/kg i.p. injection) and perfused with ice-cold phosphate buffered saline (PBS, in mM: 137 NaCl, 2.7 KCl, 4.3 Na2HPO4, 1.47 KH2PO4, pH 7.4) followed by 4% paraformaldehyde (PFA). Whole brains were dissected and post-fixed in 4% PFA for 2 h and then cryoprotected in 30% sucrose for 24–48 h at 4°C. Brains were cut into 50 μm-thick coronal sections using a freezing microtome and stored in PBS + 0.01% sodium azide at 4°C until use. Sections were mounted onto glass slides and coverslipped with antifade mounting media before imaging.
Images were acquired using an Olympus Fluoview FV1000 confocal microscope and analyzed with ImageJ software (NIH). Representative images for figures were prepared using Adobe Photoshop CC. All images meant for direct comparison were taken with the same microscope settings and uniformly processed. All image analyses were performed blinded to the experimental groups. Cell size was quantified from 20X magnification images by tracing around the soma of tdTomato+ or GFP+ cells, as appropriate for each experimental condition, and measuring the area. 20–50 randomly selected cells from 2–3 brain sections were measured and averaged for each animal. Cell placement was quantified from 10X magnification images within a region of interest (ROI) spanning from the white matter border to the pial surface. The ROI height was set to 150 μm and the length was defined by the distance from the peak of the cingulum bundle to pia. The ROI was divided into 10 equal vertical bins, and the number of tdTomato+ or GFP+ cells in each bin was counted. Data are shown as % of total tdTomato+ or GFP+ cells within the ROI. 2 brain sections were analyzed and averaged for each animal.
2.5. Statistical analysis
All statistical analyses were performed using GraphPad Prism 9 software, except for Pearson’s Chi-Squared test and the accompanying z-tests, which were performed using IBM SPSS 26.0 software. The specific tests and sample sizes (n, number of animals) are described in the results and figure legends. The significance level was set at p < 0.05. Data are presented as mean ± standard error of the mean (SEM).
3. Results
3.1. 4E-BP1CA expression in L2/3 pyramidal neurons by targeting radial glia has no effects on seizure frequency and exacerbates behavioral seizure severity in RhebCA mice
Increasing mTORC1 activity via RhebCA expression during corticogenesis in mice recapitulates human FMCD pathology, including neuronal hypertrophy and mispositioning in the cortex, and seizures (Hsieh et al., 2016; Lin et al., 2016; Nguyen et al., 2019). To examine whether embryonic 4E-BP1CA expression would prevent RhebCA-induced FMCD pathology and seizure onset, we co-electroporated RhebCA and 4E-BP1CA in E15 cortices, targeting radial glia that gives rise to L2/3 pyramidal neurons (Figure 1A). Control mice were co-electroporated with RhebCA and GFP. A tdTomato reporter was included in both groups to label targeted cells.
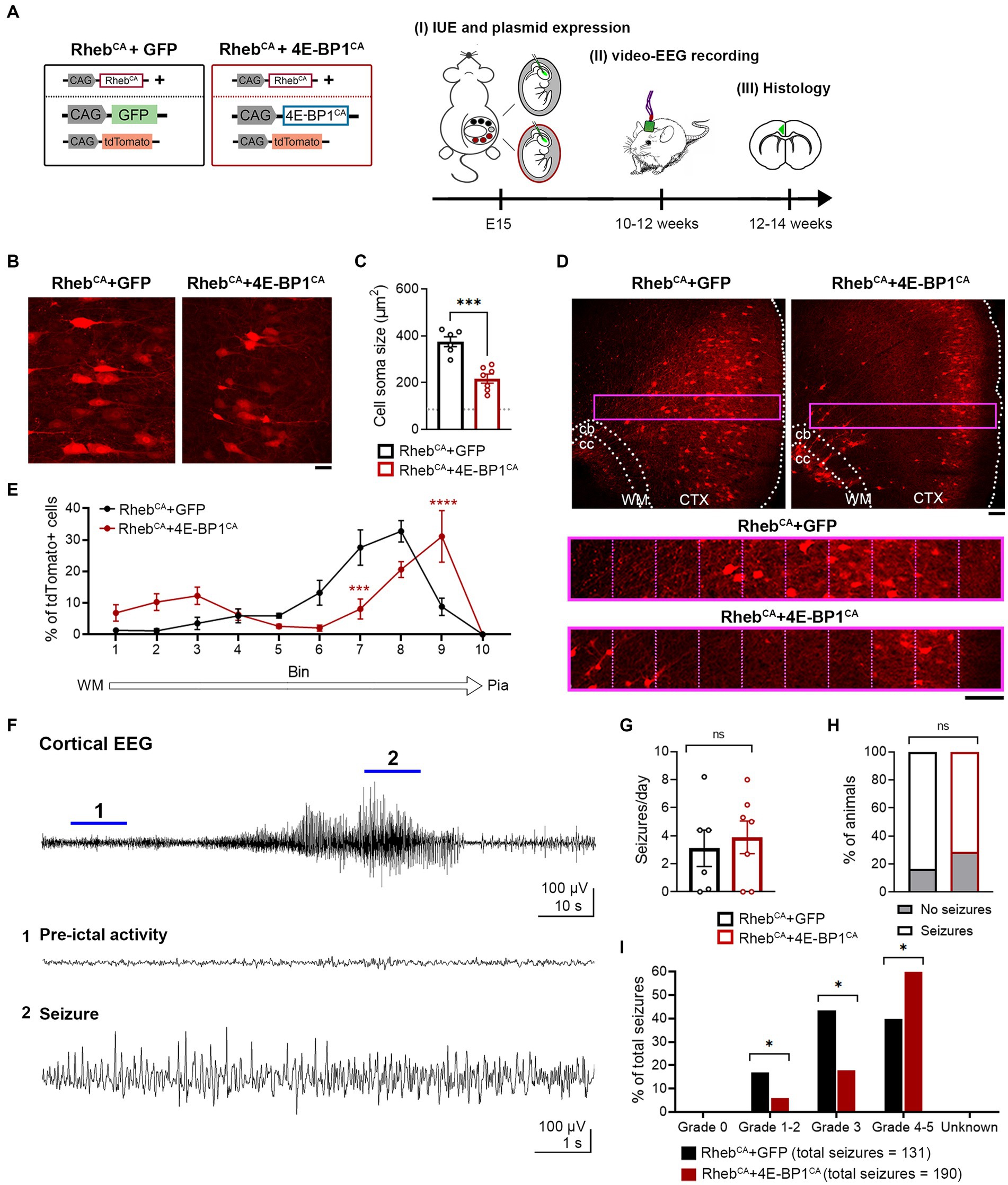
Figure 1. 4E-BP1CA expression (by embryonic targeting of radial glia) decreases neuron hypertrophy, improves cortical lamination, and exacerbates behavioral seizure severity in RhebCA mice. (A) Schematic diagram of plasmids used for IUE and experimental timeline. (B) Representative images showing tdTomato+ cells in electroporated cortices in 12-week-old RhebCA + GFP and RhebCA + 4E-BP1CA mice. Scale bar = 25 μm. (C) Quantification of tdTomato+ cell soma size. Each data point represents averaged values from 50 cells/animal. Data were analyzed using unpaired t-test. Gray dotted line shows previously reported levels for adult control mice electroporated with GPF for visual comparison (Nguyen et al., 2019). (D) Representative images showing tdTomato+ cell placement in electroporated cortices in 12-week-old RhebCA + GFP and RhebCA + 4E-BP1CA mice. A defined region of the cortex (purple rectangle, enlarged on the bottom) spanning from the WM border to pia was divided into 10 equal bins, and the % of cells in each bin was quantified to assess cell distribution. Scale bars = 100 μm. (E) Quantification of tdTomato+ cell placement in the cortex. Each data point represents averaged values from 2 brain sections/animal. Data were analyzed using two-way repeated measures ANOVA with Šídák’s post-hoc test. (F) Representative EEG trace showing a typical electrographic seizure in an 11-week-old RhebCA + GFP mouse. Expanded traces from the region indicated by the blue lines are shown at the bottom. (G) Quantification of seizure frequency. Each data point represents mean seizures/day from 7 days per animal. Data were analyzed using Mann–Whitney U test. (H) Quantification of seizure incidence. Data were analyzed using Fisher’s Exact test. (I) Quantification of behavioral seizure severity based on a modified Racine scale. Data were analyzed using Pearson’s Chi-Squared test with accompanying z-tests to compare proportions. (C,E,G–I) n = 6 RhebCA + GFP, 7 RhebCA + 4E-BP1CA mice. *p < 0.05, ***p < 0.001, ****p < 0.0001. Error bars are ± SEM. IUE, in utero electroporation; CTX, cortex; WM, white matter; cb, cingulum bundle; cc, corpus callosum.
4E-BP1CA expression significantly decreased the soma size of RhebCA neurons and increased the proportion of correctly positioned neurons in L2/3 as examined in brain sections from adult mice (Figures 1B–E). However, we observed a subset of heterotopic RhebCA + 4E-BP1CA-expressing neurons that remained in the deep cortical layers, near the cingulum bundle, and in the white matter (Figure 1D). Despite the histological improvements, the frequency and incidence of seizures, as assessed by EEG recording, were unchanged between RhebCA + 4E-BP1CA and RhebCA + GFP mice (Figures 1F–H). To determine whether the behavioral manifestations of seizures were improved, we scored the associated motor symptoms for all identified electrographic seizures using a modified Racine scale (Table 2). Surprisingly, we found a significant increase in the proportion of grade 4–5 seizures and a decrease in grades 1–2 and 3 seizures in RhebCA + 4E-BP1CA compared to RhebCA + GFP mice (Figure 1I). These findings suggest that embryonic expression of 4E-BP1CA in E15 radial glia generating L2/3 pyramidal neurons worsens RhebCA-induced seizure severity.
3.2. Conditional 4E-BP1CA expression in L2/3 pyramidal neurons by selective embryonic targeting of postmitotic migratory neurons mitigates behavioral seizure severity but not frequency in RhebCA mice
Considering that the heterotopic neurons were not prevented by 4E-BP1CA expression, we hypothesized that expression of 4E-BP1CA in radial glia may indirectly affect the migration of a subset of pyramidal neurons. In addition, we speculated that mTORC1 activation due to RhebCA may lag behind 4E-BP1CA’s effect on cap-dependent translation, resulting in an inappropriate reduction of translation prior to mTORC1 hyperactivation. To address these issues, we bypassed radial glia and selectively targeted 4E-BP1CA expression in postmitotic migratory neurons using a conditional Cre-lox system. We co-electroporated RhebCA with Cre-inducible, conditional 4E-BP1CA (c4E-BP1CA) and Cre recombinase under the control of a doublecortin (DCX) promoter (DCX-Cre) (Figure 2A). In this experimental condition, c4E-BP1CA expression is restricted to DCX+ positive migratory neurons whereas RhebCA is expressed in radial glia and subsequently neurons in the same manner as above. RhebCA is thus functionally expressed in newborn neurons before 4E-BP1CA functional expression. Control mice were co-electroporated with RhebCA, conditional DsRed (cDsRed), and DCX-Cre. A conditional GFP (cGFP) reporter was included in both groups to label targeted cells.
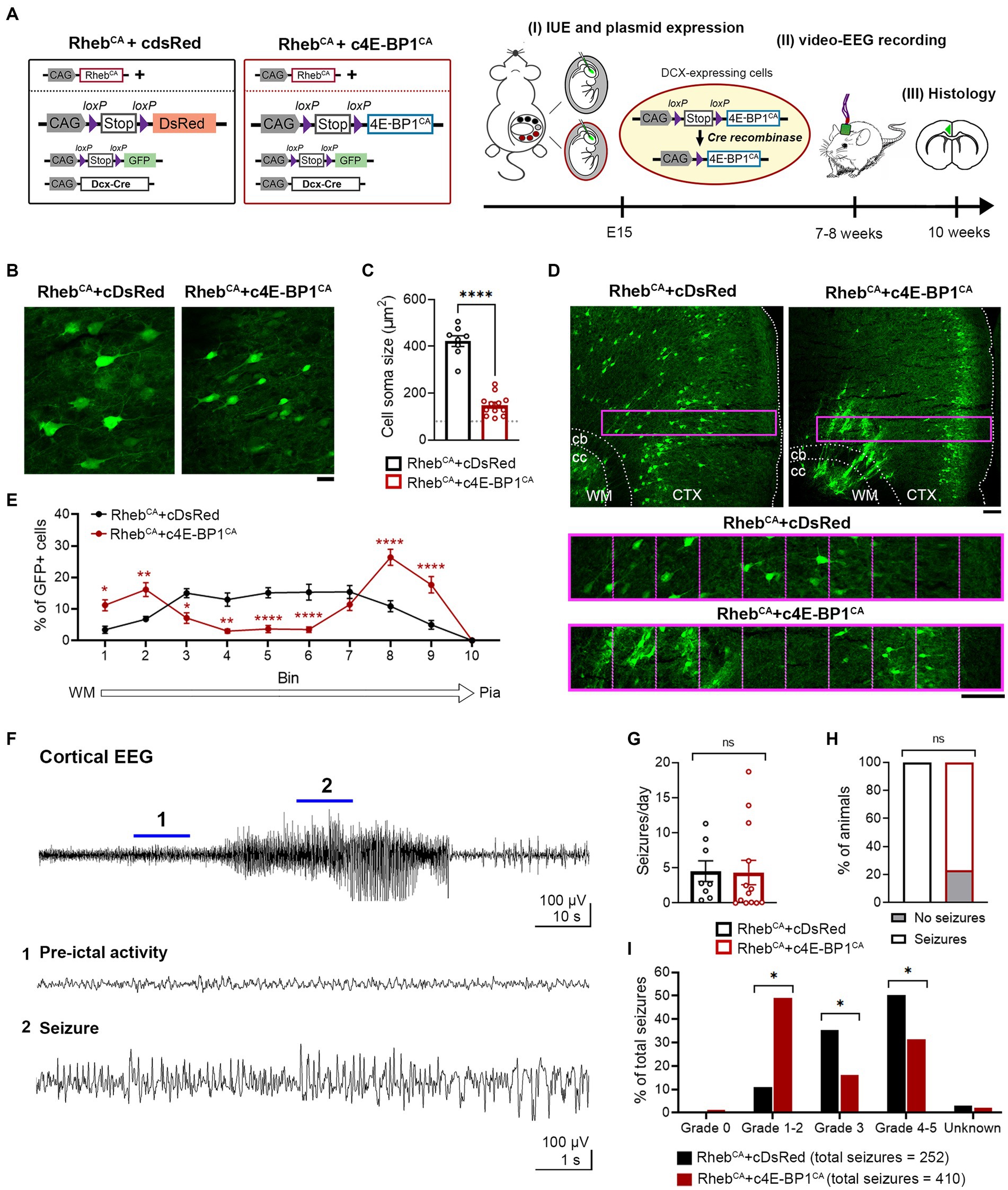
Figure 2. c4E-BP1CA expression (by selective embryonic targeting of postmitotic migratory neurons) decreases neuron hypertrophy, improves cortical lamination, and mitigates behavioral seizure severity in RhebCA mice. (A) Schematic diagram of plasmids used for IUE and experimental timeline. (B) Representative images showing GFP+ cells in electroporated cortices in 10-week-old RhebCA + cDsRed and RhebCA + c4E-BP1CA mice. Scale bar = 25 μm. (C) Quantification of GFP+ cell soma size. Each data point represents averaged values from 20 cells/animal. Data were analyzed using unpaired t-test. Gray dotted line shows previously reported levels for adult control mice electroporated with GPF for visual comaprison (Nguyen et al., 2019). (D) Representative images showing GFP+ cell placement in electroporated cortices in 10-week-old RhebCA + cDsRed and RhebCA + c4E-BP1CA mice. A defined region of the cortex (purple rectangle, enlarged on the bottom) spanning from the WM border to pia was divided into 10 equal bins, and the % of cells in each bin was quantified to assess cell distribution. Scale bars = 100 μm. (E) Quantification of GFP+ cell placement in the cortex. Each data point represents averaged values from 2 brain sections/animal. Data were analyzed using two-way repeated measures ANOVA with Šídák’s post-hoc test. (F) Representative EEG trace showing a typical electrographic seizure in a 9-week-old RhebCA + cDsRed mouse. Expanded traces from the region indicated by the blue lines are shown at the bottom. (G) Quantification of seizure frequency. Each data point represents mean seizures/day from 7 days per animal. Data were analyzed using Mann–Whitney U test. (H) Quantification of seizure incidence. Data were analyzed using Fisher’s Exact test. (I) Quantification of behavioral seizure severity based on a modified Racine scale. Data were analyzed using Pearson’s Chi-Squared test with accompanying z-tests to compare proportions. (C,E,G–I) n = 8 RhebCA + cDsRed, 12–13 RhebCA + c4E-BP1CA mice, *p < 0.05, **p < 0.01, ****p < 0.0001. Error bars are ± SEM. IUE, in utero electroporation; CTX, cortex; WM, white matter; cb, cingulum bundle; cc, corpus callosum.
c4E-BP1CA expression significantly decreased the soma size of RhebCA neurons and increased the proportion of correctly positioned neurons in L2/3 (Figures 2B–E). Similar to the 4E-BP1CA condition described above (Figure 1D), we observed a subset of heterotopic neurons in the deep cortical layers and white matter; however, this was even more prominent than in the above condition and significantly different from the control group (Figures 2D,E). c4E-BP1CA expression did not reduce the seizure frequency or incidence in RhebCA mice (Figures 2F–H). However, the behavioral symptoms associated with seizures were less severe in the RhebCA + c4E-BP1CA compared to RhebCA + cDsRed mice, with a significantly higher proportion of seizures scored at Racine grade 1–2 and a lower proportion scored at grades 3 and 4–5 (Figure 2I). These findings suggest that selective embryonic expression of 4E-BP1CA in postmitotic migrating neurons destined to L2/3 mitigates the severity of RhebCA-induced behavioral seizures despite the persistent presence of neuronal heterotopia.
4. Discussion
In this study, we examined whether embryonic expression of 4E-BP1CA prevents FMCD and seizure development in RhebCA mice with hyperactive mTORC1 signaling. We found that 4E-BP1CA expression during corticogenesis improved FMCD pathology but did not prevent seizure onset nor reduced seizure frequency in mice. These findings underscore the risks of reducing cap-dependent translation through 4E-BP1CA expression during fetal development, including in conditions where mTORC1 is hyperactivated.
Embryonic expression of 4E-BP1CA by targeting E15 radial glia or by selectively targeting postmitotic migrating neurons both led to decreased neuronal hypertrophy, consistent with previous studies (Lin et al., 2016). However, these manipulations did not fully restore the neuron soma size to the published levels for normal controls (Nguyen et al., 2019), suggesting that other molecular players may contribute to this phenotype. Embryonic expression of 4E-BP1CA also improved neuronal positioning. However, in both experimental paradigms, we observed a cluster of heterotopic neurons in the deep layer near the cingulum bundle and in the white matter, indicating that 4E-BP1CA failed to rescue these heterotopias regardless of the timing of 4E-BP1CA expression. Interestingly, these neuronal heterotopias were worse when 4E-BP1CA was selectively expressed in the migrating neurons compared to that in radial glia. These findings suggest that these heterotopias likely do not result from defects in radial glia expressing 4E-BP1CA. Why some neurons remained stuck in the deep layer and white matter when most successfully reached their proper location following 4E-BP1CA expression is unknown. Further investigation into the molecular and laminar identities of these neurons could provide insights into how they arise and why they persist after 4E-BP1CA expression.
Despite the improvements in FMCD histology, 4E-BP1CA expression targeting radial glia had no effect on seizure frequency and unexpectedly exacerbated the behavioral seizure intensity in RhebCA mice. In contrast, selective 4E-BP1CA expression targeting migrating neurons mitigated the severity of behavioral seizures although the seizure frequency remained unchanged. The failure of embryonic 4E-BP1CA expression to prevent or reduce seizures was surprising given that embryonic 4E-BP1CA expression has been shown to improve many mTORC1-induced cellular phenotypes, including somatic hypertrophy and positioning, as reported here and elsewhere (Lin et al., 2016), as well as dendritic and axonal overgrowth (Gong et al., 2015; Lin et al., 2016), and postnatal expression of 4E-BP1CA significantly reduced seizure frequency (Nguyen et al., 2022). Given that heterotopic neurons in the deep layer and white matter remained following 4E-BP1CA expression, it is possible that these contribute to seizure generation. However, we found no correlations between these neuronal heterotopias and seizure severity across individual animals, and the heterotopias were notably worse in the c4E-BP1CA condition that was associated with reduced seizure severity. Furthermore, given that such heterotopias and neuronal misplacement were present following adolescent 4E-BP1CA expression that decreased seizure frequency (Nguyen et al., 2022), the presence of these heterotopias likely does not considerably account for the persistent seizures in RhebCA mice.
Another possible explanation for the persistent seizures following 4E-BP1CA expression is that 4E-BP1 may have specific functions during embryonic cortical development (e.g., regulate the translation of specific subsets of developmentally relevant mRNAs), and altering these functions via 4E-BP1CA expression could interfere with these processes, leading to unrepressed seizures, and in some cases, exacerbation of behavioral seizure severity. In line with this idea, genetic inhibition of mTORC1, and subsequently, reduction of 4E-BP phosphorylation, via embryonic deletion of Raptor in Pten knockout mice did not suppress seizures (Chen et al., 2019) despite rescuing brain size, neuronal overgrowth, and synapse function (Chen et al., 2019; Tariq et al., 2022). One study also reported an unexpected increase in the mortality of these mice (Cullen et al., 2023). Interestingly, embryonic knockdown of eIF4E successfully reduced the seizure frequency in mice expressing mTORC1-activating mutants (Kim et al., 2019). eIF4E is a binding partner of 4E-BP1 and required for cap-dependent translation (Richter and Sonenberg, 2005). Both 4E-BP1CA expression and eIF4E knockdown reduce translation, and the reasons behind the diverging impacts on seizures are unclear. Overall, these results suggest a complicated, developmentally regulated role for 4E-BPs in mTOR-induced epilepsy.
Taken together, our study caution against 4E-BP1CA expression in embryonic life for seizure prevention and suggest a complex developmental stage-dependent role for 4E-BPs in mTORC1-induced epilepsy. Although our study shows that embryonic 4E-BP1CA expression does not prevent seizures, alternative approaches to repress translation, such as directly inhibiting the phosphorylation of endogenous 4E-BPs, may yield different results and should be further explored. Future studies aimed to dissect the functions of 4E-BPs in cortical development could shed additional light on these issues and provide insights into new treatment avenues for mTORC1-induced epilepsy.
Data availability statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding authors.
Ethics statement
The animal study was approved by Yale University Institutional Animal Care and Use Committee (IACUC). The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
LN: Conceptualization, Investigation, Formal Analysis, Visualization, Writing – original draft, Writing – review & editing, Funding acquisition. MS: Formal analysis. AB: Conceptualization, Writing – review & editing, Funding acquisition.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by National Institutes of Health (NIH) F32 HD095567 (LN), R01 NS111980 (AB).
Conflict of interest
LN and AB are co-inventors on a patent application, PCT/US2020/054007 entitled “Targeting Cap-Dependent Translation to Reduce Seizures in mTOR disorders”. 2021-04-08: Publication of WO2021067752A1.
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Baybis, M., Yu, J., Lee, A., Golden, J. A., Weiner, H., McKhann, G., et al. (2004). mTOR cascade activation distinguishes tubers from focal cortical dysplasia. Ann. Neurol. 56, 478–487. doi: 10.1002/ana.20211
Chen, C. J., Sgritta, M., Mays, J., Zhou, H., Lucero, R., Park, J., et al. (2019). Therapeutic inhibition of mTORC2 rescues the behavioral and neurophysiological abnormalities associated with Pten-deficiency. Nat. Med. 25, 1684–1690. doi: 10.1038/s41591-019-0608-y
Choi, K. M., McMahon, L. P., and Lawrence, J. C. Jr. (2003). Two motifs in the translational repressor PHAS-I required for efficient phosphorylation by mammalian target of rapamycin and for recognition by raptor. J. Biol. Chem. 278, 19667–19673. doi: 10.1074/jbc.M301142200
Crino, P. B. (2015). mTOR signaling in epilepsy: insights from malformations of cortical development. Cold Spring Harb. Perspect. Med. 5:a022442. doi: 10.1101/cshperspect.a022442
Cullen, E. R., Tariq, K., Shore, A. N., Luikart, B. W., and Weston, M. C. (2023). mTORC2 inhibition improves morphological effects of PTEN loss, but does not correct synaptic dysfunction or prevent seizures. J. Neurosci. 43, 827–845. doi: 10.1523/JNEUROSCI.1354-22.2022
Franco, S. J., Martinez-Garay, I., Gil-Sanz, C., Harkins-Perry, S. R., and Muller, U. (2011). Reelin regulates cadherin function via Dab1/Rap1 to control neuronal migration and lamination in the neocortex. Neuron 69, 482–497. doi: 10.1016/j.neuron.2011.01.003
Glenn, O. A., Cuneo, A. A., Barkovich, A. J., Hashemi, Z., Bartha, A. I., and Xu, D. (2012). Malformations of cortical development: diagnostic accuracy of fetal MR imaging. Radiology 263, 843–855. doi: 10.1148/radiol.12102492
Gong, X., Zhang, L., Huang, T., Lin, T. V., Miyares, L., Wen, J., et al. (2015). Activating the translational repressor 4E-BP or reducing S6K-GSK3beta activity prevents accelerated axon growth induced by hyperactive mTOR in vivo. Hum. Mol. Genet. 24, 5746–5758. doi: 10.1093/hmg/ddv295
Hanai, S., Sukigara, S., Dai, H., Owa, T., Horike, S. I., Otsuki, T., et al. (2017). Pathologic active mTOR mutation in brain malformation with intractable epilepsy leads to cell-autonomous migration delay. Am. J. Pathol. 187, 1177–1185. doi: 10.1016/j.ajpath.2017.01.015
Hsieh, L. S., Wen, J. H., Claycomb, K., Huang, Y., Harrsch, F. A., Naegele, J. R., et al. (2016). Convulsive seizures from experimental focal cortical dysplasia occur independently of cell misplacement. Nat. Commun. 7:11753. doi: 10.1038/ncomms11753
Kim, J. K., Cho, J., Kim, S. H., Kang, H. C., Kim, D. S., Kim, V. N., et al. (2019). Brain somatic mutations in MTOR reveal translational dysregulations underlying intractable focal epilepsy. J. Clin. Invest. 129, 4207–4223. doi: 10.1172/JCI127032
Laplante, M., and Sabatini, D. M. (2012). mTOR signaling in growth control and disease. Cells 149, 274–293. doi: 10.1016/j.cell.2012.03.017
Lerman-Sagie, T., Pogledic, I., Leibovitz, Z., and Malinger, G. (2021). A practical approach to prenatal diagnosis of malformations of cortical development. Eur. J. Paediatr. Neurol. 34, 50–61. doi: 10.1016/j.ejpn.2021.08.001
Lin, T. V., Hsieh, L., Kimura, T., Malone, T. J., and Bordey, A. (2016). Normalizing translation through 4E-BP prevents mTOR-driven cortical mislamination and ameliorates aberrant neuron integration. Proc. Natl. Acad. Sci. USA. 113, 11330–11335. doi: 10.1073/pnas.1605740113
Ma, X. M., and Blenis, J. (2009). Molecular mechanisms of mTOR-mediated translational control. Nat. Rev. Mol. Cell Biol. 10, 307–318. doi: 10.1038/nrm2672
Maehama, T., Tanaka, M., Nishina, H., Murakami, M., Kanaho, Y., and Hanada, K. (2008). RalA functions as an indispensable signal mediator for the nutrient-sensing system. J. Biol. Chem. 283, 35053–35059. doi: 10.1074/jbc.M805822200
Nguyen, L. H., Brewster, A. L., Clark, M. E., Regnier-Golanov, A., Sunnen, C. N., Patil, V. V., et al. (2015). mTOR inhibition suppresses established epilepsy in a mouse model of cortical dysplasia. Epilepsia 56, 636–646. doi: 10.1111/epi.12946
Nguyen, L. H., Mahadeo, T., and Bordey, A. (2019). mTOR hyperactivity levels influence the severity of epilepsy and associated neuropathology in an experimental model of tuberous sclerosis complex and focal cortical dysplasia. J. Neurosci. 39, 2762–2773. doi: 10.1523/Jneurosci.2260-18.2019
Nguyen, L. H., Xu, Y., Mahadeo, T., Zhang, L., Lin, T. V., Born, H. A., et al. (2022). Expression of 4E-BP1 in juvenile mice alleviates mTOR-induced neuronal dysfunction and epilepsy. Brain 145, 1310–1325. doi: 10.1093/brain/awab390
Orlova, K. A., and Crino, P. B. (2010). The tuberous sclerosis complex. Ann. N. Y. Acad. Sci. 1184, 87–105. doi: 10.1111/j.1749-6632.2009.05117.x
Richter, J. D., and Sonenberg, N. (2005). Regulation of cap-dependent translation by eIF4E inhibitory proteins. Nature 433, 477–480. doi: 10.1038/nature03205
Schalm, S. S., Fingar, D. C., Sabatini, D. M., and Blenis, J. (2003). TOS motif-mediated raptor binding regulates 4E-BP1 multisite phosphorylation and function. Curr. Biol. 13, 797–806. doi: 10.1016/s0960-9822(03)00329-4
Keywords: mTOR, 4E-BP, seizures, epilepsy, corticogenesis, cortical development, malformation of cortical development
Citation: Nguyen LH, Sharma M and Bordey A (2023) 4E-BP1 expression in embryonic postmitotic neurons mitigates mTORC1-induced cortical malformations and behavioral seizure severity but does not prevent epilepsy in mice. Front. Neurosci. 17:1257056. doi: 10.3389/fnins.2023.1257056
Edited by:
Kazuhiko Sawada, Tsukuba International University, JapanReviewed by:
Junli Zhao, Duke University, United StatesJong-Cheol Rah, Korea Brain Research Institute, Republic of Korea
Copyright © 2023 Nguyen, Sharma and Bordey. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lena H. Nguyen, bGVuYS5uZ3V5ZW5AdXRkYWxsYXMuZWR1; Angelique Bordey, YW5nZWxpcXVlLmJvcmRleUB5YWxlLmVkdQ==