 Yuan Tian
Yuan Tian Jinfang Xing1
Jinfang Xing1
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
SYSTEMATIC REVIEW article
Front. Neurosci. , 17 November 2023
Sec. Neurodegeneration
Volume 17 - 2023 | https://doi.org/10.3389/fnins.2023.1252075
Background: IGHMBP2 is a crucial gene for the development and maintenance of the nervous system, especially in the survival of motor neurons. Mutations in this gene have been associated with spinal muscular atrophy with respiratory distress type 1 (SMARD1) and Charcot-Marie-Tooth disease type 2S (CMT2S).
Methods: We conducted a systematic literature search using the PubMed database to identify studies published up to April 1st, 2023, that investigated the association between IGHMBP2 mutations and SMARD1 or CMT2S. We compared the non-truncating mutations and truncating mutations of the IGHMBP2 gene and selected high-frequency mutations of the IGHMBP2 gene.
Results: We identified 52 articles that investigated the association between IGHMBP2 mutations and SMARD1/CMT2S. We found 6 hotspot mutations of the IGHMBP2 gene. The truncating mutations in trans were all associated with SMARD1.
Conclusion: This study provides evidence that the complete LOF mechanism of the IGHMBP2 gene defect may be an important cause of SMARD1.
IGHMBP2 is a gene located on chromosome 11 that encodes the immunoglobulin mu-binding protein 2, also known as SMN-interacting protein 1 (SIP1) (Surrey et al., 2018). This protein plays a crucial role in the development and maintenance of the nervous system, particularly in the survival of motor neurons (Rzepnikowska et al., 2022). The protein contains various domains, including an ATPase domain, a RNA helicase domain, and a zinc finger domain, which are involved in RNA processing and transport and the regulation of gene expression (Rzepnikowska and Kochanski, 2021). The helicase core of the IGHMBP2 protein is comprised of four distinct regions, including two RecA-like domains (Domains 1A and 2A) with two subdomains (1B and 1C) inserted into Domain 1A (Lim et al., 2012). The regions of Domains 1A (residues 159–270 and 347–440) and 2A (residues 441–648) that are highly conserved are critical for the proper function of the IGHMBP2 protein (Saladini et al., 2020). Mutations in these regions have been associated with spinal muscular atrophy with respiratory distress type 1 (SMARD1) (Pekuz et al., 2022).
Spinal muscular atrophy with respiratory distress type 1 (SMARD1) is a rare genetic disorder characterized by progressive muscle weakness and respiratory distress in early infancy, which is caused by mutations in the IGHMBP2 gene (Perego et al., 2020). The symptoms of SMARD1 typically appear in the first few months of life and include muscle weakness, particularly in the diaphragm and other muscles involved in breathing, as well as decreased reflexes, difficulty swallowing, and impaired movement (Saeed et al., 2021). The disorder can lead to respiratory failure and death in severe cases (Bodle et al., 2021).
In recent years, increasing reports have suggested that mutations in the IGHMBP2 gene may cause CMT2S, in addition to SMARD1 (Lei et al., 2022). CMT2S is a subtype of Charcot-Marie-Tooth disease (CMT) which is a hereditary neuropathy that affects the peripheral nervous system (Chandrasekharan et al., 2022). It is characterized by muscle weakness and wasting in the distal limbs, sensory loss, and reduced or absent tendon reflexes (Xie et al., 2021). Compared to SMARD1, CMT2S has a milder phenotype and does not typically present with respiratory distress or spinal motor neuron loss. Literature has indicated that mutations in the 5′ region and the last exon of the IGHMBP2 gene are predominantly associated with CMT2, including nonsense mutations, frameshift mutations, missense mutations, and compound heterozygous mutations (Saladini et al., 2020).
Due to that different mutations in the IGHMBP2 gene can lead to either SMARD1 or CMT2S, the identification of the specific mutations in IGHMBP2 that lead to SMARD1 or CMT2S is important for clinical diagnosis, treatment, and genetic counseling. According to these, our study aims to explore the relationship between different types of IGHMBP2 gene mutations and the two distinct disease types they cause by retrieving relevant literature from the PubMed database, screening and analyzing the retrieved articles. Our goal is to identify the specific regions of the IGHMBP2 gene where mutations occur and their correlation with these two different diseases. The ultimate objective of this study is to provide more precise genetic counseling for clinical diagnosis and treatment.
We conducted a systematic literature search using the PubMed database to identify studies published up to April 1st, 2023 that investigated the association between IGHMBP2 gene mutations and Charcot-Marie-Tooth disease, spinal muscular atrophy with respiratory distress, distal hereditary motor neuronopathy, and autosomal recessive distal spinal muscular atrophy. We used the search terms “IGHMBP2” and “Charcot-Marie-Tooth disease” or “spinal muscular atrophy with respiratory distress” or “distal hereditary motor neuronopathy” or “autosomal recessive distal spinal muscular atrophy.” The final search equation was defined using the Boolean connectors “AND” and “OR,” as follows: “IGHMBP2” AND {“Charcot-Marie-Tooth disease” OR “spinal muscular atrophy with respiratory distress” OR “distal hereditary motor neuronopathy” OR “autosomal recessive distal spinal muscular atrophy”}. We excluded non-original works not related to human subjects, such as reviews, functional experiments on animals or cells, the same cases, not clearly described studies and original articles that had no information relevant to the purpose of this study. After identifying the relevant articles, we classified the pathogenicity of the IGHMBP2 gene mutations involved in the articles according to the American College of Medical Genetics and Genomics (ACMG) guidelines (Richards et al., 2015). Pathogenicity was classified as “Pathogenic (P),” “Likely pathogenic (LP),” “Uncertain significance (VUS),” “Benign (B),” or “Likely benign (LB).” Finally, these reported P/LP homozygous variants were selected for distribution analysis in the gene structure and protein functional regions. The literature search and analysis process are shown in Figure 1.

Figure 1. Flow diagram of included studies.
We calculated the percentage of IGHMBP2 gene mutations identified through the literature search that were associated with SMARD1 and CMT2S, respectively. We compared the non-truncating mutations located in RecA-like domains (Domains 1A and 2A) of the IGHMBP2 gene between the SMARD1 and CMT2S groups to determine if there were any differences. We also compared the homozygous non-truncating mutations in the IGHMBP2 gene between the SMARD1 and CMT2S groups. In addition, we compared the truncating mutations located in the last exon of the IGHMBP2 gene between the SMARD1 and CMT2S groups and assessed the homozygous truncating mutations in each group.
We selected 43 homozygous and 1 hemizygous (with heterozygous deletion of exons 6–13) IGHMBP2 gene variants classified as P/LP according to ACMG guidelines, and classified them into truncating and non-truncating variants. We analyzed the distribution of these variants in the gene structure and protein functional regions, and created separate lollipop plots for each type of variant. We performed SWISS-MODEL homology modeling and used SPDBV_4.10 software to analyze the 3D protein structure of the missense variants in these homozygous variants.
We selected IGHMBP2 gene mutations that appeared in 5 or more probands in the literature search and considered them to be high-frequency mutations. We analyzed the number of times these mutations were associated with SMARD1 and CMT2S, respectively, as well as their trans position. We then identified any patterns or relationships between these mutations and the two diseases.
In this study, statistical comparisons of data were performed using the χ2 test (p values less than 0.05 were considered statistically significant), and all data analyses were conducted using SPSS 22.0. Graphs were plotted using IBS 2.0 and Graphpad prism 9.
Among the 52 literature articles screened (Supplementary Table S1) (Robison et al., 1998; Grohmann et al., 2001, 2003; Guenther et al., 2004, 2007; Maystadt et al., 2004; Giannini et al., 2006; Wong et al., 2006; Joseph et al., 2009; AlSaman and Tomoum, 2010; Baughn et al., 2011; Pierson et al., 2011; Chalancon et al., 2012; Eckart et al., 2012; Majid et al., 2012; Messina et al., 2012; Gitiaux et al., 2013; Blaschek et al., 2014; Cottenie et al., 2014; Jedrzejowska et al., 2014; Lin et al., 2014; Litvinenko et al., 2014; Hamilton et al., 2015; Han et al., 2015; Wagner et al., 2015; Lingappa et al., 2016; Luan et al., 2016; Pedurupillay et al., 2016; San et al., 2016; Dohrn et al., 2017; Liu et al., 2017; Yuan et al., 2017; Zhang et al., 2017; Habibi et al., 2018; Kulshrestha et al., 2018; Tomaselli et al., 2018; Wu et al., 2018; Cassini et al., 2019; Kim et al., 2019; Yasui et al., 2019; Cortese et al., 2020; Tsang et al., 2020; Bodle et al., 2021; Felice et al., 2021; Saeed et al., 2021; Xie et al., 2021; Berti et al., 2022; Chandrasekharan et al., 2022; Megarbane et al., 2022; Pekuz et al., 2022; Taiana et al., 2022; Xiao et al., 2022), SMARD1 accounted for 71% and CMT2S accounted for 29% of the mutations associated with the study’s purpose (Figure 2).
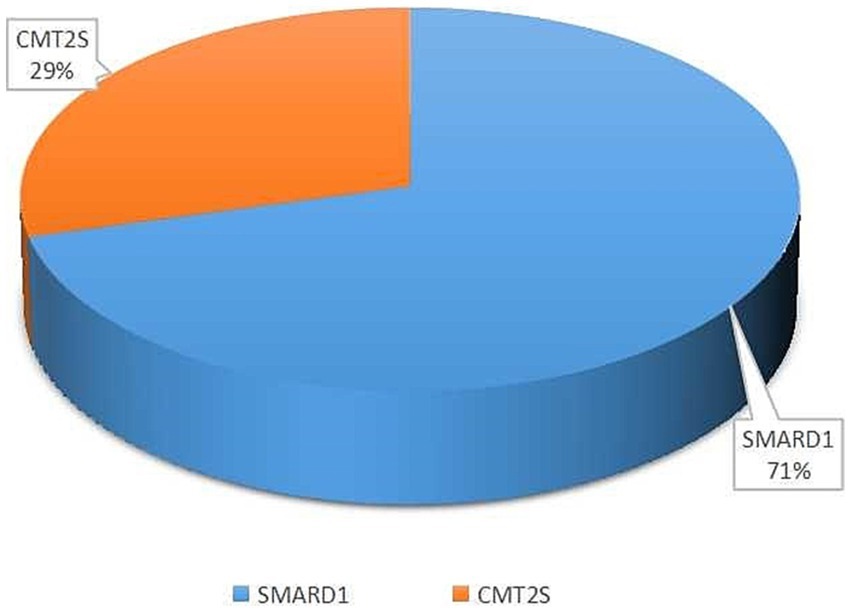
Figure 2. The percentage of SMARD1 and CMT2S within the entirety of literature data.
Among SMARD1-associated non-truncating mutations (missenese and inframe) in the IGHMBP2 gene, 90.4% (66/73) were located in RecA-like domains (Domains 1A and 2A), while in CMT2S-associated non-truncating mutations, 71.1% (27/38) were located in these domains. Statistical analysis showed a significant difference between the two diseases in terms of the RecA-like domains (Domains 1A and 2A) mutations (χ2 = 6.893, p = 0.009) (Table 1).
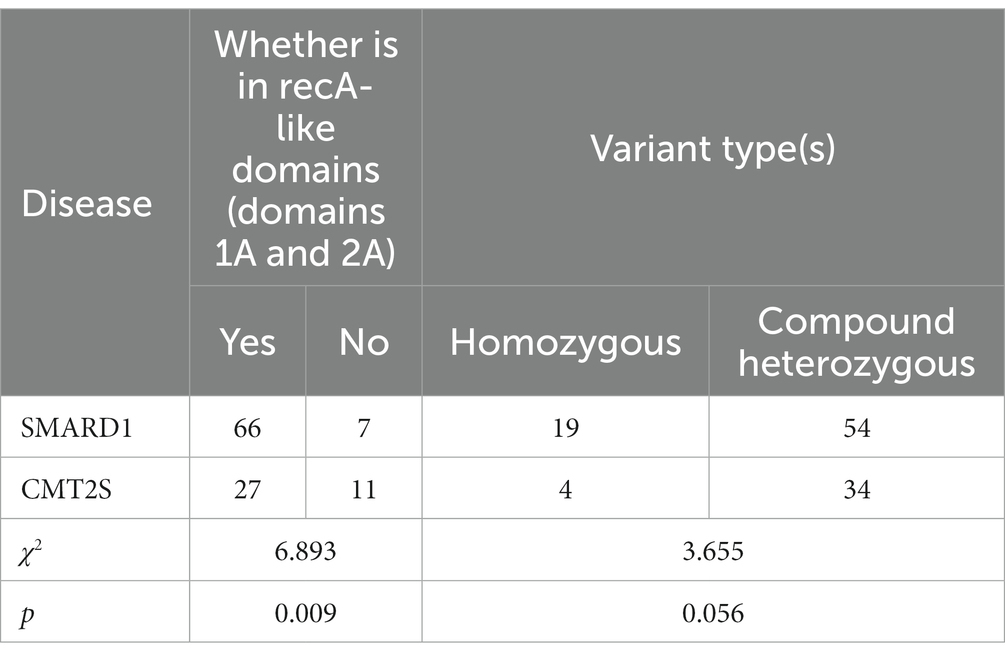
Table 1. Comparison of non-truncating IGHMBP2 gene variant positions and types between SMARD1 and CMT2S.
Among SMARD1-associated truncating mutations (nonsense, frameshift and splice) in the IGHMBP2 gene, 1.2% (1/82) were located in the last exon, while in CMT2S-associated truncating mutations, 15.4% (4/26) were located in the last exon. Statistical analysis showed a significant difference between the two diseases in terms of mutations located in the last exon (χ2 = 8.971, p = 0.012) (Table 2).
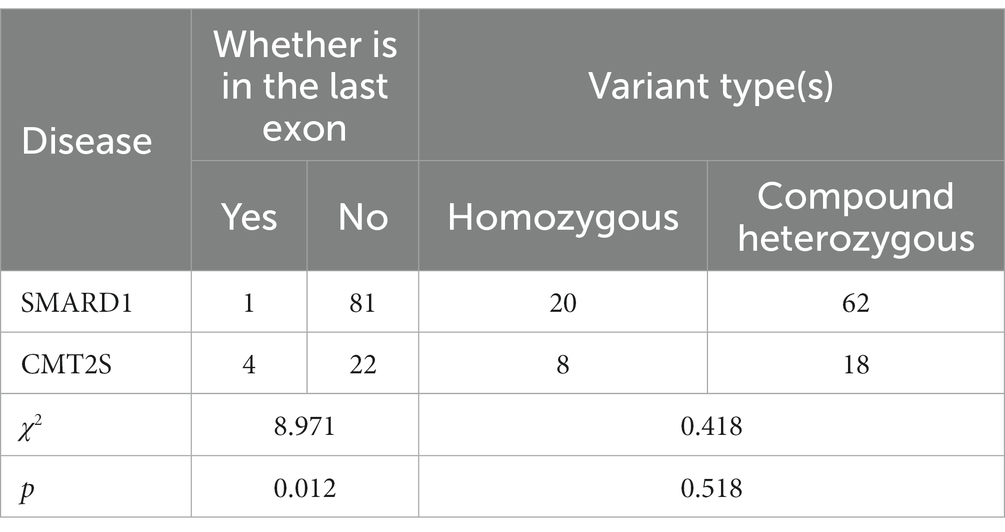
Table 2. Comparison of truncating IGHMBP2 gene variant positions and types between SMARD1 and CMT2S.
In SMARD1-associated mutations in the IGHMBP2 gene, homozygous non-truncating mutations accounted for 26.0% (19/73) of all mutations, while in CMT2S-associated non-truncating mutations, homozygous mutations accounted for 10.5% (4/38) (Table 1). Statistical analysis showed no significant difference between the two diseases in terms of homozygous mutations (χ2 = 3.655, p = 0.056) (Table 1). Besides, homozygous truncating mutations accounted for 24.4% (20/82) of all mutations, while in CMT2S-associated truncating mutations, homozygous mutations accounted for 30.8% (8/26) (Table 2). Statistical analysis showed no significant difference between the two diseases in terms of homozygous mutations (χ2 = 0.418, p = 0.518) (Table 2).
After analyzing 43 homozygous variants and 1 hemizygous variant (Supplementary Table S2) of the IGHMBP2 gene, we found that non-truncating variants located at the 5′ and 3′ ends of the gene are associated with CMT2S, while non-truncating variants associated with SMARD1 are mostly located within the two RecA-like domains (Domains 1A and 2A) with two subdomains (1B and 1C) (Figure 3A). The only exception is the p.Asp400Asn variant, which is located in Domain 1A but has been reported to be associated with CMT2S (Figure 3A). Among these homozygous truncating mutations in the IGHMBP2 gene, only the p.Lys868ProfsTer109 mutation is associated with CMT2S, while all other truncating homozygous mutations have been reported to be associated with SMARD1 (Figure 3B).

Figure 3. (A) Distribution of gene structure and protein functional regions of non-truncating mutations in IGHMBP2 gene after literature screening; (B) Gene structure and protein functional domain distribution of IGHMBP2 gene with truncating mutations identified through literature screening.
After conducting protein 3D structure analysis on the missense variants among these homozygous variants, we found that, except for the hemizygous variant p.Phe369Leu, all the homozygous variants showed changes in hydrogen bonds relative to the wild-type protein, resulting in decreased protein stability and changes in the protein 3D structure (Figure 4).

Figure 4. 3D Structure prediction of missense variants in IGHMBP2 Gene after literature screening.
Based on the identified high-frequency mutations in the IGHMBP2 gene, 6 hotspot mutations were determined: c.1488C > A (p.Cys496Ter) (Cottenie et al., 2014; San et al., 2016; Dohrn et al., 2017; Cortese et al., 2020), c.138 T > A (p.Cys46Ter) (Grohmann et al., 2003; Taiana et al., 2022), c.1478C > T (p.Thr493Ile) (Eckart et al., 2012; Hamilton et al., 2015; Pedurupillay et al., 2016), c.1738G > A (p.Val580Ile) (Wong et al., 2006), c.439C > T (p. Arg147Ter) (Jedrzejowska et al., 2014), and c.1540G > A (p.Glu514Lys) (Grohmann et al., 2001), among which c.1488C > A (p.Cys496Ter) had the highest incidence rate (13/42) (Supplementary Table S3). All of these 6 mutations exhibited phenotypic variability, with SMARD1 having a higher incidence rate (30/42) overall (Supplementary Table S3). Our analysis of the in-trans variants of these 6 hotspot mutations in terms of gene structure and protein functional domains revealed that, except for the p.Arg971GlufsTer4 mutation which is associated with CMT2S, all other truncating variants were associated with SMARD1 (Figure 5). Non-truncating mutations in these in-trans variants did not show a clear regional distribution pattern (Figure 5).
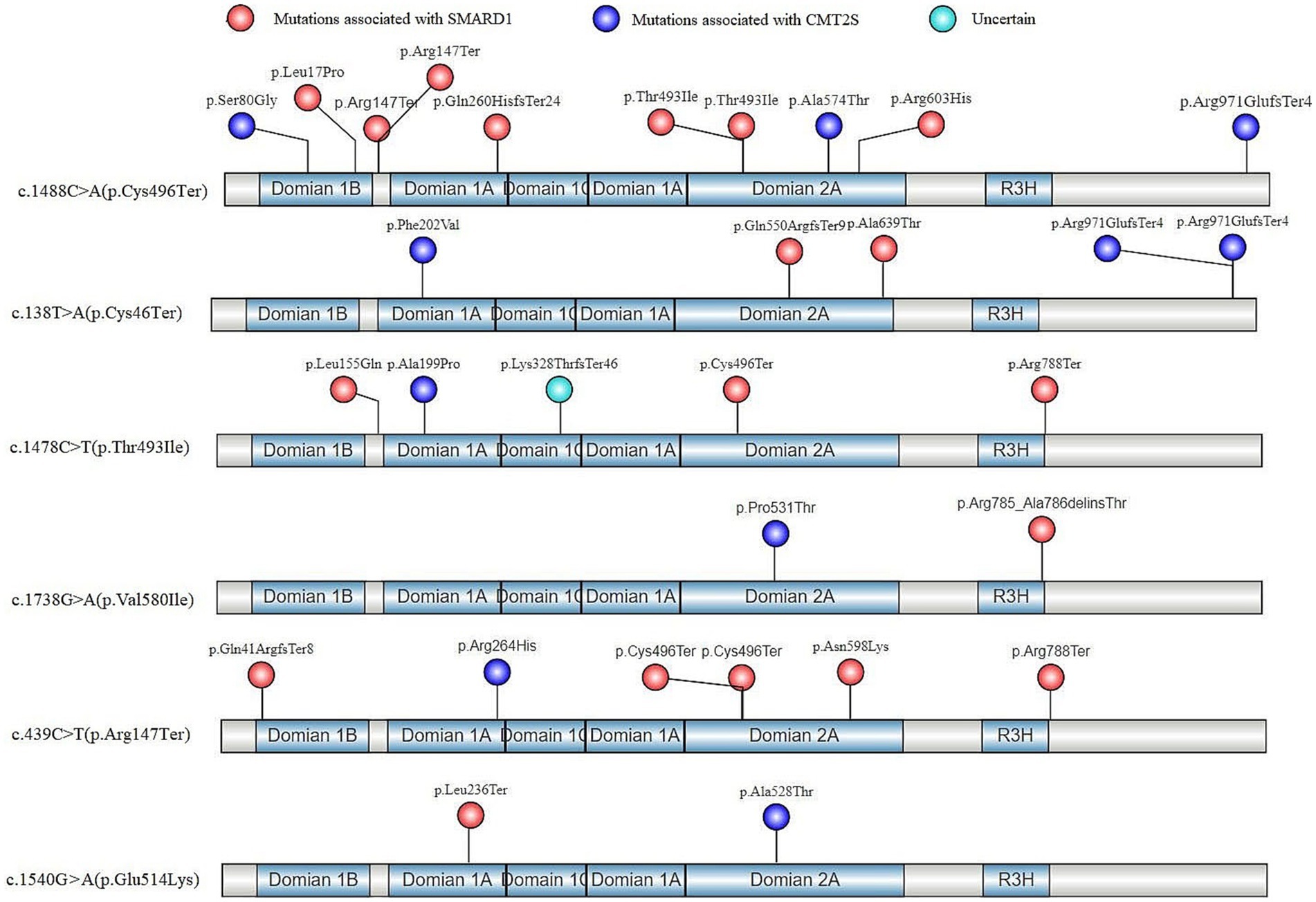
Figure 5. In-trans mutations of hotspot variants in the gene structure and protein functional regions of IGHMBP2 gene after literature screening.
IGHMBP2 gene mutations are associated with two diseases, SMARD1 (Rzepnikowska et al., 2022) and CMT2S (Martin et al., 2022), which exhibit significant differences in phenotype. Therefore, understanding the detection rate of these two diseases is important. In this study, a literature search has revealed that SMARD1 accounted for 71% of all IGHMBP2-related diseases detected, making it the most significant disease type caused by mutations in this gene.
The human IGHMBP2 gene consists of 15 exons and encodes immunoglobulin μ-binding protein 2, which contains 993 amino acids (109,149 Da) (Maystadt et al., 2004). Its function is not fully understood. The IGHMBP2 gene contains four domains: a DNA/RNA helicase domain, an R3H domain, a zinc finger domain, a DEXDc domain and an AN1-type zinc finger motif (Vadla et al., 2023). Among them, the DNA/RNA helicase domain contains two recA domains (Domains 1A and 2A) and at least seven highly conserved amino acid motifs (Lim et al., 2012). The synergistic effect of helicase and R3H domains leads to increased RNA binding affinity, thereby enhancing its ATPase activity (Rzepnikowska and Kochanski, 2021). Most of the missense mutations causing SMARD1 have been found in the helicase domain, indicating that this domain may play a central role in the pathogenesis of the disease (Viguier et al., 2019). Lim et al. (2012). analyzed seven missense mutations located in the ATPase domain through bioinformatics and found that they can affect ATPase activity by disrupting ATP binding/hydrolysis or by reducing the structural stability of the helicase motor. In this study, we compared the number of non-truncated mutations between SMARD1 and CMT2S located in the two RecA-like domains (Domains 1A and 2A) and found a statistically significant difference (χ2 = 6.893, p = 0.009). There were significantly more mutations in SMARD1 in the two RecA-like domains (Figure 1), which further confirms the important role of the two RecA-like domains in causing SMARD1.
Charcot-Marie-Tooth disease (CMT) is a series of polygenic syndromes that cause progressive length-dependent degeneration of peripheral sensory and/or motor fibers (Bolino and D'Antonio, 2023). The disease is associated with more than 80 different genes (Jani-Acsadi et al., 2015), including IGHMBP2 mutations. CMT2 cases involving IGHMBP2 mutations usually have mild myelin fiber involvement (McCray and Scherer, 2021). It is currently believed that in CMT2, mutations of the IGHMBP2 gene are mainly a combination of nonsense mutations in the 5′ region of the gene and truncating, missense or homozygous mutations in the last exon (Saladini et al., 2020). Truncating mutations in these positions can cause CMT2S because different combinations of mutations result in different amounts of residual protein (Yuan et al., 2017). In this study, we conducted a literature review and found a statistically significant difference in the number of IGHMBP2 truncating mutations located in the last exon between CMT2S and SMARD1 (χ2 = 8.971, p = 0.012), with significantly more mutations in CMT2S (Table 2). This indicates that the IGHMBP2 gene truncating mutations located in the last exon are a hotspot area for mutations leading to CMT2S.
Interestingly, we compared the number of homozygous mutations between SMARD1 and CMT2S and found that whether it was a homozygous or compound heterozygous mutation of the IGHMBP2 gene, there was no significant difference in their effect on causing SMARD1 or CMT2S, with no statistically significant differences observed (Tables 1, 2). This suggests that the heterozygosity of mutations is not a significant factor in causing SMARD1 or CMT2S. In all the homozygous variants, it was predicted that these variants caused significant 3D structural changes, leading to a decrease in protein stability. The only exception is the hemizygous variant p.Phe369Leu, which did not show any hydrogen bond changes. According to the report by Guenther et al. (2004), the p.Phe369Leu was located in the trans position of the heterozygous deletion of exons 6–13 (Guenther et al., 2004). The p.Phe369Leu mutation in the IGHMBP2 gene is classified as “likely pathogenic” according to the ACMG guidelines, with the following evidences: PM1: The variant is located in the Helicase superfamily 1/2, ATP-binding domain; PM3: The variant forms a compound heterozygous mutation with the deletion of exons 6–13, positioned in trans; PM2_Supporting: The variant is not found in population frequency databases such as gnomAD, 1,000 Genomes, and ExAC; PP3: The variant is predicted by REVEL with a score of 0.683, falling within the range of 0.644–0.773 (Pejaver et al., 2022). Based on these ACMG criterias, it is evident that the PM3 evidence, i.e., the variant being in trans with the deletion of exons 6–13, plays a crucial role in classifying p.Phe369Leu as “likely pathogenic.” Without this evidence, the variant would be classified as “variant of uncertain significance (VUS).” Moreover, according to protein 3D structure predictions, p.Phe369Leu does not show any hydrogen bond changes, suggesting that the impact of this variant on the protein’s spatial structure is limited. Therefore, the influence of p.Phe369Leu on gene function may be minimal, and the manifestation of SMARD1-related phenotypes in the patient is more likely due to the allelic variant deletion of exons 6–13. Consequently, haploinsufficiency may also be a potential mechanism for disease caused by IGHMBP2 defects.
Another noteworthy mutation combination is the compound heterozygous mutation of c.1060G ≥ A (p.Gly354Ser) and c.2356delG (p.Ala786ProfsTer45), which is present in two different cases (Supplementary Table S1), and leads to different types of diseases. In the case reported by Zhang et al. (2017), this compound heterozygous mutation caused SMARD1, while in the case reported by Tsang et al. (2020), it led to CMT2S. This indicates that there are differences in phenotypic expression in diseases related to the IGHMBP2 gene in non-homozygous mutations. However, there are also differences between these two cases. In the case reported by Zhang et al. (2017), the double heterozygous mutation of the IGHMBP2 gene is a definite compound heterozygous mutation, while in the case reported by Tsang et al. (2020), it is not clear whether the double heterozygous mutation of this gene is a compound heterozygous mutation because the c.2356delG (p.Ala786ProfsTer45) mutation has not been parentally verified, and it cannot be determined whether it is located at the trans position of the paternal c.1060G ≥ A (p.Gly354Ser) mutation. Therefore, in Tsang et al. (2020), it cannot be ruled out that this double heterozygous mutation is not a compound heterozygous mutation and that other mutations of the IGHMBP2 gene causing CMT2S may have been missed.
In our study, homozygous truncating mutations in the IGHMBP2 gene, except for the p.Lys868ProfsTer109 (Wagner et al., 2015), were found to cause SMARD1. Analysis of the gene structure and protein functional regions of in-trans variants in 6 hotspot IGHMBP2 mutations showed that all truncating mutations, except for the p.Arg971GlufsTer4 (Cottenie et al., 2014), caused SMARD1 in affected individuals. The p.Lys868ProfsTer109 and p.Arg971GlufsTer4 both results in a milder form of CMT2S, possibly because their premature termination codons (PTCs) are located in residue 977 and 975, respectively, at the 3′ end of the coding sequence, allowing for the expression of a shortened functional protein and escaping nonsense-mediated mRNA decay (NMD) of the transcripts (Lindeboom et al., 2016, 2019; Hoek et al., 2019). Therefore, we hypothesize that the complete LOF mechanism of the IGHMBP2 gene defect may be an important cause of SMARD1. However, whether LOF mutations in a single allele of the gene can cause SMARD1 is likely regulated by other complex factors.
In conclusion, this study has explored the proportion of IGHMBP2 gene variants leading to SMARD1 and CMT2S through literature search. Non-truncating variants located in two RecA-like domains (Domains 1A and 2A) of the IGHMBP2 gene were found to be the hotspots for SMARD1, while truncating variants located in the last exon of the IGHMBP gene were identified as the hotspots for CMT2S. The complete LOF mechanism of the IGHMBP2 gene defect may be an important cause of SMARD1. These findings provide further support for the diagnosis and genetic counseling of IGHMBP2-related diseases in clinical settings. However, the specific pathogenic mechanisms underlying these findings need to be investigated further in future studies.
The original contributions presented in the study are included in the article/Supplementary material, further inquiries can be directed to the corresponding author.
YT investigated and wrote the original draft. JX revised the paper. YS organized the data. EY designed the original research. All authors contributed to the article and approved the submitted version.
This study received funding from the Medical Science and Technology Joint Construction Project of Henan Province (No. LHGJ20190397).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnins.2023.1252075/full#supplementary-material
AlSaman, A., and Tomoum, H. (2010). Infantile spinal muscular atrophy with respiratory distress type 1: a case report. J. Child Neurol. 25, 764–769. doi: 10.1177/0883073809344121
Baughn, J., Gershan, W., and Rao, A. (2011). Noisy breathing and hemidiaphragm paralysis progressing to respiratory failure in an infant. Pediatr. Pulmonol. 46, 817–819. doi: 10.1002/ppul.21419
Berti, B., Buonsenso, D., De Rose, C., Ferrantini, G., De Sanctis, R., Forcina, N., et al. (2022). Point-of-care lung and diaphragm ultrasound in a patient with spinal muscular atrophy with respiratory distress type 1. J. Ultrasound 25, 395–398. doi: 10.1007/s40477-021-00584-w
Blaschek, A., Glaser, D., Kuhn, M., Schroeder, A. S., Wimmer, C., Heimkes, B., et al. (2014). Early infantile sensory-motor neuropathy with late onset respiratory distress. Neuromuscul. Disord. 24, 269–271. doi: 10.1016/j.nmd.2013.11.013
Bodle, E. E., Zhu, W., Velez-Bartolomei, F., Tesi-Rocha, A., Liu, P., and Bernstein, J. A. (2021). Combined genome sequencing and RNA analysis reveals and characterizes a deep intronic variant in IGHMBP2 in a patient with spinal muscular atrophy with respiratory distress type 1. Pediatr. Neurol. 114, 16–20. doi: 10.1016/j.pediatrneurol.2020.09.011
Bolino, A., and D'Antonio, M. (2023). Recent advances in the treatment of Charcot-Marie-Tooth neuropathies. J. Peripher. Nerv. Syst. 28, 134–149. doi: 10.1111/jns.12539
Cassini, T. A., Duncan, L., Rives, L. C., Newman, J. H., Phillips, J. A., Koziura, M. E., et al. (2019). Whole genome sequencing reveals novel IGHMBP2 variant leading to unique cryptic splice-site and Charcot-Marie-Tooth phenotype with early onset symptoms. Mol. Genet. Genom. Med. 7:e676. doi: 10.1002/mgg3.676
Chalancon, M., Debillon, T., Dieterich, K., and Commare, M. C. (2012). A rare cause of respiratory failure in infants: distal spinal-muscular atrophy 1 (DSMA1 or SMARD1). Arch. Pediatr. 19, 1082–1085. doi: 10.1016/j.arcped.2012.07.020
Chandrasekharan, S. V., Nair, S. S., Ganapathy, A., Mannan, A. U., and Sundaram, S. (2022). Charcot-Marie-Tooth disease type 2s: identical novel missense mutation of ighmbp2 gene in two unrelated families. Neurol. Sci. 43, 719–722. doi: 10.1007/s10072-021-05668-3
Cortese, A., Wilcox, J. E., Polke, J. M., Poh, R., Skorupinska, M., Rossor, A. M., et al. (2020). Targeted next-generation sequencing panels in the diagnosis of Charcot-Marie-Tooth disease. Neurology 94, e51–e61. doi: 10.1212/WNL.0000000000008672
Cottenie, E., Kochanski, A., Jordanova, A., Bansagi, B., Zimon, M., Horga, A., et al. (2014). Truncating and missense mutations in ighmbp2 cause Charcot-Marie Tooth disease type 2. Am. J. Hum. Genet. 95, 590–601. doi: 10.1016/j.ajhg.2014.10.002
Dohrn, M. F., Glockle, N., Mulahasanovic, L., Heller, C., Mohr, J., Bauer, C., et al. (2017). Frequent genes in rare diseases: panel-based next generation sequencing to disclose causal mutations in hereditary neuropathies. J. Neurochem. 143, 507–522. doi: 10.1111/jnc.14217
Eckart, M., Guenther, U. P., Idkowiak, J., Varon, R., Grolle, B., Boffi, P., et al. (2012). The natural course of infantile spinal muscular atrophy with respiratory distress type 1 (SMARD1). Pediatrics 129, e148–e156. doi: 10.1542/peds.2011-0544
Felice, K. J., Whitaker, C. H., and Khorasanizadeh, S. (2021). Diagnostic yield of advanced genetic testing in patients with hereditary neuropathies: a retrospective single-site study. Muscle Nerve 64, 454–461. doi: 10.1002/mus.27368
Giannini, A., Pinto, A. M., Rossetti, G., Prandi, E., Tiziano, D., Brahe, C., et al. (2006). Respiratory failure in infants due to spinal muscular atrophy with respiratory distress type 1. Intensive Care Med. 32, 1851–1855. doi: 10.1007/s00134-006-0346-8
Gitiaux, C., Bergounioux, J., Magen, M., Quijano-Roy, S., Blanc, T., Bonnefont, J. P., et al. (2013). Diaphragmatic weakness with progressive sensory and motor polyneuropathy: case report of a neonatal IGHMBP2-related neuropathy. J. Child Neurol. 28, 787–790. doi: 10.1177/0883073812450209
Grohmann, K., Schuelke, M., Diers, A., Hoffmann, K., Lucke, B., Adams, C., et al. (2001). Mutations in the gene encoding immunoglobulin mu-binding protein 2 cause spinal muscular atrophy with respiratory distress type 1. Nat. Genet. 29, 75–77. doi: 10.1038/ng703
Grohmann, K., Varon, R., Stolz, P., Schuelke, M., Janetzki, C., Bertini, E., et al. (2003). Infantile spinal muscular atrophy with respiratory distress type 1 (SMARD1). Ann. Neurol. 54, 719–724. doi: 10.1002/ana.10755
Guenther, U. P., Schuelke, M., Bertini, E., D'Amico, A., Goemans, N., Grohmann, K., et al. (2004). Genomic rearrangements at the ighmbp2 gene locus in two patients with smard1. Hum. Genet. 115, 319–326. doi: 10.1007/s00439-004-1156-0
Guenther, U. P., Varon, R., Schlicke, M., Dutrannoy, V., Volk, A., Hubner, C., et al. (2007). Clinical and mutational profile in spinal muscular atrophy with respiratory distress (SMARD): defining novel phenotypes through hierarchical cluster analysis. Hum. Mutat. 28, 808–815. doi: 10.1002/humu.20525
Habibi, Z. M., Eghbalkhah, A., Kamrani, K., Khosroshahi, N., Yousefimanesh, H., and Eskandarizadeh, Z. (2018). Distal spinal muscular atrophy: an overlooked etiology of weaning failure in children with respiratory insufficiency. J. Pediatr. Intensive Care 7, 159–162. doi: 10.1055/s-0037-1617434
Hamilton, M. J., Longman, C., O'Hara, A., Kirkpatrick, M., and McWilliam, R. (2015). Growing up with spinal muscular atrophy with respiratory distress (smard1). Neuromuscul. Disord. 25, 169–171. doi: 10.1016/j.nmd.2014.10.005
Han, C., Mai, J., Tian, T., He, Y., Liao, J., Wen, F., et al. (2015). Patient with spinal muscular atrophy with respiratory distress type 1 presenting initially with hypertonia. Brain and Development 37, 542–545. doi: 10.1016/j.braindev.2014.09.004
Hoek, T. A., Khuperkar, D., Lindeboom, R., Sonneveld, S., Verhagen, B., Boersma, S., et al. (2019). Single-molecule imaging uncovers rules governing nonsense-mediated MRNA decay. Mol. Cell 75, 324–339.e11. doi: 10.1016/j.molcel.2019.05.008
Jani-Acsadi, A., Ounpuu, S., Pierz, K., and Acsadi, G. (2015). Pediatric Charcot-Marie-Tooth disease. Pediatr. Clin. N. Am. 62, 767–786. doi: 10.1016/j.pcl.2015.03.012
Jedrzejowska, M., Madej-Pilarczyk, A., Fidzianska, A., Mierzewska, H., Pronicka, E., Obersztyn, E., et al. (2014). Severe phenotypes of smard1 associated with novel mutations of the IGHMBP2 gene and nuclear degeneration of muscle and Schwann cells. Eur. J. Paediatr. Neurol. 18, 183–192. doi: 10.1016/j.ejpn.2013.11.006
Joseph, S., Robb, S. A., Mohammed, S., Lillis, S., Simonds, A., Manzur, A. Y., et al. (2009). Interfamilial phenotypic heterogeneity in SMARD1. Neuromuscul. Disord. 19, 193–195. doi: 10.1016/j.nmd.2008.11.013
Kim, Y. A., Jin, H. Y., and Kim, Y. M. (2019). Diagnostic odyssey and application of targeted exome sequencing in the investigation of recurrent infant deaths in a Syrian consanguineous family: a case of spinal muscular atrophy with respiratory distress type 1. J. Korean Med. Sci. 34:e54. doi: 10.3346/jkms.2019.34.e54
Kulshrestha, R., Forrester, N., Antoniadi, T., Willis, T., Sethuraman, S. K., and Samuels, M. (2018). Charcot Marie tooth disease type 2s with late onset diaphragmatic weakness: an atypical case. Neuromuscul. Disord. 28, 1016–1021. doi: 10.1016/j.nmd.2018.09.008
Lei, L., Zhiqiang, L., Xiaobo, L., Zhengmao, H., Shunxiang, H., Huadong, Z., et al. (2022). Clinical and genetic features of Charcot-Marie-Tooth disease patients with ighmbp2 mutations. Neuromuscul. Disord. 32, 564–571. doi: 10.1016/j.nmd.2022.05.002
Lim, S. C., Bowler, M. W., Lai, T. F., and Song, H. (2012). The IGHMBP2 helicase structure reveals the molecular basis for disease-causing mutations in dmsa1. Nucleic Acids Res. 40, 11009–11022. doi: 10.1093/nar/gks792
Lin, X., Zhang, Q. J., He, J., Lin, M. T., Murong, S. X., Wang, N., et al. (2014). Variations of IGHMBP2 gene was not the major cause of Han Chinese patients with non-5q-spinal muscular atrophies. J. Child Neurol. 29:NP35-NP39. doi: 10.1177/0883073813497827
Lindeboom, R. G., Supek, F., and Lehner, B. (2016). The rules and impact of nonsense-mediated MRNA decay in human cancers. Nat. Genet. 48, 1112–1118. doi: 10.1038/ng.3664
Lindeboom, R., Vermeulen, M., Lehner, B., and Supek, F. (2019). The impact of nonsense-mediated MRNA decay on genetic disease, gene editing and cancer immunotherapy. Nat. Genet. 51, 1645–1651. doi: 10.1038/s41588-019-0517-5
Lingappa, L., Shah, N., Motepalli, A. S., and Shaik, F. (2016). Spinal muscular atrophy with respiratory distress syndrome (SMARD1): case report and review of literature. Ann. Indian Acad. Neurol. 19, 395–398. doi: 10.4103/0972-2327.168635
Litvinenko, I., Kirov, A. V., Georgieva, R., Todorov, T., Malinova, Z., Mitev, V., et al. (2014). One novel and one recurrent mutation in ighmbp2 gene, causing severe spinal muscular atrophy respiratory distress 1 with onset soon after birth. J. Child Neurol. 29, 799–802. doi: 10.1177/0883073813477203
Liu, L., Li, X., Hu, Z., Mao, X., Zi, X., Xia, K., et al. (2017). Ighmbp2-related clinical and genetic features in a cohort of Chinese Charcot-Marie-tooth disease type 2 patients. Neuromuscul. Disord. 27, 193–199. doi: 10.1016/j.nmd.2016.11.008
Luan, X., Huang, X., Liu, X., Zhou, H., Chen, S., and Cao, L. (2016). Infantile spinal muscular atrophy with respiratory distress type i presenting without respiratory involvement: novel mutations and review of the literature. Brain and Development 38, 685–689. doi: 10.1016/j.braindev.2016.02.001
Majid, A., Talat, K., Colin, L., Caroline, R., Helen, K., and Christian, D. G. (2012). Heterogeneity in spinal muscular atrophy with respiratory distress type 1. J. Pediatr. Neurosci. 7, 197–199. doi: 10.4103/1817-1745.106478
Martin, P. B., Holbrook, S. E., Hicks, A. N., Hines, T. J., Bogdanik, L. P., Burgess, R. W., et al. (2022). Clinically relevant mouse models of Charcot-Marie-Tooth type 2s. Hum. Mol. Genet. 32, 1276–1288. doi: 10.1093/hmg/ddac283
Maystadt, I., Zarhrate, M., Landrieu, P., Boespflug-Tanguy, O., Sukno, S., Collignon, P., et al. (2004). Allelic heterogeneity of smard1 at the ighmbp2 locus. Hum. Mutat. 23, 525–526. doi: 10.1002/humu.9241
McCray, B. A., and Scherer, S. S. (2021). Axonal Charcot-Marie-Tooth disease: from common pathogenic mechanisms to emerging treatment opportunities. Neurotherapeutics 18, 2269–2285. doi: 10.1007/s13311-021-01099-2
Megarbane, A., Bizzari, S., Deepthi, A., Sabbagh, S., Mansour, H., Chouery, E., et al. (2022). A 20-year clinical and genetic neuromuscular cohort analysis in Lebanon: an international effort. J. Neuromuscul. Dis. 9, 193–210. doi: 10.3233/JND-210652
Messina, M. F., Messina, S., Gaeta, M., Rodolico, C., Salpietro, D. A., Lombardo, F., et al. (2012). Infantile spinal muscular atrophy with respiratory distress type I (SMARD 1): an atypical phenotype and review of the literature. Eur. J. Paediatr. Neurol. 16, 90–94. doi: 10.1016/j.ejpn.2011.10.005
Pedurupillay, C. R., Amundsen, S. S., Baroy, T., Rasmussen, M., Blomhoff, A., Stadheim, B. F., et al. (2016). Clinical and molecular characteristics in three families with biallelic mutations in ighmbp2. Neuromuscul. Disord. 26, 570–575. doi: 10.1016/j.nmd.2016.06.457
Pejaver, V., Byrne, A. B., Feng, B. J., Pagel, K. A., Mooney, S. D., Karchin, R., et al. (2022). Calibration of computational tools for missense variant pathogenicity classification and Clingen recommendations for PP3/BP4 criteria. Am. J. Hum. Genet. 109, 2163–2177. doi: 10.1016/j.ajhg.2022.10.013
Pekuz, S., Guzin, Y., Saritas, S., Kirbiyik, O., Unalp, A., and Yilmaz, U. (2022). Spinal muscular atrophy with respiratory distress type 1 (SMARD1): a rare cause of hypotonia, diaphragmatic weakness, and respiratory failure in infants. Turk. J. Pediatr. 64, 364–374. doi: 10.24953/turkjped.2020.2012
Perego, M., Galli, N., Nizzardo, M., Govoni, A., Taiana, M., Bresolin, N., et al. (2020). Current understanding of and emerging treatment options for spinal muscular atrophy with respiratory distress type 1 (SMARD1). Cell. Mol. Life Sci. 77, 3351–3367. doi: 10.1007/s00018-020-03492-0
Pierson, T. M., Tart, G., Adams, D., Toro, C., Golas, G., Tifft, C., et al. (2011). Infantile-onset spinal muscular atrophy with respiratory distress-1 diagnosed in a 20-year-old man. Neuromuscul. Disord. 21, 353–355. doi: 10.1016/j.nmd.2011.02.005
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet. Med. 17, 405–424. doi: 10.1038/gim.2015.30
Robison, L. M., Sylvester, P. W., Birkenfeld, P., Lang, J. P., and Bull, R. J. (1998). Comparison of the effects of iodine and iodide on thyroid function in humans. J. Toxicol. Env. Health Part A 55, 93–106. doi: 10.1080/009841098158539
Rzepnikowska, W., Kaminska, J., and Kochanski, A. (2022). Validation of the pathogenic effect of ighmbp2 gene mutations based on yeast s. cerevisiae model. Int. J. Mol. Sci. 23:9913. doi: 10.3390/ijms23179913
Rzepnikowska, W., and Kochanski, A. (2021). Models for ighmbp2-associated diseases: an overview and a roadmap for the future. Neuromuscul. Disord. 31, 1266–1278. doi: 10.1016/j.nmd.2021.08.001
Saeed, M., Fawzy, W., Al-Tala, S., Magid, T. A., and Ahmed, H. (2021). Spinal muscular atrophy with respiratory distress type 1: a novel variant of ighmbp2 gene. J. Coll. Phys. Surg. 31, 1494–1496. doi: 10.29271/jcpsp.2021.12.1494
Saladini, M., Nizzardo, M., Govoni, A., Taiana, M., Bresolin, N., Comi, G. P., et al. (2020). Spinal muscular atrophy with respiratory distress type 1: clinical phenotypes, molecular pathogenesis and therapeutic insights. J. Cell. Mol. Med. 24, 1169–1178. doi: 10.1111/jcmm.14874
San, M. B., Fernandez, J. M., Navarro, C., Reparaz, A., and Teijeira, S. (2016). Spinal muscular atrophy with respiratory distress type 1 (SMARD1) report of a Spanish case with extended clinicopathological follow-up. Clin. Neuropathol. 35, 58–65. doi: 10.5414/NP300902
Surrey, V., Zoller, C., Lork, A. A., Moradi, M., Balk, S., Dombert, B., et al. (2018). Impaired local translation of beta-actin MRNA in ighmbp2-deficient motoneurons: implications for spinal muscular atrophy with respiratory distress (SMARD1). Neuroscience 386, 24–40. doi: 10.1016/j.neuroscience.2018.06.019
Taiana, M., Govoni, A., Salani, S., Kleinschmidt, N., Galli, N., Saladini, M., et al. (2022). Molecular analysis of SMARD1 patient-derived cells demonstrates that nonsense-mediated MRNA decay is impaired. J. Neurol. Neurosurg. Psychiatry 93, 908–910. doi: 10.1136/jnnp-2021-326425
Tomaselli, P. J., Horga, A., Rossor, A. M., Jaunmuktane, Z., Cortese, A., Blake, J. C., et al. (2018). IGHMBP2 mutation associated with organ-specific autonomic dysfunction. Neuromuscul. Disord. 28, 1012–1015. doi: 10.1016/j.nmd.2018.08.010
Tsang, M., Chiu, A., Kwong, B., Liang, R., Yu, M., Yeung, K. S., et al. (2020). Diagnostic value of whole-exome sequencing in Chinese pediatric-onset neuromuscular patients. Mol. Genet. Genom. Med. 8:e1205. doi: 10.1002/mgg3.1205
Vadla, G. P., Ricardez, H. S., Mao, J., Garro-Kacher, M. O., Lorson, Z. C., Rice, R. P., et al. (2023). ABT1 modifies SMARD1 pathology via interactions with ighmbp2 and stimulation of ATPase and helicase activity. JCI Insight 8:e164608. doi: 10.1172/jci.insight.164608
Viguier, A., Lauwers-Cances, V., Cintas, P., Manel, V., Peudenier, S., Desguerre, I., et al. (2019). Spinal muscular atrophy with respiratory distress type 1: a multicenter retrospective study. Neuromuscul. Disord. 29, 114–126. doi: 10.1016/j.nmd.2018.10.002
Wagner, J. D., Huang, L., Tetreault, M., Majewski, J., Boycott, K. M., Bulman, D. E., et al. (2015). Autosomal recessive axonal polyneuropathy in a sibling pair due to a novel homozygous mutation in ighmbp2. Neuromuscul. Disord. 25, 794–799. doi: 10.1016/j.nmd.2015.07.017
Wong, V. C., Chung, B. H., Li, S., Goh, W., and Lee, S. L. (2006). Mutation of gene in spinal muscular atrophy respiratory distress type i. Pediatr. Neurol. 34, 474–477. doi: 10.1016/j.pediatrneurol.2005.10.022
Wu, S., Chen, T., Li, Y., Chen, L., Xu, Q., Xiao, F., et al. (2018). An atypical phenotype of a patient with infantile spinal muscular atrophy with respiratory distress type 1 (SMARD 1). Eur. J. Med. Genet. 61, 602–606. doi: 10.1016/j.ejmg.2018.04.001
Xiao, M. J., Li, X., Li, L. F., Xie, Z. H., Zhang, Y. D., Zhang, C., et al. (2022). A case of Charcot-Marie-Tooth disease type 2s caused by mutation of ighmbp2 gene. Zhonghua Er Ke Za Zhi 60, 62–63. doi: 10.3760/cma.j.cn112140-20210825-00701
Xie, Y., Lin, Z., Liu, L., Li, X., Huang, S., Zhao, H., et al. (2021). Genotype and phenotype distribution of 435 patients with Charcot-Marie-Tooth disease from central South China. Eur. J. Neurol. 28, 3774–3783. doi: 10.1111/ene.15024
Yasui, Y., Sato, H., Niida, Y., and Kohno, M. (2019). Spinal muscular atrophy with respiratory distress type 1 associated with novel compound heterozygous mutations in IGHMBP2: differential diagnosis in a case with congenital diaphragm eventration. Congenit. Anom. 59, 22–23. doi: 10.1111/cga.12280
Yuan, J. H., Hashiguchi, A., Yoshimura, A., Yaguchi, H., Tsuzaki, K., Ikeda, A., et al. (2017). Clinical diversity caused by novel IGHMBP2 variants. J. Hum. Genet. 62, 599–604. doi: 10.1038/jhg.2017.15
Keywords: IGHMBP2 gene, spinal muscular atrophy with respiratory distress type 1, Charcot-Marie-Tooth disease, mutations, clinical diagnosis
Citation: Tian Y, Xing J, Shi Y and Yuan E (2023) Exploring the relationship between IGHMBP2 gene mutations and spinal muscular atrophy with respiratory distress type 1 and Charcot-Marie-Tooth disease type 2S: a systematic review. Front. Neurosci. 17:1252075. doi: 10.3389/fnins.2023.1252075
Edited by:
Laura Ibanez, Washington University in St. Louis, United StatesReviewed by:
Hisahide Nishio, Kobe Gakuin University, JapanCopyright © 2023 Tian, Xing, Shi and Yuan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Enwu Yuan, eXVhbmVud3VAMTYzLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.