 Yajun Shi
Yajun Shi Yan Zhao2†
Yan Zhao2† Likui Lu
Likui Lu Dongyi Yu
Dongyi Yu Miao Sun
Miao Sun
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Neurosci. , 06 July 2023
Sec. Neurodegeneration
Volume 17 - 2023 | https://doi.org/10.3389/fnins.2023.1223777
This article is part of the Research Topic Application of Gene Editing in Neurodegenerative Diseases Volume II View all 5 articles
Amyotrophic lateral sclerosis (ALS) is a deadly neurological disease with a complicated and variable pathophysiology yet to be fully understood. There is currently no effective treatment available to either slow or terminate it. However, recent advances in ALS genomics have linked genes to phenotypes, encouraging the creation of novel therapeutic approaches and giving researchers more tools to create efficient animal models. Genetically engineered rodent models replicating ALS disease pathology have a high predictive value for translational research. This review addresses the history of the evolution of gene editing tools, the most recent ALS disease models, and the application of CRISPR/Cas9 against ALS disease.
Amyotrophic lateral sclerosis (ALS), also known as Lou Gehrig’s disease, is a progressive neurodegenerative disease that destroys motor neurons (MNs) in the spinal cord, brainstem, and motor cortex. ALS within 2–5 years of the onset of symptoms, leading to muscle weakening, atrophy, paralysis, respiratory failure, and death (Marcus, 2022). Additionally, ALS is one of the most common adult motor neuron diseases, affecting 2–3 persons out of every 100,000 individuals globally (Oskarsson et al., 2018; Feldman et al., 2022). Evidence shows that the prevalence of ALS has been rising in recent years, which may be largely attributed to the advancements in clinical services that allow precise diagnosis (Logroscino et al., 2018). However, ALS is still incurable. Current medications, such as Riluzole, prolong patient survival by an average of 3 months but are only modestly beneficial, highlighting the need for solutions to treat this dangerous condition.
Most cases of ALS are sporadic and have an unknown etiology, whereas approximately 10 ~ 15% of patients have a family history of the disease, strongly indicating a genetic predisposition (Sahdeo and Goldstein, 2021). The genetic etiology of ALS has been linked to more than 50 genes that include causal or disease-modifying variations (Taylor et al., 2016; Giovannelli et al., 2023). The most commonly investigated ALS genes, including superoxide dismutase (SOD1), chromosome 9 open reading frame 72 (C9orf72), TAR DNA-binding protein 43 (TDP43), and RNA binding protein fused in sarcoma (FUS), account for roughly 75% of familial ALS cases (Owens, 2017; Fang et al., 2022). Our understanding of the pathophysiology of ALS has significantly increased due to the analysis of the molecular pathways underlying these mutant ALS genes, revealing new information about prospective therapeutic targets. Accordingly, developing models of ALS and targeting the mutant genes (i.e., gene therapy) has been investigated worldwide to translate these gene therapies into the clinical setting.
With a corrected pathogenetic mutation, gene editing offers a potential treatment for ALS, and it is now beginning to be used in clinical settings (Mullard, 2019). In particular, the genome-editing technology known as clustered regularly interspaced short palindromic repeats/CRISPR-associated system 9 (CRISPR/Cas9) has gained attention for its possible benefits in the management and treatment of ALS (Cho et al., 2013; Yamashita et al., 2013). Compared to other genome editing techniques like transcription activator-like effector nucleases (TALENs) and zinc finger nucleases (ZFNs), CRISPR/Cas9 is easy to use and reasonably priced (Yang et al., 2017; Bhardwaj et al., 2022; Meijboom et al., 2022). In this review, we emphasize the history of the emergence of gene editing tools and highlight the developments in CRISPR/Cas9 mediated gene correction and disease models gene therapy for ALS disease.
Gene editing constantly evolves to provide more precise, diverse, and large-scale implementations, allowing us to gain innovative insights into the biological field. Previously, homologous recombination (HR) and non-homologous end-joining (NHEJ) were used to carry out genetic engineering. Due to its low efficacy in many cell types and animals, we still need another alternative. Currently, there exist three main gene editing technologies: ZNFs, TALENs, and the CRISPR/Cas system, among which CRISPR/Cas system is now the most predominant technology.
Flavobacterium okeanokoites (FokI) and zinc finger structure, which promoted further development of gene editing, were discovered in the 1990s (Li et al., 1992). Zinc finger proteins region with conserved sequences are arranged in a certain order, containing three to six Cys2-His2 fingers, followed by attachment of FokI to the end of the protein. Zinc fingers are transcription factors, where each finger recognizes 3–4 bases. Ultimately, ZFNs recognized a 9–18 bp sequence (Liu et al., 1997). After ZFNs bind to DNA in DNA-binding domains, the FokI nuclease induces cleavage as a dimer, resulting in double-stranded breaks (DSB) in the target loci. Then, HR and NHEJ are activated to complete gene editing through the intracellular DNA repair mechanism (Nakashima et al., 2022). Theoretically, ZFNs recognize almost all 64 possible nucleotide triplets, but the specificity and affinity of ZNFs are still issues. Additionally, ZFNs are reported to exert a significant cytotoxic effect because of the off-target cleavages (Gupta et al., 2013).
Like ZFNs, TALENs are artificial chimeric proteins engineered by fusing a non-specific FokI endonucleases domain to a DNA-binding domain recognizing an arbitrary base sequence (Zhang et al., 2019). The TALENs are well known for their unique DNA-binding domain in their central region, conserved tandem between 7 and 34 homologously repeated amino acid residues. Two residues located at positions 12 and 13, called repeat variable di-residues (RVD), are highly variable residues responsible for recognizing a specific nucleotide (Agarwal and Gupta, 2021).
Unlike ZFNs, TALENs had the advantage that they could bind to just one nucleotide inside its DNA-binding domains rather than three, resulting in enhanced specificity and decreasing in the off-target events (Table 1). TALENs, cheaper, safer, more efficient, and easier to engineer than zinc-finger proteins, can be applied more widely to life sciences than ZFNs (Huo et al., 2019). This gene-editing tool was utilized for various cell lines, including induced pluripotent stem cells (iPSCs) (Kwon et al., 2021; Qiu et al., 2021; Siles et al., 2023), nematodes (Verdu et al., 2020, 2021), zebrafish (Zhu et al., 2021; Sertori et al., 2022), and generating knockouts in rodent models (Lee et al., 2021). Despite the improvement, simplification, and high specificity of TALENs, they have an obvious limitation: being quite large (impeding delivery). This limitation makes it difficult to deliver and express a pair of TALENs into the cells of interest because they cannot be packaged in vectors of their limited cargo capacity, such as adeno-associated viruses (AAVs), or delivered as RNA molecules (Khan, 2019; Jo et al., 2022).
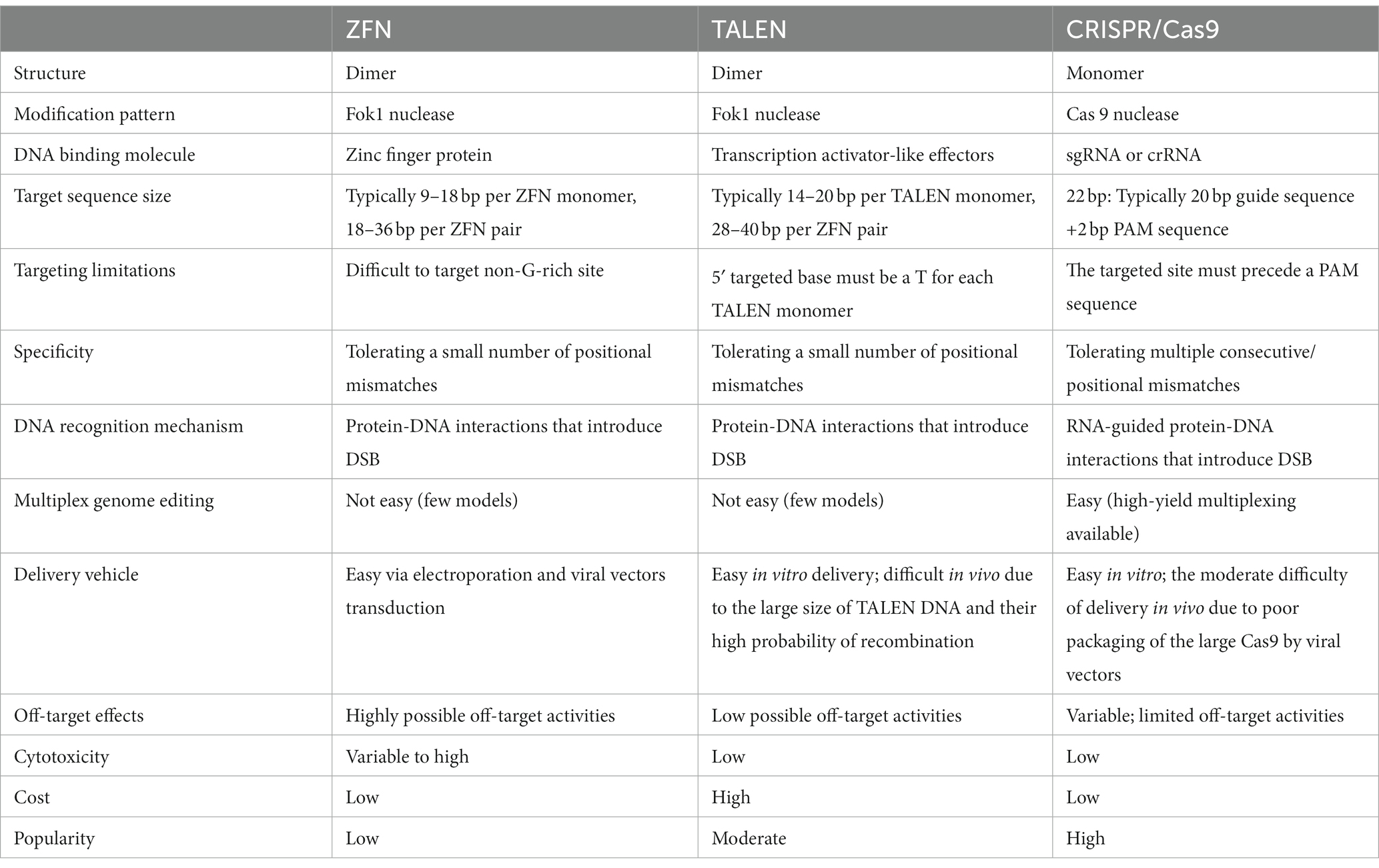
Table 1. Comparison of the three currently used gene editing platforms: ZFN, TALEN, and CRISPR/Cas9.
The CRISPR/Cas system, whose gene editing ability was confirmed in human cells in 2013 (Cho et al., 2013), is the most recent platform in genome editing. Unlike nucleases that recognize the target sequence through protein-DNA interaction, CRISPR-Cas systems target sequences through RNA and DNA base pairing (Zhang et al., 2019) (Table 1). Naturally occurring CRISPR-Cas systems can be categorized into two classes based on the Cas genes’ structural variations and their organization styles (Makarova et al., 2015). The type II CRISPR/Cas9 system, which depends on a single Cas protein from streptococcus pyogenes (SpCas9) targeting particular DNA sequences and has been adopted for gene editing applications, is the most frequently used subtype of CRISPR systems (Jiang et al., 2013). Mechanistically, CRISPR/Cas9 systems have two components: single-guide RNA (sgRNA) and Cas9 endonuclease. Usually, the sgRNA was designed near the adjacent protospacer motif (PAM) and bound to the target DNA sequence by Watson–Crick base pairing. Then Cas9 precisely cleaves the DNA to generate a DSB (Hu et al., 2018). These DSBs are commonly repaired by NHEJ or HDR, resulting in gene disruption and inactivation of the targeted gene (Lee et al., 2016) (Figure 1). The sgRNA and Cas protein-coding gene are usually delivered via transfection and packaging into AAVs or lentivirus (Kenjo et al., 2021; Ling et al., 2021; Nahmad et al., 2022).
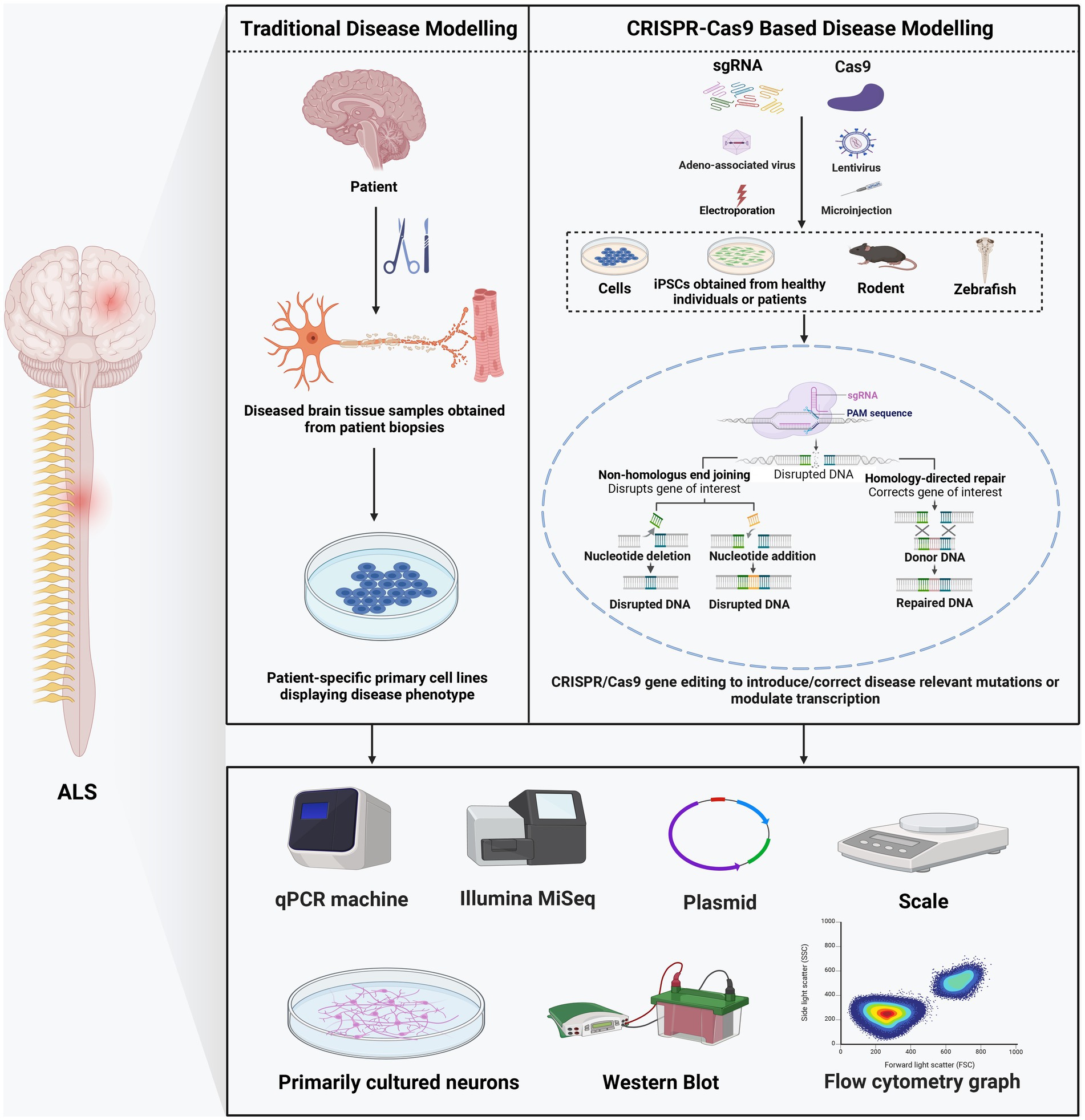
Figure 1. Comparison of traditional and CRISPR/Cas9-based disease modeling.
Although there are still several issues that remain to be resolved in CRISPR/Cas9 system, including well-known off-target effects, cytotoxicity with DNA damage, chromosomal rearrangement and frequent p53 activation (Hendriks et al., 2020; Bravo et al., 2022). However, compared to other genome editing technologies, CRISPR/Cas9 system is easier, more scalable, and more efficient (Li et al., 2020). Therefore, it is frequently used to create animal models or to treat serious, incurable human diseases (Li et al., 2021; Baskoylu et al., 2022).
Although promising, this strategy raises potential safety concerns as it relies on repairing the DSBs and NHEJ created by CRISPR/Cas9, which may cause deleterious large deletions, chromosomal rearrangements, or undesirable mutagenic outcomes (Zhao et al., 2022). To overcome this limitation and improve the safety and applicability of the CRISPR/Cas9 system, a new paradigm-shifting class of genome editing technology, CRISPR single-base editors, have recently been developed. CRISPR single-base editors may precisely and directly insert point mutations into chromosomal DNA without creating DSBs by combining the Cas9 nickase with nucleobase deaminases (Caso and Davies, 2022). To date, two main kinds of base editors have been developed: adenine base editors (ABEs), which catalyze A/T to G/C conversions, and cytosine base editors (CBEs), which catalyze the conversion of G/C base pairs to T/A base pairs (Anzalone et al., 2020). However, base editors cannot make multiple base pair insertions and deletions. When many As or Cs are present around the target site, they might inadvertently become mutated and created off-target effects. To overcome this limitation, termed prime editing, which could insert longer stretches of DNA, has been developed recently. Base editors have been applied to various cell types and organisms, including animal models of human genetic diseases (Kawamata et al., 2023).
Given that there are no ethical considerations, the experimental time is relatively short, and the expense is minimal, cell models are commonly used to explore many neurological illnesses, including ALS. Furthermore, recent advances in CRISPR/Cas9 technology allow researchers to precisely target and introduce modifications in the genomic DNA of different cell lines (Kramer et al., 2018; Karginov et al., 2023). The introduction of the CRISPR/Cas9 system has facilitated the development of numerous ALS cell models, such as HT22, iPSCs, BV2, Neuro 2a, NSC-34, hESC, and HeLa cell lines, etc. For example, Liu et al. (2022) recently used CRISPR/Cas9 gene-editing technology to create an astrocyte elevated gene-1 (AEG-1) deficient HT22 cell model that can be used to study AEG-1 related ALS. In addition, Zhang et al. (2022) used CRISPR/Cas9 to introduce a UBQLN2 P497H mutation into a healthy control iPSC line. Following this, the mutated iPSC line could differentiate into motor neurons, which can help comprehend the pathology of UBQLN2-related ALS (Zhang et al., 2022). Importantly, CRISPR-iPSC-based disease modeling could expedite research on ALS (Sen and Thummer, 2022) by targeting SOD1 (Kim et al., 2020), TDP-43 (Burkhardt et al., 2013), FUS (Wang et al., 2017), and C9ORF72 (Cali et al., 2019; Ababneh et al., 2020; Krishnan et al., 2020) genes, etc. Furthermore, Strohm et al. (2022) also made CRISPR/Cas9-engineered HeLa cells with ALS UBQLN2 mutations to determine how UBQLN2 mutations cause ALS. ALS can be caused by OPTN gene mutations, which result in cytoplasmic mislocalization, ubiquitination, and accumulation of nuclear TDP-43. Prtenjaca et al. (2022) used CRISPR/Cas9 methodology to create OPTN knockout (KO) cell lines for Neuro 2a and NSC-34 neurons and BV2 microglia to investigate the relationship between optineurin and TDP-43. Their research showed that OPTN mutations might lead to elevated levels of TDP-43 protein in microglia when optineurin is absent (Prtenjaca et al., 2022). In addition, Zhang et al. (2022) created a heterozygous FUS-Q290X knock in the human embryonic stem cell (hESC) line, which will be a valuable tool for investigating the disease mechanisms of ALS using the CRISPR/Cas9 system.
CRISPR/Cas9 technology is not only helpful in creating ALS cell models but also in creating ALS animal models. For instance, ALS can be caused by mutations in valosin-containing protein (VCP). On the other hand, there is no constitutive VCP knockout animal model due to embryonic lethality. Voisard et al. (2022) developed a constitutive CRISPR/Cas9-induced VCP knockout zebrafish model. Furthermore, Wall et al. (2021) created the Drosophila knockin models using the CRISPR/Cas9 technique to investigate VCP diseases (including multisystem proteinopathy and ALS) (Wall et al., 2021). To investigate the pathogenic effects of TP73 mutations in ALS, Russell et al. (2021) utilized CRISPR/Cas9 technology targeting TP73 in zebrafish and assessed motor neuron quantity and axonal structure. Their findings revealed that knocking out TP73 in zebrafish using CRISPR/Cas9 resulted in disrupted motor neuron development and abnormal axonal morphology, indicative of ALS pathology (Russell et al., 2021). Additionally, Bose et al. (2019) developed a TDP-43 loss-of-function model in zebrafish using the CRISPR/Cas9 system. Moreover, to examine mechanisms underlying mutations in FUS that lead to neuronal dysfunction in ALS patients, the C. elegans knockin models were generated by Baskoylu et al. (2022) using CRISPR-Cas9-mediated genome editing. Their findings suggested impaired autophagy might play a role in the neuromuscular defects and protein homeostasis associated with ALS FUS knockin animals (Baskoylu et al., 2022). As for the FUS gene, Zhang et al. (2018) developed a novel knockin rat model expressing a Fus point mutation (R521C) via CRISPR/Cas9. They discovered that FUS might regulate sleep, circadian rhythms, and learning and memory behavior (Zhang et al., 2018). Degeneration of neurons in many ALS subtypes has been linked to DNA damage and defective repair, but the underlying mechanisms are unclear. The role of TDP-43 in DNA damage response (DDR) has not been explored, despite growing evidence suggesting the involvement of other RNA/DNA binding proteins like FUS in DDR (Wang et al., 2018). Mitra et al. (2019) demonstrated that the loss of nuclear TDP-43 is linked to DNA double-strand break repair defects and DDR signaling in ALS by using CRISPR/Cas9-mediated conditional depletion of TDP-43 in C. elegans. Also, Baskoylu et al. (2018) made ALS knockin models in C. elegans by editing the endogenous Sod-1 gene with CRISPR/Cas9 and discovered that knockin models could replicate the neurotransmitter-type specificity of ALS. Using the CRISPR/Cas9 system, Kao et al. (2020) created MATR3 S85C knockin mice that mimic early-stage ALS and would be useful in future basic and preclinical research. Sporadic ALS patients were found to have mutations in the CREST gene (also called SS18L1), which acts as a calcium-regulated transcriptional activator. Loss of CREST causes neuroinflammatory responses and ALS-like motor defects in mice, as demonstrated by the work of Cheng et al. (2019), who used the CRISPR/Cas9 system to create CREST knockout and Q394X knockin mice. In addition, C9orf72-deficient mice (Sullivan et al., 2016) have been successfully established by using the CRISPR/Cas9 system due to the recent discovery of hexanucleotide repeat expansion in the C9orf72 gene as a major cause of frontotemporal lobar degeneration (FTLD) with ALS (DeJesus-Hernandez et al., 2011).
Researching the etiology and developing treatments for human diseases requires animal models. The discovery of specific gene mutations that cause neurodegenerative disorders has led to the development of numerous models in small animals with symptoms similar to those experienced by people with the diseases. Genetically modified rodents are today’s most widely used animal models because they apply to explore pathogenesis. However, most genetically modified rodent models do not exhibit overt neurodegeneration, creating difficulties in using them to rigorously test the effects of therapies on neurodegeneration (Philips and Rothstein, 2015). Recent research using CRISPR/Cas9-targeted large animals (such as pigs and monkeys) (Li et al., 2021; Yang et al., 2022) has uncovered important pathological events that resemble neurodegeneration in the patient’s brain. Still, these events could not be produced in small animal models. Injections of multiple sgRNAs are more effective than injections of a single type of sgRNA at a high concentration for achieving complete gene knockout. The complete gene knockout by the CRISPR/Cas9 system is conducive to reducing the test times of large animals and ensuring their accuracy, which is strongly supported by ethical considerations (Zuo et al., 2017). Although no ALS models in large animals have been developed so far using CRISPR/Cas9 technology, it is essential to create a large animal ALS model using this approach to gain more knowledge about the pathogenesis of ALS.
CRISPR/Cas9 system, used in cells and animal models, is a promising tool for modeling and treating genetic diseases (Table 2). Compared with cellular-level studies, animal models are more favorable for exploring the pathogenic gene and molecular pathways of diseases at the organismal level. For ALS, definite treatments are still lacking. Here, we mainly focus on CRISPR/Cas9 system applications in ALS. The great advantage of this strategy is that correction of the mutant DNA eliminates abnormal downstream pathways and is a one-time intervention theoretically. Currently, limited studies have been accomplished in the ALS field.
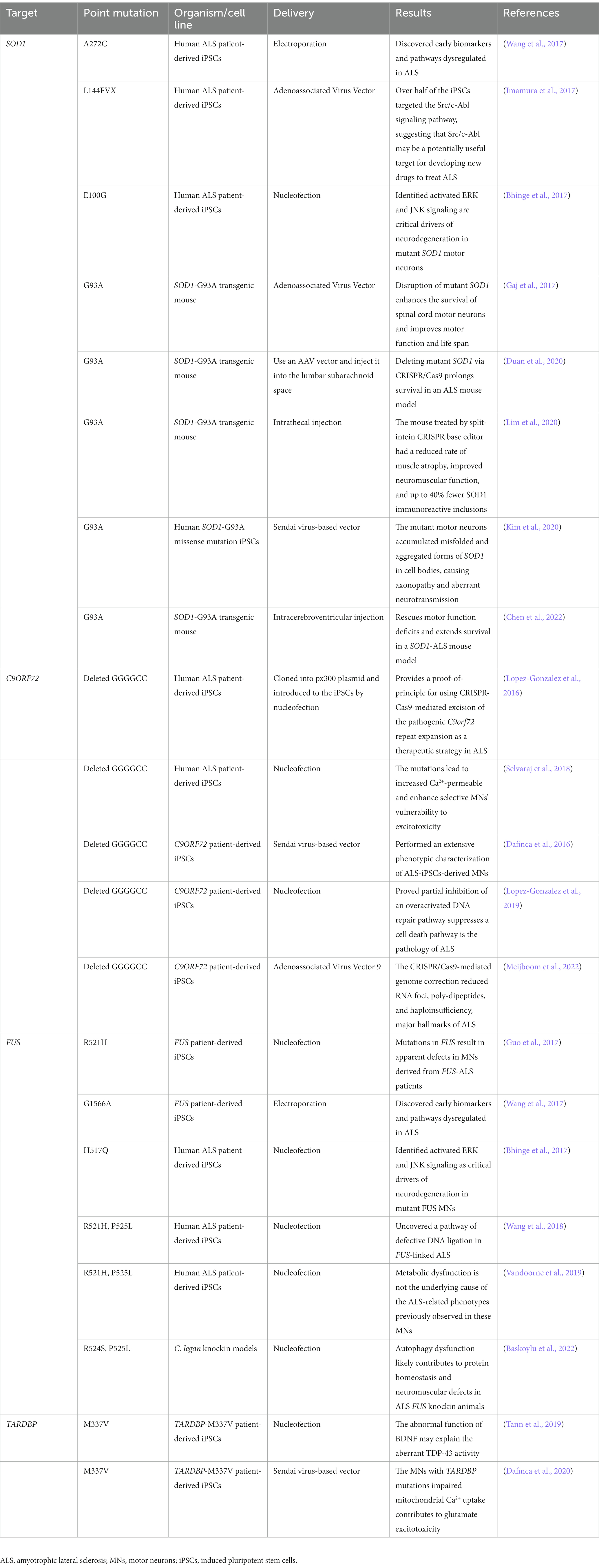
Table 2. Studies of the applications of CRISPR/Cas9-mediated genome editing in ALS.
Several cells and animal models targeted on SOD1 have been reported. A sgRNA was applied to disrupt SOD1 via the facial vein, leading to decreased expression of SOD1 protein in the spinal cord, reduced muscle atrophy, and delayed disease onset in mice models. With the improvement of motor neuron function, the survival time increased by 28–30 days (Gaj et al., 2017). Other favorable results of SOD1 decrease in the spinal cord were reported in three studies, which showed increased survivability (Lim et al., 2020; Chen et al., 2022). The CRISPR/Cas9 technology that can be used to validate the efficacy of rescuing iPSC-derived motor neuron cells from the harmful effects of the SOD1 mutation has been described in several articles (Kim et al., 2020; Deng et al., 2021). Wang et al. (2017) generated two human iPSC lines from ALS patients, SOD1+/A272C and SOD1+/C14T heterozygous disease-causing mutations. Then, they generated their respective isogenic disease-free iPSCs by CRISPR/Cas9 mediated gene correction (Wang et al., 2017). This study may help identify biomarkers and pathways for ALS and offers new evidence for ALS therapy. Additionally, a recent study showed that CRISPR/Cas9-mediated gene editing is an effective strategy to target SOD1, leading to a disease-free condition in two ALS mouse models (Deng et al., 2021). The main limitation of these studies is that the mice were treated at a young age before exhibiting symptoms of ALS. Therefore, the treatment’s effectiveness would be uncertain in older mice with ALS phenotype.
In drug screening assay, Imamura et al. (2017) used CRISPR/Cas9 mediated SOD1 corrected iPSCs lines generated from one ALS patient carrying the L144FVX mutation in the SOD1 gene and homologous arms corrected mutation as a control. The corrected cell line demonstrated a reversal of SOD1-related pathologic phenotypes, including increased motor neuron survival, reserved internal ATP level, and breakdown of misfolded SOD1 protein. They also found that bosutinib, one of the specific Src/c-Abl inhibitors, could modestly extend the survival of the SOD1 mutation mouse model of ALS, suggesting that Src/c-Abl may be a potential target for developing new drugs to treat ALS (Imamura et al., 2017).
C9orf72 mutation, leading to an expansion of GGGGCC (G4C2) hexanucleotide repeat expansion (HRE) in the first intron, is the most significant gene discovery for ALS (Gijselinck et al., 2012). The size of the hexanucleotide sequence is less than 24 repeats in a healthy person but can number in the thousands of repetitions in affected individuals (Iacoangeli et al., 2019). Multiple studies have demonstrated that the C9orf72 mutations affect subsequent protein synthesis and cause premature neuronal death by causing harmful either a gain or loss of function, such as impaired clearance of dipeptide proteins and excitotoxicity from the accumulation of glutamate receptors (Ghasemi et al., 2021; Meijboom et al., 2022). Therefore, reducing the expression of C9orf72 is a potentially promising target in halting ALS disease.
The field of gene editing, especially the CRISPR/Cas9 system, offers potential treatment modalities for ALS pathology by physically excising the repeat expansion mutation from C9orf72 at the genetic level. Meijboom et al. (2022) administer AAV9 vectors containing gRNA and Cas9 into the bilateral striatum of aged 2–3 months C9orf72-associated mice, which rescues partial ALS phenotypes. Respectively, two separate studies also reported the utility of CRISPR/Cas9 in targeting either (G4C2) repeat DNA (Pinto et al., 2017) or RNA to reduce repeat RNA transcription and decreased myotonia in the ALS mouse model. These findings are consistent with other reports demonstrating the positive benefit of excising the HRE in C9orf72 (Ababneh et al., 2020; Piao et al., 2022), indicating potentially viable strategies for therapeutic intervention.
The mutation of the FUS gene, a DNA/RNA-binding protein, in chromosome 16 was discovered as a causative factor for ALS in 2009. Serval of mutations in the FUS gene have been found in motor neurons of ALS patients, like C1574T, A1564G, G1566A, etc. (Lattante et al., 2013; Du et al., 2015), and these mutations cause up to 4% of familial ALS (Mackenzie et al., 2010). Investigating the gene editing of FUS by the CRISPR/Cas9 system will provide a platform for revealing novel therapeutic entry points.
Wang et al. (2017) first successfully corrected the FUS+/G1566A mutations in ALS iPSCs by CRISPR/Cas9 system, and the repaired ALS iPSCs possessed normal pluripotency. Furthermore, Bhinge et al. (2017) obtained iPSCs from ALS patients for recessive H517Q mutation in the FUS gene and corrected this recessive mutation to wild-type using the CRISPR/Cas9 system. Their results confirmed that both p38 and ERK kinase are activated in mutant FUS, indicating that MAPK activation is a key pathway in FUS mutation in ALS (Bhinge et al., 2017). Additionally, Guo et al. (2017) proved that the different mutations in the FUS gene of motor neurons caused typical cytoplasmic hyperexcitability and progressive axonal transport defects, which were rescued by CRISPR/Cas9-mediated genetic correction of the FUS mutation in patient-derived iPSCs. As the above results indicated, CRISPR/Cas9 system provides a platform for revealing novel therapeutic entry points.
Although the specific mechanisms by which TDP-43 protein, encoded by TARDBP, cause ALS is unknown, numerous studies have shown that a substantial fraction of ALS patients features abnormalities in TDP-43, whose function losses in the nucleus and gain of toxic in the cytoplasm (Kuo et al., 2014; De Conti et al., 2015). In addition, TDP-43 may involve stress granule formation and regulation of neurite growth (Shelkovnikova et al., 2018; Zuo et al., 2021; McMillan et al., 2023), and the reduction of cytoplasmic TDP-43 levels can rescue neurons from death (Walker et al., 2015). Brown et al. (2022) revealed evidence that TDP-43 is a promising therapeutic target for ALS.
Currently, the application of CRISPR/Cas9-mediated genetic correction targeting TARDBP gene therapy is limited. To investigate the mechanisms of TARDBP mutations in ALS, Tann et al. (2019) confirmed that hippocampal CA1-specific absence of TDP-43 enhanced the expression of Sod1 mRNA and reduced dendrite complexity and spine. They also used CRISPR/Cas9 to correct M337V in three iPSC lines from ALS patients, and then these iPSCs were differentiated into excitatory cortical/hippocampal-like neurons. Their study indicated that M337V mutation in TARDBP is a direct cause of impaired BDNF secretion, essential for neuron survival, differentiation, and synaptic plasticity (Tann et al., 2019).
Although, four therapies (riluzole, edaravone, sodium phenylbutyrate and taurursodiol, and dextromethorphan-quinidine) have been approved now, but they could not stops or reverses disease progression (Meijboom and Brown, 2022). Using ALS as the search term, 969 clinical trials were gathered from clini-caltrials.gov on 20 June 2023. Unfortunately, the bulk of ALS clinical trials have failed to show any promising outcomes during the past two decades. This is partially caused by the therapeutic agents’ low bioavailability, ineffective therapeutic delivery to the CNS, difficulties with therapeutic administration, dearth of clinically relevant disease models, lack of late diagnosis, effective biomarkers, and inadequate understanding of the underlying molecular mechanisms causing the disease (Johnson et al., 2022). Therefore, new therapies and diagnostics are urgently needed to help with therapy and early diagnosis. All the current worldwide Phase III ALS clinical trials that are being summarized in Table 3.
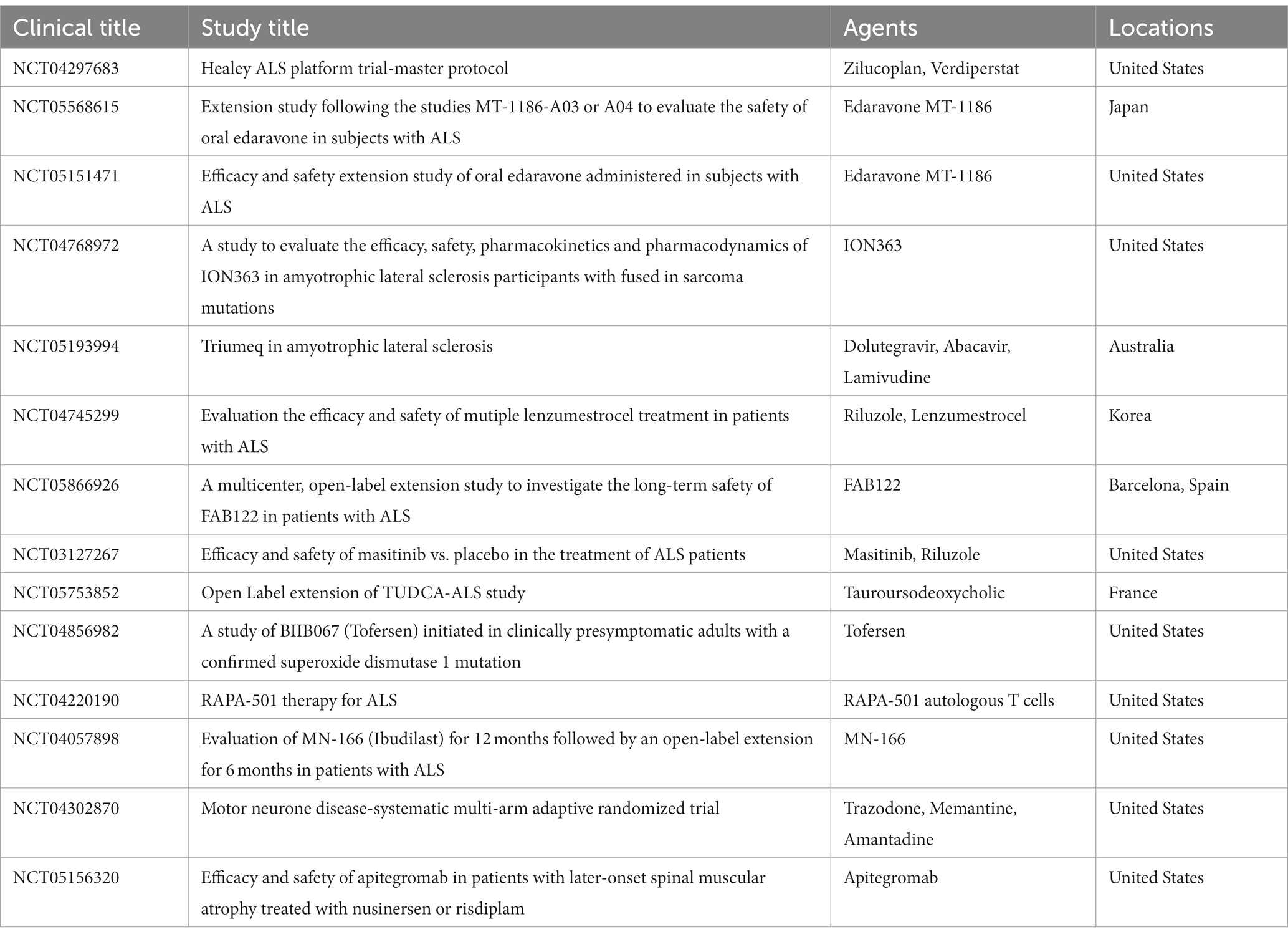
Table 3. Ongoing Phase III Clinical Trials on ALS Worldwide.
We discussed numerous recent studies that have implicated gene editing, especially CRISPR/Cas9, to investigate the pathophysiology of ALS by animal models or iPSCs in this review. There remain limitations with these approaches, such as off-target effects, limited size of delivery vehicle, and low efficiency of gene correction through the HDR mechanism (Modell et al., 2022).
Over 50 genes, in which SOD1, C9orf72, TARDBP, and FUS are commonly genetic alterations, are listed as ALS-associated genes. Studies using CRISPR/Cas9-mediated in vivo genome editing of SOD1, C9orf72, TARDBP, and FUS functions show that this technique can identify the underlying molecular mechanism and reverse the pathologic phenotype in patient-derived disease models. These studies suggest that CRISPR/Cas9 system may play a potential role in gene therapy for ALS. However, more thorough studies are still needed to investigate the various underlying mechanisms of ALS to determine the treatment for ALS.
YS, YZ, and LL wrote the manuscript with support from QG, DY, and MS. All authors contributed to the article and approved the submitted version.
This work was supported by grants from the National Key R&D Program of China (2022YFC2703700 and 2019YFA0802600) and the National Natural Science Foundation of China (81974244 and 81570960).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Ababneh, N. A., Scaber, J., Flynn, R., Douglas, A., Barbagallo, P., Candalija, A., et al. (2020). Correction of amyotrophic lateral sclerosis related phenotypes in induced pluripotent stem cell-derived motor neurons carrying a hexanucleotide expansion mutation in C9orf72 by CRISPR/Cas9 genome editing using homology-directed repair. Hum. Mol. Genet. 29, 2200–2217. doi: 10.1093/hmg/ddaa106
Agarwal, N., and Gupta, R. (2021). History, evolution and classification of CRISPR-Cas associated systems. Prog. Mol. Biol. Transl. Sci. 179, 11–76. doi: 10.1016/bs.pmbts.2020.12.012
Anzalone, A. V., Koblan, L. W., and Liu, D. R. (2020). Genome editing with CRISPR-Cas nucleases, base editors, transposases and prime editors. Nat. Biotechnol. 38, 824–844. doi: 10.1038/s41587-020-0561-9
Baskoylu, S. N., Chapkis, N., Unsal, B., Lins, J., Schuch, K., Simon, J., et al. (2022). Disrupted autophagy and neuronal dysfunction in C. elegans knockin models of FUS amyotrophic lateral sclerosis. Cell Rep. 38:110195. doi: 10.1016/j.celrep.2021.110195
Baskoylu, S. N., Yersak, J., O’Hern, P., Grosser, S., Simon, J., Kim, S., et al. (2018). Single copy/knock-in models of ALS SOD1 in C. elegans suggest loss and gain of function have different contributions to cholinergic and glutamatergic neurodegeneration. PLoS Genet. 14:e1007682. doi: 10.1371/journal.pgen.1007682
Bhardwaj, S., Kesari, K. K., Rachamalla, M., Mani, S., Ashraf, G. M., Jha, S. K., et al. (2022). CRISPR/Cas9 gene editing: new hope for Alzheimer’s disease therapeutics. J. Adv. Res. 40, 207–221. doi: 10.1016/j.jare.2021.07.001
Bhinge, A., Namboori, S. C., Zhang, X., VanDongen, A. M. J., and Stanton, L. W. (2017). Genetic correction of SOD1 mutant iPSCs reveals ERK and JNK activated AP1 as a driver of neurodegeneration in amyotrophic lateral sclerosis. Stem Cell Rep. 8, 856–869. doi: 10.1016/j.stemcr.2017.02.019
Bose, P., Armstrong, G. A. B., and Drapeau, P. (2019). Neuromuscular junction abnormalities in a zebrafish loss-of-function model of TDP-43. J. Neurophysiol. 121, 285–297. doi: 10.1152/jn.00265.2018
Bravo, J. P. K., Liu, M. S., Hibshman, G. N., Dangerfield, T. L., Jung, K., McCool, R. S., et al. (2022). Structural basis for mismatch surveillance by CRISPR-Cas9. Nature 603, 343–347. doi: 10.1038/s41586-022-04470-1
Brown, A. L., Wilkins, O. G., Keuss, M. J., Hill, S. E., Zanovello, M., Lee, W. C., et al. (2022). TDP-43 loss and ALS-risk SNPs drive mis-splicing and depletion of UNC13A. Nature 603, 131–137. doi: 10.1038/s41586-022-04436-3
Burkhardt, M. F., Martinez, F. J., Wright, S., Ramos, C., Volfson, D., Mason, M., et al. (2013). A cellular model for sporadic ALS using patient-derived induced pluripotent stem cells. Mol. Cell. Neurosci. 56, 355–364. doi: 10.1016/j.mcn.2013.07.007
Cali, C. P., Patino, M., Tai, Y. K., Ho, W. Y., McLean, C. A., Morris, C. M., et al. (2019). C9orf72 intermediate repeats are associated with corticobasal degeneration, increased C9orf72 expression and disruption of autophagy. Acta Neuropathol. 138, 795–811. doi: 10.1007/s00401-019-02045-5
Caso, F., and Davies, B. (2022). Base editing and prime editing in laboratory animals. Lab. Anim. 56, 35–49. doi: 10.1177/0023677221993895
Chen, Y. A., Kankel, M. W., Hana, S., Lau, S. K., Zavodszky, M. I., McKissick, O., et al. (2022). In vivo genome editing using novel AAV-PHP variants rescues motor function deficits and extends survival in a SOD1-ALS mouse model. Gene Ther. 30, 1–12. doi: 10.1038/s41434-022-00375-w
Cheng, C., Yang, K., Wu, X., Zhang, Y., Shan, S., Gitler, A., et al. (2019). Loss of CREST leads to neuroinflammatory responses and ALS-like motor defects in mice. Transl. Neurodegener. 8:13. doi: 10.1186/s40035-019-0152-1
Cho, S. W., Kim, S., Kim, J. M., and Kim, J. S. (2013). Targeted genome engineering in human cells with the Cas9 RNA-guided endonuclease. Nat. Biotechnol. 31, 230–232. doi: 10.1038/nbt.2507
Dafinca, R., Barbagallo, P., Farrimond, L., Candalija, A., Scaber, J., Ababneh, N. A., et al. (2020). Impairment of mitochondrial calcium buffering links mutations in C9ORF72 and TARDBP in iPS-derived motor neurons from patients with ALS/FTD. Stem Cell Rep. 14, 892–908. doi: 10.1016/j.stemcr.2020.03.023
Dafinca, R., Scaber, J., Ababneh, N., Lalic, T., Weir, G., Christian, H., et al. (2016). C9orf72 Hexanucleotide expansions are associated with altered endoplasmic reticulum calcium homeostasis and stress granule formation in induced pluripotent stem cell-derived neurons from patients with amyotrophic lateral sclerosis and frontotemporal dementia. Stem Cells 34, 2063–2078. doi: 10.1002/stem.2388
De Conti, L., Akinyi, M. V., Mendoza-Maldonado, R., Romano, M., Baralle, M., and Buratti, E. (2015). TDP-43 affects splicing profiles and isoform production of genes involved in the apoptotic and mitotic cellular pathways. Nucleic Acids Res. 43, 8990–9005. doi: 10.1093/nar/gkv814
DeJesus-Hernandez, M., Mackenzie, I. R., Boeve, B. F., Boxer, A. L., Baker, M., Rutherford, N. J., et al. (2011). Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72, 245–256. doi: 10.1016/j.neuron.2011.09.011
Deng, H. X., Zhai, H., Shi, Y., Liu, G., Lowry, J., Liu, B., et al. (2021). Efficacy and long-term safety of CRISPR/Cas9 genome editing in the SOD1-linked mouse models of ALS. Commun. Biol. 4:396. doi: 10.1038/s42003-021-01942-4
Du, Z. W., Chen, H., Liu, H., Lu, J., Qian, K., Huang, C. L., et al. (2015). Generation and expansion of highly pure motor neuron progenitors from human pluripotent stem cells. Nat. Commun. 6:6626. doi: 10.1016/j.xpro.2022.101223
Duan, W., Guo, M., Yi, L., Liu, Y., Li, Z., Ma, Y., et al. (2020). The deletion of mutant SOD1 via CRISPR/Cas9/sgRNA prolongs survival in an amyotrophic lateral sclerosis mouse model. Gene Ther. 27, 157–169. doi: 10.1038/s41434-019-0116-1
Fang, T., Je, G., Pacut, P., Keyhanian, K., Gao, J., and Ghasemi, M. (2022). Gene therapy in amyotrophic lateral sclerosis. Cells 11:2066. doi: 10.3390/cells11132066
Feldman, E. L., Goutman, S. A., Petri, S., Mazzini, L., Savelieff, M. G., Shaw, P. J., et al. (2022). Amyotrophic lateral sclerosis. Lancet 400, 1363–1380. doi: 10.1016/S0140-6736(22)01272-7
Gaj, T., Ojala, D. S., Ekman, F. K., Byrne, L. C., Limsirichai, P., and Schaffer, D. V. (2017). In vivo genome editing improves motor function and extends survival in a mouse model of ALS. Sci. Adv. 3:eaar3952. doi: 10.1126/sciadv.aar3952
Ghasemi, M., Keyhanian, K., and Douthwright, C. (2021). Glial cell dysfunction in C9orf72-related amyotrophic lateral sclerosis and frontotemporal dementia. Cells 10:249. doi: 10.3390/cells10020249
Gijselinck, I., Van Langenhove, T., van der Zee, J., Sleegers, K., Philtjens, S., Kleinberger, G., et al. (2012). A C9orf72 promoter repeat expansion in a Flanders-Belgian cohort with disorders of the frontotemporal lobar degeneration-amyotrophic lateral sclerosis spectrum: a gene identification study. Lancet Neurol. 11, 54–65. doi: 10.1016/S1474-4422(11)70261-7
Giovannelli, I., Higginbottom, A., Kirby, J., Azzouz, M., and Shaw, P. J. (2023). Prospects for gene replacement therapies in amyotrophic lateral sclerosis. Nat. Rev. Neurol. 19, 39–52. doi: 10.1038/s41582-022-00751-5
Guo, W., Naujock, M., Fumagalli, L., Vandoorne, T., Baatsen, P., Boon, R., et al. (2017). HDAC6 inhibition reverses axonal transport defects in motor neurons derived from FUS-ALS patients. Nat. Commun. 8:861. doi: 10.1038/s41467-017-00911-y
Gupta, A., Hall, V. L., Kok, F. O., Shin, M., McNulty, J. C., Lawson, N. D., et al. (2013). Targeted chromosomal deletions and inversions in zebrafish. Genome Res. 23, 1008–1017. doi: 10.1101/gr.154070.112
Hendriks, D., Clevers, H., and Artegiani, B. (2020). CRISPR-Cas tools and their application in genetic engineering of human stem cells and organoids. Cell Stem Cell 27, 705–731. doi: 10.1016/j.stem.2020.10.014
Hu, J. H., Miller, S. M., Geurts, M. H., Tang, W., Chen, L., Sun, N., et al. (2018). Evolved Cas9 variants with broad PAM compatibility and high DNA specificity. Nature 556, 57–63. doi: 10.1038/nature26155
Huo, Z., Tu, J., Xu, A., Li, Y., Wang, D., Liu, M., et al. (2019). Generation of a heterozygous p53 R249S mutant human embryonic stem cell line by TALEN-mediated genome editing. Stem Cell Res. 34:101360. doi: 10.1016/j.scr.2018.101360
Iacoangeli, A., Al Khleifat, A., Jones, A. R., Sproviero, W., Shatunov, A., Opie-Martin, S., et al. (2019). C9orf72 intermediate expansions of 24-30 repeats are associated with ALS. Acta Neuropathol. Commun. 7:115. doi: 10.1186/s40478-019-0724-4
Imamura, K., Izumi, Y., Watanabe, A., Tsukita, K., Woltjen, K., Yamamoto, T., et al. (2017). The Src/c-Abl pathway is a potential therapeutic target in amyotrophic lateral sclerosis. Sci. Transl. Med. 9:eaaf3962. doi: 10.1126/scitranslmed.aaf3962
Jiang, W., Bikard, D., Cox, D., Zhang, F., and Marraffini, L. A. (2013). RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nat. Biotechnol. 31, 233–239. doi: 10.1038/nbt.2508
Jo, S., Das, S., Williams, A., Chretien, A. S., Pagliardini, T., Le Roy, A., et al. (2022). Endowing universal CAR T-cell with immune-evasive properties using TALEN-gene editing. Nat. Commun. 13:3453. doi: 10.1038/s41467-022-30896-2
Johnson, S. A., Fang, T., De Marchi, F., Neel, D., Van Weehaeghe, D., Berry, J. D., et al. (2022). Pharmacotherapy for amyotrophic lateral sclerosis: a review of approved and upcoming agents. Drugs 82, 1367–1388. doi: 10.1007/s40265-022-01769-1
Kao, C. S., van Bruggen, R., Kim, J. R., Chen, X. X. L., Chan, C., Lee, J., et al. (2020). Selective neuronal degeneration in MATR3 S85C knock-in mouse model of early-stage ALS. Nat. Commun. 11:5304. doi: 10.1038/s41467-020-18949-w
Karginov, A. V., Tarutina, M. G., Lapteva, A. R., Pakhomova, M. D., Galliamov, A. A., Filkin, S. Y., et al. (2023). A Split-marker system for CRISPR-Cas9 genome editing in methylotrophic yeasts. Int. J. Mol. Sci. 24:8173. doi: 10.3390/ijms24098173
Kawamata, M., Suzuki, H. I., Kimura, R., and Suzuki, A. (2023). Optimization of Cas9 activity through the addition of cytosine extensions to single-guide RNAs. Nat Biomed. Eng. 7, 672–691. doi: 10.1038/s41551-023-01011-7
Kenjo, E., Hozumi, H., Makita, Y., Iwabuchi, K. A., Fujimoto, N., Matsumoto, S., et al. (2021). Low immunogenicity of LNP allows repeated administrations of CRISPR-Cas9 mRNA into skeletal muscle in mice. Nat. Commun. 12:7101. doi: 10.1038/s41467-021-26714-w
Khan, S. H. (2019). Genome-editing technologies: concept, pros, and cons of various genome-editing techniques and bioethical concerns for clinical application. Mol. Ther. Nucleic Acids 16, 326–334. doi: 10.1016/j.omtn.2019.02.027
Kim, B. W., Ryu, J., Jeong, Y. E., Kim, J., and Martin, L. J. (2020). Human motor neurons with SOD1-G93A mutation generated from CRISPR/Cas9 gene-edited iPSCs develop pathological features of amyotrophic lateral sclerosis. Front. Cell. Neurosci. 14:604171. doi: 10.3389/fncel.2020.604171
Kramer, N. J., Haney, M. S., Morgens, D. W., Jovicic, A., Couthouis, J., Li, A., et al. (2018). CRISPR-Cas9 screens in human cells and primary neurons identify modifiers of C9ORF72 dipeptide-repeat-protein toxicity. Nat. Genet. 50, 603–612. doi: 10.1038/s41588-018-0070-7
Krishnan, G., Zhang, Y., Gu, Y., Kankel, M. W., Gao, F. B., and Almeida, S. (2020). CRISPR deletion of the C9ORF72 promoter in ALS/FTD patient motor neurons abolishes production of dipeptide repeat proteins and rescues neurodegeneration. Acta Neuropathol. 140, 81–84. doi: 10.1007/s00401-020-02154-6
Kuo, P. H., Chiang, C. H., Wang, Y. T., Doudeva, L. G., and Yuan, H. S. (2014). The crystal structure of TDP-43 RRM1-DNA complex reveals the specific recognition for UG-and TG-rich nucleic acids. Nucleic Acids Res. 42, 4712–4722. doi: 10.1093/nar/gkt1407
Kwon, Y. W., Ahn, H. S., Lee, J. W., Yang, H. M., Cho, H. J., Kim, S. J., et al. (2021). HLA DR genome editing with TALENs in human iPSCs produced immune-tolerant dendritic cells. Stem Cells Int. 2021:8873383. doi: 10.1155/2021/8873383
Lattante, S., Rouleau, G. A., and Kabashi, E. (2013). TARDBP and FUS mutations associated with amyotrophic lateral sclerosis: summary and update. Hum. Mutat. 34, 812–826. doi: 10.1002/humu.22319
Lee, J., Chung, J. H., Kim, H. M., Kim, D. W., and Kim, H. (2016). Designed nucleases for targeted genome editing. Plant Biotechnol. J. 14, 448–462. doi: 10.1111/pbi.12465
Lee, J. M., Kim, U., Yang, H., Ryu, B., Kim, J., Sakuma, T., et al. (2021). TALEN-mediated generation of Nkx3.1 knockout rat model. Prostate 81, 182–193. doi: 10.1002/pros.24095
Li, L., Wu, L. P., and Chandrasegaran, S. (1992). Functional domains in Fok I restriction endonuclease. Proc. Natl. Acad. Sci. U. S. A. 89, 4275–4279. doi: 10.1073/pnas.89.10.4275
Li, H., Wu, S., Ma, X., Li, X., Cheng, T., Chen, Z., et al. (2021). Co-editing PINK1 and DJ-1 genes via adeno-associated virus-delivered CRISPR/Cas9 system in adult monkey brain elicits classical parkinsonian phenotype. Neurosci. Bull. 37, 1271–1288. doi: 10.1007/s12264-021-00732-6
Li, H., Yang, Y., Hong, W., Huang, M., Wu, M., and Zhao, X. (2020). Applications of genome editing technology in the targeted therapy of human diseases: mechanisms, advances and prospects. Signal Transduct. Target. Ther. 5:1. doi: 10.1038/s41392-019-0089-y
Lim, C. K. W., Gapinske, M., Brooks, A. K., Woods, W. S., Powell, J. E., Zeballos, C. M., et al. (2020). Treatment of a mouse model of ALS by in vivo base editing. Mol. Ther. 28, 1177–1189. doi: 10.1016/j.ymthe.2020.01.005
Ling, S., Yang, S., Hu, X., Yin, D., Dai, Y., Qian, X., et al. (2021). Lentiviral delivery of co-packaged Cas9 mRNA and a Vegfa-targeting guide RNA prevents wet age-related macular degeneration in mice. Nat. Biomed. Eng. 5, 144–156. doi: 10.1038/s41551-020-00656-y
Liu, Q., Segal, D. J., Ghiara, J. B., and Barbas, C. F. 3rd. (1997). Design of polydactyl zinc-finger proteins for unique addressing within complex genomes. Proc. Natl. Acad. Sci. U. S. A. 94, 5525–5530. doi: 10.1073/pnas.94.11.5525
Liu, K., Wan, P., Huang, Y., Wang, B., Wang, X., Zhang, R., et al. (2022). Transcriptome analysis in an AEG-1-deficient neuronal HT22 cell line. Exp. Ther. Med. 24:670. doi: 10.3892/etm.2022.11607
Logroscino, G., Piccininni, M., Marin, B., Nichols, E., Abd-Allah, F., Abdelalim, A., et al. (2018). Global, regional, and national burden of motor neuron diseases 1990-2016: a systematic analysis for the global burden of disease study 2016. Lancet Neurol. 17, 1083–1097. doi: 10.1016/S1474-4422(18)30404-6
Lopez-Gonzalez, R., Lu, Y., Gendron, T. F., Karydas, A., Tran, H., Yang, D., et al. (2016). Poly(GR) in C9ORF72-related ALS/FTD compromises mitochondrial function and increases oxidative stress and DNA damage in iPSC-derived motor neurons. Neuron 92, 383–391. doi: 10.1016/j.neuron.2016.09.015
Lopez-Gonzalez, R., Yang, D., Pribadi, M., Kim, T. S., Krishnan, G., Choi, S. Y., et al. (2019). Partial inhibition of the overactivated Ku80-dependent DNA repair pathway rescues neurodegeneration in C9ORF72-ALS/FTD. Proc. Natl. Acad. Sci. U. S. A. 116, 9628–9633. doi: 10.1073/pnas.1901313116
Mackenzie, I. R., Rademakers, R., and Neumann, M. (2010). TDP-43 and FUS in amyotrophic lateral sclerosis and frontotemporal dementia. Lancet Neurol. 9, 995–1007. doi: 10.1016/S1474-4422(10)70195-2
Makarova, K. S., Wolf, Y. I., Alkhnbashi, O. S., Costa, F., Shah, S. A., Saunders, S. J., et al. (2015). An updated evolutionary classification of CRISPR-Cas systems. Nat. Rev. Microbiol. 13, 722–736. doi: 10.1038/nrmicro3569
Marcus, R. (2022). What is amyotrophic lateral sclerosis? JAMA 328:2466. doi: 10.1001/jama.2022.19305
McMillan, M., Gomez, N., Hsieh, C., Bekier, M., Li, X., Miguez, R., et al. (2023). RNA methylation influences TDP43 binding and disease pathogenesis in models of amyotrophic lateral sclerosis and frontotemporal dementia. Mol. Cell 83, 219–36 e7. doi: 10.1016/j.molcel.2022.12.019
Meijboom, K. E., Abdallah, A., Fordham, N. P., Nagase, H., Rodriguez, T., Kraus, C., et al. (2022). CRISPR/Cas9-mediated excision of ALS/FTD-causing hexanucleotide repeat expansion in C9ORF72 rescues major disease mechanisms in vivo and in vitro. Nat. Commun. 13:6286. doi: 10.1038/s41467-022-33332-7
Meijboom, K. E., and Brown, R. H. (2022). Approaches to gene modulation therapy for ALS. Neurotherapeutics 19, 1159–1179. doi: 10.1007/s13311-022-01285-w
Mitra, J., Guerrero, E. N., Hegde, P. M., Liachko, N. F., Wang, H., Vasquez, V., et al. (2019). Motor neuron disease-associated loss of nuclear TDP-43 is linked to DNA double-strand break repair defects. Proc. Natl. Acad. Sci. U. S. A. 116, 4696–4705. doi: 10.1073/pnas.1818415116
Modell, A. E., Lim, D., Nguyen, T. M., Sreekanth, V., and Choudhary, A. (2022). CRISPR-based therapeutics: current challenges and future applications. Trends Pharmacol. Sci. 43, 151–161. doi: 10.1016/j.tips.2021.10.012
Mullard, A. (2019). First in vivo CRISPR candidate enters the clinic. Nat. Rev. Drug Discov. 18:656. doi: 10.1038/d41573-019-00139-z
Nahmad, A. D., Reuveni, E., Goldschmidt, E., Tenne, T., Liberman, M., Horovitz-Fried, M., et al. (2022). Frequent aneuploidy in primary human T cells after CRISPR-Cas9 cleavage. Nat. Biotechnol. 40, 1807–1813. doi: 10.1038/s41587-022-01377-0
Nakashima, C., Doi, H., Nakajima, S., Mashimo, T., Oga, T., Ishida-Yamamoto, A., et al. (2022). Filaggrin-deficient rats generated using zinc-finger nucleases spontaneously exhibit dry scaly skin. Allergol. Int. 71, 545–547. doi: 10.1016/j.alit.2022.04.001
Oskarsson, B., and Gendron, T. F., Staff NP (2018). Amyotrophic lateral sclerosis: An update for 2018. Mayo Clin. Proc. 93, 1617–1628. doi: 10.1016/j.mayocp.2018.04.007
Philips, T., and Rothstein, J. D. (2015). Rodent models of amyotrophic lateral sclerosis. Curr. Protoc. Pharmacol. 69, 5.67.1–5.67.21. doi: 10.1002/0471141755.ph0567s69
Piao, X., Meng, D., Zhang, X., Song, Q., Lv, H., and Jia, Y. (2022). Dual-gRNA approach with limited off-target effect corrects C9ORF72 repeat expansion in vivo. Sci. Rep. 12:5672. doi: 10.1038/s41598-022-07746-8
Pinto, B. S., Saxena, T., Oliveira, R., Mendez-Gomez, H. R., Cleary, J. D., Denes, L. T., et al. (2017). Impeding transcription of expanded microsatellite repeats by deactivated Cas9. Mol. Cell 68, 479–90 e5. doi: 10.1016/j.molcel.2017.09.033
Prtenjaca, N., Rob, M., Alam, M. S., Markovinovic, A., Stuani, C., Buratti, E., et al. (2022). Optineurin deficiency and insufficiency Lead to higher microglial TDP-43 protein levels. Int. J. Mol. Sci. 23:6829. doi: 10.3390/ijms23126829
Qiu, L., Xie, M., Zhou, M., Liu, X., Hu, Z., and Wu, L. (2021). Restoration of FVIII function and phenotypic Rescue in Hemophilia a Mice by transplantation of MSCs derived from F8-modified iPSCs. Front. Cell Dev. Biol. 9:630353. doi: 10.3389/fcell.2021.630353
Russell, K. L., Downie, J. M., Gibson, S. B., Tsetsou, S., Keefe, M. D., Duran, J. A., et al. (2021). Pathogenic effect of TP73 gene variants in people with amyotrophic lateral sclerosis. Neurology 97, e225–e235. doi: 10.1212/WNL.0000000000012285
Sahdeo, S., and Goldstein, D. B. (2021). Translating amyotrophic lateral sclerosis genes into drug development leads. Nat. Genet. 53, 1624–1626. doi: 10.1038/s41588-021-00981-1
Selvaraj, B. T., Livesey, M. R., Zhao, C., Gregory, J. M., James, O. T., Cleary, E. M., et al. (2018). C9ORF72 repeat expansion causes vulnerability of motor neurons to ca(2+)-permeable AMPA receptor-mediated excitotoxicity. Nat. Commun. 9:347. doi: 10.1038/s41467-017-02729-0
Sen, T., and Thummer, R. P. (2022). CRISPR and iPSCs: recent developments and future perspectives in neurodegenerative disease modelling, research, and therapeutics. Neurotox. Res. 40, 1597–1623. doi: 10.1007/s12640-022-00564-w
Sertori, R., Jones, R., Basheer, F., Rivera, L., Dawson, S., Loke, S., et al. (2022). Generation and characterization of a zebrafish IL-2Rgammac SCID model. Int. J. Mol. Sci. 23:2385. doi: 10.3390/ijms23042385
Shelkovnikova, T. A., Kukharsky, M. S., An, H., Dimasi, P., Alexeeva, S., Shabir, O., et al. (2018). Protective paraspeckle hyper-assembly downstream of TDP-43 loss of function in amyotrophic lateral sclerosis. Mol. Neurodegener. 13:30. doi: 10.1186/s13024-018-0263-7
Siles, L., Gaudo, P., and Pomares, E. (2023). High-efficiency CRISPR/Cas9-mediated correction of a homozygous mutation in Achromatopsia-patient-derived iPSCs. Int. J. Mol. Sci. 24:3655. doi: 10.3390/ijms24043655
Strohm, L., Hu, Z., Suk, Y., Ruhmkorf, A., Sternburg, E., Gattringer, V., et al. (2022). Multi-omics profiling identifies a deregulated FUS-MAP1B axis in ALS/FTD-associated UBQLN2 mutants. Life Sci. Alliance 5:e202101327. doi: 10.26508/lsa.202101327
Sullivan, P. M., Zhou, X., Robins, A. M., Paushter, D. H., Kim, D., Smolka, M. B., et al. (2016). The ALS/FTLD associated protein C9orf72 associates with SMCR8 and WDR41 to regulate the autophagy-lysosome pathway. Acta Neuropathol. Commun. 4:51. doi: 10.1186/s40478-016-0324-5
Tann, J. Y., Wong, L. W., Sajikumar, S., and Ibanez, C. F. (2019). Abnormal TDP-43 function impairs activity-dependent BDNF secretion, synaptic plasticity, and cognitive behavior through altered Sortilin splicing. EMBO J. 38:e100989. doi: 10.15252/embj.2018100989
Taylor, J. P., Brown, R. H. Jr., and Cleveland, D. W. (2016). Decoding ALS: from genes to mechanism. Nature 539, 197–206. doi: 10.1038/nature20413
Vandoorne, T., Veys, K., Guo, W., Sicart, A., Vints, K., Swijsen, A., et al. (2019). Differentiation but not ALS mutations in FUS rewires motor neuron metabolism. Nat. Commun. 10:4147. doi: 10.1038/s41467-019-12099-4
Verdu, S., Perez, A. J., Carrascosa, C., Barat, J. M., Talens, P., and Grau, R. (2021). Caenorhabditis elegans to model the capacity of ascorbic acid to reduce acute nitrite toxicity under different feed conditions: multivariate analytics on behavioral imaging. Int. J. Environ. Res. Public Health 18:2068. doi: 10.3390/ijerph18042068
Verdu, S., Ruiz-Rico, M., Perez, A. J., Barat, J. M., Talens, P., and Grau, R. (2020). Toxicological implications of amplifying the antibacterial activity of gallic acid by immobilisation on silica particles: a study on C. elegans. Environ. Toxicol. Pharmacol. 80:103492. doi: 10.1016/j.etap.2020.103492
Voisard, P., Diofano, F., Glazier, A. A., Rottbauer, W., and Just, S. (2022). CRISPR/Cas9-mediated constitutive loss of VCP (Valosin-containing protein) impairs Proteostasis and leads to defective striated muscle structure and function in vivo. Int. J. Mol. Sci. 23:6722. doi: 10.3390/ijms23126722
Walker, A. K., Spiller, K. J., Ge, G., Zheng, A., Xu, Y., Zhou, M., et al. (2015). Functional recovery in new mouse models of ALS/FTLD after clearance of pathological cytoplasmic TDP-43. Acta Neuropathol. 130, 643–660. doi: 10.1007/s00401-015-1460-x
Wall, J. M., Basu, A., Zunica, E. R. M., Dubuisson, O. S., Pergola, K., Broussard, J. P., et al. (2021). CRISPR/Cas9-engineered Drosophila knock-in models to study VCP diseases. Dis. Model. Mech. 14:dmm048603. doi: 10.1242/dmm.048603
Wang, H., Guo, W., Mitra, J., Hegde, P. M., Vandoorne, T., Eckelmann, B. J., et al. (2018). Mutant FUS causes DNA ligation defects to inhibit oxidative damage repair in amyotrophic lateral sclerosis. Nat. Commun. 9:3683. doi: 10.1038/s41467-018-06111-6
Wang, L., Yi, F., Fu, L., Yang, J., Wang, S., Wang, Z., et al. (2017). CRISPR/Cas9-mediated targeted gene correction in amyotrophic lateral sclerosis patient iPSCs. Protein Cell 8, 365–378. doi: 10.1007/s13238-017-0397-3
Yamashita, T., Chai, H. L., Teramoto, S., Tsuji, S., Shimazaki, K., Muramatsu, S., et al. (2013). Rescue of amyotrophic lateral sclerosis phenotype in a mouse model by intravenous AAV9-ADAR2 delivery to motor neurons. EMBO Mol. Med. 5, 1710–1719. doi: 10.1002/emmm.201302935
Yang, S., Chang, R., Yang, H., Zhao, T., Hong, Y., Kong, H. E., et al. (2017). CRISPR/Cas9-mediated gene editing ameliorates neurotoxicity in mouse model of Huntington’s disease. J. Clin. Invest. 127, 2719–2724. doi: 10.1172/JCI92087
Yang, W., Guo, X., Tu, Z., Chen, X., Han, R., Liu, Y., et al. (2022). PINK1 kinase dysfunction triggers neurodegeneration in the primate brain without impacting mitochondrial homeostasis. Protein Cell 13, 26–46. doi: 10.1007/s13238-021-00888-x
Zhang, T., Jiang, X., Xu, M., Wang, H., Sang, X., Qin, M., et al. (2018). Sleep and circadian abnormalities precede cognitive deficits in R521C FUS knockin rats. Neurobiol. Aging 72, 159–170. doi: 10.1016/j.neurobiolaging.2018.08.025
Zhang, Y., Zeng, B., Gu, A., Kang, Q., Zhao, M., Peng, G., et al. (2022). Damaged DNA is an early event of neurodegeneration in induced pluripotent stem cell-derived Motoneurons with UBQLN2(P497H). Mutat. Int. J. Mol. Sci. 23:11721. doi: 10.3390/ijms231911721
Zhang, H. X., Zhang, Y., and Yin, H. (2019). Genome editing with mRNA encoding ZFN, TALEN, and Cas9. Mol. Ther. 27, 735–746. doi: 10.1016/j.ymthe.2019.01.014
Zhang, Y., Zhu, J., Dai, Y., Wang, L., Liu, R., and Guo, X. (2022). Generation of a heterozygous FUS-Q290X knock in human embryonic stem cell line (WAe009-A-83) using CRISPR/Cas9 system. Stem Cell Res. 60:102734. doi: 10.1016/j.scr.2022.102734
Zhao, D., Jiang, G., Li, J., Chen, X., Li, S., Wang, J., et al. (2022). Imperfect guide-RNA (igRNA) enables CRISPR single-base editing with ABE and CBE. Nucleic Acids Res. 50, 4161–4170. doi: 10.1093/nar/gkac201
Zhu, P., Qiu, Q., Harris, P. C., Xu, X., and Lin, X. (2021). Mtor Haploinsufficiency ameliorates renal cysts and cilia abnormality in adult zebrafish tmem67 mutants. J. Am. Soc. Nephrol. 32, 822–836. doi: 10.1681/ASN.2020070991
Zuo, E., Cai, Y. J., Li, K., Wei, Y., Wang, B. A., Sun, Y., et al. (2017). One-step generation of complete gene knockout mice and monkeys by CRISPR/Cas9-mediated gene editing with multiple sgRNAs. Cell Res. 27, 933–945. doi: 10.1038/cr.2017.81
Keywords: amyotrophic lateral sclerosis, gene editing, CRISPR/Cas9, ALS models, gene therapeutic, clinical trials
Citation: Shi Y, Zhao Y, Lu L, Gao Q, Yu D and Sun M (2023) CRISPR/Cas9: implication for modeling and therapy of amyotrophic lateral sclerosis. Front. Neurosci. 17:1223777. doi: 10.3389/fnins.2023.1223777
Edited by:
Sen Yan, Jinan University, ChinaReviewed by:
Fang Hua, Augusta University, United StatesCopyright © 2023 Shi, Zhao, Lu, Gao, Yu and Sun. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dongyi Yu, ZG9uZ3lpX3l1QDE2My5jb20=; Miao Sun, bWlhb3N1bjMzQHN1ZGEuZWR1LmNu
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.