 Daniel Lamontagne-Kam
Daniel Lamontagne-Kam Anosha Kiran Ulfat
Anosha Kiran Ulfat Vincent Hervé
Vincent Hervé Jonathan Brouillette
Jonathan Brouillette- Department of Pharmacology and Physiology, Université de Montréal, Montréal, QC, Canada
Propagation of tau fibrils correlate closely with neurodegeneration and memory deficits seen during the progression of Alzheimer’s disease (AD). Although it is not well-established what drives or attenuates tau spreading, new studies on human brain using positron emission tomography (PET) have shed light on how tau phosphorylation, genetic factors, and the initial epicenter of tau accumulation influence tau accumulation and propagation throughout the brain. Here, we review the latest PET studies performed across the entire AD continuum looking at the impact of amyloid load on tau pathology. We also explore the effects of structural, functional, and proximity connectivity on tau spreading in a stereotypical manner in the brain of AD patients. Since tau propagation can be quite heterogenous between individuals, we then consider how the speed and pattern of propagation are influenced by the starting localization of tau accumulation in connected brain regions. We provide an overview of some genetic variants that were shown to accelerate or slow down tau spreading. Finally, we discuss how phosphorylation of certain tau epitopes affect the spreading of tau fibrils. Since tau pathology is an early event in AD pathogenesis and is one of the best predictors of neurodegeneration and memory impairments, understanding the process by which tau spread from one brain region to another could pave the way to novel therapeutic avenues that are efficient during the early stages of the disease, before neurodegeneration induces permanent brain damage and severe memory loss.
Introduction
Tau propagation across the brain has been shown to follow a stereotypical pattern in AD using histopathological staining at autopsy more than 30 years ago (Braak and Braak, 1991). It was first reported that fibrillar tau start to accumulate in the trans-entorhinal cortex and then spread to the anterior hippocampus, followed by adjacent temporal and limbic cortex, association isocortex, and ultimately to primary sensory cortex (Braak and Braak, 1991; Braak and Del Tredici, 2015; Cho et al., 2016).
Novel technologies allowing for the first time the visualization and quantification of aggregated, paired helical filament (PHF) tau in the brain of living people using positron emission tomography (PET) (Moscoso et al., 2022). Early increase in tau PET uptake was validated in the entorhinal cortex but was also found in many other regions such as the inferior temporal lobe, amygdala, banks of the superior temporal sulcus, fusiform gyrus, inferior parietal lobe, middle temporal lobe and the precuneus (Insel et al., 2020, 2023). Using novel 3D neuroimaging techniques, the noradrenergic locus coeruleus has also been shown as a very early accumulation site for trans-neuronal spreading of hyperphosphorylated tau (Gilvesy et al., 2022). Moreover, there is a growing body of evidence suggesting that substantial inter-individual variabilities in the pattern and intensity of tau signal may be more common than previously expected in affected AD brain regions (Murray et al., 2011; Scholl et al., 2017; Hanseeuw et al., 2019; Jack et al., 2019; Lowe et al., 2019; Betthauser et al., 2020; Vogel et al., 2021).
It is now widely recognized that tau can propagate, at least partly, by being secreted in an activity-dependent manner into the synaptic cleft from donor pre-synaptic neurons, and then recapture by recipient post-synaptic neurons localized in another brain region (Mohamed et al., 2013; Pooler et al., 2013; Calafate et al., 2015; Wu et al., 2016; Mudher et al., 2017; Vogel et al., 2020) (Figure 1). Tau can also be released as a result of leakage from neurodegeneration of the pre-synaptic neuron and diffuse in its close environment. Using high-resolution array tomography on post-mortem temporal and occipital cortices of AD patients, it was found recently that phosphorylated or misfolded tau, but mostly oligomeric tau accumulates in both pre- and post-synaptic terminals of the same synapses (Colom-Cadena et al., 2023), suggesting that oligomeric tau could be the main species of tau that spreads trans-synaptically. These results are in accordance with another in vitro study showing that low molecular weight tau aggregates and short fibrils (but not monomers, long fibrils, nor long tau filaments), are internalized through endocytosis and transported anterogradely and retrogradely (Wu et al., 2013). Moreover, trans-synaptic tau spreading has also been shown in various animal models overexpressing human tau or using adeno-associated virus-mediate expression of tau (de Calignon et al., 2012; Harris et al., 2012; Liu et al., 2012; Pickett et al., 2017; Wegmann et al., 2019).
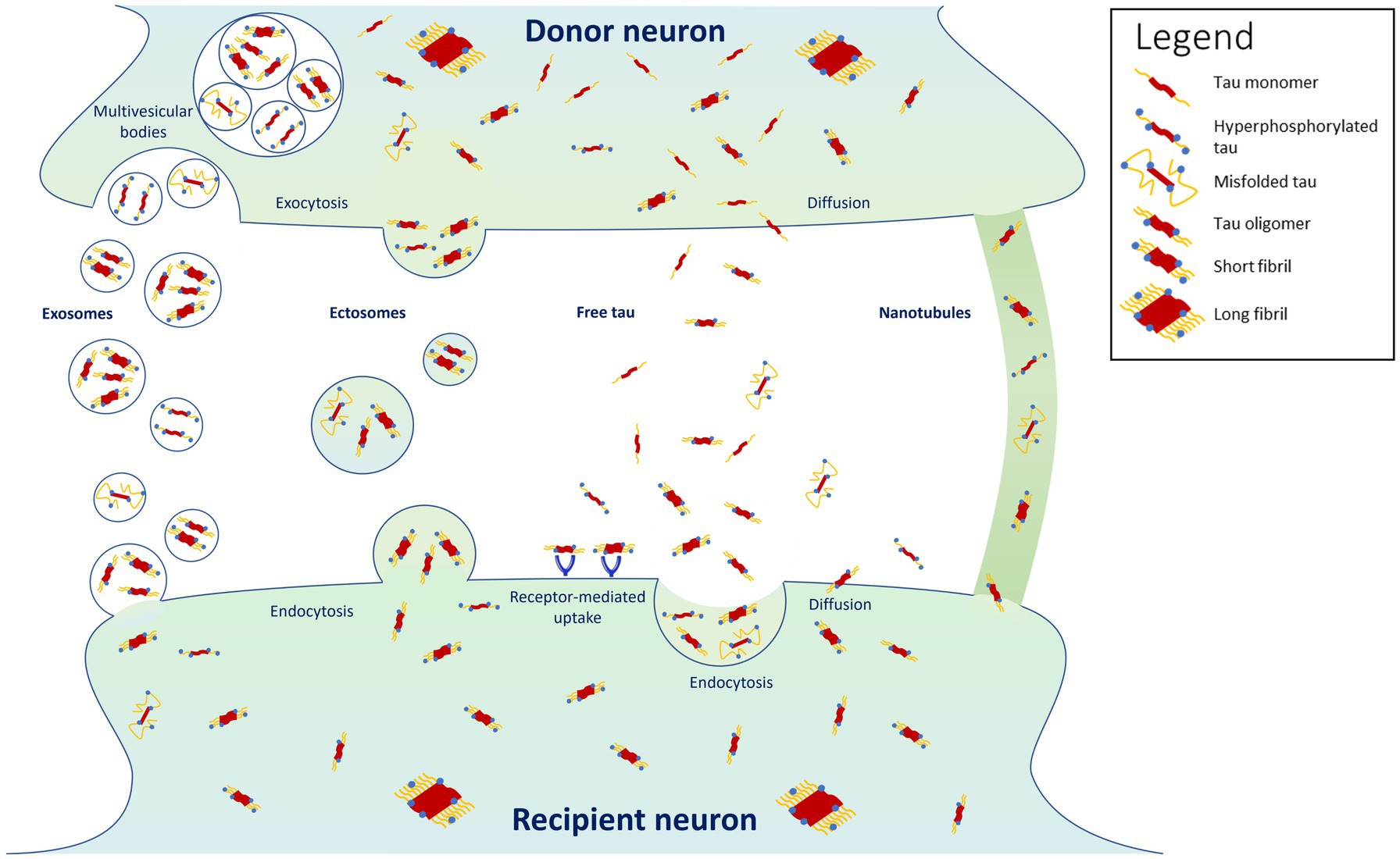
Figure 1. Proposed mechanisms for tau propagation. Highlighted are the findings that tau propagates predominantly in its oligomeric and short-fibril forms, with misfolded and hyperphosphorylated forms propagating in smaller proportions (Colom-Cadena et al., 2023). Tau monomers and long fibrils do not appear to propagate from cell to cell (Wu et al., 2013).
Propagation of tau pathology is one of the strongest predictors of progressive neurodegeneration and cognitive decline in AD (La Joie et al., 2020; Biel et al., 2021; Ossenkoppele et al., 2021b). Furthermore, elevated tau burden has been strongly associated with higher risk of progression from a clinically unimpaired to a mild cognitive impairment (MCI) status (Strikwerda-Brown et al., 2022). Thus, determining the factors that influence tau accumulation and propagation could help develop new treatments to prevent or halt the neurodegenerative process and ensuing cognitive decline that takes place in the early stages of AD. Here, we will review the latest research performed in the field of tau propagation in the human brain using neuroimaging techniques such as PET, magnetoencephalography (MEG), and functional magnetic resonance imaging (fMRI).
Impact of amyloid-beta (Aꞵ) on tau propagation
Many PET studies have consistently emphasized the critical role of Aꞵ pathology on the accumulation and spreading of tau (Wang et al., 2016; Jacobs et al., 2018; Franzmeier et al., 2020; Vogel et al., 2020; Jiang et al., 2022; Lee et al., 2022). Since Aꞵ starts to accumulate before tau in the preclinical stage of AD (Sperling et al., 2011), it is often difficult to disentangle the phenomena that are solely and specifically attributed to tau pathology. Although the interaction between Aꞵ and tau still needs to be fully determined, many studies have shown that Aꞵ greatly contribute to the deleterious effects of tau in AD brain. It was found that AD patients and cognitively normal people who tested positive for Aꞵ42 into the cerebrospinal fluid (CSF) had higher tau PET signal in the cortex and neurodegeneration in the hippocampus (Wang et al., 2016).
Higher neocortical accumulation of Aꞵ also predicted hippocampal volume loss, abnormalities in the white-matter tract that projects from the hippocampus to the posterior cingulate cortex (PCC), larger tau deposition in PCC, and faster memory decline in healthy older individuals (Jacobs et al., 2018). Other studies also found that detection of higher amyloid PET signal also extended tau PET uptake in cortical brain regions beyond the entorhinal cortex and correlates with cognitive decline (Jack et al., 2018; Pontecorvo et al., 2019; Sanchez et al., 2021). These findings in humans are consistent with animal studies showing that Aꞵ is an instigator of trans-synaptic spread of tau across the brain (de Calignon et al., 2012; Ahmed et al., 2014; Pooler et al., 2015; He et al., 2018).
Using an epidemic spreading model of tau along the AD continuum, Vogel and colleagues found that brain areas with higher Aβ burden had more tau accumulation than predicted by connectivity patterns (Vogel et al., 2020). This role of Aβ in accelerating tau spread was also observed in another study showing that individuals without amyloid plaque had almost no tangles in their brain, whereas those with Aꞵ PET uptake had more tangles at baseline and during follow-up trials (Franzmeier et al., 2020). Moreover, Aꞵ was found to interact with tau within the inferior temporal gyrus and accelerate widespread neocortical tau propagation (Lee et al., 2022). Altogether, these data indicate that temporal and spatial patterns of tau pathology depend on prior Aꞵ deposition. Although the exact mechanisms by which Aꞵ affects the accumulation and propagation of tau in distant brain regions still need to be elucidated, novel PET studies in human are giving new insights on how Aꞵ and tau pathologies are linked and influence each other in AD pathogenesis.
Effect of structural, functional, and proximity connectivity on tau spreading
Three models have been proposed as predictors for tau propagation. The functional connectivity model suggests that tau propagation is more likely to be detected in connected brain regions that fire together, whereas the structural and proximity connectivity models, respectively, propose that tau propagation relies more on direct synaptic connections or physical distance between brain regions (Schoonhoven et al., 2022).
Many cross-sectional studies have reported that the level of tau accumulation strongly correlates among regions that are functionally connected (Hoenig et al., 2018; Jacobs et al., 2018; Franzmeier et al., 2019b; Vogel et al., 2020). By combining magnetoencephalography (MEG) to measure electrical activity in the brain with positron emission tomography (PET) to evaluate tau depositions at several stages of AD, a recent study by Schoonhoven and colleagues has shown that the functional connectivity model was most accurate (r = 0.58) compared to the structural (r = 0.45) and proximity model (r = 0.44) at predicting tau propagation in the preclinical stage of AD (Schoonhoven et al., 2022). Since the prediction accuracy of the functional networks declined with AD progression, this suggests that exacerbated neuronal communication might be especially important for tau spreading in the earlier stages of the disease, before initial neuronal hyperactivity progressively switch to hypoactivity in AD (Busche and Konnerth, 2016; Hector and Brouillette, 2020).
In another study using resting-state fMRI scans to evaluate connectivity between 400 brain regions throughout the AD continuum, a strong correlation was found between functional connectivity and an increase in tangles, whereas spatial proximity was not a robust predictor of tau accumulation unless the nearby brain areas were functionally connected (Franzmeier et al., 2020). Connectivity, rather than proximity, was also identified as the primary source of tau spreading in an in vivo model of tau propagation using human P301S tau transgenic mice injected with brain extract containing tau aggregates (Ahmed et al., 2014).
These results are in line with an in vivo microdialysis study showing that higher neuronal activity induced by presynaptic glutamate release increased the level of extracellular tau in the hippocampus of wild-type mice (Yamada et al., 2014). Moreover, it was reported that higher neuronal activity induced by glutamate, (S)-AMPA or KCl depolarization increased tau release from primary mature cortical cultures, whereas inhibition of neuronal activity with tetrodotoxin impairs AMPA-mediated tau release (Pooler et al., 2013).
Although synaptic activity has been established as an important predictor of tau propagation, other studies have also highlighted the importance of the structural connectivity model to determine the pattern of tangles within the brain. In a study using resting-state fMRI, DTI, and tau PET, it was reported that anatomical connections could predict 70% of the tau PET pattern observed in the brain of MCI and AD patients, whereas the functional and proximity connectivity models, respectively, explained 58 and 48% of the tangle pattern (Vogel et al., 2020). Even though it is not clear at the moment if methodological differences between this study and others like the one by Schoonhoven et al. can explain the primary role for structural over functional connections, it seems nonetheless that both models influence tau propagation in a way that is consistent with the idea that tau spread through long-range axonal connections.
The observation that tau can propagate in both retrograde and anterograde directions of neural networks also argue in favor of an anatomical propagation of tau (Ahmed et al., 2014; Takeda et al., 2015), independent of neuronal activity that occurs unilaterally from the pre- to the post-synaptic neurons. However, it cannot be excluded that at least part of the retrograde pattern of tau propagation could be the result of residual tau that was released during synaptic activity and then taken up retroactively.
In addition to this cell-to-cell tau transmission, simple diffusion of tau release by neurons might also explain its local spread. Given that the clearing rate of tau from the extracellular space is relatively slow with a half-life of about 11 days (Yamada et al., 2014), this favors its diffusion into the interstitial fluid (ISF) within nearby brain regions. This is even more marked when neurodegeneration becomes more prominent in the late phase of the disease, when higher levels of tau leak from dying cells, and breakdowns in the glymphatic and immune system slow tau clearance (Haage and De Jager, 2022; Ishida et al., 2022). Altogether, these results suggest that tau spreading in a stereotypical manner in the brain of AD patients depends on trans-synaptic propagation especially among active neurons that fire together, and that local diffusion of tau also participate in the dispersion pattern of tau that closely correlates with progressive neurodegeneration and memory impairment seen during AD pathogenesis.
Role of starting localization of tau accumulation
Another aspect that can dictate the speed and pattern of tau propagation is the brain regions where tau starts to accumulate during AD. Although tau pathology spreads in a stereotypical pattern in AD (Braak and Braak, 1991), many studies have reported substantial individual variations in tau PET pattern, particularly in preclinical AD (Murray et al., 2011; Scholl et al., 2017; Ossenkoppele et al., 2020; Vogel et al., 2020; La Joie et al., 2021; Vogel et al., 2021; Frontzkowski et al., 2022; Young et al., 2022). By combining tau PET scans with resting-state fMRI to map brain connectivity, it was reported that tau spreads more rapidly when it is located in highly connected hub regions of the fronto-parietal association cortex compared to less connected regions in the temporo-limbic and visual cortices (Frontzkowski et al., 2022). Interestingly, AD patients with symptoms at a younger age were more likely to have tau deposition in hub regions, while participants who developed symptomatic AD at an older age had more tangles in limbic areas (Frontzkowski et al., 2022). Since hubs in the fronto-parietal network are essential for complex cognitive function (Cole et al., 2013; Zanto and Gazzaley, 2013), this could explain why stronger tau pathology in these hub regions was also associated with faster cognitive decline (Frontzkowski et al., 2022).
Different patterns of tau propagation were also observed in different subtypes of AD. Using PET scans from 1,612 participants covering the full clinical AD spectrum, tau was found to spread in four distinct spatiotemporal trajectories, including the limbic-predominant and medial temporal lobe (MTL)-sparing patterns as well as the posterior and lateral temporal patterns (Murray et al., 2011; Whitwell et al., 2012; Ferreira et al., 2020; Ossenkoppele et al., 2020; Vogel et al., 2021) (Figure 2). In early-onset AD (< 65 years), tau also tended to accumulate more in the prefrontal, premotor, and inferior parietal cortices than in late-onset AD (Scholl et al., 2017). Moreover, systematic spatiotemporal variations in tau spreading that deviate from the Braak staging system have been observed in clinical variants of AD, such as posterior cortical atrophy and logopenic primary progressive aphasia (Gorno-Tempini et al., 2011; Ossenkoppele et al., 2016; Crutch et al., 2017).
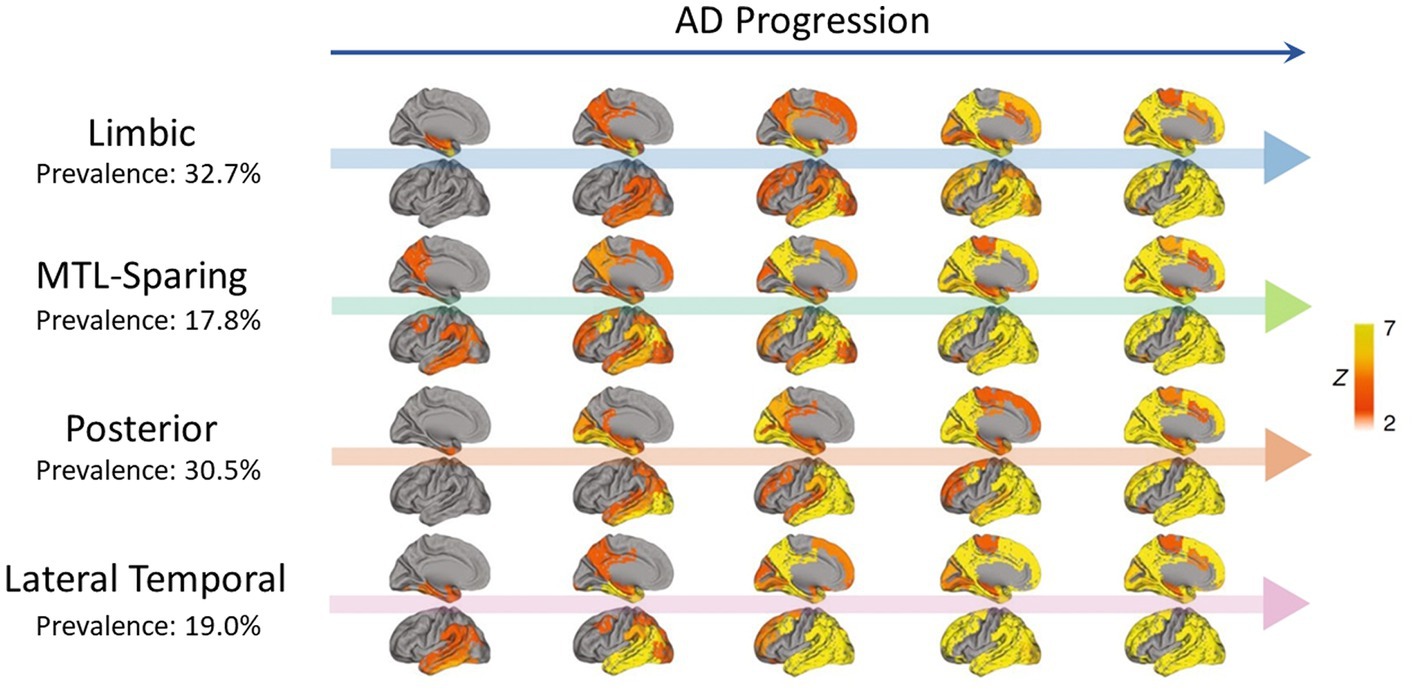
Figure 2. Representation of four distinct tau propagation patterns as determined by PET in a cross-sectional analysis (n = 2,324) (Vogel et al., 2021), with AD progression presented from left to right. The prevalence of each pattern in the analysis is indicated below the pattern names. Only the left side of the brain is shown, with the interior faces of sagittal cuts above their respective arrows. Different points of origin are highlighted in each of the defined patterns. Limbic propagation most closely resembles patterns reported by Braak and Braak (1991). Figure adapted from Vogel et al. (2021).
Tangles were also found to spread asymmetrically in vivo, with more tau pathology in the right entorhinal cortex for most participants (Vogel et al., 2020). This right-side epicenter was associated with more tau-tracer uptake on PET scans in frontal region and was mainly observed in older AD patients. Although we do not know at the moment why tau accumulation begins in distinct brain areas and follows variable spatiotemporal patterns in different people, it will be important to incorporate this heterogeneity into tau spreading models to hopefully one day be able to predict tau propagation at the individual level. Since tau pathology is the key driver of neurodegeneration and cognitive decline in AD (La Joie et al., 2020; Biel et al., 2021; Ossenkoppele et al., 2021b), understanding the cellular and molecular mechanisms underlying the different spatial patterns of tau distribution that sustained the diverse clinical manifestation of the disease could pave the way to develop new therapies that prevent cell death and memory deficits.
Genetic variants involved in tau propagation
Genetic variants could be, at least partly, a factor underpinning the variability of tangle spread in AD. Genome wide-association studies (GWAS) performed over the last few decades have underlined several genes such as APOE and BIN1 that increase the probability of developing AD (Jansen et al., 2019; Wightman et al., 2021). Although it is well-established that APOE4 is the strongest genetic predictor of sporadic AD and is associated with early amyloid deposition rate and burden (Corder et al., 1993; Ossenkoppele et al., 2015; Ridge et al., 2016; Lim et al., 2017; Ge et al., 2018; Mishra et al., 2018; Toledo et al., 2019; Insel et al., 2021), a growing body of evidences suggests that APOE4 is also involved in tau pathology (Table 1).
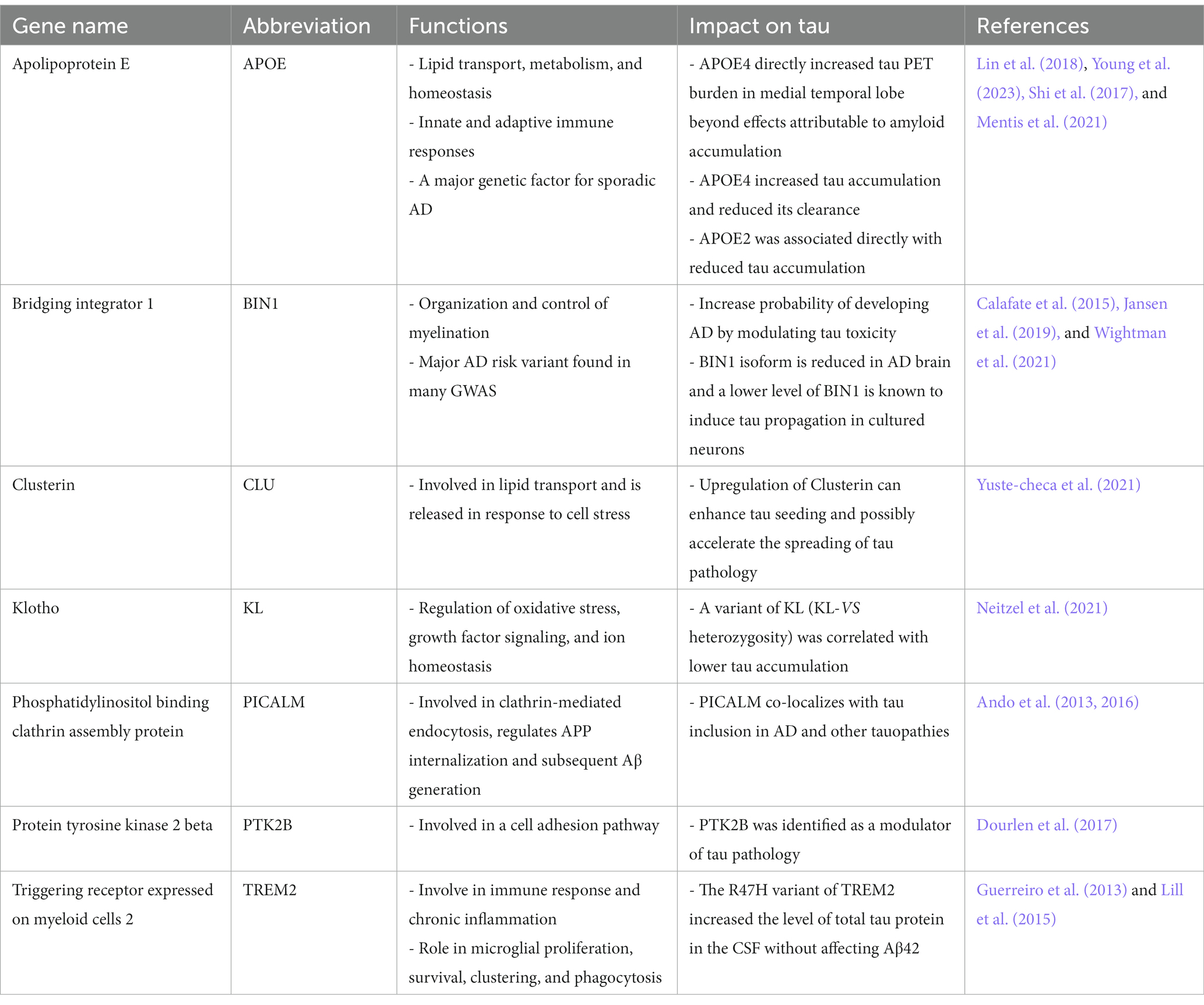
Table 1. The main genetic factors involved in the propagation of tau in AD.
In a study using preclinical AD individuals that are cognitively healthy but have abnormal amyloid level, it was found that APOE4 directly increased tau PET burden in medial temporal lobe (entorhinal cortex and amygdala) beyond effects attributable to amyloid accumulation (Young et al., 2023). APOE4 also had a deleterious influence on tau deposition in the early neocortical regions (inferior temporal, inferior parietal, precuneus), but these effects were mostly mediated by amyloid load. Conversely, APOE2 was associated directly with reduced tau accumulation in medial temporal lobe and early neocortical regions, which highlights APOE2 as a key protective variant (Young et al., 2023).
These results are consistent with previous PET reports showing that APOE4 carriers across the entire AD continuum have a more medial temporal lobe-dominant pattern of tau burden even after controlling for amyloid accumulation (Livingston et al., 2017; Mattsson et al., 2018; Therriault et al., 2020; La Joie et al., 2021; Ossenkoppele et al., 2021a). These findings are also in line with studies performed in iPSC-derived human brain cells and mouse models showing that APOE4 worsens tau deposition and neurodegeneration (Shi et al., 2017; Lin et al., 2018; Wang et al., 2018), and may have a role in reducing tau clearance through meningeal lymphosclerosis (Mentis et al., 2021; Ishida et al., 2022). However, it should be noted that other reports have not observed a significant impact of APOE4 on tau burden in clinically unimpaired individuals after adjusting for amyloid load (Lowe et al., 2018; Ramanan et al., 2019). These contrasting observations showcase that APOE4 may be an underlying factor for the accumulation of tau but not the only factor.
Another AD risk variant found in many GWAS is the gene BIN1 (i.e., bridging integrator 1), which encodes for a nucleoplasmic adaptor protein involved in many processes such as the regulation of neuronal excitability (Voskobiynyk et al., 2020), presynaptic vesicle release (De Rossi et al., 2020), and clathrin-mediated endocytosis (Calafate et al., 2016; Crotti et al., 2019). Higher gene expression of BIN1 found in AD brain has been found to increase trans-neuronal tau spreading (Chapuis et al., 2013; Calafate et al., 2016; Crotti et al., 2019), and to be associated with more pronounced tau pathology but not higher Aꞵ burden (Holler et al., 2014; Taga et al., 2020). Moreover, BIN1 rs744373 risk allele carriers were reported to have a higher tau PET signal, faster cognitive decline, and accelerated tau PET accumulation rate when Aꞵ is found at higher levels (Franzmeier et al., 2019a, 2022). Conversely, a protective variant of the klotho gene (KL-VShet) that occurs in 20–25% of the population (Dubal et al., 2014), was correlated with lower amyloid-associated tau accumulation (Neitzel et al., 2021), better cognitive performance (Deary et al., 2005; Dubal et al., 2014; Neitzel et al., 2021), and longer life expectancy (Arking et al., 2005; de Vries et al., 2017).
To have a broader view of the impact of genetic risk factors on tau pathology, Rubinski and colleagues have generated a polygenic score (PGS) from 85 GWAS single-nucleotide polymorphisms SNPs linked to AD, excluding APOE (Rubinski et al., 2023). They observed that an elevated PGS was associated with a faster rate of fibrillar tau progression and cognitive impairment, especially when amyloid plaque levels were more intense. Altogether, these results indicate that many gene risk factors have additional influences on the progression of tau pathology. Investigating the cellular and molecular pathways underpinning these effects could lead to novel therapeutic strategies to at least delay tau-dependent neurodegeneration and cognitive deficits in AD.
Tau hyperphosphorylation and spreading
Human tau protein has 85 potential phosphorylation sites, 9 of which are phosphorylated in nonpathological, soluble tau, and 45 of which have been identified as being phosphorylated in insoluble tau from AD patients (Hanger et al., 2007). The levels of phosphorylation at certain epitopes of tau also seems to have an impact on the spreading of tau fibrils. In a recent study by Pichet Binette and colleagues, it was observed that amyloid-related increases in soluble phosphorylated tau (p-tau) at epitope 217 in CSF was associated with cognitive decline and faster accumulation of tau aggregates in the early stages of AD, especially in regions that are functionally connected to areas where tau pathology started (Pichet Binette et al., 2022). However, when amyloid plaques and soluble p-tau had plateaued in later stages of the disease, soluble p-tau217 lost its effect on tangle accumulation, and cognitive deficits were more associated with the accumulation rate of insoluble tau aggregates. Thus, these results suggest that targeting soluble p-tau could be an interesting therapeutics strategy in early AD to prevent or slow down cognitive decline, the formation of insoluble tau aggregates and ensuing neurodegeneration.
To determine how amyloid plaques increase p-tau levels in the early stages of AD, Biel and colleagues (Biel et al., 2023) evaluated if tau phosphorylation was affected by the soluble fragment of triggering receptor expressed on myeloid cell 2 (sTREM2), a microglial activation marker increased in the CSF of mild AD patients that also correlates with CSF tau (Heslegrave et al., 2016; Piccio et al., 2016). They found that higher level of fibrillar Aꞵ was associated with increased CSF concentrations of sTREM2, and that sTREM2 mediated the association between fibrillar Aꞵ and the increase of CSF p-tau181 in early Aꞵ-accumulator individuals, which have high levels of Aꞵ1-42 in the CSF but subthreshold levels of Aꞵ when measured by PET. Conversely, in late Aꞵ-accumulators with high levels of Aꞵ found in both CSF and PET, higher level of sTREM2 was no longer correlated with fibrillar Aꞵ but paralleled CSF p-tau181 increases, indicating that sTREM2 is more tightly linked to soluble p-tau181 when high amounts of Aꞵ fibrils are detected in the brain.
These results are in line with a previous study showing that sTREM2 levels correlated with total and phosphorylated tau in the CSF in dominantly inherited AD approximately 5 years before the onset of symptoms (Suarez-Calvet et al., 2016). It also agrees with another report showing that amyloid-associated microglial activation correlates with both tau pathology and cognitive decline (Pascoal et al., 2021). Since plasma p-tau231 has been shown recently to be the earliest marker of amyloid aggregation (Mila-Aloma et al., 2022), it will be interesting to determine if p-tau231 affects tau phosphorylation and propagation across the entire AD spectrum.
Conclusion
Neuroimaging techniques indicate that the connectomic architecture of the brain dictates tau accumulation and spreading, which are critical for neurodegeneration and memory decline that begin in the early stages of AD. PET studies also highlight the close relationship between Aꞵ and tau, and how they drive neuropathological phenotypes observed in the entire AD continuum. Further studies are needed to decipher why tangle propagation is so heterogenous between brain regions and individuals. Finding protective factors that prevent or decrease the rate of tau deposition and spreading could also pave the way to develop therapeutic strategies against neurodegeneration and cognitive impairment in AD.
Author contributions
DL-K, AKU, VH, T-MV, and JB wrote, revised, and edited the manuscript. All authors contributed to the article and approved the submitted version.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Ahmed, Z., Cooper, J., Murray, T. K., Garn, K., McNaughton, E., Clarke, H., et al. (2014). A novel in vivo model of tau propagation with rapid and progressive neurofibrillary tangle pathology: the pattern of spread is determined by connectivity, not proximity. Acta Neuropathol. 127, 667–683. doi: 10.1007/s00401-014-1254-6
Ando, K., Brion, J. P., Stygelbout, V., Suain, V., Authelet, M., Dedecker, R., et al. (2013). Clathrin adaptor CALM/PICALM is associated with neurofibrillary tangles and is cleaved in Alzheimer's brains. Acta Neuropathol. 125, 861–878. doi: 10.1007/s00401-013-1111-z
Ando, K., Tomimura, K., Sazdovitch, V., Suain, V., Yilmaz, Z., Authelet, M., et al. (2016). Level of PICALM, a key component of clathrin-mediated endocytosis, is correlated with levels of phosphotau and autophagy-related proteins and is associated with tau inclusions in AD, PSP and pick disease. Neurobiol. Dis. 94, 32–43. doi: 10.1016/j.nbd.2016.05.017
Arking, D. E., Atzmon, G., Arking, A., Barzilai, N., and Dietz, H. C. (2005). Association between a functional variant of the KLOTHO gene and high-density lipoprotein cholesterol, blood pressure, stroke, and longevity. Circ. Res. 96, 412–418. doi: 10.1161/01.RES.0000157171.04054.30
Betthauser, T. J., Koscik, R. L., Jonaitis, E. M., Allison, S. L., Cody, K. A., Erickson, C. M., et al. (2020). Amyloid and tau imaging biomarkers explain cognitive decline from late middle-age. Brain 143, 320–335. doi: 10.1093/brain/awz378
Biel, D., Brendel, M., Rubinski, A., Buerger, K., Janowitz, D., Dichgans, M., et al. (2021). Tau-PET and in vivo Braak-staging as prognostic markers of future cognitive decline in cognitively normal to demented individuals. Alzheimers Res. Ther. 13:137. doi: 10.1186/s13195-021-00880-x
Biel, D., Suarez-Calvet, M., Hager, P., Rubinski, A., Dewenter, A., Steward, A., et al. (2023). sTREM2 is associated with amyloid-related p-tau increases and glucose hypermetabolism in Alzheimer's disease. EMBO Mol. Med. 15:e16987. doi: 10.15252/emmm.202216987
Braak, H., and Braak, E. (1991). Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 82, 239–259. doi: 10.1007/BF00308809
Braak, H., and Del Tredici, K. (2015). The preclinical phase of the pathological process underlying sporadic Alzheimer's disease. Brain 138, 2814–2833. doi: 10.1093/brain/awv236
Busche, M. A., and Konnerth, A. (2016). Impairments of neural circuit function in Alzheimer's disease. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 371:20150429. doi: 10.1098/rstb.2015.0429
Calafate, S., Buist, A., Miskiewicz, K., Vijayan, V., Daneels, G., de Strooper, B., et al. (2015). Synaptic contacts enhance cell-to-cell tau pathology propagation. Cell Rep. 11, 1176–1183. doi: 10.1016/j.celrep.2015.04.043
Calafate, S., Flavin, W., Verstreken, P., and Moechars, D. (2016). Loss of Bin1 promotes the propagation of tau pathology. Cell Rep. 17, 931–940. doi: 10.1016/j.celrep.2016.09.063
Chapuis, J., Hansmannel, F., Gistelinck, M., Mounier, A., Van Cauwenberghe, C., Kolen, K. V., et al. (2013). Increased expression of BIN1 mediates Alzheimer genetic risk by modulating tau pathology. Mol. Psychiatry 18, 1225–1234. doi: 10.1038/mp.2013.1
Cho, H., Choi, J. Y., Hwang, M. S., Kim, Y. J., Lee, H. M., Lee, H. S., et al. (2016). In vivo cortical spreading pattern of tau and amyloid in the Alzheimer disease spectrum. Ann. Neurol. 80, 247–258. doi: 10.1002/ana.24711
Cole, M. W., Reynolds, J. R., Power, J. D., Repovs, G., Anticevic, A., and Braver, T. S. (2013). Multi-task connectivity reveals flexible hubs for adaptive task control. Nat. Neurosci. 16, 1348–1355. doi: 10.1038/nn.3470
Colom-Cadena, M., Davies, C., Sirisi, S., Lee, J. E., Simzer, E. M., Tzioras, M., et al. (2023). Synaptic oligomeric tau in Alzheimer's disease - a potential culprit in the spread of tau pathology through the brain. Neuron. 10:S0896-6273(23)00305-7. doi: 10.1016/j.neuron.2023.04.020
Corder, E. H., Saunders, A. M., Strittmatter, W. J., Schmechel, D. E., Gaskell, P. C., Small, G. W., et al. (1993). Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. Science 261, 921–923. doi: 10.1126/science.8346443
Crotti, A., Sait, H. R., McAvoy, K. M., Estrada, K., Ergun, A., Szak, S., et al. (2019). BIN1 favors the spreading of tau via extracellular vesicles. Sci. Rep. 9:9477. doi: 10.1038/s41598-019-45676-0
Crutch, S. J., Schott, J. M., Rabinovici, G. D., Murray, M., Snowden, J. S., van der Flier, W. M., et al. (2017). Consensus classification of posterior cortical atrophy. Alzheimers Dement. 13, 870–884. doi: 10.1016/j.jalz.2017.01.014
de Calignon, A., Polydoro, M., Suarez-Calvet, M., William, C., Adamowicz, D. H., Kopeikina, K. J., et al. (2012). Propagation of tau pathology in a model of early Alzheimer's disease. Neuron 73, 685–697. doi: 10.1016/j.neuron.2011.11.033
De Rossi, P., Nomura, T., Andrew, R. J., Masse, N. Y., Sampathkumar, V., Musial, T. F., et al. (2020). Neuronal BIN1 regulates presynaptic neurotransmitter release and memory consolidation. Cell Rep. 30:e3527. doi: 10.1016/j.celrep.2020.02.026
de Vries, C. F., Staff, R. T., Harris, S. E., Chapko, D., Williams, D. S., Reichert, P., et al. (2017). Klotho, APOEepsilon4, cognitive ability, brain size, atrophy, and survival: a study in the Aberdeen birth cohort of 1936. Neurobiol. Aging 55, 91–98. doi: 10.1016/j.neurobiolaging.2017.02.019
Deary, I. J., Harris, S. E., Fox, H. C., Hayward, C., Wright, A. F., Starr, J. M., et al. (2005). KLOTHO genotype and cognitive ability in childhood and old age in the same individuals. Neurosci. Lett. 378, 22–27. doi: 10.1016/j.neulet.2004.12.005
Dourlen, P., Fernandez-Gomez, F. J., Dupont, C., Grenier-Boley, B., Bellenguez, C., Obriot, H., et al. (2017). Functional screening of Alzheimer risk loci identifies PTK2B as an in vivo modulator and early marker of tau pathology. Mol. Psychiatry 22, 874–883. doi: 10.1038/mp.2016.59
Dubal, D. B., Yokoyama, J. S., Zhu, L., Broestl, L., Worden, K., Wang, D., et al. (2014). Life extension factor klotho enhances cognition. Cell Rep. 7, 1065–1076. doi: 10.1016/j.celrep.2014.03.076
Ferreira, D., Nordberg, A., and Westman, E. (2020). Biological subtypes of Alzheimer disease: a systematic review and meta-analysis. Neurology 94, 436–448. doi: 10.1212/WNL.0000000000009058
Franzmeier, N., Neitzel, J., Rubinski, A., Smith, R., Strandberg, O., Ossenkoppele, R., et al. (2020). Functional brain architecture is associated with the rate of tau accumulation in Alzheimer's disease. Nat. Commun. 11:347. doi: 10.1038/s41467-019-14159-1
Franzmeier, N., Ossenkoppele, R., Brendel, M., Rubinski, A., Smith, R., Kumar, A., et al. (2022). The BIN1 rs744373 Alzheimer's disease risk SNP is associated with faster Abeta-associated tau accumulation and cognitive decline. Alzheimers Dement. 18, 103–115. doi: 10.1002/alz.12371
Franzmeier, N., Rubinski, A., Neitzel, J., and Ewers, M., Alzheimer's Disease Neuroimaging, I (2019a). The BIN1 rs744373 SNP is associated with increased tau-PET levels and impaired memory. Nat. Commun. 10:1766. doi: 10.1038/s41467-019-09564-5
Franzmeier, N., Rubinski, A., Neitzel, J., Kim, Y., Damm, A., Na, D. L., et al. (2019b). Functional connectivity associated with tau levels in ageing, Alzheimer's, and small vessel disease. Brain 142, 1093–1107. doi: 10.1093/brain/awz026
Frontzkowski, L., Ewers, M., Brendel, M., Biel, D., Ossenkoppele, R., Hager, P., et al. (2022). Earlier Alzheimer's disease onset is associated with tau pathology in brain hub regions and facilitated tau spreading. Nat. Commun. 13:4899. doi: 10.1038/s41467-022-32592-7
Ge, T., Sabuncu, M. R., Smoller, J. W., Sperling, R. A., and Mormino, E. C., Alzheimer's Disease Neuroimaging Initiative (2018). Dissociable influences of APOE epsilon4 and polygenic risk of AD dementia on amyloid and cognition. Neurology 90, e1605–e1612. doi: 10.1212/WNL.0000000000005415
Gilvesy, A., Husen, E., Magloczky, Z., Mihaly, O., Hortobagyi, T., Kanatani, S., et al. (2022). Spatiotemporal characterization of cellular tau pathology in the human locus coeruleus-pericoerulear complex by three-dimensional imaging. Acta Neuropathol. 144, 651–676. doi: 10.1007/s00401-022-02477-6
Gorno-Tempini, M. L., Hillis, A. E., Weintraub, S., Kertesz, A., Mendez, M., Cappa, S. F., et al. (2011). Classification of primary progressive aphasia and its variants. Neurology 76, 1006–1014. doi: 10.1212/WNL.0b013e31821103e6
Guerreiro, R., Wojtas, A., Bras, J., Carrasquillo, M., Rogaeva, E., Majounie, E., et al. (2013). TREM2 variants in Alzheimer's disease. N. Engl. J. Med. 368, 117–127. doi: 10.1056/NEJMoa1211851
Haage, V., and De Jager, P. L. (2022). Neuroimmune contributions to Alzheimer's disease: a focus on human data. Mol. Psychiatry 27, 3164–3181. doi: 10.1038/s41380-022-01637-0
Hanger, D. P., Byers, H. L., Wray, S., Leung, K. Y., Saxton, M. J., Seereeram, A., et al. (2007). Novel phosphorylation sites in tau from Alzheimer brain support a role for casein kinase 1 in disease pathogenesis. J. Biol. Chem. 282, 23645–23654. doi: 10.1074/jbc.M703269200
Hanseeuw, B. J., Betensky, R. A., Jacobs, H. I. L., Schultz, A. P., Sepulcre, J., Becker, J. A., et al. (2019). Association of Amyloid and tau with Cognition in preclinical Alzheimer disease: a longitudinal study. JAMA Neurol. 76, 915–924. doi: 10.1001/jamaneurol.2019.1424
Harris, J. A., Koyama, A., Maeda, S., Ho, K., Devidze, N., Dubal, D. B., et al. (2012). Human P301L-mutant tau expression in mouse entorhinal-hippocampal network causes tau aggregation and presynaptic pathology but no cognitive deficits. PLoS One 7:e45881. doi: 10.1371/journal.pone.0045881
He, Z., Guo, J. L., McBride, J. D., Narasimhan, S., Kim, H., Changolkar, L., et al. (2018). Amyloid-beta plaques enhance Alzheimer's brain tau-seeded pathologies by facilitating neuritic plaque tau aggregation. Nat. Med. 24, 29–38. doi: 10.1038/nm.4443
Hector, A., and Brouillette, J. (2020). Hyperactivity induced by soluble amyloid-beta oligomers in the early stages of Alzheimer's disease. Front. Mol. Neurosci. 13:600084. doi: 10.3389/fnmol.2020.600084
Heslegrave, A., Heywood, W., Paterson, R., Magdalinou, N., Svensson, J., Johansson, P., et al. (2016). Increased cerebrospinal fluid soluble TREM2 concentration in Alzheimer's disease. Mol. Neurodegener. 11:3. doi: 10.1186/s13024-016-0071-x
Hoenig, M. C., Bischof, G. N., Seemiller, J., Hammes, J., Kukolja, J., Onur, O. A., et al. (2018). Networks of tau distribution in Alzheimer's disease. Brain 141, 568–581. doi: 10.1093/brain/awx353
Holler, C. J., Davis, P. R., Beckett, T. L., Platt, T. L., Webb, R. L., Head, E., et al. (2014). Bridging integrator 1 (BIN1) protein expression increases in the Alzheimer's disease brain and correlates with neurofibrillary tangle pathology. J. Alzheimers Dis. 42, 1221–1227. doi: 10.3233/JAD-132450
Insel, P. S., Hansson, O., and Mattsson-Carlgren, N. (2021). Association between apolipoprotein E epsilon2 vs epsilon4, age, and beta-amyloid in adults without cognitive impairment. JAMA Neurol. 78, 229–235. doi: 10.1001/jamaneurol.2020.3780
Insel, P. S., Mormino, E. C., Aisen, P. S., Thompson, W. K., and Donohue, M. C. (2020). Neuroanatomical spread of amyloid beta and tau in Alzheimer's disease: implications for primary prevention. Brain Commun. 2:fcaa007. doi: 10.1093/braincomms/fcaa007
Insel, P. S., Young, C. B., Aisen, P. S., Johnson, K. A., Sperling, R. A., Mormino, E. C., et al. (2023). Tau positron emission tomography in preclinical Alzheimer's disease. Brain 146, 700–711. doi: 10.1093/brain/awac299
Ishida, K., Yamada, K., Nishiyama, R., Hashimoto, T., Nishida, I., Abe, Y., et al. (2022). Glymphatic system clears extracellular tau and protects from tau aggregation and neurodegeneration. J. Exp. Med. 219:e20211275. doi: 10.1084/jem.20211275
Jack, C. R., Wiste, H. J., Schwarz, C. G., Lowe, V. J., Senjem, M. L., Vemuri, P., et al. (2018). Longitudinal tau PET in ageing and Alzheimer's disease. Brain 141, 1517–1528. doi: 10.1093/brain/awy059
Jack, C. R., Wiste, H. J., Botha, H., Weigand, S. D., Therneau, T. M., Knopman, D. S., et al. (2019). The bivariate distribution of amyloid-beta and tau: relationship with established neurocognitive clinical syndromes. Brain 142, 3230–3242. doi: 10.1093/brain/awz268
Jacobs, H. I. L., Hedden, T., Schultz, A. P., Sepulcre, J., Perea, R. D., Amariglio, R. E., et al. (2018). Structural tract alterations predict downstream tau accumulation in amyloid-positive older individuals. Nat. Neurosci. 21, 424–431. doi: 10.1038/s41593-018-0070-z
Jansen, I. E., Savage, J. E., Watanabe, K., Bryois, J., Williams, D. M., Steinberg, S., et al. (2019). Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer's disease risk. Nat. Genet. 51, 404–413. doi: 10.1038/s41588-018-0311-9
Jiang, C., Wang, Q., Xie, S., Chen, Z., Fu, L., Peng, Q., et al. (2022). beta-amyloid discordance of cerebrospinal fluid and positron emission tomography imaging shows distinct spatial tau patterns. Brain Commun. 4:fcac084. doi: 10.1093/braincomms/fcac084
La Joie, R., Visani, A. V., Baker, S. L., Brown, J. A., Bourakova, V., Cha, J., et al. (2020). Prospective longitudinal atrophy in Alzheimer's disease correlates with the intensity and topography of baseline tau-PET. Sci. Transl. Med. 12:eaau5732. doi: 10.1126/scitranslmed.aau5732
La Joie, R., Visani, A. V., Lesman-Segev, O. H., Baker, S. L., Edwards, L., Iaccarino, L., et al. (2021). Association of APOE4 and clinical variability in Alzheimer disease with the pattern of tau- and amyloid-PET. Neurology 96, e650–e661. doi: 10.1212/WNL.0000000000011270
Lee, W. J., Brown, J. A., Kim, H. R., La Joie, R., Cho, H., Lyoo, C. H., et al. (2022). Regional Abeta-tau interactions promote onset and acceleration of Alzheimer's disease tau spreading. Neuron 110:e1935, 1932–1943.e5. doi: 10.1016/j.neuron.2022.03.034
Lill, C. M., Rengmark, A., Pihlstrom, L., Fogh, I., Shatunov, A., Sleiman, P. M., et al. (2015). The role of TREM2 R47H as a risk factor for Alzheimer’s disease, frontotemporal lobar degeneration, amyotrophic lateral sclerosis, and Parkinson’s disease. Alzheimers Dement. 11, 1407–1416. doi: 10.1016/j.jalz.2014.12.009
Lim, Y. Y., and Mormino, E. C., Alzheimer's Disease Neuroimaging, I (2017). APOE genotype and early beta-amyloid accumulation in older adults without dementia. Neurology 89, 1028–1034. doi: 10.1212/WNL.0000000000004336
Lin, Y. T., Seo, J., Gao, F., Feldman, H. M., Wen, H. L., Penney, J., et al. (2018). APOE4 causes widespread molecular and cellular alterations associated with Alzheimer's disease phenotypes in human iPSC-derived brain cell types. Neuron 98:e1147, 1141–1154.e7. doi: 10.1016/j.neuron.2018.05.008
Liu, L., Drouet, V., Wu, J. W., Witter, M. P., Small, S. A., Clelland, C., et al. (2012). Trans-synaptic spread of tau pathology in vivo. PLoS One 7:e31302. doi: 10.1371/journal.pone.0031302
Livingston, G., Sommerlad, A., Orgeta, V., Costafreda, S. G., Huntley, J., Ames, D., et al. (2017). Dementia prevention, intervention, and care. Lancet 390, 2673–2734. doi: 10.1016/S0140-6736(17)31363-6
Lowe, V. J., Bruinsma, T. J., Min, H. K., Lundt, E. S., Fang, P., Senjem, M. L., et al. (2018). Elevated medial temporal lobe and pervasive brain tau-PET signal in normal participants. Alzheimers Dement. 10, 210–216. doi: 10.1016/j.dadm.2018.01.005
Lowe, V. J., Bruinsma, T. J., Wiste, H. J., Min, H. K., Weigand, S. D., Fang, P., et al. (2019). Cross-sectional associations of tau-PET signal with cognition in cognitively unimpaired adults. Neurology 93, e29–e39. doi: 10.1212/WNL.0000000000007728
Mattsson, N., Ossenkoppele, R., Smith, R., Strandberg, O., Ohlsson, T., Jogi, J., et al. (2018). Greater tau load and reduced cortical thickness in APOE epsilon4-negative Alzheimer's disease: a cohort study. Alzheimers Res. Ther. 10:77. doi: 10.1186/s13195-018-0403-x
Mentis, A. A., Dardiotis, E., and Chrousos, G. P. (2021). Apolipoprotein E4 and meningeal lymphatics in Alzheimer disease: a conceptual framework. Mol. Psychiatry 26, 1075–1097. doi: 10.1038/s41380-020-0731-7
Mila-Aloma, M., Ashton, N. J., Shekari, M., Salvado, G., Ortiz-Romero, P., Montoliu-Gaya, L., et al. (2022). Plasma p-tau231 and p-tau217 as state markers of amyloid-beta pathology in preclinical Alzheimer's disease. Nat. Med. 28, 1797–1801. doi: 10.1038/s41591-022-01925-w
Mishra, S., Blazey, T. M., Holtzman, D. M., Cruchaga, C., Su, Y., Morris, J. C., et al. (2018). Longitudinal brain imaging in preclinical Alzheimer disease: impact of APOE epsilon4 genotype. Brain 141, 1828–1839. doi: 10.1093/brain/awy103
Mohamed, N. V., Herrou, T., Plouffe, V., Piperno, N., and Leclerc, N. (2013). Spreading of tau pathology in Alzheimer's disease by cell-to-cell transmission. Eur. J. Neurosci. 37, 1939–1948. doi: 10.1111/ejn.12229
Moscoso, A., Wren, M. C., Lashley, T., Arstad, E., Murray, M. E., Fox, N. C., et al. (2022). Imaging tau pathology in Alzheimer's disease with positron emission tomography: lessons learned from imaging-neuropathology validation studies. Mol. Neurodegener. 17:39. doi: 10.1186/s13024-022-00543-x
Mudher, A., Colin, M., Dujardin, S., Medina, M., Dewachter, I., Alavi Naini, S. M., et al. (2017). What is the evidence that tau pathology spreads through prion-like propagation? Acta Neuropathol. Commun. 5:99. doi: 10.1186/s40478-017-0488-7
Murray, M. E., Graff-Radford, N. R., Ross, O. A., Petersen, R. C., Duara, R., and Dickson, D. W. (2011). Neuropathologically defined subtypes of Alzheimer's disease with distinct clinical characteristics: a retrospective study. Lancet Neurol. 10, 785–796. doi: 10.1016/S1474-4422(11)70156-9
Neitzel, J., Franzmeier, N., Rubinski, A., Dichgans, M., Brendel, M., Malik, R., et al. (2021). KL-VS heterozygosity is associated with lower amyloid-dependent tau accumulation and memory impairment in Alzheimer's disease. Nat. Commun. 12:3825. doi: 10.1038/s41467-021-23755-z
Ossenkoppele, R., Jansen, W. J., Rabinovici, G. D., Knol, D. L., van der Flier, W. M., van Berckel, B. N., et al. (2015). Prevalence of amyloid PET positivity in dementia syndromes: a meta-analysis. JAMA 313, 1939–1949. doi: 10.1001/jama.2015.4669
Ossenkoppele, R., Leuzy, A., Cho, H., Sudre, C. H., Strandberg, O., Smith, R., et al. (2021a). The impact of demographic, clinical, genetic, and imaging variables on tau PET status. Eur. J. Nucl. Med. Mol. Imaging 48, 2245–2258. doi: 10.1007/s00259-020-05099-w
Ossenkoppele, R., Lyoo, C. H., Sudre, C. H., van Westen, D., Cho, H., Ryu, Y. H., et al. (2020). Distinct tau PET patterns in atrophy-defined subtypes of Alzheimer's disease. Alzheimers Dement. 16, 335–344. doi: 10.1016/j.jalz.2019.08.201
Ossenkoppele, R., Schonhaut, D. R., Scholl, M., Lockhart, S. N., Ayakta, N., Baker, S. L., et al. (2016). Tau PET patterns mirror clinical and neuroanatomical variability in Alzheimer's disease. Brain 139, 1551–1567. doi: 10.1093/brain/aww027
Ossenkoppele, R., Smith, R., Mattsson-Carlgren, N., Groot, C., Leuzy, A., Strandberg, O., et al. (2021b). Accuracy of tau positron emission tomography as a prognostic marker in preclinical and prodromal Alzheimer disease: a Head-to-Head comparison against amyloid positron emission tomography and magnetic resonance imaging. JAMA Neurol. 78, 961–971. doi: 10.1001/jamaneurol.2021.1858
Pascoal, T. A., Benedet, A. L., Ashton, N. J., Kang, M. S., Therriault, J., Chamoun, M., et al. (2021). Microglial activation and tau propagate jointly across Braak stages. Nat. Med. 27, 1592–1599. doi: 10.1038/s41591-021-01456-w
Piccio, L., Deming, Y., Del-Aguila, J. L., Ghezzi, L., Holtzman, D. M., Fagan, A. M., et al. (2016). Cerebrospinal fluid soluble TREM2 is higher in Alzheimer disease and associated with mutation status. Acta Neuropathol. 131, 925–933. doi: 10.1007/s00401-016-1533-5
Pichet Binette, A., Franzmeier, N., Spotorno, N., Ewers, M., Brendel, M., Biel, D., et al. (2022). Amyloid-associated increases in soluble tau relate to tau aggregation rates and cognitive decline in early Alzheimer's disease. Nat. Commun. 13:6635. doi: 10.1038/s41467-022-34129-4
Pickett, E. K., Henstridge, C. M., Allison, E., Pitstick, R., Pooler, A., Wegmann, S., et al. (2017). Spread of tau down neural circuits precedes synapse and neuronal loss in the rTgTauEC mouse model of early Alzheimer's disease. Synapse 71:e21965. doi: 10.1002/syn.21965
Pontecorvo, M. J., Devous, M. D., Kennedy, I., Navitsky, M., Lu, M., Galante, N., et al. (2019). A multicentre longitudinal study of flortaucipir (18F) in normal ageing, mild cognitive impairment and Alzheimer's disease dementia. Brain 142, 1723–1735. doi: 10.1093/brain/awz090
Pooler, A. M., Phillips, E. C., Lau, D. H., Noble, W., and Hanger, D. P. (2013). Physiological release of endogenous tau is stimulated by neuronal activity. EMBO Rep. 14, 389–394. doi: 10.1038/embor.2013.15
Pooler, A. M., Polydoro, M., Maury, E. A., Nicholls, S. B., Reddy, S. M., Wegmann, S., et al. (2015). Amyloid accelerates tau propagation and toxicity in a model of early Alzheimer's disease. Acta Neuropathol. Commun. 3:14. doi: 10.1186/s40478-015-0199-x
Ramanan, V. K., Castillo, A. M., Knopman, D. S., Graff-Radford, J., Lowe, V. J., Petersen, R. C., et al. (2019). Association of Apolipoprotein E varepsilon4, educational level, and sex with tau deposition and tau-mediated metabolic dysfunction in older adults. JAMA Netw. Open 2:e1913909. doi: 10.1001/jamanetworkopen.2019.13909
Ridge, P. G., Hoyt, K. B., Boehme, K., Mukherjee, S., Crane, P. K., Haines, J. L., et al. (2016). Assessment of the genetic variance of late-onset Alzheimer's disease. Neurobiol. Aging 41, 200.e13–200.e20. doi: 10.1016/j.neurobiolaging.2016.02.024
Rubinski, A., Frerich, S., Malik, R., Franzmeier, N., Ramirez, A., Dichgans, M., et al. (2023). Polygenic effect on tau pathology progression in Alzheimer's disease. Ann. Neurol. 93, 819–829. doi: 10.1002/ana.26588
Sanchez, J. S., Becker, J. A., Jacobs, H. I. L., Hanseeuw, B. J., Jiang, S., Schultz, A. P., et al. (2021). The cortical origin and initial spread of medial temporal tauopathy in Alzheimer's disease assessed with positron emission tomography. Sci. Transl. Med. 13:eabc0655. doi: 10.1126/scitranslmed.abc0655
Scholl, M., Ossenkoppele, R., Strandberg, O., Palmqvist, S., Swedish Bio, F. S., Jogi, J., et al. (2017). Distinct 18F-AV-1451 tau PET retention patterns in early- and late-onset Alzheimer's disease. Brain 140, 2286–2294. doi: 10.1093/brain/awx171
Schoonhoven, D. N., Coomans, E. M., Millan, A. P., van Nifterick, A. M., Visser, D., Ossenkoppele, R., et al. (2022). Tau protein spreads through functionally connected neurons in Alzheimer’s disease. Alzheimers Dement. 18:e066494. doi: 10.1002/alz.066494
Shi, Y., Yamada, K., Liddelow, S. A., Smith, S. T., Zhao, L., Luo, W., et al. (2017). ApoE4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy. Nature 549, 523–527. doi: 10.1038/nature24016
Sperling, R. A., Aisen, P. S., Beckett, L. A., Bennett, D. A., Craft, S., Fagan, A. M., et al. (2011). Toward defining the preclinical stages of Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 7, 280–292. doi: 10.1016/j.jalz.2011.03.003
Strikwerda-Brown, C., Hobbs, D. A., Gonneaud, J., St-Onge, F., Binette, A. P., Ozlen, H., et al. (2022). Association of Elevated Amyloid and tau Positron Emission Tomography Signal with Near-Term Development of Alzheimer disease symptoms in older adults without cognitive impairment. JAMA Neurol. 79, 975–985. doi: 10.1001/jamaneurol.2022.2379
Suarez-Calvet, M., Araque Caballero, M. A., Kleinberger, G., Bateman, R. J., Fagan, A. M., Morris, J. C., et al. (2016). Early changes in CSF sTREM2 in dominantly inherited Alzheimer's disease occur after amyloid deposition and neuronal injury. Sci. Transl. Med. 8:369ra178. doi: 10.1126/scitranslmed.aag1767
Taga, M., Petyuk, V. A., White, C., Marsh, G., Ma, Y., Klein, H. U., et al. (2020). BIN1 protein isoforms are differentially expressed in astrocytes, neurons, and microglia: neuronal and astrocyte BIN1 are implicated in tau pathology. Mol. Neurodegener. 15:44. doi: 10.1186/s13024-020-00387-3
Takeda, S., Wegmann, S., Cho, H., DeVos, S. L., Commins, C., Roe, A. D., et al. (2015). Neuronal uptake and propagation of a rare phosphorylated high-molecular-weight tau derived from Alzheimer's disease brain. Nat. Commun. 6:8490. doi: 10.1038/ncomms9490
Therriault, J., Benedet, A. L., Pascoal, T. A., Mathotaarachchi, S., Chamoun, M., Savard, M., et al. (2020). Association of Apolipoprotein E epsilon4 with medial temporal tau independent of amyloid-beta. JAMA Neurol. 77, 470–479. doi: 10.1001/jamaneurol.2019.4421
Toledo, J. B., Habes, M., Sotiras, A., Bjerke, M., Fan, Y., Weiner, M. W., et al. (2019). APOE effect on amyloid-beta PET spatial distribution, deposition rate, and cut-points. J. Alzheimers Dis. 69, 783–793. doi: 10.3233/JAD-181282
Vogel, J. W., Iturria-Medina, Y., Strandberg, O. T., Smith, R., Levitis, E., Evans, A. C., et al. (2020). Spread of pathological tau proteins through communicating neurons in human Alzheimer's disease. Nat. Commun. 11:2612. doi: 10.1038/s41467-020-15701-2
Vogel, J. W., Young, A. L., Oxtoby, N. P., Smith, R., Ossenkoppele, R., Strandberg, O. T., et al. (2021). Four distinct trajectories of tau deposition identified in Alzheimer's disease. Nat. Med. 27, 871–881. doi: 10.1038/s41591-021-01309-6
Voskobiynyk, Y., Roth, J. R., Cochran, J. N., Rush, T., Carullo, N. V., Mesina, J. S., et al. (2020). Alzheimer's disease risk gene BIN1 induces tau-dependent network hyperexcitability. elife 9:e57354. doi: 10.7554/eLife.57354
Wang, L., Benzinger, T. L., Su, Y., Christensen, J., Friedrichsen, K., Aldea, P., et al. (2016). Evaluation of tau imaging in staging Alzheimer disease and revealing interactions between beta-amyloid and Tauopathy. JAMA Neurol. 73, 1070–1077. doi: 10.1001/jamaneurol.2016.2078
Wang, C., Najm, R., Xu, Q., Jeong, D. E., Walker, D., Balestra, M. E., et al. (2018). Gain of toxic apolipoprotein E4 effects in human iPSC-derived neurons is ameliorated by a small-molecule structure corrector. Nat. Med. 24, 647–657. doi: 10.1038/s41591-018-0004-z
Wegmann, S., Bennett, R. E., Delorme, L., Robbins, A. B., Hu, M., McKenzie, D., et al. (2019). Experimental evidence for the age dependence of tau protein spread in the brain. Sci. Adv. 5:eaaw6404. doi: 10.1126/sciadv.aaw6404
Whitwell, J. L., Dickson, D. W., Murray, M. E., Weigand, S. D., Tosakulwong, N., Senjem, M. L., et al. (2012). Neuroimaging correlates of pathologically defined subtypes of Alzheimer's disease: a case-control study. Lancet Neurol. 11, 868–877. doi: 10.1016/S1474-4422(12)70200-4
Wightman, D. P., Jansen, I. E., Savage, J. E., Shadrin, A. A., Bahrami, S., Holland, D., et al. (2021). A genome-wide association study with 1,126,563 individuals identifies new risk loci for Alzheimer's disease. Nat. Genet. 53, 1276–1282. doi: 10.1038/s41588-021-00921-z
Wu, J. W., Herman, M., Liu, L., Simoes, S., Acker, C. M., Figueroa, H., et al. (2013). Small misfolded tau species are internalized via bulk endocytosis and anterogradely and retrogradely transported in neurons. J. Biol. Chem. 288, 1856–1870. doi: 10.1074/jbc.M112.394528
Wu, J. W., Hussaini, S. A., Bastille, I. M., Rodriguez, G. A., Mrejeru, A., Rilett, K., et al. (2016). Neuronal activity enhances tau propagation and tau pathology in vivo. Nat. Neurosci. 19, 1085–1092. doi: 10.1038/nn.4328
Yamada, K., Holth, J. K., Liao, F., Stewart, F. R., Mahan, T. E., Jiang, H., et al. (2014). Neuronal activity regulates extracellular tau in vivo. J. Exp. Med. 211, 387–393. doi: 10.1084/jem.20131685
Young, C. B., Johns, E., Kennedy, G., Belloy, M. E., Insel, P. S., Greicius, M. D., et al. (2023). APOE effects on regional tau in preclinical Alzheimer's disease. Mol. Neurodegener. 18:1. doi: 10.1186/s13024-022-00590-4
Young, C. B., Winer, J. R., Younes, K., Cody, K. A., Betthauser, T. J., Johnson, S. C., et al. (2022). Divergent cortical tau positron emission tomography patterns among patients with preclinical Alzheimer disease. JAMA Neurol. 79, 592–603. doi: 10.1001/jamaneurol.2022.0676
Yuste-Checa, P., Trinkaus, V. A., Riera-Tur, I., Imamoglu, R., Schaller, T. F., Wang, H., et al. (2021). The extracellular chaperone Clusterin enhances tau aggregate seeding in a cellular model. Nat. Commun. 12:4863. doi: 10.1038/s41467-021-25060-1
Keywords: Alzheimer’s disease, tau, propagation, neurodegeneration, amyloid, PET, fMRI
Citation: Lamontagne-Kam D, Ulfat AK, Hervé V, Vu T-M and Brouillette J (2023) Implication of tau propagation on neurodegeneration in Alzheimer’s disease. Front. Neurosci. 17:1219299. doi: 10.3389/fnins.2023.1219299
Edited by:
Wendy Noble, University of Exeter, United KingdomReviewed by:
Efthimios M. C. Skoulakis, Alexander Fleming Biomedical Sciences Research Center, GreeceRichard Killick, King's College London, United Kingdom
Copyright © 2023 Lamontagne-Kam, Ulfat, Hervé, Vu and Brouillette. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jonathan Brouillette, am9uYXRoYW4uYnJvdWlsbGV0dGVAdW1vbnRyZWFsLmNh