
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Neurosci., 29 June 2023
Sec. Neurodevelopment
Volume 17 - 2023 | https://doi.org/10.3389/fnins.2023.1219244
This article is part of the Research TopicSLC6A1: The Past, Present and FutureView all 8 articles
 Davide Caputo1
Davide Caputo1 Silvana Franceschetti2
Silvana Franceschetti2 Barbara Castellotti3*
Barbara Castellotti3* Elena Freri1G. Zorzi1
Elena Freri1G. Zorzi1 Veronica Saletti1
Veronica Saletti1 Laura Canafoglia4
Laura Canafoglia4 Tiziana Granata1
Tiziana Granata1We report the clinical and EEG data of two patients harboring heterozygous SLC6A1 mutations, who presented with typical absence seizures at 3 Hz spike and wave as well as with mild cognitive disability. Neuroradiological and other laboratory investigations were normal. Our observations suggest that SLC6A1 mutations can be suspected in children with typical absences as the only seizure type, especially if associated with, even mild, cognitive deficits.
The Solute Carrier Family 6 Member 1 (SLC6A1) gene encodes GAT-1, a voltage-dependent gamma-aminobutyric acid (GABA) transporter that is responsible for the re-uptake of GABA from the synapse. GABA is the principal inhibitory neurotransmitter that counterbalances neuronal excitation in the brain. Disruption of this inhibitory balance can result in seizures and other neurodevelopmental disorders (Mermer et al., 2021). To date, pathogenic variants in SLC6A1 have been associated with a complex neurodevelopmental disorder mainly characterized by mild-to-severe developmental delay or intellectual disability, epilepsy, movement disorders, and neurobehavioral and/or psychiatric manifestations (Goodspeed et al., 2023). Seizures are among the main symptoms in almost 90% of the described patients. After initial descriptions (Dikow et al., 2014; Carvill et al., 2015), two main case series (Johannesen et al., 2018; Goodspeed et al., 2020) indicated that pathogenic variants in the SLC6A1 gene are frequently associated with severe Epilepsy with Myoclonic-Atonic Seizures [EMAtS, previously termed Myoclonic-Astatic Epilepsy (MAE)], often preceded by developmental delay. Absence seizures are commonly reported, but usually in the context of MAE, and not as the only type of seizure (Carvill et al., 2015; Johannesen et al., 2018; Posar and Visconti, 2019; Goodspeed et al., 2020; Mori et al., 2023).
In this report, we describe the clinical and EEG data of two patients with mild cognitive disability in whom epilepsy presented with typical absences, completely controlled by anti-seizure medications.
Case 1 is a 20-year-old male. No familiar history of any neurological disease was reported. Pregnancy ended in cesarean section at 35 weeks of gestation due to premature membrane rupture. His Apgar score was 7 at 1′, 9 at 5′, and 10 at 10′. At the age of 2 years, the child was first evaluated for developmental delay. No signs suggestive of autism spectrum disorder were noticed. MRI and EEG were normal. Array CGH detected a 5p15.2 microdeletion inherited from his asymptomatic father. At the age of 5 years, the child began experiencing daily episodes of reduced awareness and mild oro-mandibular automatisms. Video EEG documented absence seizures at 3-Hz spike and wave discharges (Figure 1A). The EEG background activity and sleep organization were normal. Absences were controlled by combined valproic acid and ethosuximide treatment. Initial genetic screening, including karyotyping and FMR1 and SLC2A1 sequence analyses failed to detect variants. At 17 years of age, a multigene epilepsy panel (see Supplementary material) revealed a de novo variant in the SLC6A1 gene (NM_003042.3): c.1648G>A/p.Gly550Arg, (rs886042046) (Mattison et al., 2018; Trinidad et al., 2022). Segregation analysis ruled out the presence of the variant in both of the healthy parents.
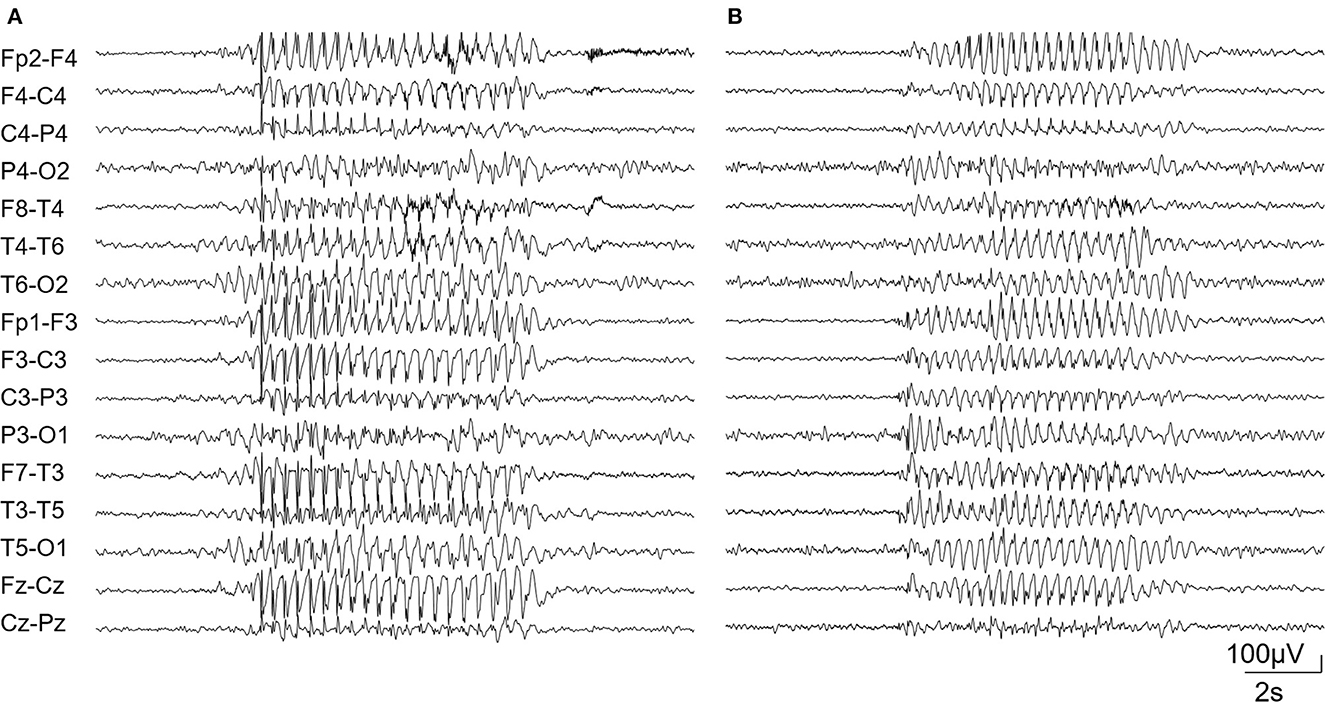
Figure 1. Diffuse bursts of the 3-Hz spike and wave complex in patient 1 (A) and 2 (B).
Case 2 is a 9-year-old boy, born at term after normal pregnancy. His Apgar score was 6 at 1′ and 9 at 5′. Family history for neurological disease was negative. Starting from the age of 12 months, a slight delay in psychomotor development was evident: the infant was able to sit unassisted at 10 months and to walk at 16 months. His Developmental Quotient, assessed using the Griffith's Scale, was 71 at the age of 12 months. At this age, the EEG was normal. The child began experiencing daily episodes of reduced awareness at the age of 5 years. EEG confirmed the presence of absence seizures associated with 3Hz spike and wave discharges (Figure 1B). Treatment with valproic acid led to seizure control. Early diagnostic investigations, including organic urinary acids, plasmatic amino acids, FMR1 analysis, array-CGH, and MRI were normal. At 6 years old, the Wechsler Intelligence Scale for Children (WISC-IV) showed a mild intellectual disability (total IQ = 67). No signs suggestive of autism spectrum disorder were noticed. At 7 years of age, a multigene epilepsy panel (see Supplementary material) showed the variant (NM_003042.3): c.1084G>A/p.Gly362Arg (rs1131691302) in the SLC6A1 gene (Johannesen et al., 2018). Variant segregation in family members was not possible. The clinical and genetic data of the two patients are summarized in Table 1.
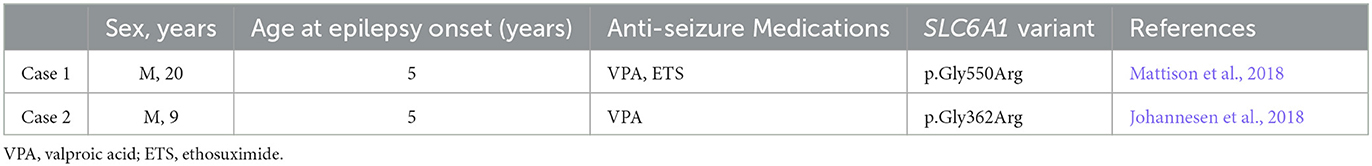
Table 1. Clinical aspects of the two patients described.
Our report shows that mutations in the SLC6A1 gene may lead to typical absence seizures associated with mild intellectual disability. The association between epilepsy and developmental delay/cognitive disability is known to occur in SCL6A1 patients; absences are the most frequently reported seizure in these patients (Johannesen et al., 2018; Goodspeed et al., 2020); however, they have been mostly reported in the context of EMAtS or in the context of epilepsy that did not fit codified epileptic syndromes.
The presence of seizures in patients carrying SLC6A1 mutations is likely related to impairment of inhibitory neurotransmission induced by the genetic anomaly. SCL6A1 encodes GAT-1, one of the major GABA transporters in the brain, responsible for the re-uptake of GABA from the synapses. This hypothesis is supported by experimental models that demonstrate dysfunctional GABA transmission in spontaneous or induced rodent models of absence epilepsy, with spike-wave discharges. Moreover, Cope et al. (2009) found that Slc6a1-knockout mice develop spike-wave discharges characteristic of absence seizures. The pathophysiological mechanism that generates spike and wave discharges involves a complex circuitry that includes the thalamus, cortex, and basal ganglia. The loss of GAT-1 function leads to higher levels of GABA in the thalamus, but not in the cortex. The resulting enhanced tonic GABA-A inhibition in both thalamo-cortical neurons (see Crunelli et al., 2020 for a review) and ventrobasal thalamo-cortical neurons is sufficient to elicit absence seizures even in wild-type rodents (Trinidad et al., 2022). The complexity of the network may explain the variable seizure phenotype and severity, ranging from pure absences to more severe EMAts encephalopathy, as well as the variable degree of cognitive impairment (Goodspeed et al., 2023).
The p.Gly550Arg variant, detected in our case 1, has been described in a patient with generalized epilepsy and absence and tonic-clonic seizures (Mattison et al., 2018). The p.Gly362Arg variant, detected in our case 2, has been reported in two cases (Johannesen et al., 2018): a 1-year old child with Lennox-Gastaut syndrome and a child with temporal lobe epilepsy. We cannot find a reason for the extreme phenotypical variability associated with SLC6A1 variants, we can only hypothesize on the influence of genetic background and epigenetic factors.
Our data suggest that comprehensive genetic evaluation, including molecular analysis for monogenic conditions associated with epilepsy, is warranted in children with typical absences associated with intellectual disability.
The original contributions presented in the study are included in the article/Supplementary material, further inquiries can be directed to the corresponding author.
Written informed consent was obtained from the individual(s), and minor(s)' legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
DC and SF contributed to the preparation of the original draft. GZ and VS were involved in the patient treatment. BC performed genetic analysis. LC, EF, and TG reviewed the manuscript. All authors contributed to the writing of the final manuscript.
This work was supported supported by the Italian Ministry of Health (RRC).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnins.2023.1219244/full#supplementary-material
Carvill, G. L., McMahon, J. M., Schneider, A., Zemel, M., Myers, C. T., Saykally, J., et al. (2015). Mutations in the GABA transporter SLC6A1 cause epilepsy with myoclonic-atonic seizures. Am. J. Hum. Genet. 96, 808–815. doi: 10.1016/j.ajhg.2015.02.016
Cope, D. W., Giovanni, D. i., Fyson, G., Orban, S. J., Errington, G., Lorincz, A. C., et al. (2009). Enhanced tonic GABA-A inhibition in typical absence epilepsy. Nat. Med. 15, 1392–1398. doi: 10.1038/nm.2058
Crunelli, V., Lorincz, M. L., McCafferty, C., Lambert, R. C., Leresche, N., Di Giovanni, G., et al. (2020). Clinical and experimental insight into pathophysiology, comorbidity and therapy of absence seizures. Brain 143, 2341–2368. doi: 10.1093/brain/awaa072
Dikow, N., Maas, B., Karch, S., Granzow, M., Janssen, J. W., Jauch, A., et al. (2014). 3p25, 3. microdeletion of GABA transporters SLC6A1 and SLC6A11 results in intellectual disability, epilepsy and stereotypic behavior. Am. J. Med. Genet. A. 164A, 3061–3068. doi: 10.1002/ajmg.a.36761
Goodspeed, K., Demarest, S., Johannesen, K., Kang, J., Lal, D., Angione, K., et al. (2023). “SLC6A1-Related Neurodevelopmental Disorder,” in GeneReviews® eds. M. P., Adam, G. M., Mirzaa, R. A., Pagon, S. E., Wallace, L. J. H., Bean, K. W., Gripp, et al. (Seattle, WA: University of Washington, Seattle) 1993–2023.
Goodspeed, K., Pérez-Palma, E., Iqbal, S., Cooper, D., Scimemi, A., Johannesen, K. M., et al. (2020). Current knowledge of SLC6A1-related neurodevelopmental disorders. Brain Commun. 2, fcaa170. doi: 10.1093/braincomms/fcaa170
Johannesen, K. M., Gardella, E., Linnankivi, T., Courage, C., Saint Martin, D. e., Lehesjoki, A., et al. (2018). Defining the phenotypic spectrum of SLC6A1 mutations. Epilepsia 59, 389–402. doi: 10.1111/epi.13986
Mattison, K. A., Butler, K. M., Inglis, G. A. S., Dayan, O., Boussidan, H., Bhambhani, V., et al. (2018). SLC6A1 variants identified in epilepsy patients reduce γ-aminobutyric acid transport. Epilepsia. 59, e135–e141. doi: 10.1111/epi.14531
Mermer, F., Poliquin, S., Rigsby, K., Rastogi, A., Shen, W., Romero-Morales, A., et al. (2021). Common molecular mechanisms of SLC6A1 variant-mediated neurodevelopmental disorders in astrocytes and neurons. Brain. 144, 2499–2512. doi: 10.1093/brain/awab207
Mori, T., Sakamoto, M., Tayama, T., Goji, A., Toda, Y., Fujita, A., et al. (2023). A case of epilepsy with myoclonic atonic seizures caused by SLC6A1 gene mutation due to balanced chromosomal translocation. Brain Develop. 23, S0387-7604(23)00044-X. doi: 10.1016/j.braindev.2023.03.001
Posar, A., and Visconti, P. (2019). Mild phenotype associated with SLC6A1 gene mutation: A case report with literature review. J. Pediatr. Neurosci. 14, 100–102. doi: 10.4103/jpn.JPN_2_19
Keywords: absence, epilepsy, SLC6A1, intellectual disability, GABA
Citation: Caputo D, Franceschetti S, Castellotti B, Freri E, Zorzi G, Saletti V, Canafoglia L and Granata T (2023) Case report: SLC6A1 mutations presenting with isolated absence seizures: description of 2 novel cases. Front. Neurosci. 17:1219244. doi: 10.3389/fnins.2023.1219244
Received: 08 May 2023; Accepted: 05 June 2023;
Published: 29 June 2023.
Edited by:
Katrine M. Johannesen, Rigshospitalet, DenmarkReviewed by:
Christian Malte Boßelmann, University of Tübingen, GermanyCopyright © 2023 Caputo, Franceschetti, Castellotti, Freri, Zorzi, Saletti, Canafoglia and Granata. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Barbara Castellotti, YmFyYmFyYS5jYXN0ZWxsb3R0aUBpc3RpdHV0by1iZXN0YS5pdA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.