 Wan Zhou
Wan Zhou Renshi Xu
Renshi Xu
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Neurosci. , 10 August 2023
Sec. Neurodegeneration
Volume 17 - 2023 | https://doi.org/10.3389/fnins.2023.1189470
Amyotrophic lateral sclerosis (ALS) is a progressive and fatal neurodegenerative disease that leads to the massive loss of motor neurons in cerebrum, brain stem and spinal cord. It affects not only motor neurons but also other neurons and glial cells, resulting in the progressive muscle atrophy, the severe disability and the eventual death due to the respiratory failure. The pathogenesis of ALS is not fully understood. Currently, several factors are considered to be involved in the pathogenesis of ALS, such as genetic factors, imbalances in protein homeostasis, RNA metabolism disorders, mitochondrial dysfunctions, glutamate-mediated excitatory toxicities and intra-neuronal material transport disorders in neurons. The study of genetic mutations related to ALS pathogenesis will link the molecular and cellular mechanisms of the disease, thus enhancing the understanding of its occurrence and progression, thereby providing new insights for the pathogenesis of ALS. This review summarizes the current insights in the molecular genetic pathogenesis of ALS.
Amyotrophic lateral sclerosis (ALS) is a rare chronic progressive motor neuron disease that impairs voluntary muscle control and movement. The main pathological features of ALS is the damage of motor neurons from motor cortex to neuromuscular system, including the defects of cerebrum, brain stem, spinal cord and neuromuscular junction, leading to the skeletal muscle decomposition and generalization, progressive atrophy (Fiscon et al., 2021). ALS is one of the most common neurodegenerative diseases with an incidence rate of 1 to 5 per 100,000 populations. Clinically, ALS is manifested by muscle weakness and atrophy, spasm, and contracture (Herrando-Grabulosa et al., 2021). The ALS progresses rapidly and the survival time after the onset is usually 3–5 years. The main cause of death is the respiratory failure due to the respiratory muscle paralysis and atrophy. ALS can be generally classified into both familial and sporadic ALS, the familial ALS accounts for about 5%–10% of ALS and involves the genetic mutations in genes such as superoxide dismutase 1 (SOD1), chromosome 9 open reading frame 72 (C9orf72), fused in sarcoma (FUS), TAR DNA-binding protein (TDP-43/TARDBP), and others. The sporadic ALS cases account for about 90%–95% of the disease, and the pathogenesis of ALS is not still completely clear (Je et al., 2021), which may be caused by genetics and related factors together (Fernández-Ruiz et al., 2021).
The pathogenesis of ALS involves in the complex processes involving the multiple genes and the multiple pathways. A variety of genes and pathways (Figure 1) are involved in the pathogenesis of ALS, such as the imbalance of protein homeostasis in neurons, the disorder of ribonucleic acid (RNA) metabolism, the mitochondrial dysfunction, the glutamate-mediated excitatory toxicity and the disturbance of intra-neuronal material transport. So far, more than 50 genes have been found to be associated with ALS (Li et al., 2023). Among them, C9orf72, SOD1, TDP-43 and FUS genes were most closely related to the pathogenesis of ALS (Kim et al., 2020). The mutations of various related genes play the different roles in the different pathways in the pathogenesis of ALS, which are interrelated and act together to lead to the occurrence and development pathogenesis of ALS.
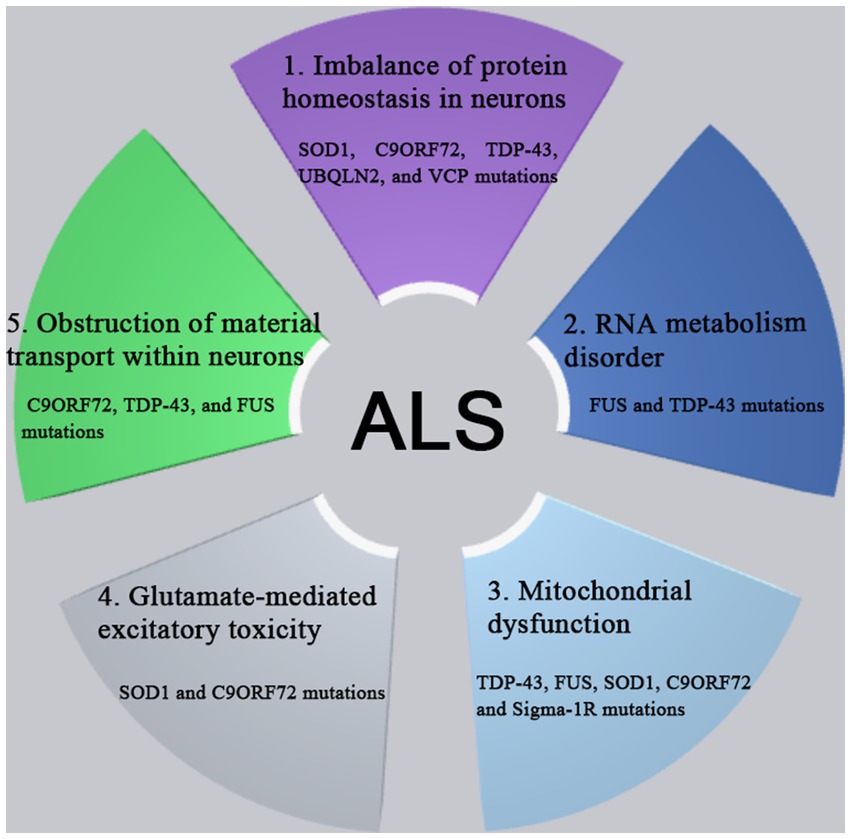
Figure 1. The pathogenesis of ALS involves complex processes with multiple genes and multiple pathways dysfunction, such as the imbalance of protein homeostasis in neurons, the disorder of RNA metabolism, the mitochondrial dysfunction, the glutamate-mediated excitatory toxicity and the disturbance of intra-neuronal material transport. So far, more than 50 genes have been found to be associated with ALS. Among them, C9orf72, SOD1, TDP-43, and FUS genes were most closely related to the pathogenesis of ALS. The mutations of various related genes play the different roles in the different pathways in the pathogenesis of ALS, which are interrelated and act together to lead to the occurrence and development pathogenesis of ALS.
Mutations in genes such as SOD1, C9orf72, TDP-43, ubiquilin-2, and valosin-containing protein (VCP) may lead to protein homeostasis imbalance through interference with the ubiquitin-proteasome system (UPS) and autophagy pathway, resulting in abnormal accumulation of toxic proteins and eventually leading to cell death (Beckers et al., 2021; Chua et al., 2022). FUS and TDP-43 are RNA-binding proteins (RBPs) associated with RNA metabolism (Hallegger et al., 2021). Mutations in these proteins cause abnormal localization and aggregation of FUS and TDP-43 in cells, affecting RNA metabolism and cellular function, ultimately leading to neurodegeneration (Ling et al., 2013). Mitochondrial dysfunction plays a crucial role in the pathogenesis of ALS as it affects multiple cellular pathways, including energy production, Ca2+ regulation, apoptosis, and protein quality control (Boyman et al., 2020). Mutations in ALS-associated genes such as TDP-43, FUS, SOD1, C9orf72, and Sigma-1 receptor (Sigma-1R) can alter mitochondrial integrity and function (Smith et al., 2019; Shoshan-Barmatz et al., 2020; Dafinca et al., 2021; Wood et al., 2021). Excitotoxicity is caused by excessive activation of glutamate receptors, inability to clear glutamate neurotransmitter, or increased postsynaptic sensitivity to glutamate. Glutamate transporters in astrocytes, such as excitatory amino acid transporters 1 (EAAT1)/glutamate–aspartate transporters (GLAST) and excitatory amino acid transporters 2 (EAAT2)/glial glutamate transporter-1 (GLT-1), maintain proper extracellular glutamate levels, preventing excitotoxicity. Aberrant post-translational modification pathways of EAAT1/GLAST and EAAT2/GLT-1 are associated with the pathogenesis of ALS (Diaper et al., 2013; Pajarillo et al., 2019; Hirschberg et al., 2021). Axonal transport pathways in motor neurons maintain their normal structure and function, and mutations in C9orf72, TDP-43, and FUS are associated with neuronal transport impairments (Burk and Pasterkamp, 2019; Pang and Hu, 2021).
At present, the study field of ALS continues to rapidly develop with the discovery of multiple disease related genes each year. The rare genes associated with the pathogenesis of ALS include glycosyltransferase 8 domain (GLT8D), carbonic anhydrase 1 (CA1) and serine palmitoyltransferase long chain base subunit 1 (SPTLC1) genes, and a series of new risk and modifying factors. A study (Cooper-Knock et al., 2019) has found a p.R92C mutation in the exon 4 of the gene encoding the glycosyltransferase GLT8D1, which is co-segregated with the disease in a family with the autosomal dominant ALS. In addition, it was discovered that the familial ALS patients exhibit a significant enrichment of additional rare deleterious variants in the exon 4 of GLT8D1, which clusters near the substrate-binding domain of the exon and reduces the enzyme activity of GLT8D1 (Cao et al., 2021; Candelise et al., 2022). More and more evidence suggests that hippocampus is involved in the pathogenesis of ALS (Machts et al., 2018), the characteristic alteration in ALS is the overall volume loss and the local atrophy of the CA1 area of hippocampus, representing the neuronal correlates of the cognitive and behavioral deficits frequently encountered in this ALS disease. Recently, it has been discovered that the mutations in the SPTLC1 subunit of serine palmitoyltransferase (SPT) catalyzes the first step of de novo synthesis of sphingolipids and leads to the childhood-onset ALS (Johnson et al., 2021; Mohassel et al., 2021). The variants in SPTLC1 shifts the usage of SPT amino acid from serine to alanine, leading to the elevated levels of deoxysphingolipids and presenting as an alternating phenotype of inherited sensory and autonomic neuropathy, and being characterized by the progressive sensory loss with the varying degrees of autonomic involvement (Mohassel et al., 2021). This article provides the current insights of molecular genetic mechanisms underlying the pathogenesis of ALS based on the currently recognized relationship between genetic genes and molecular pathways.
The maintenance of protein homeostasis is essential for cells to perform theirs functions (Figure 2). The protein imbalance can lead to the accumulation of misfolded proteins and ultimately cell death. Compared with other cell types, neurons are particularly susceptible to protein balance dysfunction (Kundra et al., 2020). The abnormal proteins accumulate excessively in neurons to form the inclusion bodies with the different characteristics and play the neurotoxic roles, resulting in the neuronal degeneration and necrosis. The degradation of abnormal proteins was mainly degraded through the intracellular protein UPS and the autophagy system. So far, more than 50 genes have been found to be associated with the pathogenesis of ALS (Li et al., 2023). And the degradation of abnormal proteins was mainly through the pathways of intracellular protein UPS and autophagy system degradation. Among more than 50 genes related to ALS, a variety of genes (SOD1, C9orf72, TDP-43, ubiquilin-2, VCP, etc.) directly affect the pathways of UPS and autophagy system (Nassif et al., 2017; Li et al., 2023).
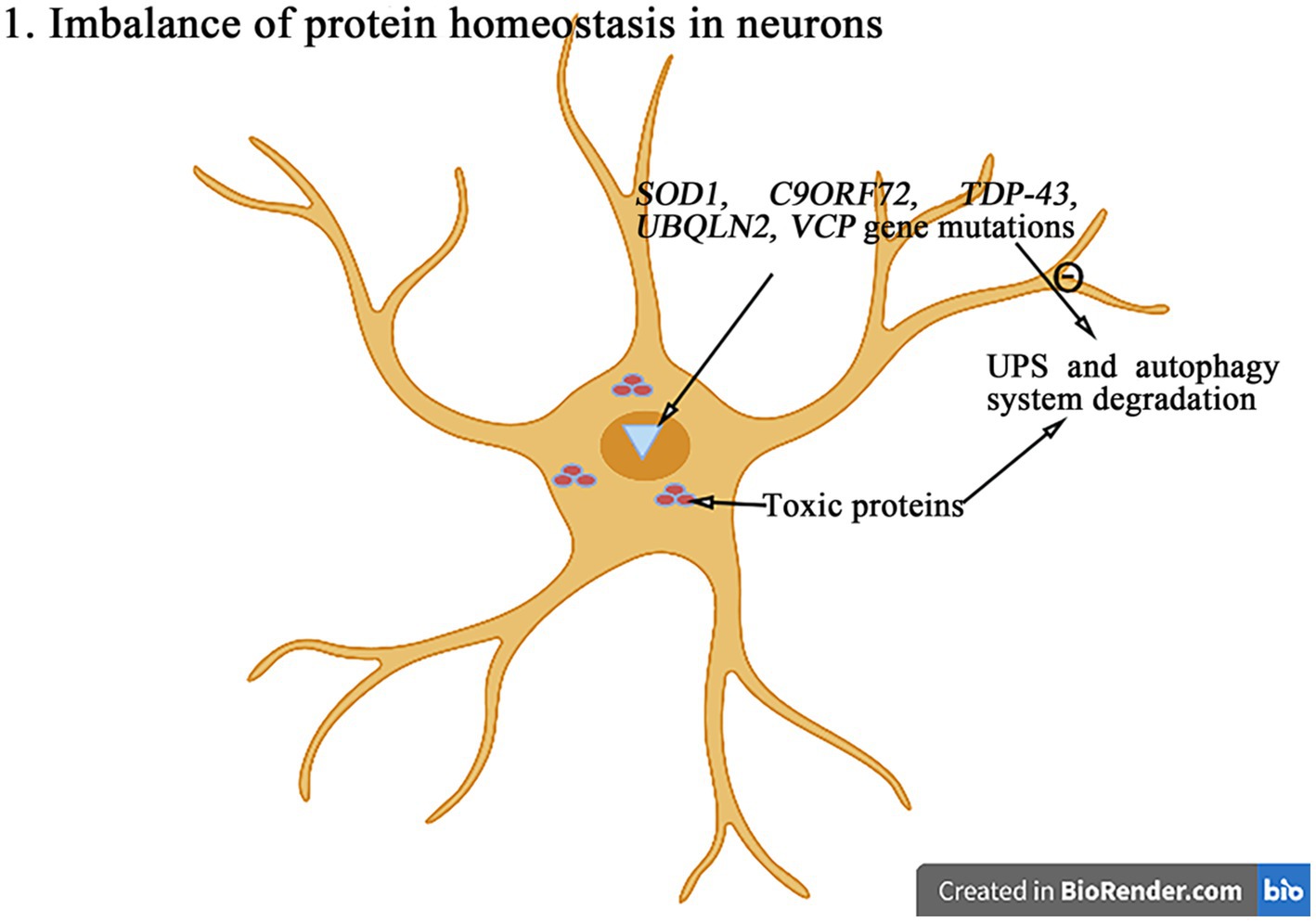
Figure 2. The mutations in genes such as SOD1, C9orf72, TDP-43, ubiquilin-2, and VCP can interfere with the UPS and autophagy degradation pathways, leading to the abnormal accumulation of toxic proteins and then leading to the protein homeostasis imbalance and ultimately inducing the cell death.
A dominant missense mutation in the gene encoding SOD1 was the first to be found to cause ALS, accounting for approximate 20% of all cases of familial ALS, in which the signature pathological feature is an insoluble SOD1 ubiquitin-positive inclusion body within motor neurons. The chaperones molecules are the facilitators of protein folding and synthesis. The chaperon-assisted proteasome degradation is a system that facilitates the degradation of abnormal proteins by bridging misfolded and aggregated proteins to heat shock protein 70 (HSP70). This system participates in maintaining the protein homeostasis and promotes the refolding or removal of potentially toxic proteins (Tedesco et al., 2022). It has been found that heat shock proteins are encapsulated in the SOD1-positive aggregates, which reduces their ability to process the misfolded proteins in ALS (Farrawell and Yerbury, 2021). In addition, these abnormally aggregated proteins induce the endoplasmic reticulum (ER) stress and activate UPS to generate a vicious circle of abnormal toxic proteins production. Autophagy also appears to play an important role in the toxicity of mutant SOD1, which may be mediated by the removal of abnormal aggregates by HSP70 and its common partner B-cell lymphoma-2 associated athanogene 5 (Tedesco et al., 2023). However, a comparison between neuroblastoma × mouse spinal cord motor neuron cell line (NSC-34) motor neuron cell lines and C2C12 muscle cell lines showed that the activation efficiency of autophagy system was reduced in NSC-34 cells in the presence of SOD1 mutant expression (Maurel et al., 2018). This further leads to excessive accumulation of mutated SOD1, creating a vicious cycle that leads to cell death.
In 2011, it was discovered that the most common genetic cause of ALS is the expansion of GGGGCC (G4C2) nucleotide repeats in an intron of C9orf72 gene. Healthy individuals have less than 30 GGGGCC repeats, but in ALS patients, GGGGCC sequences can be amplified hundreds or thousands of times. The C9orf72 mutation appears to drive the pathogenesis of ALS through the loss of normal function of C9orf72 and the acquisition of toxic effects caused by the repeated amplification (Vant Spijker and Almeida, 2023), which leads to the RNA amplification, the dipeptide repeat proteins aggregation and the C9orf72 haploid dysfunction. The RNAs containing (GGGGCC)n and the translation product the dipeptide repeat protein (GGGGCC)n are cytotoxic and can interfere with UPS and affect the protein degradation. ALS patients with C9orf72 mutations exhibit the significant UPS impairment, and their characteristic pathological features are the ubiquitin-positive neuronal inclusions (Chua et al., 2022). The proteasome inhibitors selectively induce the death of motor neurons. In the C9orf72 mutant, the proteasome subunit is significantly reduced, and the abnormal proteasome is presented in the inclusion body, suggesting that the C9orf72 mutation promotes the occurrence and development of ALS by interfering with UPS. In addition, some studies have proposed the adverse effect of the down-regulation of C9orf72 on the initiation of autophagy (Cali et al., 2019; Leskelä et al., 2019; Shi et al., 2019). The absence of C9orf72 leads to the formation of light chain 3 (LC3)-positive autophagosomes and interferes with the autophagy process (Chua et al., 2022). In addition, C9orf72 forms the complexes with Smith-Magenis syndrome chromosomal region candidate gene 8 and WD repeat-containing protein 41 and has the guanine nucleotide exchange factors activity against small guanosine triphosphateases (GTPase) (Beckers et al., 2021), which promotes guanosine triphosphate (GTP), the interconversion of GTP-Guanosine diphosphate (GDP) alters the configuration of Rab-GTPase and is involved in the formation of autophagosomes. The poor haploid function of C9orf72 negatively affects several vesicular transport pathways (Rab1A, mannose-6-phosphate receptors (M6PRs), Rab5) and further affects the Rab protein activity and autophagy. The proteins encoded by C9orf72 also interact with the Rab1A and unc-51-like autophagy activating kinase 1 complex to regulate the initiation of autophagy (Burk and Pasterkamp, 2019). Therefore, the C9orf72 gene repeat mutation might contribute to the imbalance of protein homeostasis in neurons by the degradation decrease of abnormal toxic proteins due to the dysfunction of autophagy.
The neurocytotoxicity induced by interacting the TDP-43 gene nucleotide mutation is now widely recognized as a major pathological factor in ALS. TDP-43 is a nuclear protein encoded by TDP-43 gene and involved in a variety of cellular functions, including gene transcription and RNA processing (Klim et al., 2021). The cytoplasmic TDP-43 may be involved in the accumulation of misfolded proteins in brain by inhibiting the UPS activity. The TDP-43 deposition has also been implicated in other neurodegenerative diseases, such as frontotemporal degeneration and Alzheimer’s disease. The recent studies (Yin et al., 2021) have shown that the TDP-43-induced neurotoxicity is associated with the damage of UPS. Its interaction with proteasome assemblers proteasome assembly chaperone 2 and postsynaptic density protein13 leads to impaired proteasome activity, while histone deacetylase 6 regulates TDP-43-induced the UPS damage through the autophagolysosome pathway (Lee et al., 2020). However, the molecular mechanism of TDP-43-induced neurotoxicity is largely unknown. Some studies (Lee et al., 2020) found that it may be related to the protein tyrosine kinase 2-TRAF family member associated NFkappaB activator (TANK)-binding kinase 1-sequestosome 1 axis, which regulates the UPS damage-induced neurotoxicity. Although the exact mechanisms about how to the TDP-43 gene mutation effects the abnormal protein metabolizing have not been understood up to now. According to the current studies, the mutations in TDP-43 gene may disrupt the protein homeostasis and impair the function of UPS and autophagy, which are essential for protein degradation.
Ubiquilin-2 is a protein encoded by ubiquilin-2 gene. It belongs to the ubiquilin family of proteins that have a ubiquitin-like domain at N-terminus and a ubiquitin-associated domain at C-terminus. Ubiquilin-2 binds to various ubiquitinated substrates through its C-terminal domain and directs them to UPS for the protein degradation through its N-terminal domain. Therefore, the main function of ubiquilin-2 is to transport the ubiquitinated target proteins to proteasome for degradation. Moreover, ubiquilin-2 can also function as an autophagy receptor by directly binding to microtubule-associated protein LC3, a marker of autophagosomes, through its ubiquitin-associated domain, which regulates the activity of the mechanistic target of rapamycin complex 1 and the acidification of lysosomes, and thus modulates the autophagy maturation13 (Wu et al., 2020). In the related experiments, the mutant ubiquilin-2 gene disrupts the protein homeostasis. The overexpression of this gene resulted in the motor neuron loss and the accumulation of autophagy substrates, including both sequestosome 1 and LC3-II. These effects reduce the autophagy flux and impaire the autophagic acidification (Wu et al., 2020). Moreover, the mutations in ubiquilin-2 gene weaken the interaction between ubiquilin-2 protein and HSP70, reducing the ability of UPS to clear the abnormally aggregated proteins, which in turn results in the abnormal protein aggregation (Zhang et al., 2021). Therefore, the ubiquilin-2 protein plays an important role in the regulation of neuronal protein homeostasis.
VCP gene encodes an enzyme called VCP, which belongs to the ATPase family AAA domain (ATPases associated with diverse cellular activities) family of proteins. VCP can bind to the ubiquitinated target proteins and participate in their degradation through the ubiquitin-proteasome pathway. One of VCP functions is to maintain the proteins solubility or reverse the aggregation of insoluble, misfolded proteins before they are delivered to proteasome. The most characteristic function of VCP is to participate in the ER-associated protein degradation, a system related to UPS (Wani and Weihl, 2021). According to a recent study (Ziff et al., 2022), the VCP mutation can impair the maturation and lysosomal FUSion of autophagosomes. Motor neurons and astrocytes derive from the induced pluripotent stem cells with the pathogenic VCP mutations exhibit the abnormal cytoplasmic accumulation of TDP-43 protein, ER stress, mitochondrial dysfunction and oxidative stress, leading to cell death. The presence of these mutant protein aggregates is thought to impair the proteasome and autophagy degradation pathways and may be a key mediator of protein homeostasis imbalance in the pathogenesis of ALS.
The mutations in TANK-binding kinase 1 (TBK1) gene may impair the clearance of proteins and the turnover of mitochondria, resulting in the neuronal damage and the activation of toxic protein responses. In ALS patients, the mutations in the vesicle-associated membrane protein-associated protein B (VAPB) gene induce ER stress and impair UPS, leading to the protein homeostasis reduction and the motor neuron loss. The optineurin gene regulates apoptosis by binding to the linear ubiquitin chains. In summary, the nucleotide variations in the ALS-related genes mainly affect the protein homeostasis through the ubiquitin-proteasome pathway and promote the apoptosis and autophagy pathway through impairing the mitochondria functions, which are involved in the molecular genetic pathogenesis of ALS (Maurel et al., 2018).
The molecular genetic pathogenesis of ALS is advancing with the recent discovery of many gene mutations in ALS patients, many of which encode RBPs, such as TDP-43, FUS, ataxin 2, TATA box binding protein-associated factor 15 (TAF15), ewing sarcoma breakpoint region 1 (EWSR1), heterogeneous nuclear ribonucleoprotein A1 (hnRNPA1), hnRNPA2/B1, matrin3 (MATR3) and T cell-restricted intracellular antigen-1 (TIA-1) (Figure 3). RBPs are an evolutionarily conserved group of proteins that participate in multiple steps of RNA metabolism, such as splicing, polyadenylation, mRNA stability, localization and translation (Xue et al., 2020). TDP-43 and FUS are the major pathogenic genes in familial ALS. Normally, these proteins localize to nucleus and are involved in transcription, splicing, non-coding RNA metabolism, and micro-RNA biogenesis (Mackenzie et al., 2017). Although these proteins are mainly confined to nucleus, they can be mislocalized to cytoplasm in the postmortem brain tissue of mutation carriers. This nuclear depletion of RBPs may be harmful and cause the severe transcriptome abnormalities, while the cytoplasmic aggregates may also induce toxicity. These findings highlight the important role of mutations in the different RBPs genes in the pathogenesis of ALS (Humphrey et al., 2020).
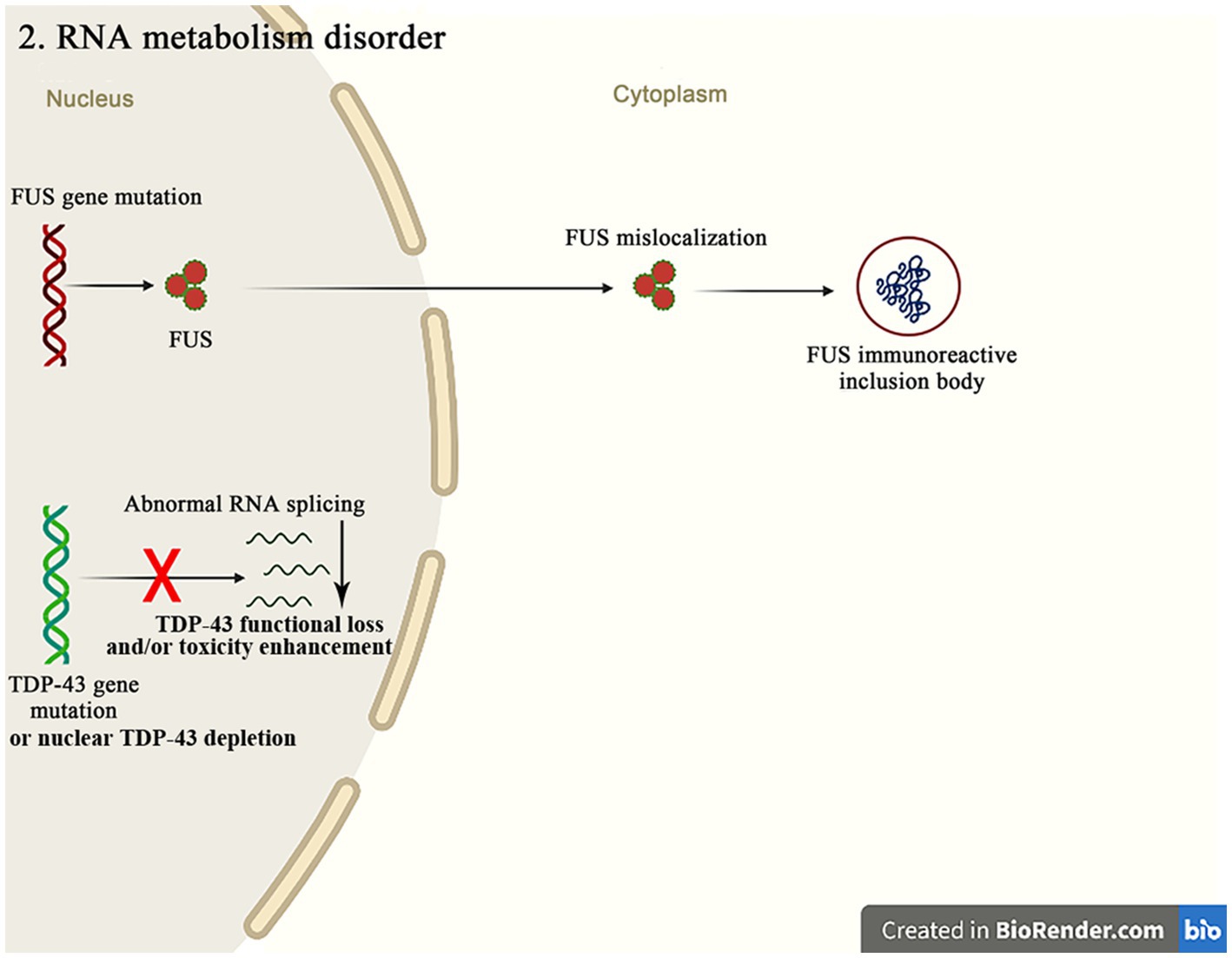
Figure 3. The FUS mutations mainly cause the mislocalization of FUS in cytoplasm, resulting in the FUS immunoreactive inclusion bodies. The abnormal RNA splicing pathway caused by the TDP-43 mutation or the nuclear TDP-43 depletion promotes the pathways of ALS occurrence through the TDP-43 functional loss and/or toxicity enhancement.
FUS protein is an RBPs that participates in the RNA metabolism and the DNA repair. It is mainly localized in nucleus and regulates transport, the local translation and stability of RNA. Specifically, FUS also binds to the RNA of many receptors and transporters involved in the glutamatergic and GABAergic signaling (Sahadevan et al., 2021). The FUS mutations predominantly affect the 3′ arginine/glycine-rich regions and the nuclear localization signal domains and are mostly missense. These mutations mainly lead to the FUS mislocalization in cytoplasm, which leads to the production of FUS immunoreactive inclusion bodies leading to the neurons degeneration in the pathogenesis of ALS. The mutant FUS toxicity may result from the irreversible formation of cytoplasmic inclusion bodies or the detergent-resistant protein aggregates, which impair the RNA processing (Humphrey et al., 2020). Therefore, both the loss of nuclear function and the increased cytotoxicity of FUS contribute to the pathogenesis of ALS. In vivo studies in mice have shown that the disease-related gene mutations in FUS alter the properties of proteins, leading to the abnormal phase transition and loss and toxic gain of function, and induce the conformational changes in the related proteins, resulting in one of the pathological markers of ALS, which is the magnocellular-like aggregation, and the further consolidation and growth of fibrous structures (Korobeynikov et al., 2022). Therefore, the FUS mutations may partly explain the molecular genetic pathogenesis of ALS.
TDP-43 is a RBPs encoded by TARDBP gene. It contains two RNA recognition motifs, a nuclear export signal and a glycine-rich region at C-terminal. TDP-43 regulates various aspects of RNA metabolism, such as transcription, the alternative splicing and the mRNA stability. It also participates in the diverse cellular processes, such as apoptosis, cell division and axon transport. TDP-43 regulates the mRNA expression of many proteins involved in the development, survival and synaptic transmission of neurons, as well as neuroplasticity in the central nervous system (CNS) (Ling, 2018). The recent studies have suggested that the TDP-43 toxicity in the molecular pathogenesis of ALS may be related to both loss of function (due to nuclear depletion of TDP-43) and gain of toxic function (due to cytoplasmic aggregation of TDP-43) (Deshaies et al., 2018). This mechanism suggests that the abnormal RNA splicing induced by the TDP-43 mutations or nuclear depletion of TDP-43 contributes to the pathogenic of ALS through the loss of function and/or gain of toxicity. Moreover, TDP-43 has been shown to act as a specific translation enhancer for several mRNAs, including DENN/MADD domain containing 4A (Dennd4a) and calmodulin binding transcription activator 1 (Camta1), which are implicated in the neurodegeneration of ALS. The A315T mutation in TDP-43 enhances the direct binding and translation of these mRNAs, leading to increase the ALS susceptibility (Neelagandan et al., 2019).
Angiotensinogen (ANG) is a stress-induced secreted ribonuclease with both nuclear and cytoplasmic activities. In the cell nucleus, ANG promotes the transcription of ribosomal RNA (Prehn and Jirström, 2020). It has found that the mutations in ANG gene disrupt the formation of RNA molecules, leading to the accumulation of toxic RNA fragments and the reduction of protein levels in motor neurons, and contributing to the development of ALS. Furthermore, ANG can promote the survival of motor neurons and prevent their degeneration, suggesting that restoring ANG levels may be a therapeutic strategy for treating ALS (Aluri et al., 2020). Overall, although the exact mechanism linking ANG and ALS is still being studied, the current research suggests that ANG may play a key role in the pathogenesis of ALS. The mutations in both hnRNPA2B1 and hnRNPA1 genes are the rare causes of both ALS and multisystem proteinopathy. Both hnRNPA1 and hnRNPA2/B1 have the similar functions in regulating the mRNA maturation, splicing, translation and stability, but they play the different roles in the transcription regulation (Kim and Taylor, 2017). HnRNPA1 is a part of RBPs complex that assembles with RNA to form ribonucleoproteins. HnRNPs are concentrated in the cell nucleus and play a role in regulating the pre-mRNA splicing, mRNA stability, transcription and translation (Beijer et al., 2021). HnRNPA2B1 is a protein involved in the RNA processing and transporting within the cell nucleus. It has found that mutations in both hnRNPA2B1 and hnRNPA1 genes lead to the abnormal accumulation of protein in motor neurons, disrupting the RNA processing and ultimately leading to the development of ALS. The knockdown of hnRNPA2/B1 also disrupts the alternative splicing and leads to impair the cognitive function (Baradaran-Heravi et al., 2020). T-cell intracellular antigen-1 (TIA-1) is a protein with multiple functions in the RNA metabolism, including the mRNA splicing, the translation inhibition and the mRNA silencing. Several missense mutations in TIA-1 gene have been identified as being associated with the pathogenesis of ALS. The TIA-1 mutations can alter the ability of protein to undergo the liquid–liquid phase separation and can damage the disassembly of stress granules (SGs) in vitro. It suggests that the TIA-1 mutations may be involved in the development of neurodegenerative diseases such as ALS (Mackenzie et al., 2017). TIA-1 can bind to the RNA molecules and form SGs in motor neurons. In ALS patients, these SGs have been found to contain the aggregated proteins and the RNA fragments, which can lead to the dysfunction and degeneration of neurons (Sekiyama et al., 2022). Ataxin-2 (ATXN2) is a RBPs that belongs to the like-Smith family and is involved in the regulation of RNA metabolism. ATXN2 plays a key role in regulating the toxicity of TDP-43 and FUS through the direct protein–protein interactions, and the reducing the levels of ATXN2 has been shown to inhibit TDP-43-mediated neurotoxicity (Becker et al., 2017). As the above mention, the gene mutations encoding many RBPs are highly associated with the pathogenesis of ALS. In addition, the dysregulation of RBPs also significantly contributes to the pathogenic mechanisms of ALS due to the impaired nuclear-cytoplasmic transport, the post-translational modifications, aggregation and the abnormal RNA sequestration.
Besides their canonical roles as the energy producers, mitochondria are also implicated in other cellular dysfunctions that contribute to the pathogenic of ALS, such as the Ca2+ homeostasis, the apoptosis regulation and the protein quality control (Figure 4). A recent study reported that mutations in 16 ALS pathogenic genes compromised the integrity of mitochondria-associated membranes and impaired the mitochondrial function (Sakai et al., 2021). The impaired mitochondrial electron transport chain function in various neurodegenerative diseases including ALS affects the oxygen consumption and the membrane potential, and causes to increase reactive oxygen species (ROS) and decrease the adenosine triphosphate (ATP) production. The excessive ROS induce the oxidative stress in cells and accelerate the neuronal degeneration and death (Delic et al., 2018). These evidences strongly suggest that the dysfunction of mitochondria is critical in the development of ALS.
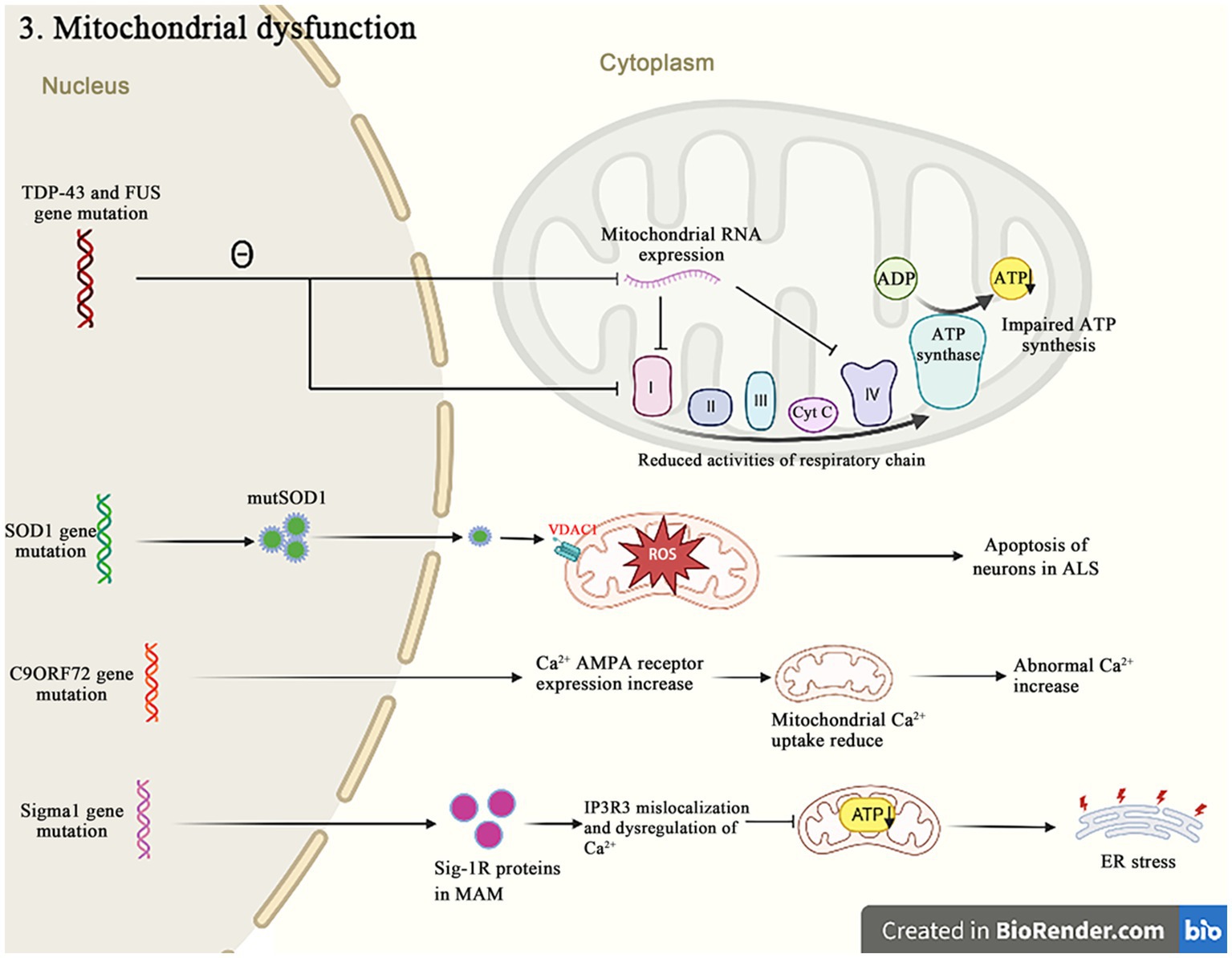
Figure 4. Both TDP-43 and FUS mutant isoforms, respectively, disrupt the mitochondrial RNA expression and interact with the mitochondrial ATP synthase catalytic subunits, resulting in the abnormal mitochondrial membrane potential, and reducing the ATP production and the oxygen consumption. The SOD1 mutations impair the ability of mitochondria to eliminate ROS. The C9orf72 mutations increase the concentration of neuronal Ca2+ influx and the overexpression of polyGR that causes the mitochondrial dysfunction by altering protein binding patterns on mitochondria. The Sigma-1R mutants cannot bind to IP3R, leading to the Ca2+ imbalance in MAM and exacerbating the related pathways of ER stress-induced neuronal death.
A growing body of evidence suggests that TDP-43 is closely related to the mitochondrial dysfunctions. When TDP-43 is mutated, the protein levels of respiratory chain complex I subunit NADH phanquinone redox enzymes 3 and 6 are affected, thereby impinging the assembly and function of complex I, resulting in the abnormal mitochondrial endometrial potential, decreasing the ATP production and the oxygen consumption (Wang et al., 2017). The TDP-43 mutation affects not only nucleus, but also mitochondria, where it suppresses the expression of mitochondrial RNA, which leads to the mitochondrial ridge damage, reduces the activities of respiratory chain complex I and IV, and impairs the ATP synthesis (Figure 5) (Shenouda et al., 2018; Wang et al., 2019). It has also been recently reported that the abrupt overexpression of TDP-43 triggers a unique self-destructive pathway of mitophagy, leading to the mitochondrial death (Gautam et al., 2019). Therefore, the TDP-43 gene nucleotide mutations participate in the pathogenesis of ALS through destroying the function and construct of mitochondria.
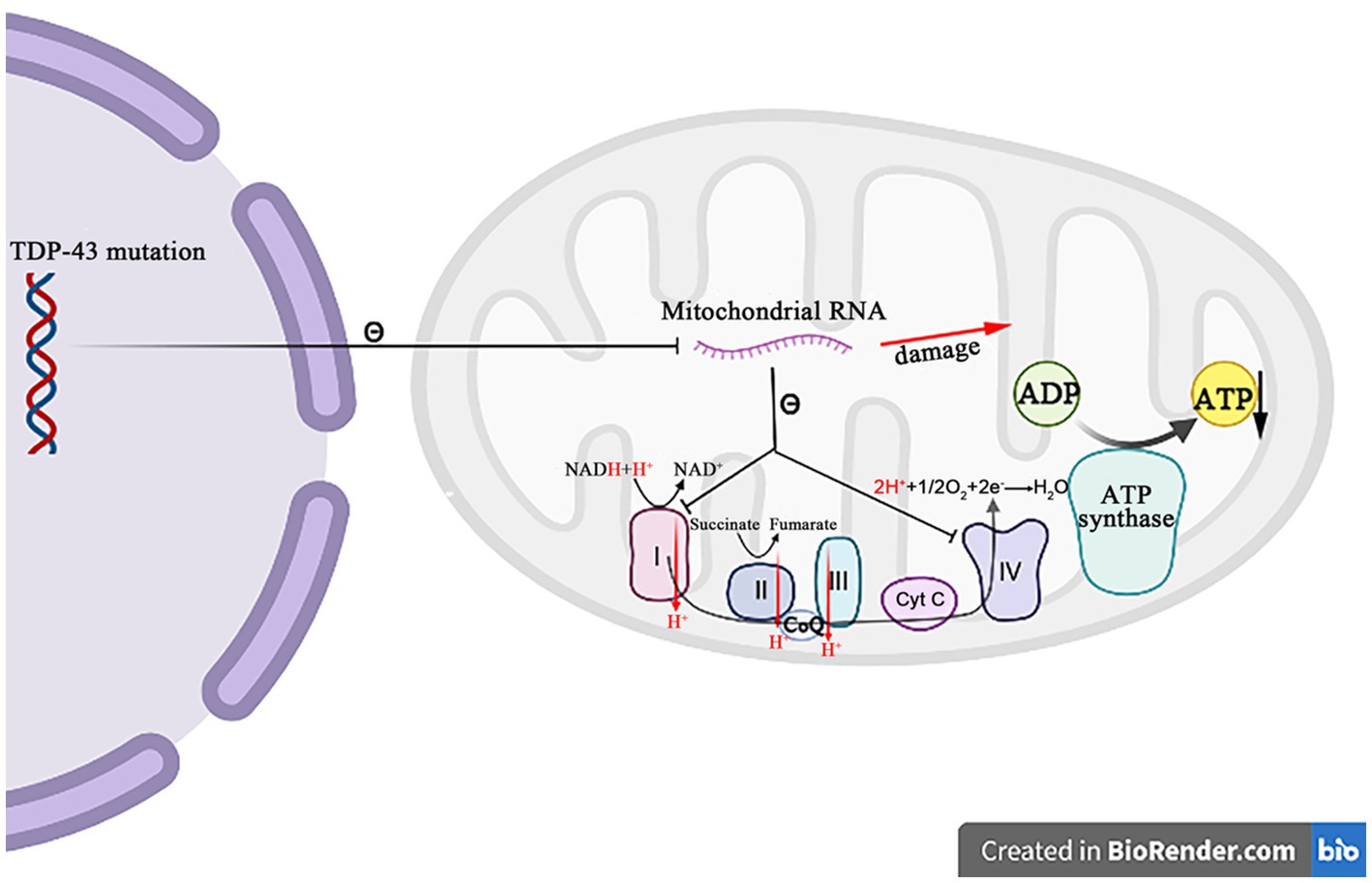
Figure 5. The mutation TDP-43 inhibits the mitochondrial RNA expression, resulting in the mitochondrial crest damage, decreasing the respiratory chain complex I and IV activity, and decreasing the ATP synthesis.
Similar to TDP-43, the FUS mutants cause the mitochondrial morphological and functional abnormalities, which has been shown that the FUS mutants can affect the mitochondrial function by disrupting the transcription of mitochondrial complex required for the ATP production. Within mitochondria, the protein encoded by the mutant FUS interacts with the mitochondrial ATP synthase catalytic subunit ATP5B, disrupting the assembly of ATP synthase complexes and reducing the mitochondrial ATP synthase activity, thereby reducing the mitochondrial ATP synthesis, inducing the mitochondrial ridge loss, and increasing the mitochondrial fragmentation (Deng et al., 2018). The FUS mutations were further found to be associated with the disruption of ER-mitochondrial association, and FUS was shown to activate glycogen synthase kinase-3beta, a regulator of ER-mitochondrial association, which inhibits the interactions between VAPB and protein tyrosine phosphatase interacting protein 51, resulting in the disruptions in cellular Ca2+ homeostasis and the mitochondrial production defect of ATP (Sakai et al., 2021). Overall, there is the substantial evidence that the FUS gene mutations alter the mitochondrial dynamics, functions and structure in the pathogenesis of ALS.
The protein encoded by SOD1 is a ubiquitous homologous dimer enzyme involved in the primary antioxidant defense of cells. The mutations in SOD1 (mutSOD1) aberrantly target the protein to the mitochondrial intermembrane space, impairing the ability of mitochondria to eliminate ROS and causing the mitochondrial damage. There are the growing evidences that the abnormal binding of mutSOD1 protein to voltage-dependent anion channel-1 (VDAC1) contributes to the progression of ALS (Shoshan-Barmatz et al., 2020). VDAC1 is an integrin of mitochondrial outer membrane that controls the metabolite flow, allows the adenosine nucleotide transport, and regulates the cellular processes such as apoptosis and the Ca2+ homeostasis. mutSOD1 specifically binds to the N-terminal domain of VDAC1, reducing its channel conductance and leading to the breakdown of mitochondrial function. In addition, it has been found that the mutSOD1 aggregates interact with the mitophagy receptors in neuronal cells, inhibiting their function as the mitochondrial flux promoters (Semmler et al., 2020). The mutations in SOD1 gene also inhibit the ATP synthesis and further inhibit the function of sodium–potassium pump, thereby impacting the electrical activity of neurons. The SOD1 gene mutations impair the mitochondrial membrane and its associated structures, leading to the imbalance of sodium–potassium and Ca2+ ion homeostasis, the dysfunction of ROS metabolism and autophagy, and ultimately increased the apoptosis of neurons in the pathogenesis of ALS.
In 2016, Benson et al. first reported the correlation between C9orf72-ALS mutation and mitochondrial dysfunction (Benson et al., 2021). The mechanism of mitochondrial dysfunction caused by the C9orf72-ALS mutation has been extensively studied. The current evidences suggest that it is mainly related to Ca2+ dysregulation and the overexpression of polyglycine-arginine (polyGR) dipeptides. In patients with C9orf72-ALS, the influx of Ca2+ into nerve cells is increased, the recovery time of intracellular Ca2+ is prolonged, and the buffering capacity of cytoplasm for Ca2+ is reduced (Candelise et al., 2022). It was found that this may be caused by increasing the expression of Ca2+AMPA receptor and reducing the production of mitochondrial Ca2+ uptake 1 and 2 Ca2+ channels, leading to the decreased uptake capacity of Ca2+ by mitochondria, which causes the abnormal increase of Ca2+ concentration in cytoplasm and causing the damage to cells. The overexpressed polyGR can lead to the mitochondrial dysfunction by changing the binding mode of proteins on mitochondria (Benson et al., 2021). Another possibility is that polyGR binds to the ATP5A1 subunit of mitochondrial complex line V and induces its degradation through ubiquitination. Such damage inhibits the redox reactions in mitochondria and leads to the lack of cell energy. The lack of energy in cell leads to a series of irreversible damage to neurons in the pathogenesis of ALS.
The recent studies have shown that Sigma-1 gene is involved in the pathogenesis of ALS. Sigma-1 gene encodes Sigma-1R protein, a molecular chaperone located in mitochondria-associated endoplasmic reticulum membranes (MAM). Sigma-1R can bind to inositol 1,4,5-trisphosphate receptor (IP3R) and facilitate Ca2+ flux into mitochondria, regulating Ca2+ homeostasis in the neuronal cytoplasm (Smith et al., 2019). The Sigma-1R mutants fail to bind IP3R, resulting in the mislocalization of IP3R3 and the dysregulation of Ca2+ in MAM. The disorders of MAM and Ca2+-related homeostasis affect the mitochondrial function and the neuronal survival (Wu et al., 2021). Moreover, it has shown that familial ALS patients exhibit a missense or non-sense mutation in the code of Sigma-1R, which replaces glutamine in the glutamate position. The expression of Sigma-1R protein in this mutant reduces the mitochondrial ATP production, inhibits the proteasome activity and leads to the mitochondrial damage, thus exacerbates the ER stress-induced neuronal death in the pathogenesis of ALS.
The mitochondrial homolog protein coiled-coil-helix-coiled-coil-helix domain containing 10 (CHCHD10) can form approximate 220 kDa complexes in the mitochondrial intermembrane space and cooperate to regulate the mitochondrial function (Burstein et al., 2018; Straub et al., 2018). It is reported that CHCHD10 regulates the mitochondrial cytochrome C oxidase activity and respiration during hypoxia (Purandare et al., 2018), and many studies suggest that CHCHD10 may play a role in the mitochondrial dynamics and cristae organization. It has been found that the mutations in CHCHD10 gene are associated with defects in the mitochondrial dynamics and cristae, leading to the accumulation of toxic waste and ultimately damaging motor neurons, resulting in the pathogenesis of ALS. The recent studies on the adolescent ALS have also found that the mutations in C19ORF12 gene can lead to a form of ALS called ALS10, which is characterized by weakness and atrophy in the lower limb muscles, as well as spasms and difficulty in movement (Kim et al., 2016). The mutations in C19ORF12 gene are rare, accounting for only a small fraction of all ALS cases. C19orf12 is a protein that localizes to both mitochondria and ER. The mutations of this protein can disrupt the lipid homeostasis in mitochondria and lead to the iron deposition in cells (Khani et al., 2019).
The accumulation of excitatory mediators and neuronal damage, a mechanism known as excitotoxicity, can result from the overactivation of glutamate receptors, the failure to clear the glutamate neurotransmitters from the synaptic cleft, or the increased post-synaptic sensitivity to glutamate (Olloquequi et al., 2018) (Figure 6). Glutamate is the main excitatory neurotransmitter in CNS, initiates a rapid signal transmission in synapses and then reuptake into the surrounding glial cells, especially astrocytes. GLAST and GLT-1 and their homologous EAAT1 and EAAT2 of astrocytes are major transporters that absorb the synaptic glutamate to maintain the optimal extracellular glutamate levels, thus, prevents the accumulation of glutamate in the synaptic cleft and the consequent excitatory toxicity. Therefore, the dysregulation of glutamate transporters in glial cells may play an important role in the excitatory toxicity and the related neuropathogenesis (Pajarillo et al., 2019). In addition, glutamate can induce the cytoplasmic Ca2+ increase, and the intracellular Ca2+ influx and accumulation are key factors causing the excitatory toxicity. Alpha-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid-type glutamate receptors (AMPAR) is a key subtype of ionic glutamate receptors that mediates the excitatory synaptic transmission in CNS and peripheral nervous system. AMPAR is an integrated transmembrane protein assembled by AMPA selective glutamate receptor (GluA1-4) subunit (also known as GRIA1-4 or GluR1-4) into a tetramer complex, in which the presence of GluA2 subunit in AMPAR prevents the influx of cations (such as Ca2+) and affects the ion permeability in neurons (Cull-Candy and Farrant, 2021).
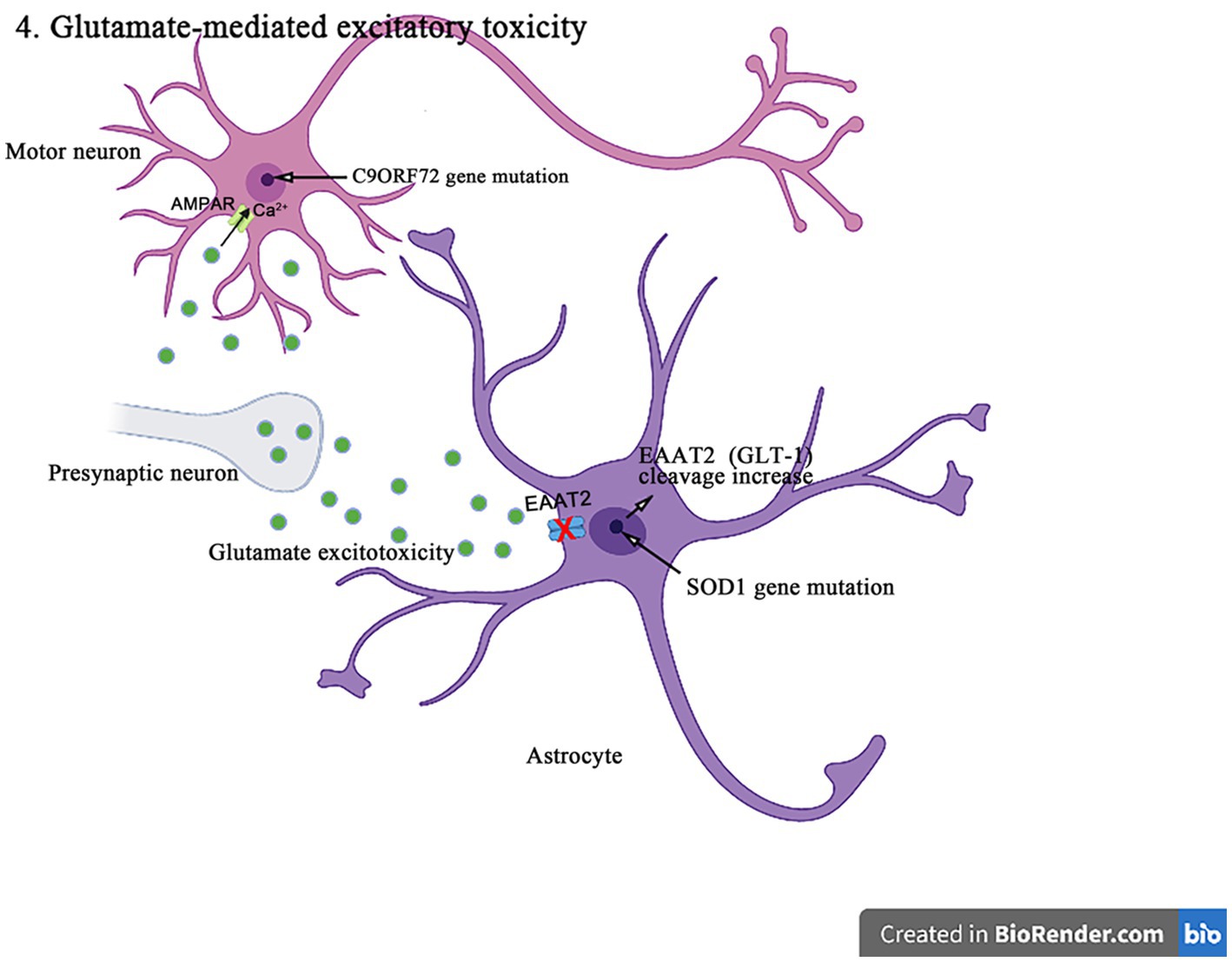
Figure 6. The SOD1 mutants enhance the caspase-3-mediated cleavage of EAAT2 (GLT-1), resulting in impairing the glutamate uptake and producing excitotoxicity in the pathogenesis of ALS. In addition, C9orf72 mutations lead to the production of toxic dipeptide repeat sequences, increasing the expression and Ca2+ permeability of GluA1, leading to excitotoxicity in motor neurons in the pathogenesis of ALS.
The increasing evidences suggest that the abnormal posttranslational modification is associated with the pathogenesis of EAAT1 (GLAST)/EAAT2 (GLT-1) dysfunction. EAAT2 (GLT-1) has a caspase-3 consensus sequence and can be activated in vitro by the caspase-3 cleavage. It has been shown that the SOD1 mutant mice enhance the caspase 3-mediated EAAT2 (GLT-1) cleavage, leading to the formation of truncated EAAT2 (GLT-1) (sumoylated EAAT2 C-terminus fragment, CTE-SUMO1). The cleavage site mutation does not affect the EAAT2 (GLT-1) activity in ALS mice, but delays the disease progression and prolongs lifespan (Rosenblum et al., 2017). In addition, the accumulation of CTE-SUMO1 in astrocytes increases toxicity, leading to the impaired growth of neurons and axons (Hirschberg et al., 2021). These findings suggest that the abnormal GLT-1 cutting impairs the glutamate uptake and contributes to the excitatory toxicity in the pathogenesis of ALS. Moreover, it was found that the histone deacetylase (HDAC) inhibitors such as a selective inhibitor of class II HDAC (MC1568), valproic acid, and sodium butyrate increased the EAAT1 (GLAST)/EAAT2 (GLT-1) expression and the glutamate uptake in both in vitro and in vivo models of ALS and Mn-induced neurotoxicity (Johnson et al., 2018). MC1568 can also regulate the EAAT2 transcription, improve the EAAT2 expression and the post-translational modification, and has been proven to have the therapeutic effects in the subclinical models of ALS (Lapucci et al., 2017).
Numerous studies have been conducted to understand the biological consequences of C9orf72 repeated amplification, one of which is the production of toxic dipeptide repeats (Gendron and Petrucelli, 2018). The recent studies have also shown that the arginine-rich dipeptide repeats (polyglycine-arginine/proline-arginine (polyGR/PR)) is one of major neurotoxicity sources. In the drosophila-related experiments (Xu and Xu, 2018), the increased levels of extracellular glutamate and intracellular Ca2+ were detected in the ventral nerve cords of GR/PR-expressing larvae, accompanied by the significant increases in the synaptosomes and the active regions of larval neuromuscular junctions. The C9orf72 mutation results in the increased expression of AMPAR subunit GluA1 accompanied by the increased permeability to Ca2+. The excessive Ca2+ load in neurons can also produce the significant cytotoxicity. The inhibiting expression of vesicular glutamate transporters in the presynaptic glutamatergic motor neurons or blocking N-methyl-d-aspartic acid receptor can effectively save the motor deficits induced by polyGR/PR and prolong lifespan, which thus indicates the mechanism of autonomic excitatory toxicity. In addition, the EAAT2 splicing may be influenced by C9orf72 dipeptide repeats and lead to the decreased uptake of glutamate by astrocytes and increased the excitatory toxicity of motor neurons (Shi et al., 2018).
In addition, the TDP-43 mutations downregulate adenosine deaminase acting on RNA editing enzyme adenosine deaminase acting on RNA 2 (ADAR2), leading to the failure of GluA2 glutamine/arginine (Q/R) site editing in GluA2 pre-mRNA (Yamashita and Kwak, 2019), which results in the significant loss of motor neurons in most sporadic ALS patients.
Motor neurons are highly dependent on the axonal transport mechanisms to maintain the normal structure and function (Figure 7). It not only determines the nutrient supply, the energy metabolism, and the signal transduction of neuronal axons but also affects various physiological functions and the homeostasis of neurons. C9orf72, TDP-43, FUS, and other gene mutations are closely related to neuronal transport disorders in the pathogenesis of ALS.
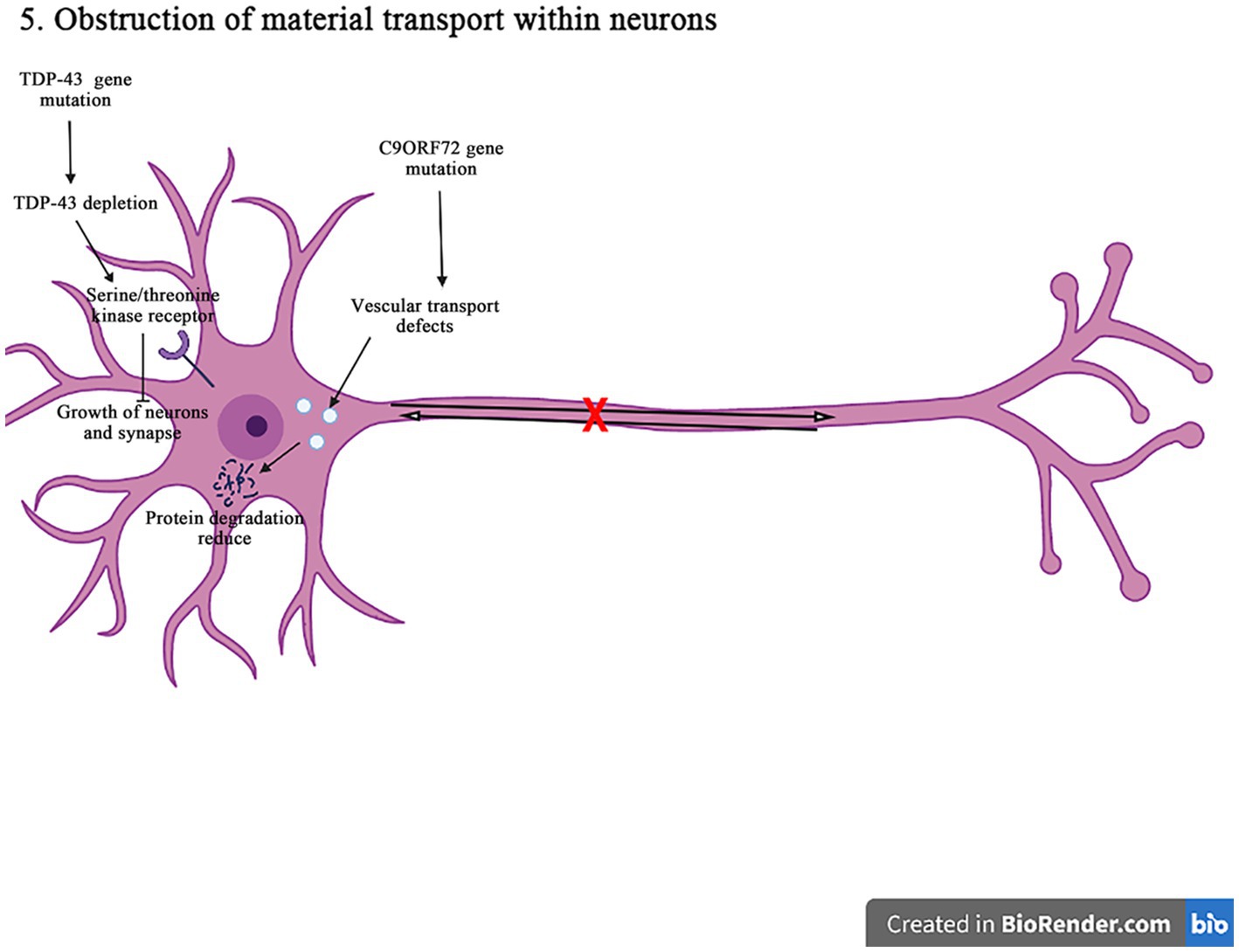
Figure 7. The C9orf72 gene mutations can affect the vesicular transport pathway, reduce the protein degradation, and cause the neurodegeneration and death of neurons. The TDP-43 gene deletion reduces the signaling pathways for the epidermal growth factor receptors and the serine/threonine kinase receptors, affecting the neuron survival and the axon innervation. In addition, it also interferences with the material circulation and target protein transport pathways on the neuronal cell membrane, ultimately damaging neurons.
The expansion of C9orf72GGGGCC nucleotide repeat can reduce the expression of C9orf72, and accumulate the RNA containing GGGGCC repeat in the nuclear foci. It can lead to the production of toxic dipeptide repeat protein, and ultimately impair the intracellular vesicle transport, resulting in decreasing the protein degradation and increasing aggregation, and inducing the neuronal degeneration and death in the pathogenesis of ALS. It has shown that in motor neurons, C9orf72 protein is localized to Rab5-positive endosomal vesicles, which can promote the role of Rab5 in vesicle transport and early endosomal maturation, assist Rab protein in GDP-GTP exchange, and improve the survival rate of neurons (Shi et al., 2018; Pang and Hu, 2021). The mutation C9orf72 inhibits the maturation of intracellular vesicles, reduces the production of lysosomes, and affects the protein degradation. In addition, the C9orf72 mutation can affect the transmembrane receptor M6PRs. It is found that the mutation C9orf72 can reduce the rate of aggregation and migration of M6PRs, which promotes the protein degradation by transporting materials to lysosomes. Therefore, the misshipment of M6PRs may lead to reduce the protein degradation in the pathogenesis of ALS (Starr and Sattler, 2018).
The disruption of receptor transport in ALS is also associated with TDP-43 gene. It is found that the knockout of TDP-43 gene leads to epidermal growth factor receptor expressed in cell membrane surface is reduced, and epidermal growth factor and epidermal growth factor receptor signaling can promote the survival of neurons, mature and grow (Chou et al., 2018). When the epidermal growth factor receptor expression is reduced on the surface of cell membrane, the circulation of cell surface receptors is impaired, thus affecting the neuronal survival and the axonal innervation. Furthermore, the TDP-43 depletion affects serine/threonine kinase receptor (bone morphogenetic proteins receptor). Bone morphogenetic proteins receptors can promote the synaptic growth through the phosphorylation of downstream mothers against decapentaplegic protein (a protein called Mad (mothers against decapentaplegic)), and the depletion of TDP-43 will lead to decrease bone morphogenetic proteins receptor signal transduction and ultimately affect the growth of neurons and synapses (Burk and Pasterkamp, 2019). The TDP-43 gene mutation can affect the circulation of materials on the cell membrane of neurons and the transport of target proteins during the degradation process, thus damaging neurons in the pathogenesis of ALS.
The SOD1 mutations gain an affinity for both actin and microtubule proteins, which impairs the neuronal growth and the mitochondrial fission (Osking et al., 2019; Muñoz-Lasso et al., 2020). The cytoplasmic inclusions of FUS prevent the ribonucleotide cargoes from reaching their normal destinations by abnormally sequestering the transport proteins and affecting the post-translational status of microtubule tracks (Yasuda et al., 2017). Kinesin family member 5A (KIF5A), a motor protein, drives the microtubule tracks to mediate the anterograde transport of mitochondria, vesicles and signaling molecules. KIF5A is expressed only in motor neurons, is critical to development and function of motor neurons, and is the only reported motor protein which mutations cause ALS (He et al., 2020; Baron et al., 2022). The reduction of KIF5A in zebrafish or rat hippocampal neurons leads to hyperexcitability, the axonal degradation and defects in the mitochondrial transport (Zhao et al., 2020). The profilin 1 (PFN1) variants (A20T, C71G, T109M, M114T, E117G, G118V, R136W, and Q139L) cause the autosomal dominant form of ALS by interfering with the actin dynamics in two opposite pathways. Firstly, PFN1 directly binds to the actin monomers, blocking the sites for new monomer addition, and thus preventing the actin filament assembly (Pimm et al., 2020; Liu et al., 2022). Secondly, PFN1 enhances the actin filament assembly by interacting with PFN, a profilamentous actin-binding protein (Schmidt et al., 2021). Taken together, these observations strongly suggest that PFN1 promotes ALS by regulating both microtubule tracks and transport machinery, such as motor proteins, dynein or spastin (Pinto-Costa et al., 2020). Therefore, understanding the underlying mechanisms of such regulation is indispensable for developing specific drug targets for the treatment of ALS.
At present, the pathogenesis of ALS is not fully understood, and the genetic factors are suggested to be closely related to the pathogenesis of ALS. The imbalance of protein homeostasis, the RNA metabolism disorder, the mitochondrial dysfunction, the glutamate-mediated excitatory toxicity and the intra-neuronal material transport disorder in neurons are currently recognized as the identifiable molecular causes of ALS pathogenesis. With the investigated progress of genetics and molecular mechanisms about the pathogenesis of ALS, a deeper understanding about the molecular genetic pathogenesis of ALS has been gained. The mutations in genes such as SOD1, C9ORF72, TDP-43, ubiquilin-2, and VCP can lead to the protein homeostasis imbalance via the following molecular and/or biological pathways in the pathogenesis of ALS. These gene mutations can interfere with UPS and autophagy degradation pathways, leading to the abnormal accumulation of toxic proteins and ultimately cell death. Both FUS and TDP-43 are RBPs associated with the RNA metabolism pathways, and their mutations can cause the FUS and TDP-43 abnormal localization and aggregation in cells, affecting both RNA metabolism and cell functions and ultimately leading to neurodegeneration in the pathogenesis of ALS. The FUS mutations mainly cause the mislocalization of FUS in cytoplasm, resulting in the FUS immunoreactive inclusion bodies. The abnormal RNA splicing pathway caused by the TDP-43 mutation or the nuclear TDP-43 depletion promotes the pathways of ALS occurrence through the TDP-43 functional loss and/or toxicity enhancement. The mitochondrial dysfunction plays a crucial role in the pathogenesis of ALS because it affects multiple cellular pathways, including the energy production and the Ca2+ regulation, apoptosis, and the protein quality control. The ALS pathogenic genes, such as TDP-43, FUS, SOD1, C9ORF72, and Sigma-1R mutations, can alter the mitochondrial integrity and function. Both TDP-43 and FUS mutant isoforms, respectively, disrupt the mitochondrial RNA expression and interact with the mitochondrial ATP synthase catalytic subunits, resulting in the abnormal mitochondrial membrane potential, reducing the ATP production and the oxygen consumption. The SOD1 mutations impair the ability of mitochondria to eliminate ROS. The C9ORF72 mutations increase the concentration of neuronal Ca2+ influx and the overexpression of polyGR that causes the mitochondrial dysfunction by altering protein binding patterns on mitochondria. Finally, the mutant Sigma-1R cannot bind to IP3R, leading to the Ca2+ imbalance in MAM and exacerbating the related pathways of ER stress-induced neuronal death. The excitotoxicity occurs due to the excessive activation of glutamate receptors, failures to clear the glutamate neurotransmitters, or increases the postsynaptic sensitivity to glutamate. Glutamate transporters in astrocytes, such as EAAT1/GLAST and EAAT2/GLT-1, maintain the optimal extracellular glutamate levels and prevent excitotoxicity. The abnormal post-translational modification pathways of EAAT1/GLAST and EAAT2/GLT-1 are associated with the pathogenesis of ALS via the following pathways. The SOD1 mutant mice enhance the caspase-3-mediated cleavage of EAAT2 (GLT-1), resulting in impairing the glutamate uptake and producing excitotoxicity in ALS patients. On the other hand, C9ORF72 mutations lead to the production of toxic dipeptide repeat sequences, increasing the expression and Ca2+ permeability of AMPA-type glutamate receptors (GluA1), leading to excitotoxicity in motor neurons. Motor neurons depend on the axonal transport pathways to maintain their normal structure and function, and C9ORF72, TDP-43, and FUS mutations are associated with the neuronal transport disorders. The C9ORF72 gene mutations can affect the vesicular transport pathway, reduce the protein degradation, and cause the neurodegeneration and death of neurons in the pathogenesis of ALS. The TDP-43 gene deletion reduces the signaling pathways for the epidermal growth factor receptors and the serine/threonine kinase receptors, affecting the neuron survival and axon innervation. In addition, it also interferences with the material circulation and target protein transport pathways on the neuronal cell membrane, ultimately damaging neurons in the pathogenesis of ALS.
In addition, the same genetic mutation may have effects on multiple disease pathways. Currently, conformational and functional variations in SOD1 caused by mutations have been suggested to confer toxicity via the interactions of several proteins and multiple mechanisms that are mutually compatible pathogenic mechanisms. These mechanisms include protein aggregation, mitochondrial dysfunction, oxidative stress, apoptosis, excitotoxicity, and ER stress, etc. (Hayashi et al., 2016; Kaur et al., 2016). TARDBP mutations are inherited in an autosomal dominant pattern. Among the various heterodimeric proteins encoded by TARDBP, TDP-43 remains the most common (Alsultan et al., 2016). TDP-43 exhibits both nuclear localization and export signals, allowing it to shuttle between the cytoplasm and the nucleus. As a regulator of gene expression, TDP-43 is involved in the regulation of thousands of genes through RNA splicing, DNA or RNA binding, and protein–protein interactions. It also plays a crucial role in several steps of RNA processing, including mRNA stability regulation, mRNA trafficking, pre-mRNA splicing, and regulation of non-coding RNA translation or function (Heyburn and Moussa, 2017). Currently, ALS-associated FUS mutations have been shown to cause toxicity by acquiring functional effects in the cytoplasm. Mutant FUS accumulates along the axons, leading to localized stress responses that inhibit local protein synthesis. This includes the suppression of RNA-encoded proteins critical for synaptic function and the inhibition of translation of axonally localized proteins. This process ultimately impairs neuronal synaptic function (López-Erauskin et al., 2020). Overexpression of mutant FUS variants, leads to mitochondrial dysfunction through a toxic gain-of-function mechanism, resulting in reduced mitochondrial membrane potential and respiratory function (Tsai et al., 2020). Additionally, the loss or mutation of C9ORF72 is associated with a range of functional impairments, including synaptic dysfunction, mitochondrial dysfunction, nucleocytoplasmic transport dysfunction, and varying levels of autophagy-lysosomal dysfunction (Beckers et al., 2021; Pang and Hu, 2021; Jiang et al., 2022). Overall, C9ORF72 plays a critical role in regulating the autophagy-lysosomal pathway, neuroinflammation, SGs formation, lipid metabolism, axonal growth and transport, as well as presynaptic and postsynaptic functions (Pang and Hu, 2021). It is believed that in the near future, the accurate pathogenesis of ALS disease will be clarified, thus alleviating the suffering of patients, extending their lives, improving their quality of life, and even cure this disease.
WZ wrote the first draft of the manuscript. RX provided critical feedback as supervisor. All authors contributed to the article and approved the submitted version.
This study was in part funded by the National Natural Science Foundation of China (30560042, 81160161, 81360198, and 82160255), Education Department of Jiangxi Province (GJJ13198 and GJJ170021), Jiangxi Provincial Department of Science and Technology (20192BAB205043) and Health and Family Planning Commission of Jiangxi Province (20181019 and 202210002).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Alsultan, A. A., Waller, R., Heath, P. R., and Kirby, J. (2016). The genetics of amyotrophic lateral sclerosis: current insights. Degener. Neurol. Neuromuscul. Dis. 6, 49–64. doi: 10.2147/DNND.S84956
Aluri, K. C., Salisbury, J. P., Prehn, J. H. M., and Agar, J. N. (2020). Loss of angiogenin function is related to earlier ALS onset and a paradoxical increase in ALS duration. Sci. Rep. 10:3715. doi: 10.1038/s41598-020-60431-6
Baradaran-Heravi, Y., Van Broeckhoven, C., and van der Zee, J. (2020). Stress granule mediated protein aggregation and underlying gene defects in the FTD-ALS spectrum. Neurobiol. Dis. 134:104639. doi: 10.1016/j.nbd.2019.104639
Baron, D. M., Fenton, A. R., Saez-Atienzar, S., Giampetruzzi, A., et al. (2022). ALS-associated KIF5A mutations abolish autoinhibition resulting in a toxic gain of function. Cell Rep. 39:110598. doi: 10.1016/j.celrep.2022.110598
Becker, L. A., Huang, B., Bieri, G., Ma, R., Knowles, D. A., Jafar-Nejad, P., et al. (2017). Therapeutic reduction of ataxin-2 extends lifespan and reduces pathology in TDP-43 mice. Nature 544, 367–371. doi: 10.1038/nature22038
Beckers, J., Tharkeshwar, A. K., and Van Damme, P. (2021). C9orf72 ALS-FTD: recent evidence for dysregulation of the autophagy-lysosome pathway at multiple levels. Autophagy 17, 3306–3322. doi: 10.1080/15548627.2021.1872189
Beijer, D., Kim, H. J., Guo, L., O’Donovan, K., Mademan, I., et al. (2021). Characterization of HNRNPA1 mutations defines diversity in pathogenic mechanisms and clinical presentation. JCI Insight 6:148363. doi: 10.1172/jci.insight.148363
Benson, B. C., Shaw, P. J., Azzouz, M., Highley, J. R., and Hautbergue, G. M. (2021). Proteinopathies as hallmarks of impaired gene expression, proteostasis and mitochondrial function in amyotrophic lateral sclerosis. Front. Neurosci. 15:783624. doi: 10.3389/fnins.2021.783624
Boyman, L., Karbowski, M., and Lederer, W. J. (2020). Regulation of mitochondrial ATP production: Ca2+ signaling and quality control. Trends Mol. Med. 26, 21–39. doi: 10.1016/j.molmed.2019.10.007
Burk, K., and Pasterkamp, R. J. (2019). Disrupted neuronal trafficking in amyotrophic lateral sclerosis. Acta Neuropathol. 137, 859–877. doi: 10.1007/s00401-019-01964-7
Burstein, S. R., Valsecchi, F., Kawamata, H., Bourens, M., Zeng, R., Zuberi, A., et al. (2018). In vitro and in vivo studies of the ALS-FTLD protein CHCHD10 reveal novel mitochondrial topology and protein interactions. Hum. Mol. Genet. 27, 160–177. doi: 10.1093/hmg/ddx397
Cali, C. P., Patino, M., Tai, Y. K., Ho, W. Y., McLean, C. A., Morris, C. M., et al. (2019). C9orf72 intermediate repeats are associated with corticobasal degeneration, increased C9orf72 expression and disruption of autophagy. Acta Neuropathol. 138, 795–811. doi: 10.1007/s00401-019-02045-5
Candelise, N., Salvatori, I., Scaricamazza, S., Nesci, V., Zenuni, H., Ferri, A., et al. (2022). Mechanistic insights of mitochondrial dysfunction in amyotrophic lateral sclerosis: an update on a lasting relationship. Meta 12:233. doi: 10.3390/metabo12030233
Cao, B., Gu, X., Wei, Q., Li, C., Chen, Y., Ou, R., et al. (2021). Mutation screening and burden analysis of GLT8D1 in Chinese patients with amyotrophic lateral sclerosis. Neurobiol. Aging 101, 298.e17–298.e21. doi: 10.1016/j.neurobiolaging.2020.10.017
Chou, C.-C., Zhang, Y., Umoh, M. E., Vaughan, S. W., Lorenzini, I., Liu, F., et al. (2018). TDP-43 pathology disrupts nuclear pore complexes and nucleocytoplasmic transport in ALS/FTD. Nat. Neurosci. 21, 228–239. doi: 10.1038/s41593-017-0047-3
Chua, J. P., De Calbiac, H., Kabashi, E., and Barmada, S. J. (2022). Autophagy and ALS: mechanistic insights and therapeutic implications. Autophagy 18, 254–282. doi: 10.1080/15548627.2021.1926656
Cooper-Knock, J., Moll, T., Ramesh, T., Castelli, L., Beer, A., Robins, H., et al. (2019). Mutations in the glycosyltransferase domain of GLT8D1 are associated with familial amyotrophic lateral sclerosis. Cell Rep. 26, 2298–2306.e5. doi: 10.1016/j.celrep.2019.02.006
Cull-Candy, S. G., and Farrant, M. (2021). Ca2+-permeable AMPA receptors and their auxiliary subunits in synaptic plasticity and disease. J. Physiol. 599, 2655–2671. doi: 10.1113/JP279029
Dafinca, R., Barbagallo, P., and Talbot, K. (2021). The role of mitochondrial dysfunction and ER stress in TDP-43 and C9ORF72 ALS. Front. Cell. Neurosci. 15:653688. doi: 10.3389/fncel.2021.653688
Delic, V., Kurien, C., Cruz, J., Zivkovic, S., Barretta, J., Thomson, A., et al. (2018). Discrete mitochondrial aberrations in the spinal cord of sporadic ALS patients. J. Neurosci. Res. 96, 1353–1366. doi: 10.1002/jnr.24249
Deng, J., Wang, P., Chen, X., Cheng, H., Liu, J., Fushimi, K., et al. (2018). FUS interacts with ATP synthase beta subunit and induces mitochondrial unfolded protein response in cellular and animal models. Proc. Natl. Acad. Sci. U. S. A. 115, E9678–E9686. doi: 10.1073/pnas.1806655115
Deshaies, J.-E., Shkreta, L., Moszczynski, A. J., Sidibé, H., Semmler, S., Fouillen, A., et al. (2018). TDP-43 regulates the alternative splicing of hnRNP A1 to yield an aggregation-prone variant in amyotrophic lateral sclerosis. Brain 141, 1320–1333. doi: 10.1093/brain/awy062
Diaper, D. C., Adachi, Y., Lazarou, L., Greenstein, M., Simoes, F. A., Di Domenico, A., et al. (2013). Drosophila TDP-43 dysfunction in glia and muscle cells cause cytological and behavioural phenotypes that characterize ALS and FTLD. Hum. Mol. Genet. 22, 3883–3893. doi: 10.1093/hmg/ddt243
Farrawell, N. E., and Yerbury, J. J. (2021). Mutant cu/Zn superoxide dismutase (A4V) turnover is altered in cells containing inclusions. Front. Mol. Neurosci. 14:771911. doi: 10.3389/fnmol.2021.771911
Fernández-Ruiz, J., de Lago, E., Rodríguez-Cueto, C., and Moro, M. A. (2021). Recent advances in the pathogenesis and therapeutics of amyotrophic lateral sclerosis. Br. J. Pharmacol. 178, 1253–1256. doi: 10.1111/bph.15348
Fiscon, G., Conte, F., Amadio, S., Volonté, C., and Paci, P. (2021). Drug repurposing: a network-based approach to amyotrophic lateral sclerosis. Neurotherapeutics 18, 1678–1691. doi: 10.1007/s13311-021-01064-z
Gautam, M., Xie, E. F., Kocak, N., and Ozdinler, P. H. (2019). Mitoautophagy: a unique self-destructive path mitochondria of upper motor neurons with TDP-43 pathology take, very early in ALS. Front. Cell. Neurosci. 13:489. doi: 10.3389/fncel.2019.00489
Gendron, T. F., and Petrucelli, L. (2018). Disease mechanisms of C9ORF72 repeat expansions. Cold Spring Harb. Perspect. Med. 8:a024224. doi: 10.1101/cshperspect.a024224
Hallegger, M., Chakrabarti, A. M., Lee, F. C. Y., Lee, B. L., Amalietti, A. G., Odeh, H. M., et al. (2021). TDP-43 condensation properties specify its RNA-binding and regulatory repertoire. Cells 184, 4680–4696.e22. doi: 10.1016/j.cell.2021.07.018
Hayashi, Y., Homma, K., and Ichijo, H. (2016). SOD1 in neurotoxicity and its controversial roles in SOD1 mutation-negative ALS. Adv Biol Regul 60, 95–104. doi: 10.1016/j.jbior.2015.10.006
He, J., Liu, X., Tang, L., Zhao, C., He, J., and Fan, D. (2020). Whole-exome sequencing identified novel KIF5A mutations in Chinese patients with amyotrophic lateral sclerosis and Charcot-Marie-tooth type 2. J. Neurol. Neurosurg. Psychiatry 91, 326–328. doi: 10.1136/jnnp-2019-320483
Herrando-Grabulosa, M., Gaja-Capdevila, N., Vela, J. M., and Navarro, X. (2021). Sigma 1 receptor as a therapeutic target for amyotrophic lateral sclerosis. Br. J. Pharmacol. 178, 1336–1352. doi: 10.1111/bph.15224
Heyburn, L., and Moussa, C. E.-H. (2017). TDP-43 in the spectrum of MND-FTLD pathologies. Mol. Cell. Neurosci. 83, 46–54. doi: 10.1016/j.mcn.2017.07.001
Hirschberg, S., Dvorzhak, A., Rasooli-Nejad, S. M. A., Angelov, S., Kirchner, M., Mertins, P., et al. (2021). Uncoupling the excitatory amino acid transporter 2 from its C-terminal interactome restores synaptic glutamate clearance at corticostriatal synapses and alleviates mutant huntingtin-induced hypokinesia. Front. Cell. Neurosci. 15:792652. doi: 10.3389/fncel.2021.792652
Humphrey, J., Birsa, N., Milioto, C., McLaughlin, M., Ule, A. M., Robaldo, D., et al. (2020). FUS ALS-causative mutations impair FUS autoregulation and splicing factor networks through intron retention. Nucl. Acids Res. 48, 6889–6905. doi: 10.1093/nar/gkaa410
Je, G., Keyhanian, K., and Ghasemi, M. (2021). Overview of stem cells therapy in amyotrophic lateral sclerosis. Neurol. Res. 43, 616–632. doi: 10.1080/01616412.2021.1893564
Jiang, L., Zhang, T., Lu, K., and Qi, S. (2022). The progress in C9orf72 research: ALS/FTD pathogenesis, functions and structure. Small GTPases 13, 56–76. doi: 10.1080/21541248.2021.1892443
Johnson, J. O., Chia, R., Miller, D. E., Li, R., Kumaran, R., Abramzon, Y., et al. (2021). Association of Variants in the SPTLC1 gene with juvenile amyotrophic lateral sclerosis. JAMA Neurol. 78, 1236–1248. doi: 10.1001/jamaneurol.2021.2598
Johnson, J., Pajarillo, E., Karki, P., Kim, J., Son, D.-S., Aschner, M., et al. (2018). Valproic acid attenuates manganese-induced reduction in expression of GLT-1 and GLAST with concomitant changes in murine dopaminergic neurotoxicity. Neurotoxicology 67, 112–120. doi: 10.1016/j.neuro.2018.05.001
Kaur, S. J., McKeown, S. R., and Rashid, S. (2016). Mutant SOD1 mediated pathogenesis of amyotrophic lateral sclerosis. Gene 577, 109–118. doi: 10.1016/j.gene.2015.11.049
Khani, M., Alavi, A., Shamshiri, H., Zamani, B., Hassanpour, H., Kazemi, M. H., et al. (2019). Mutation screening of SLC52A3, C19orf12, and TARDBP in Iranian ALS patients. Neurobiol. Aging 75, 225.e9–225.e14. doi: 10.1016/j.neurobiolaging.2018.11.003
Kim, G., Gautier, O., Tassoni-Tsuchida, E., Ma, X. R., and Gitler, A. D. (2020). ALS genetics: gains, losses, and implications for future therapies. Neuron 108, 822–842. doi: 10.1016/j.neuron.2020.08.022
Kim, J., Liao, Y.-H., Ionita, C., Bale, A. E., Darras, B., and Acsadi, G. (2016). Mitochondrial membrane protein-associated neurodegeneration mimicking juvenile amyotrophic lateral sclerosis. Pediatr. Neurol. 64, 83–86. doi: 10.1016/j.pediatrneurol.2016.08.013
Kim, H. J., and Taylor, J. P. (2017). Lost in transportation: nucleocytoplasmic transport defects in ALS and other neurodegenerative diseases. Neuron 96, 285–297. doi: 10.1016/j.neuron.2017.07.029
Klim, J. R., Pintacuda, G., Nash, L. A., Guerra San Juan, I., and Eggan, K. (2021). Connecting TDP-43 pathology with neuropathy. Trends Neurosci. 44, 424–440. doi: 10.1016/j.tins.2021.02.008
Korobeynikov, V. A., Lyashchenko, A. K., Blanco-Redondo, B., Jafar-Nejad, P., and Shneider, N. A. (2022). Antisense oligonucleotide silencing of FUS expression as a therapeutic approach in amyotrophic lateral sclerosis. Nat. Med. 28, 104–116. doi: 10.1038/s41591-021-01615-z
Kundra, R., Dobson, C. M., and Vendruscolo, M. (2020). A cell-and tissue-specific weakness of the protein homeostasis system underlies brain vulnerability to protein aggregation. iScience 23:100934. doi: 10.1016/j.isci.2020.100934
Lapucci, A., Cavone, L., Buonvicino, D., Felici, R., Gerace, E., Zwergel, C., et al. (2017). Effect of class II HDAC inhibition on glutamate transporter expression and survival in SOD1-ALS mice. Neurosci. Lett. 656, 120–125. doi: 10.1016/j.neulet.2017.07.033
Lee, S., Jeon, Y.-M., Cha, S.-J., Kim, S., Kwon, Y., Jo, M., et al. (2020). PTK2/FAK regulates UPS impairment via SQSTM1/p62 phosphorylation in TARDBP/TDP-43 proteinopathies. Autophagy 16, 1396–1412. doi: 10.1080/15548627.2019.1686729
Lee, S., Kwon, Y., Kim, S., Jo, M., Jeon, Y.-M., Cheon, M., et al. (2020). The role of HDAC6 in TDP-43-induced neurotoxicity and UPS impairment. Front. Cell Dev. Biol. 8:581942. doi: 10.3389/fcell.2020.581942
Leskelä, S., Huber, N., Rostalski, H., Natunen, T., Remes, A. M., Takalo, M., et al. (2019). C9orf72 proteins regulate autophagy and undergo autophagosomal or proteasomal degradation in a cell type-dependent manner. Cells 8:1233. doi: 10.3390/cells8101233
Li, C., Hou, Y., Wei, Q., Lin, J., Jiang, Z., Jiang, Q., et al. (2023). Mutation screening of SPTLC1 and SPTLC2 in amyotrophic lateral sclerosis. Hum. Genomics 17:28. doi: 10.1186/s40246-023-00479-3
Ling, S.-C. (2018). Synaptic paths to neurodegeneration: the emerging role of TDP-43 and FUS in synaptic functions. Neural Plast. 2018, 1–13. doi: 10.1155/2018/8413496
Ling, S.-C., Polymenidou, M., and Cleveland, D. W. (2013). Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron 79, 416–438. doi: 10.1016/j.neuron.2013.07.033
Liu, X., Pimm, M. L., Haarer, B., Brawner, A. T., and Henty-Ridilla, J. L. (2022). Biochemical characterization of actin assembly mechanisms with ALS-associated profilin variants. Eur. J. Cell Biol. 101:151212. doi: 10.1016/j.ejcb.2022.151212
López-Erauskin, J., Tadokoro, T., Baughn, M. W., Myers, B., McAlonis-Downes, M., Chillon-Marinas, C., et al. (2020). ALS/FTD-linked mutation in FUS suppresses intra-axonal protein synthesis and drives disease without nuclear loss-of-function of FUS. Neuron 106:354. doi: 10.1016/j.neuron.2020.04.006
Machts, J., Vielhaber, S., Kollewe, K., Petri, S., Kaufmann, J., and Schoenfeld, M. A. (2018). Global hippocampal volume reductions and local CA1 shape deformations in amyotrophic lateral sclerosis. Front. Neurol. 9:565. doi: 10.3389/fneur.2018.00565
Mackenzie, I. R., Nicholson, A. M., Sarkar, M., Messing, J., Purice, M. D., Pottier, C., et al. (2017). TIA1 mutations in amyotrophic lateral sclerosis and frontotemporal dementia promote phase separation and Alter stress granule dynamics. Neuron 95, 808–816.e9. doi: 10.1016/j.neuron.2017.07.025
Maurel, C., Dangoumau, A., Marouillat, S., Brulard, C., Chami, A., Hergesheimer, R., et al. (2018). Causative genes in amyotrophic lateral sclerosis and protein degradation pathways: a link to neurodegeneration. Mol. Neurobiol. 55, 6480–6499. doi: 10.1007/s12035-017-0856-0
Mohassel, P., Donkervoort, S., Lone, M. A., Nalls, M., Gable, K., Gupta, S. D., et al. (2021). Childhood amyotrophic lateral sclerosis caused by excess sphingolipid synthesis. Nat. Med. 27, 1197–1204. doi: 10.1038/s41591-021-01346-1
Muñoz-Lasso, D. C., Romá-Mateo, C., Pallardó, F. V., and Gonzalez-Cabo, P. (2020). Much more than a scaffold: cytoskeletal proteins in neurological disorders. Cells 9:358. doi: 10.3390/cells9020358
Nassif, M., Woehlbier, U., and Manque, P. A. (2017). The enigmatic role of C9ORF72 in autophagy. Front. Neurosci. 11:442. doi: 10.3389/fnins.2017.00442
Neelagandan, N., Gonnella, G., Dang, S., Janiesch, P. C., Miller, K. K., Küchler, K., et al. (2019). TDP-43 enhances translation of specific mRNAs linked to neurodegenerative disease. Nucl. Acids Res. 47, 341–361. doi: 10.1093/nar/gky972
Olloquequi, J., Cornejo-Córdova, E., Verdaguer, E., Soriano, F. X., Binvignat, O., Auladell, C., et al. (2018). Excitotoxicity in the pathogenesis of neurological and psychiatric disorders: therapeutic implications. J. Psychopharmacol. 32, 265–275. doi: 10.1177/0269881118754680
Osking, Z., Ayers, J. I., Hildebrandt, R., Skruber, K., Brown, H., Ryu, D., et al. (2019). ALS-linked SOD1 mutants enhance neurite outgrowth and branching in adult motor neurons. iScience 11, 294–304. doi: 10.1016/j.isci.2018.12.026
Pajarillo, E., Rizor, A., Lee, J., Aschner, M., and Lee, E. (2019). The role of astrocytic glutamate transporters GLT-1 and GLAST in neurological disorders: potential targets for neurotherapeutics. Neuropharmacology 161:107559. doi: 10.1016/j.neuropharm.2019.03.002
Pang, W., and Hu, F. (2021). Cellular and physiological functions of C9ORF72 and implications for ALS/FTD. J. Neurochem. 157, 334–350. doi: 10.1111/jnc.15255
Pimm, M. L., Hotaling, J., and Henty-Ridilla, J. L. (2020). Profilin choreographs actin and microtubules in cells and cancer. Int. Rev. Cell Mol. Biol. 355:5. doi: 10.1016/bs.ircmb.2020.05.005
Pinto-Costa, R., Sousa, S. C., Leite, S. C., Nogueira-Rodrigues, J., Ferreira da Silva, T., Machado, D., et al. (2020). Profilin 1 delivery tunes cytoskeletal dynamics toward CNS axon regeneration. J. Clin. Invest. 130, 2024–2040. doi: 10.1172/JCI125771
Prehn, J. H. M., and Jirström, E. (2020). Angiogenin and tRNA fragments in Parkinson’s disease and neurodegeneration. Acta Pharmacol. Sin. 41, 442–446. doi: 10.1038/s41401-020-0375-9
Purandare, N., Somayajulu, M., Hüttemann, M., Grossman, L. I., and Aras, S. (2018). The cellular stress proteins CHCHD10 and MNRR1 (CHCHD2): partners in mitochondrial and nuclear function and dysfunction. J. Biol. Chem. 293, 6517–6529. doi: 10.1074/jbc.RA117.001073
Rosenblum, L. T., Shamamandri-Markandaiah, S., Ghosh, B., Foran, E., Lepore, A. C., Pasinelli, P., et al. (2017). Mutation of the caspase-3 cleavage site in the astroglial glutamate transporter EAAT2 delays disease progression and extends lifespan in the SOD1-G93A mouse model of ALS. Exp. Neurol. 292, 145–153. doi: 10.1016/j.expneurol.2017.03.014
Sahadevan, S., Hembach, K. M., Tantardini, E., Pérez-Berlanga, M., Hruska-Plochan, M., Megat, S., et al. (2021). Synaptic FUS accumulation triggers early misregulation of synaptic RNAs in a mouse model of ALS. Nat. Commun. 12:3027. doi: 10.1038/s41467-021-23188-8
Sakai, S., Watanabe, S., Komine, O., Sobue, A., and Yamanaka, K. (2021). Novel reporters of mitochondria-associated membranes (MAM), MAMtrackers, demonstrate MAM disruption as a common pathological feature in amyotrophic lateral sclerosis. FASEB J. 35:e21688. doi: 10.1096/fj.202100137R
Schmidt, E. J., Funes, S., McKeon, J. E., Morgan, B. R., Boopathy, S., O’Connor, L. C., et al. (2021). ALS-linked PFN1 variants exhibit loss and gain of functions in the context of formin-induced actin polymerization. Proc. Natl. Acad. Sci. U. S. A. 118:e2024605118. doi: 10.1073/pnas.2024605118
Sekiyama, N., Takaba, K., Maki-Yonekura, S., Akagi, K.-I., Ohtani, Y., Imamura, K., et al. (2022). ALS mutations in the TIA-1 prion-like domain trigger highly condensed pathogenic structures. Proc. Natl. Acad. Sci. U. S. A. 119:e2122523119. doi: 10.1073/pnas.2122523119
Semmler, S., Gagné, M., Garg, P., Pickles, S. R., Baudouin, C., Hamon-Keromen, E., et al. (2020). TNF receptor-associated factor 6 interacts with ALS-linked misfolded superoxide dismutase 1 and promotes aggregation. J. Biol. Chem. 295, 3808–3825. doi: 10.1074/jbc.RA119.011215
Shenouda, M., Zhang, A. B., Weichert, A., and Robertson, J. (2018). Mechanisms associated with TDP-43 neurotoxicity in ALS/FTLD. Adv Neurobiol 20, 239–263. doi: 10.1007/978-3-319-89689-2_9
Shi, Y., Hung, S.-T., Rocha, G., Lin, S., Linares, G. R., Staats, K. A., et al. (2019). Identification and therapeutic rescue of autophagosome and glutamate receptor defects in C9ORF72 and sporadic ALS neurons. JCI Insight 4:127736. doi: 10.1172/jci.insight.127736
Shi, Y., Lin, S., Staats, K. A., Li, Y., Chang, W.-H., Hung, S.-T., et al. (2018). Haploinsufficiency leads to neurodegeneration in C9ORF72 ALS/FTD human induced motor neurons. Nat. Med. 24, 313–325. doi: 10.1038/nm.4490
Shoshan-Barmatz, V., Shteinfer-Kuzmine, A., and Verma, A. (2020). VDAC1 at the intersection of cell metabolism, apoptosis, and diseases. Biomol. Ther. 10:1485. doi: 10.3390/biom10111485
Smith, E. F., Shaw, P. J., and De Vos, K. J. (2019). The role of mitochondria in amyotrophic lateral sclerosis. Neurosci. Lett. 710:132933. doi: 10.1016/j.neulet.2017.06.052
Starr, A., and Sattler, R. (2018). Synaptic dysfunction and altered excitability in C9ORF72 ALS/FTD. Brain Res. 1693, 98–108. doi: 10.1016/j.brainres.2018.02.011
Straub, I. R., Janer, A., Weraarpachai, W., Zinman, L., Robertson, J., Rogaeva, E., et al. (2018). Loss of CHCHD10-CHCHD2 complexes required for respiration underlies the pathogenicity of a CHCHD10 mutation in ALS. Hum. Mol. Genet. 27, 178–189. doi: 10.1093/hmg/ddx393
Tedesco, B., Ferrari, V., Cozzi, M., Chierichetti, M., Casarotto, E., Pramaggiore, P., et al. (2022). The role of small heat shock proteins in protein misfolding associated motoneuron diseases. Int. J. Mol. Sci. 23:11759. doi: 10.3390/ijms231911759
Tedesco, B., Vendredy, L., Timmerman, V., and Poletti, A. (2023). The chaperone-assisted selective autophagy complex dynamics and dysfunctions. Autophagy 19, 1619–1641. doi: 10.1080/15548627.2022.2160564
Tsai, Y.-L., Coady, T. H., Lu, L., Zheng, D., Alland, I., Tian, B., et al. (2020). ALS/FTD-associated protein FUS induces mitochondrial dysfunction by preferentially sequestering respiratory chain complex mRNAs. Genes Dev. 34, 785–805. doi: 10.1101/gad.335836.119
Vant Spijker, H. M., and Almeida, S. (2023). How villains are made: the translation of dipeptide repeat proteins in C9ORF72-ALS/FTD. Gene 858:147167. doi: 10.1016/j.gene.2023.147167
Wang, W., Arakawa, H., Wang, L., Okolo, O., Siedlak, S. L., Jiang, Y., et al. (2017). Motor-coordinative and cognitive dysfunction caused by mutant TDP-43 could be reversed by inhibiting its mitochondrial localization. Mol. Ther. 25, 127–139. doi: 10.1016/j.ymthe.2016.10.013
Wang, P., Deng, J., Dong, J., Liu, J., Bigio, E. H., Mesulam, M., et al. (2019). TDP-43 induces mitochondrial damage and activates the mitochondrial unfolded protein response. PLoS Genet. 15:e1007947. doi: 10.1371/journal.pgen.1007947
Wani, A., and Weihl, C. C. (2021). Loss-of-function mutation in VCP mimics the characteristic pathology as in FTLD-TARDBP. Autophagy 17, 4502–4503. doi: 10.1080/15548627.2021.1985880
Wood, A., Gurfinkel, Y., Polain, N., Lamont, W., and Rea, S. Y. (2021). Molecular mechanisms underlying TDP-43 pathology in cellular and animal models of ALS and FTLD. Int. J. Mol. Sci. 22:4705. doi: 10.3390/ijms22094705
Wu, J. J., Cai, A., Greenslade, J. E., Higgins, N. R., Fan, C., Le, N. T. T., et al. (2020). ALS/FTD mutations in UBQLN2 impede autophagy by reducing autophagosome acidification through loss of function. Proc. Natl. Acad. Sci. U. S. A. 117, 15230–15241. doi: 10.1073/pnas.1917371117
Wu, N.-H., Ye, Y., Wan, B.-B., Yu, Y.-D., Liu, C., and Chen, Q.-J. (2021). Emerging benefits: pathophysiological functions and target drugs of the Sigma-1 receptor in neurodegenerative diseases. Mol. Neurobiol. 58, 5649–5666. doi: 10.1007/s12035-021-02524-5
Xu, W., and Xu, J. (2018). C9orf72 dipeptide repeats cause selective neurodegeneration and cell-autonomous excitotoxicity in drosophila glutamatergic neurons. J. Neurosci. 38, 7741–7752. doi: 10.1523/JNEUROSCI.0908-18.2018
Xue, Y. C., Ng, C. S., Xiang, P., Liu, H., Zhang, K., Mohamud, Y., et al. (2020). Dysregulation of RNA-binding proteins in amyotrophic lateral sclerosis. Front. Mol. Neurosci. 13:78. doi: 10.3389/fnmol.2020.00078
Yamashita, T., and Kwak, S. (2019). Cell death cascade and molecular therapy in ADAR2-deficient motor neurons of ALS. Neurosci. Res. 144, 4–13. doi: 10.1016/j.neures.2018.06.004
Yasuda, K., Clatterbuck-Soper, S. F., Jackrel, M. E., Shorter, J., and Mili, S. (2017). FUS inclusions disrupt RNA localization by sequestering kinesin-1 and inhibiting microtubule detyrosination. J. Cell Biol. 216, 1015–1034. doi: 10.1083/jcb.201608022
Yin, P., Bai, D., Zhu, L., Deng, F., Guo, X., Li, B., et al. (2021). Cytoplasmic TDP-43 impairs the activity of the ubiquitin-proteasome system. Exp. Neurol. 345:113833. doi: 10.1016/j.expneurol.2021.113833
Zhang, K., Wang, A., Zhong, K., Qi, S., Wei, C., Shu, X., et al. (2021). UBQLN2-HSP70 axis reduces poly-Gly-ala aggregates and alleviates behavioral defects in the C9ORF72 animal model. Neuron 109, 1949–1962.e6. doi: 10.1016/j.neuron.2021.04.023
Zhao, J., Fok, A. H. K., Fan, R., Kwan, P.-Y., Chan, H.-L., Lo, L. H.-Y., et al. (2020). Specific depletion of the motor protein KIF5B leads to deficits in dendritic transport, synaptic plasticity and memory. elife 9:e53456. doi: 10.7554/eLife.53456
Ziff, O. J., Clarke, B. E., Taha, D. M., Crerar, H., Luscombe, N. M., and Patani, R. (2022). Meta-analysis of human and mouse ALS astrocytes reveals multi-omic signatures of inflammatory reactive states. Genome Res. 32, 71–84. doi: 10.1101/gr.275939.121
Keywords: amyotrophic lateral sclerosis, pathogenesis, genetics, molecule, proteomics
Citation: Zhou W and Xu R (2023) Current insights in the molecular genetic pathogenesis of amyotrophic lateral sclerosis. Front. Neurosci. 17:1189470. doi: 10.3389/fnins.2023.1189470
Edited by:
Marija Cvetanovic, University of Minnesota Twin Cities, United StatesReviewed by:
Feng Wang, Emory University, United StatesCopyright © 2023 Zhou and Xu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Renshi Xu, eHVyZW5zaGlAbmN1LmVkdS5jbg==
‡ORCID: Wan Zhou orcid.org/0000-0002-3657-3594
Renshi Xu orcid.org/0000-0003-0313-3434
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.