 Zehui Li
Zehui Li Yu Cao
Yu Cao Hui Pei
Hui Pei Lina Ma
Lina Ma Yang Yang1*
Yang Yang1* Hao Li
Hao Li
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Neurosci., 07 March 2023
Sec. Neuropharmacology
Volume 17 - 2023 | https://doi.org/10.3389/fnins.2023.1158204
This article is part of the Research TopicNeuroprotection and Neurorestoration: Natural Medicinal Products in Preventing and Ameliorating Cognitive ImpairmentView all 15 articles
Alzheimer’s disease (AD) is the most common neurodegenerative disease. There are many studies targeting extracellular deposits of amyloid β-peptide (Aβ) and intracellular neurofibrillary tangles (NFTs), however, there are no effective treatments to halt the progression. Mitochondria-associated endoplasmic reticulum membranes (MAMs) have long been found to be associated with various pathogenesis hypotheses of AD, such as Aβ deposition, mitochondrial dysfunction, and calcium homeostasis. However, there is a lack of literature summarizing recent advances in the mechanism and treatment studies. Accordingly, this article reviews the latest research involving the roles of MAM structure and tethering proteins in the pathogenesis of AD and summarizes potential strategies targeting MAMs to dissect treatment perspectives for AD.
According to Alzheimer’s disease (AD) International, 55 million people worldwide suffer from dementia. The most common type of dementia is AD. The main histopathological characteristics of AD are the accumulation of extracellular deposits of amyloid β-peptide (Aβ) and intracellular neurofibrillary tangles (NFTs). However, there are other biochemical and morphological characteristics present earlier in the course of AD, such as alterations in phospholipid metabolism, the elevation of circulating cholesterol levels, aberrant calcium regulation, reduction of brain glucose levels, and mitochondrial dysfunction. Plaques and tangles have received a lot of attention because they are observable physical entities, however, the other features may be the upstream factors and should not be overlooked (Area-Gomez and Schon, 2017).
Mitochondria-associated endoplasmic reticulum membranes (MAMs) are a special subdomain of the endoplasmic reticulum (ER) that physically and biologically connects mitochondria to ER. ER-mitochondrial communication and MAM functions are increased expressively in AD (Leal et al., 2020). MAMs are generated by the ER side-by-side to the mitochondrial outer membrane linked by a series of tethering proteins (Degechisa et al., 2022). They are not the collection of membranes but the employment of the proteinaceous tethers (Rowland and Voeltz, 2012), which play a key role in many important metabolic processes including the transfer of calcium, mitochondrial dynamics, lipid synthesis, autophagy, apoptosis, and inflammation. MAM dysfunction is also central to the pathogenic mechanisms of AD, especially in the Aβ generation and deposition, mitochondrial dysfunction, imbalanced calcium homeostasis, abnormal lipids metabolism, and abnormal autophagy. This review highlights abnormal MAM structures and tethering proteins in the pathogenesis of AD from these aspects. In addition, we provide a summary of compounds and drugs that target MAM tethering proteins in AD models.
The Aβ peptide is the main component of the AD hallmark amyloid plaques, which is produced by the proteolysis of amyloid beta-precursor protein (APP) by two enzymes: β-site APP cleaving enzyme 1 (BACE1) and the γ-secretase complex. Many studies have shown that MAMs are the main site of Aβ generation. Schreiner et al. (2015) reported that Aβ may be generated directly in MAMs. APP, BACE1, and γ-secretase have all been found in MAMs (Leal et al., 2020). BACE1 cleaves APP to produce sAPPβ and C99. C99 is delivered to MAMs and cleaved to produce Aβ and APP intracellular domain (AICD) by γ-secretase (Montesinos et al., 2020). In addition, it has been reported that the main pathway for Aβ entering mitochondria is MAMs (Del Prete et al., 2017). Knockdown of mitofusin-2 (Mfn2), which is involved in MAM tethering, leads to decreased contact between mitochondria and ER, resulting in lower γ-secretase activity and decreased concentrations of intracellular and extracellular Aβ40 and Aβ42 (Leal et al., 2016). This proves that the increase of MAMs may promote mitochondrial Aβ deposition. Axonal generation of Aβ also plays a key role in AD pathology. In AD models, swollen axons contain high levels of BACE1 (Gowrishankar et al., 2017). An average of 37 ± 4% of the total Aβ40 secreted from each mouse hippocampal neuron is secreted by axons (Niederst et al., 2015). Bhattacharyya et al. showed that down-regulation of MAM assembly by silencing MAM-resident sigma-1 receptor expression resulted in reduced palmitoylated APP cleavage by BACE1, thereby decreasing Aβ generation in neuronal processes and axons (Bhattacharyya et al., 2021).
The neuronal activity must depend on the energy produced by mitochondria. Mitochondrial death can be observed before the histopathological features of AD appear. MAMs wrap around the location where mitochondria undergo fissuring by recruiting MAM protein inverted formin 2 (INF2) (Steffen and Koehler, 2018). The mitochondrial outer membrane protein FUNDC1 is a MAM protein that recruits dynamin 1 Like (DNM1L)/dynamin-related protein 1 (DRP1) to drive mitochondrial fission (Wu et al., 2016). In an AD mouse model, alterations in MAMs precede changes in mitochondrial dynamics accompanied by aberrations in mitochondrial membrane potential (MMP) and ATP production (Leal et al., 2020). Moreover, the MAMs control mitochondrial Ca2+ intake. Reduced Ca2+ intake affects mitochondrial metabolism, leading to mitochondrial dysfunction. Fernandes et al. (2021) found a decrease in close ER-mitochondria contacts, a reduction of Ca2+ transfer from ER to mitochondria, and impaired mitochondrial function in APPswe cells. γ-secretase activating protein (GSAP) fluorescence staining showed high colocalization with a MAM marker Facl4. Knockdown of GSAP significantly increased mitochondrial basal respiration and total ATP levels, which suggests that GSAP impairs mitochondria function (Xu et al., 2021). In an AD cell model, increased concentration of unprocessed C99 in the MAM region increased sphingolipid turnover and altered the lipid composition of mitochondrial membranes, which can interfere with the normal activity of the respiratory supercomplexes and thus may contribute to the bioenergetic defects in AD (Pera et al., 2017). Atlastin 2 (ATL2) is a protein associated with ER-mitochondria contacts whose expression was significantly increased both in 3 × Tg mice and AD patients. While, knockout of ATL2 rescued the elevated ER-mitochondria contacts back to normal levels, reduced the abnormally elevated mitochondrial superoxide, and significantly increased the MMP (Han et al., 2021).
Calcium signaling in neurons is essential for neurotransmission and the maintenance of synaptic plasticity (Skobeleva et al., 2022). Dysregulation of calcium homeostasis disrupts neuronal and synaptic function in AD (McDaid et al., 2020). Ca2+ homeostasis depends on the Sarcoplasmic/endoplasmic reticulum Ca2+ ATPase (SERCA) pumps to regulate Ca2+ uptake from the cytoplasm to the ER and activated inositol 1,4,5-triphosphate receptors (IP3Rs) and ryanodine receptors (RyRs) to enable Ca2+ efflux from the ER (Kawamoto et al., 2012). The IP3R–glucose-regulated protein 75 (Grp75)–voltage-dependent anion channel (VDAC) Ca2+ channeling complex was concentrated in MAMs. Up-regulation of this complex results in increased calcium content. The accumulation of mitochondrial calcium could be prohibited by the IP3R inhibitor (Chen et al., 2021b). The RyR is a Ca2+-release channel protein located in the MAMs and sarcoplasmic reticulum. The RyR3 plays a dual character in AD pathology. Deletion of RyR3 in young APP/PS1 mice increases the excitability of hippocampal neuronal networks and accelerates AD progress. However, in aged APP/PS1 mice, the deletion of RyR3 restored network excitability (Liu et al., 2014). In the 5xFAD mouse model of AD, restricting RyR2 open time blocked the excessive activity of CA1 neurons (Yao et al., 2020). A small-molecule SERCA activator can increase ER Ca2+ and has shown efficacy in APP/PS1 mice, supporting SERCA activation as a therapeutic strategy for AD (Krajnak and Dahl, 2018).
Lipid metabolism, especially cholesterol metabolism, has been implicated in the synaptic dysfunction of AD (Petrov et al., 2017). Ceramide and cholesterol have been found to increase in the brains of AD patients, normal aging mice, and neurons exposed to Aβ1–42 (Cutler et al., 2004). MAMs are transient lipid rafts that are closely related to cholesterol and phospholipid metabolism (Pera et al., 2020). It has been proven that phospholipid transport between the ER and mitochondria is dependent on membrane integrity including MAMs rather than energy or MMP (Kojima et al., 2016). MAMs are where the enzymatic activities such as acetyl-CoA acetyltransferase 1 (ACAT1) that regulate cholesterol levels reside. ACAT1 gene ablation led to the amelioration of amyloid pathology and cognitive deficits in 3 × Tg AD mice (Bryleva et al., 2010). The concentration of unprocessed C99 at the MAMs is increased in models of AD and cells from AD patients, subsequently associated with the increase in ceramide levels (Pera et al., 2017). GSAP enriched in MAMs regulates lipid homeostasis through the processing of APP (Xu et al., 2021). In a cortical impact mice model of AD, the injured cortex and hippocampus exhibited significant increases in MAM activity, phospholipid synthesis, sphingomyelinase (SMases) activity, and cholesterol turnover (Agrawal et al., 2022). When cholesterol concentrations exceed a certain threshold, SMases are activated and hydrolyze sphingomyelin (SM) to produce ceramides. The cholesterol affinity of APP is involved in limiting APP distribution, conversely, APP senses and balances the membrane cholesterol (DelBove et al., 2019). An increase in MAM-localized C99 triggers the upregulation of SMase activity. The increased cholesterol mobilization observed in AD cells may be an outcome of persistent SMase activity caused by increased MAM-C99, which disrupts cellular lipid homeostasis and causes the alterations in membrane lipid composition commonly happen in AD (Montesinos et al., 2020).
Autophagy is a degradation mechanism of cells to remove damaged and senescent organelles and maintain cellular homeostasis. Autophagy plays a vital role in the generation and clearance of Aβ. Abnormal autophagy is involved in the development of AD (Yuan et al., 2022). Activation of autophagy alleviates pathological characters and cognitive deficits in APP/PS1 mice (Yang et al., 2023). The MAMs mark the starting point of autophagosome formation. Mitochondrial fusion protein Mfn2 is a MAM protein responsible for binding the ER and mitochondria. The energy sensor AMP-activated protein kinase (AMPK) interacts with Mfn2 and phosphorylates Mfn2 and induces autophagy (Hu et al., 2021). Autophagy initiation proteins such as autophagy and beclin 1 regulator 1 (AMBRA1) and Beclin 1 are recruited to the MAMs to regulate autophagy, demonstrating that MAM raft-like microdomains play a crucial part in the autophagosome formation (Garofalo et al., 2016). The sigma-1 receptor (Sig-1R) is a receptor with molecular chaperone activity clustered on MAMs. Autophagosome clearance is impaired in Sigma-1 KO cells, possibly due to impaired autophagosome-lysosomal organelle fusion mediated by the complex formed by Sig-1R with STX17 and ATG14. STX17 and ATG14 both appear at the ER-mitochondria contact site (Yang et al., 2019). The combination of vesicle-associated membrane protein-associated protein B (VAPB) and protein tyrosine phosphatase interacting protein 51 (PTPIP51) is identified as one of the MAM tethers. Manipulation of their expression to increase and decrease ER-mitochondrial junctions had significant effects on autophagy (Gomez-Suaga et al., 2017). The decreased expression of MAM tethering protein complexes including VAPB-PTPP51, BAP31-FIS1, and Mfn2-Mfn1 leads to abnormal autophagy, which leads to the decline of cognitive ability (Liu et al., 2022). The role of MAM tethering proteins in several major pathogeneses of AD is summarized in Figure 1 schematically.
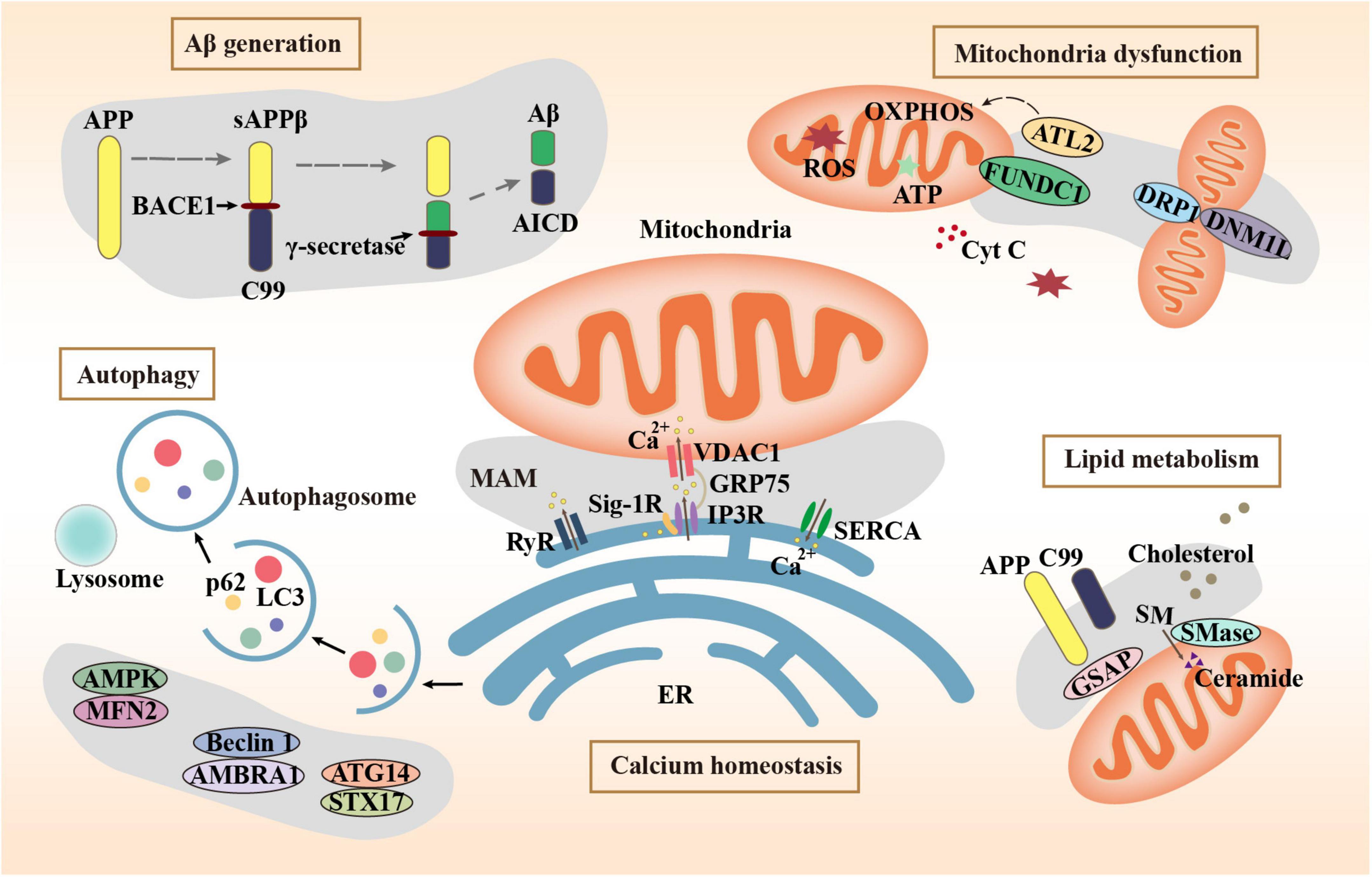
Figure 1. Mechanisms related to MAMs in AD.
The functions of MAMs are mainly exerted by its numerous tethering proteins. The abnormalities of these proteins eventually lead to the pathological processes of AD through multiple mechanisms. The expression of MAM tethering proteins and the tight connection between mitochondria and ER can be one of the hallmarks of AD progression. The regulation of these proteins to improve the corresponding phenotypes can be used as a strategy and target for AD treatment.
Knockdown of MAM protein GSAP reduced Aβ generation and plaque formation in an AD mouse model (He et al., 2010). Imatinib and imatinib methanesulfonate could prevent Aβ formation by inhibiting GSAP (He et al., 2010; Weintraub et al., 2013; Chu et al., 2014). Lithium treatment reduced abnormal IP3R-dependent ER Ca2+ signaling and enhanced synaptic plasticity in 3 × Tg AD mice (Wiseman et al., 2023). Xestospongin C can ameliorate the Ca2+ overload of primary hippocampal neurons induced by Aβ1–42 and improve the cognitive ability of APP/PS1 mice (Wang et al., 2019). Mecobalamin also reduces ER-mitochondrial calcium flux through IP3R and prevents mitochondrial dysfunction (Wang and Xu, 2019). RyR2 instability plays a vital role in the reduction of ER Ca2+ content, which alters synaptic transmission and plasticity mechanisms. While Dantrolene stabilizes RyR2 thereby reversing most AD-related phenotypes in AppNL-G-F mice (Nakamura et al., 2021). Compound 12a inhibited Ca2+ release and significantly accelerated the cognitive behavior of FAD mice in the Morris water maze test. Moreover, docking simulations testified that 12a could bind to the active site of RyR1 (Dai et al., 2021). A Sig-1R agonist (+) SKF-10,047 could significantly increase mitochondrial movement in cortical neurons of 3 × Tg mice, which might be because it leads to the removal of Sig-1R from MAMs to mitochondria. (+) SKF-10,047 also leads to an increase in the number of mitochondria (Cavendish et al., 2019). Two other Sig-1R agonists ANAVEX2-73 and PRE-084 could prevent mitochondrial respiratory dysfunction in Aβ25–35-injected mice (Lahmy et al., 2014). IRE1α inhibitor 4μ8c can reduce ER-mitochondrial association and restore the normal function of MAMs by inhibiting the expression and interaction of IP3R, Grp75, and VDAC1, thereby restoring ATP content and MMP (Chu et al., 2021). Artesunate could reverse cognitive impairment in APP/PS1 mice by regulating DRP1 to maintain mitochondrial dynamics (Qin et al., 2022). By increasing the expression of the MAM protein Mfn2, Biochanin A could reverse the imbalance of mitochondrial dynamics and abnormal mitophagy in APP/PS1 mice (Hou et al., 2022). Curcumin restores basal mitochondrial respiration and ATP production in thapsigargin-injured SH-SY5Y cells by targeting Mfn2 (Zhou et al., 2022). Besides, Myricetin, Selenomethionine, Leptin, Trans-ferulic acid, Ligustilide, and Albiflorin were also able to regulate Mfn2 and ameliorate mitochondrial dysfunction (Xu et al., 2018, 2019; Zafeer et al., 2019; Cheng et al., 2020; Zhu et al., 2020; Chen et al., 2021a; Yao et al., 2022). Cholesteryl ester produced in MAMs is involved in the pathogenesis of AD. Progesterone reduced the expression of ACAT1 through the ERK1/2 pathway, shortened the abnormally prolonged MAM length, inhibited cholesteryl ester accumulation in the cortex, and improved the cognitive function of APP/PS1 mice (Shi et al., 2021). We summarize potential compounds and drugs that exert anti-AD effects by regulating MAM tethering proteins in Table 1.
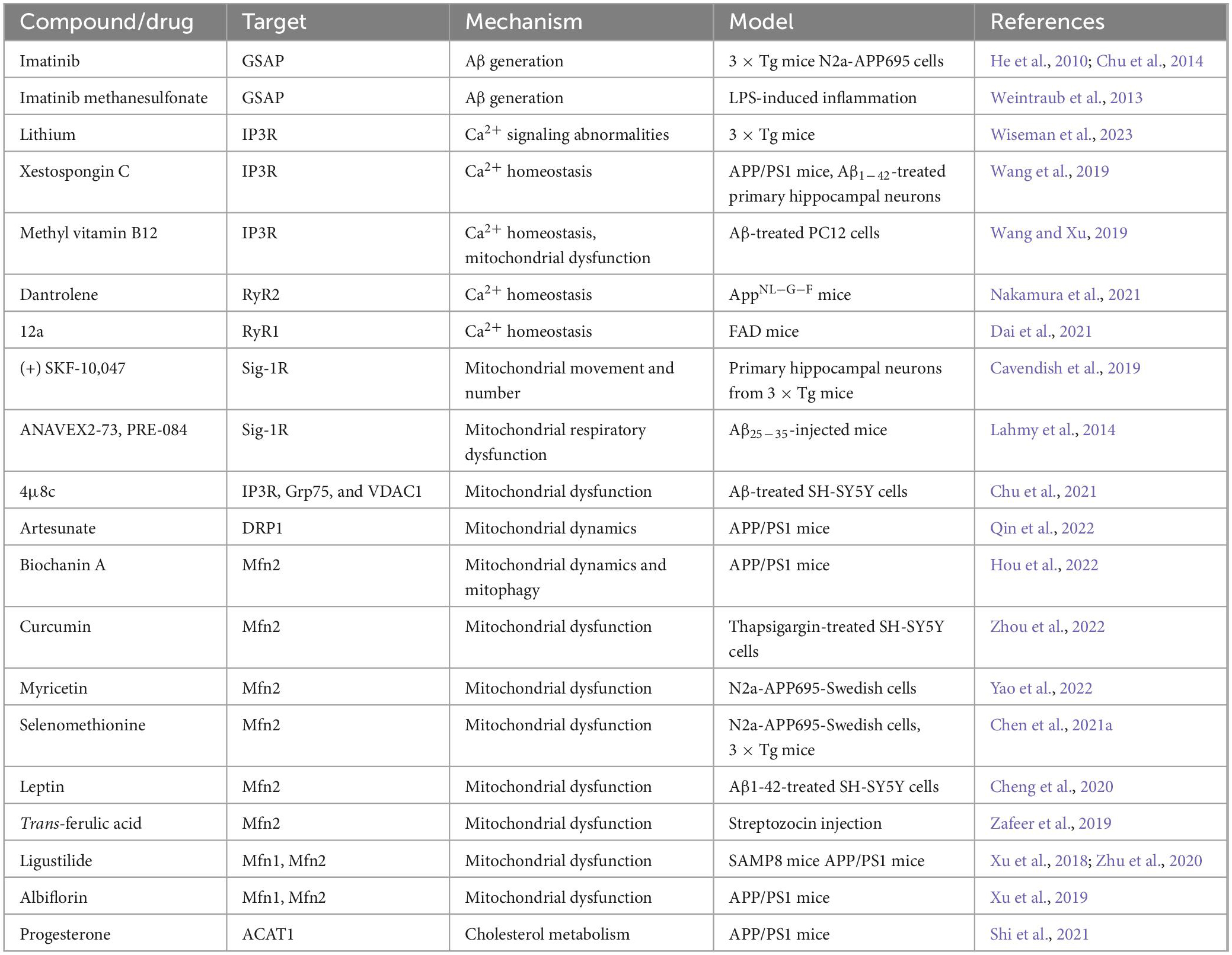
Table 1. Compounds and drugs targeting MAM tethering proteins for treating AD.
Drug development for AD has continuously been challenged. The most popular AD hypotheses are amyloid cascade, hyperphosphorylation of tau, and mitochondrial cascade. However, numerous studies targeting these hypotheses have not been able to fully elucidate the mechanism of AD or retard its progression. The interaction between different organelles, especially mitochondria and ER in cells has gradually emerged in the study of various diseases (Theurey and Rieusset, 2017; Luan et al., 2021). Cellular dysfunction in the early stage of AD, including Ca2+ homeostasis, mitochondrial dysfunction, oxidative stress, and abnormal autophagy, are all associated with MAM function (Tapella et al., 2022).
As for the amyloid cascade, MAMs are the sites where C99 is cleaved to Aβ. Inhibition of MAMs protein expression and function may reduce Aβ production and extracellular deposition. MAMs recruit proteins such as INF2 and DRP1 to participate in mitochondrial fission, thereby affecting MMP and ATP production. Upregulation of some MAM-resident proteins causes enhanced mitochondria-ER contacts, along with mitochondrial damage such as increased mitochondrial superoxide. The MAM has both protein pumps that allow Ca2+ to flow from the cytoplasm to the ER and Ca2+ to flow from the ER, thus regulating Ca2+ homeostasis. Ca2+ flux also plays a dual role in AD progression. For instance, excessive Ca2+ influx leads to increased ROS production and cell death due to caspase activation; however, attenuated Ca2+ signaling may also be detrimental to ATP production (Filadi and Greotti, 2021; Arnst et al., 2022). Modulation of MAM-resident enzymes such as ACAT1 and MAM-C99 content affects cholesterol levels and lipid homeostasis in AD, along with amyloid synthesis and synaptic transmission. MAMs mark the starting point of autophagy. Decreased expressions of MAM tethering proteins, such as Sig-1R, VAPB-PTPP51, and Mfn2-Mfn1, lead to abnormal autophagy. All these indicate that MAM is an important target that should not be ignored in the study of AD pathogenesis.
Although MAMs have been shown to be closely related to the progression of AD, the molecular pathways are still not fully understood. At present, the research targets of AD drugs targeting MAM proteins are still limited. The mechanisms of interest are mainly in Aβ production, mitochondrial function, calcium homeostasis, and lipid metabolism. Currently, there are few studies on the interaction between MAMs and tau phosphorylation. Besides, although tethering proteins are the main manifestation of MAM function, the observation of MAM structure should not be ignored despite the technical difficulties in the research of AD treatment strategies. We look forward to new studies to further explore the role of the structure and functions of MAMs in the pathogenesis and treatment of AD.
YY and HL conceived the idea. ZL wrote the manuscript. YC, HP, and LM revised the manuscript. All authors contributed to the work and approved the submitted version.
This work was supported by the Fundamental Research Funds for the Central Public Welfare Research Institutes (No. ZZ15-YQ-018), the National Natural Science Foundation of China (Nos. 82205103 and 82205233), and the CACMS Innovation Fund (No. CI2021A01401).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling editor declared a shared affiliation with the authors ZL, YC, HP, LM, and YY.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Agrawal, R. R., Larrea, D., Xu, Y., Shi, L., Zirpoli, H., Cummins, L. G., et al. (2022). Alzheimer’s-associated upregulation of mitochondria-associated ER membranes after traumatic brain injury. Cell Mol. Neurobiol. doi: 10.1007/s10571-022-01299-0 [Epub ahead of print].
Area-Gomez, E., and Schon, E. A. (2017). On the pathogenesis of Alzheimer’s disease: the MAM hypothesis. FASEB J. 31, 864–867. doi: 10.1096/fj.201601309
Arnst, N., Redolfi, N., Lia, A., Bedetta, M., Greotti, E., and Pizzo, P. (2022). Mitochondrial Ca(2+) signaling and bioenergetics in Alzheimer’s disease. Biomedicines 10:3025. doi: 10.3390/biomedicines10123025
Bhattacharyya, R., Black, S. E., Lotlikar, M. S., Fenn, R. H., Jorfi, M., Kovacs, D. M., et al. (2021). Axonal generation of amyloid-β from palmitoylated APP in mitochondria-associated endoplasmic reticulum membranes. Cell Rep. 35:109134. doi: 10.1016/j.celrep.2021.109134
Bryleva, E. Y., Rogers, M. A., Chang, C. C., Buen, F., Harris, B. T., Rousselet, E., et al. (2010). ACAT1 gene ablation increases 24(S)-hydroxycholesterol content in the brain and ameliorates amyloid pathology in mice with AD. Proc. Natl. Acad. Sci. U. S. A. 107, 3081–3086. doi: 10.1073/pnas.0913828107
Cavendish, J. Z., Sarkar, S. N., Colantonio, M. A., Quintana, D. D., Ahmed, N., White, B. A., et al. (2019). Mitochondrial movement and number deficits in embryonic cortical neurons from 3xTg-AD mice. J. Alzheimers Dis. 70, 139–151. doi: 10.3233/jad-190143
Chen, C., Chen, Y., Zhang, Z. H., Jia, S. Z., Chen, Y. B., Huang, S. L., et al. (2021a). Selenomethionine improves mitochondrial function by upregulating mitochondrial selenoprotein in a model of Alzheimer’s disease. Front. Aging Neurosci. 13:750921. doi: 10.3389/fnagi.2021.750921
Chen, L. T., Xu, T. T., Qiu, Y. Q., Liu, N. Y., Ke, X. Y., Fang, L., et al. (2021b). Homocysteine induced a calcium-mediated disruption of mitochondrial function and dynamics in endothelial cells. J. Biochem. Mol. Toxicol. 35:e22737. doi: 10.1002/jbt.22737
Cheng, Y., Buchan, M., Vitanova, K., Aitken, L., Gunn-Moore, F. J., Ramsay, R. R., et al. (2020). Neuroprotective actions of leptin facilitated through balancing mitochondrial morphology and improving mitochondrial function. J. Neurochem. 155, 191–206. doi: 10.1111/jnc.15003
Chu, B., Li, M., Cao, X., Li, R., Jin, S., Yang, H., et al. (2021). IRE1α-XBP1 affects the mitochondrial function of Aβ25-35-treated SH-SY5Y cells by regulating mitochondria-associated endoplasmic reticulum membranes. Front. Cell Neurosci. 15:614556. doi: 10.3389/fncel.2021.614556
Chu, J., Lauretti, E., Craige, C. P., and Praticò, D. (2014). Pharmacological modulation of GSAP reduces amyloid-β levels and tau phosphorylation in a mouse model of Alzheimer’s disease with plaques and tangles. J. Alzheimers Dis. 41, 729–737. doi: 10.3233/jad-140105
Cutler, R. G., Kelly, J., Storie, K., Pedersen, W. A., Tammara, A., Hatanpaa, K., et al. (2004). Involvement of oxidative stress-induced abnormalities in ceramide and cholesterol metabolism in brain aging and Alzheimer’s disease. Proc. Natl. Acad. Sci. U.S.A. 101, 2070–2075. doi: 10.1073/pnas.0305799101
Dai, B., Ma, X., Tang, Y., Xu, L., Guo, S., Chen, X., et al. (2021). Design, synthesis, and biological activity of novel semicarbazones as potent Ryanodine receptor1 inhibitors of Alzheimer’s disease. Bioorg. Med. Chem. 29:115891. doi: 10.1016/j.bmc.2020.115891
Degechisa, S. T., Dabi, Y. T., and Gizaw, S. T. (2022). The mitochondrial associated endoplasmic reticulum membranes: a platform for the pathogenesis of inflammation-mediated metabolic diseases. Immun. Inflamm. Dis. 10:e647. doi: 10.1002/iid3.647
Del Prete, D., Suski, J. M., Oulès, B., Debayle, D., Gay, A. S., Lacas-Gervais, S., et al. (2017). Localization and processing of the amyloid-β protein precursor in mitochondria-associated membranes. J. Alzheimers Dis. 55, 1549–1570. doi: 10.3233/jad-160953
DelBove, C. E., Strothman, C. E., Lazarenko, R. M., Huang, H., Sanders, C. R., and Zhang, Q. (2019). Reciprocal modulation between amyloid precursor protein and synaptic membrane cholesterol revealed by live cell imaging. Neurobiol. Dis. 127, 449–461. doi: 10.1016/j.nbd.2019.03.009
Fernandes, T., Resende, R., Silva, D. F., Marques, A. P., Santos, A. E., Cardoso, S. M., et al. (2021). Structural and functional alterations in mitochondria-associated membranes (MAMs) and in mitochondria activate stress response mechanisms in an in vitro model of Alzheimer’s disease. Biomedicines 9:881. doi: 10.3390/biomedicines9080881
Filadi, R., and Greotti, E. (2021). The yin and yang of mitochondrial Ca(2+) signaling in cell physiology and pathology. Cell Calcium 93:102321. doi: 10.1016/j.ceca.2020.102321
Garofalo, T., Matarrese, P., Manganelli, V., Marconi, M., Tinari, A., Gambardella, L., et al. (2016). Evidence for the involvement of lipid rafts localized at the ER-mitochondria associated membranes in autophagosome formation. Autophagy 12, 917–935. doi: 10.1080/15548627.2016.1160971
Gomez-Suaga, P., Paillusson, S., Stoica, R., Noble, W., Hanger, D. P., and Miller, C. C. J. (2017). The ER-mitochondria tethering complex VAPB-PTPIP51 regulates autophagy. Curr. Biol. 27, 371–385. doi: 10.1016/j.cub.2016.12.038
Gowrishankar, S., Wu, Y., and Ferguson, S. M. (2017). Impaired JIP3-dependent axonal lysosome transport promotes amyloid plaque pathology. J. Cell Biol. 216, 3291–3305. doi: 10.1083/jcb.201612148
Han, J., Park, H., Maharana, C., Gwon, A. R., Park, J., Baek, S. H., et al. (2021). Alzheimer’s disease-causing presenilin-1 mutations have deleterious effects on mitochondrial function. Theranostics 11, 8855–8873. doi: 10.7150/thno.59776
He, G., Luo, W., Li, P., Remmers, C., Netzer, W. J., Hendrick, J., et al. (2010). Gamma-secretase activating protein is a therapeutic target for Alzheimer’s disease. Nature 467, 95–98. doi: 10.1038/nature09325
Hou, Y., Zhao, W., Yu, H., Zhang, F., Zhang, H. T., and Zhou, Y. (2022). Biochanin A alleviates cognitive impairment and hippocampal mitochondrial damage in ovariectomized APP/PS1 mice. Phytomedicine 100:154056. doi: 10.1016/j.phymed.2022.154056
Hu, Y., Chen, H., Zhang, L., Lin, X., Li, X., Zhuang, H., et al. (2021). The AMPK-MFN2 axis regulates MAM dynamics and autophagy induced by energy stresses. Autophagy 17, 1142–1156. doi: 10.1080/15548627.2020.1749490
Kawamoto, E. M., Vivar, C., and Camandola, S. (2012). Physiology and pathology of calcium signaling in the brain. Front. Pharmacol. 3:61. doi: 10.3389/fphar.2012.00061
Kojima, R., Endo, T., and Tamura, Y. (2016). A phospholipid transfer function of ER-mitochondria encounter structure revealed in vitro. Sci. Rep. 6:30777. doi: 10.1038/srep30777
Krajnak, K., and Dahl, R. (2018). A new target for Alzheimer’s disease: a small molecule SERCA activator is neuroprotective in vitro and improves memory and cognition in APP/PS1 mice. Bioorg. Med. Chem. Lett. 28, 1591–1594. doi: 10.1016/j.bmcl.2018.03.052
Lahmy, V., Long, R., Morin, D., Villard, V., and Maurice, T. (2014). Mitochondrial protection by the mixed muscarinic/σ1 ligand ANAVEX2-73, a tetrahydrofuran derivative, in Aβ25-35 peptide-injected mice, a nontransgenic Alzheimer’s disease model. Front. Cell Neurosci. 8:463. doi: 10.3389/fncel.2014.00463
Leal, N. S., Dentoni, G., Schreiner, B., Naia, L., Piras, A., Graff, C., et al. (2020). Amyloid B -peptide increases mitochondria-endoplasmic reticulum contact altering mitochondrial function and autophagosome formation in Alzheimer’s disease-related models. Cells 9:2552. doi: 10.3390/cells9122552
Leal, N. S., Schreiner, B., Pinho, C. M., Filadi, R., Wiehager, B., Karlström, H., et al. (2016). Mitofusin-2 knockdown increases ER-mitochondria contact and decreases amyloid β-peptide production. J. Cell Mol. Med. 20, 1686–1695. doi: 10.1111/jcmm.12863
Liu, J., Supnet, C., Sun, S., Zhang, H., Good, L., Popugaeva, E., et al. (2014). The role of ryanodine receptor type 3 in a mouse model of Alzheimer disease. Channels 8, 230–242. doi: 10.4161/chan.27471
Liu, J., Wang, L., Ge, L., Sun, W., Song, Z., Lu, X., et al. (2022). Lanthanum decreased VAPB-PTPP51, BAP31-FIS1, and MFN2-MFN1 expression of mitochondria-associated membranes and induced abnormal autophagy in rat hippocampus. Food Chem. Toxicol. 161:112831. doi: 10.1016/j.fct.2022.112831
Luan, Y., Luan, Y., Yuan, R. X., Feng, Q., Chen, X., and Yang, Y. (2021). Structure and function of mitochondria-associated endoplasmic reticulum membranes (MAMs) and their role in cardiovascular diseases. Oxid. Med. Cell Longev. 2021:4578809. doi: 10.1155/2021/4578809
McDaid, J., Mustaly-Kalimi, S., and Stutzmann, G. E. (2020). Ca(2+) dyshomeostasis disrupts neuronal and synaptic function in Alzheimer’s disease. Cells 9:2655. doi: 10.3390/cells9122655
Montesinos, J., Pera, M., Larrea, D., Guardia-Laguarta, C., Agrawal, R. R., Velasco, K. R., et al. (2020). The Alzheimer’s disease-associated C99 fragment of APP regulates cellular cholesterol trafficking. Embo J. 39:e103791. doi: 10.15252/embj.2019103791
Nakamura, Y., Yamamoto, T., Xu, X., Kobayashi, S., Tanaka, S., Tamitani, M., et al. (2021). Enhancing calmodulin binding to ryanodine receptor is crucial to limit neuronal cell loss in Alzheimer disease. Sci. Rep. 11:7289. doi: 10.1038/s41598-021-86822-x
Niederst, E. D., Reyna, S. M., and Goldstein, L. S. (2015). Axonal amyloid precursor protein and its fragments undergo somatodendritic endocytosis and processing. Mol. Biol. Cell 26, 205–217. doi: 10.1091/mbc.E14-06-1049
Pera, M., Larrea, D., Guardia-Laguarta, C., Montesinos, J., Velasco, K. R., Agrawal, R. R., et al. (2017). Increased localization of APP-C99 in mitochondria-associated ER membranes causes mitochondrial dysfunction in Alzheimer disease. Embo J. 36, 3356–3371. doi: 10.15252/embj.201796797
Pera, M., Montesinos, J., Larrea, D., Agrawal, R. R., Velasco, K. R., Stavrovskaya, I. G., et al. (2020). MAM and C99, key players in the pathogenesis of Alzheimer’s disease. Int. Rev. Neurobiol. 154, 235–278. doi: 10.1016/bs.irn.2020.03.016
Petrov, A. M., Kasimov, M. R., and Zefirov, A. L. (2017). Cholesterol in the pathogenesis of Alzheimer’s, Parkinson’s diseases and autism: link to synaptic dysfunction. Acta Naturae 9, 26–37.
Qin, Y. R., Ma, C. Q., Jiang, J. H., Wang, D. P., Zhang, Q. Q., Liu, M. R., et al. (2022). Artesunate restores mitochondrial fusion-fission dynamics and alleviates neuronal injury in Alzheimer’s disease models. J. Neurochem. 162, 290–304. doi: 10.1111/jnc.15620
Rowland, A. A., and Voeltz, G. K. (2012). Endoplasmic reticulum-mitochondria contacts: function of the junction. Nat. Rev. Mol. Cell Biol. 13, 607–625. doi: 10.1038/nrm3440
Schreiner, B., Hedskog, L., Wiehager, B., and Ankarcrona, M. (2015). Amyloid-β peptides are generated in mitochondria-associated endoplasmic reticulum membranes. J. Alzheimers Dis. 43, 369–374. doi: 10.3233/jad-132543
Shi, W., Wu, H., Liu, S., Wu, Z., Wu, H., Liu, J., et al. (2021). Progesterone suppresses cholesterol esterification in APP/PS1 mice and a cell model of Alzheimer’s disease. Brain Res. Bull 173, 162–173. doi: 10.1016/j.brainresbull.2021.05.020
Skobeleva, K., Shalygin, A., Mikhaylova, E., Guzhova, I., Ryazantseva, M., and Kaznacheyeva, E. (2022). The STIM1/2-regulated calcium homeostasis is impaired in hippocampal neurons of the 5xFAD mouse model of Alzheimer’s disease. Int. J. Mol. Sci. 23:14810. doi: 10.3390/ijms232314810
Steffen, J., and Koehler, C. M. (2018). ER-mitochondria contacts: actin dynamics at the ER control mitochondrial fission via calcium release. J. Cell Biol. 217, 15–17. doi: 10.1083/jcb.201711075
Tapella, L., Dematteis, G., Moro, M., Pistolato, B., Tonelli, E., Vanella, V. V., et al. (2022). Protein synthesis inhibition and loss of homeostatic functions in astrocytes from an Alzheimer’s disease mouse model: a role for ER-mitochondria interaction. Cell Death Dis. 13:878. doi: 10.1038/s41419-022-05324-4
Theurey, P., and Rieusset, J. (2017). Mitochondria-associated membranes response to nutrient availability and role in metabolic diseases. Trends Endocrinol. Metab. 28, 32–45. doi: 10.1016/j.tem.2016.09.002
Wang, M., and Xu, T. (2019). Methyl B12 protects PC12 cells against cytotoxicity induced by Aβ(25-35). J. Cell Biochem. 120, 11921–11930. doi: 10.1002/jcb.28475
Wang, Z. J., Zhao, F., Wang, C. F., Zhang, X. M., Xiao, Y., Zhou, F., et al. (2019). Xestospongin C, a reversible IP3 receptor antagonist, alleviates the cognitive and pathological impairments in APP/PS1 mice of Alzheimer’s disease. J. Alzheimers Dis. 72, 1217–1231. doi: 10.3233/jad-190796
Weintraub, M. K., Bisson, C. M., Nouri, J. N., Vinson, B. T., Eimerbrink, M. J., Kranjac, D., et al. (2013). Imatinib methanesulfonate reduces hippocampal amyloid-β and restores cognitive function following repeated endotoxin exposure. Brain Behav. Immun. 33, 24–28. doi: 10.1016/j.bbi.2013.05.002
Wiseman, A. L., Briggs, C. A., Peritt, A., Kapecki, N., Peterson, D. A., Shim, S. S., et al. (2023). Lithium provides broad therapeutic benefits in an alzheimer’s disease mouse model. J. Alzheimers Dis. 91, 273–290. doi: 10.3233/jad-220758
Wu, W., Li, W., Chen, H., Jiang, L., Zhu, R., and Feng, D. (2016). FUNDC1 is a novel mitochondrial-associated-membrane (MAM) protein required for hypoxia-induced mitochondrial fission and mitophagy. Autophagy 12, 1675–1676. doi: 10.1080/15548627.2016.1193656
Xu, P., Chang, J. C., Zhou, X., Wang, W., Bamkole, M., Wong, E., et al. (2021). GSAP regulates lipid homeostasis and mitochondrial function associated with Alzheimer’s disease. J. Exp. Med. 218:e20202446. doi: 10.1084/jem.20202446
Xu, Y. J., Mei, Y., Qu, Z. L., Zhang, S. J., Zhao, W., Fang, J. S., et al. (2018). Ligustilide ameliorates memory deficiency in APP/PS1 transgenic mice via restoring mitochondrial dysfunction. Biomed. Res. Int. 2018:4606752. doi: 10.1155/2018/4606752
Xu, Y. J., Mei, Y., Shi, X. Q., Zhang, Y. F., Wang, X. Y., Guan, L., et al. (2019). Albiflorin ameliorates memory deficits in APP/PS1 transgenic mice via ameliorating mitochondrial dysfunction. Brain Res. 1719, 113–123. doi: 10.1016/j.brainres.2019.05.037
Yang, C. Z., Wang, S. H., Zhang, R. H., Lin, J. H., Tian, Y. H., Yang, Y. Q., et al. (2023). Neuroprotective effect of astragalin via activating PI3K/Akt-mTOR-mediated autophagy on APP/PS1 mice. Cell Death Discov. 9:15. doi: 10.1038/s41420-023-01324-1
Yang, H., Shen, H., Li, J., and Guo, L. W. (2019). SIGMAR1/Sigma-1 receptor ablation impairs autophagosome clearance. Autophagy 15, 1539–1557. doi: 10.1080/15548627.2019.1586248
Yao, J., Sun, B., Institoris, A., Zhan, X., Guo, W., Song, Z., et al. (2020). Limiting RyR2 open time prevents Alzheimer’s disease-related neuronal hyperactivity and memory loss but not β-amyloid accumulation. Cell Rep. 32:108169. doi: 10.1016/j.celrep.2020.108169
Yao, X., Zhang, J., Lu, Y., Deng, Y., Zhao, R., and Xiao, S. (2022). Myricetin restores Aβ-induced mitochondrial impairments in N2a-SW cells. ACS Chem. Neurosci. 13, 454–463. doi: 10.1021/acschemneuro.1c00591
Yuan, M., Wang, Y., Huang, Z., Jing, F., Qiao, P., Zou, Q., et al. (2022). Impaired autophagy in amyloid-beta pathology: a traditional review of recent Alzheimer’s research. J. Biomed. Res. 37, 30–46. doi: 10.7555/jbr.36.20220145
Zafeer, M. F., Firdaus, F., Anis, E., and Mobarak Hossain, M. (2019). Prolong treatment with Trans-ferulic acid mitigates bioenergetics loss and restores mitochondrial dynamics in streptozotocin-induced sporadic dementia of Alzheimer’s type. Neurotoxicology 73, 246–257. doi: 10.1016/j.neuro.2019.04.006
Zhou, H. Y., Sun, Y. Y., Chang, P., and Huang, H. C. (2022). Curcumin inhibits cell damage and apoptosis caused by thapsigargin-induced endoplasmic reticulum stress involving the recovery of mitochondrial function mediated by mitofusin-2. Neurotox Res. 40, 449–460. doi: 10.1007/s12640-022-00481-y
Keywords: Alzheimer’s disease, mitochondria, endoplasmic reticulum, calcium homeostasis, lipid metabolism
Citation: Li Z, Cao Y, Pei H, Ma L, Yang Y and Li H (2023) The contribution of mitochondria-associated endoplasmic reticulum membranes (MAMs) dysfunction in Alzheimer’s disease and the potential countermeasure. Front. Neurosci. 17:1158204. doi: 10.3389/fnins.2023.1158204
Received: 03 February 2023; Accepted: 23 February 2023;
Published: 07 March 2023.
Edited by:
Jianxun Liu, Xiyuan Hospital, China Academy of Chinese Medical Sciences, ChinaReviewed by:
Punya Sachdeva, Amity University, IndiaCopyright © 2023 Li, Cao, Pei, Ma, Yang and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yang Yang, eWFuZ3lhbmc4ODIxQDEyNi5jb20=; Hao Li, eHlocGxpaGFvMTk2NUAxMjYuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.