 Wujianwen Zhai
Wujianwen Zhai- Traditional Chinese Medicine Research and Development Center, Guang' Anmen Hospital, China Academy of Chinese Medical Sciences, Beijing, China
Cognitive impairment is the core precursor to dementia and other cognitive disorders. Current hypotheses suggest that they share a common pathological basis, such as inflammation, restricted neurogenesis, neuroendocrine disorders, and the destruction of neurovascular units. Fibroblast growth factors (FGFs) are cell growth factors that play essential roles in various pathophysiological processes via paracrine or autocrine pathways. This system consists of FGFs and their receptors (FGFRs), which may hold tremendous potential to become a new biological marker in the diagnosis of dementia and other cognitive disorders, and serve as a potential target for drug development against dementia and cognitive function impairment. Here, we review the available evidence detailing the relevant pathways mediated by multiple FGFs and FGFRs, and recent studies examining their role in the pathogenesis and treatment of cognitive disorders and dementia.
1. Introduction
Dementia which occurs in the late stages of the cognitive disorders is one of the most prevalent mental illnesses, particularly among the elderly (Bennett and Thomas, 2014). The primary clinical manifestation of cognitive disorders is a decline in cognitive function, accompanied by disordered thinking, memory deficits, sensory disturbances, and poor concentration, which are risk factors and precursor symptoms of dementia, including Alzheimer’s disease (AD; Anderson, 2019), vascular dementia (VaD; Dichgans and Leys, 2017), and Huntington’s disease (HD; Papoutsi et al., 2014; van der Flier et al., 2018; Iadecola et al., 2019). Recent research has shown that the pathogenesis of those diseases is associated with various pathological mechanisms, including neuroinflammation (Byers and Yaffe, 2011; Leng and Edison, 2021), oxidative stress (Luca and Luca, 2019; Manduca et al., 2020), immune dysregulation (Hayley et al., 2021), disruption of neurotransmitters (Singh, 2009), synaptic plasticity injury (Byers and Yaffe, 2011; Skaper et al., 2017), and neuroendocrine disorders (Manduca et al., 2020). FGFs are cell growth factors involved in multiple critical pathophysiologic processes in the human body via paracrine and autocrine pathways such as embryonic development and angiogenesis, neurogenesis, wound healing, and glucolipid metabolism, which need to bind fibroblast growth factors receptor (FGFR) to produce physiological effects (Hui et al., 2018). Some researchers believe that FGFs have the potential to become a novel biological marker for the diagnosis and prognosis of neurodegenerative diseases, and these results may also indicate new targets for treatment (Galvez-Contreras et al., 2016). This review focuses on how the FGF-FGFR system changes and affects the pathogenesis and treatment of cognitive disorders and other dementia. We hypothesized that maintaining a dynamic balance in the FGF-FGFR system would be beneficial for nerve repair and neuroprotection to reduce the clinical symptoms and risk of cognitive disorders.
2. Fibroblast growth factors in cognitive disorders and dementia
Patients with cognitive impairment without meeting the diagnostic criteria for dementia tend to be diagnosed by clinicians as mild cognitive impairment (MCI), while patients with concomitant cerebrovascular pathological changes or whose cognitive deficit occurs secondary to cerebrovascular disease can be diagnosed as vascular cognitive impairment. Cognitive impairment can be classified as ‘functional’ or ‘non-functional’ depending on the presence or absence of neurodegenerative changes (Ball et al., 2020). For the former, only cognitive, memory, and emotional impairments occur, which are less likely to deteriorate into dementia; for the latter, the subjective cognitive decline is accompanied by structural changes in the brain and eventually degenerates into dementia.
The pathological changes of dementia are, on the one hand, neurodegenerative changes due to senescence, such as AD, HD, frontotemporal dementia, and Parkinson’s disease dementia; on the other hand, dementia is also closely associated with cerebrovascular injury, metabolic disorder, intracranial infections, intracranial tumors, hypoxic–ischemic brain injury, which secondary to stroke, atherosclerosis, chronic renal insufficiency, diabetes, hyperlipidemia, hypertension, and other diseases. These suggest a mixed pathology for dementia. Cellular senescence apoptosis and abnormal autophagy, inflammation, oxidative stress, vascular damage, and metabolic dysfunction interact to induce neuropathic protein accumulation and morphological changes in the brain, which are further disordered in a vicious cycle of neurodegenerative disease (Gonzales et al., 2022).
The FGF-FGFR system consists of seven receptors and 22 ligands that are required to bind to their corresponding receptors via acetyl heparan sulfate (HS) cofactor or α/β-Klotho (α/β-KL) transmembrane proteins to exert their physiological effects (Beenken and Mohammadi, 2009; Xie et al., 2020). The 22 FGFs can be divided into different subgroups based on sequence homology and developmental characteristics. For example, FGF-15/19, FGF-21, and FGF-23 are members of a particular FGF type called endocrine FGFs, which require KL proteins to bind to their receptors because of their low affinity for HS (Belov and Mohammadi, 2013; Owen et al., 2015; Hui et al., 2018cc Deng et al., 2019; Chen K. et al., 2022). Except for homologous factor subfamily (FGF-11/12/13/14) and endocrine FGF subfamily, other FGFs all belong to paracrine subfamilies, including FGF-1, FGF-2, FGF-9, and FGF-17 (Deng et al., 2019). Abnormal FGF expression levels have been observed by researchers in patients with dementia and other cognitive disorders (Stopa et al., 1990; Mashayekhi et al., 2010; Hensel et al., 2016; Liang et al., 2021). Moreover, the effects of FGFs on neuromodulation and cognitive improvement have been validated in literature (Lee et al., 2011; Taliyan et al., 2019). Fibroblast growth factors in cognitive disorders and dementia is summarized in Table 1.
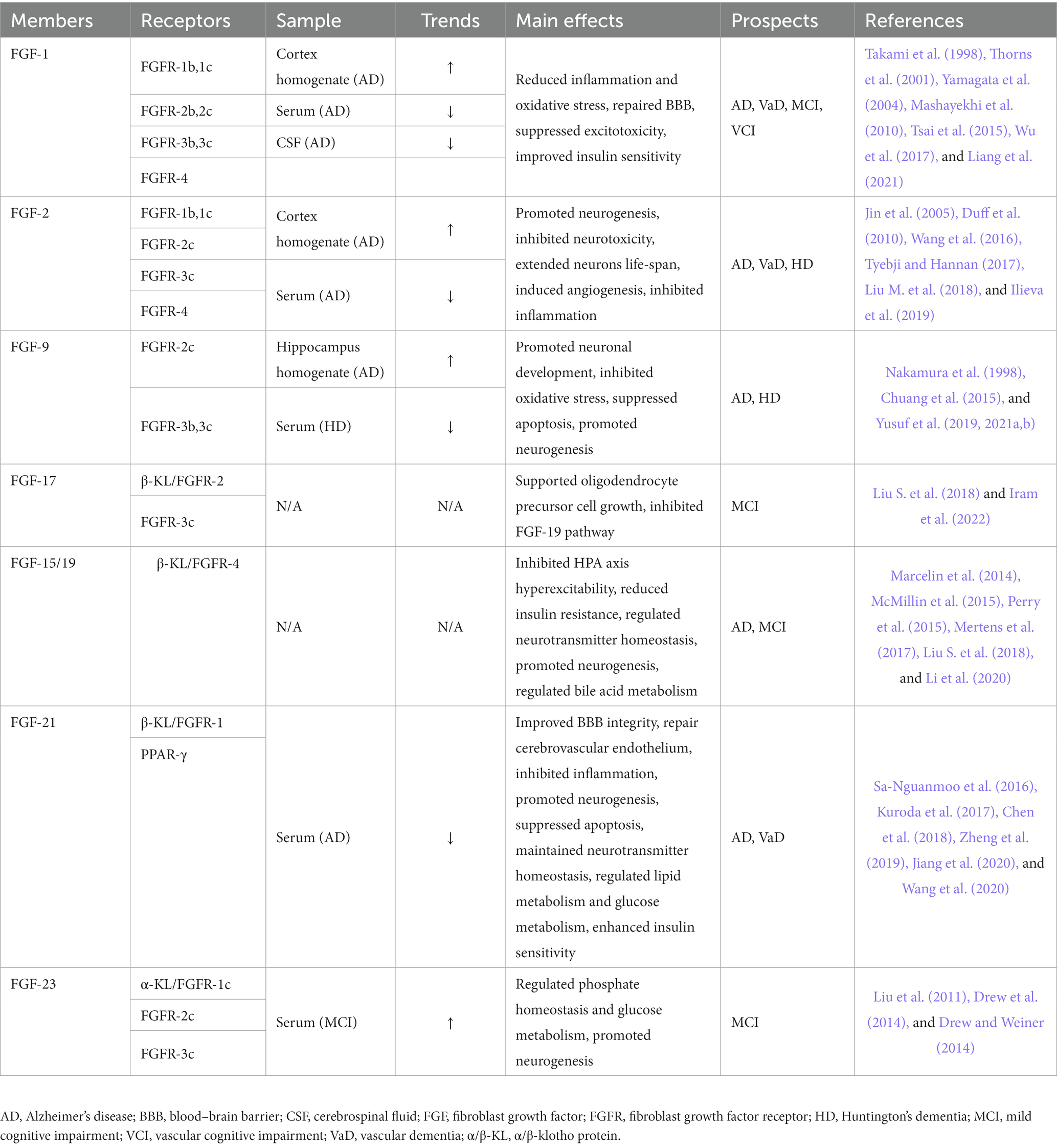
Table 1. The expressions and prospects of the FGF/FGFR system in cognitive disorders and dementia.
2.1. Paracrine fibroblast growth factors
2.1.1. FGF-1
Fibroblast growth factor-1 (FGF-1), also known as acidic fibroblast growth factor (aFGF), is secreted by meningeal cells in the ventricles of the third ventricle and is widespread in multiple tissues, including the hippocampus, pituitary gland, heart, and kidney (Beenken and Mohammadi, 2009; Mashayekhi et al., 2010). FGF-1 can modulate physiological activities such as embryonic development, angiogenesis, cell proliferation and differentiation, and adult hippocampal neurogenesis (AHN) by activating different FGFRs (Lee et al., 2011; Tsai et al., 2015; Gasser et al., 2017; Sun et al., 2021). Although there remains a paucity of studies investigating the specific role of FGF-1 in the pathological mechanisms of cognitive impairment, several lines of evidence have shown that dysregulation of FGF-1 expression is closely associated with dementia, especially Alzheimer’s disease (Takami et al., 1998; Yamagata et al., 2004).
AHN refers to the process by which neural stem cells in the hippocampus undergo symmetric or asymmetric division into neuroblasts, gradually migrate to specific regions after cell proliferation and then differentiate into new neurons and other resident brain cells. Thus, AHN is the fundamental physiological basis for neuroplasticity. After binding to FGFR-1, FGF-1 has a significant reparatory effect on damaged neurons in the cortex and hippocampus, reducing inflammatory factors secreted by microglial (MG) activation, thereby promoting neurogenesis and vascular regeneration (Tsai et al., 2015). However, there is no one-to-one correspondence between the FGFs and FGFRs. For instance, FGF-1 secreted by activated astrocytes (ASTs) induces neuroinflammation instead of neuroprotection if by binding to FGFR-2 (Lee et al., 2011). The neurovascular unit (NVU) is a microstructure composed of neurons, neuroglia, the BBB, and the extracellular matrix, and plays an essential role in maintaining nerve function. The BBB is an essential barrier, that protects the stability of the neural microenvironment and is the core of NVU coupling, providing protection against leakage of neurotoxic substances from the blood into the cerebral parenchyma. The BBB consists of cerebral microvascular endothelial cells and is connected to astrocytes, pericytes, perivascular macrophages, and basement membranes, which are hyperpermeable in depressed patients, triggering inflammation of the central nervous system (CNS) and endothelial damage to cerebral microvessels, further exacerbating neuronal injury. At the same time, FGF-1 could repair the BBB by upregulating the expression of tight junction proteins and adherens junction proteins via activating p-PI3K, PI3K, p-Akt, and Akt to suppress RhoA but activate Rac1 (Wu et al., 2017). Preliminary observations suggest that FGF-1 expression is reduced in neurons of the internal olfactory cortex in patients with AD, which inhibits the expression of calcium-binding proteins and induces overexpression of the N-methyl-D-aspartic acid receptor (NMDAR), resulting in the disruption of calcium homeostasis and glutamate-mediated excitotoxicity (Thorns and Masliah, 1999; Thorns et al., 2001). Several recent studies reported a significant increase in FGF-1 levels in both plasma and cerebrospinal fluid in AD (Mashayekhi et al., 2010; Liang et al., 2021), suggesting that there may be differences in local concentrations of FGF-1, but this remains to be verified further.
Glucose is the primary energy source of the brain (Mergenthaler et al., 2013). In addition to providing energy for neural activity, glucose indirectly regulates the transmission of signals that affect neural function (Dienel, 2012; Hossain et al., 2020). Cerebral glucose metabolism plays a crucial role in the physiological mechanisms of cognitive function and the pathomechanisms of dementia. Therefore, cerebral insulin resistance can trigger oxidative stress, mitochondrial dysfunction, inflammation, and autophagy by inducing severe dysfunction of extracellular glucose transport and intracellular glucose metabolism disorders, leading to neurodegeneration (Chen and Zhong, 2013; Butterfield and Halliwell, 2019). Notably, patients with AD or MCI exhibit a dramatic decline in cerebral glucose metabolism due to insulin resistance (Chen and Zhong, 2013; Butterfield and Halliwell, 2019). Researchers have also found that patients with dementia have significant insulin resistance in their hippocampi (Wijesekara et al., 2018; Hamer et al., 2019), suggesting that dysregulation of brain insulin signaling pathways may play an important role in cognitive dysfunction. Exogenous FGF-1 has been shown to reduce blood glucose concentrations and increase insulin sensitivity (Holmes, 2014; Suh et al., 2014; Gasser et al., 2017; Tennant et al., 2019) via modulating the Wnt/β-catenin (Sun et al., 2021) and AMPK pathways (Chen Q. et al., 2022). Thus, FGF-1 could indirectly ameliorate cognitive impairment by attenuating insulin resistance in local brain regions. The potential therapeutic mechanisms of FGF-1 are shown in Figure 1.
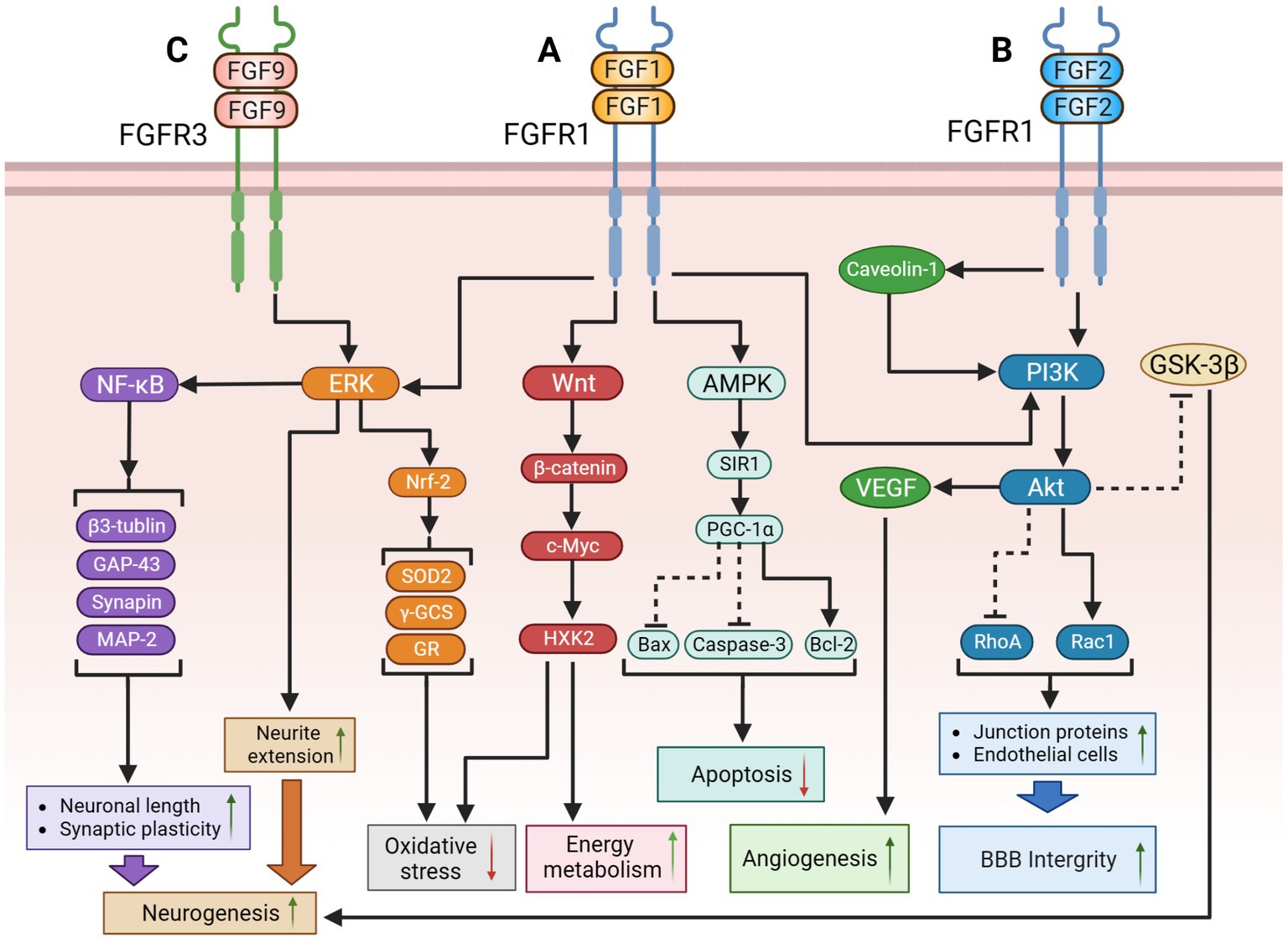
Figure 1. The potential therapeutic mechanism of paracrine FGFs (FGF-1, FGF-2, FGF-9). (A) FGF-1might regulate the following signaling pathways to improve cognition function: (1) FGF-1 activates PI3K/Akt pathway to suppress RhoA but activate Rac1, causing the upregulation of BBB integrity; (2) FGF-1 activates ERK pathway to extend neurite for promoting neurogenesis; (3) FGF-1 activates Wnt/β-catenin/c-Myc/ HXK2 pathway to improve mitochondrial energy metabolism and suppress oxidative stress; (4) FGF-1 exert the activation of the AMPK/SIRT1/PGC-1α pathway to modulate the apoptosis. (B) (1) FGF-2 might inhibit GSK-3β through PI3K/AKT pathway to promote adult hippocampal neurogenesis; FGF-2 could also repair blood–brain barrier integrity through the activation of PI3K/Akt/Rac1axis, but inhibiting RhoA; (2) Additionally, protect astrocytes and induce angiogenesis to improve cerebral blood supply by upregulating the Caveolin-1/PI3K/AKT/VEGF pathway. (C) FGF-9 might activate ERK/Nrf-2pathway to suppress oxidative stress and activate the ERK/NF-κB pathway to improve hippocampal neurogenesis. The solid black lines and arrows indicate the activation of the signaling pathway, and the dashed black lines and T-arrows indicate the inhibition of the signaling pathway. FGF, fibroblast growth factor; PI3K, phosphatidylinositol-3-hydroxy kinase; AKT, protein kinase B; Rac1, rac family small GTPase 1; RhoA, ras homolog family member A; BBB, blood–brain barrier; ERK, extracellular signal-regulated kinase; HXK2, hexokinase 2; AMPK, adenosine monophosphate activated protein kinase; SIR1, sirtuin1; PGC-1α, peroxisome proliferator-activated receptors-γ coactivator 1-α; GSK-3β, glycogen synthase kinase-3β; VEGF, vascular endothelial growth factor; Nrf-2, nuclear factor erythroid-like 2; SOD2, superoxide dismutase 2; γ-GCS, gamma-glutamylcysteine synthetase; GR, glutathione reductase; NF-κB, nuclear factor kappa-B; MAP-2, microtubule-associated protein-2; GAP-43, growth association protein-43.
2.1.2. FGF-2
FGF-2, also known as basic fibroblast growth factor (bFGF), was the first fibroblast growth factor to ever be identified. In the CNS, FGF-2 is primarily produced by ASTs and distributed throughout the cortex, hippocampus, and hypothalamus. Physiological concentrations of FGF-2 have been reported to promote proliferation, differentiation, migration, and maturation of neural stem cells and neuroglia, maintain cortical synaptic connectivity, stimulate neurogenesis, inhibit neuroinflammation, and exert tremendous neuroprotective and neurotrophic effects (Numakawa et al., 2015; Wu et al., 2017). The expression levels of FGF-2 are elevated in a compensatory manner in dementia (Stopa et al., 1990; Tennakoon et al., 2022), whereas the artificial modulation of FGF-2 levels improves cognitive function in rodents (Kiyota et al., 2011; Feng et al., 2012; Numakawa et al., 2015). In addition, FGF-2 has been shown to be a significant therapeutic target for certain anti-dementia medications, such as memantine (Namba et al., 2010).
Hippocampal neuronal cells are critical for the regulation of memory, learning, cognition, emotion, and other functions. FGF-2 has been proposed to stimulate the growth of neuronal synapses and regulate tight junction proteins in vascular endothelial cells via activating the PI3K/AKT pathway to promote AHN and protect the BBB (Wang et al., 2016; Ilieva et al., 2019). Neuroglia cells are a crucial component of the NVU, as they maintain the stability of the physiological functions of the neurons in the brain and consist of ASTs, MG, and oligodendrocytes (OLs). In HD, FGF-2 could promote the proliferation and recruitment of neuronal cells and inhibit the neurotoxicity produced by polyglutamine protein aggregation, which would prolong the lifespan of neurons (Jin et al., 2005; Duff et al., 2010; Tyebji and Hannan, 2017). ASTs, which have both an anti-inflammatory resting state and a pro-inflammatory reactive state, are the most abundant and largest neuroglia in the CNS and play a key role in neurotransmitter regulation and energy provision. In cognitive disorders, the density and number of resting ASTs decrease with the aggravation of symptoms, which leads to a decline in neurotrophic factors and nerve growth factors secreted by ASTs, while the number of reactive ASTs, which produce large amounts of inflammatory factors and neurotoxins is raised (Wang et al., 2017; Lu et al., 2020). Excessive activation of GSK-3β can induce hyperphosphorylation of tau. Furthermore, spatial memory deficits and cognitive decline in AD could be ameliorated by FGF-2 through a simultaneous decrease in amyloid-β (Aβ) and microtubule-associated protein tau while increasing the number of resting ASTs in the hippocampal dentate gyrus (Katsouri et al., 2015). A possible explanation for this result is that exogenous FGF-2 may suppress the over-activated GSK-3β-related pathway and strengthened neurogenesis (Hong et al., 2016). It could activate p-GSK-3β and GSK-3β, triggering tau hyperphosphorylation to aggravate cognitive decline, but could also inhibit GSK-3β via activating the Akt pathway to promote hippocampal neurogenesis. We assumed that these mixed results might attribute to different molecular weight isoforms of FGF-2 activating different downstream pathways. Moreover, FGF-2 could protect ASTs from ischemia–reperfusion injury, a common pathological basis of VaD, and induce angiogenesis to ameliorate chronic cerebral hypoperfusion by upregulating the Caveolin-1/PI3K/AKT/vascular endothelial growth factor (VEGF) signaling pathway (Liu M. et al., 2018).
Neuroinflammation is a critical pathological mechanism that disrupts NVU homeostasis. The levels of multiple pro-inflammatory factors observed in both the blood and cerebrospinal fluid of patients with dementia were higher than those in healthy individuals (Shen et al., 2019). For example, MG is an integral type of neuroglia for maintaining the AHN process with a pro-inflammatory phenotype M1 and an anti-inflammatory phenotype M2. Activation of M1 induces neuroinflammation, which inhibits FGF-2 expression and activates downstream ERK pathway in the hippocampal region, thereby restraining the AHN process. In contrast, exogenous FGF-2 could effectively inhibit interleukin-1β (IL-1β), interleukin-6 (IL-6), tumor growth factor-α (TNF-α) and other pro-inflammatory factors produced by M1-type MG in an attempt to improve depressive-like behaviors (Tang et al., 2017). FGF-2 may also enhance the activation of M2-type MG and its phagocytosis of Aβ, thereby promoting neurogenesis in dementia (Kiyota et al., 2011). OLs are crucial for the modulation of the cerebrovascular system and the maintenance of white matter structure and function, while FGF-2 can promoted proliferation, maturation, differentiation, and migration of OLs and suppress their apoptosis (Miyamoto et al., 2014). White matter hyperintensities (WMH) are the imaging features of white matter lesions, with several studies suggesting a clinically relevant link between WMH burden and cognitive decline in MCI and AD (Garnier-Crussard et al., 2022; Kamal et al., 2022). Researchers have also found that senescent oligodendrocyte precursor cells (OPCs) were the primary neuroglial cell expressed in neuritic plaques, which could induce cell senescence in AD, causing cognitive decline (Zhang et al., 2019). Therefore, modulation of neuroglial cells (OLs, ASTs, and MG) via the FGF-2 pathway may play a role in the treatment of cerebral white matter lesions and VaD via angiotensin-converting enzyme II (Wakayama et al., 2021). The current application of FGF-2 is mainly in synthetic recombinant human-derived FGF-2 which has several limitations such as short half-life in the blood, inability to completely cross the BBB, or side effects on the vascular system (Bogousslavsky et al., 2002). However, novel synthetic compounds such as SUN11602 have been proposed, which mimic the structure of FGF-2, and could potentially avoid the aforementioned defects and exert neuroprotective effects of FGF-2 to a certain extent (Ogino et al., 2014; Ardizzone et al., 2022).
In recent decades, several researchers have observed opposing trends in the changes of FGF-2 in serum and brain tissue in dementia. For example, Katsouri et al. (2015) found that the concentration of FGF-2 in the frontal cortical homogenates of patients with AD was decreased, rather than increased, contrary to previous studies (Stopa et al., 1990). Additionally, the original hypothesis assumed that FGF-2 attracts neurons into plaques in AD (Cummings et al., 1993); however, recent studies have argued that FGF-2 confers neuroprotective effects (Feng et al., 2012; Katsouri et al., 2015). We speculate that discrepancies could be explained with the following reasons: (1) Numerous cells in the peripheral system, such as blood cells, bone marrow stromal cells, and smooth muscle cells, can also secrete FGF-2. Therefore, serum FGF-2 levels might be affected by other systemic diseases in ways that truly reflect the level of FGF-2 in the CNS. (2) Subjects in the discussed studies were not receiving uniform treatment, which may increase the uptake of FGF-2 into damaged neuronal cells, resulting in a relative decrease in serum FGF-2 levels. (3) An increase in serum FGF-2 levels could also be a result of compensatory secretion to repair nerve injury. (4) There are at least six different molecular weight isoforms of FGF-2 (Chlebova et al., 2009), which may have different physiological roles, but routine detection could be unable to show the differences between the isomers (Jiang et al., 2007; Chen X. et al., 2019). Therefore, despite ample evidence supporting the key role of FGF-2 in cognitive impairment and dementia, it still fails to become an independent clinical diagnosis or assessment indicator. These points made above are also the common challenges we face during the study of the FGF family members. The potential therapeutic mechanisms of FGF-2 are shown in Figure 1.
2.1.3. FGF-9
FGF-9 is synthesized by neurons of the CNS which binds to FGFR-2, FGFR-3, but not FGFR1 and FGFR4, playing a prominent role in angiogenesis, neurogenesis, cellular differentiation and cardiac development (Hecht et al., 1995; Reuss and von Bohlen und Halbach, 2003; Wang S. et al., 2018). Nakamura et al. (1998) found that high levels of FGF-9 expression in the hippocampus of patients with AD promoted the activation of the pro-inflammatory phenotype of ASTs around senile plaques, thereby exacerbating cognitive impairment. Alternatively the expression level in the CNS of FGF-9 may decrease in patients with HD. Previous results of Chuang et al. (2015) which have identified prominent FGF-9 and FGFR-3 expression in primary neuron-enriched cultures, suggesting the FGF-9 effects are cell-type specific in brain, may be a likely explanation for the different expression trends of FGF-9 in AD and HD.
It has been shown that exogenous FGF-9 may promote neuronal development and synaptic growth in striatal cell models of HD in response to anti-oxidant and anti-apoptotic effects via extracellular signaling that modulates the ERK/NF-κB pathway (Yusuf et al., 2019, 2021a,b), which provides a novel insight into the treatment of cognitive decline due to HD. FGF-9 activates Nrf-2 to upregulate transcription factors (SOD2, γ-GCS, and GR) for suppressing oxidative stress, through the activation of the ERK pathway. Additionally, FGF-9 could also activate the ERK/NF-κB pathway to upregulate β-tubulin, MAP-2, GAP-43, and synapsin to improve the neuronal length and synaptic plasticity. These studies suggest that FGF-9 overexpression in AD might be a compensatory mechanism for neuroprotection.
In addition, a negative correlation was found between FGF-9 and adiponectin (ADPN). ADPN is a pleiotropic adipocyte-secreting hormone with neurotrophic effects. Animals knocked out of the ADPN gene could present depressive-like behavior and cognitive deficits (You et al., 2021). Researchers have found that the Chinese herbal extract carnosic acid could simultaneously reduce FGF-9 levels and raise ADPN levels both in mice’s serum and hippocampal tissues, thereby ameliorating depression-like symptoms (Azhar et al., 2021; Wang et al., 2021). Interestingly, Carnosic acid could also alleviate AD-induced cognitive decline by inhibiting neuroinflammation, ameliorating cholinergic deficits, and enhancing energy metabolism (Chen Y. et al., 2022; Yi-Bin et al., 2022). We supposed that FGF-9 may also participate in carnosic acid’s anti-dementia mechanism of action, but it requires further study. The potential therapeutic mechanisms of FGF-9 are shown in Figure 1.
2.1.4. FGF-17
FGF-17 is mainly expressed in the cerebrospinal fluid, plasma, and cortical neurons. It binds mainly to FGFR-3 to regulate downstream signaling, fulfilling vital roles in embryonic development, cell proliferation, and early neurogenesis, which declines during senescence (Hoshikawa et al., 1998; Machado et al., 2015; Han et al., 2019; Sathyan et al., 2020; Iram et al., 2022). Iram et al. (2022) recently observed that the growth of OPCs was suppressed after blocking the FGF-17/FGFR-3 pathway, which impaired hippocampal function in mice and provoked cognitive impairments and memory loss. Furthermore, they proved that exogenous injection of FGF-17 could enhance cognitive and memory performance in older mice. This finding is contrary to a previous study by Scearce-Levie et al. (2008) which found that young mice did not show obvious symptoms of cognitive impairment after knocking out the FGF-17 gene. These differences can be attributed to the fact that the subjects were in different physiological states that were influenced by their age. Besides, FGF-17 has been found to bind to the β-KL/FGFR-2 homodimer in the hypothalamus and show antagonize FGF-15/19, which blocked the insulin signaling pathway regulated by FGF-15/19 and triggered a decrease in glucose tolerance (Liu S. et al., 2018). The following may indicate that FGF-17 exerts distinct effects when bound to different receptors separately in the hippocampus or hypothalamus. Therefore, the therapeutic potential and side effects of FGF-17 remain to be studied over a long period of time. The potential therapeutic mechanisms of FGF-17 are shown in Figure 2.
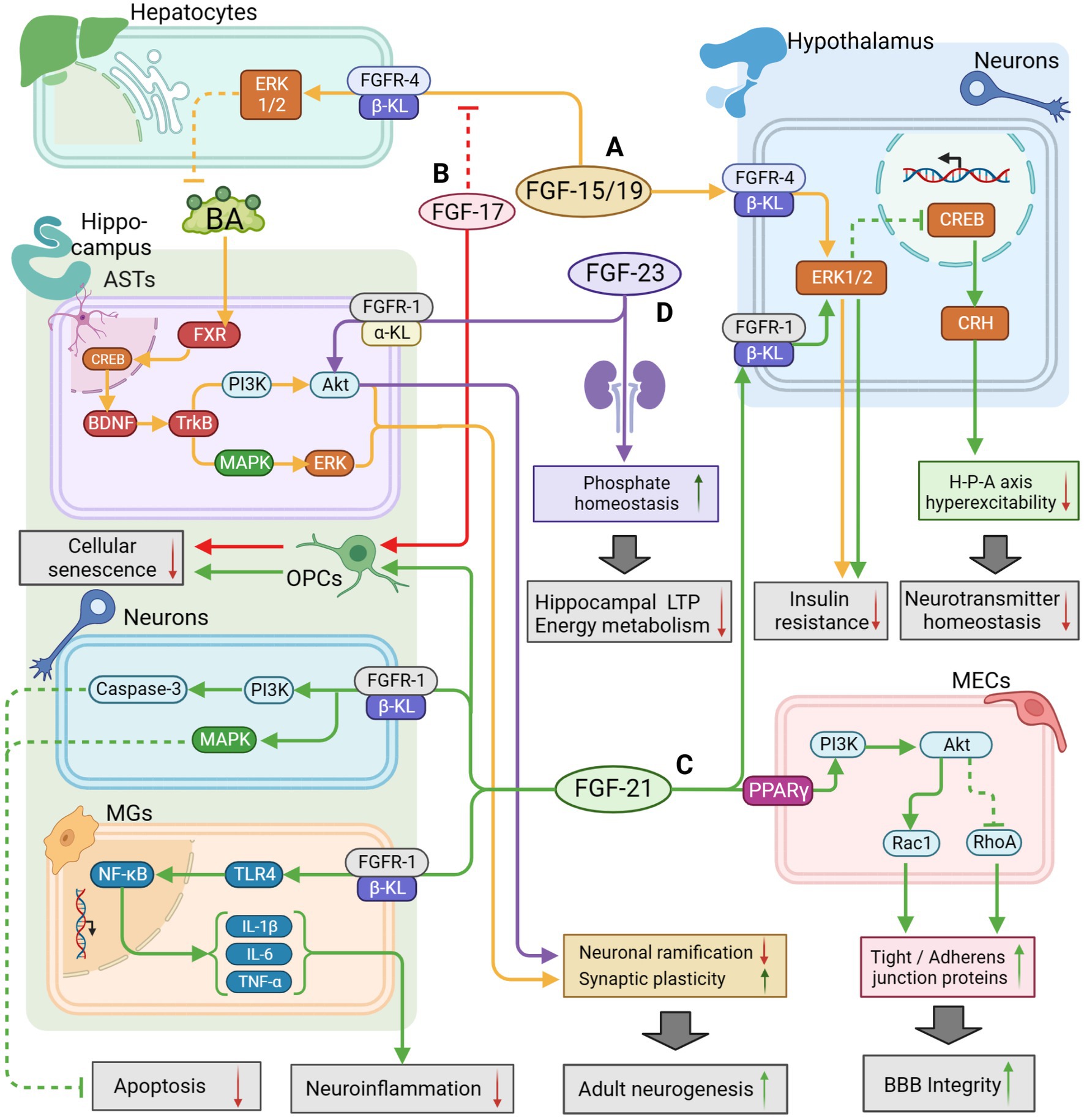
Figure 2. The potential therapeutic mechanism of endocrine FGFs (FGF15/19, FGF21, FGF23) and FGF17. (A) FGF-15/19/FGFR4/β-KL might activate the ERK1/2 pathway in hepatocytes to regulate BA metabolism for activating BDNF/TrkB/PI3K pathway and BDNF/TrkB/MAPK pathway in hippocampal ASTs, leading to improvement of adult neurogenesis; FGF15/19 could also penetrate the BBB to inhibit insulin resistance by activating the ERK1/2 pathway in hypothalamus. (B) FGF-17 could enhance the growth of OPCs to defer cell aging and suppress FGF-15/19 pathway as a competitive inhibitor. (C) FGF-21 could enhance BBB integrity by activating PI3K/Akt pathway in MECs, and reduce neuroinflammation through the suppression of TLR4/ NF-κB pathway in MGs; FGF-21 could also inhibit apoptosis by activating PI3K/Caspase-3 pathway and MAPK pathway in neurons; FGF-21 could also regulate HPA axis function by activating ERK/CREB pathway to maintain neurotransmitter balance; Additionally, FGF-21 might defer OPCs aging for repairing cognitive function. (D) FGF-23/FGFR-1/α-KL might directly activate the AKT pathway to inhibit neuronal ramification but maintain synaptic plasticity; FGF-23 expressed in the kidney could also indirectly regulate phosphate homeostasis to promote LTP and mitochondrial energy metabolism for improvement of cognitive function. The solid arrows indicate the activation of the signaling pathway, and the dashed T-arrows indicate the inhibition of the signaling pathway. The yellow lines represent the FGF-15/19 pathway, the red lines represent the FGF-17 pathway, the green lines represent the FGF-21 pathway and the purple lines represent the FGF-23 pathway. FGF, fibroblast growth factor; BA, bile acid; FXR, farnesoid X receptor; GR, glucocorticoid receptors; KL, klotho protein; BBB, blood–brain barrier; BDNF, Brain Derived Neurotrophic Factor; PI3K, phosphatidylinositol-3-hydroxy kinase; Akt, protein kinase B; TrkB, tyrosine kinase receptor B; ERK, extracellular signal-regulated kinase; MAPK, mitogen-activated protein kinase; CREB, cAMP-response element binding protein; TLR4, toll like receptor-4; NF-κB, nuclear factor kappa-B; OPCs, oligodendrocyte precursor cells; ASTs, astrocytes; MECs, microvascular endothelial cells; LTP, long-time potentiation.
2.2. Endocrine fibroblast growth factors
FGF-19, FGF-21, and FGF-23 are members of the same subgroup, have low affinity for HS, and require the activation of corresponding receptors by binding to the KL transmembrane protein, which causes them to diffuse into the bloodstream and exert hormone-like regulatory effects readily. Therefore, they are also known as endocrine-FGFs. FGF-19 from intestinal epithelial cells and hepatocytes could restrain the synthesis of primary bile acids and alter the ratio of primary to secondary bile acids, thus indirectly regulating lipid metabolism and bile acid signaling (Owen et al., 2015; Al-Aqil et al., 2018). FGF-21, produced mainly by hepatocytes and adipocytes, is a vital regulator for maintaining the homeostasis of lipid, glucose, and energy metabolism (Kharitonenkov et al., 2005; Owen et al., 2015). FGF-23, synthesized in osteoblasts and osteocytes, is highly expressed in the kidney and parathyroid glands and regulates the dynamic balance of phosphate, calcium, and vitamin D metabolism (Cunningham et al., 2011; Vervloet, 2019). The above three FGFs have been shown to affect metabolism in the CNS and have a relationship with cognitive disorders and dementia (Hsuchou et al., 2013; Hensel et al., 2016; Li, 2019; Jiang et al., 2020).
2.2.1. FGF-15/19
Researchers often refer to FGF-19 as FGF15/19, since FGF-19 in both humans and rats is expressed in mice as the homologous protein FGF-15. This protein is synthesized primarily in the enterocyte at the end of the ileum via the farnesoid X receptor (FXR) -related pathway and is expressed in the small intestine, liver, gallbladder, kidney, brain, and other tissues (Rysz et al., 2015). In tissues with rich β-KL, for example, the liver, FGF-15/19 binds to β-Kloth and activates FGFR-1, FGFR-2, FGFR-3, and FGFR-4, whereas, in tissues relatively deficient in β-KL like the brain, FGF-15/19 only binds to FGFR-4 (Nakamura et al., 2011). FGF-15/19 could stimulate the development of the heart and brain in the embryonic stage, regulate the production and circulation of bile acids (BAs) and promote glucolipid metabolism mainly in the liver at maturity; recently, it has also been found to have an insulin-like effect in the CNS, with the potential to tune sleep, cognition, and sensory functions (Hsuchou et al., 2013; Giacomini et al., 2016).
Due to the significant neuromodulatory, protective and nutritional effects of insulin on the brain, insulin resistance in the brain might exacerbate Aβ deposition, tau hyperphosphorylation, and vascular inflammation, causing cognitive impairments in AD and VaD (Hotamisligil, 2006; Kellar and Craft, 2020). Hypothalamic–pituitary–adrenal (HPA) axis hyperfunction is prevalent in dementia and other cognitive disorders, which might be the reason for higher adrenocorticotropic hormone (ACTH) and glucocorticoid (GC) levels, resulting in insulin resistance. Perry et al. (2015) have shown that intracerebroventricular injection of exogenous FGF-19 may improve insulin sensitivity by reducing serum ACTH and GC levels through inhibition of the HPA axis. This effect may arise from the inhibition of agouti-related protein/neuropeptide-Y (AGRP/NPY) neurons and the activation of the ERK1/2 pathway in the hypothalamus (Marcelin et al., 2014; Liu S. et al., 2018). In addition, recent studies have identified a link between the HPA axis dysfunction and cognitive decline in MCI and AD (Csernansky et al., 2006; Canet et al., 2019). Researchers have observed that the HPA axis dysfunction might emerge in the early stages of cognitive deficits, leading to structural damage in the hippocampus and cortex and NVU destabilization through the elevation of cortisol and norepinephrine (NE) levels with concomitant glucocorticoid receptor disruption and neurotoxicity (Popp et al., 2015; Canet et al., 2019). This finding was supported by Csernansky et al. (2006), Lara et al. (2013), and Wang L. Y. et al. (2018). Thus, the FGF-15/19 pathway might become a novel therapeutic target to maintain HPA axis function for drug discovery.
Furthermore, FGF-15/19 could affect NVU homeostasis by modulating BA production and circulation. The FXR for BA is found in neurons and ASTs in the hypothalamus and hippocampus of both humans and rodents (Mano et al., 2004; Huang et al., 2016; He et al., 2021), while physiological concentrations of BA can penetrate the BBB and suppress hyperexcitability of the HPA axis by activating glucocorticoid receptors in the hypothalamus and protect NVU by activating the BDNF–TrkB pathway (McMillin et al., 2015; Mertens et al., 2017; Li et al., 2020). Brain-derived neurotrophic factor (BDNF) is widely distributed in the CNS, especially in the cerebral cortex and hippocampus, with a significant role in the survival and maintenance of neurons. BDNF regulates synaptic plasticity via the autocrine pathway and modulates the presynaptic gamma-aminobutyric acid system via the paracrine pathway. BAs regulated by FGF-15/19 could enter hippocampal astrocytes and bind to FXR binding to promote BDNF synthesis, activate TrkB/PI3K/AKT pathway and TrkB/MAPK/ERK pathway, thus promoting hippocampal neurogenesis. Al-Aqil et al. (2018) observed that a high dosage of GC could elevate BA levels and downregulate FGF-15 expression in the serum and liver of mice. A recent metabolomics study identified that the serum concentrations of cholic acid (CA) and lithocholic acid (LCA) were significantly lower in patients with AD than in normal individuals (Pan et al., 2017). In contrast, the levels of CA, lithocholic acid, deoxycholic acid (DCA), chenodeoxycholic acid (CDCA), taurodeoxycholic acid (TDCA), and glycinodeoxycholic acid (GDCA) in serum, cerebrospinal fluid, and brain tissue have increased observably (MahmoudianDehkordi et al., 2019; Baloni et al., 2020). Changes in the ratio of secondary to primary BA have also been shown to be in close correlation with cognitive decline. For instance, the proportion changes of GDCA to CA shows a positive relationship with the amount of Aβ deposition in the cerebrospinal fluid in animal models of AD, while the proportion of TDCA and GDCA to CA is negatively correlated with glucose levels in the prefrontal cortex and hippocampus volume (MahmoudianDehkordi et al., 2019; Nho et al., 2019). Meanwhile, mounting evidence suggests that anti-dementia effects could be achieved with some common medicines used to treat gallstones, sclerosing cholangitis, sloughy hepatitis, and other cholestatic diseases. For example, tauroursodeoxycholic acid (TUDCA) has been insulin resistance shown to possess antidepressant efficacy and effectively inhibit Aβ deposition in AD (Lo et al., 2013; Lu et al., 2018; Cheng et al., 2019); Additionally, obeticholic acid, a synthetic FXR agonist that was developed based on the structure of CDCA, has been shown to enhance memory and cognition by maintaining BBB permeability and retarding neuronal degeneration (Gee et al., 2022). These findings suggest that modulation of BA-related pathways has the potential to be a new approach to improve mood and cognitive function and defer neuronal degeneration. In physiological conditions, FGF-15/19 activates the ERK1/2 pathway to inhibit cholesterol 7α-hydroxylase from preventing BA synthesis in order to bring BA concentration into equilibrium. In cases of dementia, disrupted endogenous glucocorticoids in the serum or disordered gut microbiota could restrict FGF-15/19 synthesis following FXR activation and counterbalance the inhibitory effect of FGF-15/19 on BA, thereby perturbing the balance of blood and tissue concentrations of BA. As a result, excessive BA would enter the CNS through circulation and cross the BBB, producing cytotoxicity, lysing the membranes of neuronal and endothelial cells, further disrupting NVU homeostasis. The following would aggravate the structural damage or lead to dysfunction in the hippocampus, hypothalamus, and other key regions, which would create a vicious cycle of cognitive impairment and morphological damage. Thus, FGF-15/19 may have an indirect regulatory role in the pathological changes seen in dementia by regulating glucose metabolism, HPA axis function, and BA homeostasis in critical brain regions. Although there is still a lack of direct evidence for changes in FGF-15/19 levels in dementia or other cognitive disorders, the above studies suggest a promising new therapeutic method for modulating BA synthesis, metabolism, and component ratios through the FGF-15/19 pathway to improve cognitive function. The potential therapeutic mechanisms of FGF-15/19 are shown in Figure 2.
2.2.2. FGF-21
Fibroblast growth factor 21 (FGF-21) is primarily synthesized in the liver and adipose tissue, which is distributed throughout the bone, muscle, heart, kidney, and brain. FGF-21 is regulated by peroxisome proliferator-activated receptor-α (PPAR-α) and binds mainly to PPAR-γ, FGFR-1, and other receptors (Kuroda et al., 2017). FGF-21 has been proven to enhance glucose tolerance and insulin sensitivity, inhibit lipid synthesis and exert anti-inflammatory, antioxidant, and anti-apoptotic effects (Hui et al., 2018; Dolegowska et al., 2019). It could also penetrate the BBB to bind to FGFR-1 in the CNS and exert potent neuroprotective effects (Kuroda et al., 2017; Jiang et al., 2020). Researchers have currently identified that FGF-21 exerts neuroprotective effects through the following five significant pathways. (1) Protection of the BBB integrity: the FGF-21/β-KL/FGFR-1 pathway is automatically activated after cerebral microvascular injury or focal ischemia to mitigate neural and vascular endothelial damage. In addition, FGF-21 protects BBB integrity through the activation of PPAR-γ/PI3K/AKT/Rac1 pathway in cerebral microvascular endothelial cells and upregulation of tight junction proteins and adherent junction proteins expressions (Chen et al., 2018; Jiang et al., 2020). (2) Inhibition of neuroinflammation: FGF-21 could inhibit the pro-inflammatory phenotype of MG and NF-κB signaling pathway by suppressing the expression of pro-inflammatory factors such as IL-1β, IL-6, and TNF-α, in a way that nerve cells are protected from the damage caused by inflammation (Wang et al., 2020). (3) Promotion of neurogenesis: FGF-21 could activate PI3K/Caspase-3 signaling pathway and alleviate the apoptosis of neuronal cells (Zheng et al., 2019). In addition, FGF-21 may enhance hippocampal synaptic plasticity, increase dendritic spine density, promote restoration of mitochondrial function in the brain tissue, and inhibit apoptosis (Sa-Nguanmoo et al., 2016). Furthermore, since OPCs are widely distributed in the CNS, OLs differentiated from OPCs could be a crucial link between neural regeneration and myelin restoration. More specially, Kuroda et al. (2017) showed that the regulation of OPCs proliferation and differentiation by the FGF-21/β-KL/FGFR-1 pathway would be beneficial for neuroprotection. (4) Neurotransmitter regulation: FGF-21 could activate the HPA axis through the ERK/CREB pathway and induce the expression of corticotropin-releasing hormone (CRH) and ACTH, thereby regulating the secretion of serum corticosterone (Kuroda et al., 2017).
Researchers have gradually identified a close link between lipid metabolism disorders and dementia, especially AD. Disturbed lipid metabolism leads to aberrant levels and types of lipids such as fats, cholesterol, fatty acids, lipoproteins, and phospholipids. These abnormal changes affect the gut microbiota, brain-gut peptides, and neurotransmitter signaling and cause BBB disruption, mitochondrial dysfunction, oxidative stress, and inflammation, which eventually combine to cause a decline in synaptic plasticity and cognitive impairment (Kao et al., 2020). Lipidomics is a novel technique for researching the mechanism of lipid metabolism, which Akyol et al. (2021) have utilized to compare the biochemical profiles of brain tissues from patients with different degrees of AD. They found that different subgenera of lipids in AD were significantly disturbed, including neutral lipids, glycerolipids, glycerophospholipids, and sphingolipids (Akyol et al., 2021).
Interestingly, serum FGF-21 levels were reduced in both animal models and patients of AD but increased following cognitive improvement (Tournissac et al., 2019; Conte et al., 2021). Recombinant human FGF-21 could reduce the concentrations of total cholesterol, low-density lipoprotein, and high-density lipoprotein, inhibit neuroinflammation, and correct cognitive decline in cognitive impairment caused by hyperlipidaemia (Wang Q. et al., 2018). This result may be attributed to the capacity of FGF21 to regulate the lipolytic signaling pathway, or insulin signaling pathway, in hepatocytes (Gimeno and Moller, 2014). Researchers also found that the administration of exogenous FGF-21 suppressed the expression of β-site amyloid precursor protein cleaving enzyme1 (Bace1), reduced Aβ deposition, and improved manifestations of dementia by inhibiting neuroinflammation through the TLR4/NF-κB signaling pathway, which could also restrain apoptosis via the MAPK signaling pathway (Amiri et al., 2018; Chen S. et al., 2019; Taliyan et al., 2019). These suggest a correlation between FGF-21 and AD, which makes it a potential biomarker for exploring new biomarkers, developing new drug targets, and providing new directions for in-depth exploration of AD metabolic mechanisms. Besides, since FGF21 could improve neuronal metabolism and energy supply in the CNS, enhance neuronal plasticity, and repair cerebrovascular endothelium to relieve symptoms of cognitive impairment, developing new drugs based on FGF-21 might be the most suitable choice to explore a new therapy for VaD. The potential therapeutic mechanisms of FGF-21 are shown in Figure 2.
2.2.3. FGF-23
FGF-23 is a bone-released endocrine growth factor and a member of the FGF-19 subgroup. FGF-23 is synthesized by osteoblasts and osteoclasts that are distributed primarily in the kidney, cortex, hippocampus, hypothalamus, thyroid gland, and bone, but also to a small degree in the spleen, liver, and other tissues (Hensel et al., 2016; Kuro-O, 2021; Ursem et al., 2021). FGF-23 primarily binds to α-KL/FGFR-1 to exert physiological effects, including regulating phosphate homeostasis and glucose metabolism, promoting neurogenesis, and maintaining emotional and cognitive functions (Beenken and Mohammadi, 2009; Kuro-O, 2021). FGF-23 overexpression in the serum may induce impaired hippocampal long-time potentiation (LTP) and reduce hippocampal adenosine-triphosphate (ATP) content with cognitive and memory decline, especially in people with chronic kidney disease (Liu et al., 2011; Drew et al., 2014; Drew and Weiner, 2014). Although Zhu et al. (2018) arrived at the opposite conclusion as they observed no significant differences between the levels of serum FGF-23 in individuals with mild cognitive impairment and healthy individuals, however, they assayed FGF-23 in a different way compared to previous experiments, which may explain the discrepancy in the results. The difference might also be attributed to the fact that FGF-23, like FGF-21, is affected by sex, and it is also possible that FGF-23 levels in the cerebrospinal fluid and blood are different. FGF-23 knockout mice also exhibited cognitive impairment in several in vivo studies, which may be associated with dysregulation of phosphate homeostasis and cytotoxicity (Laszczyk et al., 2019). As for the in vitro studies, the FGF-23/α-KL/FGFR-1 pathway was shown to increase hippocampal neuronal synaptic density but inhibits neuronal ramification via activating the downstream Akt signaling pathway, leading to memory deficits (Hensel et al., 2016; Zhu et al., 2018). These difference between animal and cellular experiments suggest that FGF-23 may have bidirectional modulatory effects on cognitive function.
On the other hand, intracranial atherosclerosis may trigger endothelial injury in blood vessels, induce neuroinflammation, and thus damage the structure and function of key brain regions where emotion and cognition are regulated, such as the hippocampus (Castello et al., 2022). In the vascular system, FGF-23 could bind directly to FGFR-2 or FGFR-3 without α-KL to induce vascular calcification (Vervloet, 2019). Thus, FGF-23 is highly associated with atherosclerosis and is a significant risk factor for atherosclerosis and stroke (Fakhri et al., 2014; Chang et al., 2020; Zheng et al., 2020). In other words, FGF-23 may indirectly trigger cognitive impairment by aggravating cerebrovascular damage. The potential therapeutic mechanisms of FGF-23 are shown in Figure 2.
3. Fibroblast growth factor receptors in cognitive disorders and dementia
The four main fractions of the transmembrane receptor tyrosine kinase FGFR, including FGFR-1, FGFR-2, FGFR-3, and FGFR-4, can mediate the signaling of FGFs via HS or KL-dependent pathways. Although all FGFRs are widely distributed in the CNS, existing evidence suggests that the main receptor that has been observed to change significantly in cognitive disorders is FGFR-1 (Goswami et al., 2013).
3.1. FGFRs
FGFR-1 is predominantly expressed in the hippocampus, and mediates downstream pathways by inhibiting neuroinflammation and maintaining LTP to protect learning and cognitive abilities (Rajendran et al., 2021). FGF-2, FGF-9, and FGF-22 can bind directly to FGFR-1 in the CNS. For example, the BBB protective effect on the BBB exhibited by FGF-2 is achieved by activating FGFR-1 (Lin et al., 2018), whereas the involvement of β-KL is required for binding FGF-21 and FGF-23 to FGFR-1, as mentioned above. Alternatively, dramatic AHN restriction, diminished amplitude of LTP, and hypomnesia could be observed in FGFR-1 knockout mice, and these changes were reversed with FGFR-1 agonists (Zhao et al., 2007; Pereda-Pérez et al., 2019). A compensatory increase in FGFR-3 expression can be observed in ASTs in the vicinity of “senile plaques” in AD patients (Ferrer and Martí, 1998). Moreover, FGF-2 binding to FGFR-3 could activate the anti-inflammatory phenotype of MG, inhibit excitatory toxicity, and exert neuroprotective effects through regulation of the ERK1/2 signaling pathway (Noda et al., 2014). Although the expression of FGFR-4 is low in the CNS and mainly concentrated in specific brain areas, especially the hypothalamus, FGF-15/19 could only inhibit the HPA axis and exert insulin-like effects by specifically binding to FGFR-4 due to the relatively low number of β-KL in the hypothalamus. Experiments by Ryan et al. (2013) showed that FGF-15/19/β-KL/FGFR-4 in the hypothalamus plays an integral role in regulating glucose metabolism throughout the body. As previously mentioned, abnormal glucose metabolism is strongly associated with the development of cognitive impairment and dementia. FGFRs demonstrate high homology and overlapping recognizability, which means there is no corresponding relationship between FGFRs and FGFs. Thus, it is difficult to study the changes in FGFRs. However, the future progressive exploration of the correspondence between FGFs and FGFRs and their downstream pathways will have profound implications for the study and application of the FGFs/FGFRs system in the field of neuroscience.
3.2. Co-receptor KL protein
Endocrine FGFs have a poor affinity to HS, and all have to form different homodimers by binding the co-receptor KL protein to the corresponding FGFR to activate the downstream pathways, such as FGF-19/β-KL/FGFR-4, FGF-21/β-KL/FGFR-1, or FGF-23/α-KL/FGFR-1. Subsequently, endocrine FGFs exert beneficial cognitive effects by inhibiting the HPA axis activation, suppressing neuroinflammation, facilitating BA metabolism, enhancing insulin sensitivity, and promoting neurogenesis. KL is divided into three categories according to the protein-coding genes, namely low molecular weight α-KL, high molecular weight β-KL, and γ-KL (Massó et al., 2015; Zhou et al., 2015). The α-KL can be further divided into transmembrane (m-KL), free, and secretory type, with the latter two referred to as soluble KL (s-KL), which is one of the hot spots in current dementia research. Researchers have only observed γ-KL in brown adipose tissue and eyeballs and have not yet found an association with the FGFs/FGFRs system (Zhang et al., 2017; Ma et al., 2021). Although much evidence suggests that α-KL has essential effects on cognition and memory, these studies have focused on s-KL rather than m-KL as a ligand for FGF-23 (Cararo-Lopes et al., 2017; Li et al., 2017; Zhao et al., 2020; Tank et al., 2021). These two α-KL have different structural features and physiological roles. However, recent studies have also identified, that hydrolyzed m-KL is a significant source of s-KL (Chen et al., 2007). β-KL protein is mainly distributed in the liver, gallbladder, kidney, brain, and other tissues, synthesized by ependymal cells in the hippocampus in CNS, and excreted into the cerebrospinal fluid, where it can exert antioxidant, anti-inflammatory, and neuroprotective effects (Paroni et al., 2019). These effects may be attributed directly to inhibiting neuroinflammation, reducing oxidative stress, and correcting vascular endothelial dysfunction or indirectly by activating pathways related to endocrine FGFs (Gold et al., 2013).
4. Discussion
FGFs and their receptors constitute a complex network of endocrine functions, and the dynamic balance between FGFs and their receptors may be a critical link that our understanding of cognitive regulation. When the FGFs/FGFRs system is out of balance, pathological changes such as disturbance of glucose metabolism disorder, neuroinflammation, hyperfunction of the HPA axis, disruption of the BBB, reduced neuroplasticity, inhibition of neurogenesis, and apoptosis may occur in the body, which in turn can affect the structure and function of the cerebral cortex, hippocampus, hypothalamus, pituitary gland, and other tissues, resulting in cognitive decline. Therefore, maintaining the stability of the FGF-FGFR system might have the potential to be a new therapeutic strategy for retarding neurodegeneration and improving cognitive function. Endocrine FGFs could be distributed through the blood circulation to multiple organs throughout the body, so they can exert a comprehensive neuroprotective effect by intervening in multiple systems. Besides, The physiological characteristics of endocrine FGFs allow them to penetrate the BBB to act on the brain parenchyma and exert neuroprotective effects easier than paracrine FGFs, including FGF-1, FGF-2, and FGF-9, via multiple metabolic pathways.
In recent years, FGFs-based drugs have been widely used in the clinical treatment of wound healing, osteoarthritis, malignancies, hepatitis, diabetes, cardiovascular disease and cerebrovascular disease (Zhang and Li, 2016; Sanyal et al., 2019; Abou-Alfa et al., 2020; Chen K. et al., 2022; Subbiah et al., 2022). Although the relevance of the FGF-FGFR system to neurodegenerative diseases has been identified, the application of FGF-based drugs remains in vivo and in vivo experiments which are still far from clinical application for treating cognitive disorders. Moreover, the use of FGFs as biomarkers for detecting neurodegenerative diseases is also limited by many factors. Although researchers are still unable to elucidate the specific mechanisms of the FGFs/FGFRs system, they have noted the trends and therapeutic potential of multiple FGFs in various diseases and have discovered or developed drugs, such as synthetic human recombinant FGFs (Bogousslavsky et al., 2002), synthetic analogs of FGFs (Sanyal et al., 2019; Ardizzone et al., 2022), molecular inhibitors or agonists of FGFs/FGFRs (Bahleda et al., 2020; van Brummelen et al., 2020; Subbiah et al., 2022), or extracts of natural herbal medicines (Hong et al., 2016; Yuan et al., 2019; Cheng et al., 2021; Wang et al., 2021) that could effectively modulate this system. In the current literature review, we found that the difficulties faced by previous studies on the application of FGFs/FGFRs systems in treatment and diagnose mainly focus on the following four aspects (Figure 3): (1) Different FGFs isoforms: some FGFs have different molecular weight isoforms with different physiological effects, but their individual microscopic expression changes cannot be reflected by the overall changes, for example, High molecular weight (HMW) FGF-2 and Light molecular weight (LMW) FGF-2 (Chlebova et al., 2009; Katsouri et al., 2015; Chen X. et al., 2019). (2) Inhomogeneous space distribution of FGFs: the spatial distribution of FGFs in serum, cerebrospinal fluid and brain parenchyma is inhomogeneous, because some FGFs cannot completely penetrate the BBB. The following may obscure particular properties of FGFs in various tissues under pathological state, and render the statistical assessment their heterogeneity inaccurate (Katsouri et al., 2015; Liu et al., 2017; Chang et al., 2018; Tennakoon et al., 2022). (3) Correspondence between FGFs and FGFRs: single FGF could bind to different FGFRs in different microenvironments and regulate different signaling pathways, thus exerting distinct physiological or pathological effects, due to the overlapping recognition of FGFRs and discrepancy in expression sites (Lee et al., 2011; Noda et al., 2014; Lin et al., 2018; Vervloet, 2019). (4) Competitive inhibition between FGFs: FGFs are highly homologous, and subtypes with similar physiological structures may compete with one another for the same receptors, thereby inhibiting the regulation of downstream signaling pathways such as FGF-17 and FGF-15/19 (Liu S. et al., 2018), FGF-2 and FGF-9 (Aurbach et al., 2015).
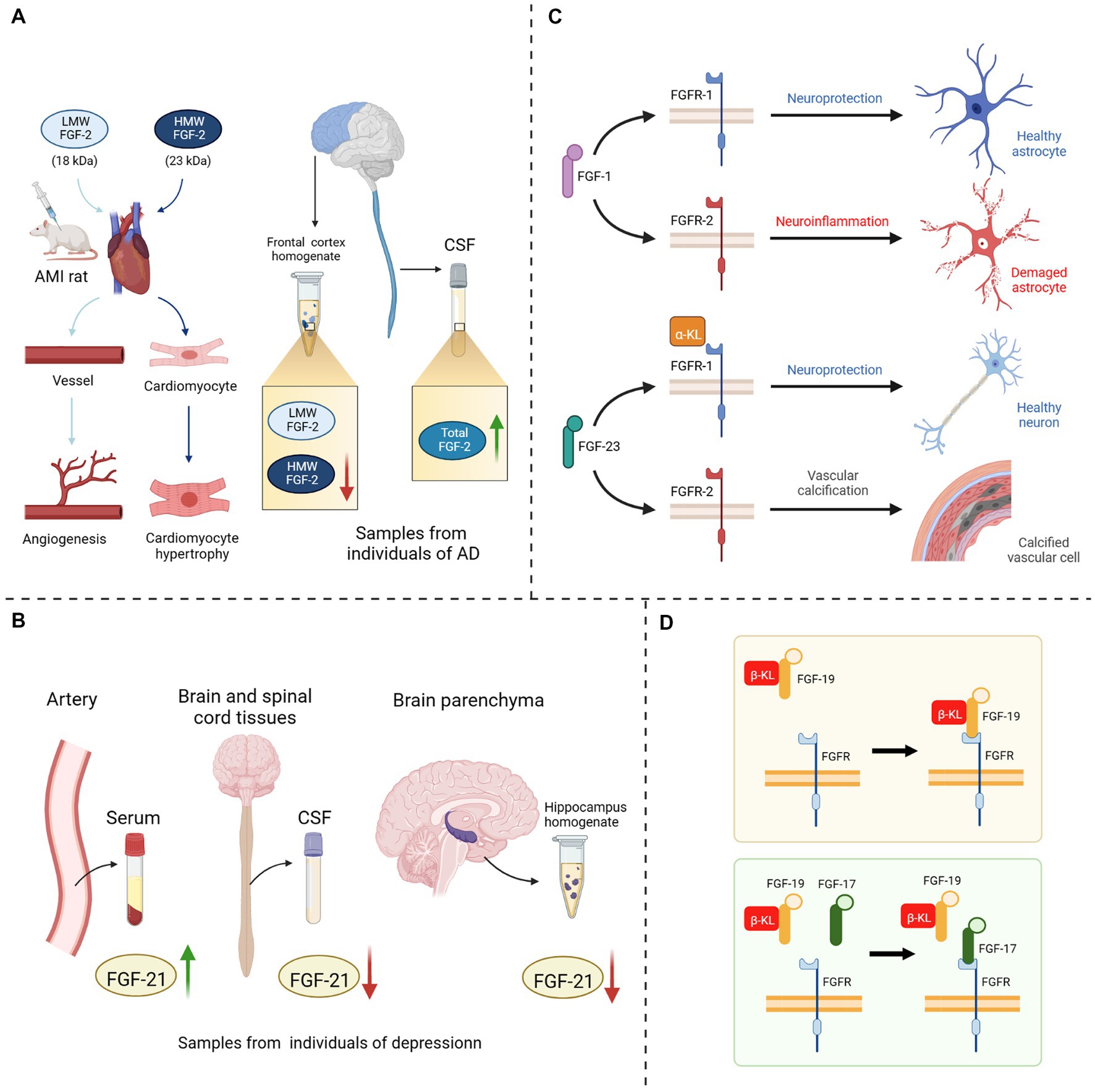
Figure 3. Four potential explanations for the hampered FGF in-depth study. (A) Different isoforms of FGF-2: injection of HMW FGF-2 into AMI rats could induce cardiomyocyte hypertrophy, whereas injection of LMW FGF-2 only promoted angiogenesis in rat heart; A decrease in HMW FGF-2 expression could be detected in the anterior cortical homogenate of AD patients, while the expression level of LMW FGF-2 remained unchanged, and the level of total FGF-2 in the CSF was significantly increased. (B) Inhomogeneous space distribution of FGF21: the expression level of FGF-21 was elevated in the serum of depressed patients, while decreased in both CSF and hippocampus homogenate. (C) Correspondence between FGFs and FGFRs: when FGF-1 bound to FGFR-1, it could exert neuroprotective effects, while binding to FGFR-2 could provoke neuroinflammation; The binding of FGF-23 to α-KL/FGFR-1 protects neuronal cells, on the contrary it directly binds to FGFR-2 to induce vascular calcification. (D) Competitive inhibition between FGF17 and FGF19: FGF-17 could bind to the β-KL/FGFR-2 homodimer and shown antagonism against FGF-19. LMW, light molecular weight; HMW, high molecular weight; AMI, acute myocardial infarction; CSF, cerebrospinal fluid; AD, Alzheimer’s disease; FGF, fibroblast growth factor; FGFR, fibroblast growth factor receptor; α/β-KL, α/β-klotho protein.
5. Conclusion
In summary, despite the induced cell proliferation, survival and differentiation, angiogenesis promotion, inflammation inhibition, immune and metabolic modulation, and antioxidant effects possessed by the FGF/FGFR system have been applied in the treatment of tumors, trauma, and renal, hepatic, and cardiovascular diseases, there is still a substantial unexplored gap area, especially for the modulatory effects and diagnostic value of central nervous system diseases and psychiatric disorders. Considering the extensive range of actions and the many targets of the FGF/FGFR system intervention, we should also be aware of the potential risk associated with its long-term or high-dose use, such as adverse effects, resistance, and addiction, when used to treat cognitive or mood disorders. Here, we propose to conduct more clinical and basic studies in combination with new technologies to investigate the structural and functional characteristics of the FGFs/FGFRs system and its specific mechanism in cognitive disorders and dementia to find new biomarkers and therapeutic approaches for these diseases.
Author contributions
WZ and TZ conceived the topic and drafted the manuscript. YJ helped WZ to draw the figures. SH, MX, and JP revised the draft. All authors contributed to the article and approved the submitted version.
Funding
This research was supported by National Natural Science Foundation of China (NSFC-81573790, NSFC-81603443, and NSFC-82204922) and the Fundamental Research Funds for the Central public welfare research institutes of China (no. ZZ15-XY-LCQ-04).
Acknowledgments
We would like to thank Editage (www.editage.cn) for English language editing and BioRender (www.biorender.com) for Excellent drawing tool. All the figures in the text are created with BioRender.com.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Abou-Alfa, G. K., Sahai, V., Hollebecque, A., Vaccaro, G., Melisi, D., Al-Rajabi, R., et al. (2020). Pemigatinib for previously treated, locally advanced or metastatic cholangiocarcinoma: a multicentre, open-label, phase 2 study. Lancet Oncol. 21, 671–684. doi: 10.1016/S1470-2045(20)30109-1
Akyol, S., Ugur, Z., Yilmaz, A., Ustun, I., Gorti, S. K. K., Oh, K., et al. (2021). Lipid profiling of Alzheimer's disease brain highlights enrichment in glycerol(phospho)lipid, and sphingolipid metabolism. Cells 10:2591. doi: 10.3390/cells10102591
Al-Aqil, F. A., Monte, M. J., Peleteiro-Vigil, A., Briz, O., Rosales, R., González, R., et al. (2018). Interaction of glucocorticoids with FXR/FGF19/FGF21-mediated ileum-liver crosstalk. Biochimica et biophysica acta. Mol Basis Dis 1864, 2927–2937. doi: 10.1016/j.bbadis.2018.06.003
Amiri, M., Braidy, N., and Aminzadeh, M. (2018). Protective effects of fibroblast growth factor 21 against amyloid-Beta-induced toxicity in SH-SY5Y cells. Neurotox. Res. 34, 574–583. doi: 10.1007/s12640-018-9914-2
Anderson, N. D. (2019). State of the science on mild cognitive impairment (MCI). CNS Spectr. 24, 78–87. doi: 10.1017/S1092852918001347
Ardizzone, A., Bova, V., Casili, G., Filippone, A., Campolo, M., Lanza, M., et al. (2022). SUN11602, a bFGF mimetic, modulated neuroinflammation, apoptosis and calcium-binding proteins in an in vivo model of MPTP-induced nigrostriatal degeneration. J. Neuroinflammation 19:107. doi: 10.1186/s12974-022-02457-3
Aurbach, E. L., Inui, E. G., Turner, C. A., Hagenauer, M. H., Prater, K. E., Li, J. Z., et al. (2015). Fibroblast growth factor 9 is a novel modulator of negative affect. Proc. Natl. Acad. Sci. U. S. A. 112, 11953–11958. doi: 10.1073/pnas.1510456112
Azhar, M., Zeng, G., Ahmed, A., Dar Farooq, A., Choudhary, M. I., De-Jiang, J., et al. (2021). Carnosic acid ameliorates depressive-like symptoms along with the modulation of FGF9 in the hippocampus of middle carotid artery occlusion-induced Sprague Dawley rats. Phytother. Res. 35, 384–391. doi: 10.1002/ptr.6810
Bahleda, R., Meric-Bernstam, F., Goyal, L., Tran, B., He, Y., Yamamiya, I., et al. (2020). Phase I, first-in-human study of futibatinib, a highly selective, irreversible FGFR1-4 inhibitor in patients with advanced solid tumors. Ann. Oncol. 31, 1405–1412. doi: 10.1016/j.annonc.2020.06.018
Ball, H. A., McWhirter, L., Ballard, C., Bhome, R., Blackburn, D. J., Edwards, M. J., et al. (2020). Functional cognitive disorder: dementia's blind spot. Brain: a. J. Neurol. 143, 2895–2903. doi: 10.1093/brain/awaa224
Baloni, P., Funk, C. C., Yan, J., Yurkovich, J. T., Kueider-Paisley, A., Nho, K., et al. (2020). Metabolic network analysis reveals altered bile acid synthesis and metabolism in Alzheimer's disease. Cell Rep Med 1:100138. doi: 10.1016/j.xcrm.2020.100138
Beenken, A., and Mohammadi, M. (2009). The FGF family: biology, pathophysiology and therapy. Nat. Rev. Drug Discov. 8, 235–253. doi: 10.1038/nrd2792
Belov, A. A., and Mohammadi, M. (2013). Molecular mechanisms of fibroblast growth factor signaling in physiology and pathology. Cold Spring Harb. Perspect. Biol. 5:a015958. doi: 10.1101/cshperspect.a015958
Bennett, S., and Thomas, A. J. (2014). Depression and dementia: cause, consequence or coincidence? Maturitas 79, c–190. doi: 10.1016/j.maturitas.2014.05.009
Bogousslavsky, J., Victor, S. J., Salinas, E. O., Pallay, A., Donnan, G. A., Fieschi, C., et al. (2002). Fiblast (trafermin) in acute stroke: results of the European-Australian phase II/III safety and efficacy trial. Cerebrovasc. Dis. 14, 239–251. doi: 10.1159/000065683
Butterfield, D. A., and Halliwell, B. (2019). Oxidative stress, dysfunctional glucose metabolism and Alzheimer disease. Nat. Rev. Neurosci. 20, 148–160. c
Byers, A. L., and Yaffe, K. (2011). Depression and risk of developing dementia. Nat. Rev. Neurol. 7, 323–331. doi: 10.1038/nrneurol.2011.60
Canet, G., Hernandez, C., Zussy, C., Chevallier, N., Desrumaux, C., and Givalois, L. (2019). Is AD a stress-related disorder? Focus on the HPA Axis and its promising therapeutic targets. Front. Aging Neurosci. 11:269. doi: 10.3389/fnagi.2019.00269
Cararo-Lopes, M. M., Mazucanti, C. H. Y., Scavone, C., Kawamoto, E. M., and Berwick, D. C. (2017). The relevance of α-KLOTHO to the central nervous system: some key questions. Ageing Res. Rev. 36, 137–148. doi: 10.1016/j.arr.2017.03.003
Castello, J. P., Pasi, M., Kubiszewski, P., Abramson, J. R., Charidimou, A., Kourkoulis, C., et al. (2022). Cerebral small vessel disease and depression among intracerebral hemorrhage survivors. Stroke 53, 523–531. doi: 10.1161/STROKEAHA.121.035488
Chang, H. H., Chen, P. S., Cheng, Y. W., Wang, T. Y., Yang, Y. K., and Lu, R. B. (2018). FGF21 is associated with metabolic effects and treatment response in depressed bipolar II disorder patients treated with valproate. Int. J. Neuropsychopharmacol. 21, 319–324. doi: 10.1093/ijnp/pyx093
Chang, Y., Kim, J., Woo, H. G., Ryu, D. R., Oh, H. J., and Song, T. J. (2020). Plasma fibroblast growth factor 23 concentration is associated with intracranial cerebral atherosclerosis in acute ischemic stroke patients. J. Clin. Neurol. 16, 29–36. doi: 10.3988/jcn.2020.16.1.29
Chen, Q., Chen, X., Jia, Z., Du, Y., Zhang, S., Xu, W., et al. (2022). ∆nFGF1 protects-cells against high glucose-induced apoptosis via the AMPK/SIRT1/PGC-1 Axis. Oxid. Med. Cell. Longev. 2022:1231970. doi: 10.1155/2022/1231970
Chen, S., Chen, S. T., Sun, Y., Xu, Z., Wang, Y., Yao, S. Y., et al. (2019). Fibroblast growth factor 21 ameliorates neurodegeneration in rat and cellular models of Alzheimer's disease. Redox Biol. 22:101133. doi: 10.1016/j.redox.2019.101133
Chen, J., Hu, J., Liu, H., Xiong, Y., Zou, Y., Huang, W., et al. (2018). FGF21 protects the blood-brain barrier by upregulating PPARgamma via FGFR1/beta-klotho after traumatic brain injury. J. Neurotrauma 35, 2091–2103. doi: 10.1089/neu.2017.5271
Chen, X., Li, Z., Cheng, Y., Kardami, E., and Loh, Y. P. (2019). Low and high molecular weight FGF-2 have differential effects on astrocyte proliferation, but are both protective against Aβ-induced cytotoxicity. Front. Mol. Neurosci. 12:328. doi: 10.3389/fnmol.2019.00328
Chen, C.-D., Podvin, S., Gillespie, E., Leeman, S. E., and Abraham, C. R. (2007). Insulin stimulates the cleavage and release of the extracellular domain of klotho by ADAM10 and ADAM17. Proc. Natl. Acad. Sci. U. S. A. 104, 19796–19801. doi: 10.1073/pnas.0709805104
Chen, K., Rao, Z., Dong, S., Chen, Y., Wang, X., Luo, Y., et al. (2022). Roles of the fibroblast growth factor signal transduction system in tissue injury repair. Burns Trauma 10:tkac005. doi: 10.1093/burnst/tkac005
Chen, Y., Wang, Y., Qin, Q., Zhang, Y., Xie, L., Xiao, J., et al. (2022). Carnosic acid ameliorated Aβ-mediated (amyloid-β peptide) toxicity, cholinergic dysfunction and mitochondrial defect in of Alzheimer's model. Food Funct. 13, 4624–4640. doi: 10.1039/d1fo02965g
Chen, Z., and Zhong, C. (2013). Decoding Alzheimer's disease from perturbed cerebral glucose metabolism: implications for diagnostic and therapeutic strategies. Prog. Neurobiol. 108, 21–43. doi: 10.1016/j.pneurobio.2013.06.004
Cheng, J., Chen, M., Wan, H.-Q., Chen, X.-Q., Li, C.-F., Zhu, J.-X., et al. (2021). Paeoniflorin exerts antidepressant-like effects through enhancing neuronal FGF-2 by microglial inactivation. J. Ethnopharmacol. 274:114046. doi: 10.1016/j.jep.2021.114046
Cheng, L., Huang, C., and Chen, Z. (2019). Tauroursodeoxycholic acid ameliorates lipopolysaccharide-induced depression like behavior in mice via the inhibition of Neuroinflammation and Oxido-Nitrosative stress. Pharmacology 103, 93–100. doi: 10.1159/000494139
Chlebova, K., Bryja, V., Dvorak, P., Kozubik, A., Wilcox, W. R., and Krejci, P. (2009). High molecular weight FGF2: the biology of a nuclear growth factor. Cell. Mol. Life Sci. 66, c. doi: 10.1007/s00018-008-8440-4
Chuang, J.-I., Huang, J.-Y., Tsai, S.-J., Sun, H. S., Yang, S.-H., Chuang, P.-C., et al. (2015). FGF9-induced changes in cellular redox status and HO-1 upregulation are FGFR-dependent and proceed through both ERK and AKT to induce CREB and Nrf2 activation. Free Radic. Biol. Med. 89, 274–286. doi: 10.1016/j.freeradbiomed.2015.08.011
Conte, M., Sabbatinelli, J., Chiariello, A., Martucci, M., Santoro, A., Monti, D., et al. (2021). Disease-specific plasma levels of mitokines FGF21, GDF15, and Humanin in type II diabetes and Alzheimer's disease in comparison with healthy aging. GeroScience 43, 985–1001. doi: 10.1007/s11357-020-00287-w
Csernansky, J. G., Dong, H., Fagan, A. M., Wang, L., Xiong, C., Holtzman, D. M., et al. (2006). Plasma cortisol and progression of dementia in subjects with Alzheimer-type dementia. Am. J. Psychiatry 163, 2164–2169. doi: 10.1176/ajp.2006.163.12.2164
Cummings, B. J., Su, J. H., and Cotman, C. W. (1993). Neuritic involvement within bFGF immunopositive plaques of Alzheimer's disease. Exp. Neurol. 124, 315–325.
Cunningham, J., Locatelli, F., and Rodriguez, M. (2011). Secondary hyperparathyroidism: pathogenesis, disease progression, and therapeutic options. Clin. J. Am. Soc. Nephrol. 6, 913–921. doi: 10.2215/CJN.06040710
Deng, Z., Deng, S., Zhang, M. R., and Tang, M. M. (2019). Fibroblast growth factors in depression. Front. Pharmacol. 10:60. doi: 10.3389/fphar.2019.00060
Dichgans, M., and Leys, D. (2017). Vascular Cognitive Impairment. Circ. Res. 120, 573–591. doi: 10.1161/CIRCRESAHA.116.308426
Dienel, G. A. (2012). Fueling and imaging brain activation. ASN Neuro 4:e00093. doi: 10.1042/AN20120021
Dolegowska, K., Marchelek-Mysliwiec, M., Nowosiad-Magda, M., Slawinski, M., and Dolegowska, B. (2019). FGF19 subfamily members: FGF19 and FGF21. J. Physiol. Biochem. 75, 229–240. doi: 10.1007/s13105-019-00675-7
Drew, D. A., Tighiouart, H., Scott, T. M., Lou, K. V., Fan, L., Shaffi, K., et al. (2014). FGF-23 and cognitive performance in hemodialysis patients. Hemodialysis international. Int Symp Home Hemodial 18, 78–86. doi: 10.1111/hdi.12100
Drew, D. A., and Weiner, D. E. (2014). Cognitive impairment in chronic kidney disease: keep vascular disease in mind. Kidney Int. 85, 505–507. doi: 10.1038/ki.2013.437
Duff, K., Paulsen, J., Mills, J., Beglinger, L. J., Moser, D. J., Smith, M. M., et al. (2010). Mild cognitive impairment in prediagnosed Huntington disease. Neurology 75, 500–507. doi: 10.1212/WNL.0b013e3181eccfa2
Fakhri, H., Ricken, R., Adli, M., Fajol, A., Walter, M., Föller, M., et al. (2014). Impact of lithium treatment on FGF-23 serum concentrations in depressive patients. J. Clin. Psychopharmacol. 34, 745–747. doi: 10.1097/JCP.0000000000000163
Feng, C., Zhang, C., Shao, X., Liu, Q., Qian, Y., Feng, L., et al. (2012). Enhancement of nose-to-brain delivery of basic fibroblast growth factor for improving rat memory impairments induced by co-injection of β-amyloid and ibotenic acid into the bilateral hippocampus. Int. J. Pharm. 423, 226–234. doi: 10.1016/j.ijpharm.2011.12.008
Ferrer, I., and Martí, E. (1998). Distribution of fibroblast growth factor receptor-1 (FGFR-1) and FGFR-3 in the hippocampus of patients with Alzheimer's disease. Neurosci. Lett. 240, 139–142.
Galvez-Contreras, A. Y., Campos-Ordonez, T., Lopez-Virgen, V., Gomez-Plascencia, J., Ramos-Zuniga, R., and Gonzalez-Perez, O. (2016). Growth factors as clinical biomarkers of prognosis and diagnosis in psychiatric disorders. Cytokine Growth Factor Rev. 32, 85–96. doi: 10.1016/j.cytogfr.2016.08.004
Garnier-Crussard, A., Bougacha, S., Wirth, M., Dautricourt, S., Sherif, S., Landeau, B., et al. (2022). White matter hyperintensity topography in Alzheimer's disease and links to cognition. Alzheimers Dement. 18, 422–433. doi: 10.1002/alz.12410
Gasser, E., Moutos, C. P., Downes, M., and Evans, R. M. (2017). FGF1-a new weapon to control type 2 diabetes mellitus. Nat. Rev. Endocrinol. 13, 599–609. doi: 10.1038/nrendo.2017.78
Gee, L. M. V., Barron-Millar, B., Leslie, J., Richardson, C., Zaki, M. Y. W., Luli, S., et al. (2022). Anti-Cholestatic therapy with Obeticholic acid improves short-term memory in bile duct ligated mice. Am. J. Pathol. 193, 11–26. doi: 10.1016/j.ajpath.2022.09.005
Giacomini, A., Chiodelli, P., Matarazzo, S., Rusnati, M., Presta, M., and Ronca, R. (2016). Blocking the FGF/FGFR system as a two-compartment antiangiogenic/antitumor approach in cancer therapy. Pharmacol. Res. cc. doi: 10.1016/j.phrs.2016.03.024
Gimeno, R. E., and Moller, D. E. (2014). FGF21-based pharmacotherapy--potential utility for metabolic disorders. Trends Endocrinol. Metab. 25, 303–311. doi: 10.1016/j.tem.2014.03.001
Gold, P. W., Licinio, J., and Pavlatou, M. G. (2013). Pathological parainflammation and endoplasmic reticulum stress in depression: potential translational targets through the CNS insulin, klotho and PPAR-gamma systems. Mol. Psychiatry 18, 154–165. doi: 10.1038/mp.2012.167
Gonzales, M. M., Garbarino, V. R., Pollet, E., Palavicini, J. P., Kellogg, D. L., Kraig, E., et al. (2022). Biological aging processes underlying cognitive decline and neurodegenerative disease. J. Clin. Invest. 132:e158453. doi: 10.1172/JCI158453
Goswami, D. B., Jernigan, C. S., Chandran, A., Iyo, A. H., May, W. L., Austin, M. C., et al. (2013). Gene expression analysis of novel genes in the prefrontal cortex of major depressive disorder subjects. Prog. Neuropsychopharmacol. Biol. Psychiatry 43, 126–133. doi: 10.1016/j.pnpbp.2012.12.010
Hamer, J. A., Testani, D., Mansur, R. B., Lee, Y., Subramaniapillai, M., and McIntyre, R. S. (2019). Brain insulin resistance: a treatment target for cognitive impairment and anhedonia in depression. Exp. Neurol. 315, 1–8. doi: 10.1016/j.expneurol.2019.01.016
Han, K.-H., Kim, M.-H., Jeong, G.-J., Kim, A.-K., Chang, J. W., and Kim, D.-I. (2019). FGF-17 from hypoxic human Wharton's jelly-derived mesenchymal stem cells is responsible for maintenance of cell proliferation at late passages. Int J Stem Cells 12, 279–290. doi: 10.15283/ijsc18042
Hayley, S., Hakim, A. M., and Albert, P. R. (2021). Depression, dementia and immune dysregulation. Brain 144, 746–760. doi: 10.1093/brain/awaa405
He, H., Chen, Z., Chen, D., Lu, X., Huang, C., and Chen, J. (2021). Identification of the expression of farnesoid X receptor in astrocytes. Neuroreport 32, 1216–1222. doi: 10.1097/WNR.0000000000001717
Hecht, D., Zimmerman, N., Bedford, M., Avivi, A., and Yayon, A. (1995). Identification of fibroblast growth factor 9 (FGF9) as a high affinity, heparin dependent ligand for FGF receptors 3 and 2 but not for FGF receptors 1 and 4. Growth Factors 12, 223–233.
Hensel, N., Schön, A., Konen, T., Lübben, V., Förthmann, B., Baron, O., et al. (2016). Fibroblast growth factor 23 signaling in hippocampal cells: impact on neuronal morphology and synaptic density. J. Neurochem. 137, 756–769. doi: 10.1111/jnc.13585
Holmes, D. (2014). Therapy: FGF1 restores blood glucose levels and insulin sensitivity in diabetic mice. Nat. Rev. Endocrinol. 10:576. doi: 10.1038/nrendo.2014.129
Hong, X.-P., Chen, T., Yin, N.-N., Han, Y.-M., Yuan, F., Duan, Y.-J., et al. (2016). Puerarin ameliorates D-galactose induced enhanced hippocampal neurogenesis and tau hyperphosphorylation in rat brain. J. Alzheimers Dis. 51, 605–617. doi: 10.3233/JAD-150566
Hoshikawa, M., Ohbayashi, N., Yonamine, A., Konishi, M., Ozaki, K., Fukui, S., et al. (1998). Structure and expression of a novel fibroblast growth factor, FGF-17, preferentially expressed in the embryonic brain. Biochem. Biophys. Res. Commun. 244, 187–191.
Hossain, M. S., Oomura, Y., Fujino, T., and Akashi, K. (2020). Glucose signaling in the brain and periphery to memory. Neurosci. Biobehav. Rev. 110, 100–113. doi: 10.1016/j.neubiorev.2019.03.018
Hotamisligil, G. S. (2006). Inflammation and metabolic disorders. Nature 444, 860–867. doi: 10.1038/nature05485
Hsuchou, H., Pan, W., and Kastin, A. J. (2013). Fibroblast growth factor 19 entry into brain. Fluids Barriers CNS 10:32. doi: 10.1186/2045-8118-10-32
Huang, C., Wang, J., Hu, W., Wang, C., Lu, X., Tong, L., et al. (2016). Identification of functional farnesoid X receptors in brain neurons. FEBS Lett. 590, 3233–3242. doi: 10.1002/1873-3468.12373
Hui, Q., Jin, Z., Li, X., Liu, C., and Wang, X. (2018). FGF family: from drug development to clinical application. Int. J. Mol. Sci. 19:1875. doi: 10.3390/ijms19071875
Iadecola, C., Duering, M., Hachinski, V., Joutel, A., Pendlebury, S. T., Schneider, J. A., et al. (2019). Vascular cognitive impairment and dementia: JACC scientific expert panel. J. Am. Coll. Cardiol. 73, 3326–3344. doi: 10.1016/j.jacc.2019.04.034
Ilieva, M., Nielsen, T. T., Michel, T., and Pankratova, S. (2019). FGF2 and dual agonist of NCAM and FGF receptor 1, Enreptin, rescue neurite outgrowth loss in hippocampal neurons expressing mutated huntingtin proteins. J. Neural Transm. 126, 1493–1500. doi: 10.1007/s00702-019-02073-1
Iram, T., Kern, F., Kaur, A., Myneni, S., Morningstar, A. R., Shin, H., et al. (2022). Young CSF restores oligodendrogenesis and memory in aged mice via Fgf17. Nature 605, 509–515. doi: 10.1038/s41586-022-04722-0
Jiang, Z.-S., Jeyaraman, M., Wen, G.-B., Fandrich, R. R., Dixon, I. M. C., Cattini, P. A., et al. (2007). High-but not low-molecular weight FGF-2 causes cardiac hypertrophy in vivo; possible involvement of cardiotrophin-1. J. Mol. Cell. Cardiol. 42, 222–233. doi: 10.1016/j.yjmcc.2006.09.002
Jiang, Y., Lin, L., Liu, N., Wang, Q., Yuan, J., Li, Y., et al. (2020). FGF21 protects against aggravated blood-brain barrier disruption after ischemic focal stroke in diabetic db/db male mice via cerebrovascular PPARgamma activation. Int. J. Mol. Sci. 21:824. doi: 10.3390/ijms21030824
Jin, K., LaFevre-Bernt, M., Sun, Y., Chen, S., Gafni, J., Crippen, D., et al. (2005). FGF-2 promotes neurogenesis and neuroprotection and prolongs survival in a transgenic mouse model of Huntington's disease. Proc. Natl. Acad. Sci. U. S. A. 102, 18189–18194. doi: 10.1073/pnas.0506375102
Kamal, F., Morrison, C., Maranzano, J., Zeighami, Y., and Dadar, M. (2022). Topographical differences in white matter hyperintensity burden and cognition in aging, MCI, and AD. GeroScience. 45, 1–16. doi: 10.1007/s11357-022-00665-6
Kao, Y.-C., Ho, P.-C., Tu, Y.-K., Jou, I. M., and Tsai, K.-J. (2020). Lipids and Alzheimer's disease. Int. J. Mol. Sci. 21:1505. doi: 10.3390/ijms21041505
Katsouri, L., Ashraf, A., Birch, A. M., Lee, K. K. L., Mirzaei, N., and Sastre, M. (2015). Systemic administration of fibroblast growth factor-2 (FGF2) reduces BACE1 expression and amyloid pathology in APP23 mice. Neurobiol. Aging 36, 821–831. doi: 10.1016/j.neurobiolaging.2014.10.004
Kellar, D., and Craft, S. (2020). Brain insulin resistance in Alzheimer's disease and related disorders: mechanisms and therapeutic approaches. Lancet Neurol. 19, 758–766. doi: 10.1016/S1474-4422(20)30231-3
Kharitonenkov, A., Shiyanova, T. L., Koester, A., Ford, A. M., Micanovic, R., Galbreath, E. J., et al. (2005). FGF-21 as a novel metabolic regulator. J. Clin. Invest. 115, 1627–1635. doi: 10.1172/JCI23606
Kiyota, T., Ingraham, K. L., Jacobsen, M. T., Xiong, H., and Ikezu, T. (2011). FGF2 gene transfer restores hippocampal functions in mouse models of Alzheimer's disease and has therapeutic implications for neurocognitive disorders. Proc. Natl. Acad. Sci. U. S. A. 108, E1339–E1348. doi: 10.1073/pnas.1102349108
Kuroda, M., Muramatsu, R., Maedera, N., Koyama, Y., Hamaguchi, M., Fujimura, H., et al. (2017). Peripherally derived FGF21 promotes remyelination in the central nervous system. J. Clin. Invest. 127, 3496–3509. doi: 10.1172/JCI94337
Kuro-O, M. (2021). Aging and FGF23-klotho system. Vitam. Horm. 115, 317–332. doi: 10.1016/bs.vh.2020.12.013
Lara, V. P., Caramelli, P., Teixeira, A. L., Barbosa, M. T., Carmona, K. C., Carvalho, M. G., et al. (2013). High cortisol levels are associated with cognitive impairment no-dementia (CIND) and dementia. Clin Chim Acta Int J Clin Chem 423, 18–22. doi: 10.1016/j.cca.2013.04.013
Laszczyk, A. M., Nettles, D., Pollock, T. A., Fox, S., Garcia, M. L., Wang, J., et al. (2019). FGF-23 deficiency impairs hippocampal-dependent cognitive function. ENeuro 6, e0469–18. doi: 10.1523/ENEURO.0469-18.2019
Lee, M., Kang, Y., Suk, K., Schwab, C., Yu, S., and McGeer, P. L. (2011). Acidic fibroblast growth factor (FGF) potentiates glial-mediated neurotoxicity by activating FGFR2 IIIb protein. J. Biol. Chem. 286, 41230–41245. doi: 10.1074/jbc.M111.270470
Leng, F., and Edison, P. (2021). Neuroinflammation and microglial activation in Alzheimer disease: where do we go from here? Nature reviews. Neurology 17, 157–172. doi: 10.1038/s41582-020-00435-y
Li, Q., Vo, H. T., Wang, J., Fox-Quick, S., Dobrunz, L. E., and King, G. D. (2017). Klotho regulates CA1 hippocampal synaptic plasticity. Neuroscience 347, 123–133. doi: 10.1016/j.neuroscience.2017.02.006
Li, C., Wang, X., Yan, J., Cheng, F., Ma, X., Chen, C., et al. (2020). Cholic acid protects in vitro neurovascular units against oxygen and glucose deprivation-induced injury through the BDNF-TrkB signaling pathway. Oxid. Med. Cell. Longev. 2020:1201624. doi: 10.1155/2020/1201624
Liang, C.-S., Tsai, C.-L., Lin, G.-Y., Lee, J.-T., Lin, Y.-K., Chu, C.-S., et al. (2021). Better identification of cognitive decline with Interleukin-2 than with amyloid and tau protein biomarkers in amnestic mild cognitive impairment. Front. Aging Neurosci. 13:670115. doi: 10.3389/fnagi.2021.670115
Lin, L., Wang, Q., Qian, K., Cao, Z., Xiao, J., Wang, X., et al. (2018). bFGF protects against oxygen glucose deprivation/Reoxygenation-induced endothelial monolayer permeability via S1PR1-dependent mechanisms. Mol. Neurobiol. 55, 3131–3142. doi: 10.1007/s12035-017-0544-0
Liu, P., Chen, L., Bai, X., Karaplis, A., Miao, D., and Gu, N. (2011). Impairment of spatial learning and memory in transgenic mice overexpressing human fibroblast growth factor-23. Brain Res. 1412, 9–17. doi: 10.1016/j.brainres.2011.07.028
Liu, S., Marcelin, G., Blouet, C., Jeong, J. H., Jo, Y.-H., Schwartz, G. J., et al. (2018). A gut-brain axis regulating glucose metabolism mediated by bile acids and competitive fibroblast growth factor actions at the hypothalamus. Molec Metab 8, 37–50. doi: 10.1016/j.molmet.2017.12.003
Liu, Y., Wang, M., Tan, X., Wang, X., Yang, X., Xiao, J., et al. (2017). Negative correlation between cerebrospinal fluid FGF21 levels and BDI scores in male Chinese subjects. Psychiatry Res. 252, 111–113. doi: 10.1016/j.psychres.2017.01.075
Liu, M., Wu, Y., Liu, Y., Chen, Z., He, S., Zhang, H., et al. (2018). Basic fibroblast growth factor protects astrocytes against ischemia/reperfusion injury by upregulating the Caveolin-1/VEGF signaling pathway. J. Mol. Neurosci. 64, 211–223. doi: 10.1007/s12031-017-1023-9
Lo, A. C., Callaerts-Vegh, Z., Nunes, A. F., Rodrigues, C. M. P., and D'Hooge, R. (2013). Tauroursodeoxycholic acid (TUDCA) supplementation prevents cognitive impairment and amyloid deposition in APP/PS1 mice. Neurobiol. Dis. 50, 21–29. doi: 10.1016/j.nbd.2012.09.003
Lu, Y., Sareddy, G. R., Wang, J., Zhang, Q., Tang, F.-L., Pratap, U. P., et al. (2020). Neuron-derived estrogen is critical for astrocyte activation and neuroprotection of the ischemic brain. J. Neurosci. 40, 7355–7374. doi: 10.1523/JNEUROSCI.0115-20.2020
Lu, X., Yang, R.-R., Zhang, J.-L., Wang, P., Gong, Y., Hu, W.-F., et al. (2018). Tauroursodeoxycholic acid produces antidepressant-like effects in a chronic unpredictable stress model of depression via attenuation of neuroinflammation, oxido-nitrosative stress, and endoplasmic reticulum stress. Fundam. Clin. Pharmacol. 32, 363–377. doi: 10.1111/fcp.12367
Luca, M., and Luca, A. (2019). Oxidative stress-related endothelial damage in vascular depression and vascular cognitive impairment: beneficial effects of aerobic physical exercise. Oxid. Med. Cell. Longev. 2019:8067045. doi: 10.1155/2019/8067045
Ma, Z., Li, J., Jiang, H., and Chu, Y. (2021). Expression of α-klotho is downregulated and associated with oxidative stress in the Lens in Streptozotocin-induced diabetic rats. Curr. Eye Res. 46, 482–489. doi: 10.1080/02713683.2020.1805768
Machado, M. F., Caixeta, E. S., Sudiman, J., Gilchrist, R. B., Thompson, J. G., Lima, P. F., et al. (2015). Fibroblast growth factor 17 and bone morphogenetic protein 15 enhance cumulus expansion and improve quality of in vitro-produced embryos in cattle. Theriogenology 84, 390–398. doi: 10.1016/j.theriogenology.2015.03.031
MahmoudianDehkordi, S., Arnold, M., Nho, K., Ahmad, S., Jia, W., Xie, G., et al. (2019). Altered bile acid profile associates with cognitive impairment in Alzheimer's disease-an emerging role for gut microbiome. Alzheimers Dement. 15, 76–92. doi: 10.1016/j.jalz.2018.07.217
Manduca, J. D., Thériault, R.-K., and Perreault, M. L. (2020). Glycogen synthase kinase-3: the missing link to aberrant circuit function in disorders of cognitive dysfunction? Pharmacol. Res. 157:104819. doi: 10.1016/j.phrs.2020.104819
Mano, N., Goto, T., Uchida, M., Nishimura, K., Ando, M., Kobayashi, N., et al. (2004). Presence of protein-bound unconjugated bile acids in the cytoplasmic fraction of rat brain. J. Lipid Res. 45, 295–300. doi: 10.1194/jlr.M300369-JLR200
Marcelin, G., Jo, Y.-H., Li, X., Schwartz, G. J., Zhang, Y., Dun, N. J., et al. (2014). Central action of FGF19 reduces hypothalamic AGRP/NPY neuron activity and improves glucose metabolism. Molec metab 3, 19–28. doi: 10.1016/j.molmet.2013.10.002
Mashayekhi, F., Hadavi, M., Vaziri, H. R., and Naji, M. (2010). Increased acidic fibroblast growth factor concentrations in the serum and cerebrospinal fluid of patients with Alzheimer's disease. J. Clin. Neurosci. 17, 357–359. doi: 10.1016/j.jocn.2009.05.037
Massó, A., Sánchez, A., Gimenez-Llort, L., Lizcano, J. M., Cañete, M., García, B., et al. (2015). Secreted and transmembrane αKlotho isoforms have different Spatio-temporal profiles in the brain during aging and Alzheimer's disease progression. PLoS One 10:e0143623. doi: 10.1371/journal.pone.0143623
McMillin, M., Frampton, G., Quinn, M., Divan, A., Grant, S., Patel, N., et al. (2015). Suppression of the HPA Axis during cholestasis can be attributed to hypothalamic bile acid signaling. Mol. Endocrinol. 29, 1720–1730. doi: 10.1210/me.2015-1087
Mergenthaler, P., Lindauer, U., Dienel, G. A., and Meisel, A. (2013). Sugar for the brain: the role of glucose in physiological and pathological brain function. Trends Neurosci. 36, 587–597. doi: 10.1016/j.tins.2013.07.001
Mertens, K. L., Kalsbeek, A., Soeters, M. R., and Eggink, H. M. (2017). Bile acid signaling pathways from the enterohepatic circulation to the central nervous system. Front. Neurosci. 11:617. doi: 10.3389/fnins.2017.00617
Miyamoto, N., Pham, L.-D. D., Seo, J. H., Kim, K.-W., Lo, E. H., and Arai, K. (2014). Crosstalk between cerebral endothelium and oligodendrocyte. Cell. Mol. Life Sci. 71, 1055–1066. doi: 10.1007/s00018-013-1488-9
Nakamura, S., Arima, K., Haga, S., Aizawa, T., Motoi, Y., Otsuka, M., et al. (1998). Fibroblast growth factor (FGF)-9 immunoreactivity in senile plaques. Brain Res. 814, 222–225.
Nakamura, M., Uehara, Y., Asada, M., Honda, E., Nagai, N., Kimata, K., et al. (2011). Sulfated glycosaminoglycans are required for specific and sensitive fibroblast growth factor (FGF) 19 signaling via FGF receptor 4 and betaKlotho. J. Biol. Chem. 286, 26418–26423. doi: 10.1074/jbc.M111.251140
Namba, T., Yabe, T., Gonda, Y., Ichikawa, N., Sanagi, T., Arikawa-Hirasawa, E., et al. (2010). Pigment epithelium-derived factor up-regulation induced by memantine, an N-methyl-D-aspartate receptor antagonist, is involved in increased proliferation of hippocampal progenitor cells. Neuroscience 167, 372–383. doi: 10.1016/j.neuroscience.2010.01.033
Nho, K., Kueider-Paisley, A., MahmoudianDehkordi, S., Arnold, M., Risacher, S. L., Louie, G., et al. (2019). Altered bile acid profile in mild cognitive impairment and Alzheimer's disease: relationship to neuroimaging and CSF biomarkers. Alzheimers Dement. 15, 232–244. doi: 10.1016/j.jalz.2018.08.012
Noda, M., Takii, K., Parajuli, B., Kawanokuchi, J., Sonobe, Y., Takeuchi, H., et al. (2014). FGF-2 released from degenerating neurons exerts microglial-induced neuroprotection via FGFR3-ERK signaling pathway. J. Neuroinflammation 11:76. doi: 10.1186/1742-2094-11-76
Numakawa, T., Nakajima, S., Yamamoto, N., Ooshima, Y., Odaka, H., Hashido, K., et al. (2015). Basic fibroblast growth factor induces miR-134 upregulation in astrocyte for cell maturation. Biochem. Biophys. Res. Commun. 456, 465–470. doi: 10.1016/j.bbrc.2014.11.108
Ogino, R., Murayama, N., Noshita, T., Takemoto, N., Toba, T., Oka, T., et al. (2014). SUN11602 has basic fibroblast growth factor-like activity and attenuates neuronal damage and cognitive deficits in a rat model of Alzheimer's disease induced by amyloid β and excitatory amino acids. Brain Res. 1585, 159–166. doi: 10.1016/j.brainres.2014.08.023
Owen, B. M., Mangelsdorf, D. J., and Kliewer, S. A. (2015). Tissue-specific actions of the metabolic hormones FGF15/19 and FGF21. Trends Endocrinol. Metab. 26, 22–29. doi: 10.1016/j.tem.2014.10.002
Pan, X., Elliott, C. T., McGuinness, B., Passmore, P., Kehoe, P. G., Hölscher, C., et al. (2017). Metabolomic profiling of bile acids in clinical and experimental samples of Alzheimer's disease. Metabolites 7:28. doi: 10.3390/metabo7020028
Papoutsi, M., Labuschagne, I., Tabrizi, S. J., and Stout, J. C. (2014). The cognitive burden in Huntington's disease: pathology, phenotype, and mechanisms of compensation. Mov. Disord. 29, 673–683. doi: 10.1002/mds.25864
Paroni, G., Panza, F., De Cosmo, S., Greco, A., Seripa, D., and Mazzoccoli, G. (2019). Klotho at the edge of Alzheimer's disease and senile depression. Mol. Neurobiol. 56, 1908–1920. doi: 10.1007/s12035-018-1200-z
Pereda-Pérez, I., Valencia, A., Baliyan, S., Núñez, Á., Sanz-García, A., Zamora, B., et al. (2019). Systemic administration of a fibroblast growth factor receptor 1 agonist rescues the cognitive deficit in aged socially isolated rats. Neurobiol. Aging 78, 155–165. doi: 10.1016/j.neurobiolaging.2019.02.011
Perry, R. J., Lee, S., Ma, L., Zhang, D., Schlessinger, J., and Shulman, G. I. (2015). FGF1 and FGF19 reverse diabetes by suppression of the hypothalamic-pituitary-adrenal axis. Nat. Commun. 6:6980. doi: 10.1038/ncomms7980
Popp, J., Wolfsgruber, S., Heuser, I., Peters, O., Hüll, M., Schröder, J., et al. (2015). Cerebrospinal fluid cortisol and clinical disease progression in MCI and dementia of Alzheimer's type. Neurobiol. Aging 36, 601–607. doi: 10.1016/j.neurobiolaging.2014.10.031
Rajendran, R., Rajendran, V., Giraldo-Velasquez, M., Megalofonou, F. F., Gurski, F., Stadelmann, C., et al. (2021). Oligodendrocyte-specific deletion of FGFR1 reduces cerebellar inflammation and neurodegeneration in MOG35-55-induced EAE. Int. J. Mol. Sci. 22:9495. doi: 10.3390/ijms22179495
Reuss, B., and von Bohlen und Halbach, O. (2003). Fibroblast growth factors and their receptors in the central nervous system. Cell Tissue Res. 313, 139–157. doi: 10.1007/s00441-003-0756-7
Ryan, K. K., Kohli, R., Gutierrez-Aguilar, R., Gaitonde, S. G., Woods, S. C., and Seeley, R. J. (2013). Fibroblast growth factor-19 action in the brain reduces food intake and body weight and improves glucose tolerance in male rats. Endocrinology 154, 9–15. doi: 10.1210/en.2012-1891
Rysz, J., Gluba-Brzózka, A., Mikhailidis, D. P., and Banach, M. (2015). Fibroblast growth factor 19-targeted therapies for the treatment of metabolic disease. Expert Opin. Investig. Drugs 24, 603–610. doi: 10.1517/13543784.2015.1006357
Sa-Nguanmoo, P., Tanajak, P., Kerdphoo, S., Satjaritanun, P., Wang, X., Liang, G., et al. (2016). FGF21 improves cognition by restored synaptic plasticity, dendritic spine density, brain mitochondrial function and cell apoptosis in obese-insulin resistant male rats. Horm. Behav. 85, 86–95. doi: 10.1016/j.yhbeh.2016.08.006
Sanyal, A., Charles, E. D., Neuschwander-Tetri, B. A., Loomba, R., Harrison, S. A., Abdelmalek, M. F., et al. (2019). Pegbelfermin (BMS-986036), a PEGylated fibroblast growth factor 21 analogue, in patients with non-alcoholic steatohepatitis: a randomised, double-blind, placebo-controlled, phase 2a trial. Lancet 392, 2705–2717. doi: 10.1016/S0140-6736(18)31785-9
Sathyan, S., Ayers, E., Gao, T., Weiss, E. F., Milman, S., Verghese, J., et al. (2020). Plasma proteomic profile of age, health span, and all-cause mortality in older adults. Aging Cell 19:e13250. doi: 10.1111/acel.13250
Scearce-Levie, K., Roberson, E. D., Gerstein, H., Cholfin, J. A., Mandiyan, V. S., Shah, N. M., et al. (2008). Abnormal social behaviors in mice lacking Fgf17. Genes Brain Behav. 7, 344–354. doi: 10.1111/j.1601-183X.2007.00357.x
Shen, X.-N., Niu, L.-D., Wang, Y.-J., Cao, X.-P., Liu, Q., Tan, L., et al. (2019). Inflammatory markers in Alzheimer's disease and mild cognitive impairment: a meta-analysis and systematic review of 170 studies. J. Neurol. Neurosurg. Psychiatry 90, 590–598. doi: 10.1136/jnnp-2018-319148
Skaper, S. D., Facci, L., Zusso, M., and Giusti, P. (2017). Synaptic plasticity, dementia and Alzheimer disease. CNS Neurol. Disord. Drug Targets 16, 220–233. doi: 10.2174/1871527316666170113120853
Stopa, E. G., Gonzalez, A. M., Chorsky, R., Corona, R. J., Alvarez, J., Bird, E. D., et al. (1990). Basic fibroblast growth factor in Alzheimer's disease. Biochem. Biophys. Res. Commun. 171, 690–696.
Subbiah, V., Iannotti, N. O., Gutierrez, M., Smith, D. C., Féliz, L., Lihou, C. F., et al. (2022). FIGHT-101, a first-in-human study of potent and selective FGFR 1-3 inhibitor pemigatinib in pan-cancer patients with FGF/FGFR alterations and advanced malignancies. Ann. Oncol. 33, 522–533. doi: 10.1016/j.annonc.2022.02.001
Suh, J. M., Jonker, J. W., Ahmadian, M., Goetz, R., Lackey, D., Osborn, O., et al. (2014). Endocrinization of FGF1 produces a neomorphic and potent insulin sensitizer. Nature 513, 436–439. doi: 10.1038/nature13540
Sun, J., Huang, X., Niu, C., Wang, X., Li, W., Liu, M., et al. (2021). aFGF alleviates diabetic endothelial dysfunction by decreasing oxidative stress via Wnt/β-catenin-mediated upregulation of HXK2. Redox Biol. 39:101811. doi: 10.1016/j.redox.2020.101811
Takami, K., Matsuo, A., Terai, K., Walker, D. G., McGeer, E. G., and McGeer, P. L. (1998). Fibroblast growth factor receptor-1 expression in the cortex and hippocampus in Alzheimer's disease. Brain Res. 802, 89–97.
Taliyan, R., Chandran, S. K., and Kakoty, V. (2019). Therapeutic approaches to Alzheimer's type of dementia: a focus on FGF21 mediated neuroprotection. Curr. Pharm. Des. 25, 2555–2568. doi: 10.2174/1381612825666190716101411
Tang, M.-M., Lin, W.-J., Zhang, J.-T., Zhao, Y.-W., and Li, Y.-C. (2017). Exogenous FGF2 reverses depressive-like behaviors and restores the suppressed FGF2-ERK1/2 signaling and the impaired hippocampal neurogenesis induced by neuroinflammation. Brain Behav. Immun. 66, 322–331. doi: 10.1016/j.bbi.2017.05.013
Tank, R., Ward, J., Celis-Morales, C., Smith, D. J., Flegal, K. E., and Lyall, D. M. (2021). Testing for interactions between APOE and klotho genotypes on cognitive, dementia, and brain imaging metrics in UK biobank. J. Alzheimers Dis. 83, 51–55. doi: 10.3233/JAD-210181
Tennakoon, A., Katharesan, V., Musgrave, I. F., Koblar, S. A., Faull, R. L. M., Curtis, M. A., et al. (2022). Normal aging, motor neurone disease, and Alzheimer's disease are characterized by cortical changes in inflammatory cytokines. J. Neurosci. Res. 100, 653–669. doi: 10.1002/jnr.24996
Tennant, K. G., Lindsley, S. R., Kirigiti, M. A., True, C., and Kievit, P. (2019). Central and peripheral Administration of Fibroblast Growth Factor 1 improves pancreatic islet insulin secretion in diabetic mouse models. Diabetes 68, 1462–1472. doi: 10.2337/db18-1175
Thorns, V., Licastro, F., and Masliah, E. (2001). Locally reduced levels of acidic FGF lead to decreased expression of 28-kda calbindin and contribute to the selective vulnerability of the neurons in the entorhinal cortex in Alzheimer's disease. Neuropathology 21, 203–211. doi: 10.1046/j.1440-1789.2001.00399.x
Thorns, V., and Masliah, E. (1999). Evidence for neuroprotective effects of acidic fibroblast growth factor in Alzheimer disease. J. Neuropathol. Exp. Neurol. 58, 296–306.
Tournissac, M., Bourassa, P., Martinez-Cano, R. D., Vu, T.-M., Hébert, S. S., Planel, E., et al. (2019). Repeated cold exposures protect a mouse model of Alzheimer's disease against cold-induced tau phosphorylation. Molec Metab 22, 110–120. doi: 10.1016/j.molmet.2019.01.008
Tsai, M. J., Tsai, S. K., Huang, M. C., Liou, D. Y., Huang, S. L., Hsieh, W. H., et al. (2015). Acidic FGF promotes neurite outgrowth of cortical neurons and improves neuroprotective effect in a cerebral ischemic rat model. Neuroscience 305, 238–247. doi: 10.1016/j.neuroscience.2015.07.074
Tyebji, S., and Hannan, A. J. (2017). Synaptopathic mechanisms of neurodegeneration and dementia: insights from Huntington's disease. Prog. Neurobiol. 153, 18–45. doi: 10.1016/j.pneurobio.2017.03.008
Ursem, S. R., Diepenbroek, C., Bacic, V., Unmehopa, U. A., Eggels, L., Maya-Monteiro, C. M., et al. (2021). Localization of fibroblast growth factor 23 protein in the rat hypothalamus. Eur. J. Neurosci. 54, 5261–5271. doi: 10.1111/ejn.15375
van Brummelen, E. M. J., Levchenko, E., Dómine, M., Fennell, D. A., Kindler, H. L., Viteri, S., et al. (2020). A phase Ib study of GSK3052230, an FGF ligand trap in combination with pemetrexed and cisplatin in patients with malignant pleural mesothelioma. Invest. New Drugs 38, 457–467. doi: 10.1007/s10637-019-00783-7
van der Flier, W. M., Skoog, I., Schneider, J. A., Pantoni, L., Mok, V., Chen, C. L. H., et al. (2018). Vascular cognitive impairment. Nat. Rev. Dis. Primers. 4:18003. doi: 10.1038/nrdp.2018.3
Vervloet, M. (2019). Renal and extrarenal effects of fibroblast growth factor 23. Nat. Rev. Nephrol. 15, 109–120. doi: 10.1038/s41581-018-0087-2
Wakayama, K., Shimamura, M., Yoshida, S., Hayashi, H., Ju, N., Nakagami, H., et al. (2021). Prevention of vascular dementia via immunotherapeutic blockade of renin-angiotensin system in a rat model. Brain Res. 1772:147667. doi: 10.1016/j.brainres.2021.147667
Wang, Z.-G., Cheng, Y., Yu, X.-C., Ye, L.-B., Xia, Q.-H., Johnson, N. R., et al. (2016). bFGF protects against blood-brain barrier damage through junction protein regulation via PI3K-Akt-Rac1 pathway following traumatic brain injury. Mol. Neurobiol. 53, 7298–7311. doi: 10.1007/s12035-015-9583-6
Wang, Q., Jie, W., Liu, J.-H., Yang, J.-M., and Gao, T.-M. (2017). An astroglial basis of major depressive disorder? An overview. Glia 65, 1227–1250. doi: 10.1002/glia.23143
Wang, S., Li, Y., Jiang, C., and Tian, H. (2018). Fibroblast growth factor 9 subfamily and the heart. Appl. Microbiol. Biotechnol. 102, 605–613. doi: 10.1007/s00253-017-8652-3
Wang, L. Y., Raskind, M. A., Wilkinson, C. W., Shofer, J. B., Sikkema, C., Szot, P., et al. (2018). Associations between CSF cortisol and CSF norepinephrine in cognitively normal controls and patients with amnestic MCI and AD dementia. Int. J. Geriatr. Psychiatry 33, 763–768. doi: 10.1002/gps.4856
Wang, X.-Q., Tang, Y.-H., Zeng, G.-R., Wu, L.-F., Zhou, Y.-J., Cheng, Z.-N., et al. (2021). Carnosic acid alleviates depression-like behaviors on chronic mild stressed mice via PPAR-γ-dependent regulation of ADPN/FGF9 pathway. Psychopharmacology (Berl) 238, 501–516. doi: 10.1007/s00213-020-05699-2
Wang, Q., Yuan, J., Yu, Z., Lin, L., Jiang, Y., Cao, Z., et al. (2018). FGF21 attenuates high-fat diet-induced cognitive impairment via metabolic regulation and anti-inflammation of obese mice. Mol. Neurobiol. 55, 4702–4717. doi: 10.1007/s12035-017-0663-7
Wang, X., Zhu, L., Hu, J., Guo, R., Ye, S., Liu, F., et al. (2020). FGF21 attenuated LPS-induced depressive-like behavior via inhibiting the inflammatory pathway. Front. Pharmacol. 11:154. doi: 10.3389/fphar.2020.00154
Wijesekara, N., Gonçalves, R. A., De Felice, F. G., and Fraser, P. E. (2018). Impaired peripheral glucose homeostasis and Alzheimer's disease. Neuropharmacology 136, 172–181. doi: 10.1016/j.neuropharm.2017.11.027
Wu, F. C. Z., Tang, C., Zhang, J., Cheng, L., Zuo, H., Zhang, H., et al. (2017). Acid fibroblast growth factor preserves blood-brain barrier integrity by activating the PI3K-Akt-Rac1 pathway and inhibiting RhoA following traumatic brain injury. Am. J. Transl. Res. 9, 910–925.
Xie, Y., Su, N., Yang, J., Tan, Q., Huang, S., Jin, M., et al. (2020). FGF/FGFR signaling in health and disease. Signal Transduct. Target. Ther. 5:181. doi: 10.1038/s41392-020-00222-7
Yamagata, H., Chen, Y., Akatsu, H., Kamino, K., Ito, J.-I., Yokoyama, S., et al. (2004). Promoter polymorphism in fibroblast growth factor 1 gene increases risk of definite Alzheimer's disease. Biochem. Biophys. Res. Commun. 321, 320–323. doi: 10.1016/j.bbrc.2004.06.142
Yi-Bin, W., Xiang, L., Bing, Y., Qi, Z., Fei-Tong, J., Minghong, W., et al. (2022). Inhibition of the CEBPβ-NFκB interaction by nanocarrier-packaged Carnosic acid ameliorates glia-mediated neuroinflammation and improves cognitive function in an Alzheimer's disease model. Cell Death Dis. 13:318. doi: 10.1038/s41419-022-04765-1
You, J., Sun, L., Wang, J., Sun, F., Wang, W., Wang, D., et al. (2021). Role of adiponectin-notch pathway in cognitive dysfunction associated with depression and in the therapeutic effect of physical exercise. Aging Cell 20:e13387. doi: 10.1111/acel.13387
Yuan, H., Ni, X., Zheng, M., Han, X., Song, Y., and Yu, M. (2019). Effect of catalpol on behavior and neurodevelopment in an ADHD rat model. Biomed. Pharmacother. 118:109033. doi: 10.1016/j.biopha.2019.109033
Yusuf, I. O., Chen, H.-M., Cheng, P.-H., Chang, C.-Y., Tsai, S.-J., Chuang, J.-I., et al. (2019). Fibroblast growth factor 9 activates anti-oxidative functions of Nrf2 through ERK signalling in striatal cell models of Huntington's disease. Free Radic. Biol. Med. 130, 256–266. doi: 10.1016/j.freeradbiomed.2018.10.455
Yusuf, I. O., Chen, H.-M., Cheng, P.-H., Chang, C.-Y., Tsai, S.-J., Chuang, J.-I., et al. (2021a). FGF9 induces neurite outgrowth upon ERK signaling in knock-in striatal Huntington's disease cells. Life Sci. 267:118952. doi: 10.1016/j.lfs.2020.118952
Yusuf, I. O., Chen, H.-M., Cheng, P.-H., Chang, C.-Y., Tsai, S.-J., Chuang, J.-I., et al. (2021b). Fibroblast growth factor 9 stimulates neuronal length through NF-kB signaling in striatal cell Huntington's disease models. Mol. Neurobiol. 58, 2396–2406. doi: 10.1007/s12035-020-02220-w
Zhang, P., Kishimoto, Y., Grammatikakis, I., Gottimukkala, K., Cutler, R. G., Zhang, S., et al. (2019). Senolytic therapy alleviates Aβ-associated oligodendrocyte progenitor cell senescence and cognitive deficits in an Alzheimer's disease model. Nat. Neurosci. 22, 719–728. doi: 10.1038/s41593-019-0372-9
Zhang, J., and Li, Y. (2016). Therapeutic uses of FGFs. Semin. Cell Dev. Biol. 53, 144–154. doi: 10.1016/j.semcdb.2015.09.007
Zhang, Y., Wang, L., Wu, Z., Yu, X., Du, X., and Li, X. (2017). The expressions of klotho family genes in human ocular tissues and in anterior Lens capsules of age-related cataract. Curr. Eye Res. 42, 871–875. doi: 10.1080/02713683.2016.1259421
Zhao, M., Li, D., Shimazu, K., Zhou, Y.-X., Lu, B., and Deng, C.-X. (2007). Fibroblast growth factor receptor-1 is required for long-term potentiation, memory consolidation, and neurogenesis. Biol. Psychiatry 62, 381–390. doi: 10.1016/j.biopsych.2006.10.019
Zhao, Y., Zeng, C.-Y., Li, X.-H., Yang, T.-T., Kuang, X., and Du, J.-R. (2020). Klotho overexpression improves amyloid-β clearance and cognition in the APP/PS1 mouse model of Alzheimer's disease. Aging Cell 19:e13239. doi: 10.1111/acel.13239
Zheng, K., Lin, L., Cui, P., Liu, T., Chen, L., Yang, C., et al. (2020). Association of Fibroblast Growth Factor 23 with ischemic stroke and its subtypes: a Mendelian randomization study. Front. Genet. 11:608517. doi: 10.3389/fgene.2020.608517
Zheng, W., Matei, N., Pang, J., Luo, X., Song, Z., Tang, J., et al. (2019). Delayed recanalization at 3 days after permanent MCAO attenuates neuronal apoptosis through FGF21/FGFR1/PI3K/Caspase-3 pathway in rats. Exp. Neurol. 320:113007. doi: 10.1016/j.expneurol.2019.113007
Zhou, X., Chen, K., Lei, H., and Sun, Z. (2015). Klotho gene deficiency causes salt-sensitive hypertension via monocyte chemotactic protein-1/CC chemokine receptor 2-mediated inflammation. J. Am. Soc. Nephrol. 26, 121–132. doi: 10.1681/ASN.2013101033
Zhu, B., Jin, L.-N., Shen, J.-Q., Liu, J.-F., Jiang, R.-Y., Yang, L., et al. (2018). Differential expression of serum biomarkers in hemodialysis patients with mild cognitive decline: a prospective single-center cohort study. Sci. Rep. 8:12250. doi: 10.1038/s41598-018-29760-5
Glossary
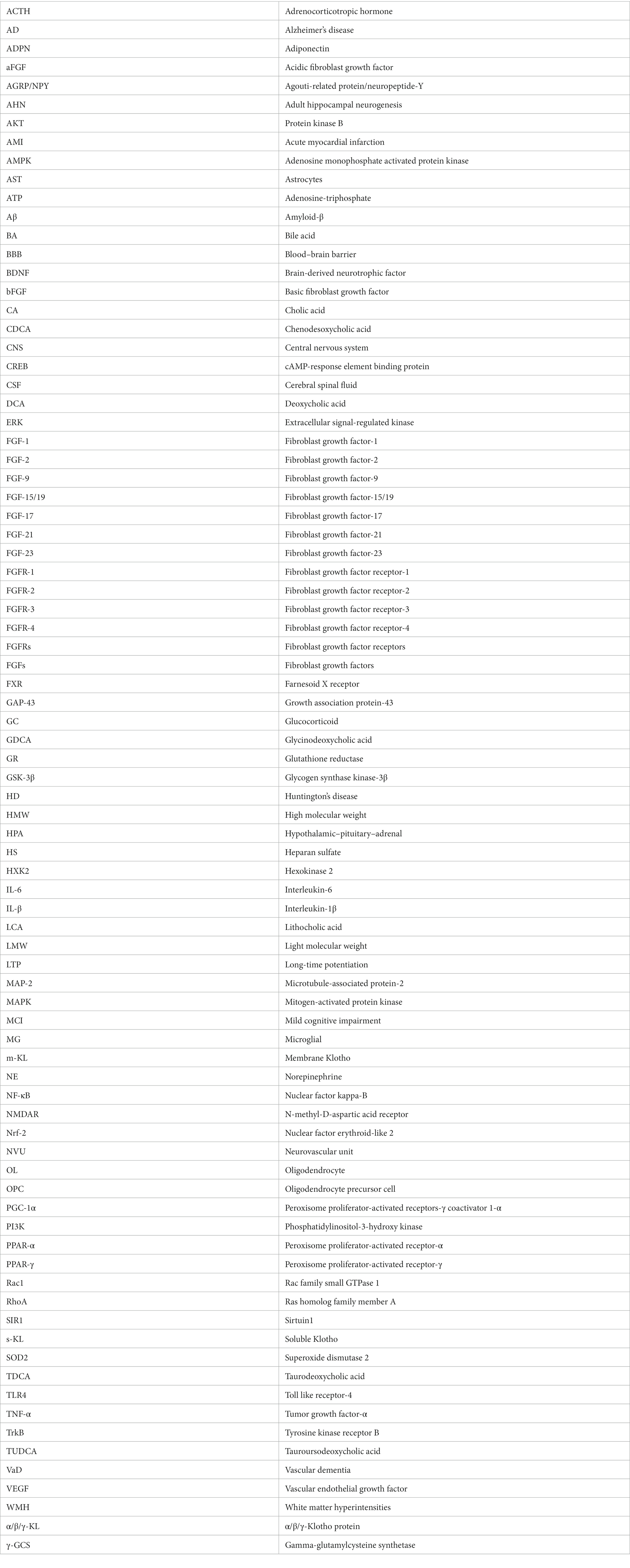
Keywords: fibroblast growth factor, fibroblast growth factor receptor, dementia, cognitive disorders, Alzheimer’s disease, neurodegenerative diseases
Citation: Zhai W, Zhang T, Jin Y, Huang S, Xu M and Pan J (2023) The fibroblast growth factor system in cognitive disorders and dementia. Front. Neurosci. 17:1136266. doi: 10.3389/fnins.2023.1136266
Edited by:
Jianxun Liu, China Academy of Chinese Medical Sciences, ChinaReviewed by:
Zequn Yin, Hefei University of Technology, ChinaKe-Yang Chen, Wenzhou Medical University, China
Copyright © 2023 Zhai, Zhang, Jin, Huang, Xu and Pan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shijing Huang, hsjgam2878@163.com; Manman Xu, xummjournal@163.com; Juhua Pan, journal5739@163.com
†These authors have contributed equally to this work and share first authorship