 Mahmoud B. Maina
Mahmoud B. Maina
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Neurosci. , 22 March 2023
Sec. Neurodegeneration
Volume 17 - 2023 | https://doi.org/10.3389/fnins.2023.1132670
This article is part of the Research Topic The Biochemistry of Amyloids in Neurodegenerative Diseases, Volume II View all 15 articles
Oxidative stress is a significant source of damage that accumulates during aging and contributes to Alzheimer’s disease (AD) pathogenesis. Oxidation of proteins can give rise to covalent links between adjacent tyrosines known as dityrosine (DiY) cross-linking, amongst other modifications, and this observation suggests that DiY could serve as a biomarker of accumulated oxidative stress over the lifespan. Many studies have focused on understanding the contribution of DiY to AD pathogenesis and have revealed that DiY crosslinks can be found in both Aβ and tau deposits – the two key proteins involved in the formation of amyloid plaques and tau tangles, respectively. However, there is no consensus yet in the field on the impact of DiY on Aβ and tau function, aggregation, and toxicity. Here we review the current understanding of the role of DiY on Aβ and tau gathered over the last 20 years since the first observation, and discuss the effect of this modification for Aβ and tau aggregation, and its potential as a biomarker for AD.
Dityrosine (DiY) is a covalent cross-link formed by ortho-ortho coupling between two tyrosine residues between carbons in the phenol ring (Figure 1). The general mechanism of its formation begins with the generation of a tyrosyl radical generated due to the removal of the hydrogen atom from the hydroxyl group on the phenoxy ring (Atwood et al., 2004; DiMarco and Giulivi, 2007). The two tyrosyl radicals then undergo radical isomerization followed by diradical coupling, and finally enolization resulting in DiY (Correia et al., 2012). The tyrosyl radical also has the capacity to react with another tyrosine to form tri-tyrosine and other high-order structures (Partlow et al., 2016).
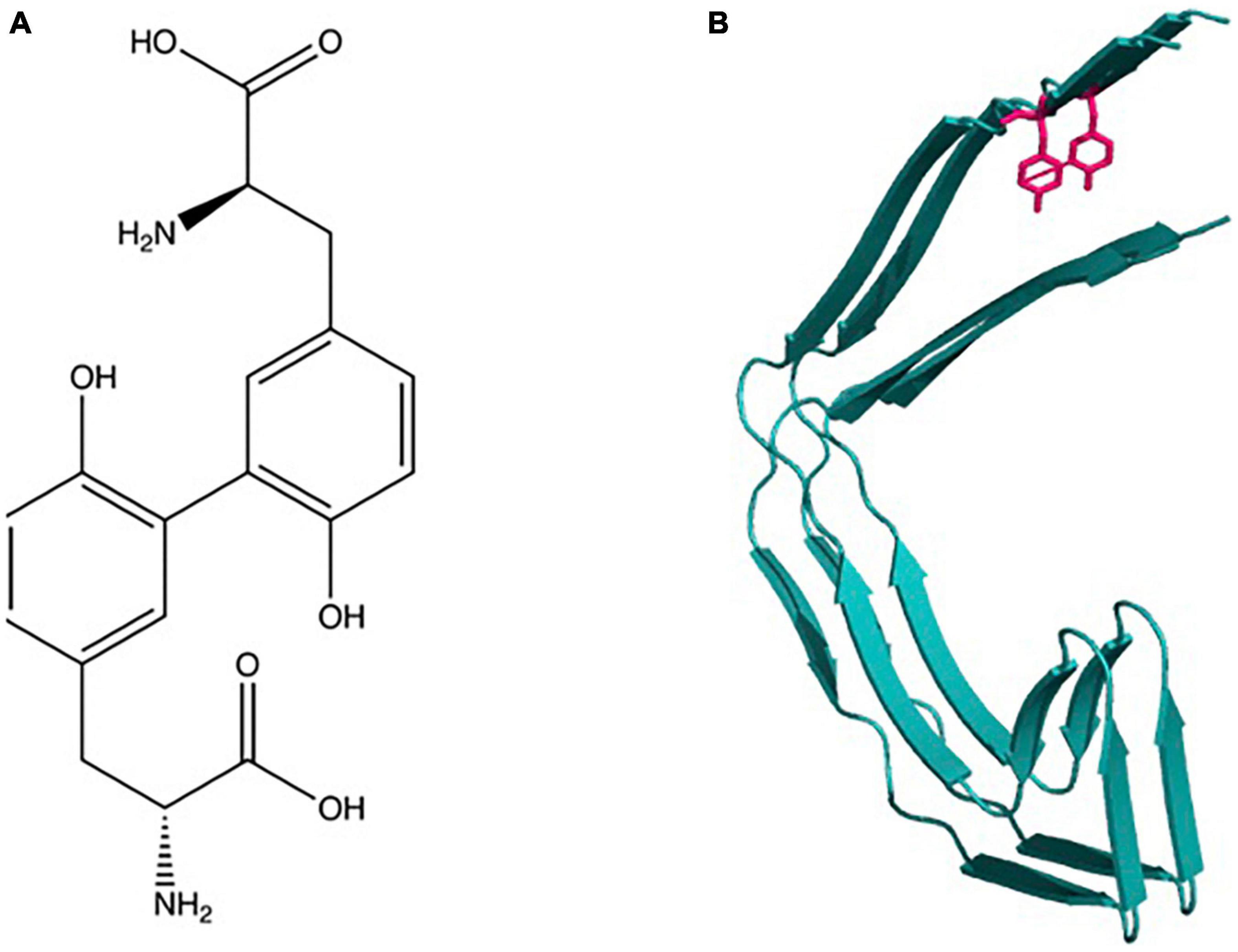
Figure 1. Dityrosine: (A) Chemical structure and molecular formula of dityrosine produced from two tyrosine amino acids. (B) Schematic shows depiction of crosslinking of two β-sheet rich protein molecules.
Dityrosine was initially recognized by Gross and Sizer (1959) generated by peroxidase oxidation of tyrosine in the presence of H2O2. Previous work had established that peroxidase enzymes could catalyze the oxidation of phenols and aromatic amines by hydrogen peroxide (Elliott, 1932). Later, it was revealed that the diphenyl formation mechanism involved free radical generation as intermediates (Waters, 1952). Based on these observations, Gross and Sizer (1959) suggested that DiY formation was achieved through the generation of tyrosyl radical as an intermediate. Several studies revealed a native role of DiY in natural elastic materials and invertebrate tissues. DiY has been identified within resilin, the rubber-like protein found in arthropods (Andersen, 1964). This was the first documentation of the natural occurrence of this cross-linker in proteins and it was suggested to stabilize resilin through the formation of a stable three-dimensional network (Andersen, 1964). It was subsequently reported that DiY cross-links occur naturally in several elastic and structural proteins, including elastin, fibroin, keratin, cuticlin, and collagen (LaBella et al., 1967; Raven et al., 1971; Fujimoto, 1975; Waykole and Heidemann, 1976). In these proteins, DiY cross-links can contribute to increased mechanical strength and insolubility (Skaff et al., 2005).
Dityrosine has been shown to play a protective or stabilizing role in many proteins (Bailey, 1991; Kanwar and Balasubramanian, 1999). DiY has been found in fungal cell wall proteins (Smail et al., 1995) and in the fertilization envelope of the sea urchin egg where it may regulate the production of a hard fertilization membrane that blocks the entry of additional sperm (Foerder and Shapiro, 1977). In the large roundworm of the pig (Ascaris suum) it forms part of the structural components of the cuticle (Fetterer et al., 1993), and is involved in hardening of the mosquito egg chorion (Li et al., 1996). DiY has been suggested to contribute to spores’ resistance to lytic enzymes (Briza et al., 1990) and has been suggested to be involved in the biosynthesis of thyroxine and melanin (Bayse et al., 1972). For a detailed review of DiY in natural materials and mammalian tissues, the reader is referred to Partlow et al. (2016).
Oxidative environments can lead to the cross-linking of other amino acids as well as tyrosine and have been suggested to be associated with biological dysfunction. These include cross-links that form between Cys and Tyr, Trp, Lys, Ser, Phe; between Tyr and Gly, His, Trp and between His and His, Arg, Lys, and finally Trp-Trp. The most commonly reported cross-link in proteins is DiY (see Fuentes-Lemus et al., 2021 for extensive review of detailed chemical mechanisms). In this review, we focus on the literature relating to DiY formation in Alzheimer’s disease (AD) proteins Amyloid-β and Tau.
Alzheimer’s disease, Parkinson’s disease (PD), and other neurodegenerative diseases are classified as misfolding diseases reflecting the characteristic amyloid fibril pathology. Each disease is characterized by one or more proteins that form amyloid fibrils and in the case of Alzheimer’s disease, Aβ and Tau fibrils accumulate extracellularly and intracellularly, respectively. DiY cross-links in proteins have also been implicated in many diseases, including AD and PD (Souza et al., 2000; Atwood et al., 2004; Al-Hilaly et al., 2013, 2016, 2019), cystic fibrosis (Van Der Vliet et al., 2000), atherosclerosis (Leeuwenburgh et al., 1996), cataracts in the eye lens (Bodaness and Zigler, 1983; Wells-Knecht et al., 1993),, and acute myocardial infarction (Mayer et al., 2014). For example, DiY can be generated in vitro in samples of Aβ (Galeazzi et al., 1999; Atwood et al., 2004; Al-Hilaly et al., 2013; Maina et al., 2020b), α-synuclein (Souza et al., 2000; Al-Hilaly et al., 2016), and Tau (Reynolds et al., 2005; Maina et al., 2021, 2022b). However, there remains more to learn about the specific role played by this modification in these aggregation-prone proteins. Here, we review the studies that have explored the potential roles of DiY on Aβ, and Tau in the pathogenesis of AD.
In his 1907 article describing the disease, Alzheimer’s post-mortem analysis of Auguste Deter, revealed intracellular neurofibrillary tangles and “minute miliary foci” deposited extracellularly as amyloid plaques (Alzheimer et al., 1995). This finding triggered a strong interest in understanding the biochemistry of these plaques and tangles, leading to the discovery that the plaques are predominantly made up of an amino acid peptide of about 40–42 residues and 4.2 kDa, now called Aβ (Glenner and Wong, 1984). The tangles were shown to be comprised of a hyperphosphorylated form of the microtubule-associated protein tau (Grundke-Iqbal et al., 1986a; Wood et al., 1986). Aβ peptide is synthesized from the processing of a single-pass integral membrane protein called amyloid precursor protein (APP) encoded by a gene located on chromosome 21, which has 18 exons, of which, exon 16 and 17 encode the Aβ peptide (Yoshikai et al., 1990). The processing of APP by β secretase 1 or β-site APP cleaving enzyme 1 (BACE 1) and γ-secretase (comprised of a complex including presenilin 1 and 2) leads to the generation of species of Aβ peptide (Figure 2).
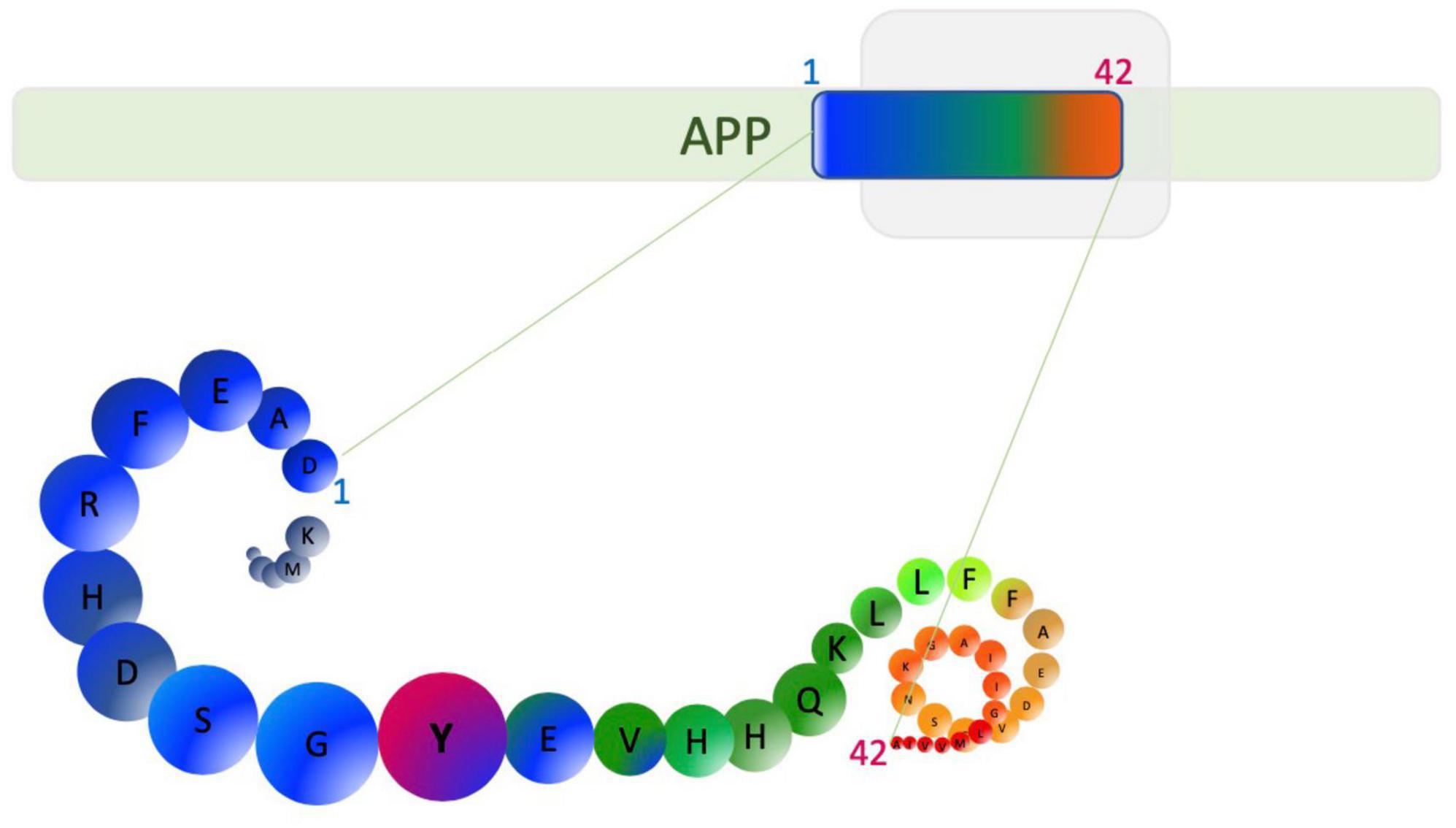
Figure 2. Aβ1–42 generation from amyloid precursor protein (APP). β-secretase cleavage beside aspartic acid and cleavage by γ-secretase can generate Aβ1–42 as well as other alternative length Aβ peptides. Amino acids are depicted as circles. Tyrosine (Y10) is able to form dityrosine cross-links and is highlighted in pink.
The important role played by Aβ in AD pathogenesis led to the amyloid cascade hypothesis (Hardy and Higgins, 1992), based on the observation that early onset, familial forms of AD are associated with mutations in APP or presenilin 1 or 2 and that all appear to be associated with Aβ generation, deposition, or aggregation propensity (Levy et al., 1990; Van Broeckhoven et al., 1990; Chartier-Harlin et al., 1991; Goate et al., 1991; Hardy, 1991). The updated amyloid cascade hypothesis suggests that aggregation of Aβ from monomers to dimers, oligomers, fibrils, and eventually to plaques subsequently drives downstream changes, such as tau phosphorylation, cell loss, and dementia (Selkoe and Hardy, 2016). The amyloid cascade hypothesis has been supported by biomarker studies which show that changes in CSF levels of Aβ and its deposition into plaques appear decades before the onset of dementia (Jack et al., 2013).
Many pieces of evidence have subsequently shown that the oligomeric form of Aβ is the most toxic species, not fibrils, even though a consensus is lacking about the exact structure and composition of these soluble species (Benilova et al., 2012). Accumulated evidence shows that Aβ oligomers disrupt cellular function in cultured cells and animal models (Lambert et al., 1998; Lacor et al., 2007; Reddy and Beal, 2008; Wu et al., 2010; Li et al., 2011; Soura et al., 2012; Zhang et al., 2014; Fuchsberger et al., 2016; Marshall et al., 2016, 2020; Selkoe and Hardy, 2016; Biasetti et al., 2023). In the double-transgenic APPswe-Tau mouse, neuronal loss and activated astrocytes in the entorhinal cortex and the CA1 hippocampal subfield were found to correlate with the burden of Aβ oligomers (DaRocha-Souto et al., 2011). In human AD, soluble Aβ also correlates positively with the severity of dementia (McLean et al., 1999; Walsh and Selkoe, 2007). With the onset of the accumulation of Aβ oligomers, the novel AD mouse model—PS1V97L-Tg expressing the human PS gene with the V97L mutation, show synaptic alteration, tau hyperphosphorylation, and glial activation, hence supporting an early role for this Aβ species and their role in neurotoxicity (Zhang et al., 2014). Aβ oligomers alone have been shown to impair learning and memory in the pond snail Lymnaea stagnalis (Marshall et al., 2016; Ford et al., 2017). Collectively, these studies support the deleterious role of soluble Aβ, rather than fibrils (Zhu et al., 2011; Selkoe and Hardy, 2016). The role of the insoluble fibrils and plaques remains debated but it has been suggested that they may contain around them an equilibrium of both toxic oligomers and inert fibrils which may “spillover” to surrounding tissues to cause neuronal damage, and/or they may mediate toxicity by triggering neuroinflammation (Benilova et al., 2012).
Amyloid beta is known to become increasingly stable and insoluble as it self-assembles and is deposited in amyloid plaques in brain tissue that are highly protease resistant (Kheterpal et al., 2001). Aβ oligomers and fibrils formed in vitro and extracted from tissue show SDS-stability and resistance to proteolytic degradation (Wang et al., 1999; Walsh and Selkoe, 2007; Rambaran and Serpell, 2008; Shankar et al., 2008; Masters and Selkoe, 2012). However, such SDS resistance could arise from a number of different modifications, and/or experimental artifacts (Jomova et al., 2010; Sitkiewicz et al., 2014). The stability of the Aβ assemblies could arise from either the formation of a strong ionic complex or the formation of covalent cross-links, which may include transglutaminase (TGase)-induced cross-linking between the glutamyl side chains and the ε-amino groups of lysine; cross-linking generated by 4-hydroxynonenal (4-HNE) and DiY cross-linking (Siegel et al., 2007; Wilhelmus et al., 2009; Roberts et al., 2012; Al-Hilaly et al., 2013; Sitkiewicz et al., 2014).
Multiple studies suggest that DiY formation through Tyrosine (Y10) of Aβ could provide the observed stability of Aβ. DiY cross-linked Aβ has been found in the AD brain (de la Torre et al., 2018), around Aβ plaques and shown to occur in vitro (Al-Hilaly et al., 2013). We have also demonstrated that DiY cross-linked Aβ could be internalized into cells and found around amyloid plaques in the human AD brain, suggesting it may play a role in disease pathogenesis or as a marker of the disease process (Al-Hilaly et al., 2013). To understand the specific contribution of DiY cross-linking on Aβ and its contribution to AD, several studies have studied the influence of this cross-linking on Aβ assembly. In vitro, DiY formation has been primarily generated via metal-catalyzed oxidation (MCO), photooxidation, and enzyme-catalyzed mechanisms (Partlow et al., 2016). To induce DiY, copper has been mostly employed for MCO, ultra-violet light (UV) for photooxidation, and peroxidase for enzyme-catalyzed reactions (Yoburn et al., 2003; Atwood et al., 2004; Maina et al., 2020b). Early work from Galeazzi et al. (1999) showed that in vitro oxidation using H2O2/peroxidase induces DiY cross-linking on Aβ42 and suggested that this would promote Aβ aggregation in AD. This was the first evidence to suggest that DiY formation may play a role in stabilizing Aβ assemblies in AD.
Among the many processes that could lead to the formation of DiY, transition metal catalysis is particularly very relevant in the context of AD (Yoburn et al., 2003; Atwood et al., 2004). About 400 μM copper, 100 μM zinc, and 100 μM iron have been found around amyloid plaques, suggesting increased concentrations of these metals could be implicated in the pathogenesis of AD (Lovell et al., 1998; Suh et al., 2000; Bush, 2003). Many studies have demonstrated that copper Cu2+ ions coordinate Aβ via the three histidine residues; His6, His13, His14, and Tyr10 (Curtain et al., 2001; Atwood et al., 2004; Tickler et al., 2005). Coordination of Cu2+ ions to Aβ at these residues places Y10 in close proximity to the redox-active copper ion (Sarell et al., 2009; Viles, 2012; Gu et al., 2018). The Y10 has been shown to play a critical role in facilitating Aβ/Cu mediated H2O2 production (Barnham et al., 2004). Thus, it has been suggested that copper interactions with Aβ could be responsible for causing DiY cross-linking. This would be facilitated by the presence of H2O2 in the milieu which can be produced through Aβ’s ability to reduce Cu2+ (Yoburn et al., 2003; Atwood et al., 2004; Barnham et al., 2004). Indeed, Barnham et al. (2004) revealed that Aβ peptide coordinates Cu2+ to form an Aβ-Cu2+ complex, which in turn leads to the production of H2O2 catalytically in the presence of a reducing substrate such as ascorbate and through this process, tyrosine radicals are generated that are later coupled and result in Aβ aggregation. Atwood et al. (2004) showed that the incubation of Aβ with Cu2+ at a concentration lower than that in amyloid plaques led to the formation DiY cross-links on Aβ and the appearance of SDS-resistance Aβ oligomers, which is a characteristic feature of the neurotoxic Aβ extracted from the AD brain (Walsh and Selkoe, 2007). The authors also revealed that the addition of H2O2 to the reaction significantly promoted DiY formation compared to incubation with Cu2+ only, indicating that DiY formation becomes more favorable in an increased oxidative environment (Atwood et al., 2004). Smith et al. (2006) also demonstrated that the generation of the Aβ toxic species is modulated by both the Cu2+ concentration and the ability to form intermolecular histidine bridges.
There is an extensive literature on the impact of DiY cross-linking on Aβ assembly (Urbanc, 2021). Most of the early studies link DiY formation with Aβ aggregation (Galeazzi et al., 1999; Yoburn et al., 2003; Atwood et al., 2004; Zhang et al., 2017) or serves as a seed that promotes further aggregation (Ono et al., 2009). However, in the early in vitro studies in which DiY formation on Aβ was established, the aggregation of Aβ was not followed over time, especially using techniques like Thioflavine T(Th-T) fluorescence assay, to study the typical aggregation kinetics of Aβ. Collectively, recent literature suggests that DiY formation significantly stabilizes Aβ assemblies (Williams et al., 2016; Maina et al., 2020b), slows Aβ aggregation to fibrils (Al-Hilaly et al., 2013; Kok et al., 2013; O’Malley et al., 2014, 2016), or inhibits assembly into fibrils (Gu et al., 2018; Vázquez et al., 2019; Maina et al., 2020b). Some studies have linked DiY cross-linked Aβ with toxicity (Barnham et al., 2004; O’Malley et al., 2014; Williams et al., 2016) and others have shown that DiY inhibits self-assembly, and as a result, reduces or prevents Aβ toxicity (Maina et al., 2020b). To fully understand the exact roles of DiY on Aβ, it is thus important to consider the preparation method, buffer and temperature conditions, and Aβ sequence carefully since environmental conditions will have a significant effect on the eventual structures and the formation of any intermediate species (Table 1).

Table 1. Different oxidation conditions used in producing dityrosine in Aβ.
Based on these studies (Table 1), it is difficult to compare the levels of DiY produced by the different conditions reported in the literature. This may partly be responsible for the difference in the outcome of DiY cross-linking from the different studies. It is also likely that the dosage of DiY formed per condition would be critical for the assembly of Aβ. This may explain why in some studies, the DiY cross-linked Aβ assembled to fibrils, albeit in a significantly slower fashion (Kok et al., 2013; O’Malley et al., 2014). While in other studies, DiY cross-linking completely inhibited the formation of such higher assemblies (Gu et al., 2018; Maina et al., 2020b).
Moreover, the cross-linking timing may be another factor that would impact the assembly of Aβ. Indeed, it was suggested that DiY cross-linking inhibits primary nucleation and subsequently retards fibril elongation and fibril–fibril interactions (O’Malley et al., 2014). We also showed that DiY cross-linking before the onset of aggregation leads to the rapid self-association of the random-coil rich monomeric assemblies of Aβ into stable amorphous-like structures that never assemble into amyloid fibrils (Maina et al., 2020b). Cross-linking after the onset of aggregation also significantly inhibited Aβ elongations into fibrils and stabilized the intermediate Aβ species (Maina et al., 2020b). Based on our work, it appears like the self-association of Aβ assemblies caused by the DiY cross-linking depletes the pool of available monomers that are needed for the primary and secondary nucleation.
The available data in the literature is yet to clearly define whether DiY cross-linking on Aβ leads to a deleterious effect. This is specifically reflected in the data on toxicity experiments with DiY cross-linked Aβ (Table 1). Some of the studies that suggested that DiY cross-linked Aβ42 dimers and oligomers were toxic did not fully characterize the self-assembly property of the Aβ (Barnham et al., 2004; Davis et al., 2011). Other studies have shown that dimers, trimers, and tetramers stabilized by Aβ oxidation using photo-induced cross-linking of unmodified proteins (PICUP) are toxic to cultured cells (Ono et al., 2009; Jana et al., 2016; Cline et al., 2019). Work from the Walsh group suggested that the toxicity of the DiY cross-linked assemblies was not due to the dimers but the higher assemblies formed by the dimers (O’Malley et al., 2014). Our recent series of work has made a strong case for self-assembly process as a critical driver of Aβ toxicity (Marshall et al., 2016, 2020; Vadukul et al., 2017, 2020; Maina et al., 2020b). The DiY cross-linked dimers reported by O’Malley et al. (2014, 2016) retained their self-assembly properties, albeit at a slow rate. These DiY cross-linked dimers were reported to self-associate to form larger structures and generate aggregates that potently block LTP (O’Malley et al., 2014). In another study, it was shown that toxic DiY cross-linked Aβ assemblies retain their self-assembling property, but at a significantly reduced speed that results in the formation of long-lived, soluble oligomeric aggregates (Kok et al., 2013). This has led to the assumption that DiY cross-linking may stabilize toxic oligomers and prolong their toxicity (Kok et al., 2013; O’Malley et al., 2016; Williams et al., 2016). However, our recent work revealed that DiY cross-linking trapping of Aβ oligomers, a mixture of oligomers and protofibrils and fibrils in a non-assembling conformation renders these assemblies non-toxic to differentiated neuroblastoma cells (Maina et al., 2020b). While non-oxidized Aβ was more toxic during its self-assembling (oligomeric) state compared to when the assembly plateaus (Maina et al., 2020b). We have previously designed a variant form of Aβ that lacks the propensity to aggregate due to F19S and G37D substitutions in Aβ42 (Marshall et al., 2016). Interestingly, the DiY cross-linked non-assembling variant Aβ showed no toxicity to cells (Maina et al., 2020b). Our work suggested that the self-assembly process, not individual Aβ assemblies or DiY cross-linking was responsible for the Aβ toxicity. If DiY Aβ is genuinely toxic, then it may depend on the DiY dose. However, none of the previous studies that evaluated DiY Aβ toxicity except (Maina et al., 2020b) quantified the level of DiY formed on Aβ. As a result, discrepancies may also arise from the differences in the amount of Aβ DiY cross-links used in toxicity assays.
At this stage, it is difficult to conclude the exact role of DiY cross-linking on Aβ aggregation or toxicity in a physiological environment and clearly more studies are needed. This raises the need for maintaining a closely similar method of preparations for Aβ experiments, as the method of peptide preparation, peptide concentration, time and aggregation, and model system used may play a huge role in determining the role of DiY on Aβ toxicity (Cecchi et al., 2008; Krishtal et al., 2015; Jana et al., 2016; Kaniyappan et al., 2017).
On the other hand, DiY cross-linking on existing fibrils might stabilize and help to maintain insoluble Aβ species found in plaques in the AD brain (Wang et al., 1999; Rambaran and Serpell, 2008; Masters and Selkoe, 2012). We have shown previously that DiY antibody colocalizes with Aβ fibrils in human AD plaques (Al-Hilaly et al., 2013). Overall, more work is needed to fully understand the role of DiY on Aβ species in vitro and in vivo. However, it is also worth noting that many modifications may occur on Aβ e.g., oxidation of histidine, lysine, and methionine35 of Aβ (Palmblad et al., 2002; Kowalik-Jankowska et al., 2004; Ali et al., 2005), which may also affect the Aβ differently (Smith et al., 2007a). Ultimately, studies that define these different modifications and specifically isolate the contribution of DiY would provide the much-needed data to support the interpretation of the studies generated thus far in the literature.
Tau is a small molecular weight protein with the capacity to promote microtubule assembly in vitro (Weingarten et al., 1975). It is found in both neuronal and non-neuronal eukaryotic cells, but predominantly in neurons (Loomis et al., 1990; Stoothoff and Johnson, 2005; Rossi et al., 2008; Martin et al., 2011). Tau is well-known for its role in stabilizing the microtubules through their nucleation and elongation (Cleveland et al., 1977). However, recent evidence suggest that tau is a multifunctional protein, playing a role in many cellular compartments, including the synapse and nucleus (Buée et al., 2000; Dixit et al., 2008; Ittner et al., 2010; Martin et al., 2011; Bukar Maina et al., 2016; Maina et al., 2018a,b). Tau is a product of the microtubule-associated protein gene, located on chromosome 17q21.1 (Neve et al., 1986; Andreadis et al., 1992; Andreadis, 2005; Figure 3). A complex post-transcriptional processing of the tau transcript results in a less abundant 2kb tau transcript which encodes for a tau mainly targeted to the nucleus (Wang et al., 1993); 6kb transcript which tau predominantly directed to the soma/axons in the central nervous system (CNS) (Andreadis, 2005; Liu and Götz, 2013); and 8/9 kb transcript producing a tau preferentially expressed in the retina and peripheral nervous system (PNS) and with an apparent molecular weight of about 110–120 kDa, often called high molecular weight tau (Georgieff et al., 1993; Nunez and Fischer, 1997).
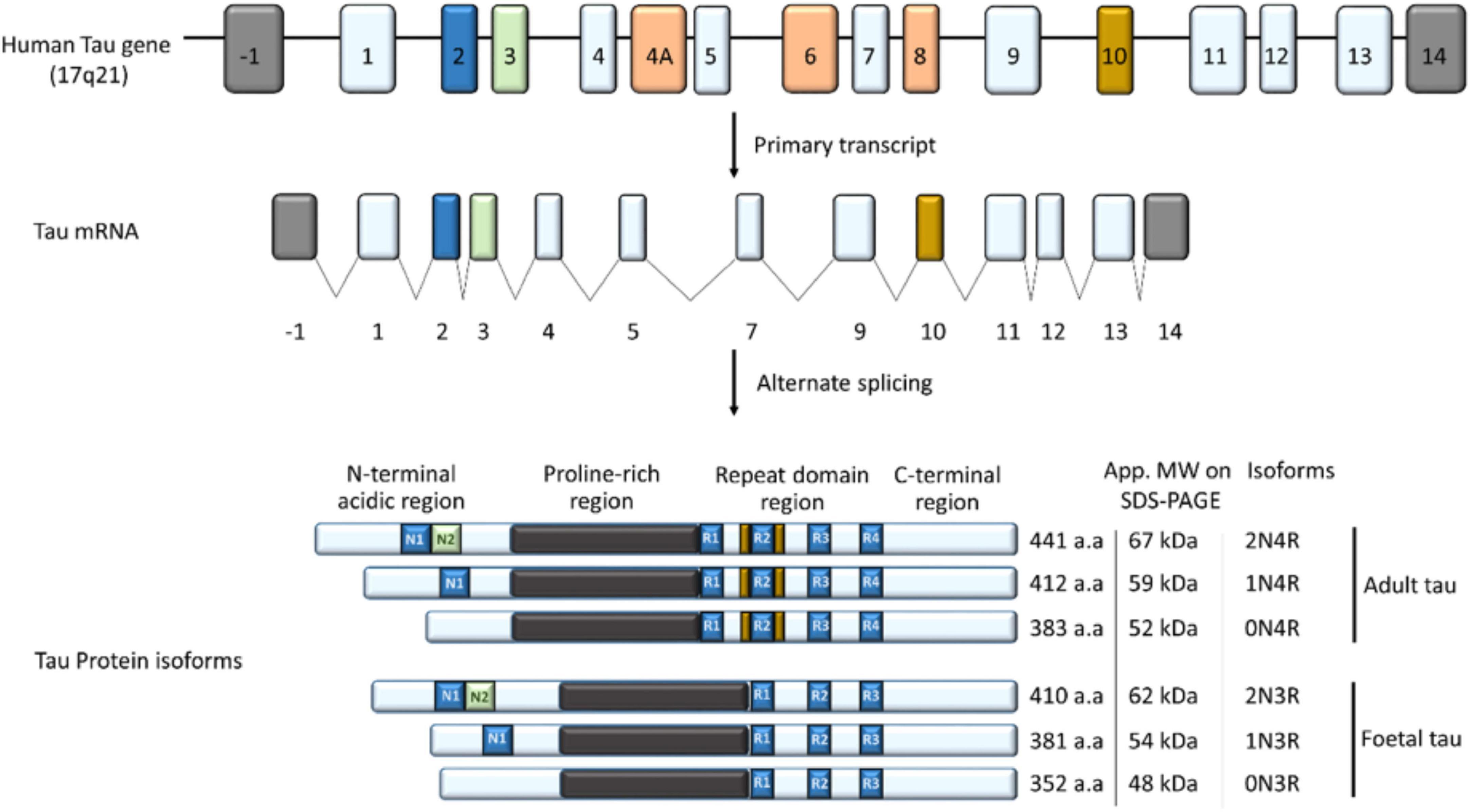
Figure 3. The tau gene has 16 exons; exon 1, 4, 5, 7, 9, 11, 12, and 13 (light blue) are constitutively transcribed in the CNS (Martin et al., 2011). Exon 4A, 6, and 8 (orange) are rarely expressed in the brain but included in mRNA of most peripheral tissues, while exon 14 forms part of the 3′ untranslated region of the tau mRNA (Andreadis, 2005; Connell et al., 2005). Alternate splicing of exon 2 (blue), 3 (Green), and 10 (Yellow) in the CNS generates the widely known six isoforms of tau; 352–441 amino acids in length and 48–67 kDa on SDS-PAGE (Martin et al., 2011). Depending on the inclusion and/or exclusion of exon 2, 3, and 10, tau has zero, one or two (0/1/2) N-terminal inserts and three or four (3R/4R) microtubule binding repeats, leading to the six isoforms of tau in the CNS. Structurally, tau is subdivided into an N-terminal acidic region; proline-rich region/domain (PRD), repeat domain region, and a C-terminal region.
The alternate splicing of exon 2, 3, and 10 of the Tau gene generates the six major isoforms of tau in the CNS with three (3R) or four (4R) microtubule-binding repeats on its C-terminal (3R) (Figure 3; Buée et al., 2000; Martin et al., 2011). Structurally, tau is subdivided into four regions; an N-terminal acidic region, a proline-rich domain (PRD), microtubule-binding repeat domain region (MBD), and a C-terminal region, and the epitopes across these areas vary depending on the tau isoform (Buée et al., 2000; Martin et al., 2011; Figure 3).
Physiologically, Tau is a monomeric protein that exists in solution in a random-coil conformation (Schweers et al., 1994). Thus, its aggregation is believed to be a sign of pathology thought to be driven by changes in its conformational state, moving from a random coil to the amyloid cross-β structure (von Bergen et al., 2005). This pathological folding of Tau is evident in a group of diseases collectively called tauopathies, which include AD, Pick’s disease, Huntington’s disease, and Fronto-Temporal Dementia with Parkinsonism linked to Chromosome 17 with MAPT mutations (FTDP-17T) (Oakley et al., 2020). For example, in AD, tau misfolds into paired helical filaments (PHFs) and straight filaments (SFs) and these filaments accumulate in the cell bodies of neurons as neurofibrillary tangles (NFT) (Grundke-Iqbal et al., 1986b; Wischik et al., 1988b). NFT burden correlates with the extent of AD pathology (Arriagada et al., 1992) and provides a reliable staging of the disease process (Braak and Braak, 1991). The precise mechanism involved in tau filament formation is still not fully understood. However, it involves the aggregation of Tau monomers to dimers, oligomers, some of which convert to fibrils, and eventually to PHFs and SFs (Cowan and Mudher, 2013). Many post-translational modifications have been proposed as key molecular events in the abnormal tau aggregation leading to the formation of PHFs. Tau can undergo glycosylation, glycation, prolyl-isomerization, polyamination, nitration, oxidation, ubiquitination, sumoylation phosphorylation (Martin et al., 2011; Guo et al., 2017), and truncation (Wischik et al., 1988b; Wang et al., 2007).
Interest in understanding Tau physiology and pathology has resulted in the development of many in vitro models over the past decades. These models include the full-length Tau (T40, 2N4R), dGAE (Wischik et al., 1988b), K18, and K19 (Mukrasch et al., 2005; Oakley et al., 2020). The T40 tau which has 441 amino acid residues contains five tyrosine residues, located at positions 18, 29, 197, 310, and 394. However, the model peptides, dGAE, K18, and K19 contain only the tyrosine residue at position 310. Y310 is part of the aggregation prone hexapeptide 306VQIVYK311 which is thought to be important for Tau assembly in vitro (von Bergen et al., 2000; Ganguly et al., 2015). Unlike Aβ, there is a significant lack of research on the role of DiY formation on tau and its contribution to tau pathology. This raises the question as to whether the conclusions drawn from research using Aβ also apply to Tau. Few of the early studies in this area were from the Binder’s laboratory on T40 tau, which represents the full-length tau carrying five tyrosine residues. Specifically, this showed that peroxynitrite oxidation results in the oligomerization of soluble T40 tau via the cross-linking of the tyrosine residues independent of disulfide bonds (Reynolds et al., 2005). However, it was not clear whether DiY formed on tau filaments and the consequence on its aggregation or stability. Tau filaments are usually induced in vitro with the help of heparin, arachidonic acid, or other polyanionic molecules (Goedert et al., 1996; Kampers et al., 1996; Pérez et al., 1996; Wilson and Binder, 1997; Friedhoff et al., 1998). Thus, the same group further showed that oxidative stress induced by peroxynitrite on arachidonic acid-induced filaments of T40 tau results in the formation of DiY on the tau filaments. Their work showed that the DiY cross-linking is associated with the stabilization of the pre-formed tau filaments (Reynolds et al., 2006). Although these studies provided the initial evidence that DiY forms in tau, it remained unclear how DiY cross-linking influences tau oligomer formation and aggregation into filaments and NFTs, especially in the absence of additives.
We recently established a truncated tau fragment corresponding to residues Ile297–Glu391, called dGAE, as an excellent model of studying tau assembly in vitro (Al-Hilaly et al., 2017, 2020). This tau fragment was first isolated from the proteolytically stable core of paired-helical filaments (PHFs) (Wischik et al., 1988b) and more recently was found to overlap with the region identified in the PHF and SF core by cryogenic electron microscopy (CryoEM) (Fitzpatrick et al., 2017; Figure 4). Our work showed that without the addition of exogenous additives dGAE can self-assemble to form filaments in vitro that structurally resemble PHF (Al-Hilaly et al., 2017, 2020; Lutter et al., 2022) and more recently, CryoEM structure determination showed the capacity for tau297–391 to form a filament structure identical to those derived from AD brain (Lövestam et al., 2022). Recently, we used MCO and UV oxidation to induce DiY cross-linking in dGAE, showing that oxidation facilitates the assembly of soluble dGAE into T22 antibody-positive tau oligomers which do not elongate into fibrils (Maina et al., 2020a). Different preparations of tau oligomers produced in vitro have been described to possess a β-sheet conformation and to induce toxicity in culture (Chen et al., 2009; Lasagna-Reeves et al., 2010; Flach et al., 2012; Tian et al., 2013). Interestingly, we found that the DiY cross-linked tau oligomers formed by dGAE lacked β-sheet structure, instead remained in a random-coil conformation, and were not acutely toxic to differentiated neuroblastoma cells after 3 days of culture (Maina et al., 2020a). This supports the work from the Binder’s group on T40 tau (Reynolds et al., 2005), in showing that DiY cross-linking promotes the formation of tau oligomers. Our work specifically suggests that the oligomers are incapable of further elongation into fibrils and are non-toxic.

Figure 4. Cryo-electron microscopy structure of paired helical filaments (Fitzpatrick et al., 2017) 5O3L.ENT showing position of tyrosine10 in magenta. Left side view, right panel top view.
Furthermore, we showed that dGAE fibrils formed DiY to a lower extent than the oligomers and other smaller assemblies (Maina et al., 2022a). The reduced ability for these fibrils to form DiY could be due to the reduced accessibility to the tyrosine residue in dGAE (Y310) in the fibrils, unlike in prefibrillar assemblies. Y310 is buried in one of the eight β-sheets of the tau molecule that run along the length of the protofilament, adopting a C-shaped architecture in AD and other tauopathies (Shi et al., 2021; Lövestam et al., 2022; Figure 4). Given the DiY’s ability to enhance protein stability, DiY cross-linked oligomers and fibrils would be expected to have enhanced stability and increased insolubility, similar to tau assemblies extracted from AD brain that show increased insolubility and resistance to proteolytic degradation (Wischik et al., 1988a,b). PHFs are specifically highly insoluble in SDS and sarcosyl (Kondo et al., 1988; Greenberg and Davies, 1990; Lee et al., 1991; Miao et al., 2019). Previous work from the Binder’s group showed that DiY cross-linked full length tau fibrils have increased SDS stability (Reynolds et al., 2006). We also recently showed that DiY cross-linked dGAE oligomers and especially fibrils, display a significantly reduced SDS and heat solubility (Maina et al., 2022a). The fibrils are particularly difficult to resuspend in solution, are heat and SDS-insoluble and transmission electron microscopy show that they maintain strong lateral association (Maina et al., 2022a). These studies suggest that indeed, DiY could promote the stabilization of tau assemblies in vivo. This begs the question as to whether DiY forms on tau assemblies in vivo? It is known that exposure to reactive oxygen species (ROS), aging, nitrogen dioxide, and lipid hydroperoxides can result in DiY formation (Giulivi and Davies, 1993, 1994; Kato et al., 1994). Oxidative stress is believed to accumulate early on in AD, increasing with the disease pathology (Nunomura et al., 2001; Butterfield and Halliwell, 2019). Thus, such oxidative environment that occurs in vivo in AD would favor DiY formation. Indeed, we recently showed that tau oligomers and fibrils extracted from the AD brain are DiY cross-linked (Maina et al., 2022a). However, whether DiY cross-linked tau oligomers also fail to elongate to fibrils in vivo is a question of future research. Nonetheless, available data suggests that the DiY cross-linking will promote the characteristic insoluble feature of AD fibrils. There is a significant lack of research on the role of DiY on tau and its implications. More research is needed to fully understand its relevance to AD.
The chemical stability of DiY could serve as a suitable biomarker for oxidative stress. DiY remains unchanged by exposure to oxygen and low or high pH (DiMarco and Giulivi, 2007). Furthermore, it is highly resistant to acid hydrolysis and proteases (Amado et al., 1984; Giulivi and Davies, 1994). DiY is also not incorporated into de novo synthesis of proteins (Eiserich et al., 1999), indicating that the level of DiY will reflect the oxidative damage to endogenous proteins (DiMarco and Giulivi, 2007). In this regard, there has been a strong interest since the 1990s on the possibility of using DiY as a marker of oxidative stress in different diseases. In 1993, DiY was considered an index of organismal oxidative stress (Giulivi and Davies, 1993). In this work, DiY production was shown in red blood cells challenged with the continuous flux of hydrogen peroxide. In the same year, Heinecke et al. (1993a) also showed that DiY could be formed via myeloperoxidase–hydrogen peroxide reaction in human neutrophils and macrophages. For this reason, they suggested that DiY could serve as a suitable marker of radical damage in phagocyte-rich inflammatory lesions in vivo (Heinecke et al., 1993b).
In the context of aging, it was found that DiY cross-linked proteins of mouse cardiac and skeletal muscle increased with age (Leeuwenburgh et al., 1997). Leeuwenburgh et al. (1999b) similarly showed that an increase in DiY could be detected through urine assays in aging rats. Such an increase was mirrored by a similar rise in rat skeletal muscle (Leeuwenburgh et al., 1999b). The authors suggested that urine assays of DiY could be used as a non-invasive method of estimating oxidative stress in vivo (Leeuwenburgh et al., 1999a,b). Kato et al. (1998) used immunohistochemistry in the human brain tissue to reveal the presence of DiY in lipofuscin pigments from aged human brains. Their work showed a significant elevation of DiY cross-links with age, suggesting a role for it in aging and lipofuscin accumulation (Kato et al., 1998). High levels of DiY was seen in lipofuscin pigments in AD human brains (Al-Hilaly et al., 2019).
In the context of neurodegenerative diseases, increased levels of DiY has been shown in the PD mouse model of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) injection (Pennathur et al., 1999). In this model, DiY and nitrotyrosine were specifically increased in the striatum and midbrain, but not in brain regions resistant to MPTP. The authors suggested that oxidative species, including hydroxyl radicals, tyrosyl radicals or peroxynitrite might mediate the damage caused by MPTP to dopaminergic neurons (Pennathur et al., 1999). Similarly, Hensley et al. (1998) measured DiY and nitrotyrosine level in the AD brain regions compared to controls. They revealed that DiY and nitrotyrosine are significantly increased in the hippocampus and neocortical regions of the AD brain and ventricular cerebrospinal fluid compared to levels in controls (Hensley et al., 1998). Their work provided evidence of the suitability of DiY as a biomarker for AD although the sensitivity and reproducibility of the method they utilized—HPLC with electrochemical array detection (HPLC-ECD), has been questioned by others (Duncan, 2003). However, another report using HPLC with fluorescence detection in 63 confirmed AD patient plasma samples revealed that DiY levels were significantly increased compared to controls (Polidori et al., 2004). A similar increase, albeit not significant compared to controls, was observed in the plasma samples from vascular dementia patients (Polidori et al., 2004). DiY cross-linking can result in Aβ dimer formation (Galeazzi et al., 1999; Yoburn et al., 2003; Al-Hilaly et al., 2013). Interestingly, an increase in the plasma level of Aβ dimers was recently shown in AD, serving as a potential biomarker (Villemagne et al., 2010). This further suggests a potential role DiY cross-linking that could help in AD biomarker discovery.
Using immunogold labeling, we have previously shown an increased co-localization of DiY antibody and Aβ antibody in Aβ plaques (Al-Hilaly et al., 2013). Using the same method, we showed an increase in DiY labeling in the CSF samples from AD patients compared to age-matched controls (Al-Hilaly et al., 2013). Although more studies are needed to establish DiY as a biomarker for AD, these studies suggest its utility as a potential biomarker. It would be interesting if future studies investigate whether an increase in DiY shows a disease-dependent rise in the CSF from AD patients. Given the recent promise in plasma-based markers for AD (Karikari et al., 2020a,b; Rodriguez et al., 2020; Suárez-Calvet et al., 2020; Thijssen et al., 2020; O’Connor et al., 2021), it would be useful to expand previous studies on DiY detection in plasma from AD patients (Polidori et al., 2004), to examine whether DiY levels could predict disease progression. This could especially be promising, giving that DiY is a general marker of protein oxidation, which is established to substantially increase in AD (Conrad et al., 2000; Butterfield and Kanski, 2001; Nunomura et al., 2001; Castegna et al., 2003; Polidori et al., 2004; Butterfield and Halliwell, 2019). As such, the DiY could be on multiple proteins, not just Aβ and Tau, which would increase its detection with disease progression.
There are many unanswered questions about the role of DiY on Aβ and Tau which would only become clearer in the future. One of the burning questions is whether DiY cross-linking is good or bad in AD? Given the well-established role of DiY in tissue stability, it may serve to strengthen proteins or tissue following injury in AD. What is DiY’s in vivo impact on Aβ and Tau for disease development or progression? Multiple groups have tried to address some of these questions, but the results have been variable, partly due to the differences in the peptide model used, methods of peptide preparation, peptide concentration and aggregation, and experiment duration. Importantly, many oxidative modifications can co-occur on both Aβ and Tau which may also affect them differently. Ultimately, studies that define these different modifications and specifically isolate the contribution of DiY would provide the much-needed data to support the interpretation of the studies generated thus far in the literature. Together, this will be essential for understanding the specific role of DiY on Aβ and tau in AD and its utility for bio-marker or drug discovery.
All authors listed have made a substantial, direct, and intellectual contribution to the work, and approved it for publication.
This work was funded by Alzheimer’s Society UK (AS-PG-16b-010).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Al-Hilaly, Y. K., Foster, B., Biasetti, L., Lutter, L., Pollack, S., Rickard, J., et al. (2020). Tau (297-391) forms filaments that structurally mimic the core of paired helical filaments in Alzheimer’s disease brain. FEBS Lett. 594, 944–950. doi: 10.1002/1873-3468.13675
Al-Hilaly, Y. K., Mohammed, A. H., Thorpe, J. R., and Serpell, L. C. (2019). The involvement of dityrosine crosslinks in lipofuscin accumulation in Alzheimer’s disease. J. Phys. Conf. Ser. 1294:062107.
Al-Hilaly, Y., Biasetti, L., Blakeman, B., Pollack, S., Zibaee, S., Abdul-Sada, A., et al. (2016). The involvement of dityrosine crosslinking in α-synuclein assembly and deposition in Lewy Bodies in Parkinson’s disease. Sci. Rep. 6:39171. doi: 10.1038/srep39171
Al-Hilaly, Y., Pollack, S., Vadukul, D., Citossi, F., Rickard, J., Simpson, M., et al. (2017). Alzheimer’s disease-like paired helical filament assembly from truncated tau protein is independent of disulfide crosslinking. J. Mol. Biol. 429, 3650–3665. doi: 10.1016/j.jmb.2017.09.007
Al-Hilaly, Y., Williams, T., Stewart-Parker, M., Ford, L., Skaria, E., Cole, M., et al. (2013). A central role for dityrosine crosslinking of Amyloid-β in Alzheimer’s disease. Acta Neuropathol. Commun. 1:83. doi: 10.1186/2051-5960-1-83
Ali, F., Leung, A., Cherny, R., Mavros, C., Barnham, K., Separovic, F., et al. (2006). Dimerisation of N-acetyl-L-tyrosine ethyl ester and Abeta peptides via formation of dityrosine. Free Radic. Res. 40, 1–9. doi: 10.1080/10715760500329721
Ali, F., Separovic, F., Barrow, C., Cherny, R., Fraser, F., Bush, A., et al. (2005). Methionine regulates copper/hydrogen peroxide oxidation products of Abeta. J. Pept. Sci. 11, 353–360. doi: 10.1002/psc.626
Alzheimer, A., Stelzmann, R., Schnitzlein, H., and Murtagh, F. (1995). An English translation of Alzheimer’s 1907 paper, “uber eine eigenartige erkankung der hirnrinde”. Clin. Anat. 8, 429–431. doi: 10.1002/ca.980080612
Amado, R., Aeschbach, R., and Neukom, H. (1984). Dityrosine – Invitro production and characterization. Methods Enzymol. 107, 377–388.
Andersen, S. (1964). The cross-links in resilin identified as dityrosine and trityrosine. Biochim. Biophys. Acta 93, 213–215. doi: 10.1016/0304-4165(64)90289-2
Andreadis, A. (2005). Tau gene alternative splicing: Expression patterns, regulation and modulation of function in normal brain and neurodegenerative diseases. Biochim. Biophys. Acta 1739, 91–103. doi: 10.1016/j.bbadis.2004.08.010
Andreadis, A., Brown, W., and Kosik, K. (1992). Structure and novel exons of the human tau gene. Biochemistry 31, 10626–10633. doi: 10.1021/bi00158a027
Arriagada, P., Growdon, J., Hedley-Whyte, E., and Hyman, B. (1992). Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer’s disease. Neurology 42, 631–639. doi: 10.1212/wnl.42.3.631
Atwood, C., Perry, G., Zeng, H., Kato, Y., Jones, W., Ling, K., et al. (2004). Copper mediates dityrosine cross-linking of Alzheimer’s amyloid-beta. Biochemistry 43, 560–568. doi: 10.1021/bi0358824
Bailey, A. (1991). The chemistry of natural enzyme-induced cross-links of proteins. Amino Acids 1, 293–306. doi: 10.1007/BF00813999
Barnham, K., Haeffner, F., Ciccotosto, G., Curtain, C., Tew, D., Mavros, C., et al. (2004). Tyrosine gated electron transfer is key to the toxic mechanism of Alzheimer’s disease beta-amyloid. FASEB J. 18, 1427–1429. doi: 10.1096/fj.04-1890fje
Bayse, G. S., Michaels, A. W., and Morrison, M. (1972). The peroxidase-catalyzed oxidation of tyrosine. Biochim. Biophys. Acta 284, 34–42.
Benilova, I., Karran, E., and De Strooper, B. (2012). The toxic Abeta oligomer and Alzheimer’s disease: An emperor in need of clothes. Nat. Neurosci. 15, 349–357. doi: 10.1038/nn.3028
Biasetti, L., Rey, S., Fowler, M., Ratnayaka, A., Fennell, K., Smith, C., et al. (2023). Elevated amyloid beta disrupts the nanoscale organization and function of synaptic vesicle pools in hippocampal neurons. Cereb. Cortex 33, 1263–1276. doi: 10.1093/cercor/bhac134
Bodaness, R. S., and Zigler, J. S. Jr. (1983). The rapid H2O2-mediated nonphotodynamic crosslinking of lens crystallins generated by the heme-undecapeptide from cytochrome C: Potential implications for cataractogenesis in man. Biochem. Biophys. Res. Commun. 113, 592–597. doi: 10.1016/0006-291x(83)91767-9
Braak, H., and Braak, E. (1991). Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 82, 239–259. doi: 10.1007/BF00308809
Briza, P., Ellinger, A., Winkler, G., and Breitenbach, M. (1990). Characterization of a Dl-dityrosine-containing macromolecule from yeast ascospore walls. J. Biol. Chem. 265, 15118–15123.
Buée, L., Bussière, T., Buée-Scherrer, V., Delacourte, A., and Hof, P. (2000). Tau protein isoforms, phosphorylation and role in neurodegenerative disorders. Brain Res. Brain Res. Rev. 33, 95–130. doi: 10.1016/s0165-0173(00)00019-9
Bukar Maina, M., Al-Hilaly, Y., and Serpell, L. (2016). Nuclear tau and its potential role in Alzheimer’s disease. Biomolecules 6:9. doi: 10.3390/biom6010009
Bush, A. (2003). Copper, zinc, and the metallobiology of Alzheimer disease. Alzheimer Dis. Assoc. Disord. 17, 147–150. doi: 10.1097/00002093-200307000-00005
Butterfield, D. A., and Halliwell, B. (2019). Oxidative stress, dysfunctional glucose metabolism and Alzheimer disease. Nat. Rev. Neurosci. 20, 148–160.
Butterfield, D. A., and Kanski, J. (2001). Brain protein oxidation in age-related neurodegenerative disorders that are associated with aggregated proteins. Mech. Ageing Dev. 122, 945–962.
Castegna, A., Thongboonkerd, V., Klein, J., Lynn, B., Markesbery, W., and Butterfield, D. (2003). Proteomic identification of nitrated proteins in Alzheimer’s disease brain. J. Neurochem. 85, 1394–1401. doi: 10.1046/j.1471-4159.2003.01786.x
Cecchi, C., Pensalfini, A., Liguri, G., Baglioni, S., Fiorillo, C., Guadagna, S., et al. (2008). Differentiation increases the resistance of neuronal cells to amyloid toxicity. Neurochem. Res. 33, 2516–2531. doi: 10.1007/s11064-008-9627-7
Chartier-Harlin, M., Crawford, F., Houlden, H., Warren, A., Hughes, D., Fidani, L., et al. (1991). Early-onset Alzheimer’s disease caused by mutations at codon 717 of the beta-amyloid precursor protein gene. Nature 353, 844–846. doi: 10.1038/353844a0
Chen, L., Wei, Y., Wang, X., and He, R. D. - (2009). Ribosylated Tau forms globular aggregates with high cytotoxicity. Cell. Mol. Life Sci. 66, 2559–2571. doi: 10.1007/s00018-009-0058-7
Cleveland, D. W., Hwo, S. Y., and Kirschner, M. W. (1977). Purification of tau, a microtubule-associated protein that induces assembly of microtubules from purified tubulin. J. Mol. Biol. 116, 207–225. doi: 10.1016/0022-2836(77)90213-3
Cline, E., Das, A., Bicca, M., Mohammad, S., Schachner, L., Kamel, J., et al. (2019). A novel crosslinking protocol stabilizes amyloid β oligomers capable of inducing Alzheimer’s-associated pathologies. J. Neurochem. 148, 822–836. doi: 10.1111/jnc.14647
Connell, J., Rodriguez-Martin, T., Gibb, G., Kahn, N., Grierson, A., Hanger, D., et al. (2005). Quantitative analysis of tau isoform transcripts in sporadic tauopathies. Brain Res. Mol. Brain Res. 137, 104–109. doi: 10.1016/j.molbrainres.2005.02.014
Conrad, C., Marshall, P., Talent, J., Malakowsky, C., Choi, J., and Gracy, R. (2000). Oxidized proteins in Alzheimer’s plasma. Biochem. Biophys. Res. Commun. 275, 678–681. doi: 10.1006/bbrc.2000.3356
Correia, M., Neves-Petersen, M., Jeppesen, P., Gregersen, S., and Petersen, S. B. (2012). UV-light exposure of insulin: Pharmaceutical implications upon covalent insulin dityrosine dimerization and disulphide bond photolysis. PLoS One 7:e50733. doi: 10.1371/journal.pone.0050733
Cowan, C. M., and Mudher, A. (2013). Are tau aggregates toxic or protective in tauopathies? Front. Neurol. 4:114. doi: 10.3389/fneur.2013.00114
Curtain, C., Ali, F., Volitakis, I., Cherny, R., Norton, R., Beyreuther, K., et al. (2001). Alzheimer’s disease amyloid-beta binds copper and zinc to generate an allosterically ordered membrane-penetrating structure containing superoxide dismutase-like subunits. J. Biol. Chem. 276, 20466–20473. doi: 10.1074/jbc.M100175200
DaRocha-Souto, B., Scotton, T., Coma, M., Serrano-Pozo, A., Hashimoto, T., Serenó, L., et al. (2011). Brain oligomeric β-amyloid but not total amyloid plaque burden correlates with neuronal loss and astrocyte inflammatory response in amyloid precursor protein/tau transgenic mice. J. Neuropathol. Exp. Neurol. 70, 360–376. doi: 10.1097/NEN.0b013e318217a118
Davis, R., Marsden, I., Maloney, M., Minamide, L., Podlisny, M., Selkoe, D., et al. (2011). Amyloid beta dimers/trimers potently induce cofilin-actin rods that are inhibited by maintaining cofilin-phosphorylation. Mol. Neurodegener. 6:10. doi: 10.1186/1750-1326-6-10
de la Torre, A. V., Gay, M., Vilaprinyó-Pascual, S., Mazzucato, R., Serra-Batiste, M., Vilaseca, M., et al. (2018). Direct evidence of the presence of cross-linked Aβ dimers in the brains of Alzheimer’s disease patients. Anal. Chem. 90, 4552–4560. doi: 10.1021/acs.analchem.7b04936
DiMarco, T., and Giulivi, C. (2007). Current analytical methods for the detection of dityrosine, a biomarker of oxidative stress, in biological samples. Mass Spectrom. Rev. 26, 108–120. doi: 10.1002/mas.20109
Dixit, R., Ross, J., Goldman, Y., and Holzbaur, E. (2008). Differential regulation of dynein and kinesin motor proteins by tau. Science 319, 1086–1089. doi: 10.1126/science.1152993
Duncan, M. (2003). A review of approaches to the analysis of 3-nitrotyrosine. Amino Acids 25, 351–361. doi: 10.1007/s00726-003-0022-z
Eiserich, J. P., Estevez, A. G., Bamberg, T. V., Ye, Y. Z., Chumley, P. H., Beckman, J. S., et al. (1999). Microtubule dysfunction by posttranslational nitrotyrosination of alpha-tubulin: A nitric oxide-dependent mechanism of cellular injury. Proc. Natl. Acad. Sci. U.S.A. 96: 6365–6370. doi: 10.1073/pnas.96.11.6365
Elliott, K. (1932). Milk peroxidase: Its preparation, properties, and action with H(2)O(2) on metabolites. With a method for determining small amounts of H(2)O(2) in complex mixtures. Biochem. J. 26, 10–24. doi: 10.1042/bj0260010
Fetterer, R. H., Rhoads, M. L., and Urban, J. F. Jr. (1993). Synthesis of tyrosine-derived cross-links in Ascaris suum cuticular proteins. J. Parasitol. 79, 160–166.
Fitzpatrick, A., Falcon, B., He, S., Murzin, A., Murshudov, G., Garringer, H., et al. (2017). Cryo-EM structures of tau filaments from Alzheimer’s disease. Nature 547, 185–190. doi: 10.1038/nature23002
Flach, K., Hilbrich, I., Schiffmann, A., Gärtner, U., Krüger, M., Leonhardt, M., et al. (2012). Tau oligomers impair artificial membrane integrity and cellular viability. J. Biol. Chem. 287, 43223–43233. doi: 10.1074/jbc.M112.396176
Foerder, C. A., and Shapiro, B. M. (1977). Release of ovoperoxidase from sea urchin eggs hardens the fertilization membrane with tyrosine crosslinks. Proc. Natl. Acad. Sci. U.S.A. 74, 4214–4218. doi: 10.1073/pnas.74.10.4214
Ford, L., Crossley, M., Vadukul, D., Kemenes, G., and Serpell, L. (2017). Structure-dependent effects of amyloid-β on long-term memory in Lymnaea stagnalis. FEBS Lett. 591, 1236–1246. doi: 10.1002/1873-3468.12633
Friedhoff, P., Schneider, A., Mandelkow, E., and Mandelkow, E. (1998). Rapid assembly of Alzheimer-like paired helical filaments from microtubule-associated protein tau monitored by fluorescence in solution. Biochemistry 37, 10223–10230. doi: 10.1021/bi980537d
Fuchsberger, T., Martínez-Bellver, S., Giraldo, E., Teruel-Martí, V., Lloret, A., and Viña, J. (2016). Aβ induces excitotoxicity mediated by APC/C-Cdh1 depletion that can be prevented by glutaminase inhibition promoting neuronal survival. Sci. Rep. 6:31158. doi: 10.1038/srep31158
Fuentes-Lemus, E., Hägglund, P., López-Alarcón, C., and Davies, M. (2021). Oxidative crosslinking of peptides and proteins: Mechanisms of formation, detection, characterization and quantification. Molecules 27:15. doi: 10.3390/molecules27010015
Fujimoto, D. (1975). Occurrence of dityrosine in cuticlin, a structural protein from ascaris cuticle. Compar. Biochem. Physiol. B Biochem. Mol. Biol. 51, 205–207.
Galeazzi, L., Ronchi, P., Franceschi, C., and Giunta, S. (1999). In vitro peroxidase oxidation induces stable dimers of β-amyloid (1-42) through dityrosine bridge formation. Amyloid 6, 7–13. doi: 10.3109/13506129908993282
Ganguly, P., Do, T., Larini, L., LaPointe, N., Sercel, A., Shade, M., et al. (2015). Tau assembly: The dominant role of PHF6 (VQIVYK) in microtubule binding region repeat R3. J. Phys. Chem. B 119, 4582–4593. doi: 10.1021/acs.jpcb.5b00175
Georgieff, I. S., Liem, R. K., Couchie, D., Mavilia, C., Nunez, J., and Shelanski, M. L. (1993). Expression of high molecular weight tau in the central and peripheral nervous systems. J. Cell Sci. 105, 729–737.
Giulivi, C., and Davies, K. J. (1993). Dityrosine and tyrosine oxidation products are endogenous markers for the selective proteolysis of oxidatively modified red blood cell hemoglobin by (the 19 S) proteasome. J. Biol. Chem. 268, 8752–8759.
Giulivi, C., and Davies, K. J. (1994). Dityrosine: A marker for oxidatively modified proteins and selective proteolysis. Methods Enzymol. 233, 363–371.
Glenner, G. G., and Wong, C. W. (1984). Alzheimer’s disease: Initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem. Biophys. Res. Commun. 120, 885–890.
Goate, A., Chartier-Harlin, M., Mullan, M., Brown, J., Crawford, F., Fidani, L., et al. (1991). Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer’s disease. Nature 349, 704–706. doi: 10.1038/349704a0
Goedert, M., Jakes, R., Spillantini, M. G., Hasegawa, M., Smith, M. J., and Crowther, R. A. (1996). Assembly of microtubule-associated protein tau into Alzheimer-like filaments induced by sulphated glycosaminoglycans. Nature 383, 550–553.
Greenberg, S. G., and Davies, P. (1990). A preparation of Alzheimer paired helical filaments that displays distinct tau proteins by polyacrylamide gel electrophoresis. Proc. Natl. Acad. Sci. U.S.A. 87, 5827–5831. doi: 10.1073/pnas.87.15.5827
Gross, A. J., and Sizer, I. W. (1959). The oxidation of tyramine, tyrosine, and related compounds by peroxidase. J. Biol. Chem. 234, 1611–1614. doi: 10.1016/S0021-9258(18)70059-8
Grundke-Iqbal, I., Iqbal, K., Quinlan, M., Tung, Y., Zaidi, M., and Wisniewski, H. (1986a). Microtubule-associated protein tau. A component of Alzheimer paired helical filaments. J. Biol. Chem. 261, 6084–6089.
Grundke-Iqbal, I., Iqbal, K., Tung, Y. C., Quinlan, M., Wisniewski, H. M., and Binder, L. I. (1986b). Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc. Natl. Acad. Sci. U.S.A. 83, 4913–4917.
Gu, M., Bode, D., and Viles, J. (2018). Copper redox cycling inhibits Aβ fibre formation and promotes fibre fragmentation, while generating a dityrosine Aβ dimer. Sci. Rep. 8:16190. doi: 10.1038/s41598-018-33935-5
Guo, T., Noble, W., and Hanger, D. P. (2017). Roles of tau protein in health and disease. Acta Neuropathol. 133, 665–704.
Hardy, J., and Higgins, G. (1992). Alzheimer’s disease: The amyloid cascade hypothesis. Science 256, 184–185.
Heinecke, J., Li, W., Daehnke, H., and Goldstein, J. (1993a). Dityrosine, a specific marker of oxidation, is synthesized by the myeloperoxidase-hydrogen peroxide system of human neutrophils and macrophages. J. Biol. Chem. 268, 4069–4077.
Heinecke, J., Li, W., Francis, G., and Goldstein, J. (1993b). Tyrosyl radical generated by myeloperoxidase catalyzes the oxidative cross-linking of proteins. J. Clin. Invest. 91, 2866–2872. doi: 10.1172/JCI116531
Hensley, K., Maidt, M., Yu, Z., Sang, H., Markesbery, W., and Floyd, R. (1998). Electrochemical analysis of protein nitrotyrosine and dityrosine in the Alzheimer brain indicates region-specific accumulation. J. Neurosci. 18, 8126–8132. doi: 10.1523/JNEUROSCI.18-20-08126.1998
Ittner, L., Ke, Y., Delerue, F., Bi, M., Gladbach, A., van Eersel, J., et al. (2010). Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer’s disease mouse models. Cell 142, 387–397. doi: 10.1016/j.cell.2010.06.036
Jack, C., Knopman, D., Jagust, W., Petersen, R., Weiner, M., Aisen, P., et al. (2013). Tracking pathophysiological processes in Alzheimer’s disease: An updated hypothetical model of dynamic biomarkers. Lancet Neurol. 12, 207–216. doi: 10.1016/S1474-4422(12)70291-0
Jana, M., Cappai, R., Pham, C., and Ciccotosto, G. (2016). Membrane-bound tetramer and trimer Aβ oligomeric species correlate with toxicity towards cultured neurons. J. Neurochem. 136, 594–608. doi: 10.1111/jnc.13443
Jomova, K., Vondrakova, D., Lawson, M., and Valko, M. (2010). Metals, oxidative stress and neurodegenerative disorders. Mol. Cell. Biochem. 345, 91–104. doi: 10.1007/s11010-010-0563-x
Kampers, T., Friedhoff, P., Biernat, J., Mandelkow, E., and Mandelkow, E. (1996). RNA stimulates aggregation of microtubule-associated protein tau into Alzheimer-like paired helical filaments. FEBS Lett. 399, 344–349. doi: 10.1016/s0014-5793(96)01386-5
Kaniyappan, S., Chandupatla, R. R., Mandelkow, E. M., and Mandelkow, E. (2017). Extracellular low-n oligomers of tau cause selective synaptotoxicity without affecting cell viability. Alzheimers Dement. 13, 1270–1291. doi: 10.1016/j.jalz.2017.04.002
Kanwar, R., and Balasubramanian, D. (1999). Structure and stability of the dityrosine-linked dimer of gamma B-crystallin. Exp. Eye Res. 68, 773–784. doi: 10.1006/exer.1999.0669
Karikari, T., Benedet, A., Ashton, N., Lantero Rodriguez, J., Snellman, A., Suárez-Calvet, M., et al. (2020a). Diagnostic performance and prediction of clinical progression of plasma phospho-tau181 in the Alzheimer’s disease neuroimaging initiative. Mol. Psychiatry 26, 429–442. doi: 10.1038/s41380-020-00923-z
Karikari, T., Pascoal, T., Ashton, N., Janelidze, S., Benedet, A., Rodriguez, J., et al. (2020b). Blood phosphorylated tau 181 as a biomarker for Alzheimer’s disease: A diagnostic performance and prediction modelling study using data from four prospective cohorts. Lancet Neurol. 19, 422–433. doi: 10.1016/S1474-4422(20)30071-5
Kato, Y., Maruyama, W., Naoi, M., Hashizume, Y., and Osawa, T. (1998). Immunohistochemical detection of dityrosine in lipofuscin pigments in the aged human brain. Febs Lett. 439, 231–234. doi: 10.1016/s0014-5793(98)01372-6
Kato, Y., Uchida, K., and Kawakishi, S. (1994). Aggregation of collagen exposed to UVA in the presence of riboflavin: A plausible role of tyrosine modification. Photochem. Photobiol. 59, 343–349. doi: 10.1111/j.1751-1097.1994.tb05045.x
Kheterpal, I., Williams, A., Murphy, C., Bledsoe, B., and Wetzel, R. (2001). Structural features of the Abeta amyloid fibril elucidated by limited proteolysis. Biochemistry 40, 11757–11767. doi: 10.1021/bi010805z
Kok, W. M., Cottam, J. M., Ciccotosto, G. D., Miles, L. A., Karas, J. A., Scanlon, D. B., et al. (2013). Synthetic dityrosine-linked β-amyloid dimers form stable, soluble, neurotoxic oligomers. Chem. Sci. 4, 4449–4454.
Kondo, J., Honda, T., Mori, H., Hamada, Y., Miura, R., Ogawara, M., et al. (1988). The carboxyl third of tau is tightly bound to paired helical filaments. Neuron 1, 827–834. doi: 10.1016/0896-6273(88)90130-4
Kowalik-Jankowska, T., Ruta, M., Wiśniewska, K., Łankiewicz, L., and Dyba, M. (2004). Products of Cu(Ii)-catalyzed oxidation in the presence of hydrogen peroxide of the 1-10, 1-16 fragments of human and mouse beta-amyloid peptide. J. Inorg. Biochem. 98, 940–950. doi: 10.1016/j.jinorgbio.2004.03.001
Krishtal, J., Bragina, O., Metsla, K., Palumaa, P., and Tõugu, V. (2015). Toxicity of amyloid beta 1-40 and 1-42 on Sh-Sy5Y cell line. Springerplus 4:19.
LaBella, F., Keeley, F., Vivian, S., and Thornhill, D. (1967). Evidence for dityrosine in elastin. Biochem. Biophys. Res. Commun. 26, 748–753. doi: 10.1016/s0006-291x(67)80137-2
Lacor, P., Buniel, M., Furlow, P., Clemente, A., Velasco, P., Wood, M., et al. (2007). Abeta oligomer-induced aberrations in synapse composition, shape, and density provide a molecular basis for loss of connectivity in Alzheimer’s disease. J. Neurosci. 27, 796–807. doi: 10.1523/JNEUROSCI.3501-06.2007
Lambert, M., Barlow, A., Chromy, B., Edwards, C., Freed, R., Liosatos, M., et al. (1998). Diffusible, nonfibrillar ligands derived from Abeta1-42 are potent central nervous system neurotoxins. Proc. Natl. Acad. Sci. U.S.A. 95, 6448–6453. doi: 10.1073/pnas.95.11.6448
Lasagna-Reeves, C., Castillo-Carranza, D., Guerrero-Muoz, M., Jackson, G., and Kayed, R. (2010). Preparation and characterization of neurotoxic tau oligomers. Biochemistry 49, 10039–10041. doi: 10.1021/bi1016233
Lee, V., Balin, B., Otvos, L., and Trojanowski, J. (1991). A68: A major subunit of paired helical filaments and derivatized forms of normal Tau. Science 251, 675–678. doi: 10.1126/science.1899488
Leeuwenburgh, C., Hansen, P. A., Holloszy, J. O., and Heinecke, J. W. (1999b). Oxidized amino acids in the urine of aging rats: Potential markers for assessing oxidative stress in vivo. Am. J. Physiol. 276, R128–R135. doi: 10.1152/ajpregu.1999.276.1.R128
Leeuwenburgh, C., Hansen, P. A., Holloszy, J. O., and Heinecke, J. W. (1999a). Hydroxyl radical generation during exercise increases mitochondrial protein oxidation and levels of urinary dityrosine. Free Radic. Biol. Med. 27, 186–192. doi: 10.1016/s0891-5849(99)00071-4
Leeuwenburgh, C., Hazen, S. L., Pennathur, S., and Heinecke, J. W. (1996). Massive increase in protein-bound dityrosine in Ldl isolated from human atherosclerotic aorta: Implications for the role of tyrosyl radical in atherogenesis. Circulation 94, 2332–2332.
Leeuwenburgh, C., Wagner, P., Holloszy, J., Sohal, R., and Heinecke, J. (1997). Caloric restriction attenuates dityrosine cross-linking of cardiac and skeletal muscle proteins in aging mice. Arch. Biochem. Biophys. 346, 74–80. doi: 10.1006/abbi.1997.0297
Levy, E., Carman, M., Fernandez-Madrid, I., Power, M., Lieberburg, I., van Duinen, S., et al. (1990). Mutation of the Alzheimer’s disease amyloid gene in hereditary cerebral hemorrhage, Dutch type. Science 248, 1124–1126. doi: 10.1126/science.2111584
Li, J., Hodgeman, B., and Christensen, B. (1996). Involvement of peroxidase in chorion hardening in Aedes aegypti. Insect. Biochem. Mol. Biol. 26, 309–317. doi: 10.1016/0965-1748(95)00099-2
Li, S., Jin, M., Koeglsperger, T., Shepardson, N. E., Shankar, G. M., and Selkoe, D. J. (2011). Soluble Abeta oligomers inhibit long-term potentiation through a mechanism involving excessive activation of extrasynaptic Nr2B-containing Nmda receptors. J. Neurosci. 31, 6627–6638.
Liu, C., and Götz, J. (2013). Profiling murine Tau with 0N, 1N and 2N isoform-specific antibodies in brain and peripheral organs reveals distinct subcellular localization, with the 1N isoform being enriched in the nucleus. PLoS One 8:e84849. doi: 10.1371/journal.pone.0084849
Loomis, P., Howard, T., Castleberry, R., and Binder, L. (1990). Identification of nuclear tau isoforms in human neuroblastoma cells. Proc. Natl. Acad. Sci. U.S.A. 87, 8422–8426. doi: 10.1073/pnas.87.21.8422
Lovell, M., Robertson, J., Teesdale, W., Campbell, J., and Markesbery, W. (1998). Copper, iron and zinc in Alzheimer’s disease senile plaques. J. Neurol. Sci. 158, 47–52. doi: 10.1016/s0022-510x(98)00092-6
Lövestam, S., Koh, F., van Knippenberg, B., Kotecha, A., Murzin, A., Goedert, M., et al. (2022). Assembly of recombinant tau into filaments identical to those of Alzheimer’s disease and chronic traumatic encephalopathy. Elife 11:e76494. doi: 10.7554/eLife.76494
Lutter, L., Al-Hilaly, Y., Serpell, C., Tuite, M., Wischik, C., Serpell, L., et al. (2022). Structural identification of individual helical amyloid filaments by integration of cryo-electron microscopy-derived maps in comparative morphometric atomic force microscopy image analysis. J. Mol. Biol. 434:167466. doi: 10.1016/j.jmb.2022.167466
Maina, M. B., Bailey, L. J., Doherty, A. J., and Serpell, L. C. (2018a). The involvement of Aβ42 and tau in nucleolar and protein synthesis machinery dysfunction. Front. Cell. Neurosci. 12:220. doi: 10.3389/fncel.2018.00220
Maina, M. B., Bailey, L., Wagih, S., Biasetti, L., Pollack, S., Quinn, J., et al. (2018b). The involvement of tau in nucleolar transcription and the stress response. Acta Neuropathol. Commun. 6:70. doi: 10.1186/s40478-018-0565-6
Maina, M., Burra, G., Al-Hilaly, Y., Mengham, K., Fennell, K., and Serpell, L. (2020b). Metal- and UV- catalyzed oxidation results in trapped Amyloid-β intermediates revealing that self-assembly is required for Aβ-induced cytotoxicity. iScience 23:101537. doi: 10.1016/j.isci.2020.101537
Maina, M., Al-Hilaly, Y., Burra, G., Rickard, J., Harrington, C., Wischik, C., et al. (2020a). Oxidative stress conditions result in trapping of PHF-core tau (297-391) intermediates. bioRxiv [Preprint]. bioRxiv: 2020.12.07.414532. doi: 10.3390/cells10030703
Maina, M., Al-Hilaly, Y., Burra, G., Rickard, J., Harrington, C., Wischik, C., et al. (2021). Oxidative stress conditions result in trapping of PHF-core tau (297-391) intermediates. Cells 10:703.
Maina, M., Al-Hilaly, Y., Oakley, S., Burra, G., Khanom, T., Biasetti, L., et al. (2022b). Dityrosine cross-links are present in Alzheimer’s disease-derived tau oligomers and paired helical filaments (PHF) which promotes the stability of the PHF-core tau (297-391) in vitro. bioRxiv [Preprint]. bioRxiv: 2022.05.28.493839. doi: 10.1016/j.jmb.2022.167785
Maina, M., Al-Hilaly, Y., Oakley, S., Burra, G., Khanom, T., Biasetti, L., et al. (2022a). Dityrosine cross-links are present in Alzheimer’s disease-derived tau oligomers and paired helical filaments (PHF) which promotes the stability of the PHF-core tau (297-391) in vitro. J. Mol. Biol. 434:167785.
Marshall, K. E., Vadukul, D. M., Staras, K., and Serpell, L. C. (2020). Misfolded amyloid-beta-42 impairs the endosomal-lysosomal pathway. Cell. Mol. Life Sci. 77, 5031–5043. doi: 10.1007/s00018-020-03464-4
Marshall, K., Vadukul, D., Dahal, L., Theisen, A., Fowler, M., Al-Hilaly, Y., et al. (2016). A critical role for the self-assembly of Amyloid-β1-42 in neurodegeneration. Sci. Rep. 6:30182. doi: 10.1038/srep30182
Martin, L., Latypova, X., and Terro, F. (2011). Post-translational modifications of tau protein: Implications for Alzheimer’s disease. Neurochem. Int. 58, 458–471.
Masters, C. L., and Selkoe, D. J. (2012). Biochemistry of amyloid β-protein and amyloid deposits in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2:a006262.
Mayer, F., Pröpper, S., and Ritz-Timme, S. (2014). Dityrosine, a protein product of oxidative stress, as a possible marker of acute myocardial infarctions. Int. J. Legal Med. 128, 787–794. doi: 10.1007/s00414-014-1015-z
McLean, C., Cherny, R., Fraser, F., Fuller, S., Smith, M., Beyreuther, K., et al. (1999). Soluble pool of Abeta amyloid as a determinant of severity of neurodegeneration in Alzheimer’s disease. Ann. Neurol. 46, 860–866.
Miao, J., Shi, R., Li, L., Chen, F., Zhou, Y., Tung, Y., et al. (2019). Pathological tau from Alzheimer’s brain induces site-specific hyperphosphorylation and SDS- and reducing agent-resistant aggregation of tau in vivo. Front. Aging Neurosci. 11:34. doi: 10.3389/fnagi.2019.00034
Mukrasch, M. D., Biernat, J., Von Bergen, M., Griesinger, C., Mandelkow, E., and Zweckstetter, M. (2005). Sites of tau important for aggregation populate {beta}-structure and bind to microtubules and polyanions. J. Biol. Chem. 280, 24978–24986. doi: 10.1074/jbc.M501565200
Neve, R., Harris, P., Kosik, K., Kurnit, D., and Donlon, T. (1986). Identification of cDNA clones for the human microtubule-associated protein tau and chromosomal localization of the genes for tau and microtubule-associated protein 2. Brain Res. 387, 271–280. doi: 10.1016/0169-328x(86)90033-1
Nunez, J., and Fischer, I. (1997). Microtubule-associated proteins (Maps) in the peripheral nervous system during development and regeneration. J. Mol. Neurosci. 8, 207–222.
Nunomura, A., Perry, G., Aliev, G., Hirai, K., Takeda, A., Balraj, E., et al. (2001). Oxidative damage is the earliest event in Alzheimer disease. J. Neuropathol. Exp. Neurol. 60, 759–767. doi: 10.1093/jnen/60.8.759
O’Connor, A., Karikari, T., Poole, T., Ashton, N., Lantero Rodriguez, J., Khatun, A., et al. (2021). Plasma phospho-tau181 in presymptomatic and symptomatic familial Alzheimer’s disease: A longitudinal cohort study. Mol. Psychiatry 26, 5967–5976. doi: 10.1038/s41380-020-0838-x
O’Malley, T., Oktaviani, N., Zhang, D., Lomakin, A., O’Nuallain, B., Linse, S., et al. (2014). Aβ dimers differ from monomers in structural propensity, aggregation paths and population of synaptotoxic assemblies. Biochem. J. 461, 413–426. doi: 10.1042/BJ20140219
O’Malley, T., Witbold, W., Linse, S., and Walsh, D. (2016). The aggregation paths and products of Aβ42 dimers are distinct from those of the Aβ42 monomer. Biochemistry 55, 6150–6161. doi: 10.1021/acs.biochem.6b00453
Oakley, S., Maina, M., Marshall, K., Al-Hilaly, Y., Harrington, C., Wischik, C., et al. (2020). Tau filament self-assembly and structure: Tau as a therapeutic target. Front. Neurol. 11:590754. doi: 10.3389/fneur.2020.590754
Ono, K., Condron, M. M., and Teplow, D. B. (2009). Structure-neurotoxicity relationships of amyloid beta-protein oligomers. Proc. Natl. Acad. Sci. U.S.A. 106, 14745–14750. doi: 10.1073/pnas.0905127106
Palmblad, M., Westlind-Danielsson, A., and Bergquist, J. (2002). Oxidation of methionine 35 attenuates formation of amyloid beta -peptide 1-40 oligomers. J. Biol. Chem. 277, 19506–19510. doi: 10.1074/jbc.M112218200
Partlow, B. P., Applegate, M. B., Omenetto, F. G., and Kaplan, D. L. (2016). Dityrosine cross-linking in designing biomaterials. ACS Biomater. Sci. Eng. 2, 2108–2121.
Pennathur, S., Jackson-Lewisu, V., Przedborskiu, S., and Heinecke, J. (1999). Marked elevations of 3-nitrotyrosine and dityrosine in mice with Mptp-induced Parkinson’s disease. Free Radic. Biol. Med. 27, S141–S141.
Pérez, M., Valpuesta, J., Medina, M., Montejo de Garcini, E., and Avila, J. (1996). Polymerization of tau into filaments in the presence of heparin: The minimal sequence required for tau-tau interaction. J. Neurochem. 67, 1183–1190. doi: 10.1046/j.1471-4159.1996.67031183.x
Polidori, M., Mattioli, P., Aldred, S., Cecchetti, R., Stahl, W., Griffiths, H., et al. (2004). Plasma antioxidant status, immunoglobulin g oxidation and lipid peroxidation in demented patients: Relevance to Alzheimer disease and vascular dementia. Dement. Geriatr. Cogn. Disord. 18, 265–270. doi: 10.1159/000080027
Rambaran, R. N., and Serpell, L. C. (2008). Amyloid fibrils: Abnormal protein assembly. Prion 2, 112–117.
Raven, D. J., Earland, C., and Little, M. (1971). Occurrence of dityrosine in tussah silk fibroin and keratin. Biochim. Biophys. Acta 251:96. doi: 10.1016/0005-2795(71)90065-1
Reddy, P. H., and Beal, M. F. (2008). Amyloid beta, mitochondrial dysfunction and synaptic damage: Implications for cognitive decline in aging and Alzheimer’s disease. Trends Mol. Med. 14, 45–53.
Reynolds, M. R., Berry, R. W., and Binder, L. I. (2005). Site-specific nitration and oxidative dityrosine bridging of the tau protein by peroxynitrite: Implications for Alzheimer’s disease. Biochemistry 44, 1690–1700. doi: 10.1021/bi047982v
Reynolds, M., Lukas, T., Berry, R., and Binder, L. (2006). Peroxynitrite-mediated tau modifications stabilize preformed filaments and destabilize microtubules through distinct mechanisms. Biochemistry 45, 4314–4326. doi: 10.1021/bi052142h
Roberts, B. R., Ryan, T. M., Bush, A. I., Masters, C. L., and Duce, J. A. (2012). The role of metallobiology and amyloid-beta peptides in Alzheimer’s disease. J. Neurochem. 120, 149–166.
Rodriguez, J. L., Karikari, T., Suárez-Calvet, M., Troakes, C., King, A., Emersic, A., et al. (2020). Plasma p-tau181 accurately predicts Alzheimer’s disease pathology at least 8 years prior to post-mortem and improves the clinical characterisation of cognitive decline. Acta Neuropathol. 140, 267–278. doi: 10.1007/s00401-020-02195-x
Rossi, G., Dalprà, L., Crosti, F., Lissoni, S., Sciacca, F., Catania, M., et al. (2008). A new function of microtubule-associated protein tau: Involvement in chromosome stability. Cell Cycle 7, 1788–1794. doi: 10.4161/cc.7.12.6012
Sarell, C., Syme, C., Rigby, S., and Viles, J. (2009). Copper(II) binding to amyloid-beta fibrils of Alzheimer’s disease reveals a picomolar affinity: Stoichiometry and coordination geometry are independent of Abeta oligomeric form. Biochemistry 48, 4388–4402. doi: 10.1021/bi900254n
Schweers, O., Schönbrunn-Hanebeck, E., Marx, A., and Mandelkow, E. (1994). Structural studies of tau protein and Alzheimer paired helical filaments show no evidence for beta-structure. J. Biol. Chem. 269, 24290–24297.
Selkoe, D. J., and Hardy, J. (2016). The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 8, 595–608.
Shankar, G., Li, S., Mehta, T., Garcia-Munoz, A., Shepardson, N., Smith, I., et al. (2008). Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat. Med. 14, 837–842. doi: 10.1038/nm1782
Shi, Y., Zhang, W., Yang, Y., Murzin, A., Falcon, B., Kotecha, A., et al. (2021). Structure-based classification of tauopathies. bioRxiv [Preprint]. bioRxiv: 2021.05.28.446130.
Siegel, S., Bieschke, J., Powers, E., and Kelly, J. (2007). The oxidative stress metabolite 4-hydroxynonenal promotes Alzheimer protofibril formation. Biochemistry 46, 1503–1510. doi: 10.1021/bi061853s
Sitkiewicz, E., Olędzki, J., Poznański, J., and Dadlez, M. (2014). Di-tyrosine cross-link decreases the collisional cross-section of aβ peptide dimers and trimers in the gas phase: An ion mobility study. PLoS One 9:e100200. doi: 10.1371/journal.pone.0100200
Skaff, O., Jolliffe, K., and Hutton, C. (2005). Synthesis of the side chain cross-linked tyrosine oligomers dityrosine, trityrosine, and pulcherosine. J. Org. Chem. 70, 7353–7363. doi: 10.1021/jo051076m
Smail, E., Briza, P., Panagos, A., and Berenfeld, L. (1995). Candida albicans cell walls contain the fluorescent cross-linking amino acid dityrosine. Infect. Immun. 63, 4078–4083. doi: 10.1128/iai.63.10.4078-4083.1995
Smith, D. G., Ciccotosto, G., Tew, D., Fodero-Tavoletti, M., Johanssen, T., Masters, C., et al. (2007b). Concentration dependent Cu2+ induced aggregation and dityrosine formation of the Alzheimer’s disease amyloid-beta peptide. Biochemistry 46, 2881–2891. doi: 10.1021/bi0620961
Smith, D. G., Cappai, R., and Barnham, K. J. (2007a). The redox chemistry of the Alzheimer’s disease amyloid β peptide. Biochim. Biophys. Acta 1768, 1976–1990.
Smith, D., Smith, D., Curtain, C., Boas, J., Pilbrow, J., Ciccotosto, G., et al. (2006). Copper-mediated amyloid-beta toxicity is associated with an intermolecular histidine bridge. J. Biol. Chem. 281, 15145–15154. doi: 10.1074/jbc.M600417200
Soura, V., Stewart-Parker, M., Williams, T., Ratnayaka, A., Atherton, J., Gorringe, K., et al. (2012). Visualization of co-localization in Aβ42-administered neuroblastoma cells reveals lysosome damage and autophagosome accumulation related to cell death. Biochem. J. 441, 579–590. doi: 10.1042/BJ20110749
Souza, J. M., Giasson, B. I., Chen, Q., Lee, V. M., and Ischiropoulos, H. (2000). Dityrosine cross-linking promotes formation of stable alpha -synuclein polymers. Implication of nitrative and oxidative stress in the pathogenesis of neurodegenerative synucleinopathies. J. Biol. Chem. 275, 18344–18349. doi: 10.1074/jbc.M000206200
Stoothoff, W. H., and Johnson, G. V. W. (2005). Tau phosphorylation: Physiological and pathological consequences. Biochim. Biophys. Acta 1739, 280–297.
Suárez-Calvet, M., Karikari, T. K., Ashton, N. J., Lantero Rodríguez, J., Milà-Alomà, M., Gispert, J. D., et al. (2020). Novel tau biomarkers phosphorylated at T181, T217 or T231 rise in the initial stages of the preclinical Alzheimer’s continuum when only subtle changes in Aβ pathology are detected. EMBO Mol. Med. 12:e12921.
Suh, S., Jensen, K., Jensen, M., Silva, D., Kesslak, P., Danscher, G., et al. (2000). Histochemically-reactive zinc in amyloid plaques, angiopathy, and degenerating neurons of Alzheimer’s diseased brains. Brain Res. 852, 274–278. doi: 10.1016/s0006-8993(99)02096-x
Thijssen, E., La Joie, R., Wolf, A., Strom, A., Wang, P., Iaccarino, L., et al. (2020). Diagnostic value of plasma phosphorylated tau181 in Alzheimer’s disease and frontotemporal lobar degeneration. Nat. Med. 26, 387–397. doi: 10.1038/s41591-020-0762-2
Tian, H., Davidowitz, E., Lopez, P., Emadi, S., Moe, J., and Sierks, M. (2013). Trimeric tau is toxic to human neuronal cells at low nanomolar concentrations. Int. J. Cell Biol. 2013:260787. doi: 10.1155/2013/260787
Tickler, A., Smith, D., Ciccotosto, G., Tew, D., Curtain, C., Carrington, D., et al. (2005). Methylation of the imidazole side chains of the Alzheimer disease amyloid-beta peptide results in abolition of superoxide dismutase-like structures and inhibition of neurotoxicity. J. Biol. Chem. 280, 13355–13363. doi: 10.1074/jbc.M414178200
Urbanc, B. (2021). Cross-linked amyloid β-protein oligomers: A missing link in Alzheimer’s disease pathology? J. Phys. Chem. B. 125, 1307–1316. doi: 10.1021/acs.jpcb.0c07716
Vadukul, D., Gbajumo, O., Marshall, K., and Serpell, L. (2017). Amyloidogenicity and toxicity of the reverse and scrambled variants of amyloid-β 1-42. FEBS Lett. 591, 822–830. doi: 10.1002/1873-3468.12590
Vadukul, D., Maina, M., Franklin, H., Nardecchia, A., Serpell, L., and Marshall, K. (2020). Internalisation and toxicity of amyloid-β 1-42 are influenced by its conformation and assembly state rather than size. FEBS Lett. 594, 3490–3503. doi: 10.1002/1873-3468.13919
Van Broeckhoven, C., Haan, J., Bakker, E., Hardy, J., Van Hul, W., Wehnert, A., et al. (1990). Amyloid beta protein precursor gene and hereditary cerebral hemorrhage with amyloidosis (Dutch). Science 248, 1120–1122. doi: 10.1126/science.1971458
Van Der Vliet, A., Nguyen, M., Shigenaga, M., Eiserich, J., Marelich, G., and Cross, C. (2000). Myeloperoxidase and protein oxidation in cystic fibrosis. Am. J. Physiol. Lung Cell Mol. Physiol. 279, L537–L546. doi: 10.1152/ajplung.2000.279.3.L537
Vázquez, G., Caballero, A., Kokinda, J., Hijano, A., Sabaté, R., and Gamez, P. (2019). Copper, dityrosine cross-links and amyloid-β aggregation. J. Biol. Inorg. Chem. 24, 1217–1229. doi: 10.1007/s00775-019-01734-6
Viles, J. H. (2012). Metal ions and amyloid fiber formation in neurodegenerative diseases. Copper, zinc and iron in Alzheimer’s, Parkinson’s, and prion diseases. Coord. Chem. Rev. 256, 2271–2284.
Villemagne, V., Perez, K., Pike, K., Kok, W., Rowe, C., White, A., et al. (2010). Blood-borne amyloid-beta dimer correlates with clinical markers of Alzheimer’s disease. J. Neurosci. 30, 6315–6322. doi: 10.1523/JNEUROSCI.5180-09.2010
von Bergen, M., Barghorn, S., Biernat, J., Mandelkow, E. M., and Mandelkow, E. (2005). Tau aggregation is driven by a transition from random coil to beta sheet structure. Biochim. Biophys. Acta Mol. Basis Dis. 1739, 158–166. doi: 10.1016/j.bbadis.2004.09.010
von Bergen, M., Friedhoff, P., Biernat, J., Heberle, J., Mandelkow, E., and Mandelkow, E. (2000). Assembly of tau protein into Alzheimer paired helical filaments depends on a local sequence motif ((306)VQIVYK(311)) forming beta structure. Proc. Natl. Acad. Sci. U.S.A. 97, 5129–5134. doi: 10.1073/pnas.97.10.5129
Walsh, D. M., and Selkoe, D. J. (2007). A beta oligomers – A decade of discovery. J. Neurochem. 101, 1172–1184.
Wang, J., Dickson, D. W., Trojanowski, J. Q., and Lee, V. M. (1999). The levels of soluble versus insoluble brain Abeta distinguish Alzheimer’s disease from normal and pathologic aging. Exp. Neurol. 158, 328–337. doi: 10.1006/exnr.1999.7085
Wang, Y. P., Biernat, J., Pickhardt, M., Mandelkow, E., and Mandelkow, E. M. (2007). Stepwise proteolysis liberates tau fragments that nucleate the Alzheimer-like aggregation of full-length tau in a neuronal cell model. Proc. Natl. Acad. Sci. U.S.A. 104, 10252–10257. doi: 10.1073/pnas.0703676104
Wang, Y., Loomis, P., Zinkowski, R., and Binder, L. (1993). A novel tau transcript in cultured human neuroblastoma cells expressing nuclear tau. J. Cell Biol. 121, 257–267. doi: 10.1083/jcb.121.2.257
Weingarten, M., Lockwood, A., Hwo, S., and Kirschner, M. (1975). A protein factor essential for microtubule assembly. Proc. Natl. Acad. Sci. U.S.A. 72, 1858–1862. doi: 10.1073/pnas.72.5.1858
Wells-Knecht, M., Huggins, T., Dyer, D., Thorpe, S., and Baynes, J. (1993). Oxidized amino acids in lens protein with age. Measurement of o-tyrosine and dityrosine in the aging human lens. J. Biol. Chem. 268, 12348–12352.
Wilhelmus, M., Grunberg, S., Bol, J., van Dam, A., Hoozemans, J., Rozemuller, A., et al. (2009). Transglutaminases and transglutaminase-catalyzed cross-links colocalize with the pathological lesions in Alzheimer’s disease brain. Brain Pathol. 19, 612–622. doi: 10.1111/j.1750-3639.2008.00197.x
Williams, T. L., Serpell, L. C., and Urbanc, B. (2016). Stabilization of native amyloid β-protein oligomers by copper and hydrogen peroxide induced cross-linking of unmodified proteins (Chicup). Biochim. Biophys. Acta 1864, 249–259. doi: 10.1016/j.bbapap.2015.12.001
Wilson, D. M., and Binder, L. I. (1997). Free fatty acids stimulate the polymerization of tau and amyloid beta peptides. In vitro evidence for a common effector of pathogenesis in Alzheimer’s disease. Am. J. Pathol. 150, 2181–2195.
Wischik, C., Novak, M., Thøgersen, H., Edwards, P., Runswick, M., Jakes, R., et al. (1988b). Isolation of a fragment of tau derived from the core of the paired helical filament of Alzheimer disease. Proc. Natl. Acad. Sci. U.S.A. 85, 4506–4510. doi: 10.1073/pnas.85.12.4506
Wischik, C., Novak, M., Edwards, P., Klug, A., Tichelaar, W., and Crowther, R. (1988a). Structural characterization of the core of the paired helical filament of Alzheimer disease. Proc. Natl. Acad. Sci. U.S.A. 85, 4884–4888. doi: 10.1073/pnas.85.13.4884
Wood, J. G., Suzanne, S. M., Nancy, J. P., and Binder, L. I. (1986). Microtubule-associated protein tau (tau) is a major antigenic component of paired helical filaments in Alzheimer disease. Proc. Natl. Acad. Sci. U.S.A. 83, 4044–4048. doi: 10.1073/pnas.83.11.4044
Wu, H., Hudry, E., Hashimoto, T., Kuchibhotla, K., Rozkalne, A., Fan, Z., et al. (2010). Amyloid beta induces the morphological neurodegenerative triad of spine loss, dendritic simplification, and neuritic dystrophies through calcineurin activation. J. Neurosci. 30, 2636–2649. doi: 10.1523/JNEUROSCI.4456-09.2010
Yoburn, J., Tian, W., Brower, J., Nowick, J., Glabe, C., and Van Vranken, D. (2003). Dityrosine cross-linked Abeta peptides: Fibrillar beta-structure in Abeta(1-40) is conducive to formation of dityrosine cross-links but a dityrosine cross-link in Abeta(8-14) does not induce beta-structure. Chem. Res. Toxicol. 16, 531–535. doi: 10.1021/tx025666g
Yoshikai, S., Sasaki, H., Doh-Ura, K., Furuya, H., and Sakaki, Y. (1990). Genomic organization of the human amyloid beta-protein precursor gene. Gene 87, 257–263.
Zhang, S., Fox, D. M., and Urbanc, B. (2017). Insights into formation and structure of Aβ oligomers cross-linked via tyrosines. J. Phys. Chem. B 121, 5523–5535. doi: 10.1021/acs.jpcb.7b02495
Zhang, Y., Lu, L., Jia, J., Jia, L., Geula, C., Pei, J., et al. (2014). A lifespan observation of a novel mouse model: In vivo evidence supports aβ oligomer hypothesis. PLoS One 9:e85885. doi: 10.1371/journal.pone.0085885
Keywords: Alzheimer’s disease, amyloid-beta, tau, oxidative, dityrosine
Citation: Maina MB, Al-Hilaly YK and Serpell LC (2023) Dityrosine cross-linking and its potential roles in Alzheimer’s disease. Front. Neurosci. 17:1132670. doi: 10.3389/fnins.2023.1132670
Received: 27 December 2022; Accepted: 01 March 2023;
Published: 22 March 2023.
Edited by:
Jinghui Luo, Paul Scherrer Institut (PSI), SwitzerlandReviewed by:
Sean Chia, Bioprocessing Technology Institute (A*STAR), SingaporeCopyright © 2023 Maina, Al-Hilaly and Serpell. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Louise C. Serpell, L.C.Serpell@sussex.ac.uk
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.