
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
BRIEF RESEARCH REPORT article
Front. Neurosci. , 24 May 2023
Sec. Neurodegeneration
Volume 17 - 2023 | https://doi.org/10.3389/fnins.2023.1095761
This article is part of the Research Topic Prodromal Stage of Neurodegenerative Proteinopathies: From Bench to Bedside View all 5 articles
 Victoria Morley1
Victoria Morley1 Karamjit Singh Dolt1
Karamjit Singh Dolt1 Carlos J. Alcaide-Corral2
Carlos J. Alcaide-Corral2 Tashfeen Walton2Christophe Lucatelli2
Tashfeen Walton2Christophe Lucatelli2 Tomoji Mashimo3
Tomoji Mashimo3 Adriana A. S. Tavares2*
Adriana A. S. Tavares2* Tilo Kunath1*
Tilo Kunath1*Parkinson’s disease (PD) is a neurodegenerative condition with several major hallmarks, including loss of substantia nigra neurons, reduction in striatal dopaminergic function, and formation of α-synuclein-rich Lewy bodies. Mutations in SNCA, encoding for α-synuclein, are a known cause of familial PD, and the G51D mutation causes a particularly aggressive form of the condition. CRISPR/Cas9 technology was used to introduce the G51D mutation into the endogenous rat SNCA gene. SNCAG51D/+ and SNCAG51D/G51D rats were born in Mendelian ratios and did not exhibit any severe behavourial defects. L-3,4-dihydroxy-6-18F-fluorophenylalanine (18F-DOPA) positron emission tomography (PET) imaging was used to investigate this novel rat model. Wild-type (WT), SNCAG51D/+ and SNCAG51D/G51D rats were characterized over the course of ageing (5, 11, and 16 months old) using 18F-DOPA PET imaging and kinetic modelling. We measured the influx rate constant (Ki) and effective distribution volume ratio (EDVR) of 18F-DOPA in the striatum relative to the cerebellum in WT, SNCAG51D/+ and SNCAG51D/G51D rats. A significant reduction in EDVR was observed in SNCAG51D/G51D rats at 16 months of age indicative of increased dopamine turnover. Furthermore, we observed a significant asymmetry in EDVR between the left and right striatum in aged SNCAG51D/G51D rats. The increased and asymmetric dopamine turnover observed in the striatum of aged SNCAG51D/G51D rats reflects one aspect of prodromal PD, and suggests the presence of compensatory mechanisms. SNCAG51D rats represent a novel genetic model of PD, and kinetic modelling of 18F-DOPA PET data has identified a highly relevant early disease phenotype.
Parkinson’s disease (PD) is a common progressive neurodegenerative condition affecting ~1% of people over the age of 60 (Reeve et al., 2014). The condition results in deficits of motor function including abnormal posture and gait, tremor, and rigidity (Gelb et al., 1999; Lees et al., 2009). Characteristic neuropathological findings include the loss of neurons from the substantia nigra pars compacta (SNpc) which project axons to the striatum where dopamine (DA) is released (Bernheimer et al., 1973). The pathological hallmark of PD is the formation of Lewy bodies and Lewy neurites, complex intracellular inclusions abundant in α-synuclein protein (Gibb and Lees, 1988; Spillantini et al., 1997). Cases of familial PD have been shown to result from mutations in SNCA, encoding α-synuclein, and the G51D mutation (c.152 G > A) has been identified to cause an early onset and aggressive form of PD with associated dementia and multiple system atrophy (Kiely et al., 2013; Lesage et al., 2013).
It has been extensively reported that PD patients have a decreased number of tyrosine hydroxylase (TH) positive neurons in the striatum when compared to controls, and this occurs when the condition has significantly progressed (Huot et al., 2007; Kordower et al., 2013). However, prior to significant neuronal loss, earlier deficits in presynaptic dopaminergic function in the striatum of PD patients can be investigated using positron emission tomography (PET) and the radiotracer L-3,4-dihydroxy-6-18F-fluorophenylalanine (18F-DOPA) (Garnett et al., 1983; Nahmias et al., 1985). 18F-DOPA is metabolized in dopaminergic nerve terminals by the enzyme aromatic L-amino acid decarboxylase (AADC) to produce 18F-DA, which is incorporated into synaptic vesicles and then released into the synaptic cleft following neuronal stimulation (Firnau et al., 1987). In patients with PD, dopaminergic function in the striatum is impaired, and the influx rate constant (Ki) of 18F-DOPA is significantly decreased in the caudate and putamen (Brooks et al., 1990; Snow et al., 1993; Holthoff-Detto et al., 1997). Dopamine turnover can be measured with extended scanning time, and the effective distribution volume ratio (EDVR) of 18F-DOPA can be calculated (Sossi et al., 2001). A decreased EDVR in the striatum indicates an increase in dopamine turnover, and this was observed in newly diagnosed PD patients relative to healthy controls prior to a decrease in Ki (Sossi et al., 2002). An increase in dopamine turnover was also observed in asymptomatic LRRK2 mutation carriers before any evidence of changes in Ki (Sossi et al., 2010). This has been hypothesized to be a compensatory mechanism in early PD potentially due to upregulation of AADC decarboxylase activity (Lee et al., 2000; Adams et al., 2005). The rat is easily amenable to 18F-DOPA PET imaging and correlation with numbers of surviving dopaminergic neurons in the 6-hydroxy-dopamine (6-OHDA) lesion rat model of PD has been demonstrated (Kyono et al., 2011). Furthermore, in vivo dopaminergic compensation has been reported in this rat 6-OHDA lesion model of PD (Sossi et al., 2009). The MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) lesion model in monkeys have also identified a very close correlation with 18F-DOPA uptake in the striatum, and multiple measures of parkinsonism, include symptoms on the Kurlan scale, striatal dopamine content, and markers of dopaminergic neurons, such as tyrosine hydroxylase (Blesa et al., 2012). PET imaging with 18F-DOPA has also been applied to genetic rodent models of PD. Weaver mutant mice have a point mutation in the GIRK2 potassium channel and 18F-DOPA PET imaging non-invasively identified a dopaminergic deficit in the striatum and this deficit worsened with increasing age (Sharma and Ebadi, 2005; Sharma et al., 2006).
Genetic mouse and rat models of PD have been generated using different approaches with variable success. Although the majority of transgenic and knock-out/knock-in models are in the mouse, there have been several reported rat models. One transgenic rat model has been produced with a human SNCA construct with two PD mutations (A30P and A53T) under control of the rat TH promoter, and the major deficits reported in this model were olfactory (Lelan et al., 2011). A bacterial artificial chromosome (BAC) transgenic rat model expressing human SNCA with the E46K mutation identified a trend for decreased immunostaining for TH in the striatum and α-synuclein aggregates were identified in neuronal processes in the striatum (Cannon et al., 2013). A spontaneous non-coding SNCA mutation in a Sprague–Dawley rat colony was found to significantly increase SNCA expression in the brain, caused accumulation of nitrosylated and phosphorylated α-synuclein protein, and lead to a significant impairment of dopamine release in the striatum (Stoica et al., 2012; Guatteo et al., 2017). The most representative genetic rat model of PD to date used a human BAC transgene to over-express WT human SNCA (BAC-hSNCA) (Nuber et al., 2013). Transgenic rats were observed to have significantly decreased expression of TH in the striatum as well as significantly decreased striatal dopamine levels in aged BAC-hSNCA rats compared with healthy controls (Nuber et al., 2013). Furthermore, neuritic α-synuclein pathology was identified in the striatum of aged transgenic rats and significant motor deficits were observed from 12 months of age (Nuber et al., 2013). A subsequent study found that BAC-hSNCA rats have a significant early-onset (4 months) serotonergic deficit leading to anxiety-like phenotypes prior to the onset of motor deficits (Kohl et al., 2016).
To date the majority of genetic rat models of PD have been generated by the random insertion of a transgene or the knock-out of a PD-related gene (Creed and Goldberg, 2018). Replicating the precise single amino acid substitution observed in PD kindreds has become possible in the rat with Clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated protein 9 (Cas9) genome engineering (Yoshimi et al., 2014). Here, we used CRISPR/Cas9 and a donor oligonucleotide in rat zygotes to mutate glycine-51 to aspartic acid (G51D) in rat SNCA exon 3 to produce a novel SNCAG51D rat model of PD. Striatal dopamine metabolism and turnover were investigated in SNCAG51D/+ and SNCAG51D/G51D rats over the course of ageing using 18F-DOPA PET imaging and kinetic modelling.
CRISPR/Cas9 constructs used were the gRNA_Cloning vector (a gift from George Church, Addgene plasmid # 418241; RRID:Addgene_41824) and humanized Cas9 nuclease vector (a gift from George Church, Addgene plasmid # 418152; RRID:Addgene_41815) (Mali et al., 2013). The gRNA sequence was designed, synthesized, and inserted into the gRNA_Cloning vector into the AflII site. The gRNA sequence is: 5′-GTCGTTCATGGAGTGACAAC-3′. The gRNA and Cas9 vectors were each linearized with XhoI and in vitro transcribed using a MessageMAXT7 ARCA-Capped Message Transcription Kit (CELLSCRIPT, Madison, WI, USA). A 80-nt ssDNA mutant donor oligonucleotide with the desired 2-bp mutation (GGA to GAT) was synthesized. The sequence of the ssDNA donor is: 5′-CAATTCTTTTTTTAGGTTCCAAAACTAAGGAGGGAGTCGTTCATGatGTGACAACAGGTAAGCTCTGTTGTCTTTTATCC-3′. The gRNA, Cas9 mRNA, and ssDNA donor oligonucleotide were co-injected into male pronuclei of F344/Stm rat zygotes. Eleven (11) founder rats were screened by Sanger sequencing. Five (5) founders had mutations in exon 3 of SNCA, and one founder had the desired GGA to GAT (G51D) mutation (Supplementary Figure S1). This founder was mated to produce F1 progeny, and the mutation transmitted through the germ-line as determined by Sanger sequencing of genomic DNA extracted from ear notches (Figure 1A). Since the mutation introduced a new BspHI restriction site (Figure 1B), subsequent genotyping was performed by SNCA exon 3 PCR (forward primer 5′-TGGTGGCTGTTTGTCTTCTG-3′ and reverse primer 5′-TCCTCTGAAGACAATGGCTTTT-3′), a BspHI restriction enzyme digest, and agarose gel electrophoresis (Figure 1C).
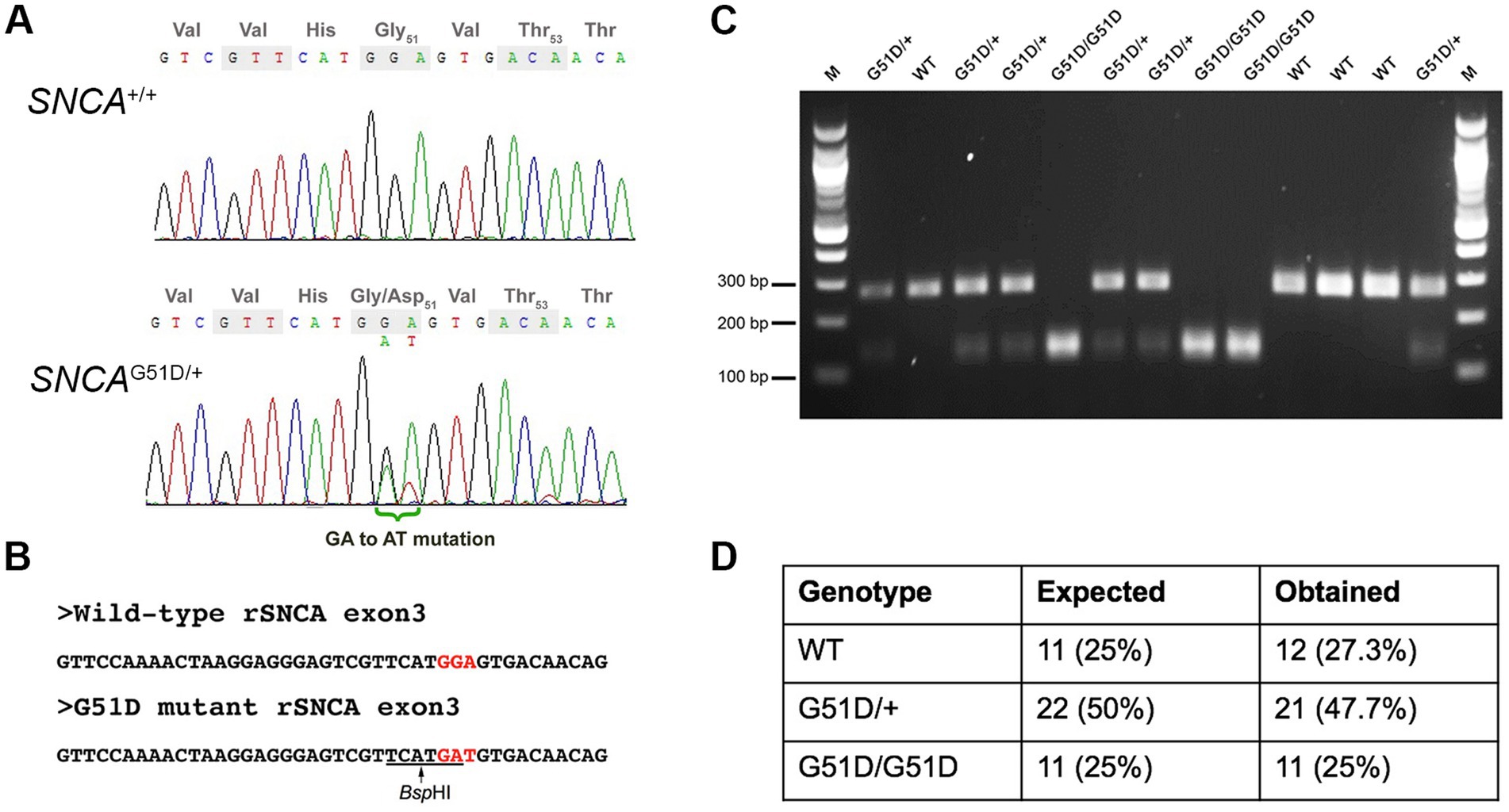
Figure 1. Genotyping of SNCAG51D rats. (A) Sanger sequencing of rat SNCA exon 3 of a WT and an SNCAG51D/+ F1 rat revealed a heterozygous mutation (GGA/GAT) at codon 51. (B) The G51D mutation generated a new BspHI restriction site (TCATGA) in exon 3. (C) SNCA exon 3 PCR and BspHI restriction enzyme digest can reliably genotype WT, SNCAG51D/+ and SNCAG51D/G51D rats. (D) 44 neonatal (P2, P3) pups from five (5) SNCAG51D/+ X SNCAG51D/+ crosses were genotyped, and Mendelian ratios were observed.
The breeding and maintenance of wild-type (WT), SNCAG51D/+ and SNCAG51D/G51D rats, and the in vivo experiments conducted were approved by the UK Home Office under project licence PC6C08D7D. WT, SNCAG51D/+ and SNCAG51D/G51D rats were analyzed over the course of ageing at 5, 11, and 16 months using 18F-DOPA PET imaging and no rats were re-used for repeat scanning (n = 4 per genotype per age-group, for a total of 36 rats).
18F-DOPA was produced using a multi-step nucleophilic fluorination pathway, and used the TRACERlab MX synthesiser and cassette with the nucleophilic precursor to 18F-DOPA (ABX Advanced Biochemical Compounds, PEDP-0062-H), with the final product formulated in PBS (Martin et al., 2013).
Imaging was conducted under general anesthesia and used Isoflurane in 0.6 L/min O₂ and 0.4 L/min N2O. 10 mg/kg Carbidopa (Sigma-Aldrich, C1335), then 10 mg/kg Entacapone (Sigma-Aldrich, SML-0654) were injected intravenously to prevent the peripheral metabolism of 18F-DOPA by AADC and catechol-O-methyltransferase (COMT), respectively (Walker et al., 2013a). Thirty minutes after injection of Carbidopa and Entacapone, 18.5 +/− 7.1 MBq (mean +/− SD) of 18F-DOPA was injected as a bolus via the tail vein. Activity in the empty syringe was measured, with the activity injected calculated and decay corrected. Dynamic PET imaging lasted 2 h.
Images were obtained using the nanoScan PET/CT scanner (Mediso Medical Imaging Systems, Budapest, Hungary), with PET/CT data acquired using Nucline™ v2.01 acquisition software. Prior to radiotracer injection a scout view CT image was acquired. Dynamic PET imaging commenced upon injection of 18F-DOPA and used a coincidence mode 1–5 and coincidence time window of 5 ns. After PET imaging, CT data was acquired (trajectory semi-circular, maximum field of view, 480 projections, 55 kVp and 300 ms, binning 1:4) and used for attenuation correction of PET data.
The PET data that was reconstructed and extended from the olfactory bulbs to the caudal border of the heart. Reconstruction was 3D, dynamic and used the TeraTomo3D reconstruction method. Coincidence mode was 1–3, voxel size 0.4 mm × 0.4 mm × 0.4 mm, used 4 iterations and 6 subsets, and was corrected for scatter, attenuation and randoms. Data was reconstructed into frames comprising 6 frames of 30 s, 3 frames of 60 s, 2 frames of 120 s and 22 frames of 300 s. All PET imaging data is freely accessible.3
Images were analyzed using PMOD 3.409 software (PMOD Technologies LLC) and a hand-drawn template. Volumes of interest (VOIs) comprised the left and right striatum, whole striatum, and the cerebellum which was a reference region for non-specific uptake. The same VOI template was used for all rats and were only moved into position over the respective anatomical areas. PET images for display purposes and to aid with VOI drawing were obtained by averaging over 0–120 min of emission data then a 1 mm × 1 mm × 1 mm Gaussian filter was applied. Standardized uptake value (SUV) images were produced using rat body weight and the activity injected. Similarly, SUV TACs were calculated as follow SUV (g/mL) = activity concentration in the target VOI (kBq/mL)/[decay corrected amount of 18F-DOPA injected (MBq)/weight of the rat (kg)].
Kinetic modelling used the Patlak reference tissue model and Logan reference tissue model to determine the Ki of 18F-DOPA in the striatum and the distribution volume ratio (DVR) of 18F-DOPA in the striatum relative to the cerebellum, respectively (Logan, 2000). The cerebellum is used as a reference brain region since dopamine receptor binding in rat cerebellum is much lower than in striatum and equivalent to background (Kuhar et al., 1978), and PET imaging in humans demonstrated the cerebellum is devoid of any dopaminergic signals (Wong et al., 1986). Furthermore, PD patients do not exhibit any Lewy pathology in the cerebellum (Devi et al., 2008). The Patlak reference tissue model used 60 min of data and a t* of 10 min in accordance with previous studies in Sprague Dawley rats (Kyono et al., 2011). This is a graphical analysis method that models the irreversible uptake of radiotracers (Patlak and Blasberg, 1985). In contrast, reversible tracer uptake was analyzed with the Logan reference tissue model using 120 min of data and t* of 30 min, which were parameters optimized from our WT F344 rat data and these differ from other parameters used to analyze PET data from Sprague Dawley rats (Walker et al., 2013a). The Logan reference tissue model was also used to determine the effective DVR (EDVR) of 18F-DOPA which involved subtracting the TAC (kBq/mL) for the cerebellum from the TACs for the striatum before analysis (Walker et al., 2013a). EDVR can be used to estimate the effective dopamine turnover (Sossi et al., 2002). Differences in EDVR between left and right striatum were also investigated by calculating asymmetry in EDVR which = (EDVR contralateral − EDVR ipsilateral)/EDVR contralateral (Walker et al., 2013b). All Ki, DVR, EDVR, and asymmetry EDVR data for each individual rat is supplied in Supplementary Tables 1, 2.
18F-DOPA PET data from WT, SNCAG51D/+, and SNCAG51D/G51D rats are presented where appropriate as the mean +/− standard deviation. Ki, DVR, and EDVR results from age-matched WT, SNCAG51D/+ and SNCAG51D/G51D rats were analyzed using a 1-way ANOVA with Tukey’s multiple comparison test. Left–right EDVR asymmetry results were investigated for statistical significance using a paired t test. Although the sample size is small for each experimental variable (n = 4 per genotype per time-point), quantile-quantile plots of the Ki, DVR, and EDVR data-sets indicated the data is normally distributed.
The G51D mutation in PD patients is due to a single nucleotide change (c.152 G > A) that mutates the Gly-51 codon (GGT) to GAT, encoding for aspartic acid. The rat SNCA Gly-51 codon is GGA, which requires a 2-nucleotide mutation to produce the Asp-51 codon GAT. Cas9 mRNA, gRNA and ssDNA donor oligonucleotide with GAT codon were co-injected into F344/Stm rat zygotes. Eleven founder rats were screened by Sanger sequencing and 5 F0 rats had mutations in SNCA, but only one founder had the desired GGA to GAT (G51D) mutation in exon 3 of SNCA (Supplementary Figure S1). This F0 founder rat was bred, and F1 animals with the G51D mutation were identified by Sanger sequencing confirming the mutation could transmit through the germ-line (Figure 1A). All subsequent progeny from the sequence-confirmed F1 rats were PCR-genotyped taking advantage of a new BspHI restriction site introduced by the G51D mutation (Figures 1B,C). Heterozygous SNCAG51D/+ matings produced progeny in Mendelian ratios suggesting the mutation did not reduce embryonic or fetal viability (Figure 1D).
The methods used for the reconstruction of in vivo PET imaging experiments were optimized since the rat striatum is a small structure and the optimal acquisition and reconstruction of PET data is scanner dependent. A homogenous solution of 18F-FDG was used in a National Electrical Manufacturers Association (NEMA) NU-4 mouse image quality (IQ) phantom (Harteveld et al., 2011). Imaging data was acquired and reconstructed using variations of several parameters (Supplementary Figure S2). The most promising reconstruction scenario used iterative methods and employed 4 iterations and 6 subsets (4i6S) at a normal resolution and a coincidence mode of 1–3. The in vivo imaging and kinetic modelling methods were optimized using data obtained from preliminary experiments using two WT F344 rats (Figure 2). Time activity curves (TACs) and standardized uptake values (SUV) were calculated and plotted for striatum and cerebellum (Figures 2B,C). The cerebellum was used as a reference region since it lacks AADC expression (Baker et al., 1991), and dopamine receptor binding in rat cerebellum is similar to background (Kuhar et al., 1978). Plotting the SUV ratio (SUVr) of the striatum and cerebellum for both rats showed the data was consistent and that the activity reached a pseudo-equilibrium between 50 and 85 min (Figure 2D). This data indicated that methods used for kinetic modelling in recent PET imaging studies of models of PD were optimal (Kyono et al., 2011). Kinetic modeling using Patlak and Logan graphical analysis used data collected over 60 and 180 min, respectively (Figure 3).
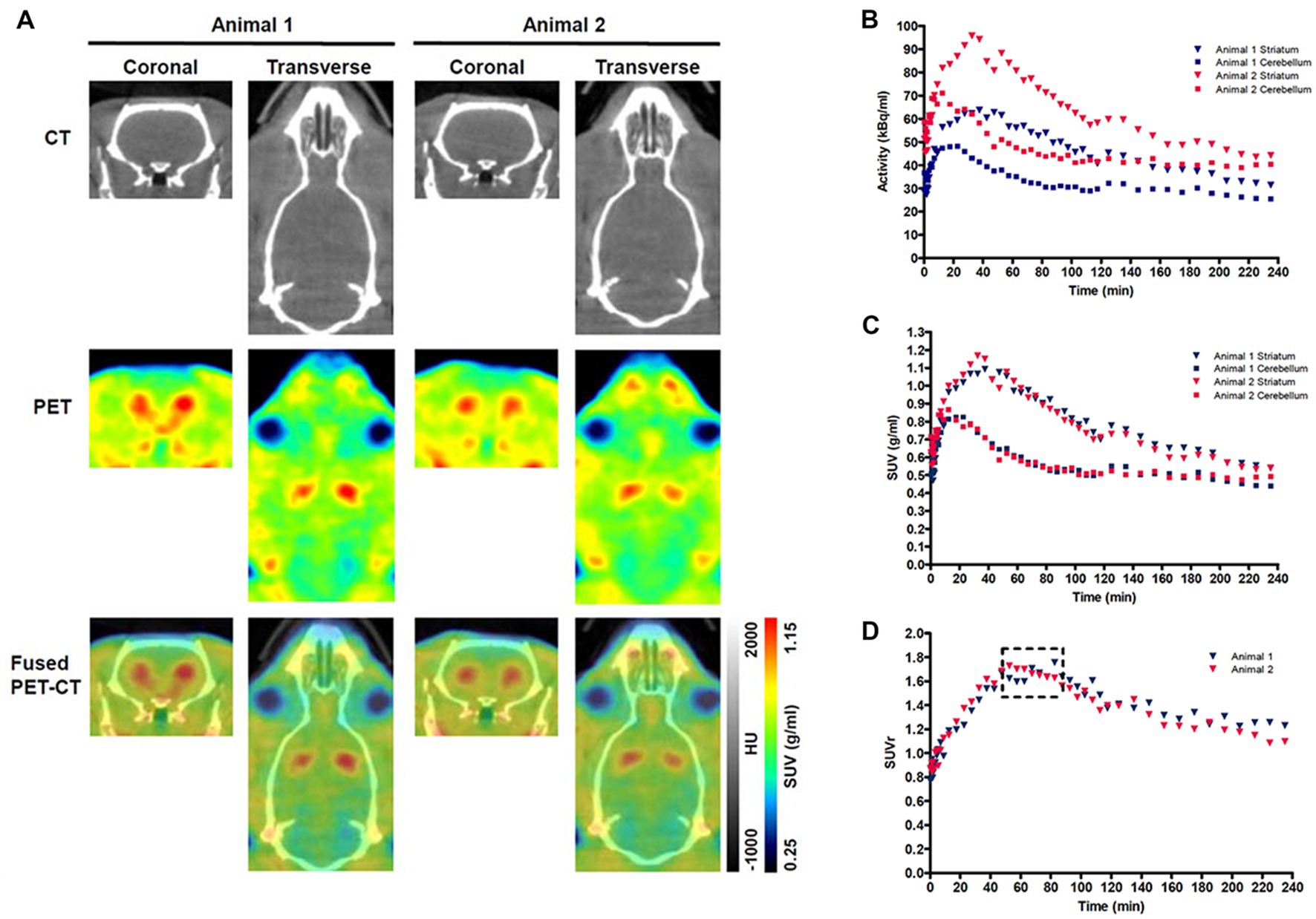
Figure 2. Representative PET-CT images and Standardized uptake value (SUV) time activity curves (TACs) for wild-type (WT) rats. (A) PET-CT images of two WT rats are shown in coronal and transverse planes. 18F-DOPA PET data averaged over frames 1–33 and smoothed using a 1 mm × 1 mm × 1 mm Gaussian filter. HU-Hounsfield Units. Specific activity (B), SUV TAC data (C), and SUV ratio (SUVr) data (D) for the specific uptake of 18F-DOPA into the striatum relative to the cerebellum is shown for two WT rats. The dashed box indicates the phase of pseudo-equilibrium (50–85 min).
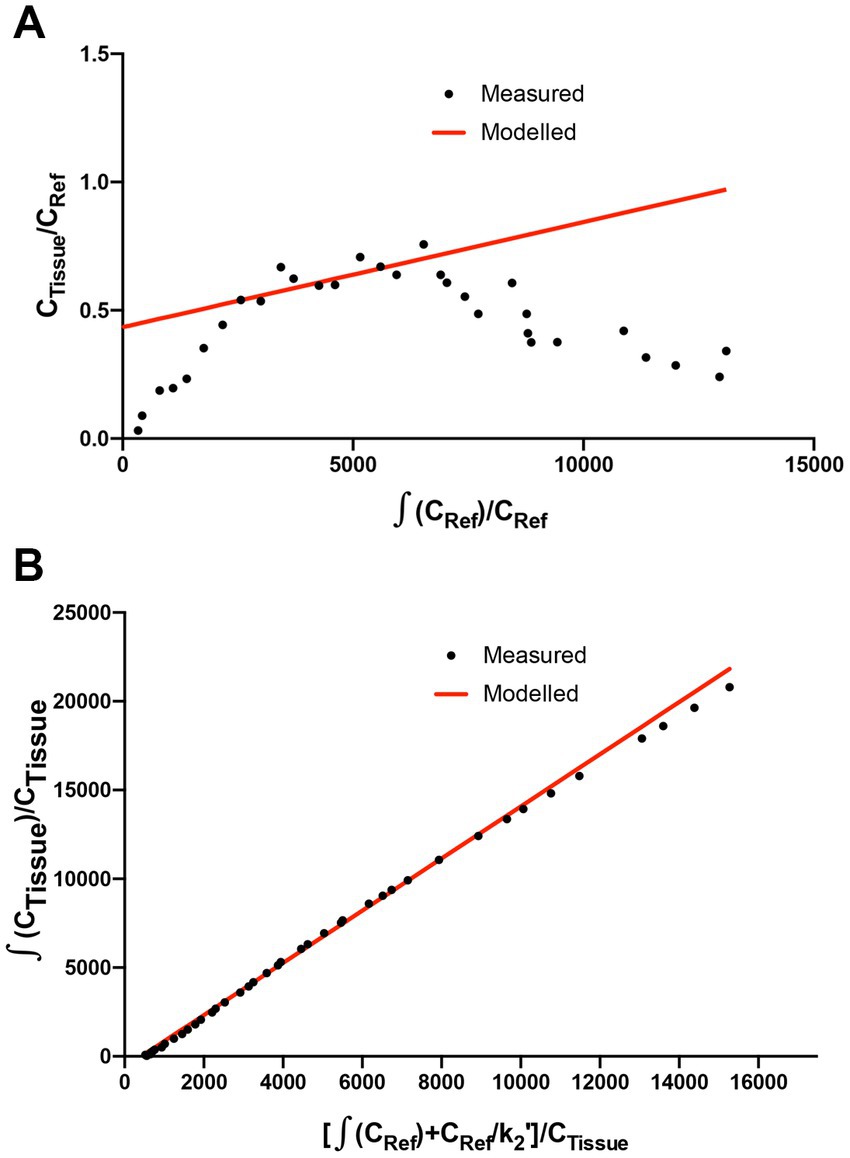
Figure 3. Patlak and Logan analysis for WT rats. (A) Patlak graphical analysis of 60 min of data (t* of 10 min) from the whole striatum of a WT rat relative to cerebellum. (B) Logan graphical analysis of 180 min of data (t* of 130 min) from the whole striatum of a WT rat.
WT, SNCAG51D/+ and SNCAG51D/G51D rats were subjected to 18F-DOPA PET at 5, 11, and 16 months of age using the conditions optimized above. A total of 36 rats were scanned with 4 rats per genotype per age-group, and no rats were re-scanned for longitudinal studies. Data was analyzed to determine SUV TACs from WT, SNCAG51D/+ and SNCAG51D/G51D rats (Figure 4A), and plotted to show the specific uptake of 18F-DOPA into the striatum compared with the cerebellum.
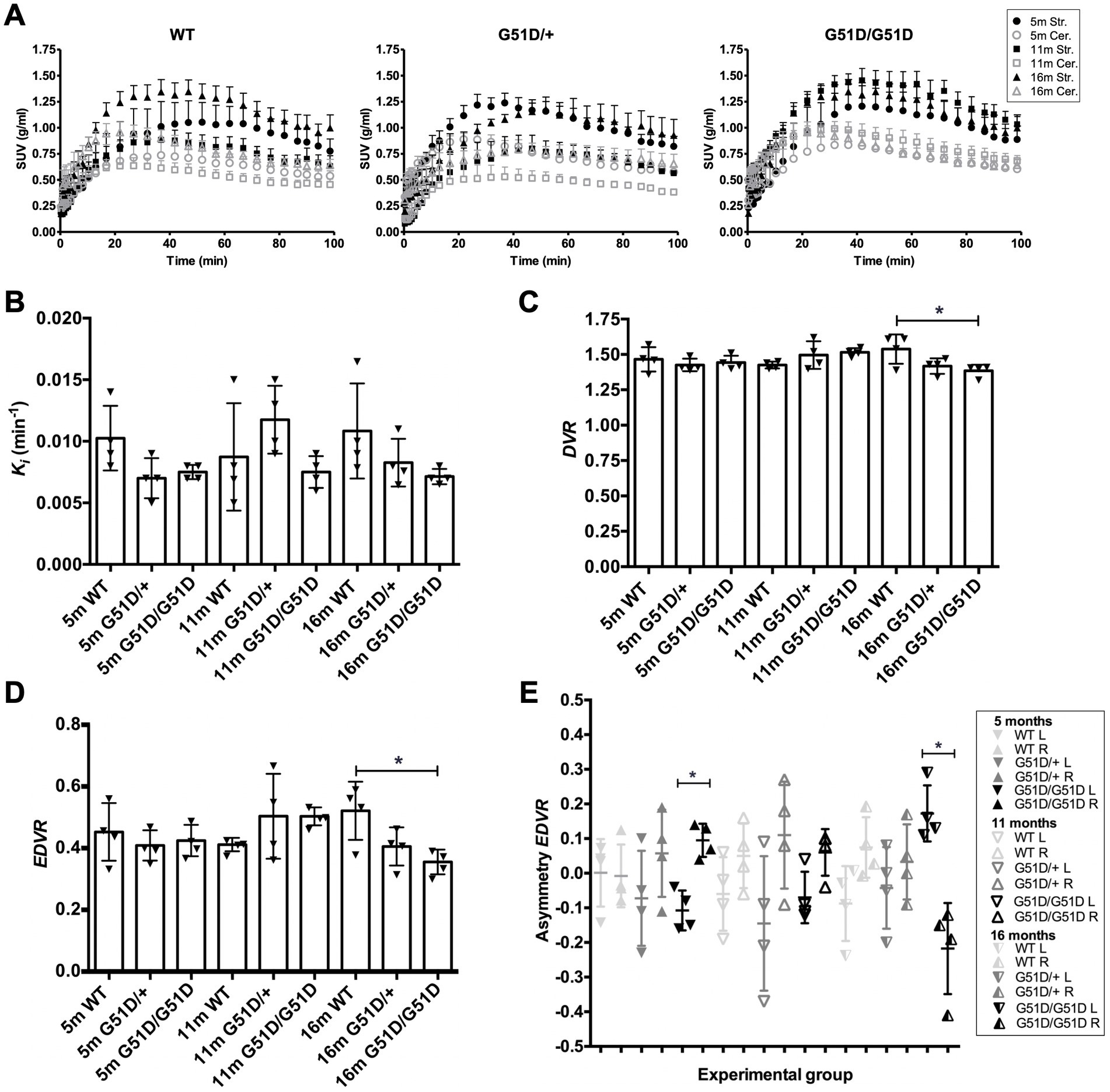
Figure 4. Kinetic modelling of 18F-DOPA PET data from WT, SNCAG51D/+ and SNCAG51D/G51D rats at 5, 11, and 16 months of age. (A) Standardized uptake value (SUV) time activity curves (TACs) for all rat genotypes—WT, SNCAG51D/+, and SNCAG51D/G51D rats, n = 4 per genotype per age-group. m-months old, Str.-Striatum, Cer.-Cerebellum. (B) The mean Ki in the striatum of 5–16 month old WT, SNCAG51D/+ and SNCAG51D/G51D rats. (C) There was a significant decrease in mean DVR of 18F-DOPA in 16 month old SNCAG51D/G51D rats when compared with age-matched WT rats (*p < 0.05), and (D) also a significant decrease in EDVR of 18F-DOPA in 16 months SNCAG51D/G51D rats compared with age-matched WT rats (*p < 0.05). (E) Mean left–right asymmetry in EDVR was significantly different in 5 month old and 16 month old SNCAG51D/G51D rats. Data shows the mean, standard deviation and individual data-points. n = 4 per genotype per age-group. Paired t test. *p < 0.05 L-left striatum, R-right striatum, m-months old.
The mean Ki values of 18F-DOPA in 5–16 month old SNCAG51D/+ and SNCAG51D/G51D rats compared with age-matched WT rats were not significantly different (Figure 4B). The mean DVR and EDVR of 18F-DOPA in the striatum relative to the cerebellum was significantly decreased in 16 month old rats compared to WT rats, but these differences were not observed at 5 and 11 months of age (Figures 4C,D) (Supplementary Table 1). The EDVR of 18F-DOPA is the ratio of the distribution volumes of 18F-DOPA in the specific and precursor compartments reduced by the factor k2/(k2 + k3), and since EDVR is estimated to be the inverse of effective dopamine turnover (Sossi et al., 2002), the results indicate an increase in mean dopamine turnover in 16 month SNCAG51D/G51D rats compared with age-matched WT rats. Since early Parkinson’s often presents with an asymmetric dopaminergic deficit (Djaldetti et al., 2006), we investigated this phenotype in our rat model. Interestingly, the mean EDVR of 18F-DOPA in the left and right striatum of 5 month old and 16 month old SNCAG51D/G51D rats were significantly different, but this was not observed in age-matched SNCAG51D/+ and WT rats (Figure 4E). The EDVR asymmetry in SNCAG51D/G51D rats was between −0.4 and 0.3, while the normal range determined for Sprague Dawley rats is between −0.1 and 1.0 (Walker et al., 2013b).
The aim of the study was to characterize a novel SNCAG51D rat model of Parkinson’s disease using 18F-DOPA PET imaging. Experiments were conducted over an ageing time-course, since phenotypes were anticipated to worsen with time. In patients with PD, striatal dopaminergic function decreases prior to degeneration of nerve terminals (Morrish et al., 1995, 1998; Nurmi et al., 2001).
18F-DOPA PET imaging data indicated no significant differences in mean Ki of 18F-DOPA in the striatum of ageing SNCAG51D/+ and SNCAG51D/G51D rats compared with age-matched WT rats (Figure 4B). However, there was a significant decreased mean DVR and EDVR of 18F-DOPA in the striatum of 16 month old SNCAG51D/G51D rats compared with age-matched WT rats (Figures 4C,D), which indicates increased dopamine turnover in the aged SNCAG51D/G51D rats.
In early PD the EDVR of 18F-DOPA in the striatum relative to the cerebellum is significantly decreased compared with healthy controls, which indicates increased effective dopamine turnover and this is likely due to a compensatory change in response to striatal dysfunction (Sossi et al., 2002). This data is supported by results from 18F-DOPA PET imaging studies of asymptomatic MPTP lesioned monkeys which have also implicated increased effective dopamine turnover as a compensatory mechanism (Doudet et al., 1998). The significantly decreased mean EDVR in 16 month old SNCAG51D/G51D rats may indicate a compensatory change in dopaminergic function in the striatum of SNCAG51D/G51D rats following putative striatal dysregulation.
In PD the Ki of 18F-DOPA in the striatum is significantly decreased compared with healthy controls (Brooks et al., 1990; Burn et al., 1994; Holthoff-Detto et al., 1997; Rinne et al., 2000), with Ki in the putamen reaching 57%–80% of normal levels before symptoms of PD develop (Morrish et al., 1995, 1998; Nurmi et al., 2001). The lack of change in mean Ki in the striatum of SNCAG51D/+ and SNCAG51D/G51D rats suggests any potential phenotypes are being sufficiently compensated for in this model and reflects what is observed in patients with prodromal PD. In chemical lesion models of PD, TH has been found to be significantly increased in the striatum post lesioning, a compensatory mechanism in response to nerve damage, which either results in increased TH protein expression or involves morphological changes such as the expansion of nerve terminals (Blanchard et al., 1995; Bezard and Gross, 1998). Compensatory changes in dopaminergic terminals have also been shown to involve the upregulation of AADC activity and the downregulation of DATs (Lee et al., 2000; Adams et al., 2005). In 6-OHDA lesion models of PD, PET imaging studies have identified a complex relationship between the kref of 18F-DOPA and the binding potential of 11C-DTBZ (denervation severity) (Walker et al., 2013a).
Results measuring asymmetry in the EDVR of 18F-DOPA showed decreased mean EDVR in the left striatum when compared to the right in 5 out of 6 groups of rats, with greatest asymmetry in 16 month old SNCAG51D/G51D rats (Figure 4E). Reasons that may explain the predilection for the left side could be due to the unique anatomy of the blood vessel supply to the brain in F344 rats which may result in non-uniform perfusion in this strain of rat (Iwasaki et al., 1995).
SNCAG51D rats were generated using CRISPR/Cas9 technology to model the G51D α-synuclein mutation in humans. The G51D model has good construct validity, and mutant α-synuclein is expressed from the endogenous rat locus. SNCAG51D rats have the potential to model the widespread neurological abnormalities found in PD and may be a more representative model of PD than the focal 6-OHDA lesion model, which has previously been studied using 18F-DOPA PET imaging (Kyono et al., 2011; Walker et al., 2013a). Since rodent models of human disease are almost always less severe, we expected the SNCAG51D rats to have phenotypes similar to early stages of the disease (Petrucci et al., 2016). SNCAG51D may exhibit a subtle phenotype in rats due to protective factors such as β-synuclein which have been shown to the ameliorate PD-like phenotypes in mice (Hashimoto et al., 2001; Fan et al., 2006). Furthermore, rodents may lack key triggers or cellular components necessary for exhibiting a full PD phenotype. Differences in the SNCA sequence between rodents and humans may explain some of the difficulties in modelling genetic PD, since rodents, indeed most vertebrates, encode a threonine at position 53 which is a cause of PD in humans, and human A53T mice have demonstrated exaggerated motor deficits and α-synuclein pathology following removal of endogenous mouse α-synuclein (Cabin et al., 2005). The SNCAG51D rat model can be used to investigate sensitivity to potential environmental causes of Parkinson’s. Indeed, the E46K α-synuclein BAC transgenic rat model exhibited a significantly increased sensitivity to rotenone (Cannon et al., 2013).
SNCAG51D/G51D rats show a significant increase in dopamine turnover in the striatum at 16 months of age, but not a significant decrease in Ki – dopamine synthesis and storage. These findings mimic one component of the early stages of Parkinson’s, and may reflect the compensatory changes in the dopaminergic system observed in humans. SNCAG51D rats represent an interesting model of early PD pathophysiology, and provide a tractable platform for investigating additional genetic or environmental triggers of Parkinson’s.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary material.
The animal study was reviewed and approved by UK Home Office under project licence PC6C08D7D.
VM, AT, and TK devised the study. TM (Kyoto University) generated the SNCAG51D rat model. KD (University of Edinburgh) established genotyping of SNCAG51D rats, maintained the rat colony, and conducted preliminary studies. TW and CL (University of Edinburgh) synthesized the 18F-DOPA radiotracer, and CA-C (University of Edinburgh) provided assistance during in vivo 18F-DOPA PET imaging experiments. VM and AT designed and conducted PET experiments and data analysis. VM and TK wrote the manuscript and AT edited the manuscript. All authors contributed to the article and approved the submitted version.
This work was funded by a Carnegie Trust PhD Scholarship to VM, Parkinson’s UK Senior Fellowship to TK, and a Wellcome Trust ISSF award to TK and AT.
The authors would like to thank Yayoi Kunihiro for assistance in generation of the SNCAG51D rat model.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnins.2023.1095761/full#supplementary-material
Adams, J. R., van Netten, H., Schulzer, M., Mak, E., McKenzie, J., Strongosky, A., et al. (2005). PET in LRRK2 mutations: comparison to sporadic Parkinson's disease and evidence for presymptomatic compensation. Brain 128, 2777–2785. doi: 10.1093/brain/awh607
Baker, H., Abate, C., Szabo, A., and Joh, T. H. (1991). Species-specific distribution of aromatic L-amino acid decarboxylase in the rodent adrenal gland, cerebellum, and olfactory bulb. J. Comp. Neurol. 305, 119–129. doi: 10.1002/cne.903050111
Bernheimer, H., Birkmayer, W., Hornykiewicz, O., Jellinger, K., and Seitelberger, F. (1973). Brain dopamine and the syndromes of Parkinson and Huntington. Clinical, morphological and neurochemical correlations. J. Neurol. Sci. 20, 415–455. doi: 10.1016/0022-510X(73)90175-5
Bezard, E., and Gross, C. E. (1998). Compensatory mechanisms in experimental and human parkinsonism: towards a dynamic approach. Prog. Neurobiol. 55, 93–116. doi: 10.1016/S0301-0082(98)00006-9
Blanchard, V., Chritin, M., Vyas, S., Savasta, M., Feuerstein, C., Agid, Y., et al. (1995). Long-term induction of tyrosine hydroxylase expression: compensatory response to partial degeneration of the dopaminergic nigrostriatal system in the rat brain. J. Neurochem. 64, 1669–1679. doi: 10.1046/j.1471-4159.1995.64041669.x
Blesa, J., Pifl, C., Sánchez-González, M. A., Juri, C., García-Cabezas, M. A., Adánez, R., et al. (2012). The nigrostriatal system in the presymptomatic and symptomatic stages in the MPTP monkey model: a PET, histological and biochemical study. Neurobiol. Dis. 48, 79–91. doi: 10.1016/j.nbd.2012.05.018
Brooks, D. J., Ibanez, V., Sawle, G. V., Quinn, N., Lees, A. J., Mathias, C. J., et al. (1990). Differing patterns of striatal 18F-dopa uptake in Parkinson's disease, multiple system atrophy, and progressive supranuclear palsy. Ann. Neurol. 28, 547–555. doi: 10.1002/ana.410280412
Burn, D. J., Sawle, G. V., and Brooks, D. J. (1994). Differential diagnosis of Parkinson's disease, multiple system atrophy, and Steele-Richardson-Olszewski syndrome: discriminant analysis of striatal 18F-dopa PET data. J. Neurol. Neurosurg. Psychiatry 57, 278–284. doi: 10.1136/jnnp.57.3.278
Cabin, D. E., Gispert-Sanchez, S., Murphy, D., Auburger, G., Myers, R. R., and Nussbaum, R. L. (2005). Exacerbated synucleinopathy in mice expressing A53T SNCA on a SNCA null background. Neurobiol. Aging 26, 25–35. doi: 10.1016/j.neurobiolaging.2004.02.026
Cannon, J. R., Geghman, K. D., Tapias, V., Sew, T., Dail, M. K., Li, C., et al. (2013). Expression of human E46K-mutated α-synuclein in BAC-transgenic rats replicates early-stage Parkinson's disease features and enhances vulnerability to mitochondrial impairment. Exp. Neurol. 240, 44–56. doi: 10.1016/j.expneurol.2012.11.007
Creed, R. B., and Goldberg, M. S. (2018). New developments in genetic rat models of Parkinson's disease. Mov. Disord. 33, 717–729. doi: 10.1002/mds.27296
Devi, L., Raghavendran, V., Prabhu, B. M., Avadhani, N. G., and Anandatheerthavarada, H. K. (2008). Mitochondrial import and accumulation of alpha-synuclein impair complex I in human dopaminergic neuronal cultures and Parkinson disease brain. J. Biol. Chem. 283, 9089–9100. doi: 10.1074/jbc.M710012200
Djaldetti, R., Ziv, I., and Melamed, E. (2006). The mystery of motor asymmetry in Parkinson’s disease. Lancet Neurol 5, 796–823.
Doudet, D. J., Chan, G. L., Holden, J. E., McGeer, E. G., Aigner, T. A., Wyatt, R. J., et al. (1998). 6-[18F]Fluoro-L-DOPA PET studies of the turnover of dopamine in MPTP-induced parkinsonism in monkeys. Synapse 29, 225–232. doi: 10.1002/(SICI)1098-2396(199807)29:3<225::AID-SYN4>3.0.CO;2-8
Fan, Y., Limprasert, P., Murray, I. V. J., Smith, A. C., Lee, V. M. Y., Trojanowski, J. Q., et al. (2006). β-Synuclein modulates α-synuclein neurotoxicity by reducing α-synuclein protein expression. Hum. Mol. Genet. 15, 3002–3011. doi: 10.1093/hmg/ddl242
Firnau, G., Sood, S., Chirakal, R., Nahmias, C., and Garnett, E. S. (1987). Cerebral metabolism of 6-[18F]fluoro-L-3,4-dihydroxyphenylalanine in the primate. J. Neurochem. 48, 1077–1082. doi: 10.1111/j.1471-4159.1987.tb05629.x
Garnett, E. S., Firnau, G., and Nahmias, C. (1983). Dopamine visualized in the basal ganglia of living man. Nature 305, 137–138. doi: 10.1038/305137a0
Gelb, D. J., Oliver, E., and Gilman, S. (1999). Diagnostic criteria for Parkinson disease. Arch. Neurol. 56, 33–39. doi: 10.1001/archneur.56.1.33
Gibb, W. R., and Lees, A. J. (1988). The relevance of the Lewy body to the pathogenesis of idiopathic Parkinson's disease. J. Neurol. Neurosurg. Psychiatry 51, 745–752. doi: 10.1136/jnnp.51.6.745
Guatteo, E., Rizzo, F. R., Federici, M., Cordella, A., Ledonne, A., Latini, L., et al. (2017). Functional alterations of the dopaminergic and glutamatergic systems in spontaneous α-synuclein overexpressing rats. Exp. Neurol. 287, 21–33. doi: 10.1016/j.expneurol.2016.10.009
Harteveld, A. A., Meeuwis, A. P. W., Disselhorst, J. A., Slump, C. H., Oyen, W. J. G., Boerman, O. C., et al. (2011). Using the NEMA NU 4 PET image quality phantom in multipinhole small-animal SPECT. J. Nucl. Med. 52, 1646–1653. doi: 10.2967/jnumed.110.087114
Hashimoto, M., Rockenstein, E., Mante, M., Mallory, M., and Masliah, E. (2001). β-Synuclein inhibits α-synuclein aggregation: a possible role as an anti-parkinsonian factor. Neuron 32, 213–223. doi: 10.1016/S0896-6273(01)00462-7
Holthoff-Detto, V. A., Kessler, J., Herholz, K., Bönner, H., Pietrzyk, U., Würker, M., et al. (1997). Functional effects of striatal dysfunction in Parkinson disease. Arch. Neurol. 54, 145–150. doi: 10.1001/archneur.1997.00550140025008
Huot, P., Lévesque, M., and Parent, A. (2007). The fate of striatal dopaminergic neurons in Parkinson“s disease and Huntington”s chorea. Brain 130, 222–232. doi: 10.1093/brain/awl332
Iwasaki, H., Ohmachi, Y., Kume, E., and Krieglstein, J. (1995). Strain differences in vulnerability of hippocampal neurons to transient cerebral ischaemia in the rat. Int. J. Exp. Pathol. 76, 171–178.
Kiely, A. P., Asi, Y. T., Kara, E., Limousin, P., Ling, H., Lewis, P., et al. (2013). α-Synucleinopathy associated with G51D SNCA mutation: a link between Parkinson's disease and multiple system atrophy? Acta Neuropathol. 125, 753–769. doi: 10.1007/s00401-013-1096-7
Kohl, Z., Ben Abdallah, N., Vogelgsang, J., Tischer, L., Deusser, J., Amato, D., et al. (2016). Severely impaired hippocampal neurogenesis associates with an early serotonergic deficit in a BAC α-synuclein transgenic rat model of Parkinson’s disease. Neurobiol. Dis. 85, 206–217. doi: 10.1016/j.nbd.2015.10.021
Kordower, J. H., Olanow, C. W., Dodiya, H. B., Chu, Y., Beach, T. G., Adler, C. H., et al. (2013). Disease duration and the integrity of the nigrostriatal system in Parkinson's disease. Brain 136, 2419–2431. doi: 10.1093/brain/awt192
Kuhar, M. J., Murrin, L. C., Malouf, A. T., and Klemm, N. (1978). Dopamine receptor binding in vivo: the feasibility of autoradiographic studies. Life Sci. 22, 203–210. doi: 10.1016/0024-3205(78)90538-6
Kyono, K., Takashima, T., Katayama, Y., Kawasaki, T., Zochi, R., Gouda, M., et al. (2011). Use of [18F]FDOPA-PET for in vivo evaluation of dopaminergic dysfunction in unilaterally 6-OHDA-lesioned rats. EJNMMI Res. 1:25. doi: 10.1186/2191-219X-1-25
Lee, C. S., Samii, A., Sossi, V., Ruth, T. J., Schulzer, M., Holden, J. E., et al. (2000). In vivo positron emission tomographic evidence for compensatory changes in presynaptic dopaminergic nerve terminals in Parkinson's disease. Ann. Neurol. 47, 493–503. doi: 10.1002/1531-8249(200004)47:4<493::AID-ANA13>3.0.CO;2-4
Lees, A. J., Hardy, J., and Revesz, T. (2009). Parkinson's disease. Lancet 373, 2055–2066. doi: 10.1016/S0140-6736(09)60492-X
Lelan, F., Boyer, C., Thinard, R., Remy, S., Usal, C., Tesson, L., et al. (2011). Effects of human α-Synuclein A53T-A30P mutations on SVZ and local olfactory bulb cell proliferation in a transgenic rat model of Parkinson disease. Parkinson's Dis. 2011, 1–11. doi: 10.4061/2011/987084
Lesage, S., Anheim, M., Letournel, F., Bousset, L., Honoré, A., Rozas, N., et al. (2013). G51D α-synuclein mutation causes a novel parkinsonian-pyramidal syndrome. Ann. Neurol. 73, 459–471. doi: 10.1002/ana.23894
Logan, J. (2000). Graphical analysis of PET data applied to reversible and irreversible tracers. Nucl. Med. Biol. 27, 661–670. doi: 10.1016/S0969-8051(00)00137-2
Mali, P., Yang, L., Esvelt, K. M., Aach, J., Guell, M., DiCarlo, J. E., et al. (2013). RNA-guided human genome engineering via Cas9. Science 339, 823–826. doi: 10.1126/science.1232033
Martin, R., Baumgart, D., Hübner, S., Jüttler, S., Saul, S., Clausnitzer, A., et al. (2013). Automated nucleophilic one-pot synthesis of 18F-L-DOPA with high specific activity using the GE TRACERlab MXFDG. J. Label. Compd. Radiopharm. 56:S126. doi: 10.1002/jlcr.3054
Morrish, P. K., Rakshi, J. S., Bailey, D. L., Sawle, G. V., and Brooks, D. J. (1998). Measuring the rate of progression and estimating the preclinical period of Parkinson's disease with [18F] dopa PET. J. Neurol. Neurosurg. Psychiatry 64, 314–319. doi: 10.1136/jnnp.64.3.314
Morrish, P. K., Sawle, G. V., and Brooks, D. J. (1995). Clinical and [18F] dopa PET findings in early Parkinson's disease. J. Neurol. Neurosurg. Psychiatry 59, 597–600. doi: 10.1136/jnnp.59.6.597
Nahmias, C., Garnett, E. S., Firnau, G., and Lang, A. (1985). Striatal dopamine distribution in parkinsonian patients during life. J. Neurol. Sci. 69, 223–230. doi: 10.1016/0022-510X(85)90135-2
Nuber, S., Harmuth, F., Kohl, Z., Adame, A., Trejo, M., Schönig, K., et al. (2013). A progressive dopaminergic phenotype associated with neurotoxic conversion of α-synuclein in BAC-transgenic rats. Brain 136, 412–432. doi: 10.1093/brain/aws358
Nurmi, E., Ruottinen, H. M., Bergman, J., Haaparanta, M., Solin, O., Sonninen, P., et al. (2001). Rate of progression in Parkinson's disease: a 6-[18F]fluoro-L-dopa PET study. Mov. Disord. 16, 608–615. doi: 10.1002/mds.1139
Patlak, C. S., and Blasberg, R. G. (1985). Graphical evaluation of blood-to-brain transfer constants from multiple-time uptake data. Generalizations. J. Cereb. Blood Flow Metab. 5, 584–590. doi: 10.1038/jcbfm.1985.87
Petrucci, S., Ginevrino, M., and Valente, E. M. (2016). Phenotypic spectrum of α-synuclein mutations: new insights from patients and cellular models. Parkinsonism Relat. Disord. 22, S16–S20. doi: 10.1016/j.parkreldis.2015.08.015
Reeve, A., Simcox, E., and Turnbull, D. (2014). Ageing and Parkinson's disease: why is advancing age the biggest risk factor? Ageing Res. Rev. 14, 19–30. doi: 10.1016/j.arr.2014.01.004
Rinne, J. O., Portin, R., Ruottinen, H., Nurmi, E., Bergman, J., Haaparanta, M., et al. (2000). Cognitive impairment and the brain dopaminergic system in Parkinson disease: [18F] fluorodopa positron emission tomographic study. Arch. Neurol. 57, 470–475. doi: 10.1001/archneur.57.4.470
Sharma, S. K., and Ebadi, M. (2005). Distribution kinetics of 18F-DOPA in weaver mutant mice. Brain Res. Mol. Brain Res. 139, 23–30. doi: 10.1016/j.molbrainres.2005.05.018
Sharma, S. K., El Refaey, H., and Ebadi, M. (2006). Complex-1 activity and 18F-DOPA uptake in genetically engineered mouse model of Parkinson’s disease and the neuroprotective role of coenzyme Q10. Brain Res. Bull. 70, 22–32. doi: 10.1016/j.brainresbull.2005.11.019
Snow, B. J., Tooyama, I., McGeer, E. G., Yamada, T., Calne, D. B., Takahashi, H., et al. (1993). Human positron emission tomographic [18F] fluorodopa studies correlate with dopamine cell counts and levels. Ann. Neurol. 34, 324–330. doi: 10.1002/ana.410340304
Sossi, V., de la Fuente-Fernández, R., Holden, J. E., Doudet, D. J., McKenzie, J., Stoessl, A. J., et al. (2002). Increase in dopamine turnover occurs early in Parkinson's disease: evidence from a new modeling approach to PET 18 F-fluorodopa data. J. Cereb. Blood Flow Metab. 22, 232–239. doi: 10.1097/00004647-200202000-00011
Sossi, V., de la Fuente-Fernández, R., Nandhagopal, R., Schulzer, M., McKenzie, J., Ruth, T. J., et al. (2010). Dopamine turnover increases in asymptomatic LRRK2 mutations carriers. Mov. Disord. 25, 2717–2723. doi: 10.1002/mds.23356
Sossi, V., Dinelle, K., Topping, G. J., Holden, J. E., Doudet, D., Schulzer, M., et al. (2009). Dopamine transporter relation to levodopa-derived synaptic dopamine in a rat model of Parkinson's: an in vivo imaging study. J. Neurochem. 109, 85–92. doi: 10.1111/j.1471-4159.2009.05904.x
Sossi, V., Doudet, D. J., and Holden, J. E. (2001). A reversible tracer analysis approach to the study of effective dopamine turnover. J. Cereb. Blood Flow Metab. 21, 469–476. doi: 10.1097/00004647-200104000-00015
Spillantini, M. G., Schmidt, M. L., Lee, V. M. Y., Trojanowski, J. Q., Jakes, R., and Goedert, M. (1997). α-Synuclein in Lewy bodies. Nature 388, 839–840. doi: 10.1038/42166
Stoica, G., Lungu, G., Bjorklund, N. L., Taglialatela, G., Zhang, X., Chiu, V., et al. (2012). Potential role of α-synuclein in neurodegeneration: studies in a rat animal model. J. Neurochem. 122, 812–822. doi: 10.1111/j.1471-4159.2012.07805.x
Walker, M. D., Dinelle, K., Kornelsen, R., Lee, A., Farrer, M. J., Stoessl, A. J., et al. (2013a). Measuring dopaminergic function in the 6-OHDA-lesioned rat: a comparison of PET and microdialysis. EJNMMI Res. 3, 69–11. doi: 10.1186/2191-219X-3-69
Walker, M. D., Dinelle, K., Kornelsen, R., McCormick, S., Mah, C., Holden, J. E., et al. (2013b). In-vivo measurement of LDOPA uptake, dopamine reserve and turnover in the rat brain using [18F] FDOPA PET. J. Cereb. Blood Flow Metab. 33, 59–66. doi: 10.1038/jcbfm.2012.120
Wong, D. F., Gjedde, A., and Wagner, H. N. (1986). Quantification of neuroreceptors in the living human brain. I. Irreversible binding of ligands. J. Cereb. Blood Flow Metab. 6, 137–146. doi: 10.1038/jcbfm.1986.27
Keywords: Parkinson’s, G51D α-synuclein mutation, rat model, PET imaging, kinetic modeling
Citation: Morley V, Dolt KS, Alcaide-Corral CJ, Walton T, Lucatelli C, Mashimo T, Tavares AAS and Kunath T (2023) In vivo18F-DOPA PET imaging identifies a dopaminergic deficit in a rat model with a G51D α-synuclein mutation. Front. Neurosci. 17:1095761. doi: 10.3389/fnins.2023.1095761
Edited by:
Yashar Zeighami, McGill University, CanadaReviewed by:
Melissa Crabbé, Belgian Nuclear Research Centre, BelgiumCopyright © 2023 Morley, Dolt, Alcaide-Corral, Walton, Lucatelli, Mashimo, Tavares and Kunath. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Adriana A. S. Tavares, YWRyaWFuYS50YXZhcmVzQGVkLmFjLnVr; Tilo Kunath, dGlsby5rdW5hdGhAZWQuYWMudWs=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.