 Elisabetta Mormone
Elisabetta Mormone Eugenio Luigi Iorio
Eugenio Luigi Iorio Lucrezia Abate1
Lucrezia Abate1 Carlo Rodolfo
Carlo Rodolfo
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Neurosci., 01 February 2023
Sec. Neurodegeneration
Volume 17 - 2023 | https://doi.org/10.3389/fnins.2023.1073689
This article is part of the Research TopicRegenerative Medicine in Neurodegenerative Diseases and Aging: Challenging the Redox HomeostasisView all 5 articles
Since the discovery of Neural Stem Cells (NSCs) there are still mechanism to be clarified, such as the role of mitochondrial metabolism in the regulation of endogenous adult neurogenesis and its implication in neurodegeneration. Although stem cells require glycolysis to maintain their stemness, they can perform oxidative phosphorylation and it is becoming more and more evident that mitochondria are central players, not only for ATP production but also for neuronal differentiation’s steps regulation, through their ability to handle cellular redox state, intracellular signaling, epigenetic state of the cell, as well as the gut microbiota-brain axis, upon dietary influences. In this scenario, the 8-oxoguanine DNA glycosylase (OGG1) repair system would link mitochondrial DNA integrity to the modulation of neural differentiation. On the other side, there is an increasing interest in NSCs generation, from induced pluripotent stem cells, as a clinical model for neurodegenerative diseases (NDs), although this methodology still presents several drawbacks, mainly related to the reprogramming process. Indeed, high levels of reactive oxygen species (ROS), associated with telomere shortening, genomic instability, and defective mitochondrial dynamics, lead to pluripotency limitation and reprogramming efficiency’s reduction. Moreover, while a physiological or moderate ROS increase serves as a signaling mechanism, to activate differentiation and suppress self-renewal, excessive oxidative stress is a common feature of NDs and aging. This ROS-dependent regulatory effect might be modulated by newly identified ROS suppressors, including the NAD+-dependent deacetylase enzymes family called Sirtuins (SIRTs). Recently, the importance of subcellular localization of NAD synthesis has been coupled to different roles for NAD in chromatin stability, DNA repair, circadian rhythms, and longevity. SIRTs have been described as involved in the control of both telomere’s chromatin state and expression of nuclear gene involved in the regulation of mitochondrial gene expression, as well as in several NDs and aging. SIRTs are ubiquitously expressed in the mammalian brain, where they play important roles. In this review we summarize the current knowledge on how SIRTs-dependent modulation of mitochondrial metabolism could impact on neurogenesis and neurodegeneration, focusing mainly on ROS function and their role in SIRTs-mediated cell reprogramming and telomere protection.
Adult neurogenesis (AN) can be defined as “the birth and development of new neurons in adulthood” (Just et al., 2022). The discovery of the existence of neural progenitor cells (NPCs) in the mice brain, paved the way to the identification of specific brain areas, containing cells with a neurogenic potential, termed neural stem cells (NSCs) (Reynolds and Weiss, 1992). A better understanding of the mechanisms controlling adult neurogenesis could be the key for the treatment of a wide range of neurodegenerative, neuropsychiatric, and metabolic disease (Just et al., 2022). In the adult mammalian brain, NSCs mainly reside in the subventricular zone (SVZ) located in the wall of the lateral ventricles, and the sub-granular zone of the dentate gyrus (DG), in the hippocampus (Llorente et al., 2022). NSCs are capable to self-renew and, when needed, to differentiate into the diverse cell types of the nervous system. In the last years, extensive research focused on the understanding and the possible manipulation of the molecular pathways preserving NSCs function, as well as on their usage in transplantation, to restore cognitive and behavioral deficits, in preclinical models of neurodegenerative diseases (NDs). Although many progresses have been made in the field, some aspects still need to be clarified, such as the role of mitochondrial metabolism in the regulation of endogenous adult neurogenesis and neurodegeneration. In addition, considering the limited migratory capacity and the low availability of NSCs for allogenic transplantation, the interest is moving toward the definition of a suitable protocol for generating them from induced pluripotent stem cells (iPSCs). Cell reprogramming could be a powerful technique for the regenerative medicine filed, although still characterized by several drawbacks generated during the reprogramming process, such as the telomere shortening. Moreover, the transcription factors used in somatic cells reprogramming protocols may alter the genomic contents, leading to genetic mutations (Chen et al., 2019), as well as to impact on mitochondrial dynamics, resulting in excessive mitochondrial fission and ROS production, thus greatly limiting pluripotency and reprogramming efficiency. Furthermore, excessive ROS production might induce apoptosis in the transplanted cells (Sart et al., 2014), although recent studies challenged this dogma by demonstrating a physiological role for ROS in the regulation of stem cell fate decision (Sart et al., 2015). Indeed, iPSCs proliferation and differentiation are actively controlled by mild levels of ROS (e.g., 1.8-fold the normal level) (Lee et al., 2010). All these aspects, including genomic instability and impaired mitochondrial dynamics, should be addressed to exploit cell reprograming for clinical research as well as to comply efficiency and safety concerns.
Recent studies showed the Sirtuins (SIRTs) protein family as involved in the modulation of a variety of cellular processes, associated with antioxidant (AOX) and redox signaling. In detail, SIRT1, SIRT3, and SIRT5 protect the cell from ROS; SIRT2, SIRT6, and SIRT7 modulate key oxidative stress genes and mechanisms; whereas SIRT4 induces ROS production and performs antioxidative roles, as well (Singh et al., 2018). In addition, SIRTs are involved in the control of genomic instability (Shin et al., 2018). Despite the remarkable number of works about the role of metabolic switch in neurogenesis regulation, it is not completely understood how SIRTs might regulate neurogenesis through mitochondria metabolism (Tay et al., 2021), particularly through ROS and NAD system, as well as their role in telomere length and genomic stability maintenance, during cell reprogramming.
The purpose of this review is to summarize the current knowledge about the emerging role of SIRTs, with a focus on SIRT1, SIRT2, and SIRT3, as regulators of neurogenesis through metabolic modulation and ROS signaling, in NDs and aging as well as their role in the cell reprogramming. We think that genetic, hormonal, and drug manipulation of NSCs mitochondria, may be useful to prolong NSCs longevity and stability prior to their clinical usage, or even to improve their endogenous function. Therefore, a better understanding of the molecular mechanisms underlying neurogenesis and cellular reprogramming may be of help to identify new potential therapeutic targets.
Mitochondria are essential organelles for cell’s life and death, as they not only provide ATP but are central in the modulation of several cellular pathways, from Ca2+ signaling to apoptosis. Alteration of mitochondrial functions is a common feature of both apoptosis and autophagy. During apoptosis, mitochondria integrate intrinsic and extrinsic death signals, with the loss of mitochondrial membrane potential (MMP) and the permeability transition pore considered as a latest executioner event associated with the release, from the mitochondrial intermembrane space, of cofactors required for caspases’ activation (Wang and Youle, 2009). MMP loss could also be a signal for the induction of mitochondrial fission and subsequent elimination of the damaged organelles, through a specialized form of autophagy, termed mitophagy (Mormone et al., 2006; Li et al., 2021). Functional versatility of mitochondria is matched by a complex morphology as they not only display a complex ultrastructure, but also appear interconnected and networked (Anesti and Scorrano, 2006). In humans, the shape of both individual mitochondria and the mitochondrial network depends upon fission and fusion processes, which are tightly regulated by the so called “mitochondria-shaping” proteins (Dhingra and Kirshenbaum, 2014), able to promote fission (Drp1, Fis1), fusion (Mfn1/2, Opa1), and transport (Miro, Milton) of the organelles (Detmer and Chan, 2007). The mitochondrial network is an extremely dynamic structure subjected to continuous changes during cell-cycle progression and cell division. The importance of mitochondrial dynamics is further substantiated by the observation that mutations in mitochondria-shaping proteins can result into NDs (Panchal and Tiwari, 2019), like autosomal-dominant optic atrophy and Charcot-Marie-Tooth (Carelli et al., 2009). In addition, mitochondrial dysfunction, caused by excessive oxidative stress, depletion of cellular energy levels, and defective protein production, associate with dopaminergic neurons loss in Parkinson’s disease (PD). Sporadic PD etiopathogenesis is complex, comprising both environmental and genetic factors, able to affect mitochondrial life cycle, bioenergetic capacity, quality control, dynamic changes (fusion and fission), subcellular distribution, as well as cell death pathways regulation (Fukae et al., 2007; Prasuhn et al., 2020). In addition, in a mouse model of Alzheimer’s disease (AD), it has been reported that the expression of the mitochondrial transport and dynamics regulator Miro2, which is degraded through PINK1/Parkin-dependent mitophagy (Saotome et al., 2008; Shlevkov et al., 2016), was decreased in Nestin-positive cells of the hippocampus (Woo et al., 2021). Indeed, in cultured adult hippocampus-derived NSCs of normal mice, Miro2 downregulation results into excessive mitochondrial fission, increased ROS production, and autophagic cell death, rescued by Miro2 over-expression and pharmaceutical inhibition of Drp1 activity (Mdivi-1) (Woo et al., 2021). In addition, both α-Synuclein (α-SYN) and amyloid-β (Aβ), responsible for neurotoxic protein aggregates accumulation in PD and AD, impair mitochondrial respiration (Manczak et al., 2006; Reeve et al., 2015), suggesting that mitochondrial defects also contributed to abnormal adult hippocampal neurogenesis (AHN) (Zhang et al., 2019; Amber et al., 2020). Taken together, this evidence supports the idea that mitochondrial-associated abnormalities of adult hippocampal neurogenesis, contribute to cognitive and psychiatric disturbances in neurodegenerative illnesses. In Huntington’s disease (HD), the degenerative process relies on both the acquisition of toxic function by mutated huntingtin as well as on the loss of protection exerted by the wild type protein, leading to diverse cellular alterations. Indeed, mitochondria impairment and increased oxidative stress result into cell death induction, by apoptosis and/or autophagy (Mormone et al., 2006; Duan et al., 2014; Panchal and Tiwari, 2019). In vitro and in vivo observations for Amyotrophic Lateral Sclerosis (ALS), suggested that mutation of genes associated with the disease (SOD1, TDP-43, FUS, and TAF15), can alter mitochondrial dynamics and induce oxidative stress (Panchal and Tiwari, 2019; Kodavati et al., 2020), coupled to the nuclear accumulation of the nuclear factor erythroid 2-related factor 2 (NRF2), a master regulator of detoxification, AOX, and anti-inflammatory mechanisms (Obrador et al., 2020).
Multiple sclerosis (MS) is a chronic demyelinating disease of complex etiology affecting the CNS, where oligodendrocytes act as myelination cells, in which a role for mitochondrial dysfunction preceding the axonal damage has been suggested (Heidker et al., 2017; Wentling et al., 2019). Indeed, it has been suggested that inflammatory demyelination could result into neurodegeneration through different mechanisms including energy depletion, due to mitochondrial dysfunction and/or hypoxia related processes, activated microglia, oxidative stress, activated astrocytes, iron accumulation, Wallerian degeneration, and apoptosis. Hence, while the primary therapeutic approach is still directed against the immune system, new experimental protocols, aiming to lessen or delay MS progression, are focused on neuronal and glial metabolism support and/or remyelination promotion (Heidker et al., 2017).
Mitochondrial dependent generation of ROS proved to be a common feature of NDs (Liu et al., 2017) and related to neuronal injury and pathological progression (Ismail et al., 2020). A better understanding of the molecular pathways controlled by mitochondrial metabolism, through oxidative stress, would be useful to design new therapeutic approaches targeting specific proteins or molecules.
Energy requirements of brain cells are quite diverse, with neurons relying on mitochondrial-based oxidative phosphorylation (OXPHOS) and glia cells mostly on glycolysis (Lopez-Fabuel et al., 2016). SGZ and SVZ resident NSCs, preferentially rely on aerobic glycolysis, while their more differentiated progeny generates ATP mainly by OXPHOS (Rafalski et al., 2012; Zheng et al., 2016; Lorenz and Prigione, 2017). Proteomic analysis of cultured NSCs, derived at different ages, revealed that the main age-related alterations were found in glycolysis, fatty acid and propanoate metabolism, protein ubiquitination, valine, leucine, and isoleucine degradation pathways (Castiglione et al., 2017). For a long time, stem cells have been considered to rely only on glycolysis to fulfill their energy requirements, mainly because of a combination of their peculiar cellular demands and the hypoxic microenvironment where they reside. Nevertheless, it has become recently clear that stem cells are indeed capable of performing OXPHOS, even though glycolysis is critical for stemness maintenance, rather than being an adaptation to their environment (Maffezzini et al., 2020). In vivo, NPCs differentiation may be induced by the activation of a transcriptional program through NRF2, responsible for redox signaling genes expression, thereby supporting neuronal differentiation by protecting against ROS toxic insults (Lopez-Fabuel et al., 2016). Indeed, the differentiation from a pluripotent progenitor cell into a neuron is characterized by the reduced expression of glycolysis-related proteins [e.g., Hexokinase 2 (HK2) and Lactate Dehydrogenase isoform A (LDHA)], coupled to the activation of the Pyruvate Kinase PKM2, to its constitutively active isoform PKM1, and the upregulation of OXPHOS-related genes (Zheng et al., 2016). In vitro cultured mice SVZ-derived NSCs showed the upregulation of different energy metabolism-associated genes, such as: the Insulin-like growth factor binding protein 3 (IGFBP3), Enolase I, and Cytochrome c oxidase subunit VIIa, when compared to hematopoietic (HSCs) and embryonic stem cells (ESCs) (Ivanova et al., 2002); hypoxia-inducible factor (HIF1α), Acetyl-coenzyme A transporter, and IGFBP3, when compared to differentiated cells of the lateral ventricle (Ramalho-Santos et al., 2002; Bonnert et al., 2006). In addition, also cultured postnatal NSCs showed increased expression of metabolic genes, such as Acetyl-coenzyme A synthetase 1, Enolase I, and Pyruvate dehydrogenase, as compared to differentiating neural cells (Geschwind et al., 2001; Karsten et al., 2003), although other metabolic genes (e.g., Glucose-6-phosphate dehydrogenase) were upregulated during differentiation (Gurok et al., 2004).
Energy metabolism regulation is a crucial player in stem cell fate determination and the right balance between stem cell quiescence, multipotency, and differentiation relies on the reversible switching between aerobic and anaerobic metabolism (Beckervordersandforth, 2017). Rodents subjected to calorie restriction (CR), a regimen of calorie reduction without malnutrition, displayed an increased numbers of newly produced neural cells in the SGZ, coupled to increased expression of the brain-derived neurotrophic factor (BDNF) (Lee et al., 2000); while in models of diet-induced obesity and diabetes, adult neurogenesis results to be impaired (Pani, 2015). Furthermore, aberrant adult neurogenesis has been reported in mice models for nutrient-triggered signals impairment, thus confirming that nutrient-regulated switches influence NSCs fate decisions. The transition between quiescent and activated states is a critical step, as the required cell cycle entry is a major energetic commitment (Cavallucci et al., 2016; Wentling et al., 2019). For this reason, nutrient-responsive pathways and transducers, such as the growth differentiation factor-11 (GDF11), the insulin-IGF cascade, the AMPK/mTOR axis, and the transcription regulators CREB and SIRT1 have been included, alongside the canonical “developmental” signals (e.g., Notch and Wnt), in the molecular networks controlling NSCs self-renewal, migration, and differentiation, in response to local and systemic inputs (Figure 1). In the context of metabolic diseases as well as in aging, many of these nutrient-related cascades prove to be dysregulated, thus suggesting a possible explanation/contribution to both impaired neurogenesis and the cognition defects observed (Katsimpardi et al., 2014; Fidaleo et al., 2017). Recently, a role for short chain fatty acids (SCFAs) in adult neurogenesis has been proposed. SCFAs are the major product of fiber fermentation in the large intestine and, after entering systemic circulation, could directly impact on mitochondrial metabolism in diverse body’s tissues, including the brain (van de Wouw et al., 2018; Silva et al., 2020). Specific diet regimens could increase SCFAs levels which in turn could trigger mitochondrial biogenesis and premature differentiation of NSCs, through a ROS- and ERK1/2-dependent mechanism, thus resulting into a rapid depletion of the NSCs pool (Ribeiro et al., 2020). Therefore, nutrients are necessary to trigger neurogenesis but chronic overnutrition and/or metabolic imbalances, leading to an impaired nutrient signaling in the brain (Soto et al., 2018), could potentially result into NSCs exhaustion, thus accelerating brain aging, and altering neurobehaviors (Soto et al., 2018; Ribeiro et al., 2020). These observations suggest the existence of a new role for mitochondria, as mediators of the gut microbiota-brain axis, able to respond to dietary influences. Of note, the role of nutrients in NSCs activation is not in conflict with the established notion that CR enhances mammalian neurogenesis, as previously discussed. In fact, starvation/refeeding cycles are likely to synchronize NSCs, optimizing their refeeding response and re-entry into a quiescent state. Intriguingly, feeding cycles are in strict relationship with the circadian clock (Asher and Schibler, 2011) and a NSCs quiescence maintenance defect has been reported in mice model devoid of key molecular clock proteins (Bouchard-Cannon et al., 2013). Indeed, when normal human fibroblasts were synced in vitro, by means of two different protocols, rhythmic oscillations of mitochondrial respiration and glycolytic activity have been observed (Pacelli et al., 2019). Conversely, PD patients-derived fibroblasts, carrying Parkin gene mutations, showed a severe dampening of the bioenergetic oscillatory patterns, associated with a dysregulation of core clock genes expression, which was also confirmed in iPSCs and in the induced neural stem cells (iNSCs). These results highlight the existence of a reciprocal interplay between mitochondrial energy metabolism and clockwork machinery, pointing to a Parkin-dependent mechanism of regulation, and a greater level of complexity in PD pathophysiology, that could eventually be a common feature of other NDs (Pacelli et al., 2019). This evidence agrees with those emphasizing the conserved nature of diurnal variation of redox control in eukaryotes, where key cellular activities depend upon the coordination between the NAD-dependent control of metabolism and the NADPH/H2O2 control of the redox proteome spatiotemporal organization (Jones and Sies, 2015). Several evidence suggest that energy metabolism and nutrient sensing are the major physiological determinants of NSCs fate, as well as modulators of the entire neurogenic process. Nevertheless, the molecular pathways underlying metabolic regulation of neurogenesis, are still poorly understood. Their full comprehension, as well as their interplay with novel dietary and/or pharmacological approaches, aiming at improving neurogenic activity and delaying its age-related decline, may be of help in the prevention of neurodegenerative disorders and brain aging (Cavallucci et al., 2016).
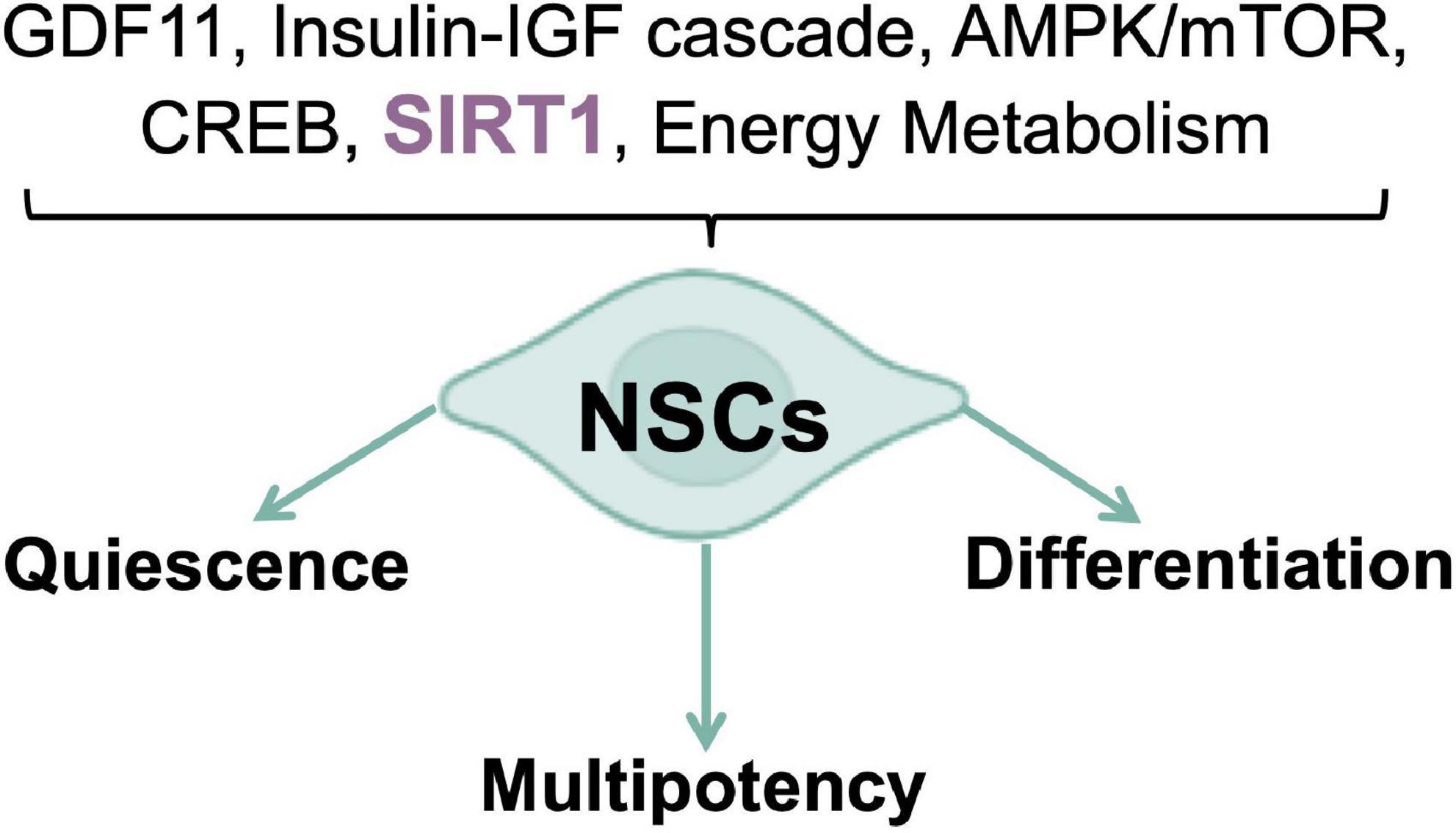
Figure 1. Nutrient pathways control of NSCs fate. Nutrient sensing and energy metabolism are the major physiological determinants of neurogenic processes.
Oxidative eustress has been defined as a “low physiological exposure to prooxidants,” characterized by the involvement of specific redox signaling targets, while oxidative distress as a “supraphysiological exposure to prooxidants,” with unspecific targets, disrupted redox signaling, and damaging molecule modifications (Lushchak and Storey, 2021; Sies, 2021). Enzymatical or non-enzymatical ROS generation could occur in different cellular compartments, such as mitochondria, peroxisome, endoplasmic reticulum (ER), and lysosome (Malard et al., 2021). Nevertheless, approximately 90% of cellular ROS are generated by the mitochondrial Electron Transport Chain (ETC) during ATP production (Nissanka and Moraes, 2018). In fact, about 0.1–0.2% of the O2 used by mitochondria is thought to generate ROS, through the premature electron flow deriving from NADH or FADH2 to O2, mainly through electron transport chain complexes I and III (Chen et al., 2003; Tahara et al., 2009). ROS-dependent generation of O2-derived reactive species is initiated by the conversion of O2 to O2.– and the formation of H2O2, hydroxyl radicals OH., OH–, and many other derivative species (Olson et al., 2017). It is very unlikely that O2.– can play a role in physiology, especially considering its very short lifetime (approximately 10–9 s), although it can possibly induce oxidative damage in the neighboring area. Therefore, superoxide dismutase 1 (SOD1) is more likely to act as a signaling rather than antioxidative enzyme, because it converts O2.– to the more stable H2O2, which can be transported as a signaling molecule. However, H2O2 is dangerous for the cell, as by the Fenton reaction it could generate hydroxyl anion, the most toxic form of ROS (Angelova and Abramov, 2018). These activation/deactivation cycles of the H2O2 metabolism support complex time-dependent processes in the life cycle of cells and organisms, as described by third principle of the redox code (Jones and Sies, 2015). In mitochondria, O2.– generation from O2 depends upon the activity of different proteins, such as glycerol 3-phosphate dehydrogenase and 2-oxoglutarate dehydrogenase. Other sources of ROS are: (i) the peroxisomes, through the activity of fatty acid β-oxidation, iron-Sculpture flavin hydroxylase xanthine oxidoreductase (XOR), and urate oxidase (UO), in the matrix, and by electron chain, in the membrane; (ii) the ER, by nicotinamide adenine dinucleotide phosphate (NADPH) oxidases (NOXs) and dual oxidases, and the microsomal monooxygenase (MMO) system, which contains cytochrome P450 oxidase (Malard et al., 2021). In addition, non-home iron containing dioxygenases, lipoxygenases, and cyclooxygenases also generate O2.– from polyunsaturated fatty acids (PUFAs) (Snezhkina et al., 2019). O2.– reduces key transition metal ions iron Fe3+, copper Cu2+, and manganese Mn3+, which generate O2 (Hayyan et al., 2016). Under physiological conditions, ROS levels are tightly regulated by the ROS scavenging system, through the activity of antioxidant enzymes that can neutralize ROS by directly reacting with as well as accepting electrons from them. Glutathione Peroxidases (GPXs), through GSH/GSSG metabolism, and Peroxiredoxin (PRDX) generate H2O from H2O2 (Angelova and Abramov, 2018), with a circadian variation (Jones and Sies, 2015). Other two important antioxidant defense enzymes are Catalase and the home containing enzyme Myeloperoxidase (MPO). Catalase, the most abundant peroxisomal antioxidant enzyme, dismutates H2O2 to O2 and H2O (Ogino et al., 2001; Olson et al., 2017), while MPO catalysis the oxidation of Cl– by H2O2 to hypochlorous acid (HOCl) (Garai et al., 2017). In addition, H2O2 reacts with transition metal ions Fe2+ or Cu+ to generate OH., OH–, and the corresponding reactive Fe3+ or Cu2+ in the Fenton reaction (Kajarabille and Latunde-Dada, 2019).
Proteins belonging to the NOX family are the second major source of intracellular ROS, and this enzymatic ROS synthesis contributes toward the maintenance of physiological ROS levels, accordingly to cellular demands (Bedard and Krause, 2007). In cultured hippocampal neurons, NOX enzymes are responsible for the generation of almost 45% of intracellular hydrogen peroxide, thus proving the key role of these enzymes’ activity on the redox state of the cell (Bedard and Krause, 2007). The NOX-produced superoxide anion is rapidly converted, either spontaneously or enzymatically (by SOD), into hydrogen peroxide, which plays an important role in killing pathogens, although excess superoxide can lead to oxidative stress and cell damage. ROS accumulation leads to oxidative stress, which produces adverse effects on multiple cellular components, including proteins, lipids, and nucleotides. ROS generation in the mitochondrial matrix leads to damage of mitochondrial proteins, membranes, and DNA. Additionally, ROS can impair ATP synthesis thus impacting on metabolic functions, such as fatty acid oxidation, TCA cycle, urea cycle, amino acid, and home synthesis (Zorov et al., 2014). Moreover, oxidative damage can result in cytochrome c release, through the mitochondrial permeability transition pore, thus leading to apoptosis induction (Rao et al., 2014). We can thus assume that ROS levels act as a function of mitochondrial respiration and that multiple factors, such as oxygen availability, NADH, FADH2, and ubiquinone redox states, antioxidant factors activity, mitochondrial morphology, as well as mutations in OXPHOS subunits, could influence ROS levels as well (Maffezzini et al., 2020).
The brain is one of the most metabolically active among all organs, highly susceptible to stresses, specifically to oxidative distress, due to the multifaceted roles of ROS, especially superoxide anion O2.– and H2O2, in redox signaling. Indeed, dopamine metabolism, via monoamine oxidases (MAO), and OXPHOS are important sources for H2O2 generation, that could behave as a signaling molecule. In addition, the brain shows low endogenous antioxidant defense, as compared to other organs, mainly due to its reduced glutathione (GSH) content and low catalase. Further, enriched unsaturated lipids, such as omega-3 docosahexaenoic acid, cause distress due to their susceptibility to lipid peroxidation related to cell death, including ferroptosis (Cobley et al., 2018). Finally, brain susceptibility is region- and time-dependent, based on the condition that induces oxidative distress and the endogenous antioxidant capacity. In fact, while many neuronal subtypes can cope with oxidative stress rise, selected neuronal populations showed a higher susceptibility to ROS increase (Mattson and Magnus, 2006; Wang and Michaelis, 2010). The four areas of the brain most susceptible to oxidative stress are the cerebral cortex, the hippocampus, the striatum, and the cerebellum (Malard et al., 2021). These observations agree also with the evidence that oxidative stress and poor antioxidant defense, underlying the deleterious effects of vitamin B12 deficiency in mice model, would act on the expression of histone modifying enzymes that act on the behavior (Ghosh et al., 2020).
Even though high ROS levels associate with cellular dysfunction, it is now evident that ROS are necessary for some physiological cellular functions (Sart et al., 2015; Nissanka and Moraes, 2018). Indeed, ROS act as second messengers by modulating cytokines and growth factors, whose activity regulates classical signaling cascades under both physiological and stress-related conditions, such as ERK, JNK, MAPK, and JAK/STAT pathways (Simon et al., 1998). Furthermore, the regulatory effect of ROS might be modulated not only by classical ROS-scavenging enzymes, such as SOD, catalase, peroxiredoxins (PRX), thioredoxin (TRX), glutathione peroxidase (GPX), reductase (GR), and transferases (GST), but also by newly identified ROS suppressors, including PTEN-induced putative kinase 1 (PINK1) and SIRT1 (Bigarella et al., 2014; Xavier et al., 2016).
Tissue development and regeneration rely on the balance between stem cell self-renewal vs. differentiation, and several reports highlighted mitochondria as a key player, with functional alterations of the organelle leading to stem cell abnormalities (Llorens-Bobadilla et al., 2015; Shin et al., 2015; Stoll et al., 2015; Xie et al., 2016; Zheng et al., 2016; Khacho et al., 2019). The hGFAP-SDHD mouse model, bearing homozygous deletion of the succinate dehydrogenase subunit D gene (SDHD) restricted to the cells of glial fibrillary acidic protein lineage, showed that the genetic mitochondrial damage did not alter the generation, maintenance, or multipotency of glia-like NSCs. However, a differentiation impairment of neurons and oligodendrocytes, but not of astrocytes, has been observed alongside with extensive brain atrophy (Diaz-Castro et al., 2015). Another study proved the Nestin-Cdk5-Drp1 axis as a negative modulator of OXPHOS, which is indispensable for neural stem/progenitor cell maintenance (Wang J. et al., 2018), and it has also been reported that, during embryonic neurogenesis, NSCs’ mitochondrial morphology acts as an upstream regulatory mechanism for stem cell fate decisions (Rodolfo et al., 2016; Bueler, 2021). Indeed, enhanced mitochondrial fusion promotes NSCs self-renewal, while mitochondrial fragmentation commits NSCs to neuronal differentiation and maturation (Figures 2A, C). Khacho et al. (2016) observed a reduction of uncommitted Sox2+ NSCs and immature DCX+ neurons in the DG, when the mitochondrial fusion proteins MFN1/2 were knocked out in adult hippocampal NSCs. Indeed, when MFN2, OPA-1, and Drp1 were downregulated in Sox2+ NSCs, they observed aberrant mitochondrial dynamics as well as an impairment of NSCs self-renewal and fate decisions, linked with changes in ROS signaling but not in ATP levels. Mitochondrial dynamics also seem to regulate neurogenesis in the adult SVZ, as suggested by chemical inhibition (with Mdivi-1) of Drp1 in SVZ-derived neurosphere cultures, which results in a reduction of both NSCs migration form neurosphere as well as their differentiation into neurons (Detmer and Chan, 2007). This evidence showed that mitochondrial morphology changes could regulate mitochondrial metabolism and ROS generation, whereby the commitment of NSCs to a progenitor fate is mediated by a physiological increase in mitochondrial ROS (mtROS), associated with mitochondrial fragmentation (Khacho and Slack, 2018; Bueler, 2021; Ozgen et al., 2022). Mitochondrial fragmentation fulfils a biological role to regulate neuronal development (Khacho and Slack, 2018) and mtROS would act as a rheostat to direct gene expression changes regulating cell fate decisions (Maryanovich and Gross, 2013). Indeed, the physiological mtROS increase functions as a signaling mechanism to activate a cascade of events leading to the stabilization of the master redox regulator protein NRF2, whose translocation into the nucleus mediate the transcription of genes responsible for differentiation induction and self-renewal suppression (Khacho et al., 2016). On the other hand, ROS production and excessive fission are responsible for neurodegeneration and are detrimental for neurogenesis (Rodolfo et al., 2016; Bueler, 2021; Figure 2B). In this scenario, Sirtuins may act as potential modulators of specific gene activation, leading to neural differentiation. In fact, oxidative stress, or a general alteration of cellular redox homeostasis, impacts on SIRTs activity at different levels: (i) by inducing or repressing SIRTs genes expression; (ii) by posttranslational oxidative modifications of SIRTs; (iii) by altering SIRTs-protein interactions; (iv) by changing NAD+ levels (Santos et al., 2016). Moreover, recent evidence highlights a molecular linkage between mitochondrial DNA (mtDNA) integrity and the modulation of neural differentiation, suggesting another way by which ROS can modulate stem cells differentiation. Many pathological insults can affect mtDNA integrity, but ROS-dependent oxidative damage is the most discussed (Nissanka and Moraes, 2018). It has been reported that increased mtDNA mutation loads correlate with a reduction of NSCs population in the SVZ of adult mice, reduced self-renewal capacity, and decreased mitochondrial respiration (Ahlqvist et al., 2012; Nissanka and Moraes, 2018; Figure 3). Moreover, during brain repair, mtDNA damage was shown to favor NSCs differentiation into astrocytes, and to affect mitochondrial DNA transcription, and replication (Xavier et al., 2016). Cells developed different strategies to abolish the deleterious consequences of ROS on DNA. Indeed, a multienzymes repair cascade, known as the base excision repair (BER), leads to mtDNA repair and replication. In this cascade, OGG1 activity triggering, allow for the identification and elimination of several base lesions, including 8-oxoguanine, one of the most abundant genomic base modifications generated by reactive oxygen and nitrogen species. OGG1 acts as a transcription modulator, which can control transcription factor homing, induce allosteric transition of G-quadruplex structure, or recruit chromatin remodelers (Wang R. et al., 2018). In particular, OGG1 activation in the mitochondria results in the induction of the mitochondrial DNA polymerase, Pol γ (Xavier et al., 2016). Experiments in mice showed OGG1 as essential for the repair of mtDNA damage and NSCs viability, upon mitochondrial oxidative stress (Wang et al., 2011). In fact, differentiating neural cells from ogg1–/– mice spontaneously accumulate mtDNA damage and shift their fate toward an astrocytic lineage. Interestingly, these events are associated with SIRT1 enzymatic activation, due to an increased NAD+/NADH ratio, similarly to what observed in wild type NSCs subjected to mtDNA damaging insults. Instead, antioxidant administration reversed mtDNA damage accumulation and increased neurogenesis in ogg1–/– cells. Moreover, the expression of a mitochondrially targeted human OGG1 in ogg1–/– NSCs results in the protection from mtDNA damage during differentiation, and increased neurogenesis (Wang et al., 2011; Figure 3). Similarly, it has been reported that AOX halted the neurogenic to gliogenic lineage shift during NSCs differentiation, by strongly reducing ROS generation and nuclear translocation of NRF2 and SIRT1 (Santos et al., 2013). This evidence would suggest that ROS-sensitive SIRTs activity could be modulated by alterations of the mitochondrial respiratory chain, in response to mtDNA damage, thus impacting on NSCs fate at different levels.
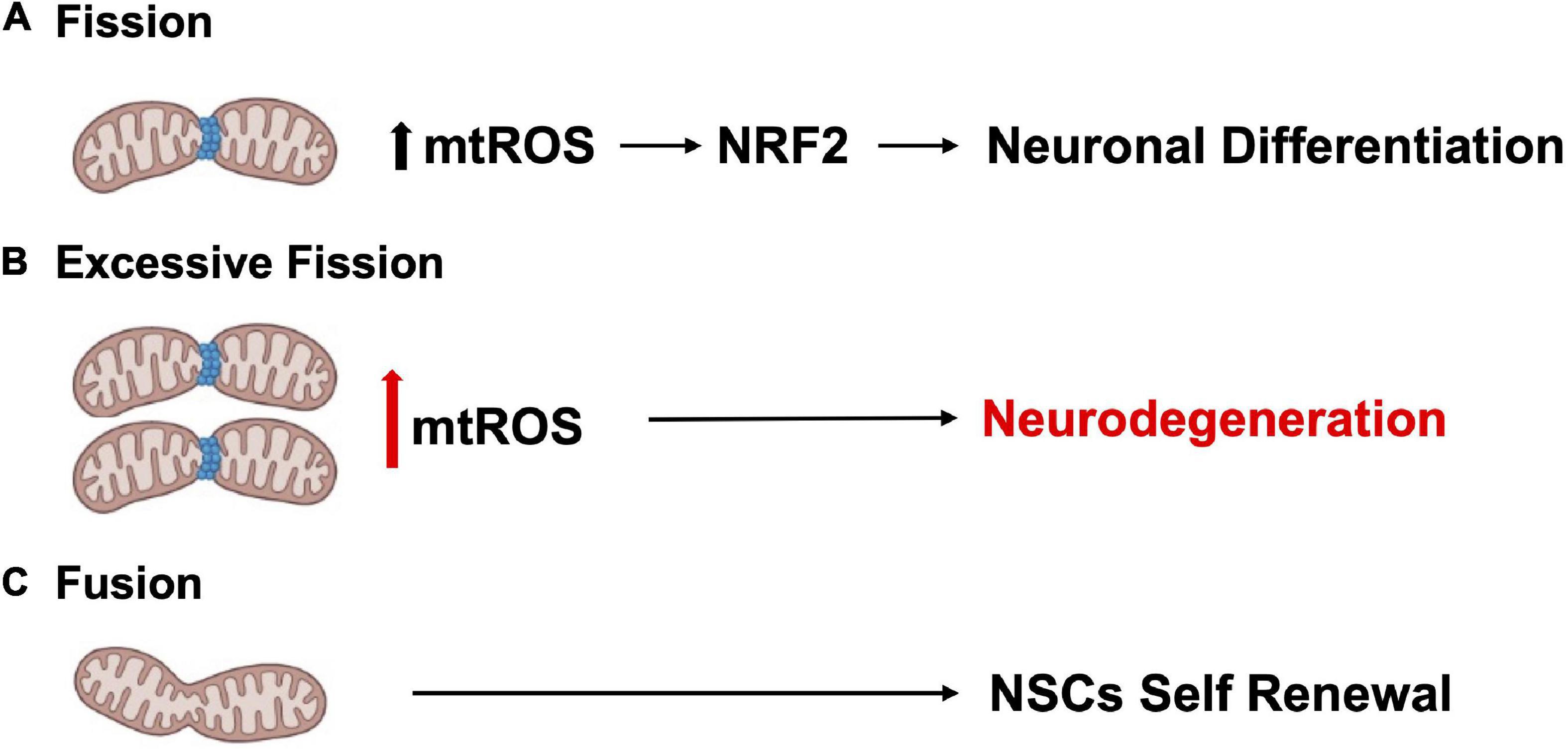
Figure 2. Mitochondrial dynamics regulate neurogenesis. (A) Mitochondrial fission promotes neuronal differentiation. (B) Excessive fission and ROS production contributes to neurodegeneration. (C) Enhanced mitochondrial fusion promotes NSCs self-renewal.
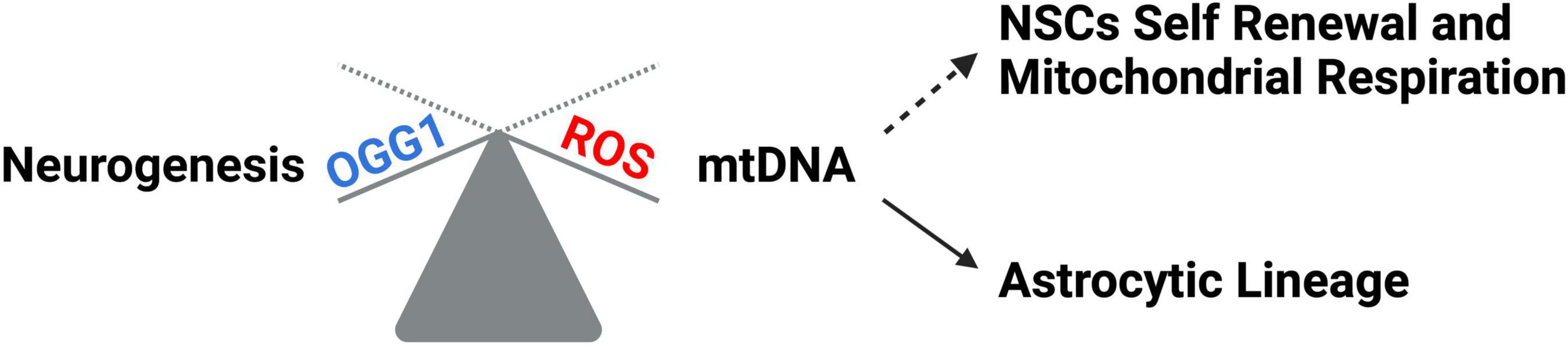
Figure 3. mtDNA impact on neural differentiation. ROS increase leads to mtDNA damage which correlates with NSCs self-renewal reduction, decreased mitochondrial respiration, and a shift toward the astrocytic lineage. OGG1 repair system protects NSCs from ROS-dependent mtDNA damage, thus favoring neuronal differentiation.
Oxygen levels play a significant role in the molecular mechanisms that guide stem cells to either differentiate or renew, by directly influencing enzymatic reactions and by regulating specific gene expression profiles, through transcription factors such as HIF1α (Mootha and Chinnery, 2018). Energy generation in NSCs preferentially relies on aerobic glycolysis, through HK2 and the pentose phosphate pathway, and may be due to multiple factors, such as their location within a hypoxic niche, the low energy requirements of quiescence, and the need to minimize mtROS-dependent oxidative stress, as shown in HSCs (Kunisaki et al., 2013). The differentiation of ESCs into neural progenitors is regulated by the eicosanoid pathway and by fatty acid metabolism (Yanes et al., 2010). Indeed, stem cells’ mitochondria are relatively metabolically inactive, in terms of ATP production, when compared with more differentiated cells. Nonetheless, functional mitochondria are still required for proper adult stem cells maintenance (Bigarella et al., 2014). Stem cells maintain low basal levels of ROS, which preserves stem cell potential by maintaining an appropriate balance between stem cell quiescence, differentiation, and self-renewal. The oxidative stress response mediated by Forkhead box O 3 (FOXO3) becomes rapidly deactivated upon NSCs differentiation, suggesting that mitochondrial oxidation-induced ROS are required in neural progenitors (Shyh-Chang et al., 2013). In fact, in NSCs and HSCs ROS reduction below the basal level is associated with reduced regenerative potential, characterized by impaired proliferation, differentiation, and self-renewal (Collins et al., 2018; Tan and Suda, 2018). However, high levels of H2O2 are required to maintain regular NSCs and progenitor cells’ self-renewal, a process related to Doublecortin (DCX), Nestin, and FOXO proteins (Le Belle et al., 2011). Conversely, ROS accumulation leads to loss of quiescence and induction of senescence, via p38-MAPK activation, leading to stem cell exhaustion and impaired regenerative potential, which could be reverted by administration of the antioxidant N-acetylcysteine (NAC) (Ito et al., 2006; Paik et al., 2009; Takubo et al., 2010; Ahlqvist et al., 2012; Borodkina et al., 2014; Garcia-Prat et al., 2016; Shaban et al., 2017; Tan and Suda, 2018). Further, accumulation of ROS to high levels ultimately results in cell death (Sart et al., 2015; Tan and Suda, 2018). On the other hand, it has been reported that p53-dependent mtROS reduction impacts on the neural differentiation potential, by favoring neuronal rather than astroglial conversion (Xavier et al., 2014). In the SGZ, transient oxidative distress, stimulates the expression of oxidation-responsive genes, which in turn drive neurogenesis by promoting NSCs and progenitor cells differentiation (Walton et al., 2012). However, a recent study in which mice NPCs from hippocampus were FACS-sorted accordingly to their ROS levels, and subjected to staminal markers and transcriptome analyses, unexpectedly showed that the cells with the highest ROS levels were quiescent NSCs (qNSCs). Shifts to lower ROS content primed NPCs to a subsequent state transition, showing that lower ROS levels correlated with increased expression of proliferation and differentiation genes. In addition, NOX2 was not necessary for NPCs proliferation under physiological conditions, even if it has been reported that a transient NOX2-dependent ROS burst promotes exercise-induced recruitment of qNSCs to proliferation (Adusumilli et al., 2021). While this evidence seems to be in contrast with previous ones, they may not be mutually exclusive, because the transient nature of ROS and ROS signals likely triggers cell transitions without substantially altering ROS levels in the next cell type, especially if the ROS burst also activates anti-oxidative genes (Bueler, 2021). Interestingly, from the antioxidant defense side, in vivo experiments in mice deficient for cytoplasmic (SOD1) and mitochondrial SOD (SOD2), showed reduced adult hippocampal neurogenesis, in favor of an increased generation of new-born astrocytes (Garcia-Prat et al., 2016). In addition, NSCs and NPCs proliferation were increased by the accumulation of catalase in mitochondria (Liao et al., 2013). Finally, MPO inhibition increases adult neurogenesis through, at least, DCX and SOX2 stimulation (Yu et al., 2018). ROS behavior during neurogenesis, is like that described in the Hekimi’s Mitohormesis theory (Hekimi et al., 2011), which states that cellular insults trigger protective stress response, where ROS would act as secondary messenger. Several key transcription factors and signaling pathways, including NOTCH and WNT/β-catenin, NRF2, p53, PI3K/AKT, and pERK1/2 (Bouchard-Cannon et al., 2013), are involved in the ROS-dependent modulation of both adult and embryonic neurogenesis. Therefore, ROS can act as signaling molecules to modulate the stress response pathway and small increases can extend lifespan. However, at a certain point this age-dependent damage would increase past the threshold where ROS signaling is sustained and maladaptive (Figure 4).
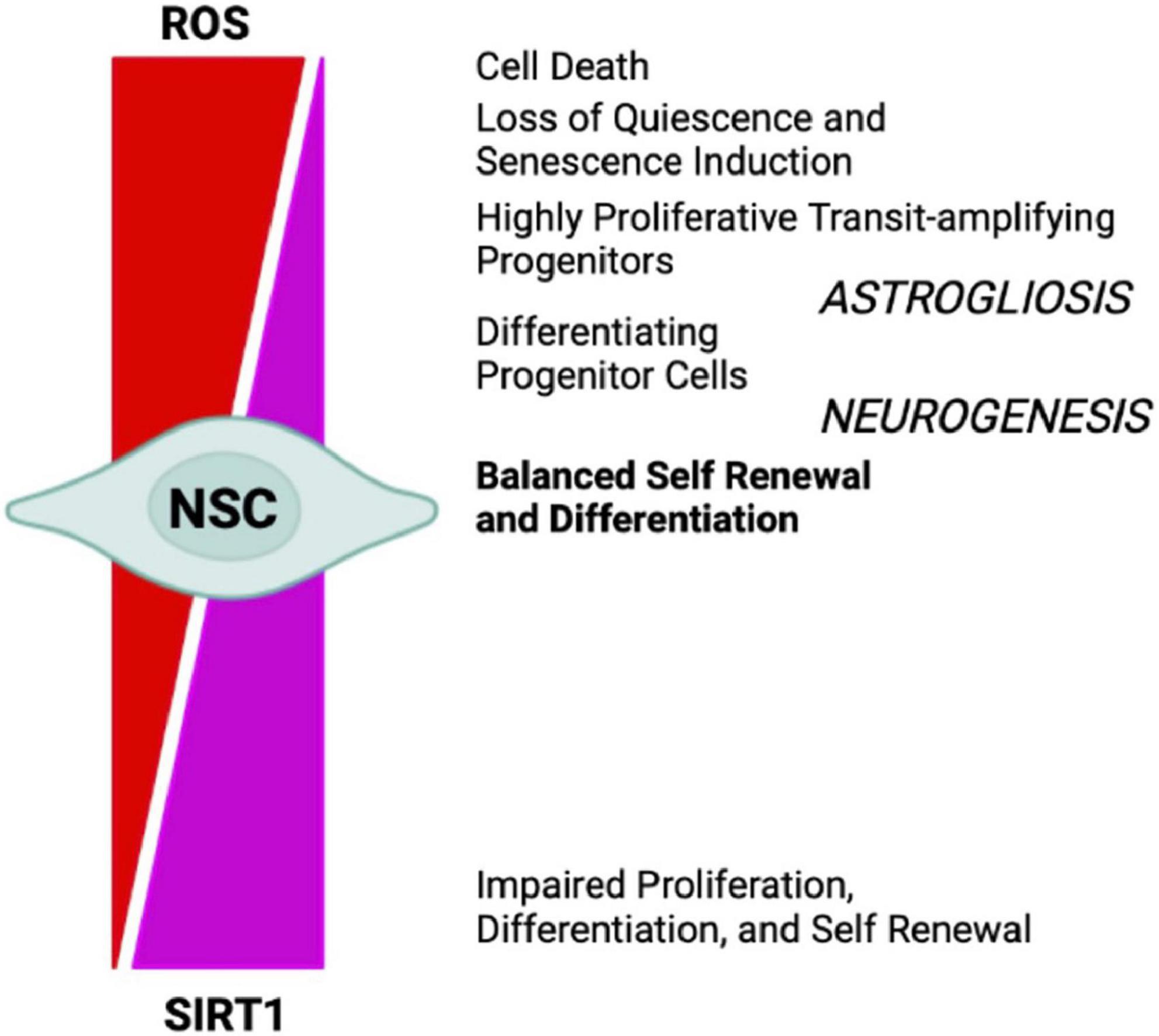
Figure 4. NSCs cell-fate decision depends upon redox balance and SIRTs activity. High oxidizing conditions favor NSCs differentiation into astrocytes (astrogliosis), senescence, and cell death, whereas mild oxidative/reducing conditions favor neuron formation (neurogenesis), through a complex regulatory network modulated by SIRT1 expression and activity.
It has been shown that mild oxidative stress triggers SIRT1 activation and subsequent HES-1 mediated transcriptional inhibition of MASH1, leading to increased astrogliogenesis (Prozorovski et al., 2008). Additionally, mouse models of advanced aging, bearing a deletion in the clock gene BMAL1, show high levels of ROS and SIRT1 expression in the brain, coupled with a reduction of hippocampal adult neurogenesis in favor of an increased production of astrocytes (Ali et al., 2015). SIRT1 activation is crucial for mitochondrial homeostasis, as it regulates the expression of OXPHOS enzymes as well as of the peroxisome proliferator-activated receptor γ (PPARγ) coactivator-1a (PGC-1α), which is crucial for mitochondrial gene expression (Lin J. et al., 2005). NAD+/NADH ratio, which is in turn a measure of cellular redox status (Jones and Sies, 2015), plays a role in regulating stem cell fate through SIRTs activity (Haigis and Sinclair, 2010; Imai and Guarente, 2014, 2016). During aging, the oxidative stress increase determines a NAD+ depletion that negatively impacts on mitochondria (Du et al., 2003). In vivo mice model of chronic cerebral hypoperfusion, showed that NAD+ improved cognitive function and reduced neuroinflammation in association to mitochondrial protection and ROS inhibition through the activation of SIRT1/PGC-1α pathway (Zhao et al., 2021). Moreover, NAD+ levels depend upon the energetic level of the cells, increasing during CR and decreasing under conditions of high-energy load, such as high-fat diet. Interestingly, CR decreases oxidative stress leading to increased NAD+ levels and improving mitochondrial function, by the SIRT3-mediated increase of SOD2 activity (Qiu et al., 2010). Therefore, NAD plays a role in the mito-nuclear protein imbalance, which has been described as a conserved mechanism in the regulation of energy metabolism (Houtkooper et al., 2013). Thus, as SIRTs enzymatic activity depends upon NAD+ levels they could act as metabolic sensors coupling cellular metabolic status to regulatory responses (Nemoto et al., 2004; Canto and Auwerx, 2009; Peng et al., 2010; Khoury et al., 2018). It has been shown that ROS levels also influence the oxidation state of cysteine (Cys)-containing redox sensors, following a diurnal variation (Blanco et al., 2007), which alter the activity and localization of these proteins, thereby regulating NSCs state and fate. SIRT1 contains critical cysteine residues vulnerable to oxidation, whose alteration decreases enzyme’s activity and favors its degradation (Cai et al., 2012; Chen et al., 2012). Summarizing, pathological ROS balance alteration as well as their age-associated physiological accumulation, may affect SIRTs expression and/or activity and in turn NSCs fate decisions. It would be of outmost importance to better clarify the exact molecular mechanisms linking SIRTs enzymatic activity and ROS-dependent modulation of the correct lineage specification and/or aging in NSCs (Figure 5).
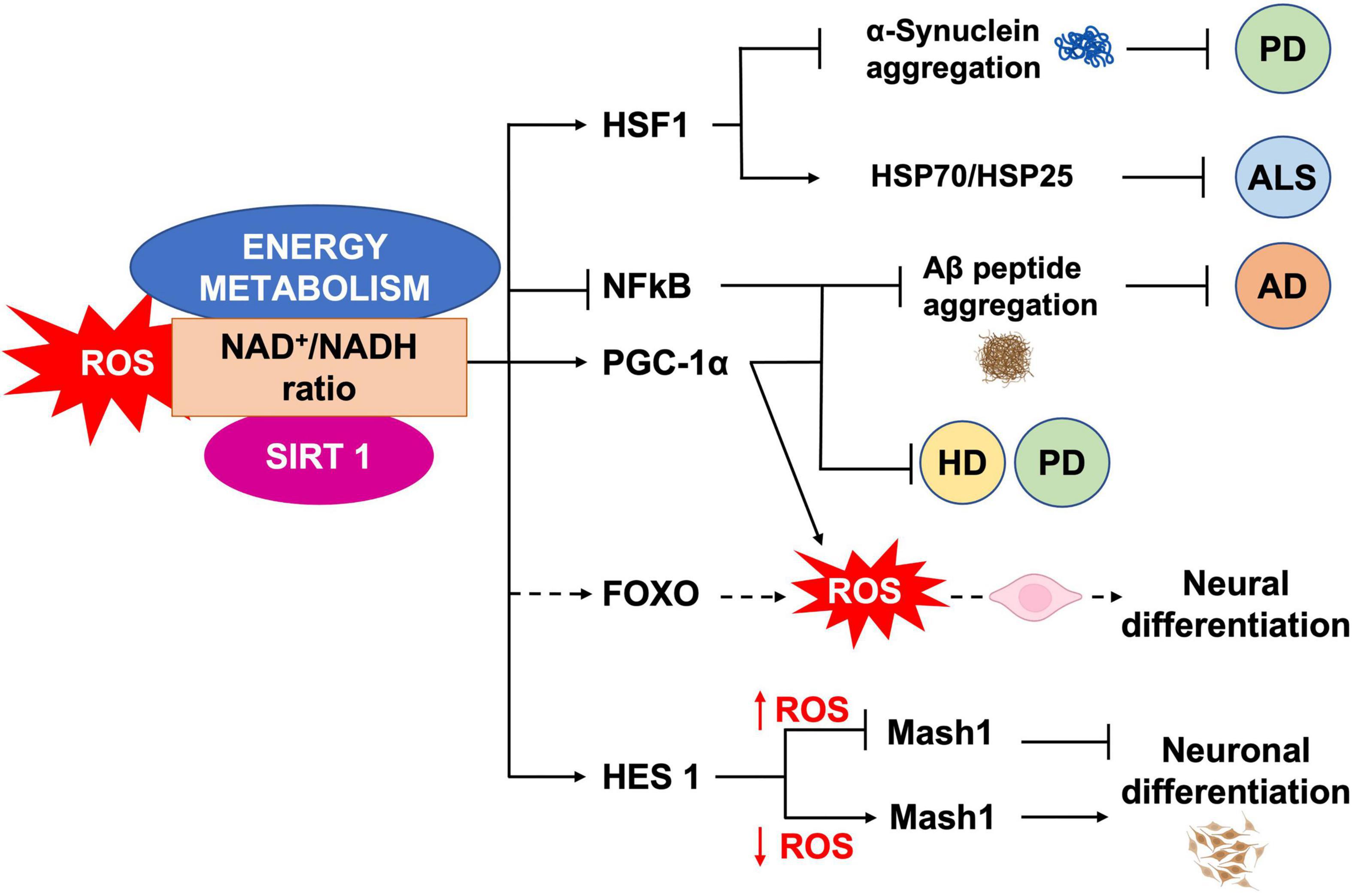
Figure 5. ROS, SIRT1 and energy metabolism interplay. NAD+/NADH ratio is a measure of the cellular redox status, and plays a role in NSCs fate regulation, through SIRT1 activity. SIRT1 regulates neural differentiation as well as cellular pathways related to NDs, by regulating the activity of different transcription factors.
In mammals, beside the above described SIRT1, seven SIRTs (SIRT1-7) have been identified, all possessing a highly conserved central nicotinamide adenine dinucleotide (NAD+)-binding site and a conserved catalytic domain. SIRT1 mainly localizes in the nucleus; SIRT2 in the cytoplasm, where primarily targets tubulin, PEPCK and FOXO1; SIRT3, SIRT4, and SIRT5 in the mitochondria, targeting various oxidative phosphorylation enzyme complexes and SODs; SIRT6 and SIRT7 in the nucleus, with SIRT6 targeting histone H3, PARP-1, and p65 (Walton et al., 2012; Maissan et al., 2021; Figure 6). Recently, SIRTs have been reported to also modulate neurodegeneration and toxicity associated with different proteins such as α-synuclein (α-SYN), huntingtin (HTT), TAU, or amyloid-beta (Aβ) peptide (Donmez, 2012). The possibility to direct NPCs differentiation may be useful to protect brain against inflammatory diseases, such as MS, which involves astrogliosis. In addition, the ability to precisely direct NPCs differentiation toward neurons may provide new therapeutic options for stroke, spinal cord injury, and age-related cognitive conditions, characterized by neuronal loss, such as AD and PD (Watroba et al., 2017). Aging is the major risk factor for NDs development and aging stem cells lose their ability to produce NPCs as well as their differentiation capacity, as described above. The conspicuous presence of SIRTs in the brain and the importance of their role in mitochondrial metabolism regulation, suggest this proteins family as a good target candidate for therapeutic protocols, aiming to control neurogenesis and NDs (Figure 5). SIRTs-dependent modulation of pluripotency and differentiation is not entirely understood, but it has been shown that SIRT1, 2, 3, 6, and 7 were involved in the modulation of these processes (Karuppagounder et al., 2015; Correia et al., 2017). In this review, we mainly focused on SIRT1 role in CNS, as this is the most studied member of the family (Mishra et al., 2021). In vitro and in vivo observations showed SIRT1 as required for proper differentiation of both ESCs and adult stem cells. Indeed, SIRT1 expression, which is higher in ESCs, decreases during differentiation, through a miRNA-mediated post-transcriptional regulation (Saunders et al., 2010; Lee et al., 2019). In addition, oxidative stress and inflammation can promote NPCs differentiation toward the astrocyte lineage, through a SIRT1-dependent regulation of the MASH1 promoter1. It has been shown that the redox state does affect NPCs cell-fate decision in vitro, with oxidizing conditions favoring differentiation into astrocytes, whereas reducing conditions favor neuron formation (Libert et al., 2008). In fact, administration of the SIRT1-activating compound resveratrol to NPCs mimicked oxidizing conditions and increased NPCs differentiation toward astrocytes, through a SIRT1-dependent mechanism (Figure 4). Under oxidizing conditions, SIRT1 and the hairy and enhancer of split 1 (HES1) form a complex that binds to and deacetylates histones at the MASH1 promoter, while recruiting co-repressors, such as TLE1 (Libert et al., 2008). Together, these events cause a down-regulation of MASH1 expression and block neuronal differentiation. The HES1-SIRT1 complex is not detected under reducing conditions, where HES1 could recruit transcription activators, such as CREB binding protein (CBP), to the MASH1 promoter, driving NPCs differentiation toward a neuronal fate (Prozorovski et al., 2008). In a recent work, it has been reported that extracellular glucose, through the coordinated action of CREB and SIRT1, could modulate HES1 expression in NSCs and NPCs. Indeed, excess glucose reduced CREB-activated HES1 expression and resulted in impaired cell proliferation. Moreover, CREB-deficient NSCs expanded poorly in vitro and did not respond to glucose availability. Elevated glucose levels also promoted SIRT1-dependent repression of the HES1 promoter. Conversely, in low glucose, CREB replaced SIRT1 on the chromatin associated with the HES1 promoter, enhancing HES1 expression and cell proliferation. Thus, the glucose-regulated antagonism between CREB and SIRT1 for HES1 transcription participates in the metabolic regulation of neurogenesis (Fusco et al., 2016). These works suggested that NSCs proliferative potential is subject to tight intrinsic regulation; therefore, a better knowledge of SIRTs role in the modulation of intracellular pathways controlling cell cycling would be of importance for the reactivation of latent NSCs populations, to engage endogenous neurogenesis, as a treatment for different NDs. In vivo and in vitro AD models showed that SIRT1 could exert a protective action toward neuronal damage (Julien et al., 2009). Indeed, SIRT1 has been shown to directly deacetylate TAU protein, thus increasing its susceptibility to degradation and preventing neurofibrillary tangles formation (Min et al., 2010; Xu et al., 2018). Moreover, autophagy-dependent Aβ degradation may also be related to SIRT1 activity (Park et al., 2016), as SIRT1 activation/overexpression has been reported to interfere with microglia-mediated Aβ toxicity, through its ability to inhibit NF-κB signaling (Chen et al., 2005). SIRT1 can also deacetylate PGC-1α, thus increasing its transcriptional regulation activity. In fact, deacetylated PGC-1α can instill transcriptional repression of β-secretase, which in turn can reduce Aβ production levels and senile plaque accumulation (Xu et al., 2018). These results are corroborated by a systematic review where it has been shown the protective role of resveratrol in AD patients (Buglio et al., 2022). Recently, the analyses of PD animal and cellular models, showed that SIRT1 overexpression was able to suppress α-SYN aggregates formation, through the activation of the molecular chaperones, driven by Heat Shock Factor 1 (HSF1) deacetylation (Donmez et al., 2012). Resveratrol was shown to have a protective effect against α-SYN-induced toxicity in SK-N-BE cells (Albani et al., 2009), and another study showed that resveratrol administration results into increased PGC-1α transcription and improved mitochondrial function, through the AMPK-SIRT1-PGC-1α signaling pathway (Ferretta et al., 2014). In PD mouse models, treatment with resveratrol and the polyphenol Epigallocatechin gallate (ECGC), results in the protection against toxicity through an up-regulation of PGC-1α, via SIRT1 activity (Xu et al., 2018). In addition, CR or 2-deoxy-D-glucose (2-DG) administration were found to reduce dopaminergic neurons loss in mice as well as to improve motor function (Duan et al., 2003), corroborating the involvement of SIRT1 in the longevity-modulating role of the insulin/IGF signaling (IIS) under CR (Holzenberger et al., 2003; Suh et al., 2008; Deelen et al., 2013). SIRT1 has a neuroprotective role also in HD (Duan, 2013; Smith et al., 2014). In fact, it has been observed that mutant HTT reduces SIRT1 activity, impairing its positive role in neuronal survival, probably due to the structural similarity between mutated HTT and SIRTs-interacting transcription factors (Naia and Rego, 2015). A recent study showed that, in mouse, SIRT1 improved survival, neuropathology, and the expression of brain-derived neurotrophic factor (BDNF), which requires the presence of CREB-regulated transcription coactivator 1 (Jeong et al., 2011). In HD knock in mice model, PGC-1α is repressed by mutant HTT and PGC-1α knockout exacerbates neurodegeneration and motor abnormalities. Conversely, PGC-1α expression ameliorates mitochondrial dysfunction and rescued neuronal toxicity induced by mutant HTT. In this context, SIRT1 ablation exacerbates neurodegeneration, whereas SIRT1 overexpression improves motor functions and rescued brain atrophy. As already described, PGC-1α is a transcriptional coactivator regulating several key mitochondrial processes, among which mitochondrial biogenesis and oxidative phosphorylation. Accordingly, SIRT1 protection against HD-related neurodegeneration is, at least partially, related to prevention of mitochondrial function impairment, through PGC-1α activation (Rodgers et al., 2005; Min et al., 2013). Several studies, using resveratrol administration, indicated that SIRT1 may be protective also in tissue culture and mouse models of ALS, by promoting neuronal survival. Indeed, increased SIRT1 expression levels have been reported for different brain regions in SOD1G93A transgenic mice, suggesting a role for SIRT1 in the motor functions in ALS, although the mechanisms and functional implications of this increased SIRT1 expression still require elucidation (Lee J. et al., 2012). One proposed mechanism is that SIRT1-dependent deacetylation of HSF1, results in an increased expression of molecular chaperones, like HSP70 and HSP25, that help to maintain intracellular protein homeostasis, thus reducing motor neurons toxicity (Watanabe et al., 2014); in another mechanism, SIRT1 activation results in an increased mitochondrial biogenesis, through PGC-1α and MFN2 (Min et al., 2010; Imai and Guarente, 2014, 2016; Park et al., 2016). Other works revealed that, in the ventral spinal cord, resveratrol protective effects were associated with increased expression and activation of SIRT1 and AMPK, resulting in the normalization of the autophagic flux and, more importantly, in an increased mitochondrial biogenesis (Mancuso et al., 2014). Finally, in the spinal cord of wild type mice, SIRT1 expression decreases during aging. Mouse models, either overexpressing or lacking SIRT1 in motor neurons, showed that SIRT1 slows age-related degeneration of motor neurons’ presynaptic sites at neuromuscular junctions (NMJs) (Herskovits et al., 2018).
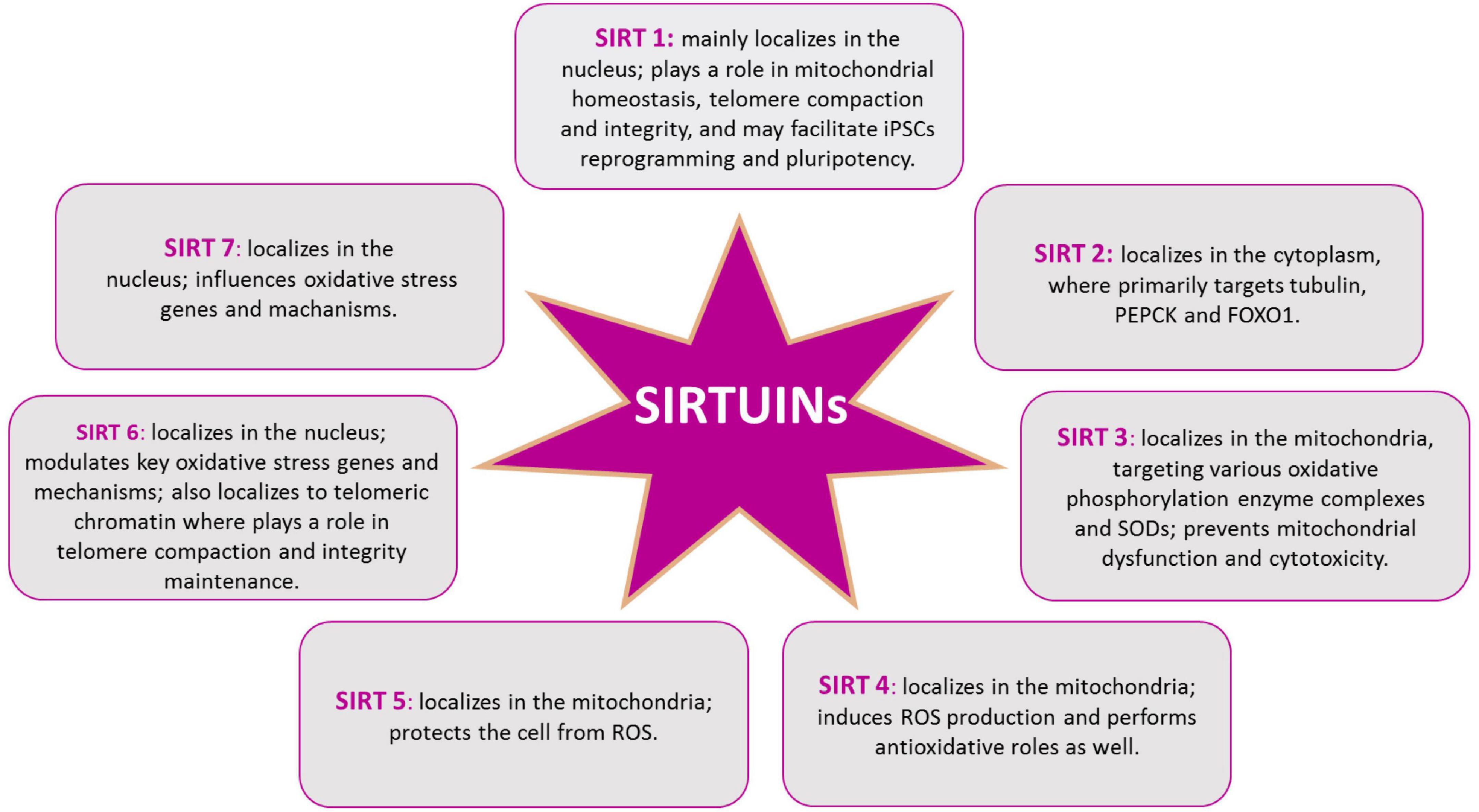
Figure 6. SIRTs protein family. Intracellular distribution of SIRTs in the cytoplasm, the mitochondria, and the nucleus, allows them to play different roles in cellular redox homeostasis.
SIRT2 is abnormally overexpressed in AD, and it is responsible for tubulin deacetylation, leading to microtubule destabilization, TAU dissociation from microtubules, and its subsequent oligomerization and aggregate formation (Silva et al., 2017). In two mouse models, SIRT2 small molecule inhibitors have been shown to reduce Aβ load and led to cognitive improvement (Biella et al., 2016). In PD, SIRT2 inhibition reduces α-SYN aggregation and toxicity, by modifying its acetylation levels (de Oliveira et al., 2017). Instead, the potential role of SIRT2 in aging is suggested by the association found between human longevity and a polymorphism in the probably regulatory elements of the SIRT2 gene (Crocco et al., 2016).
SIRT3 reacts to nutritional status and mediates some of the beneficial effect of CR, including many of the CR-induced transcriptional changes, as observed in the suppression of cochlear neurons degeneration (Someya et al., 2010). SIRT3 is upregulated when ROS are pharmacologically augmented in neuronal culture and in human AD brains, while is reduced in cells expressing mutant HTT (Weir et al., 2012). On the other hand, small molecules mediated SIRT3 upregulation decreases ROS levels and prevents mitochondrial dysfunction and cytotoxicity induced by mutant HTT (Fu et al., 2012). It has been also suggested that SIRT3 plays a role in ALS, as its overexpression protects against SOD1G93A-induced mitochondrial fragmentation and neuronal cell death (Song et al., 2013), in agreement with other works showing SIRT3 protection against aging-linked apoptosis in mice and excitotoxic insults in cultured neurons (Lee J. et al., 2012), although the exact mechanism remains elusive. It has been proposed that Cyclophilin D (CYPD), a component of the mitochondrial permeability transition pore, could be involved. Indeed, CYPD reduction delays motor neuron cell death and extends the lifespan of SOD1G93A mice (Martin et al., 2009). Notably, CYPD is a SIRT3 substrate, and SIRT3-dependent deacetylation inhibits CYPD function, prevents mitochondrial permeability transition and age-related cardiac hypertrophy (Hafner et al., 2010). In the same work, it has been shown that PGC-1α, similarly to SIRT3, is able to restore mitochondrial dynamics and cell viability of mutant SOD1G93A neurons (Song et al., 2013) and PGC-1α promotes SIRT3 expression (Kong et al., 2010). SIRT3 was also shown to physically interact with the long chain acyl-CoA dehydrogenase (LCAD) in NSCs and to require its activation to prevent age-impaired neurogenesis in mice (Santos et al., 2021). The repertoire of SIRT3 interacting partners suggest further aspects of its role also in longevity. In fact, SIRT3 deacetylation also supports the stability and activity of OGG1, thus protecting mtDNA from the accumulation of the mutagenic damage produced by 8-oxoguanine (Cheng et al., 2013); deacetylates the DNA repair regulator protein Ku70 (Sundaresan et al., 2008); binds the heat shock protein HSP70, resulting in an increased nuclear presence (Law et al., 2009). Summarizing, these interactions are potentially linked to the mechanisms of age-related neurodegeneration (Jesko et al., 2017).
Alzheimer’s disease patients showed decreased expression of SIRT6 and mice lacking SIRT6 showed TAU protein stabilization and increased phosphorylation, via the activation of the glycogen synthase kinase 3 (GSK3) (Kaluski et al., 2017). Like SIRT1 and SIRT3, also SIRT6 plays a role in CR (Kanfi et al., 2008). In cells under H2O2-induced oxidative stress, the suppression of SIRT6 protein levels mediates premature senescence-like phenotype (Liu et al., 2014). In turn, SIRT6 levels restoration rescues several senescence linked traits, through the modulation of IIS-mTOR signaling and restores the DNA base excision repair efficiency in human foreskin fibroblasts from aged donors (Takasaka et al., 2014; Xu et al., 2015). The links among SIRT6, DNA repair, and aging also extend to telomere maintenance. Indeed, SIRT6 localizes to telomeric chromatin where it facilitates the binding of Werner Syndrome (WS) protein, a DNA helicase crucial for genome stability. Accordingly, SIRT6 deficiency leads to replicative senescence and telomere dysfunction, resembling the WS pathology (Michishita et al., 2008). Moreover, to mitigate aging and oxidative stress SIRT6 interacts with several crucial pathways of transcriptional regulation as NRF-2 and NF-κB (Kawahara et al., 2009; Pan et al., 2016).
Lastly, insufficient data are available on mitochondrial SIRT4, SIRT5, and SIRT7 dysregulation in NDs and aging. In conclusion, although until now relatively little is known about the role of all SIRTs in neurogenesis and age-associated neurodegenerative diseases, it is becoming clear that this protein family plays a role in adult neurogenesis, controlled by mitochondrial metabolism and ROS, as well as in the antioxidative defense in the aging brain and in the aging-related CNS diseases (Singh et al., 2018; Figure 7).
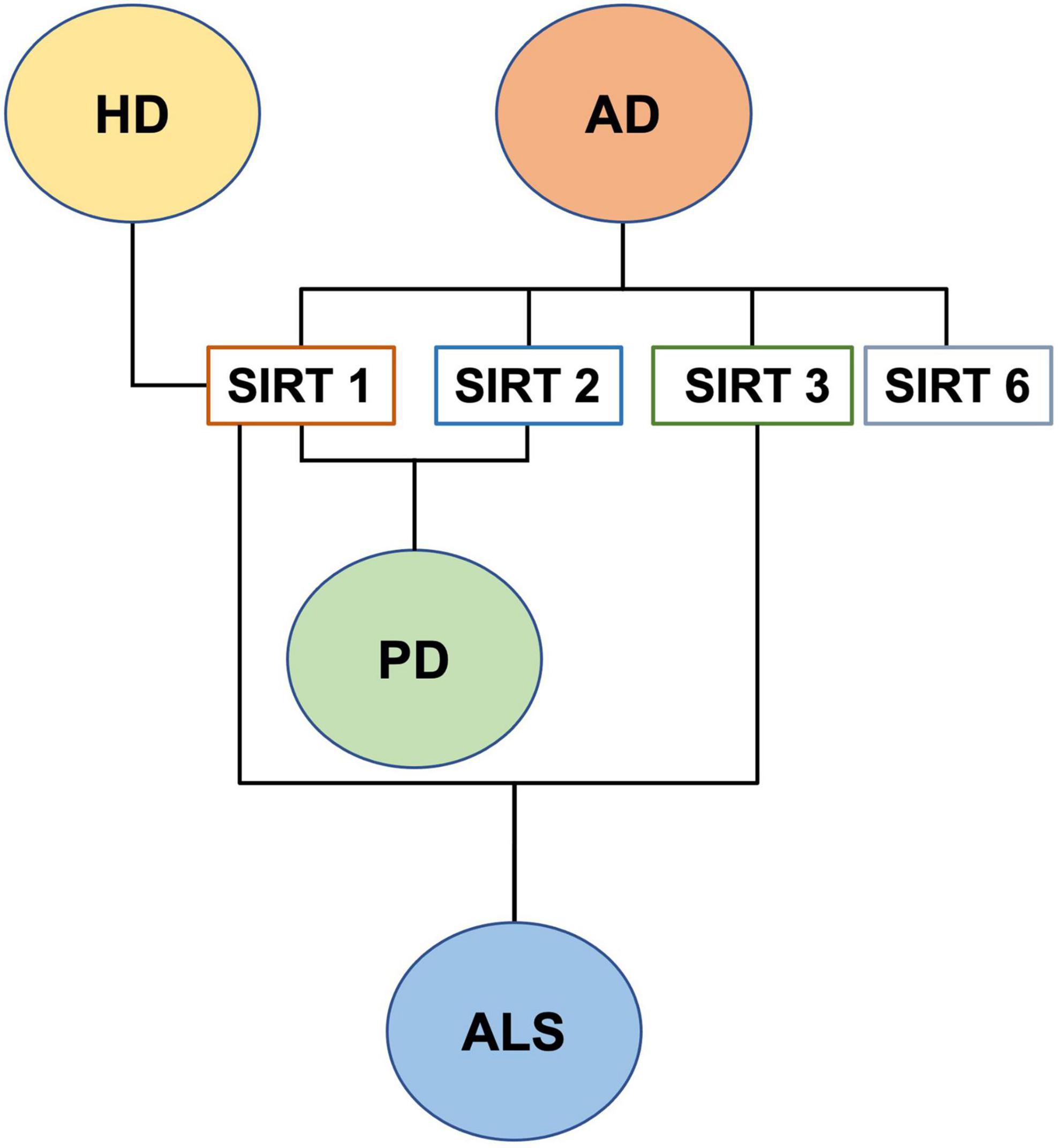
Figure 7. SIRTs and neurodegenerative diseases.
Telomeres are chromosome termini structures consisting of tandem DNA nucleotide repeats and the shelterin complex, a six-protein complex comprising TRF1, TRF2, POT1, TIN2, TPP1, and RAP1 (de Lange, 2018). It has been reported that in NSCs, isolated from the subventricular zone (SVZ) of telomerase-deficient adult mice, telomere attrition dramatically impairs in vitro proliferation. In addition, NSCs with short telomere, showed upregulation of p53 expression, in agreement with the importance of p53 pathways in the telomere damage response in mice (Ferron et al., 2004). SIRT1 overexpressing mice showed increased health span and longer telomeres, as compared to both wt and SIRT1-deficient mice, the latter showing even shorter telomeres. SIRT1 overexpression prevents telomere shortening, as the mice grew older, through the stimulation of the telomerase enzyme, whose activity is the major contributor to telomere production (Palacios et al., 2010). Moreover, SIRT1 may also influence a second telomeres maintenance pathway, called alternative lengthening of telomeres (ALT). In fact, SIRT1 overexpression increased the amount of homologous recombination, a key step in the ALT pathway, all along the chromosomes and at chromosome ends. Accordingly, SIRT1-deficient cells showed increased damage at their chromosome ends (Palacios et al., 2010) and SIRT1-overexpressing mice stay healthier for longer (Short, 2010). Notably, it has been also shown that SIRT1 binds to the elongated telomeres of differentiated cells reprogrammed into an embryonic stem cell-like state (Palacios et al., 2010). Resveratrol and progesterone can mediate telomerase activity in self-renewing human cells, with resveratrol activating, and progesterone inactivating the enzyme. Nevertheless, a direct connection between telomerase activity modulation and SIRT1 activity has not been shown yet (Pearce et al., 2008; Koziel et al., 2011). A study reported longer leukocyte telomere length (LTL) in PD patients (Pearce et al., 2008) while others, examining LTL in psychological stress, cognitive impairment, and dementia, found shorter LTL associated with these conditions (Eitan et al., 2014). In a recent study, whole genome sequencing of ALS patient’s leukocyte-derived DNA, revealed longer telomeres, in agreement with observations in PD patients (Al Khleifat et al., 2019). Another work showed a trend for longer telomeres in microglia from human post-mortem brain tissue with ALS (Linkus et al., 2016). However, the same authors found that knocking out telomerase in SOD1G93A-transgenic mice accelerated the ALS phenotype, concluding that telomerase dysfunction might contribute to the age-related risk for this disease. As discussed above, during normal aging as well as in NDs an increase of oxidative stress in neurons and glial cells is observed, but it is still unknown neither if oxidative stress causes telomere erosion, nor if ROS-induced telomere shortening in neurons and glia is a causal or contributing factor to NDs. As reviewed by Eitan et al. (2014), short telomeres in immune cells, astrocytes, and neurons could enhance oxidative stress-dependent senescence as well as the associated secretion of pro-inflammatory mediators (senescence-associated secretory phenotype), that may enhance disease progression. Moreover, in vitro cultured neurons showed that telomere damage can trigger cell death (Jurk et al., 2012; Stephenson et al., 2018) while telomerase activation may reduce neuronal vulnerability (Cheng et al., 2007; Eitan et al., 2012). In aged primary cells, increased ROS, caused by progressive mitochondrial failure, is concomitant with telomere shortening (Sanderson and Simon, 2017) and ROS neutralization does not restore mitochondrial function but still inhibits telomere shortening, thus suggesting ROS as the main player in telomere shortening (Billard and Poncet, 2019). One possible explanation for this ROS-dependent event relies on the presence of telomeric GGG repeats. ROS influence the GGG repeats by generating stretches of 8-oxoguanine, especially difficult to repair. The presence of unrepaired single or tandem 8-oxoguanine drastically inhibits TRF1 and TRF2 binding, thus impairing telomerase recruitment and contributing to telomere deprotection and shortening. Indeed, when oxidative stress is combined with telomerase inhibitors, it results into faster telomere shortening, only in oxidative damage repair deficient cells (von Zglinicki, 2002; Billard and Poncet, 2019). To counteract the deleterious telomeric consequences of ROS production, cells exploit OGG1 activity, which plays an important role also at telomeres, beside the mitochondrial one already described above. In fact, it has been reported that OGG1 depletion results into chronic replication stress and an increased telomere loss (Billard and Poncet, 2019). As OGG1 activity modulation depends upon SIRT1-dependent deacetylation, it is reasonable to hypothesize that SIRT1 plays a role in the repair of telomeric 8-oxoG in hippocampal cells (Sarga et al., 2013; Figure 8). SIRT1 also plays a role in telomere compaction and integrity, as showed by the attenuation of telomere shortening during aging, in SIRT1 gain of function mouse model, and by the reduction of H3K9me3 and fragile telomeres, in SIRT1 depletion model (Palacios et al., 2010). Finally, SIRT6 is responsible for H3K9 deacetylation at telomeres, and its depletion leads to replication defects, unrepaired DNA damage, and an accelerated aging phenotype. Moreover, SIRT6 facilitate the binding of the DNA helicase WRN, crucial for genome stability, to telomeres while SIRT6 deficiency leads to replicative senescence and telomere dysfunction, resembling the pathology seen in WS (Michishita et al., 2008). To mitigate aging and oxidative stress, SIRT6 interacts with several crucial pathways of transcriptional regulation, such as NRF-2 and NF-κB (Kawahara et al., 2009; Pan et al., 2016). All these data confirm the interplay between ROS levels and SIRTs also in the modulation and protection of telomere length, a key factor for NSCs self-renewing, protection from NDs onset, and brain aging.
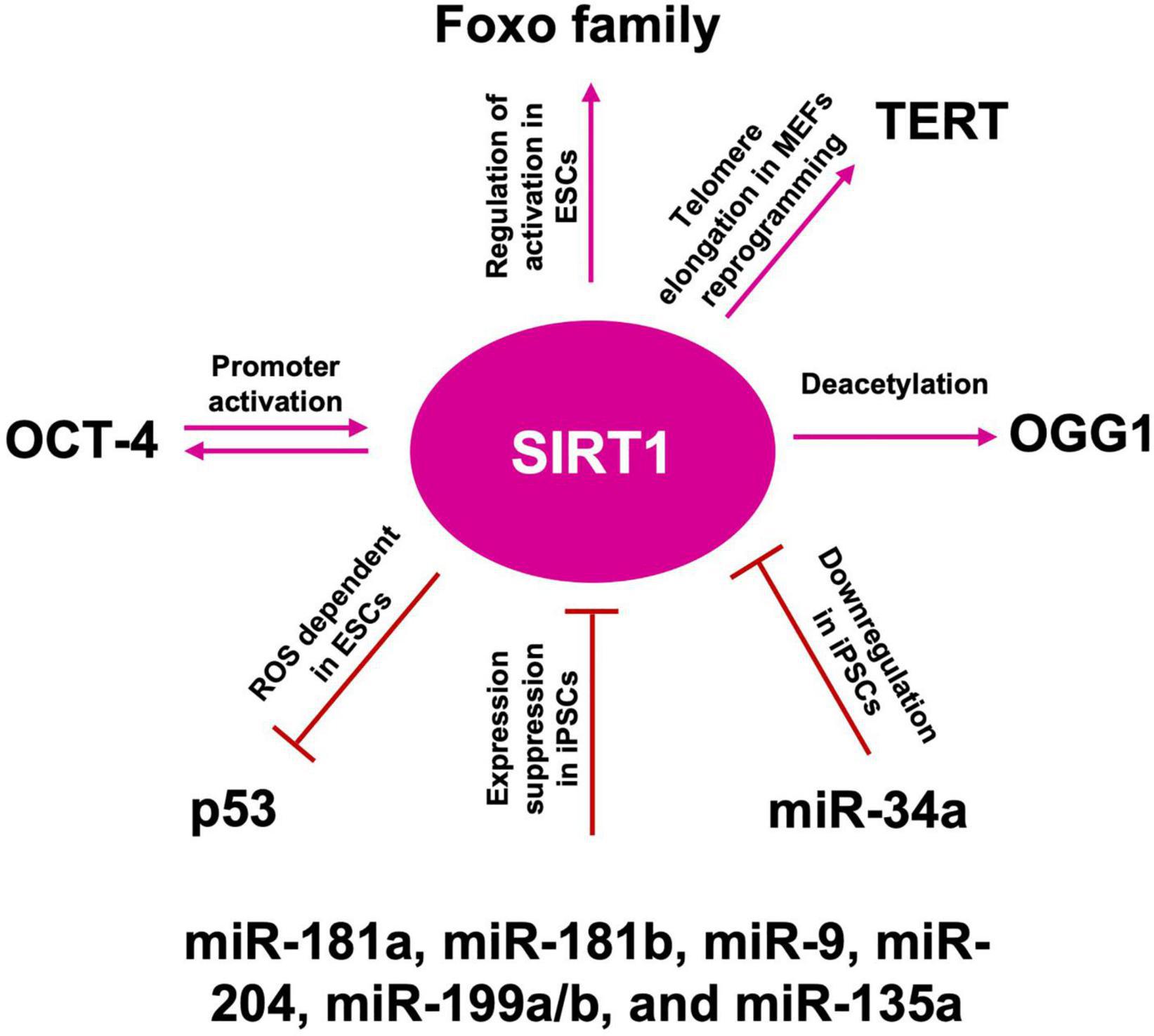
Figure 8. SIRT1 role in telomere length maintenance and cellular reprogramming. SIRT1: acts on OGG1 deacetylation, preserving telomere repairing; activates TERT transcription promoting telomere elongation; acts by blocking p53 nuclear translocation through ROS favoring the reprogramming; mutually interacts with OCT-4 in the cellular reprogramming process; activates FOXO family of transcription factors, which are required to maintain pluripotency; miR-181a, miR-181b, miR-9, miR-204, miR-199a/b, and miR-135a suppress SIRT1 expression, facilitating iPSCs reprogramming and pluripotency; downregulated by miR-34a promotes iPSCs differentiation into NSCs.
Somatic cells reprogramming, by the forced expression of Yamanaka factors (OCT-4, KL-4, SOX-2, and c-MYC), enables the generation of iPSCs, displaying ESCs-like properties (Takahashi et al., 2007). After reprogramming, also somatic mitochondria can revert to an ESC-like state in terms of morphology, cellular distribution, and rate of biogenesis (Prigione et al., 2010). Both iPSCs and ESCs are characterized by a low redox status and by the capability to repair their DNA, following oxidative damage (Armstrong et al., 2010). However, the iPSCs reprogramming protocol, through viral transduction, is associated with high ROS generation, leading to oxidative damage, impaired cell survival, and increased genetic aberrations. Administration of AOX, such as NAC or vitamin C, improves reprogramming efficiency and reduces genetic abnormalities (Ji et al., 2014), thus validating the paradigm that also during the cell reprogramming, balanced levels of oxidative phosphorylation must be maintained on the route to pluripotency (Skvortsova et al., 2022). Telomere elongation is an iPSCs hallmark, thus the role of SIRTs in pluripotency maintenance has been extensively investigated. As described above, in iPSCs SIRT1 is recruited to the telomeres and binds the telomeric repeats (Palacios et al., 2010). In murine embryonic fibroblasts (MEFs) reprogramming, SIRT1 is required for efficient post-reprogramming telomere elongation, through a c-MYC-dependent regulation of the mTERT gene, and SIRT1-deficient iPSCs accumulate chromosomal aberrations and show a de-repression of telomeric heterochromatin (De Bonis et al., 2014). Moreover, it has been shown that SIRT1 plays a role in the maintenance of iPSCs also after the acquisition of pluripotency (Zhang et al., 2014). In late passages iPSCs, SIRT1 slows down c-MYC degradation, thus ensuring enough binding to the TERT promoter and increasing TERT transcription and expression (Wu et al., 1999; Figure 8).
It has been reported that reprogramming, by classic Yamanaka factors, of human dermal fibroblasts (HDFs) from older human subjects, was more difficult than those of youngers, but that could be improved by SIRT6 expression. As of today, little is known about the molecular mechanism of SIRT6 regulation (Sharma et al., 2013). Several studies showed p53 as a negative regulator of reprogramming and that reprogramming efficiency could be ameliorated by p53 pathway’s inhibition (Gong et al., 2016; Ong and Ramasamy, 2018). In wild type mouse ESCs (mESCs), following DNA damage, p53 binding to Nanog promoter inhibits expression and results in pluripotency loss and differentiation (Lin T. et al., 2005). In wt mESCs endogenous ROS could trigger apoptosis, through mitochondrial translocation of p53 and BAX, while in SIRT1–/– mESCs they promote nuclear p53 translocation and Nanog inhibition. Hence, ROS-dependent SIRT1 activation, acts by blocking p53 nuclear translocation (Han et al., 2008), thus modulating gene expression under ROS control, confirming the role of ROS as signaling molecules (Figure 8). Jang et al. (2017) showed that SIRT1 depletion in human ESCs (hESCs) results in p53 hyperacetylation and a dramatic reduction of DNA repair proteins, thus favoring DNA damage accumulation. Nevertheless, SIRT1 role as well as its interplay with different transcription factors are still under debate. In fact, even if it has been reported that OCT-4 could directly interact with and activate the SIRT1 promoter, thus in turn inactivating p53 through SIRT1-dependent deacetylation (Zhang et al., 2014), other studies showed that SIRT1 and OCT-4, along with SOX2, co-occupy the same distal enhancer region at the OCT-4 promoter, and cells lacking SIRT1 showed hyper-acetylation of OCT-4 (Williams et al., 2016). There are also controversial reports about a lack of binding between SIRT1 and the OCT-4 promoter (Chen et al., 2014), and other suggesting that SOX2 and SIRT1 interaction requires OCT-4 (Mu et al., 2015). Nevertheless, a key role for SIRT1 is suggested by the observation that SIRT1–/– MEFs exhibited decreased iPSCs reprogramming efficiency, a defect that could be rescued by SIRT1 overexpression. In hESCs, SIRT1 also regulates the activation of the FOXO family of transcription factors, which are required to maintain pluripotency, by directly regulating the expression levels of OCT-4, Nanog, and SOX-2 (Zhang et al., 2011). In addition, SIRT1 downregulation has been observed during mouse iPSCs differentiation into NSCs (Hu et al., 2014). Furthermore, miRNA such as miR-181a, miR-181b, miR-9, miR-204, miR-199a/b, and miR-135a have been shown to suppress SIRT1 expression, suggesting a new strategy in the regulation of somatic cells reprogramming toward iPSCs (Hsu et al., 2018). Indeed, SIRT1 may facilitate iPSCs reprogramming and pluripotency, through the miR-34a-SIRT1-p53 axis, as SIRT1 downregulation by miR-34a, results in the inhibition of MEFs-derived iPSC formation, suggesting a possible involvement in iPSCs differentiation into NSCs. These results indicate that the early stage SIRT1 repression may contribute to the initiation of NSCs/NPCs differentiation from ESCs and iPSCs and explain, at least partially, the developmental defects observed in the CNS of SIRT1 deficient mice (Lee Y. et al., 2012; Figure 8). Despite these data highlight that the SIRT1-p53 regulatory axis plays a role also in cellular reprogramming, the real extent of SIRTs involvement in cell reprogramming needs to be further investigated. Moreover, further understanding of SIRTs involvement in age-related mitochondrial alteration, like ROS levels increase, could also help to modulate, or ameliorate stem cell reprogramming, through the usage of SIRTs modulators, such as resveratrol, or NAD+ modulators, as bioarginine.
The scientific evidence we reviewed here, suggested that, in the CNS, Sirtuins build up a connection between epigenetic and metabolism, by acting as modulators in a complex molecular network, under the direct and indirect ROS control, to determine NSCs fate, reprogramming, and aging, through mitochondria regulation and telomere protection. Indeed, oxidative stress, or a general disruption of redox cellular homeostasis, can affect SIRTs expression levels and activity, leading to a modulation of the balance between stem cell quiescence, self-renewal, and differentiation (Figure 9). Moreover, in physiological aging as well as NDs, neurons and glial cells are characterized by increased oxidative stress and several evidence suggest a key role for SIRTs in the antioxidative defense in the brain (Figure 10). However, it is still unknown whether oxidative stress can cause telomere erosion in CNS cell populations, nor if ROS-induced telomere shortening could be a causal or contributing factor to NDs. Nevertheless, SIRT1 and SIRT6 proved to play a role in telomere compaction and integrity maintenance. As we discussed, one possible explanation of ROS-dependent telomere shortening relies on the presence of GGG telomeric repeats, which are particularly sensitive ROS target sites, for the generation of 8-oxoguanine stretches, which are especially difficult to repair. The presence of unrepaired single or tandem 8-oxoguanine drastically impairs the recruitment of telomerase, thereby contributing to telomere deprotection and shortening observed in aging and NDs. To counteract this deleterious ROS-dependent effect, OGG1 activity plays an important role both at telomeres and during the NSCs differentiation. In fact, OGG1 depletion results into chronic replication stress and an accelerated telomere loss, as well as in a shift of NSCs differentiation toward the astrocytic lineage. ogg1–/– mice differentiating neural cells spontaneously accumulate mtDNA damage coupled to a NAD+/NADH ratio increase, which in turn leads to SIRT1 activation. SIRT1 activation, caused by oxidizing conditions, inhibits MASH1 expression and blocks neuronal differentiation but favors a shift toward an astrocytic lineage. This evidence suggests that SIRTs, being dependent upon NAD+ levels, could act as metabolic sensors able to couple cellular metabolic status to a specific regulatory response. Moreover, ROS-dependent oxidation of cysteine-containing redox sensors, such as SIRT1, impairs their activity and localization, thereby regulating NSCs state and fate. Finally, the role of SIRT1 in the regulation of ROS-controlled gene expression during cellular reprogramming, corroborate the importance of this protein family in the correct NSCs lineage specification, both in vitro and in vivo.
Figure 9. Energy metabolism regulation in NSCs fate determination. Imbalance of redox cellular homeostasis, can affect SIRTs expression levels and activity, leading to a modulation of the balance between stem cell quiescence, self-renewal, and differentiation.
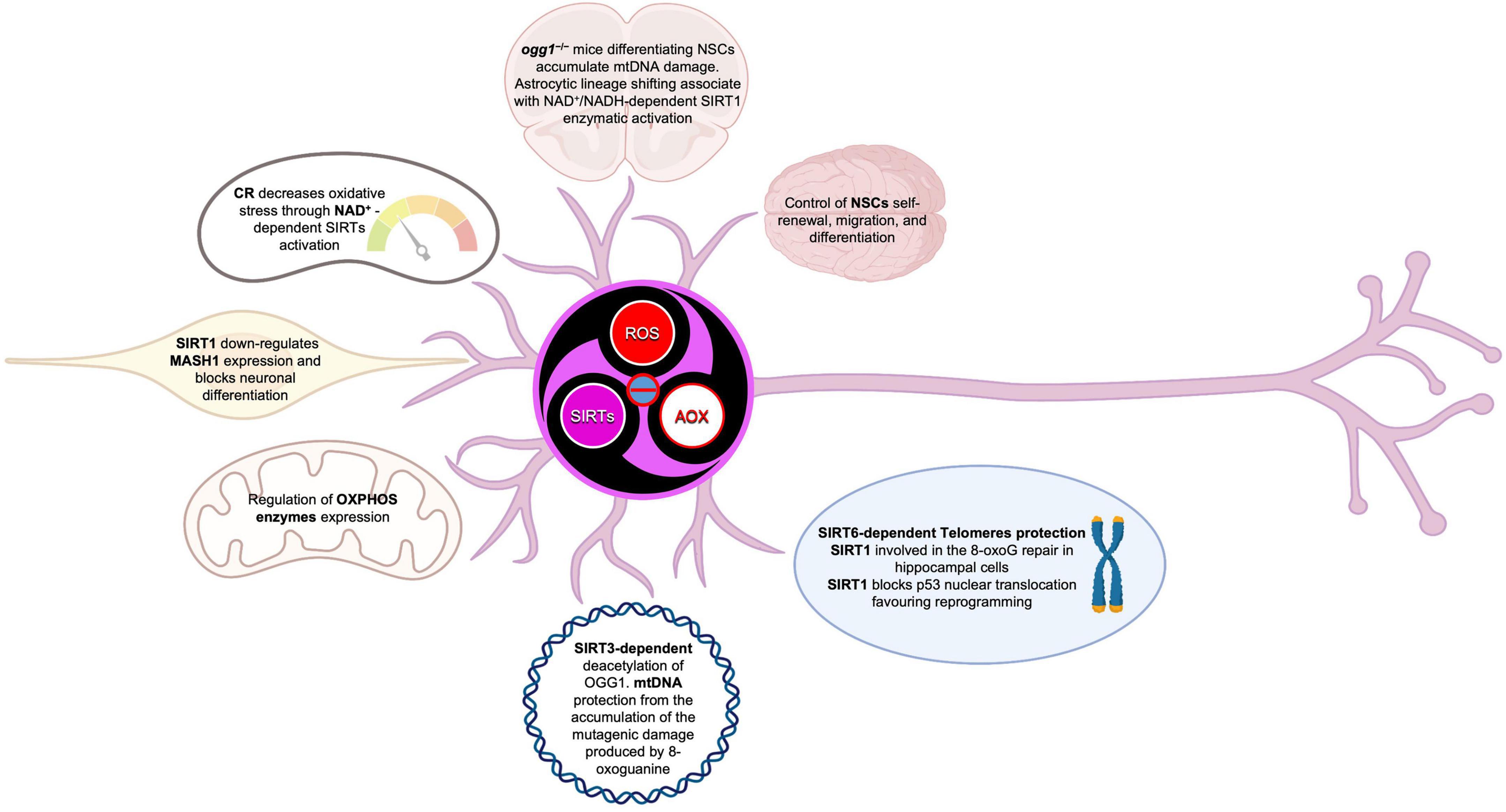
Figure 10. Sirtuins in the brain build up a connection between epigenetic and metabolism. SIRTs act as modulators in a complex molecular network, under direct and indirect ROS control. SIRTs-ROS/AOX balance affects NSCs fate, reprogramming, and aging, mainly through mitochondria physiology regulation and telomere protection.
Therefore, increased oxidative stress impact on mitochondrial physiology, by generating mtDNA damage and/or electron transport chain impairment, as well as on plasma cysteine homeostasis, thus further increasing ROS generation. Indeed, SIRTs have been shown to be involved not only in the regulation of antioxidative enzymes expression and activity, but also in the production of pro-oxidants, which, through the alteration of the NAD+/NADH ratio, affect SIRTs activity in a feedback loop that helps prevent the cell from entering or maintaining a state of oxidative stress.
We think that a deeper understanding of the molecular mechanism underlying the ROS-dependent regulation of SIRTs activity, as a response to cellular redox homeostasis alterations, would be of great help in the modulation of both iPSCs reprogramming and NSCs differentiation fate, as well as for a more detailed comprehension of NDs, aging, and some behavioral anomalies associated with nutrient deficiency. Moreover, a better knowledge of activation/deactivation cycles of H2O2 production and responsive redox protein systems, will help the understanding of the redox biology of neurogenesis.
EM and EI: conceptualization. EM, EI, and LA: literature search. EM and CR: writing. EM, EI, CR, and LA: review and editing. All authors contributed to the manuscript and approved the submitted version.
This work has been funded by the Italian Minister of Health, Ricerca Corrente 2022–2024.
The Figures 1–3, 5, 9, 10 were created with BioRender.com.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Adusumilli, V., Walker, T., Overall, R., Klatt, G., Zeidan, S., Zocher, S., et al. (2021). ROS dynamics delineate functional states of hippocampal neural stem cells and link to their activity-dependent exit from quiescence. Cell Stem Cell 28, 300–314.e6. doi: 10.1016/j.stem.2020.10.019
Ahlqvist, K., Hamalainen, R., Yatsuga, S., Uutela, M., Terzioglu, M., Gotz, A., et al. (2012). Somatic progenitor cell vulnerability to mitochondrial DNA mutagenesis underlies progeroid phenotypes in Polg mutator mice. Cell Metab. 15, 100–109. doi: 10.1016/j.cmet.2011.11.012
Al Khleifat, A., Iacoangeli, A., Shatunov, A., Fang, T., Sproviero, W., Jones, A., et al. (2019). Telomere length is greater in ALS than in controls: A whole genome sequencing study. Amyotroph. Lateral Scler. Frontotemporal Degener. 20, 229–234. doi: 10.1080/21678421.2019.1586951
Albani, D., Polito, L., Batelli, S., De Mauro, S., Fracasso, C., Martelli, G., et al. (2009). The SIRT1 activator resveratrol protects SK-N-BE cells from oxidative stress and against toxicity caused by alpha-synuclein or amyloid-beta (1-42) peptide. J. Neurochem. 110, 1445–1456.
Ali, A., Schwarz-Herzke, B., Stahr, A., Prozorovski, T., Aktas, O., and von Gall, C. (2015). Premature aging of the hippocampal neurogenic niche in adult Bmal1-deficient mice. Aging 7, 435–449. doi: 10.18632/aging.100764
Amber, S., Sumera, Mirza, F., Asif, M., Hassan, D., Ahmed, T., et al. (2020). Amyloid-beta induced neurotoxicity impairs cognition and adult hippocampal neurogenesis in a mouse model for Alzheimer’s disease. Curr. Alzheimer Res. 17, 1033–1042. doi: 10.2174/1567205017666201224162730
Anesti, V., and Scorrano, L. (2006). The relationship between mitochondrial shape and function and the cytoskeleton. Biochim. Biophys. Acta 1757, 692–699.
Angelova, P., and Abramov, A. (2018). Role of mitochondrial ROS in the brain: From physiology to neurodegeneration. FEBS Lett. 592, 692–702.
Armstrong, L., Tilgner, K., Saretzki, G., Atkinson, S., Stojkovic, M., Moreno, R., et al. (2010). Human induced pluripotent stem cell lines show stress defense mechanisms and mitochondrial regulation similar to those of human embryonic stem cells. Stem Cells 28, 661–673. doi: 10.1002/stem.307
Asher, G., and Schibler, U. (2011). Crosstalk between components of circadian and metabolic cycles in mammals. Cell Metab. 13, 125–137.
Beckervordersandforth, R. (2017). Mitochondrial metabolism-mediated regulation of adult neurogenesis. Brain Plast. 3, 73–87. doi: 10.3233/BPL-170044
Bedard, K., and Krause, K. (2007). The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 87, 245–313.
Biella, G., Fusco, F., Nardo, E., Bernocchi, O., Colombo, A., Lichtenthaler, S., et al. (2016). Sirtuin 2 Inhibition improves cognitive performance and acts on amyloid-beta protein precursor processing in two Alzheimer’s disease mouse models. J. Alzheimers Dis. 53, 1193–1207. doi: 10.3233/JAD-151135
Bigarella, C., Liang, R., and Ghaffari, S. (2014). Stem cells and the impact of ROS signaling. Development 141, 4206–4218.
Billard, P., and Poncet, D. (2019). Replication stress at telomeric and mitochondrial DNA: Common origins and consequences on ageing. Int. J. Mol. Sci. 20:4959. doi: 10.3390/ijms20194959
Blanco, R., Ziegler, T., Carlson, B., Cheng, P., Park, Y., Cotsonis, G., et al. (2007). Diurnal variation in glutathione and cysteine redox states in human plasma. Am. J. Clin. Nutr. 86, 1016–1023.
Bonnert, T., Bilsland, J., Guest, P., Heavens, R., McLaren, D., Dale, C., et al. (2006). Molecular characterization of adult mouse subventricular zone progenitor cells during the onset of differentiation. Eur. J. Neurosci. 24, 661–675.
Borodkina, A., Shatrova, A., Abushik, P., Nikolsky, N., and Burova, E. (2014). Interaction between ROS dependent DNA damage, mitochondria and p38 MAPK underlies senescence of human adult stem cells. Aging 6, 481–495. doi: 10.18632/aging.100673
Bouchard-Cannon, P., Mendoza-Viveros, L., Yuen, A., Kaern, M., and Cheng, H. (2013). The circadian molecular clock regulates adult hippocampal neurogenesis by controlling the timing of cell-cycle entry and exit. Cell Rep. 5, 961–973. doi: 10.1016/j.celrep.2013.10.037
Bueler, H. (2021). Mitochondrial and autophagic regulation of adult neurogenesis in the healthy and diseased brain. Int. J. Mol. Sci. 22:3342. doi: 10.3390/ijms22073342
Buglio, D., Marton, L., Laurindo, L., Guiguer, E., Araujo, A., Buchaim, R., et al. (2022). The role of resveratrol in mild cognitive impairment and alzheimer’s disease: A systematic review. J. Med. Food 25, 797–806. doi: 10.1089/jmf.2021.0084
Cai, W., Ramdas, M., Zhu, L., Chen, X., Striker, G., and Vlassara, H. (2012). Oral advanced glycation endproducts (AGEs) promote insulin resistance and diabetes by depleting the antioxidant defenses AGE receptor-1 and sirtuin 1. Proc. Natl. Acad. Sci. U.S.A. 109, 15888–15893. doi: 10.1073/pnas.1205847109
Canto, C., and Auwerx, J. (2009). Caloric restriction, SIRT1 and longevity. Trends Endocrinol. Metab. 20, 325–331.
Carelli, V., La Morgia, C., Valentino, M., Barboni, P., Ross-Cisneros, F., and Sadun, A. (2009). Retinal ganglion cell neurodegeneration in mitochondrial inherited disorders. Biochim. Biophys. Acta 1787, 518–528.
Castiglione, F., Ferro, M., Mavroudakis, E., Pellitteri, R., Bossolasco, P., Zaccheo, D., et al. (2017). NMR metabolomics for stem cell type discrimination. Sci. Rep. 7:15808. doi: 10.1038/s41598-017-16043-8
Cavallucci, V., Fidaleo, M., and Pani, G. (2016). Neural stem cells and nutrients: poised between quiescence and exhaustion. Trends Endocrinol. Metab. 27, 756–769. doi: 10.1016/j.tem.2016.06.007
Chen, J., Xavier, S., Moskowitz-Kassai, E., Chen, R., Lu, C., Sanduski, K., et al. (2012). Cathepsin cleavage of sirtuin 1 in endothelial progenitor cells mediates stress-induced premature senescence. Am. J. Pathol. 180, 973–983. doi: 10.1016/j.ajpath.2011.11.033
Chen, J., Zhou, Y., Mueller-Steiner, S., Chen, L., Kwon, H., Yi, S., et al. (2005). SIRT1 protects against microglia-dependent amyloid-beta toxicity through inhibiting NF-kappaB signaling. J. Biol. Chem. 280, 40364–40374. doi: 10.1074/jbc.M509329200
Chen, Q., Vazquez, E., Moghaddas, S., Hoppel, C., and Lesnefsky, E. (2003). Production of reactive oxygen species by mitochondria: Central role of complex III. J. Biol. Chem. 278, 36027–36031.
Chen, X., Sun, K., Jiao, S., Cai, N., Zhao, X., Zou, H., et al. (2014). High levels of SIRT1 expression enhance tumorigenesis and associate with a poor prognosis of colorectal carcinoma patients. Sci. Rep. 4:7481.
Chen, Z., Pietrobon, A., Julian, L., and Stanford, W. (2019). “Somatic cell epigenetic reprogramming: Molecular mechanisms and therapeutic potential,” in Epigenetics and regeneration, ed. D. Palacios (Cambridge, MA: Academic Press), 165–196.
Cheng, A., Shin-ya, K., Wan, R., Tang, S., Miura, T., Tang, H., et al. (2007). Telomere protection mechanisms change during neurogenesis and neuronal maturation: Newly generated neurons are hypersensitive to telomere and DNA damage. J. Neurosci. 27, 3722–3733. doi: 10.1523/JNEUROSCI.0590-07.2007
Cheng, Y., Ren, X., Gowda, A., Shan, Y., Zhang, L., Yuan, Y., et al. (2013). Interaction of Sirt3 with OGG1 contributes to repair of mitochondrial DNA and protects from apoptotic cell death under oxidative stress. Cell Death Dis. 4:e731. doi: 10.1038/cddis.2013.254
Cobley, J., Fiorello, M., and Bailey, D. (2018). 13 reasons why the brain is susceptible to oxidative stress. Redox Biol. 15, 490–503.
Collins, S., Tumpach, C., Groveman, B., Drew, S., and Haigh, C. (2018). Prion protein cleavage fragments regulate adult neural stem cell quiescence through redox modulation of mitochondrial fission and SOD2 expression. Cell. Mol. Life Sci. 75, 3231–3249. doi: 10.1007/s00018-018-2790-3
Correia, M., Perestrelo, T., Rodrigues, A., Ribeiro, M., Pereira, S., Sousa, M., et al. (2017). Sirtuins in metabolism, stemness and differentiation. Biochim. Biophys. Acta Gen. Subj. 1861, 3444–3455.
Crocco, P., Montesanto, A., Passarino, G., and Rose, G. (2016). Polymorphisms falling within putative miRNA Target Sites in the 3’UTR Region of SIRT2 and DRD2 genes are correlated with human longevity. J. Gerontol. A Biol. Sci. Med. Sci. 71, 586–592. doi: 10.1093/gerona/glv058
De Bonis, M., Ortega, S., and Blasco, M. (2014). SIRT1 is necessary for proficient telomere elongation and genomic stability of induced pluripotent stem cells. Stem Cell Rep. 2, 690–706. doi: 10.1016/j.stemcr.2014.03.002
de Oliveira, R., Vicente Miranda, H., Francelle, L., Pinho, R., Szego, E., Martinho, R., et al. (2017). The mechanism of sirtuin 2-mediated exacerbation of alpha-synuclein toxicity in models of Parkinson disease. PLoS Biol. 15:e2000374. doi: 10.1371/journal.pbio.2000374
Deelen, J., Uh, H., Monajemi, R., van Heemst, D., Thijssen, P., Bohringer, S., et al. (2013). Gene set analysis of GWAS data for human longevity highlights the relevance of the insulin/IGF-1 signaling and telomere maintenance pathways. Age 35, 235–249. doi: 10.1007/s11357-011-9340-3
Detmer, S., and Chan, D. (2007). Functions and dysfunctions of mitochondrial dynamics. Nat. Rev. Mol. Cell Biol. 8, 870–879.
Dhingra, R., and Kirshenbaum, L. (2014). Regulation of mitochondrial dynamics and cell fate. Circ. J. 78, 803–810.
Diaz-Castro, B., Pardal, R., Garcia-Flores, P., Sobrino, V., Duran, R., Piruat, J., et al. (2015). Resistance of glia-like central and peripheral neural stem cells to genetically induced mitochondrial dysfunction–differential effects on neurogenesis. EMBO Rep. 16, 1511–1519. doi: 10.15252/embr.201540982
Donmez, G. (2012). The neurobiology of sirtuins and their role in neurodegeneration. Trends Pharmacol. Sci. 33, 494–501.
Donmez, G., Arun, A., Chung, C., McLean, P., Lindquist, S., and Guarente, L. (2012). SIRT1 protects against alpha-synuclein aggregation by activating molecular chaperones. J. Neurosci. 32, 124–132.
Du, L., Zhang, X., Han, Y., Burke, N., Kochanek, P., Watkins, S., et al. (2003). Intra-mitochondrial poly(ADP-ribosylation) contributes to NAD+ depletion and cell death induced by oxidative stress. J. Biol. Chem. 278, 18426–18433. doi: 10.1074/jbc.M301295200
Duan, W. (2013). Targeting sirtuin-1 in huntington’s disease: Rationale and current status. CNS Drugs 27, 345–352. doi: 10.1007/s40263-013-0055-0
Duan, W., Guo, Z., Jiang, H., Ware, M., and Mattson, M. (2003). Reversal of behavioral and metabolic abnormalities, and insulin resistance syndrome, by dietary restriction in mice deficient in brain-derived neurotrophic factor. Endocrinology 144, 2446–2453. doi: 10.1210/en.2002-0113
Duan, W., Jiang, M., and Jin, J. (2014). Metabolism in HD: Still a relevant mechanism? Mov. Disord. 29, 1366–1374. doi: 10.1002/mds.25992
Eitan, E., Hutchison, E., and Mattson, M. (2014). Telomere shortening in neurological disorders: An abundance of unanswered questions. Trends Neurosci. 37, 256–263. doi: 10.1016/j.tins.2014.02.010
Eitan, E., Tichon, A., Gazit, A., Gitler, D., Slavin, S., and Priel, E. (2012). Novel telomerase-increasing compound in mouse brain delays the onset of amyotrophic lateral sclerosis. EMBO Mol. Med. 4, 313–329. doi: 10.1002/emmm.201200212
Ferretta, A., Gaballo, A., Tanzarella, P., Piccoli, C., Capitanio, N., Nico, B., et al. (2014). Effect of resveratrol on mitochondrial function: Implications in parkin-associated familiar Parkinson’s disease. Biochim. Biophys. Acta 1842, 902–915. doi: 10.1016/j.bbadis.2014.02.010
Ferron, S., Mira, H., Franco, S., Cano-Jaimez, M., Bellmunt, E., Ramirez, C., et al. (2004). Telomere shortening and chromosomal instability abrogates proliferation of adult but not embryonic neural stem cells. Development 131, 4059–4070. doi: 10.1242/dev.01215
Fidaleo, M., Cavallucci, V., and Pani, G. (2017). Nutrients, neurogenesis and brain ageing: From disease mechanisms to therapeutic opportunities. Biochem. Pharmacol. 141, 63–76. doi: 10.1016/j.bcp.2017.05.016
Fu, J., Jin, J., Cichewicz, R., Hageman, S., Ellis, T., Xiang, L., et al. (2012). trans-(-)-epsilon-Viniferin increases mitochondrial sirtuin 3 (SIRT3), activates AMP-activated protein kinase (AMPK), and protects cells in models of huntington disease. J. Biol. Chem. 287, 24460–24472. doi: 10.1074/jbc.M112.382226
Fukae, J., Mizuno, Y., and Hattori, N. (2007). Mitochondrial dysfunction in Parkinson’s disease. Mitochondrion 7, 58–62.
Fusco, S., Leone, L., Barbati, S., Samengo, D., Piacentini, R., Maulucci, G., et al. (2016). A CREB-Sirt1-Hes1 circuitry mediates neural stem cell response to glucose availability. Cell Rep. 14, 1195–1205. doi: 10.1016/j.celrep.2015.12.092
Garai, D., Rios-Gonzalez, B., Furtmuller, P., Fukuto, J., Xian, M., Lopez-Garriga, J., et al. (2017). Mechanisms of myeloperoxidase catalyzed oxidation of H2S by H2O2 or O2 to produce potent protein Cys-polysulfide-inducing species. Free Radic. Biol. Med. 113, 551–563.
Garcia-Prat, L., Martinez-Vicente, M., Perdiguero, E., Ortet, L., Rodriguez-Ubreva, J., Rebollo, E., et al. (2016). Autophagy maintains stemness by preventing senescence. Nature 529, 37–42.
Geschwind, D., Ou, J., Easterday, M., Dougherty, J., Jackson, R., Chen, Z., et al. (2001). A genetic analysis of neural progenitor differentiation. Neuron 29, 325–339.
Ghosh, S., Sinha, J., Khandelwal, N., Chakravarty, S., Kumar, A., and Raghunath, M. (2020). Increased stress and altered expression of histone modifying enzymes in brain are associated with aberrant behaviour in vitamin B12 deficient female mice. Nutr. Neurosci. 23, 714–723. doi: 10.1080/1028415X.2018.1548676
Gong, L., Pan, X., Chen, H., Rao, L., Zeng, Y., Hang, H., et al. (2016). p53 isoform Delta133p53 promotes efficiency of induced pluripotent stem cells and ensures genomic integrity during reprogramming. Sci. Rep. 6:37281. doi: 10.1038/srep37281
Gurok, U., Steinhoff, C., Lipkowitz, B., Ropers, H., Scharff, C., and Nuber, U. (2004). Gene expression changes in the course of neural progenitor cell differentiation. J. Neurosci. 24, 5982–6002.
Hafner, A., Dai, J., Gomes, A., Xiao, C., Palmeira, C., Rosenzweig, A., et al. (2010). Regulation of the mPTP by SIRT3-mediated deacetylation of CypD at lysine 166 suppresses age-related cardiac hypertrophy. Aging 2, 914–923. doi: 10.18632/aging.100252
Haigis, M., and Sinclair, D. (2010). Mammalian sirtuins: Biological insights and disease relevance. Annu. Rev. Pathol. 5, 253–295.
Han, M., Song, E., Guo, Y., Ou, X., Mantel, C., and Broxmeyer, H. (2008). SIRT1 regulates apoptosis and Nanog expression in mouse embryonic stem cells by controlling p53 subcellular localization. Cell Stem Cell 2, 241–251. doi: 10.1016/j.stem.2008.01.002
Hayyan, M., Hashim, M., and AlNashef, I. (2016). Superoxide ion: Generation and chemical implications. Chem. Rev. 116, 3029–3085.
Heidker, R., Emerson, M., and LeVine, S. (2017). Metabolic pathways as possible therapeutic targets for progressive multiple sclerosis. Neural Regen. Res. 12, 1262–1267.
Hekimi, S., Lapointe, J., and Wen, Y. (2011). Taking a “good” look at free radicals in the aging process. Trends Cell Biol. 21, 569–576. doi: 10.1016/j.tcb.2011.06.008
Herskovits, A., Hunter, T., Maxwell, N., Pereira, K., Whittaker, C., Valdez, G., et al. (2018). SIRT1 deacetylase in aging-induced neuromuscular degeneration and amyotrophic lateral sclerosis. Aging Cell 17:e12839. doi: 10.1111/acel.12839
Holzenberger, M., Dupont, J., Ducos, B., Leneuve, P., Geloen, A., Even, P., et al. (2003). IGF-1 receptor regulates lifespan and resistance to oxidative stress in mice. Nature 421, 182–187.
Houtkooper, R., Mouchiroud, L., Ryu, D., Moullan, N., Katsyuba, E., Knott, G., et al. (2013). Mitonuclear protein imbalance as a conserved longevity mechanism. Nature 497, 451–457.
Hsu, Y., Wu, Y., Tsai, C., and Wei, Y. (2018). Current understanding and future perspectives of the roles of sirtuins in the reprogramming and differentiation of pluripotent stem cells. Exp. Biol. Med. 243, 563–575. doi: 10.1177/1535370218759636
Hu, B., Guo, Y., Chen, C., Li, Q., Niu, X., Guo, S., et al. (2014). Repression of SIRT1 promotes the differentiation of mouse induced pluripotent stem cells into neural stem cells. Cell. Mol. Neurobiol. 34, 905–912. doi: 10.1007/s10571-014-0071-8
Imai, S., and Guarente, L. (2016). It takes two to tango: NAD(+) and sirtuins in aging/longevity control. NPJ Aging Mech. Dis. 2:16017. doi: 10.1038/npjamd.2016.17
Imai, S., and Guarente, L. N. A. D. (2014). + and sirtuins in aging and disease. Trends Cell Biol. 24, 464–471.
Ismail, H., Shakkour, Z., Tabet, M., Abdelhady, S., Kobaisi, A., Abedi, R., et al. (2020). Traumatic brain injury: Oxidative stress and novel anti-oxidants such as mitoquinone and edaravone. Antioxidants 9:943. doi: 10.3390/antiox9100943
Ito, K., Hirao, A., Arai, F., Takubo, K., Matsuoka, S., Miyamoto, K., et al. (2006). Reactive oxygen species act through p38 MAPK to limit the lifespan of hematopoietic stem cells. Nat. Med. 12, 446–451. doi: 10.1038/nm1388
Ivanova, N., Dimos, J., Schaniel, C., Hackney, J., Moore, K., and Lemischka, I. R. (2002). A stem cell molecular signature. Science 298, 601–604.
Jang, J., Huh, Y., Cho, H., Lee, B., Park, J., Hwang, D., et al. (2017). SIRT1 enhances the survival of human embryonic stem cells by promoting DNA repair. Stem Cell Rep. 9, 629–641. doi: 10.1016/j.stemcr.2017.06.001
Jeong, H., Cohen, D., Cui, L., Supinski, A., Savas, J., Mazzulli, J., et al. (2011). Sirt1 mediates neuroprotection from mutant huntingtin by activation of the TORC1 and CREB transcriptional pathway. Nat. Med. 18, 159–165. doi: 10.1038/nm.2559
Jesko, H., Wencel, P., Strosznajder, R., and Strosznajder, J. (2017). Sirtuins and their roles in brain aging and neurodegenerative disorders. Neurochem. Res. 42, 876–890.
Ji, J., Sharma, V., Qi, S., Guarch, M., Zhao, P., Luo, Z., et al. (2014). Antioxidant supplementation reduces genomic aberrations in human induced pluripotent stem cells. Stem Cell Rep. 2, 44–51. doi: 10.1016/j.stemcr.2013.11.004
Julien, C., Tremblay, C., Emond, V., Lebbadi, M., Salem, N. Jr., Bennett, D., et al. (2009). Sirtuin 1 reduction parallels the accumulation of tau in Alzheimer disease. J. Neuropathol. Exp. Neurol. 68, 48–58. doi: 10.1097/NEN.0b013e3181922348
Jurk, D., Wang, C., Miwa, S., Maddick, M., Korolchuk, V., Tsolou, A., et al. (2012). Postmitotic neurons develop a p21-dependent senescence-like phenotype driven by a DNA damage response. Aging Cell 11, 996–1004. doi: 10.1111/j.1474-9726.2012.00870.x
Just, N., Chevillard, P., and Migaud, M. (2022). Imaging and spectroscopic methods to investigate adult neurogenesis in vivo: New models and new avenues. Front. Neurosci. 16:933947. doi: 10.3389/fnins.2022.933947
Kajarabille, N., and Latunde-Dada, G. (2019). Programmed cell-death by ferroptosis: Antioxidants as mitigators. Int. J. Mol. Sci. 20:4968.
Kaluski, S., Portillo, M., Besnard, A., Stein, D., Einav, M., Zhong, L., et al. (2017). Neuroprotective functions for the histone deacetylase SIRT6. Cell Rep. 18, 3052–3062.
Kanfi, Y., Shalman, R., Peshti, V., Pilosof, S., Gozlan, Y., Pearson, K., et al. (2008). Regulation of SIRT6 protein levels by nutrient availability. FEBS Lett. 582, 543–548.
Karsten, S., Kudo, L., Jackson, R., Sabatti, C., Kornblum, H., and Geschwind, D. (2003). Global analysis of gene expression in neural progenitors reveals specific cell-cycle, signaling, and metabolic networks. Dev. Biol. 261, 165–182. doi: 10.1016/s0012-1606(03)00274-4
Karuppagounder, S., Kumar, A., Shao, D., Zille, M., Bourassa, M., Caulfield, J., et al. (2015). Metabolism and epigenetics in the nervous system: Creating cellular fitness and resistance to neuronal death in neurological conditions via modulation of oxygen-, iron-, and 2-oxoglutarate-dependent dioxygenases. Brain Res. 1628, 273–287. doi: 10.1016/j.brainres.2015.07.030
Katsimpardi, L., Litterman, N., Schein, P., Miller, C., Loffredo, F., Wojtkiewicz, G., et al. (2014). Vascular and neurogenic rejuvenation of the aging mouse brain by young systemic factors. Science 344, 630–634. doi: 10.1126/science.1251141
Kawahara, T., Michishita, E., Adler, A., Damian, M., Berber, E., Lin, M., et al. (2009). SIRT6 links histone H3 lysine 9 deacetylation to NF-kappaB-dependent gene expression and organismal life span. Cell 136, 62–74. doi: 10.1016/j.cell.2008.10.052
Khacho, M., and Slack, R. (2018). Mitochondrial dynamics in the regulation of neurogenesis: From development to the adult brain. Dev. Dyn. 247, 47–53. doi: 10.1002/dvdy.24538
Khacho, M., Clark, A., Svoboda, D., Azzi, J., MacLaurin, J., Meghaizel, C., et al. (2016). Mitochondrial dynamics impacts stem cell identity and fate decisions by regulating a nuclear transcriptional program. Cell Stem Cell 19, 232–247. doi: 10.1016/j.stem.2016.04.015
Khacho, M., Harris, R., and Slack, R. (2019). Mitochondria as central regulators of neural stem cell fate and cognitive function. Nat. Rev. Neurosci. 20, 34–48. doi: 10.1038/s41583-018-0091-3
Khoury, N., Koronowski, K., Young, J., and Perez-Pinzon, M. (2018). The NAD(+)-dependent family of sirtuins in cerebral ischemia and preconditioning. Antioxid. Redox Signal. 28, 691–710. doi: 10.1089/ars.2017.7258
Kodavati, M., Wang, H., and Hegde, M. (2020). Altered mitochondrial dynamics in motor neuron disease: An emerging perspective. Cells 9:1065. doi: 10.3390/cells9041065
Kong, X., Wang, R., Xue, Y., Liu, X., Zhang, H., Chen, Y., et al. (2010). Sirtuin 3, a new target of PGC-1alpha, plays an important role in the suppression of ROS and mitochondrial biogenesis. PLoS One 5:e11707. doi: 10.1371/journal.pone.0011707
Koziel, J., Fox, M., Steding, C., Sprouse, A., and Herbert, B. (2011). Medical genetics and epigenetics of telomerase. J. Cell. Mol. Med. 15, 457–467.
Kunisaki, Y., Bruns, I., Scheiermann, C., Ahmed, J., Pinho, S., Zhang, D., et al. (2013). Arteriolar niches maintain haematopoietic stem cell quiescence. Nature 502, 637–643.
Law, I., Liu, L., Xu, A., Lam, K., Vanhoutte, P., Che, C., et al. (2009). Identification and characterization of proteins interacting with SIRT1 and SIRT3: Implications in the anti-aging and metabolic effects of sirtuins. Proteomics 9, 2444–2456. doi: 10.1002/pmic.200800738
Le Belle, J., Orozco, N., Paucar, A., Saxe, J., Mottahedeh, J., Pyle, A., et al. (2011). Proliferative neural stem cells have high endogenous ROS levels that regulate self-renewal and neurogenesis in a PI3K/Akt-dependant manner. Cell Stem Cell 8, 59–71. doi: 10.1016/j.stem.2010.11.028
Lee, J., Duan, W., Long, J., Ingram, D., and Mattson, M. (2000). Dietary restriction increases the number of newly generated neural cells, and induces BDNF expression, in the dentate gyrus of rats. J. Mol. Neurosci. 15, 99–108. doi: 10.1385/JMN:15:2:99
Lee, J., Shin, J., Park, B., Kim, G., Kim, J., Kang, K., et al. (2012). Region-specific changes in the immunoreactivity of SIRT1 expression in the central nervous system of SOD1(G93A) transgenic mice as an in vivo model of amyotrophic lateral sclerosis. Brain Res. 1433, 20–28.
Lee, S., Lee, J., Lee, H., and Min, K. (2019). Sirtuin signaling in cellular senescence and aging. BMB Rep. 52, 24–34.
Lee, S., Lee, Y., and Han, H. (2010). Effect of arachidonic acid on hypoxia-induced IL-6 production in mouse ES cells: Involvement of MAPKs, NF-kappaB, and HIF-1alpha. J. Cell. Physiol. 222, 574–585. doi: 10.1002/jcp.21973
Lee, Y., Peng, Q., Fong, S., Chen, A., Lee, K., Ng, E., et al. (2012). Sirtuin 1 facilitates generation of induced pluripotent stem cells from mouse embryonic fibroblasts through the miR-34a and p53 pathways. PLoS One 7:e45633. doi: 10.1371/journal.pone.0045633
Li, W., He, P., Huang, Y., Li, Y., Lu, J., Li, M., et al. (2021). Selective autophagy of intracellular organelles: Recent research advances. Theranostics 11, 222–256.
Liao, A., Craver, B., Tseng, B., Tran, K., Parihar, V., Acharya, M., et al. (2013). Mitochondrial-targeted human catalase affords neuroprotection from proton irradiation. Radiat. Res. 180, 1–6. doi: 10.1667/RR3339.1
Libert, S., Cohen, D., and Guarente, L. (2008). Neurogenesis directed by Sirt1. Nat. Cell Biol. 10, 373–374.
Lin, J., Handschin, C., and Spiegelman, B. (2005). Metabolic control through the PGC-1 family of transcription coactivators. Cell Metab. 1, 361–370.
Lin, T., Chao, C., Saito, S., Mazur, S., Murphy, M., Appella, E., et al. (2005). p53 induces differentiation of mouse embryonic stem cells by suppressing Nanog expression. Nat. Cell Biol. 7, 165–171.
Linkus, B., Wiesner, D., Messner, M., Karabatsiakis, A., Scheffold, A., Rudolph, K., et al. (2016). Telomere shortening leads to earlier age of onset in ALS mice. Aging 8, 382–393. doi: 10.18632/aging.100904
Liu, R., Liu, H., Ha, Y., Tilton, R., and Zhang, W. (2014). Oxidative stress induces endothelial cell senescence via downregulation of Sirt6. Biomed. Res. Int. 2014:902842.
Liu, Z., Zhou, T., Ziegler, A., Dimitrion, P., and Zuo, L. (2017). Oxidative stress in neurodegenerative diseases: From molecular mechanisms to clinical applications. Oxid. Med. Cell. Longev. 2017:2525967.
Llorens-Bobadilla, E., Zhao, S., Baser, A., Saiz-Castro, G., Zwadlo, K., and Martin-Villalba, A. (2015). Single-cell transcriptomics reveals a population of dormant neural stem cells that become activated upon brain injury. Cell Stem Cell 17, 329–340. doi: 10.1016/j.stem.2015.07.002
Llorente, V., Velarde, P., Desco, M., and Gomez-Gaviro, M. (2022). Current understanding of the neural stem cell niches. Cells 11:3002.
Lopez-Fabuel, I., Le Douce, J., Logan, A., James, A., Bonvento, G., Murphy, M., et al. (2016). Complex I assembly into supercomplexes determines differential mitochondrial ROS production in neurons and astrocytes. Proc. Natl. Acad. Sci. U.S.A. 113, 13063–13068. doi: 10.1073/pnas.1613701113
Lorenz, C., and Prigione, A. (2017). Mitochondrial metabolism in early neural fate and its relevance for neuronal disease modeling. Curr. Opin. Cell Biol. 49, 71–76. doi: 10.1016/j.ceb.2017.12.004
Lushchak, V., and Storey, K. (2021). Oxidative stress concept updated: Definitions, classifications, and regulatory pathways implicated. EXCLI J. 20, 956–967. doi: 10.17179/excli2021-3596
Maffezzini, C., Calvo-Garrido, J., Wredenberg, A., and Freyer, C. (2020). Metabolic regulation of neurodifferentiation in the adult brain. Cell. Mol. Life Sci. 77, 2483–2496.
Maissan, P., Mooij, E., and Barberis, M. (2021). Sirtuins-mediated system-level regulation of mammalian tissues at the interface between metabolism and cell cycle: A systematic review. Biology 10:194. doi: 10.3390/biology10030194
Malard, E., Valable, S., Bernaudin, M., Peres, E., and Chatre, L. (2021). The reactive species interactome in the brain. Antioxid. Redox Signal. 35, 1176–1206.
Mancuso, R., del Valle, J., Modol, L., Martinez, A., Granado-Serrano, A., Ramirez-Nunez, O., et al. (2014). Resveratrol improves motoneuron function and extends survival in SOD1(G93A) ALS mice. Neurotherapeutics 11, 419–432. doi: 10.1007/s13311-013-0253-y
Manczak, M., Anekonda, T., Henson, E., Park, B., Quinn, J., and Reddy, P. (2006). Mitochondria are a direct site of a beta accumulation in Alzheimer’s disease neurons: Implications for free radical generation and oxidative damage in disease progression. Hum. Mol. Genet. 15, 1437–1449. doi: 10.1093/hmg/ddl066
Martin, L., Gertz, B., Pan, Y., Price, A., Molkentin, J., and Chang, Q. (2009). The mitochondrial permeability transition pore in motor neurons: Involvement in the pathobiology of ALS mice. Exp. Neurol. 218, 333–346. doi: 10.1016/j.expneurol.2009.02.015
Maryanovich, M., and Gross, A. (2013). A ROS rheostat for cell fate regulation. Trends Cell Biol. 23, 129–134.
Mattson, M., and Magnus, T. (2006). Ageing and neuronal vulnerability. Nat. Rev. Neurosci. 7, 278–294.
Michishita, E., McCord, R., Berber, E., Kioi, M., Padilla-Nash, H., Damian, M., et al. (2008). SIRT6 is a histone H3 lysine 9 deacetylase that modulates telomeric chromatin. Nature 452, 492–496. doi: 10.1038/nature06736
Min, S., Cho, S., Zhou, Y., Schroeder, S., Haroutunian, V., Seeley, W., et al. (2010). Acetylation of tau inhibits its degradation and contributes to tauopathy. Neuron 67, 953–966.
Min, S., Sohn, P., Cho, S., Swanson, R., and Gan, L. (2013). Sirtuins in neurodegenerative diseases: An update on potential mechanisms. Front. Aging Neurosci. 5:53. doi: 10.3389/fnagi.2013.00053
Mishra, P., Mittal, A., Kalonia, H., Madan, S., Ghosh, S., Sinha, J., et al. (2021). SIRT1 promotes neuronal fortification in neurodegenerative diseases through attenuation of pathological hallmarks and enhancement of cellular lifespan. Curr. Neuropharmacol. 19, 1019–1037. doi: 10.2174/1570159X18666200729111744
Mootha, V., and Chinnery, P. (2018). Oxygen in mitochondrial disease: Can there be too much of a good thing? J. Inherit. Metab. Dis. 41, 761–763. doi: 10.1007/s10545-018-0210-3
Mormone, E., Matarrese, P., Tinari, A., Cannella, M., Maglione, V., Farrace, M., et al. (2006). Genotype-dependent priming to self- and xeno-cannibalism in heterozygous and homozygous lymphoblasts from patients with huntington’s disease. J. Neurochem. 98, 1090–1099. doi: 10.1111/j.1471-4159.2006.03998.x
Mu, W., Wang, Y., Xu, P., Hao, D., Liu, X., Wang, T., et al. (2015). Sox2 deacetylation by Sirt1 is involved in mouse somatic reprogramming. Stem Cells 33, 2135–2147. doi: 10.1002/stem.2012
Naia, L., and Rego, A. (2015). Sirtuins: Double players in huntington’s disease. Biochim. Biophys. Acta 1852, 2183–2194. doi: 10.1016/j.bbadis.2015.07.003
Nemoto, S., Fergusson, M., and Finkel, T. (2004). Nutrient availability regulates SIRT1 through a forkhead-dependent pathway. Science 306, 2105–2108. doi: 10.1126/science.1101731
Nissanka, N., and Moraes, C. (2018). Mitochondrial DNA damage and reactive oxygen species in neurodegenerative disease. FEBS Lett. 592, 728–742.
Obrador, E., Salvador, R., Lopez-Blanch, R., Jihad-Jebbar, A., Valles, S., and Estrela, J. (2020). Oxidative stress, neuroinflammation and mitochondria in the pathophysiology of amyotrophic lateral sclerosis. Antioxidants 9:901.
Ogino, K., Kodama, N., Nakajima, M., Yamada, A., Nakamura, H., Nagase, H., et al. (2001). Catalase catalyzes nitrotyrosine formation from sodium azide and hydrogen peroxide. Free Radic. Res. 35, 735–747.
Olson, K., Gao, Y., DeLeon, E., Arif, M., Arif, F., Arora, N., et al. (2017). Catalase as a sulfide-sulfur oxido-reductase: An ancient (and modern?) regulator of reactive sulfur species (RSS). Redox Biol. 12, 325–339. doi: 10.1016/j.redox.2017.02.021
Ong, A., and Ramasamy, T. (2018). Role of Sirtuin1-p53 regulatory axis in aging, cancer and cellular reprogramming. Ageing Res. Rev. 43, 64–80. doi: 10.1016/j.arr.2018.02.004
Ozgen, S., Krigman, J., Zhang, R., and Sun, N. (2022). Significance of mitochondrial activity in neurogenesis and neurodegenerative diseases. Neural Regen. Res. 17, 741–747.
Pacelli, C., Rotundo, G., Lecce, L., Menga, M., Bidollari, E., Scrima, R., et al. (2019). Parkin mutation affects clock gene-dependent energy metabolism. Int. J. Mol. Sci. 20:2772. doi: 10.3390/ijms20112772
Paik, J., Ding, Z., Narurkar, R., Ramkissoon, S., Muller, F., Kamoun, W., et al. (2009). FoxOs cooperatively regulate diverse pathways governing neural stem cell homeostasis. Cell Stem Cell 5, 540–553. doi: 10.1016/j.stem.2009.09.013
Palacios, J., Herranz, D., De Bonis, M., Velasco, S., Serrano, M., and Blasco, M. (2010). SIRT1 contributes to telomere maintenance and augments global homologous recombination. J. Cell Biol. 191, 1299–1313. doi: 10.1083/jcb.201005160
Pan, H., Guan, D., Liu, X., Li, J., Wang, L., Wu, J., et al. (2016). SIRT6 safeguards human mesenchymal stem cells from oxidative stress by coactivating NRF2. Cell Res. 26, 190–205. doi: 10.1038/cr.2016.4
Panchal, K., and Tiwari, A. (2019). Mitochondrial dynamics, a key executioner in neurodegenerative diseases. Mitochondrion 47, 151–173.
Pani, G. (2015). Neuroprotective effects of dietary restriction: Evidence and mechanisms. Semin. Cell Dev. Biol. 40, 106–114.
Park, S., Lee, H., Lee, W., Shin, H., Kim, H., Hong, K., et al. (2016). Cilostazol modulates autophagic degradation of beta-amyloid peptide via SIRT1-Coupled LKB1/AMPKalpha signaling in neuronal cells. PLoS One 11:e0160620. doi: 10.1371/journal.pone.0160620
Pearce, V., Sherrell, J., Lou, Z., Kopelovich, L., Wright, W., and Shay, J. (2008). Immortalization of epithelial progenitor cells mediated by resveratrol. Oncogene 27, 2365–2374. doi: 10.1038/sj.onc.1210886
Peng, C., Chang, Y., Kao, C., Tseng, L., Wu, C., Chen, Y., et al. (2010). SirT1–a sensor for monitoring self-renewal and aging process in retinal stem cells. Sensors 10, 6172–6194. doi: 10.3390/s100606172
Prasuhn, J., Davis, R., and Kumar, K. (2020). Targeting mitochondrial impairment in parkinson’s disease: challenges and opportunities. Front. Cell Dev. Biol. 8:615461. doi: 10.3389/fcell.2020.615461
Prigione, A., Fauler, B., Lurz, R., Lehrach, H., and Adjaye, J. (2010). The senescence-related mitochondrial/oxidative stress pathway is repressed in human induced pluripotent stem cells. Stem Cells 28, 721–733. doi: 10.1002/stem.404
Prozorovski, T., Schulze-Topphoff, U., Glumm, R., Baumgart, J., Schroter, F., Ninnemann, O., et al. (2008). Sirt1 contributes critically to the redox-dependent fate of neural progenitors. Nat. Cell Biol. 10, 385–394. doi: 10.1038/ncb1700
Qiu, X., Brown, K., Hirschey, M., Verdin, E., and Chen, D. (2010). Calorie restriction reduces oxidative stress by SIRT3-mediated SOD2 activation. Cell Metab. 12, 662–667. doi: 10.1016/j.cmet.2010.11.015
Rafalski, V., Mancini, E., and Brunet, A. (2012). Energy metabolism and energy-sensing pathways in mammalian embryonic and adult stem cell fate. J. Cell Sci. 125, 5597–5608. doi: 10.1242/jcs.114827
Ramalho-Santos, M., Yoon, S., Matsuzaki, Y., Mulligan, R., and Melton, D. (2002). “Stemness”: Transcriptional profiling of embryonic and adult stem cells. Science 298, 597–600.
Rao, V., Carlson, E., and Yan, S. (2014). Mitochondrial permeability transition pore is a potential drug target for neurodegeneration. Biochim. Biophys. Acta 1842, 1267–1272.
Reeve, A., Ludtmann, M., Angelova, P., Simcox, E., Horrocks, M., Klenerman, D., et al. (2015). Aggregated alpha-synuclein and complex I deficiency: Exploration of their relationship in differentiated neurons. Cell Death Dis. 6:e1820. doi: 10.1038/cddis.2015.166
Reynolds, B., and Weiss, S. (1992). Generation of neurons and astrocytes from isolated cells of the adult mammalian central nervous system. Science 255, 1707–1710.
Ribeiro, M., Santos, A., Afonso, M., Rodrigues, P., Sa Santos, S., Castro, R., et al. (2020). Diet-dependent gut microbiota impacts on adult neurogenesis through mitochondrial stress modulation. Brain Commun. 2:fcaa165. doi: 10.1093/braincomms/fcaa165
Rodgers, J., Lerin, C., Haas, W., Gygi, S., Spiegelman, B., and Puigserver, P. (2005). Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature 434, 113–118.
Rodolfo, C., Di Bartolomeo, S., and Cecconi, F. (2016). Autophagy in stem and progenitor cells. Cell. Mol. Life Sci. 73, 475–496.
Sanderson, S., and Simon, A. (2017). In aged primary T cells, mitochondrial stress contributes to telomere attrition measured by a novel imaging flow cytometry assay. Aging Cell 16, 1234–1243. doi: 10.1111/acel.12640
Santos, D., Santos, M., Moreira, R., Sola, S., and Rodrigues, C. (2013). Synthetic condensed 1,4-naphthoquinone derivative shifts neural stem cell differentiation by regulating redox state. Mol. Neurobiol. 47, 313–324. doi: 10.1007/s12035-012-8353-y
Santos, L., Escande, C., and Denicola, A. (2016). Potential modulation of sirtuins by oxidative stress. Oxid. Med. Cell. Longev. 2016:9831825.
Santos, S., Moreira, J., Costa, M., Rodrigues, R., Sebastiao, A., Xapelli, S., et al. (2021). The mitochondrial antioxidant sirtuin3 cooperates with lipid metabolism to safeguard neurogenesis in aging and depression. Cells 11:90. doi: 10.3390/cells11010090
Saotome, M., Safiulina, D., Szabadkai, G., Das, S., Fransson, A., Aspenstrom, P., et al. (2008). Bidirectional Ca2+-dependent control of mitochondrial dynamics by the Miro GTPase. Proc. Natl. Acad. Sci. U.S.A. 105, 20728–20733. doi: 10.1073/pnas.0808953105
Sarga, L., Hart, N., Koch, L., Britton, S., Hajas, G., Boldogh, I., et al. (2013). Aerobic endurance capacity affects spatial memory and SIRT1 is a potent modulator of 8-oxoguanine repair. Neuroscience 252, 326–336. doi: 10.1016/j.neuroscience.2013.08.020
Sart, S., Ma, T., and Li, Y. (2014). Preconditioning stem cells for in vivo delivery. BioRes. Open Access. 3, 137–149.
Sart, S., Song, L., and Li, Y. (2015). Controlling redox status for stem cell survival, expansion, and differentiation. Oxid. Med. Cell. Longev. 2015:105135. doi: 10.1155/2015/105135
Saunders, L., Sharma, A., Tawney, J., Nakagawa, M., Okita, K., Yamanaka, S., et al. (2010). miRNAs regulate SIRT1 expression during mouse embryonic stem cell differentiation and in adult mouse tissues. Aging 2, 415–431. doi: 10.18632/aging.100176
Shaban, S., El-Husseny, M., Abushouk, A., Salem, A., Mamdouh, M., and Abdel-Daim, M. (2017). Effects of antioxidant supplements on the survival and differentiation of stem cells. Oxid. Med. Cell. Longev. 2017:5032102.
Sharma, A., Diecke, S., Zhang, W., Lan, F., He, C., Mordwinkin, N., et al. (2013). The role of SIRT6 protein in aging and reprogramming of human induced pluripotent stem cells. J. Biol. Chem. 288, 18439–18447. doi: 10.1074/jbc.M112.405928
Shin, J., Berg, D., Zhu, Y., Shin, J., Song, J., Bonaguidi, M., et al. (2015). Single-cell RNA-Seq with waterfall reveals molecular cascades underlying adult neurogenesis. Cell Stem Cell 17, 360–372. doi: 10.1016/j.stem.2015.07.013
Shin, J., Kim, J., Park, H., and Kim, J. (2018). Investigating the role of sirtuins in cell reprogramming. BMB Rep. 51, 500–507.
Shlevkov, E., Kramer, T., Schapansky, J., LaVoie, M., and Schwarz, T. (2016). Miro phosphorylation sites regulate Parkin recruitment and mitochondrial motility. Proc. Natl. Acad. Sci. U.S.A. 113, E6097–E6106. doi: 10.1073/pnas.1612283113
Shyh-Chang, N., Daley, G., and Cantley, L. (2013). Stem cell metabolism in tissue development and aging. Development 140, 2535–2547.
Sies, H. (2021). Oxidative eustress: On constant alert for redox homeostasis. Redox Biol. 41:101867. doi: 10.1016/j.redox.2021.101867
Silva, D., Esteves, A., Oliveira, C., and Cardoso, S. (2017). Mitochondrial metabolism power SIRT2-dependent deficient traffic causing Alzheimer’s-disease related pathology. Mol. Neurobiol. 54, 4021–4040. doi: 10.1007/s12035-016-9951-x
Silva, Y., Bernardi, A., and Frozza, R. (2020). The role of short-chain fatty acids from gut microbiota in gut-brain communication. Front. Endocrinol. 11:25. doi: 10.3389/fendo.2020.00025
Simon, A., Rai, U., Fanburg, B., and Cochran, B. (1998). Activation of the JAK-STAT pathway by reactive oxygen species. Am. J. Physiol. 275:C1640–C1652.
Singh, C., Chhabra, G., Ndiaye, M., Garcia-Peterson, L., Mack, N., and Ahmad, N. (2018). The role of sirtuins in antioxidant and redox signaling. Antioxid. Redox Signal. 28, 643–661.
Skvortsova, E., Nazarov, I., Tomilin, A., and Sinenko, S. (2022). Dual mode of mitochondrial ROS action during reprogramming to pluripotency. Int. J. Mol. Sci. 23:10924. doi: 10.3390/ijms231810924
Smith, M., Syed, A., Lukacsovich, T., Purcell, J., Barbaro, B., Worthge, S., et al. (2014). A potent and selective sirtuin 1 inhibitor alleviates pathology in multiple animal and cell models of huntington’s disease. Hum. Mol. Genet. 23, 2995–3007. doi: 10.1093/hmg/ddu010
Snezhkina, A., Kudryavtseva, A., Kardymon, O., Savvateeva, M., Melnikova, N., Krasnov, G., et al. (2019). ROS generation and antioxidant defense systems in normal and malignant cells. Oxid. Med. Cell. Longev. 2019:6175804.
Someya, S., Yu, W., Hallows, W., Xu, J., Vann, J., Leeuwenburgh, C., et al. (2010). Sirt3 mediates reduction of oxidative damage and prevention of age-related hearing loss under caloric restriction. Cell 143, 802–812. doi: 10.1016/j.cell.2010.10.002
Song, W., Song, Y., Kincaid, B., Bossy, B., and Bossy-Wetzel, E. (2013). Mutant SOD1G93A triggers mitochondrial fragmentation in spinal cord motor neurons: Neuroprotection by SIRT3 and PGC-1alpha. Neurobiol. Dis. 51, 72–81. doi: 10.1016/j.nbd.2012.07.004
Soto, M., Herzog, C., Pacheco, J., Fujisaka, S., Bullock, K., Clish, C., et al. (2018). Gut microbiota modulate neurobehavior through changes in brain insulin sensitivity and metabolism. Mol. Psychiatry 23, 2287–2301. doi: 10.1038/s41380-018-0086-5
Stephenson, J., Nutma, E., van der Valk, P., and Amor, S. (2018). Inflammation in CNS neurodegenerative diseases. Immunology 154, 204–219.
Stoll, E., Makin, R., Sweet, I., Trevelyan, A., Miwa, S., Horner, P., et al. (2015). Neural stem cells in the adult subventricular zone oxidize fatty acids to produce energy and support neurogenic activity. Stem Cells 33, 2306–2319. doi: 10.1002/stem.2042
Suh, Y., Atzmon, G., Cho, M., Hwang, D., Liu, B., Leahy, D., et al. (2008). Functionally significant insulin-like growth factor i receptor mutations in centenarians. Proc. Natl. Acad. Sci. U.S.A. 105, 3438–3442.
Sundaresan, N., Samant, S., Pillai, V., Rajamohan, S., and Gupta, M. (2008). SIRT3 is a stress-responsive deacetylase in cardiomyocytes that protects cells from stress-mediated cell death by deacetylation of Ku70. Mol. Cell Biol. 28, 6384–6401. doi: 10.1128/MCB.00426-08
Tahara, E., Navarete, F., and Kowaltowski, A. (2009). Tissue-, substrate-, and site-specific characteristics of mitochondrial reactive oxygen species generation. Free Radic. Biol. Med. 46, 1283–1297. doi: 10.1016/j.freeradbiomed.2009.02.008
Takahashi, K., Tanabe, K., Ohnuki, M., Narita, M., Ichisaka, T., Tomoda, K., et al. (2007). Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131, 861–872.
Takasaka, N., Araya, J., Hara, H., Ito, S., Kobayashi, K., Kurita, Y., et al. (2014). Autophagy induction by SIRT6 through attenuation of insulin-like growth factor signaling is involved in the regulation of human bronchial epithelial cell senescence. J. Immunol. 192, 958–968. doi: 10.4049/jimmunol.1302341
Takubo, K., Goda, N., Yamada, W., Iriuchishima, H., Ikeda, E., Kubota, Y., et al. (2010). Regulation of the HIF-1alpha level is essential for hematopoietic stem cells. Cell Stem Cell 7, 391–402.
Tan, D., and Suda, T. (2018). Reactive Oxygen species and mitochondrial homeostasis as regulators of stem cell fate and function. Antioxid. Redox Signal. 29, 149–168.
Tay, E., Chia, K., and Ong, D. (2021). Epigenetic plasticity and redox regulation of neural stem cell state and fate. Free Radic. Biol. Med. 170, 116–130. doi: 10.1016/j.freeradbiomed.2021.02.030
van de Wouw, M., Boehme, M., Lyte, J., Wiley, N., Strain, C., O’Sullivan, O., et al. (2018). Short-chain fatty acids: Microbial metabolites that alleviate stress-induced brain-gut axis alterations. J. Physiol. 596, 4923–4944. doi: 10.1113/JP276431
Walton, N., Shin, R., Tajinda, K., Heusner, C., Kogan, J., Miyake, S., et al. (2012). Adult neurogenesis transiently generates oxidative stress. PLoS One 7:e35264. doi: 10.1371/journal.pone.0035264
Wang, C., and Youle, R. (2009). The role of mitochondria in apoptosis*. Annu. Rev. Genet. 43, 95–118.
Wang, J., Huang, Y., Cai, J., Ke, Q., Xiao, J., Huang, W., et al. (2018). A nestin-cyclin-dependent kinase 5-dynamin-related protein 1 axis regulates neural stem/progenitor cell stemness via a metabolic shift. Stem Cells 36, 589–601. doi: 10.1002/stem.2769
Wang, R., Hao, W., Pan, L., Boldogh, I., and Ba, X. (2018). The roles of base excision repair enzyme OGG1 in gene expression. Cell. Mol. Life Sci. 75, 3741–3750.
Wang, W., Esbensen, Y., Kunke, D., Suganthan, R., Rachek, L., Bjoras, M., et al. (2011). Mitochondrial DNA damage level determines neural stem cell differentiation fate. J. Neurosci. 31, 9746–9751.
Wang, X., and Michaelis, E. (2010). Selective neuronal vulnerability to oxidative stress in the brain. Front. Aging Neurosci. 2:12. doi: 10.3389/fnagi.2010.00012
Watanabe, S., Ageta-Ishihara, N., Nagatsu, S., Takao, K., Komine, O., Endo, F., et al. (2014). SIRT1 overexpression ameliorates a mouse model of SOD1-linked amyotrophic lateral sclerosis via HSF1/HSP70i chaperone system. Mol. Brain 7:62. doi: 10.1186/s13041-014-0062-1
Watroba, M., Dudek, I., Skoda, M., Stangret, A., Rzodkiewicz, P., and Szukiewicz, D. (2017). Sirtuins, epigenetics and longevity. Ageing Res. Rev. 40, 11–19.
Weir, H., Murray, T., Kehoe, P., Love, S., Verdin, E., O’Neill, M., et al. (2012). CNS SIRT3 expression is altered by reactive oxygen species and in Alzheimer’s disease. PLoS One 7:e48225. doi: 10.1371/journal.pone.0048225
Wentling, M., Lopez-Gomez, C., Park, H., Amatruda, M., Ntranos, A., Aramini, J., et al. (2019). A metabolic perspective on CSF-mediated neurodegeneration in multiple sclerosis. Brain 142, 2756–2774. doi: 10.1093/brain/awz201
Williams, E., Taylor, A., Bell, E., Lim, R., Kim, D., and Guarente, L. (2016). Sirtuin 1 promotes deacetylation of Oct4 and maintenance of naive pluripotency. Cell Rep. 17, 809–820. doi: 10.1016/j.celrep.2016.09.046
Woo, H., Park, S., Kim, H., Jung, M., Pack, C., Park, J., et al. (2021). miR-351-5p/Miro2 axis contributes to hippocampal neural progenitor cell death via unbalanced mitochondrial fission. Mol. Ther. Nucleic Acids 23, 643–656. doi: 10.1016/j.omtn.2020.12.014
Wu, K., Grandori, C., Amacker, M., Simon-Vermot, N., Polack, A., Lingner, J., et al. (1999). Direct activation of TERT transcription by c-MYC. Nat. Genet. 21, 220–224. doi: 10.1038/6010
Xavier, J., Morgado, A., Sola, S., and Rodrigues, C. (2014). Mitochondrial translocation of p53 modulates neuronal fate by preventing differentiation-induced mitochondrial stress. Antioxid. Redox Signal. 21, 1009–1024. doi: 10.1089/ars.2013.5417
Xavier, J., Rodrigues, C., and Sola, S. (2016). Mitochondria: Major regulators of neural development. Neuroscientist 22, 346–358.
Xie, Z., Jones, A., Deeney, J., Hur, S., and Bankaitis, V. (2016). Inborn errors of long-chain fatty acid beta-oxidation link neural stem cell self-renewal to autism. Cell Rep. 14, 991–999. doi: 10.1016/j.celrep.2016.01.004
Xu, J., Jackson, C., Khoury, N., Escobar, I., and Perez-Pinzon, M. (2018). Brain SIRT1 mediates metabolic homeostasis and neuroprotection. Front. Endocrinol. 9:702. doi: 10.3389/fendo.2018.00702
Xu, Z., Zhang, L., Zhang, W., Meng, D., Zhang, H., Jiang, Y., et al. (2015). SIRT6 rescues the age related decline in base excision repair in a PARP1-dependent manner. Cell Cycle 14, 269–276. doi: 10.4161/15384101.2014.980641
Yanes, O., Clark, J., Wong, D., Patti, G., Sanchez-Ruiz, A., Benton, H., et al. (2010). Metabolic oxidation regulates embryonic stem cell differentiation. Nat. Chem. Biol. 6, 411–417.
Yu, G., Liang, Y., Zheng, S., and Zhang, H. (2018). Inhibition of myeloperoxidase by N-Acetyl lysyltyrosylcysteine amide reduces oxidative stress-mediated inflammation, neuronal damage, and neural stem cell injury in a murine model of stroke. J. Pharmacol. Exp. Ther. 364, 311–322.
Zhang, X., Anwar, S., Kim, Y., Brown, J., Comte, I., Cai, H., et al. (2019). The A30P alpha-synuclein mutation decreases subventricular zone proliferation. Hum. Mol. Genet. 28, 2283–2294. doi: 10.1093/hmg/ddz057
Zhang, X., Yalcin, S., Lee, D., Yeh, T., Lee, S., Su, J., et al. (2011). FOXO1 is an essential regulator of pluripotency in human embryonic stem cells. Nat. Cell Biol. 13, 1092–1099. doi: 10.1038/ncb2293
Zhang, Z., Chung, S., Xu, Z., and Xu, Y. (2014). Oct4 maintains the pluripotency of human embryonic stem cells by inactivating p53 through Sirt1-mediated deacetylation. Stem Cells 32, 157–165. doi: 10.1002/stem.1532
Zhao, Y., Zhang, J., Zheng, Y., Zhang, Y., Zhang, X., Wang, H., et al. (2021). NAD(+) improves cognitive function and reduces neuroinflammation by ameliorating mitochondrial damage and decreasing ROS production in chronic cerebral hypoperfusion models through Sirt1/PGC-1alpha pathway. J. Neuroinflamm. 18:207. doi: 10.1186/s12974-021-02250-8
Zheng, X., Boyer, L., Jin, M., Mertens, J., Kim, Y., Ma, L., et al. (2016). Metabolic reprogramming during neuronal differentiation from aerobic glycolysis to neuronal oxidative phosphorylation. eLife 5:e13374.
Keywords: NSCs, neurogenesis, mitochondria, reactive oxygen species (ROS), Sirtuins (SIRTs), neurodegenerative diseases (NDs), 8-oxoguanine DNA glycosylase (OGG1)
Citation: Mormone E, Iorio EL, Abate L and Rodolfo C (2023) Sirtuins and redox signaling interplay in neurogenesis, neurodegenerative diseases, and neural cell reprogramming. Front. Neurosci. 17:1073689. doi: 10.3389/fnins.2023.1073689
Received: 18 October 2022; Accepted: 13 January 2023;
Published: 01 February 2023.
Edited by:
Wendy Noble, King’s College London, United KingdomReviewed by:
Virve Cavallucci, Agostino Gemelli University Polyclinic (IRCCS), ItalyCopyright © 2023 Mormone, Iorio, Abate and Rodolfo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Elisabetta Mormone,  ZS5tb3Jtb25lQG9wZXJhcGFkcmVwaW8uaXQ=; ZW1vcm1vbmVAeWFob28uY29t; Carlo Rodolfo, Y2FybG8ucm9kb2xmb0B1bmlyb21hMi5pdA==
ZS5tb3Jtb25lQG9wZXJhcGFkcmVwaW8uaXQ=; ZW1vcm1vbmVAeWFob28uY29t; Carlo Rodolfo, Y2FybG8ucm9kb2xmb0B1bmlyb21hMi5pdA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.