 Deepak Kunhi Valappil
Deepak Kunhi Valappil Sangeeta Nath
Sangeeta Nath
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Neurosci., 09 September 2022
Sec. Neurodegeneration
Volume 16 - 2022 | https://doi.org/10.3389/fnins.2022.936897
This article is part of the Research TopicPrion and prion-like proteins in neurodegenerative diseasesView all 6 articles
Progressive development of pathology is one of the major characteristic features of neurodegenerative diseases. Alzheimer’s disease (AD) is the most prevalent among them. Extracellular amyloid-β (Aβ) plaques and intracellular tau neurofibrillary tangles are the pathological phenotypes of AD. However, cellular and animal studies implicate tau as a secondary pathology in developing AD while Aβ aggregates is considered as a trigger point. Interaction of Aβ peptides with plasma membrane (PM) seems to be a promising site of involvement in the events that lead to AD. Aβ binding to the lipid membranes initiates formation of oligomers of Aβ species, and these oligomers are known as primary toxic agents for neuronal toxicities. Once initiated, neuropathological toxicities spread in a “prion-like” fashion probably through the mechanism of intercellular transfer of pathogenic aggregates. In the last two decades, several studies have demonstrated neuron-to-neuron transfer of neurodegenerative proteins including Aβ and tau via exosomes and tunneling nanotubes (TNTs), the two modes of long-range intercellular transfer. Emerging pieces of evidence indicate that molecular pathways related to the biogenesis of exosomes and TNTs interface with endo-lysosomal pathways and cellular signaling in connection to vesicle recycling-imposed PM and actin remodulation. In this review, we discuss interactions of Aβ aggregates at the membrane level and its implications in intercellular spread of pathogenic aggregates. Furthermore, we hypothesize how spread of pathogenic aggregates contributes to complex molecular events that could regulate pathological and synaptic changes related to AD.
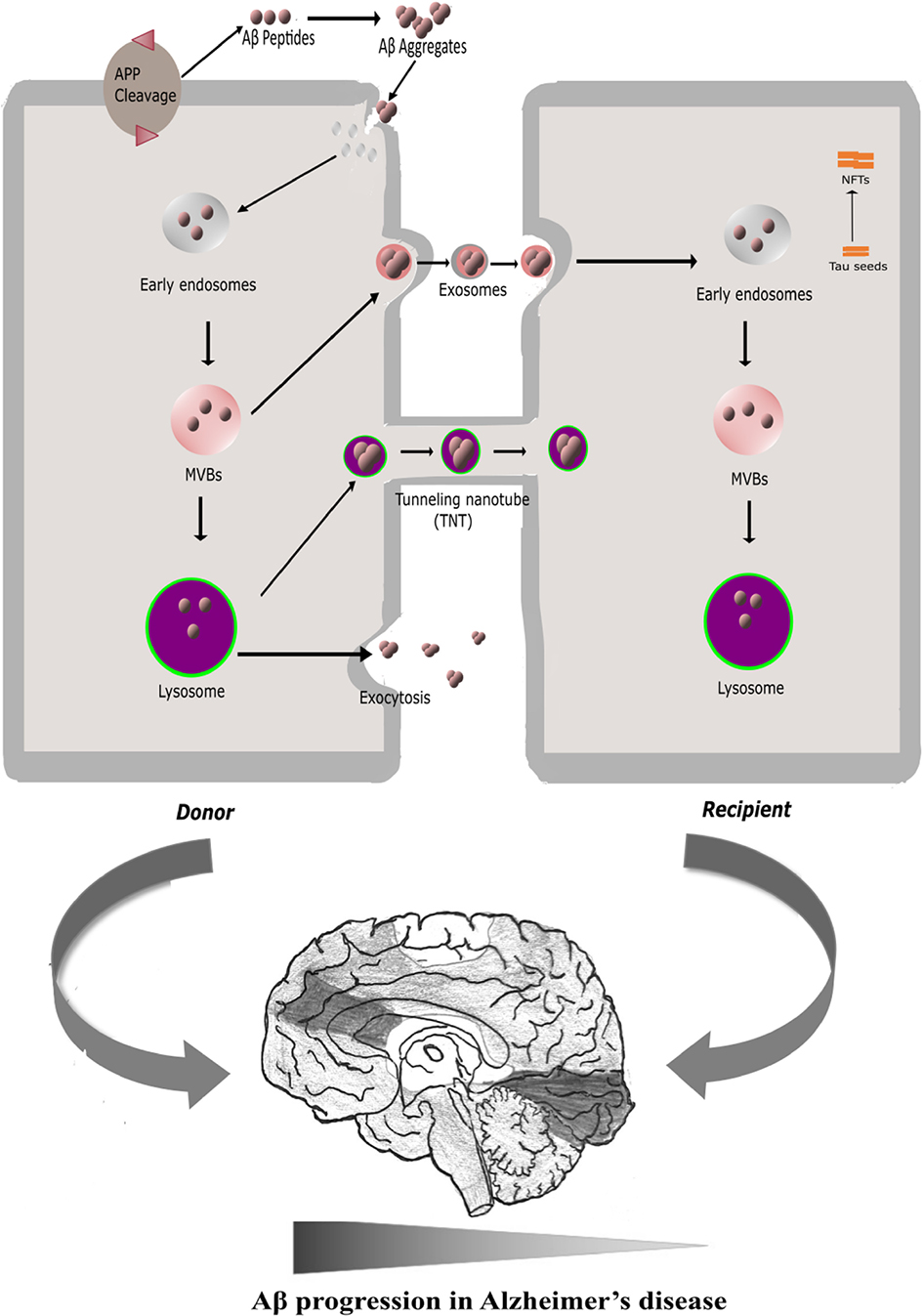
GRAPHICAL ABSTRACT. APP (amyloid precursor protein) cleavage at plasma membrane (PM) generates Aβ peptides extracellularly. Interaction of Aβ peptides with PM promotes toxic amyloid aggregates. PM damage induced by toxic aggregates facilitate internalization and accumulation of Aβ to Multi Vesicular Bodies (MVB) and lysosomes, and proteotoxicity stress could subsequently modulate intercellular spreading of pathogenic aggregates in AD via exosome release and TNTs (tunneling nanotubes). Progression of Aβ toxicities may facilitate tau toxicities and formation of Neuro Fibrillary Tangles (NFT) in AD.
Extracellular Aβ plaques and intracellular tau neurofibrillary tangles (NFTs) are the pathological phenotypes of AD. APP (amyloid precursor protein) cleavage at plasma membrane (PM) of neuronal cells results in amyloid-β (Aβ) peptide generation extracellularly, following which these peptide aggregate and gradually form fibrils or plaques (Ito et al., 2011; Zhang et al., 2011). Later, several studies have stated that the soluble oligomers are explicitly neurotoxic and intraneuronal Aβ accumulation acts as a disease driver (McLean et al., 1999; LaFerla et al., 2007; Gouras et al., 2010). However, contributions of extracellular deposits and intraneuronal accumulation of Aβ in AD pathogenesis is complex and contradictions were reported (McLean et al., 1999; LaFerla et al., 2007; Gouras et al., 2010). The study (Bloom, 2014) has shown that intraneuronal Aβ accumulation plays no significant role to form NFTs. On the other hand, several studies have shown that human subjects with amyloid deposits and senile plaques in the brain display no cognitive impairment (Andersen et al., 2017; Kametani and Hasegawa, 2018; Morris et al., 2018). Clinicopathologic studies on human subjects found that cognitive symptoms vary between patients with temporal patterns of deposits (Garcia-Marin et al., 2009; Blazquez-Llorca et al., 2010; Nelson et al., 2012). Studies have also shown that, neuropathological changes in brain areas vary within the same patient (Garcia-Marin et al., 2009; Blazquez-Llorca et al., 2010; Nelson et al., 2012). The spreading of neuropathology and cognitive impairment follows different temporal patterns depending on the different regions of the brain from which the pathologies have begun to spread (Braak and Braak, 1991; Thal et al., 2002; Jack et al., 2019).
Mutations that enhance the aggregation propensity of Aβ or changes its production, processing, and clearance are implicated in AD (Oddo et al., 2006). The membrane environment can enhance the transition of monomeric Aβ to the toxic β-sheet conformation, which acts as a nucleation location for rapid self-aggregation, to form pre-fibrillar oligomers and eventually the fibrils, depending on the lipid component inside the membrane (Yasumoto et al., 2019). Oligomers induce a toxic cascade involving PM damage (Yasumoto et al., 2019), lipid oxidation, ion-permeable membrane pores, as a result of which internalization takes place in the vesicles of endo-lysosomal pathways (Sengupta et al., 2016; Julien et al., 2018; Dilna et al., 2021).
Another pathological feature of AD is the endo-lysosomal dysfunction, which is induced by toxic Aβ aggregates (Murphy and LeVine, 2010; Whyte et al., 2017). This dysfunction alters the trafficking of multivesicular bodies (MVBs) to lysosomes or to PM (Baixauli et al., 2014), thereby, enhances exosome release. In 1980s, exosomes were identified in the extracellular space as one of the subtypes of extracellular vesicles (EVs) (Johnstone et al., 1987). Studies have shown that exosomes are key players in the transport of molecules and cell-to-cell spreading of pathology in neurodegenerative diseases (Howitt and Hill, 2016). Exosomes isolated from the brain of AD patients contained elevated levels of Aβ oligomers, which vehicle the spread of Aβ pathology from one neuron-to-another (Sardar Sinha et al., 2018). Similarly, tau also spreads via exosomes and elevated levels of tau within exosomes were also detected in the cerebrospinal fluid (CSF) of AD patients (Wang et al., 2017).
Recent discoveries revealed that cells establish essential long-range intercellular communications by tunneling nanotubes (TNTs) (Raghavan et al., 2021). TNTs are long-range intercellular membrane-actin conduits (Rustom, 2004), reported to be responsible for the direct transfer of organelles, neurodegenerative aggregates (Victoria and Zurzolo, 2017), pathogens like viruses (Jansens et al., 2020), and RNA between neighboring cells (Haimovich et al., 2021). TNT is defined as an open-ended actin membrane conduit that transfer organelles and pathogens directly between distant cells (Rustom, 2004). Cell-to-cell spreading of prion via TNTs was first demonstrated in 2009 (Gousset and Zurzolo, 2009). Then, studies have shown that almost all known neurodegenerative proteins (including Aβ and tau) use TNTs as their mode of intercellular transport to spread aggregates (Victoria and Zurzolo, 2017; Dilna et al., 2021; Zhang et al., 2021). Recently Dilna and collaborators (Dilna et al., 2021) have shown that cells repair PM perforation induced by oligomers of Aβ1–42, which promotes biogenesis of TNTs and cell-to-cell transfer. Thus, we discuss here toxic amyloid aggregates induced membrane disorder to subsequent endo-lysosomal dysfunctions and how they modulate various cellular signaling and intercellular spreading of pathogenic aggregates in AD (Graphical Abstract).
Amyloid-β peptides are generated from the transmembrane protein APP, by the sequential cleavage of β and γ secretases. Processing of transmembrane domain of APP by secretases produces N-terminal soluble domains sAPPα and sAPPβ, Aβ peptides and C-terminal fragments (Westmark, 2013). C-terminal cleavage between 40 and 42 hydrophobic residues is responsible for strong hydrophobicity in Aβ peptides (Nixon, 2017). The hydrophobic region is crucial in initiating the process of aggregation. The membrane environment can enhance the transition of hydrophobic Aβ peptides readily to the toxic β-sheet structure conformation, which acts as nucleation locations for rapid self-aggregation (Ito et al., 2011; Cline et al., 2018). Oligomerization may happen in several stages, the transformation of non-toxic monomers to oligomers and protofibrils can trigger neuronal damage (Hashimoto et al., 2003; Zempel et al., 2010). Aβ interacts with membrane and alters membrane compositions by binding to the surface and cell surface receptors such as APP, NMDAR and mGLuR5. Clustering of Aβ reduces lateral diffusion in the membranes and increases accumulation on excitatory synapses, resulting in synaptic dysfunction and LTD (long-term depression) (Westmark, 2013). Aβ increases membrane depolarization and Ca2+ influx (Blanchard et al., 2002). Aβ can also pervade the membrane and exert a wide range of toxicities to neurons, like functional alterations of neurons, dystrophy, neuronal loss, as well as synaptic dysfunctions (Niu et al., 2018). Pore/ion channel formation (Cline et al., 2018) and lipid extraction (Arispe et al., 1993; Delgado et al., 2016) are the major mechanisms which are present in the literature to explain the penetrating ability of Aβ.
A leaky PM may be the result of a variety of events, like mechanical stress or pore-forming toxin attacks (Cooper and McNeil, 2015; Jimenez and Perez, 2017). It has already been shown that Aβ can form ion-permeable channels in a synthetic membrane. Aβ can induce a response to membrane repair similar to that induced by bacterial pore-forming toxin produced by B. thuringensis (Julien et al., 2018). The effects of Aβ oligomers, specifically tau hyperphosphorylation (Bilousova et al., 2016), can be mimicked by exposure to pore-forming toxins (Julien et al., 2018). Aβ oligomers induced PM damage and subsequently PM repair through coupled lysosomal exocytosis and endocytosis has recently been demonstrated (Dilna et al., 2021).
ESCRT (Endosomal Sorting Complex Required for Transportation) involved in the biogenesis of MVB, has also been shown to have a role in the PM repair (Jimenez et al., 2014). Aβ disrupts the PM integrity of the membranes which lack ESCRT-III machineries (Fruhmann et al., 2018). ESCRTs help in the APP sorting and accumulation of Aβ in MVBs (Edgar et al., 2015). Uptake of the exogenous Aβ oligomers leads to accumulation and enlargement of MVBs (Willén et al., 2017). The MVBs either are ubiquitinated for degradation, or targeted by lysosomes for their intracellular degradation, where they can be processed into perinuclear MVB, that are eliminated by exocytosis (Draeger et al., 2014). Recent studies have shown that ESCRT-III is not only involved in PM repair but also plays a significant role in early endo-lysosomal membrane repair (Radulovic et al., 2018).
The intra-vesicular Aβ pool is generated from the secretase enzymes and APP, which are exclusively present in the lumen of the endo-lysosomal vesicles (Cataldo et al., 1997; Willén et al., 2017; Colacurcio et al., 2018). Accumulation of toxic pool of Aβ1–42 is evident in the endo-lysosomal vesicles when compared with Aβ1–40. Internalization of exogenous Aβ takes place by a mechanism of membrane stress sensitive, Rho GTPase regulated actin mediated endocytosis in neuronal cells (Wesén et al., 2017; Dilna et al., 2021). In primary neurons Aβ internalization takes place by a sphingolipid and cholesterol sensitive lipid rafts mediated dynamin dependent but clathrin independent pathway (Saavedra et al., 2007; Marshall et al., 2020). Internalization results in the selective accumulation of toxic pools of Aβ1–42 (Wesén et al., 2017). This could be a result of the non-degradable toxic pool that ultimately ends up accumulating in endo-lysosomal vesicles (Nath et al., 2012). Moreover, during PM repair toxin pores endocytose and traffic to lumen of MVBs and ultimately to lysosomes for degradation (Corrotte and Castro-Gomes, 2019).
Prion-like cell-to-cell propagation of neurodegenerative proteins has been proposed since the discovery of prions. Li et al. (2008) has shown the trans-neuronal propagation of α-synuclein from grafted Lewy body neurons. Subsequently, several researchers demonstrated neuron to neuron transfer of neurodegenerative proteins α-synuclein (Desplats et al., 2009), tau (Clavaguera et al., 2009), polyglutamine (Ren et al., 2009), superoxide dismutase-1 (Ilieva et al., 2009), TDP-43 (Furukawa et al., 2011), and Aβ (Nath et al., 2012).
Aggregation-prone neurodegenerative proteins involved in AD, Parkinson’s disease (PD), Huntington disease (HD), Creutzfeldt-Jakob disease (CJD), and amyotrophic lateral sclerosis (ALS) were found to be contained within exosomes isolated from CSF and blood of patients with these conditions (Brettschneider et al., 2015). Prion-like proteins can be released by exosomes and internalized by the neighboring cells. This was proposed for Aβ (Rajendran et al., 2006), tau (Wang et al., 2017); α-synuclein (Fevrier et al., 2004; Danzer et al., 2007; Saman et al., 2012); superoxide dismutase (SOD-1) (Münch et al., 2011) and polyglutamine aggregates (Ren et al., 2009).
Spread of pathology via exosomes in neurodegenerative diseases has been widely studied, but the mechanisms by which neurodegenerative proteins propagate from one neuron to its neighboring one, is not completely studied yet. The recent studies of direct cell-to-cell transfer of prion-like proteins using TNT conduits have opened up a new avenue (Gousset et al., 2009). TNTs have been implicated in the intercellular transfer of almost all neurodegenerative proteins including, α-synuclein (Abounit et al., 2016; Valdinocci et al., 2021), mutant Huntington line (mHtt) (Costanzo et al., 2013), tau (Tardivel et al., 2016) and Aβ (Wang et al., 2011; Dilna et al., 2021; Zhang et al., 2021). It was shown that, the intracellular accumulation of neurodegenerative aggregates can induce biogenesis of TNTs and thereby the accumulated aggregates propagate from a cell to its neighboring one (Costanzo and Zurzolo, 2013; Abounit et al., 2016; Zhu et al., 2018).
The pathological progression of neurodegenerative disorders has been attributed to the prion-like self-propagation of toxic aggregates following the route of anatomically connected neurons (Prusiner, 2013). The transmission of AD pathology between neurons has been suggested (Lewis et al., 1987) since long, to explain the spread of pathology through anatomically connected brain regions. Inoculation of brain extract from AD patients manifests propagation of AD-like pathology in the brain of transgenic APP mice, but the propagation is not noticed in the non-transgenic control mice (Kane et al., 2000; Meyer-Luehmann, 2006). In addition, researchers have shown the propagation of synaptic dysfunction trans-synaptically from Aβ overexpressed neuron to its neighboring one (Harris et al., 2010; Wei et al., 2010).
The cell-to-cell transfer of tau (Clavaguera et al., 2009) and Aβ aggregates (Nath et al., 2012; Domert et al., 2014) have been demonstrated in in vitro neuronal models. Aβ induced pathology may directly or indirectly drive tau-mediated neurotoxicity and NFT formation (Blurton-Jones and Laferla, 2006). However, tau aggregates can propagate and induce other pathologies, independent of Aβ pathology (Victoria and Zurzolo, 2017). Therefore, understanding the neuron-to-neuron transfer of Aβ aggregates is highly demanding in the context of AD.
Exosomes isolated from AD patients were reported to be a potential vehicle which propagate Aβ aggregates from neuron-to-neuron (Rajendran et al., 2006; Sardar Sinha et al., 2018). Aβ oligomers when microinjected in a single hippocampal primary neuron spread through the neuronal path over time (Nath et al., 2012). A recent study has shown that crosstalk between astrocytes and glial co-culture via networks of TNTs resulted in increased degradation of Aβ aggregates via constant exchange of their internalized Aβ pools (Rostami et al., 2021). Studies have shown that extracellularly applied Aβ oligomers are internalized in the cell and then transferred between neuronal cells by lysosomal vesicles through TNTs (Wang et al., 2011; Dilna et al., 2021). It was also shown that, non-degradable aggregates transfer efficiently between neurons (Nath et al., 2012; Domert et al., 2014; Lim and Yue, 2015) and probably dominates the spreading through neuronal pathway over astrocytes or glial cells. A recent study has shown that tau and Aβ aggregates internalized from extracellular milieu transfer faster between cells through TNTs, compared to the intracellularly produced aggregates (Zhang et al., 2021).
Alterations in the endo-lysosomal pathway is another pathological hallmark of neurodegenerative diseases. Endo-lysosomal dysfunction regulates the trafficking of MVBs to lysosomes, PM (Baixauli et al., 2014) and regulate the release of exosomes. MVBs generate small intraluminal vesicles due to the inward budding of late endosomes. These bodies contain proteins, mRNA and lipids, which would either fuse with lysosomes for degradation or get released extracellularly after fusing with PM (Piper and Katzmann, 2007). The intracellular accumulation of amyloidogenic proteins aggravates endosomal abnormalities, lysosomal membrane damage and impair degradative capacity (Gauthier et al., 2017). Diminished degradative dysfunction can also induce a compensatory increase of exosome release (Alvarez-Erviti et al., 2011; Mathews and Levy, 2019).
Swollen lysosomes with accumulated Aβ aggregates were found at the axonal terminal surrounding the amyloid plaques in the AD brain (Domert et al., 2014; Gowrishankar et al., 2015). Large pools of Aβ accumulation were detected in both MVBs and lysosomes (Willén et al., 2017). MVBs accumulated with Aβ could enhance the spread of Aβ through exosome release (Rajendran et al., 2006). Exosomes isolated from the brain of AD patients contained high levels of Aβ (Sardar Sinha et al., 2018). The lysosomal stress associated with accumulation of amyloidogenic proteins contributes to biogenesis of TNTs (Abounit et al., 2016). Most of the studies have shown that appearance of TNTs is predominantly seen in case of cellular stress and diseased conditions. Studies indicated that the pathology associated with endo-lysosomal toxicities, vesicle recycling, and fusion with PM is involved in the formation of TNTs (Kimura et al., 2013; Victoria and Zurzolo, 2017).
Extracellularly applied Aβ oligomers induce PM damage and trigger the PM repair process via coupled endocytosis followed by lysosomal exocytosis (Julien et al., 2018). Our recent study found that Aβ oligomers induced PM damage. Subsequently, Ca2+ dependent membrane repair via lysosomal exocytosis instigates the biogenesis of TNT-like membrane nanotubes and cell-to-cell transfer of Aβ oligomers (Dilna et al., 2021). Similarly, extracellularly applied tau aggregates can induce the formation of TNTs in neuronal cells and tau proteins were found inside the TNTs (Abounit et al., 2016; Tardivel et al., 2016). The means of two extracellular release routes, lysosomal exocytosis and exosome release share a common interface within the endo-lysosomal system, but biogenesis of the two follows distinct pathways. Neurodegenerative aggregates induced alterations in endo-lysosomal pathway play a significant role to modulate the release of extracellular vesicles (Willén et al., 2017; Bécot et al., 2020).
Ras-related protein-A (RalA, small GTPase) promotes the biogenesis of TNTs by interacting with M-sec, a protein of exocyst complex (Hase et al., 2009). The release of exosomes and docking of MVBs are regulated by Rab 11 in a Ca+2 dependent manner (Escudero et al., 2014). Rab 11 plays an important role in vesicle recycling, inhibition of the same would interfere with membrane recycling and reduce the formation of TNTs (Zhu et al., 2018). Neurodegenerative proteins such as Aβ, α-synuclein, and mHtt, perturb Ca2+ homeostasis, and can deregulate lysosomal exocytosis (Freeman et al., 2013). Increased lysosomal exocytosis enhances the release of exosomes, probably by altering the preferential docking of MVBs to lysosomes, than fusing with PM (van de Vlekkert et al., 2019).
Coordinated assembly and disassembly of actin filaments play a crucial role in regulated exocytosis. Actin cytoskeleton regulates integration of vesicles with PM and pre-fusion events in neurons, neuroendocrine, endocrine, and hematopoietic cells and regulates dynamics of extracellular vesicles release (Porat-Shliom et al., 2013). In other secretory cells, where large cargo molecules undergo slow exocytosis, actin plays a major role in post-fusion events by providing structural support to PM via curvature induced expulsion of large cargo (Porat-Shliom et al., 2013). Exosome biogenesis and sequential regulation can be controlled either in the endosomal pathway or at secretion. This coordination is achieved with the help of two small GTPases RhoA and Cdc42 (Chi et al., 2013). Cdc42, the effector molecule of p21-activated kinase (PAK), stimulates regulated exocytosis by activating actin polymerization, by modulating the downstream signals N-Wasp (Wiskott-Aldrich Syndrom Protein) and Arp2/3 (Gasman et al., 2003).
Interestingly, recent studies have shown that the Rho family of GTPases, Rac1, and Cdc42, play an important role in TNT formation in immune cells (Hanna et al., 2017). Similarly the endocytosis of HIV (human immunodeficiency virus) and HSV (herpes simplex virus) viruses via PAK dependent route induces the biogenesis of TNTs (Van den Broeke et al., 2009; Mukerji et al., 2012; Jacob et al., 2015; Jansens et al., 2020). Furthermore, cofilin an actin-binding protein, a downstream signaling cascade of PAK1, modulates F-actin polymerization and biogenesis of TNTs (Dagar et al., 2021). In our recent study, we have observed that the Aβ induced membrane damage instigates PAK1 dependent endocytosis and coupled lysosomal exocytosis to repair the membrane. The consequence of the repair mechanism promotes the formation of TNTs (Dilna et al., 2021).
Amyloid-β modulates activity of PAK pathways in several ways. Alteration of PAK pathways and consequent actin remodulations can lead to deep changes in neuronal health (Civiero and Greggio, 2018). However, role of PAK in AD pathogenesis is not clear. Aβ has been shown to modulate cofilin regulated actin polymerization through both PAK dependent and independent pathways. Aβ aggregate induced synaptic rigidity via cofilin pathology was reported by many studies (Kang and Woo, 2019). Presence of cofilin-actin rods/aggregates is a salient feature of AD (Bamburg and Bloom, 2009; Kang and Woo, 2019). Aβ aggregates intervene with various surface receptor mediated signaling cascades and promotes cofilin-actin rod formation by regulating the dephosphorylation of cofilin. Slingshot1 (SSH-1) is one of the conserved isoform of cofilin phosphatase, and Aβ induced activation escapes 14-3-3ζ mediated inhibition of SSH-1, which dephosphorylates cofilin.
On the other hand, Aβ aggregates modulate LIM kinase mediated inactivation/phosphorylation of cofilin, either by activating or inhibiting Rac/Pak signaling. PAK1, a Cdc42 GTP-bound effector, is a LIM kinase activator, that holds cofilin in an inactive state; hence, PAK1 depletion can lead to over-activation of cofilin and synaptic dysfunction due to excessive actin dynamics (Bamburg and Bloom, 2009). On contrary, cognitive deficits observed in AD models with Aβ-induced altered postsynaptic PAK levels and massive loss of postsynaptic protein Drebrin, could be prevented by an anti-Aβ antibody and/or by in vivo or in vitro PAK overexpression (Ma et al., 2008; Mokhtar et al., 2013). In addition, studies have shown that reduction of PAK1 in the cytosolic fraction, occurs due to aberrant activation and translocation of PAK to the membrane cytoskeletal fractions in the AD brain (Ma et al., 2008; Civiero and Greggio, 2018). From the literature it is not clear, how the role of activated PAK1 and its distributions in the cellular compartments affects the development of AD pathology.PAK1 plays an important role in cytoskeleton outgrowth and actin polarization in developing neurites (Daniels, 1998). Developing neurites connect to the neighboring astrocytes via TNT-like structures by gap junction mediated electrical coupling (Wang et al., 2012). In a cellular model of AD, we have recently shown that oligomeric Aβ1–42 induced phosphorylated active PAK1, promotes biogenesis of TNT-like structure for intercellular transfer of Aβ aggregates (Dilna et al., 2021). Role of cofilin has recently been reported in the biogenesis of TNT. A recent study by Dagar et al. (2021); suggests that, in addition to cofilin-regulated actin modulation, the exocyst complex protein, M-sec dependent recycling of membrane needs to function in a cohort, as a necessary step in the biogenesis of TNTs. RNA-binding protein nucleolin interacts with M-sec, while nucleolin regulates 14-3-3ζ mRNA, which is also required for the formation of TNT. On the other hand, Cdc42 and Rac1 (effector molecules of PAK) partially colocalize with exocyst complex and M-sec (Vitale et al., 2005; Lopez et al., 2008; Rondaij et al., 2008). However, further studies need to understand how oAβ induced TNT biogenesis via PAK1 activation pathway could contribute to development and progression of AD pathology. The role of regulatory signaling pathways of cofilin in TNT formation and AD is summarized here in the Figure 1 schematically.
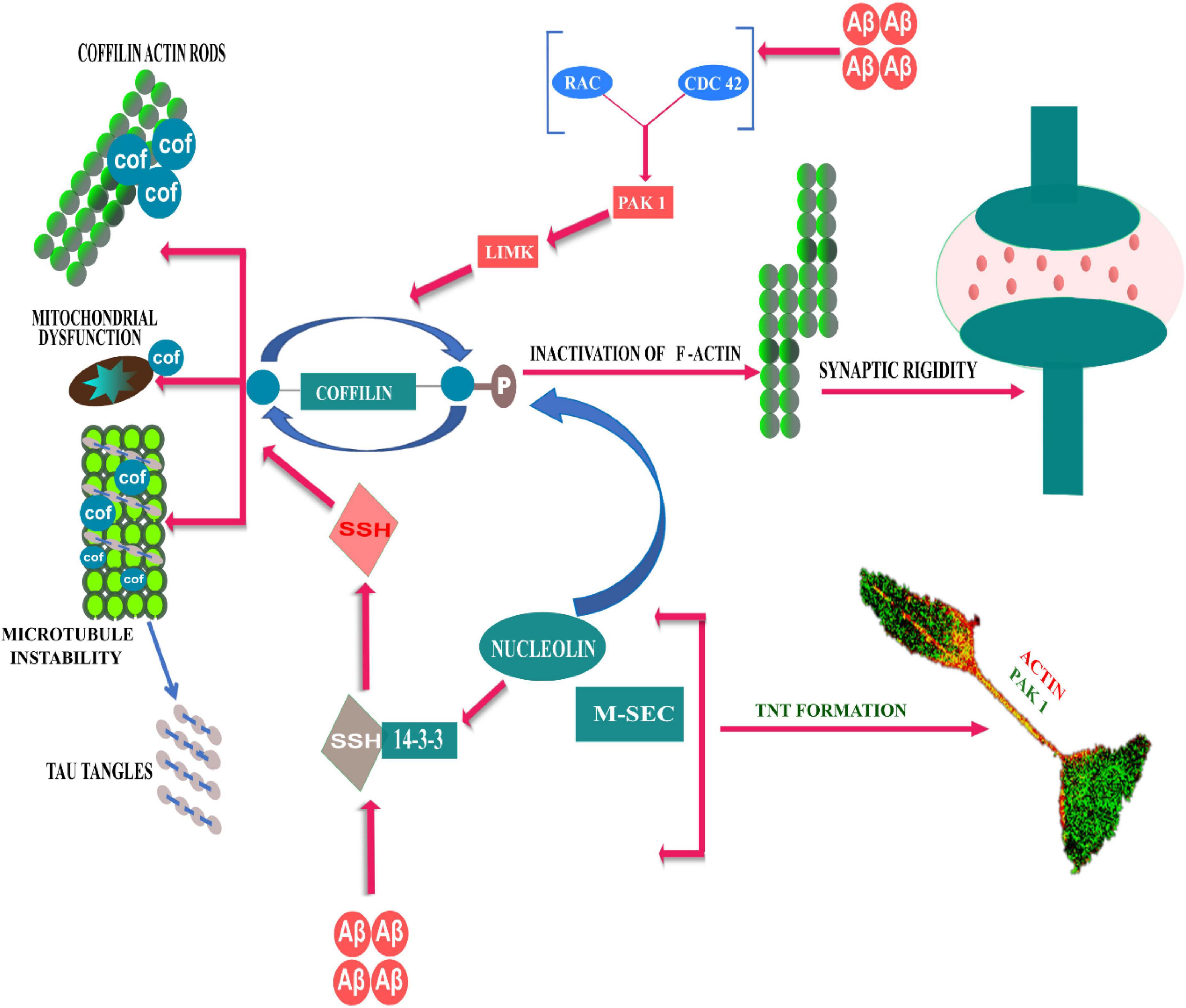
Figure 1. Schematic representative diagram to demonstrate Rho GTPases and actin cytoskeleton reorganization dynamics in TNT formation and AD development. Aβ modulates cofilin regulated actin polymerization through both PAK dependent and independent pathways. Slingshot1 (SSH-1) is one of the conserved isoform of cofilin phosphatase, and Aβ induced activation escapes 14-3-3ζ mediated inhibition of SSH-1, which dephosphorylates cofilin and resulting in cofilin-actin rod formation. On the other hand, Aβ aggregates can modulate LIM kinase mediated phosphorylation of cofilin via PAK kinase pathways (downstream of Rac/Cdc42), which can lead to over-activation of cofilin and synaptic dysfunction due to excessive actin dynamics. RNA-binding protein nucleolin interacts to M-sec, while nucleolin regulates 14-3-3ζ mRNA. The signaling axis of nucleolin and 14-3-3ζ mRNA phosphorylates cofilin to induce formation of TNTs.
Since decades, Aβ induced alterations and related toxicities at the membrane level are considered as a central cause for initiation of AD pathogenesis. Interestingly, Aβ induced membrane damage and repair, can direct endo-lysosomal accumulation to cell-to-cell spread of pathogenic aggregates via exosomes and/or tunneling nanotubes, the two long range intercellular modes of transfer. Spread of pathogenic aggregates could contribute to complex molecular events related to neuropathological and synaptic changes in AD.
Cells repair PM perforation induced by Aβ oligomers via Ca+2 dependent lysosomal exocytosis and coupled endocytosis via PAK1 dependent actin remodulation and this promotes biogenesis of TNTs and cell-to-cell transfer (Dilna et al., 2021). PM perforation by Aβ oligomers generates influx of intracellular Ca+2 and leads to synaptic dysfunction (Peters et al., 2016). Additionally, Aβ mediated alterations of PAK and cofilin phosphorylation play a vital role in synaptic dysfunction which leads to AD development. Interaction of Ca2+ with the membrane is a well-known phenomenon of membrane fusion, lysosomal exocytosis, and exosome release (Melcrová et al., 2016). Overall, the stresses which are induced by the PM damage and intracellular endo-lysosomal impairment aggravate the biogenesis of both exosomes and TNTs, and subsequently cell-to-cell transfer (Victoria and Zurzolo, 2017). Exosomes have also been demonstrated as a potential mediator to induce the formation of TNTs (Ady et al., 2014).
Alzheimer’s disease is categorically a disease characterized by degeneration of neurons, whereas astroglial cells and microenvironment of neurons play an important role in the disease progression. Several studies have shown that increased reactive astrocytes in the AD brain facilitate clearance of plaques and Aβ deposits (Frost and Li, 2017). The crosstalk between neurons and astroglial cells facilitates long-range intercellular transfer of neurodegenerative aggregates probably that helps to enhance cellular clearance of toxic burdens (Rostami et al., 2021; Scheiblich et al., 2021). On contrary, studies have also shown that reactive astrocytes enhance APP processing and as a result increased production of Aβ (Frost and Li, 2017). Reactive microglia have also been proposed to enhance propagation of tau pathology (Maphis et al., 2015; Hopp et al., 2018). Tau toxicity is also considered as one of the causatives of neurodegeneration. However, primary role of Aβ induced tau toxicity in AD pathology is debated. Several studies have shown that tau pathology can develop independent of Aβ induced toxicities and Aβ plays no role in developing tauopathies (Bloom, 2014).
In this context, it has to be mentioned here that AD confronts major challenges of variability in pathogenesis between patients. Variability of neurotoxicity in relation to membrane interactions with Aβ aggregates, have been studied a lot (Niu et al., 2018). Many diverse and complex interactions that exist between complex composition of membranes and transient Aβ aggregates are difficult to mimic and poorly understood. Thereby, to unfold the exact mechanism of progressive spreading in relation to Aβ aggregates induced membrane interactions and intercellular transfers, causes of variabilities need to be addressed. Nevertheless, revealing of the plausible mechanism of progressive spreading of AD pathology via intercellular transfer in association to PM damage, endo-lysosomal accumulation and actin remodulation may open up new inference in AD research.
SN conceived the idea for the review. DV, NM, AD, and SN researched the literatures and wrote the manuscript. DV and NM drew the figures. All authors contributed to the article and approved the submitted version.
We thank the Intramural fund of Manipal Academy of Higher Education, Manipal, India, the Indian Council of Medical Research of India (#5/4-5/Ad hoc/Neuro/216/2020-NCD-I), and the Science and Engineering Research Board of India for the SERB-SRG (#SRG/2021/001315).
DV thanks Manipal Academy of higher education for the TMA Pai fellowship.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abounit, S., Wu, J. W., Duff, K., Victoria, G. S., and Zurzolo, C. (2016). Tunneling nanotubes: A possible highway in the spreading of tau and other prion-like proteins in neurodegenerative diseases. Prion 10, 344–351. doi: 10.1080/19336896.2016.1223003
Ady, J. W., Desir, S., Thayanithy, V., Vogel, R. I., Moreira, A. L., Downey, R. J., et al. (2014). Intercellular communication in malignant pleural mesothelioma: Properties of tunneling nanotubes. Front. Physiol. 5:400. doi: 10.3389/fphys.2014.00400
Alvarez-Erviti, L., Seow, Y., Schapira, A. H., Gardiner, C., Sargent, I. L., Wood, M. J. A., et al. (2011). Lysosomal dysfunction increases exosome-mediated alpha-synuclein release and transmission. Neurobiol. Dis. 42, 360–367. doi: 10.1016/j.nbd.2011.01.029
Andersen, J. V., Christensen, S. K., Aldana, B. I., Nissen, J. D., Tanila, H., and Waagepetersen, H. S. (2017). Alterations in Cerebral Cortical Glucose and Glutamine Metabolism Precedes Amyloid Plaques in the APPswe/PSEN1dE9 Mouse Model of Alzheimer’s Disease. Neurochem. Res. 42, 1589–1598. doi: 10.1007/s11064-016-2070-2
Arispe, N., Rojas, E., and Pollard, H. B. (1993). Alzheimer disease amyloid beta protein forms calcium channels in bilayer membranes: Blockade by tromethamine and aluminum. Proc. Natl. Acad. Sci. U. S. A. 90, 567–571.
Baixauli, F., López-Otín, C., and Mittelbrunn, M. (2014). Exosomes and Autophagy: Coordinated Mechanisms for the Maintenance of Cellular Fitness. Front. Immunol. 5:403. doi: 10.3389/fimmu.2014.00403
Bamburg, J. R., and Bloom, G. S. (2009). Cytoskeletal pathologies of Alzheimer disease. Cell Motil. Cytoskelet. 66, 635–649.
Bécot, A., Volgers, C., and van Niel, G. (2020). Transmissible Endosomal Intoxication: A Balance between Exosomes and Lysosomes at the Basis of Intercellular Amyloid Propagation. Biomedicines 8:272. doi: 10.3390/biomedicines8080272
Bilousova, T., Miller, C. A., Poon, W. W., Vinters, H. V., Corrada, M., Kawas, C., et al. (2016). Synaptic Amyloid-β Oligomers Precede p-Tau and Differentiate High Pathology Control Cases. Am. J. Pathol. 186, 185–198. doi: 10.1016/j.ajpath.2015.09.018
Blanchard, B. J., Thomas, V. L., and Ingram, V. M. (2002). Mechanism of membrane depolarization caused by the Alzheimer Abeta1-42 peptide. Biochem. Biophys. Res. Commun. 293, 1197–1203.
Blazquez-Llorca, L., Garcia-Marin, V., and Defelipe, J. (2010). Pericellular innervation of neurons expressing abnormally hyperphosphorylated tau in the hippocampal formation of Alzheimer’s disease patients. Front. Neuroanat. 4:20. doi: 10.3389/fnana.2010.00020
Bloom, G. S. (2014). Amyloid-β and tau: The trigger and bullet in Alzheimer disease pathogenesis. JAMA Neurol. 71, 505–508. doi: 10.1001/jamaneurol.2013.5847
Blurton-Jones, M., and Laferla, F. M. (2006). Pathways by which Abeta facilitates tau pathology. Curr. Alzheimer Res. 3, 437–448.
Braak, H., and Braak, E. (1991). Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 82, 239–259. doi: 10.1007/BF00308809
Brettschneider, J., Del, T. K., Lee, V. M., and Trojanowski, J. Q. (2015). Spreading of pathology in neurodegenerative diseases: A focus on human studies. Nat. Rev. Neurosci. 16, 109–120.
Cataldo, A. M., Barnett, J. L., Pieroni, C., and Nixon, R. A. (1997). Increased Neuronal Endocytosis and Protease Delivery to Early Endosomes in Sporadic Alzheimer’s Disease: Neuropathologic Evidence for a Mechanism of Increased β-Amyloidogenesis. J. Neurosci. 17, 6142–6151. doi: 10.1523/JNEUROSCI.17-16-06142.1997
Chi, X., Wang, S., Huang, Y., Stamnes, M., and Chen, J. L. (2013). Roles of rho GTPases in intracellular transport and cellular transformation. Int. J. Mol. Sci. 14, 7089–7108.
Civiero, L., and Greggio, E. (2018). PAKs in the brain: Function and dysfunction. Biochim. Biophys. Acta Mol. Basis Dis. 1864, 444–453.
Clavaguera, F., Bolmont, T., Crowther, R. A., Abramowski, D., Frank, S., Probst, A., et al. (2009). Transmission and spreading of tauopathy in transgenic mouse brain. Nat. Cell Biol. 11, 909–913.
Cline, E. N., Bicca, M. A., Viola, K. L., and Klein, W. L. (2018). The Amyloid-β Oligomer Hypothesis: Beginning of the Third Decade. J. Alzheimers Dis. 64, S567–S610.
Colacurcio, D. J., Pensalfini, A., Jiang, Y., and Nixon, R. A. (2018). Dysfunction of autophagy and endosomal-lysosomal pathways: Roles in pathogenesis of Down syndrome and Alzheimer’s Disease. Free Radic. Biol. Med. 114, 40–51. doi: 10.1016/j.freeradbiomed.2017.10.001
Cooper, S. T., and McNeil, P. L. (2015). Membrane Repair: Mechanisms and Pathophysiology. Physiol. Rev. 95, 1205–1240.
Corrotte, M., and Castro-Gomes, T. (2019). Lysosomes and plasma membrane repair. Curr. Top. Membr. 84, 1–16.
Costanzo, M., Abounit, S., Marzo, L., Danckaert, A., Chamoun, Z., Roux, P., et al. (2013). Transfer of polyglutamine aggregates in neuronal cells occurs in tunneling nanotubes. J. Cell Sci. 126, 3678–3685. doi: 10.1242/jcs.126086
Costanzo, M., and Zurzolo, C. (2013). The cell biology of prion-like spread of protein aggregates: Mechanisms and implication in neurodegeneration. Biochem. J. 452, 1–17.
Dagar, S., Pushpa, K., Pathak, D., Samaddar, S., Saxena, A., Banerjee, S., et al. (2021). Nucleolin regulates 14-3-3ζ mRNA and promotes cofilin phosphorylation to induce tunneling nanotube formation. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 35:e21199. doi: 10.1096/fj.202001152R
Daniels, R. H. (1998). Membrane targeting of p21-activated kinase 1(PAK1) induces neurite outgrowth from PC12 cells. EMBO J. 17, 754–764.
Danzer, K. M., Haasen, D., Karow, A. R., Moussaud, S., Habeck, M., Giese, A., et al. (2007). Different Species of -Synuclein Oligomers Induce Calcium Influx and Seeding. J. Neurosci. 27, 34. doi: 10.1523/JNEUROSCI.2617-07.2007
Delgado, D. A., Doherty, K., Cheng, Q., Kim, H., Xu, D., Dong, H., et al. (2016). Distinct Membrane Disruption Pathways Are Induced by 40-Residue β-Amyloid Peptides. J. Biol. Chem. 291, 12233–12244. doi: 10.1074/jbc.M116.720656
Desplats, P., Lee, H. J., Bae, E. J., Patrick, C., Rockenstein, E., Crews, L., et al. (2009). Inclusion formation and neuronal cell death through neuron-to-neuron transmission of -synuclein. Proc. Natl. Acad. Sci. U. S. A. 106, 13010–13015.
Dilna, A., Deepak, K. V., Damodaran, N., Kielkopf, C. S., Kagedal, K., Ollinger, K., et al. (2021). Amyloid-β induced membrane damage instigates tunneling nanotube-like conduits by p21-activated kinase dependent actin remodulation. Biochim. Biophys. Acta Mol. Basis Dis. 1867:166246. doi: 10.1016/j.bbadis.2021.166246
Domert, J., Rao, S. B., Agholme, L., Brorsson, A. C., Marcusson, J., Hallbeck, M., et al. (2014). Spreading of amyloid-β peptides via neuritic cell-to-cell transfer is dependent on insufficient cellular clearance. Neurobiol. Dis. 65, 82–92. doi: 10.1016/j.nbd.2013.12.019
Draeger, A., Schoenauer, R., Atanassoff, A. P., Wolfmeier, H., and Babiychuk, E. B. (2014). Dealing with damage: Plasma membrane repair mechanisms. Biochimie 107, 66–72.
Edgar, J. R., Willén, K., Gouras, G. K., and Futter, C. E. (2015). ESCRTs regulate amyloid precursor protein sorting in multivesicular bodies and intracellular beta amyloid accumulation. J. Cell Sci. 128, 2520–2528.
Escudero, C. A., Lazo, O. M., Galleguillos, C., Parraguez, J. I., Lopez-Verrilli, M. A., Cabeza, C., et al. (2014). The p75 neurotrophin receptor evades the endolysosomal route in neuronal cells, favouring multivesicular bodies specialised for exosomal release. J. Cell Sci. 127, 1966–1979. doi: 10.1242/jcs.141754
Fevrier, B., Vilette, D., Archer, F., Loew, D., Faigle, W., Vidal, M., et al. (2004). Cells release prions in association with exosomes. Proc. Natl. Acad. Sci. U. S. A. 101, 9683–9688.
Freeman, D., Cedillos, R., Choyke, S., Lukic, Z., McGuire, K., Marvin, S., et al. (2013). Alpha-Synuclein Induces Lysosomal Rupture and Cathepsin Dependent Reactive Oxygen Species Following Endocytosis. PLoS One 8:e62143. doi: 10.1371/journal.pone.0062143
Frost, G. R., and Li, Y. M. (2017). The role of astrocytes in amyloid production and Alzheimer’s disease. Open Biol. 7:170228.
Fruhmann, G., Marchal, C., Vignaud, H., Verduyckt, M., Talarek, N., De Virgilio, C., et al. (2018). The Impact of ESCRT on Aβ(1-42) Induced Membrane Lesions in a Yeast Model for Alzheimer’s Disease. Front. Mol. Neurosci. 11:406. doi: 10.3389/fnmol.2018.00406
Furukawa, Y., Kaneko, K., Watanabe, S., Yamanaka, K., and Nukina, N. A. (2011). Seeding Reaction Recapitulates Intracellular Formation of Sarkosyl-insoluble Transactivation Response Element (TAR) DNA-binding Protein-43 Inclusions. J. Biol. Chem. 286, 18664–18672. doi: 10.1074/jbc.M111.231209
Garcia-Marin, V., Blazquez-Llorca, L., Rodriguez, J. R., Boluda, S., Muntane, G., Ferrer, I., et al. (2009). Diminished perisomatic GABAergic terminals on cortical neurons adjacent to amyloid plaques. Front. Neuroanat. 3:28. doi: 10.3389/neuro.05.028.2009
Gasman, S., Chasserot-Golaz, S., Malacombe, M., Way, M., and Bader, M. F. (2003). Regulated exocytosis in neuroendocrine cells: A role for subplasmalemmal Cdc42/N-WASP-induced actin filaments. Mol. Biol. Cell 15, 520–531. doi: 10.1091/mbc.e03-06-0402
Gauthier, S. A., Pérez-González, R., Sharma, A., Huang, F. K., Alldred, M. J., Pawlik, M., et al. (2017). Enhanced exosome secretion in Down syndrome brain - a protective mechanism to alleviate neuronal endosomal abnormalities. Acta Neuropathol. Commun. 5:65. doi: 10.1186/s40478-017-0466-0
Gouras, G. K., Tampellini, D., Takahashi, R. H., and Capetillo-Zarate, E. (2010). Intraneuronal β-amyloid accumulation and synapse pathology in Alzheimer’s disease. Acta Neuropathol. 119, 523–541.
Gousset, K., Schiff, E., Langevin, C., Marijanovic, Z., Caputo, A., Browman, D. T., et al. (2009). Prions hijack tunnelling nanotubes for intercellular spread. Nat. Cell Biol. 11, 328–336.
Gousset, K., and Zurzolo, C. (2009). Tunnelling nanotubes: A highway for prion spreading? Prion 3, 94–98.
Gowrishankar, S., Yuan, P., Wu, Y., Schrag, M., Paradise, S., Grutzendler, J., et al. (2015). Massive accumulation of luminal protease-deficient axonal lysosomes at Alzheimer’s disease amyloid plaques. Proc. Natl. Acad. Sci. U. S. A. 112, E3699–E3708. doi: 10.1073/pnas.1510329112
Haimovich, G., Dasgupta, S., and Gerst, J. E. (2021). RNA transfer through tunneling nanotubes. Biochem. Soc. Trans. 49, 145–160.
Hanna, S. J., McCoy-Simandle, K., Miskolci, V., Guo, P., Cammer, M., Hodgson, L., et al. (2017). The Role of Rho-GTPases and actin polymerization during Macrophage Tunneling Nanotube Biogenesis. Sci. Rep. 7:8547. doi: 10.1038/s41598-017-08950-7
Harris, J. A., Devidze, N., Verret, L., Ho, K., Halabisky, B., Thwin, M. T., et al. (2010). Transsynaptic Progression of Amyloid-β-Induced Neuronal Dysfunction within the Entorhinal-Hippocampal Network. Neuron 68, 428–441. doi: 10.1016/j.neuron.2010.10.020
Hase, K., Kimura, S., Takatsu, H., Ohmae, M., Kawano, S., Kitamura, H., et al. (2009). M-Sec promotes membrane nanotube formation by interacting with Ral and the exocyst complex. Nat. Cell Biol. 11, 1427–1432.
Hashimoto, M., Rockenstein, E., Crews, L., and Masliah, E. (2003). Role of protein aggregation in mitochondrial dysfunction and neurodegeneration in Alzheimer’s and Parkinson’s diseases. Neuromol. Med. 4, 21–36.
Hopp, S. C., Lin, Y., Oakley, D., Roe, A. D., DeVos, S. L., Hanlon, D., et al. (2018). The role of microglia in processing and spreading of bioactive tau seeds in Alzheimer’s disease. J. Neuroinflammation 15:269. doi: 10.1186/s12974-018-1309-z
Howitt, J., and Hill, A. F. (2016). Exosomes in the Pathology of Neurodegenerative Diseases. J. Biol. Chem. 291, 26589–26597.
Ilieva, H., Polymenidou, M., and Cleveland, D. W. (2009). Non–cell autonomous toxicity in neurodegenerative disorders: ALS and beyond. J. Cell Biol. 187, 761–772.
Ito, M., Johansson, J., Strömberg, R., and Nilsson, L. (2011). Unfolding of the amyloid β-peptide central helix: Mechanistic insights from molecular dynamics simulations. PLoS One 6:e17587. doi: 10.1371/journal.pone.0017587
Jack, C. R. J., Wiste, H. J., Therneau, T. M., Weigand, S. D., Knopman, D. S., Mielke, M. M., et al. (2019). Associations of Amyloid, Tau, and Neurodegeneration Biomarker Profiles With Rates of Memory Decline Among Individuals Without Dementia. JAMA 321, 2316–2325.
Jacob, T., Broeke, C. V. D., Waesberghe, C. V., Troys, L. V., and Favoreel, H. W. (2015). Pseudorabies virus US3 triggers RhoA phosphorylation to reorganize the actin cytoskeleton. J. Gen. Virol. 96, 2328–2335. doi: 10.1099/vir.0.000152
Jansens, R. J. J., Tishchenko, A., and Favoreel, H. W. (2020). Bridging the Gap: Virus Long-Distance Spread via Tunneling Nanotubes. J. Virol. 94, e02120–19. doi: 10.1128/JVI.02120-19
Jimenez, A. J., Maiuri, P., Lafaurie-Janvore, J., Divoux, S., Piel, M., and Perez, F. (2014). ESCRT machinery is required for plasma membrane repair. Science 343:1247136.
Jimenez, A. J., and Perez, F. (2017). Plasma membrane repair: The adaptable cell life-insurance. Curr. Opin. Cell Biol. 47, 99–107. doi: 10.1016/j.ceb.2017.03.011
Johnstone, R. M., Adam, M., Hammond, J. R., Orr, L., and Turbide, C. (1987). Vesicle formation during reticulocyte maturation. Association of plasma membrane activities with released vesicles (exosomes). J. Biol. Chem. 262, 9412–9420.
Julien, C., Tomberlin, C., Roberts, C. M., Akram, A., Stein, G. H., Silverman, M. A., et al. (2018). In vivo induction of membrane damage by β-amyloid peptide oligomers. Acta Neuropathol. Commun. 6:131.
Kametani, F., and Hasegawa, M. (2018). Reconsideration of Amyloid Hypothesis and Tau Hypothesis in Alzheimer’s Disease. Front. Neurosci. 12:25. doi: 10.3389/fnins.2018.00025
Kane, M. D., Lipinski, W. J., Callahan, M. J., Bian, F., Durham, R. A., Schwarz, R. D., et al. (2000). Evidence for Seeding of β-Amyloid by Intracerebral Infusion of Alzheimer Brain Extracts in β-Amyloid Precursor Protein-Transgenic Mice. J. Neurosci. 20, 3606–3611.
Kang, D. E., and Woo, J. A. (2019). Cofilin, a Master Node Regulating Cytoskeletal Pathogenesis in Alzheimer’s Disease. J. Alzheimers Dis. 72, S131–S144. doi: 10.3233/JAD-190585
Kimura, S., Hase, K., and Ohno, H. (2013). The molecular basis of induction and formation of tunneling nanotubes. Cell Tissue Res. 352, 67–76.
LaFerla, F. M., Green, K. N., and Oddo, S. (2007). Intracellular amyloid-β in Alzheimer’s disease. Nat. Rev. Neurosci. 8, 499–509.
Lewis, D., Campbell, M., Terry, R., and Morrison, J. (1987). Laminar and regional distributions of neurofibrillary tangles and neuritic plaques in Alzheimer’s disease: A quantitative study of visual and auditory cortices. J. Neurosci. 7, 1799–1808. doi: 10.1523/JNEUROSCI.07-06-01799.1987
Li, J. Y., Englund, E., Holton, J. L., Soulet, D., Hagell, P., Lees, A. J., et al. (2008). Lewy bodies in grafted neurons in subjects with Parkinson’s disease suggest host-to-graft disease propagation. Nat. Med. 14, 501–503. doi: 10.1038/nm1746
Lim, J., and Yue, Z. (2015). Neuronal Aggregates: Formation, Clearance, and Spreading. Dev. Cell 32, 491–501.
Lopez, J. A., Kwan, E. P., Xie, L., He, Y., James, D. E., and Gaisano, H. Y. (2008). The RalA GTPase is a central regulator of insulin exocytosis from pancreatic islet beta cells. J. Biol. Chem. 283, 17939–17945. doi: 10.1074/jbc.M800321200
Ma, Q. L., Yang, F., Calon, F., Ubeda, O. J., Hansen, J. E., Weisbart, R. H., et al. (2008). p21-activated Kinase-aberrant Activation and Translocation in Alzheimer Disease Pathogenesis. J. Biol. Chem. 283, 14132–14143. doi: 10.1074/jbc.M708034200
Maphis, N., Xu, G., Kokiko-Cochran, O. N., Jiang, S., Cardona, A., Ransohoff, R. M., et al. (2015). Reactive microglia drive tau pathology and contribute to the spreading of pathological tau in the brain. Brain 138, 1738–1755. doi: 10.1093/brain/awv081
Marshall, K. E., Vadukul, D. M., Staras, K., and Serpell, L. C. (2020). Misfolded amyloid-β-42 impairs the endosomal–lysosomal pathway. Cell Mol. Life Sci. 77, 5031–5043. doi: 10.1007/s00018-020-03464-4
Mathews, P. M., and Levy, E. (2019). Exosome Production Is Key to Neuronal Endosomal Pathway Integrity in Neurodegenerative Diseases. Front. Neurosci. 13:1347. doi: 10.3389/fnins.2019.01347
McLean, C. A., Cherny, R. A., Fraser, F. W., Fuller, S. J., Smith, M. J., Beyreuther, K., et al. (1999). Soluble pool of Abeta amyloid as a determinant of severity of neurodegeneration in Alzheimer’s disease. Ann. Neurol. 46, 860–866.
Melcrová, A., Pokorna, S., Pullanchery, S., Kohagen, M., Jurkiewicz, P., Hof, M., et al. (2016). The complex nature of calcium cation interactions with phospholipid bilayers. Sci. Rep. 6:38035.
Meyer-Luehmann, M. (2006). Exogenous Induction of Cerebral -Amyloidogenesis Is Governed by Agent and Host. Science 313, 1781–1784. doi: 10.1126/science.1131864
Mokhtar, S. H., Bakhuraysah, M. M., Cram, D. S., and Petratos, S. (2013). The Beta-Amyloid Protein of Alzheimer’s Disease: Communication Breakdown by Modifying the Neuronal Cytoskeleton. Int. J. Alzheimers Dis. 2013:910502. doi: 10.1155/2013/910502
Morris, G. P., Clark, I. A., and Vissel, B. (2018). Questions concerning the role of amyloid-β in the definition, aetiology and diagnosis of Alzheimer’s disease. Acta Neuropathol. 136, 663–689. doi: 10.1007/s00401-018-1918-8
Mukerji, J., Olivieri, K. C., Misra, V., Agopian, K. A., and Gabuzda, D. (2012). Proteomic analysis of HIV-1 Nef cellular binding partners reveals a role for exocyst complex proteins in mediating enhancement of intercellular nanotube formation. Retrovirology 9:33. doi: 10.1186/1742-4690-9-33
Münch, C., O’Brien, J., and Bertolotti, A. (2011). Prion-like propagation of mutant superoxide dismutase-1 misfolding in neuronal cells. Proc. Natl. Acad. Sci. U. S. A. 108, 3548–3553.
Murphy, M. P., and LeVine, H. III (2010). Alzheimer’s disease and the amyloid-beta peptide. J. Alzheimers Dis. 19, 311–323.
Nath, S., Agholme, L., Kurudenkandy, F. R., Granseth, B., Marcusson, J., and Hallbeck, M. (2012). Spreading of Neurodegenerative Pathology via Neuron-to-Neuron Transmission of -Amyloid. J. Neurosci. 32:26.
Nelson, P. T., Alafuzoff, I., Bigio, E. H., Bouras, C., Braak, H., Cairns, N. J., et al. (2012). Correlation of Alzheimer disease neuropathologic changes with cognitive status: A review of the literature. J. Neuropathol. Exp. Neurol. 71, 362–381. doi: 10.1097/NEN.0b013e31825018f7
Niu, Z., Zhang, Z., Zhao, W., and Yang, J. (2018). Interactions between amyloid β peptide and lipid membranes. Biochim. Biophys. Acta Biomembr. 1860, 1663–1669.
Nixon, R. A. (2017). Amyloid precursor protein and endosomal-lysosomal dysfunction in Alzheimer’s disease: Inseparable partners in a multifactorial disease. FASEB J. 31, 2729–2743. doi: 10.1096/fj.201700359
Oddo, S., Caccamo, A., Smith, I. F., Green, K. N., and LaFerla, F. M. (2006). A dynamic relationship between intracellular and extracellular pools of Abeta. Am. J. Pathol. 168, 184–194. doi: 10.2353/ajpath.2006.050593
Peters, C., Bascuñán, D., Opazo, C., and Aguayo, L. G. (2016). Differential Membrane Toxicity of Amyloid-β Fragments by Pore Forming Mechanisms. J. Alzheimers Dis. 51, 689–699. doi: 10.3233/JAD-150896
Piper, R. C., and Katzmann, D. J. (2007). Biogenesis and function of multivesicular bodies. Annu. Rev. Cell Dev. Biol. 23, 519–547.
Porat-Shliom, N., Milberg, O., Masedunskas, A., and Weigert, R. (2013). Multiple roles for the actin cytoskeleton during regulated exocytosis. Cell. Mol. Life Sci. 70, 2099–2121.
Prusiner, S. B. (2013). Biology and Genetics of Prions Causing Neurodegeneration. Annu. Rev. Genet. 47, 601–623.
Radulovic, M., Schink, K. O., Wenzel, E. M., Nähse, V., Bongiovanni, A., Lafont, F., et al. (2018). ESCRT-mediated lysosome repair precedes lysophagy and promotes cell survival. EMBO J. 37:e99753. doi: 10.15252/embj.201899753
Raghavan, A., Rao, P., Neuzil, J., Pountney, D. L., and Nath, S. (2021). Oxidative stress and Rho GTPases in the biogenesis of tunnelling nanotubes: Implications in disease and therapy. Cell. Mol. Life Sci. 79:36. doi: 10.1007/s00018-021-04040-0
Rajendran, L., Honsho, M., Zahn, T. R., Keller, P., Geiger, K. D., Verkade, P., et al. (2006). Alzheimer’s disease beta-amyloid peptides are released in association with exosomes. Proc. Natl. Acad. Sci. U. S. A. 103, 11172–11177.
Ren, P. H., Lauckner, J. E., Kachirskaia, I., Heuser, J. E., Melki, R., and Kopito, R. R. (2009). Cytoplasmic penetration and persistent infection of mammalian cells by polyglutamine aggregates. Nat. Cell. Biol. 11, 219–225. doi: 10.1038/ncb1830
Rondaij, M. G., Bierings, R., van Agtmaal, E. L., Gijzen, K. A., Sellink, E., Kragt, A., et al. (2008). Guanine exchange factor RalGDS mediates exocytosis of Weibel-Palade bodies from endothelial cells. Blood 112, 56–63.
Rostami, J., Mothes, T., Kolahdouzan, M., Eriksson, O., Moslem, M., Bergström, J., et al. (2021). Crosstalk between astrocytes and microglia results in increased degradation of α-synuclein and amyloid-β aggregates. J. Neuroinflammation 18:124. doi: 10.1186/s12974-021-02158-3
Rustom, A. (2004). Nanotubular Highways for Intercellular Organelle Transport. Science 303, 1007–1010. doi: 10.1126/science.1093133
Saavedra, L., Mohamed, A., Ma, V., Kar, S., and de Chaves, E. P. (2007). Internalization of β-Amyloid Peptide by Primary Neurons in the Absence of Apolipoprotein E. J. Biol. Chem. 282, 35722–35732. doi: 10.1074/jbc.M701823200
Saman, S., Kim, W., Raya, M., Visnick, Y., Miro, S., Saman, S., et al. (2012). Exosome-associated Tau Is Secreted in Tauopathy Models and Is Selectively Phosphorylated in Cerebrospinal Fluid in Early Alzheimer Disease. J. Biol. Chem. 287, 3842–3849. doi: 10.1074/jbc.M111.277061
Sardar Sinha, M., Ansell-Schultz, A., Civitelli, L., Hildesjö, C., Larsson, M., Lannfelt, L., et al. (2018). Alzheimer’s disease pathology propagation by exosomes containing toxic amyloid-beta oligomers. Acta Neuropathol. 136, 41–56. doi: 10.1007/s00401-018-1868-1
Scheiblich, H., Dansokho, C., Mercan, D., Schmidt, S. V., Bousset, L., Wischhof, L., et al. (2021). Microglia jointly degrade fibrillar alpha-synuclein cargo by distribution through tunneling nanotubes. Cell 184, 5089–5106.e21. doi: 10.1016/j.cell.2021.09.007
Sengupta, U., Nilson, A. N., and Kayed, R. (2016). The Role of Amyloid-β Oligomers in Toxicity, Propagation, and Immunotherapy. Ebiomedicine 6, 42–49.
Tardivel, M., Bégard, S., Bousset, L., Dujardin, S., Coens, A., Melki, R., et al. (2016). Tunneling nanotube (TNT)-mediated neuron-to neuron transfer of pathological Tau protein assemblies. Acta Neuropathol. Commun. 4:117. doi: 10.1186/s40478-016-0386-4
Thal, D. R., Rüb, U., Orantes, M., and Braak, H. (2002). Phases of A beta-deposition in the human brain and its relevance for the development of AD. Neurology 58, 1791–1800.
Valdinocci, D., Kovarova, J., Neuzil, J., and Pountney, D. L. (2021). Alpha-Synuclein Aggregates Associated with Mitochondria in Tunnelling Nanotubes. Neurotox. Res. 39, 429–443. doi: 10.1007/s12640-020-00285-y
van de Vlekkert, D., Demmers, J., Nguyen, X. X., Campos, Y., Machado, E., Annunziata, I., et al. (2019). Excessive exosome release is the pathogenic pathway linking a lysosomal deficiency to generalized fibrosis. Sci. Adv. 5:eaav3270. doi: 10.1126/sciadv.aav3270
Van den Broeke, C., Radu, M., Deruelle, M., Nauwynck, H., Hofmann, C., Jaffer, Z. M., et al. (2009). Alphaherpesvirus US3-mediated reorganization of the actin cytoskeleton is mediated by group A p21-activated kinases. Proc. Natl. Acad. Sci. U. S. A. 106, 8707–8712. doi: 10.1073/pnas.0900436106
Victoria, G. S., and Zurzolo, C. (2017). The spread of prion-like proteins by lysosomes and tunneling nanotubes: Implications for neurodegenerative diseases. J. Cell. Biol. 216, 2633–2644. doi: 10.1083/jcb.201701047
Vitale, N., Mawet, J., Camonis, J., Regazzi, R., Bader, M. F., and Chasserot-Golaz, S. (2005). The Small GTPase RalA controls exocytosis of large dense core secretory granules by interacting with ARF6-dependent phospholipase D1. J. Biol. Chem. 280, 29921–29928. doi: 10.1074/jbc.M413748200
Wang, X., Bukoreshtliev, N. V., and Gerdes, H.-H. (2012). Developing Neurons Form Transient Nanotubes Facilitating Electrical Coupling and Calcium Signaling with Distant Astrocytes. PLoS One 7:e47429. doi: 10.1371/journal.pone.0047429
Wang, Y., Balaji, V., Kaniyappan, S., Krüger, L., Irsen, S., Tepper, K., et al. (2017). The release and trans-synaptic transmission of Tau via exosomes. Mol. Neurodegener. 12:5. doi: 10.1186/s13024-016-0143-y
Wang, Y., Cui, J., Sun, X., and Zhang, Y. (2011). Tunneling-nanotube development in astrocytes depends on p53 activation. Cell Death Differ. 18, 732–742. doi: 10.1038/cdd.2010.147
Wei, W., Nguyen, L. N., Kessels, H. W., Hagiwara, H., Sisodia, S., and Malinow, R. (2010). Amyloid beta from axons and dendrites reduces local spine number and plasticity. Nat. Neurosci. 13, 190–196. doi: 10.1038/nn.2476
Wesén, E., Jeffries, G. D. M., Matson Dzebo, M., and Esbjörner, E. K. (2017). Endocytic uptake of monomeric amyloid-β peptides is clathrin- and dynamin-independent and results in selective accumulation of Aβ(1–42) compared to Aβ(1–40). Sci. Rep. 7:2021.
Westmark, C. J. (2013). What’s hAPPening at synapses? The role of amyloid β-protein precursor and β-amyloid in neurological disorders. Mol. Psychiatry 18, 425–434. doi: 10.1038/mp.2012.122
Whyte, L. S., Lau, A. A., Hemsley, K. M., Hopwood, J. J., and Sargeant, T. J. (2017). Endo-lysosomal and autophagic dysfunction: A driving factor in Alzheimer’s disease? J. Neurochem. 140, 703–717. doi: 10.1111/jnc.13935
Willén, K., Edgar, J. R., Hasegawa, T., Tanaka, N., Futter, C. E., and Gouras, G. K. (2017). Aβ accumulation causes MVB enlargement and is modelled by dominant negative VPS4A. Mol. Neurodegener. 12:61. doi: 10.1186/s13024-017-0203-y
Yasumoto, T., Takamura, Y., Tsuji, M., Watanabe-Nakayama, T., Imamura, K., Inoue, H., et al. (2019). High molecular weight amyloid β(1-42) oligomers induce neurotoxicity via plasma membrane damage. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 33, 9220–9234.
Zempel, H., Thies, E., Mandelkow, E., and Mandelkow, E. M. (2010). Abeta oligomers cause localized Ca(2+) elevation, missorting of endogenous Tau into dendrites, Tau phosphorylation, and destruction of microtubules and spines. J. Neurosci. 30, 11938–11950. doi: 10.1523/JNEUROSCI.2357-10.2010
Zhang, K., Sun, Z., Chen, X., Zhang, Y., Guo, A., and Zhang, Y. (2021). Intercellular transport of Tau protein and β-amyloid mediated by tunneling nanotubes. Am. J. Transl. Res. 13, 12509–12522.
Zhang, Y., Thompson, R., Zhang, H., and Xu, H. (2011). APP processing in Alzheimer’s disease. Mol. Brain 4:3.
Keywords: Alzheimer’s disease, neurodegenerative diseases, prion-like propagation, exosomes, tunneling nanotubes (TNTs), intercellular communication, amyloid-β, tau
Citation: Valappil DK, Mini NJ, Dilna A and Nath S (2022) Membrane interaction to intercellular spread of pathology in Alzheimer’s disease. Front. Neurosci. 16:936897. doi: 10.3389/fnins.2022.936897
Received: 05 May 2022; Accepted: 24 August 2022;
Published: 09 September 2022.
Edited by:
Antonella Borreca, Institute of Neuroscience, National Research Council (CNR), ItalyReviewed by:
Diana Furcila, Camilo José Cela University, SpainCopyright © 2022 Valappil, Mini, Dilna and Nath. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sangeeta Nath, c2FuZ2VldGEubmF0aEBtYW5pcGFsLmVkdQ==; orcid.org/0000-0003-0050-0606
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.