 Clíona Farrell
Clíona Farrell Paige Mumford
Paige Mumford Frances K. Wiseman
Frances K. Wiseman
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Neurosci., 07 June 2022
Sec. Neurogenomics
Volume 16 - 2022 | https://doi.org/10.3389/fnins.2022.909669
This article is part of the Research TopicCurrent Advances in the Study of Down Syndrome: from Development to AgingView all 9 articles
There are an estimated 6 million people with Down syndrome (DS) worldwide. In developed countries, the vast majority of these individuals will develop Alzheimer's disease neuropathology characterized by the accumulation of amyloid-β (Aβ) plaques and tau neurofibrillary tangles within the brain, which leads to the early onset of dementia (AD-DS) and reduced life-expectancy. The mean age of onset of clinical dementia is ~55 years and by the age of 80, approaching 100% of individuals with DS will have a dementia diagnosis. DS is caused by trisomy of chromosome 21 (Hsa21) thus an additional copy of a gene(s) on the chromosome must cause the development of AD neuropathology and dementia. Indeed, triplication of the gene APP which encodes the amyloid precursor protein is sufficient and necessary for early onset AD (EOAD), both in people who have and do not have DS. However, triplication of other genes on Hsa21 leads to profound differences in neurodevelopment resulting in intellectual disability, elevated incidence of epilepsy and perturbations to the immune system. This different biology may impact on how AD neuropathology and dementia develops in people who have DS. Indeed, genes on Hsa21 other than APP when in three-copies can modulate AD-pathogenesis in mouse preclinical models. Understanding this biology better is critical to inform drug selection for AD prevention and therapy trials for people who have DS. Here we will review rodent preclinical models of AD-DS and how these can be used for both in vivo and ex vivo (cultured cells and organotypic slice cultures) studies to understand the mechanisms that contribute to the early development of AD in people who have DS and test the utility of treatments to prevent or delay the development of disease.
Down syndrome (DS) is caused by trisomy of human chromosome 21 (Hsa21). The condition is associated with intellectual disability, craniofacial dysmorphology, increased risk of congenital heart defects, and disorders of the immune system (Antonarakis et al., 2020), however, these features occur with variable levels of penetrance and severity between individuals who have DS (Wiseman et al., 2009). DS is also a significant genetic risk factor for the development of Alzheimer's disease (AD) (Wiseman et al., 2015; Strydom et al., 2018). DS occurs in 1–700 to 1–1,000 live births, and recently there has been an increase in life expectancy for people who have DS due to improved access and advances in medical care in developed countries (Wu and Morris, 2013; Glasson et al., 2016; de Graaf et al., 2017). The increased life expectancy of people with DS means that more people than ever with DS will develop AD within their lifetime; in the UK, dementia is now a leading cause of mortality for adults who have DS (Hithersay et al., 2019).
AD is a neurodegenerative condition and the leading cause of dementia worldwide. Early-onset AD (EOAD), occurring before 65-years, accounts for <5% of AD cases and is caused by mutations in the amyloid precursor protein gene (APP), or in the presenilin genes which encode subunits of the γ-secretase enzyme which proteolytically cleaves APP to form amyloid-β (Aβ) (Mendez, 2017). Alternatively, late-onset AD (LOAD) accounts for the majority of AD cases in the general population. Although many risk loci associated with the immune system, lipid metabolism and others have been identified for LOAD, these are not sufficient or necessary to cause AD (Kunkle et al., 2019).
Pathologically, both EOAD and LOAD are associated with the hallmark protein aggregates of AD, including the accumulation of amyloid plaques, composed of the APP cleavage product Aβ, and neurofibrillary tau tangles (NFTs), which precede the development of neuronal loss and brain atrophy. The first phase of AD is considered to be largely asymptomatic and is associated with the deposition of Aβ, first in diffuse plaques, followed by the formation of dense core plaques starting in the temporal cortices (Davidson et al., 2018). Aβ is formed by the sequential cleavage of APP by β-secretase to form a membrane-bound fragment β-C-Terminal fragment (β-CTF) which is then further cleaved by γ-secretase to generate Aβ. This process is thought to occur largely in neuronal endosomes (Das et al., 2016; Chen et al., 2017). In AD, two major isoforms of the Aβ peptide, Aβ40 and Aβ42, are formed, with Aβ42 being more prone to aggregate. Peptide monomers can form higher order oligomers, fibrils, and ultimately extracellular plaques in AD (Sideris et al., 2021; Yang et al., 2022). Following Aβ deposition, NFTs composed of hyperphosphorylated mis-folded tau protein form intracellularly. NFT pathology has been linked to the first clinical symptoms of AD, whereby mild cognitive impairment (MCI) occurs (Parnetti et al., 2012; Betthauser et al., 2020). NFT pathology occurs early in the entorhinal cortex and spreads to other regions of the brain in a defined temporal-spatial pattern which can be classified by Braak tangle staging (Braak and Braak, 1991; Braak et al., 2006). Neuroinflammation is thought to contribute to synapse loss and neuronal degradation in AD, whereby microglia, the resident brain macrophages, change their activation state and function in response to protein aggregates to prune synapses, release pro-inflammatory mediators and no longer undertake key homeostatic functions (Heneka et al., 2015; Hong et al., 2016; Lučiunaite et al., 2020; Leng and Edison, 2021). Together, these processes of protein aggregation and neuroinflammation lead to cell loss and brain atrophy resulting in the clinical manifestations of dementia.
Phenotypes associated with DS arise due to the aberrant dosage of genes encoded on Hsa21. People with DS have three-copies of APP, which is necessary for the development of AD in those individuals (Prasher et al., 1998; Korbel et al., 2009; Wiseman et al., 2015; Doran et al., 2017). Moreover, duplication of the APP locus, in the absence of DS, is a rare cause of EOAD (Rovelet-Lecrux et al., 2006, 2007; Sleegers et al., 2006; Hooli et al., 2012; McNaughton et al., 2012). Thus, three-copies of APP are sufficient and necessary to drive AD pathogenesis both in people who have and don't have DS. Consistent with this, a third copy of APP causes a 1.5-fold or higher level of full-length APP and its cleavage products in the adult brain of people that have DS and in preclinical mouse models (Cheon et al., 2008; Lana-Elola et al., 2021). However, from what age and in which cell types this gene is dosage-sensitive is unclear, as a previous report suggested that APP protein levels are not raised in the young adult brain in a preclinical model of DS (Choi et al., 2009). APP is central in the study of AD-DS, however, three-copies of other Hsa21 genes modulate aspects of AD pathology in preclinical animal models (Sheppard et al., 2012; García-Cerro et al., 2017; Naert et al., 2018; Wiseman et al., 2018; Tosh et al., 2021).
AD pathology in DS broadly progresses in the same temporal pattern as EOAD and LOAD, with Aβ accumulation occurring first, followed by the progression of NFT pathology and neuronal loss (Davidson et al., 2018; Wegiel et al., 2022). However, AD pathology onset occurs decades earlier in people with DS than in the general population (Wiseman et al., 2015; Davidson et al., 2018; Fortea et al., 2021). The first site of Aβ accumulation in AD-DS in the brain is in intracellular neuronal endosomes, which undergo structural changes and enlargement in DS (Gyure et al., 2001; Mori et al., 2002). This accumulation has been observed during fetal development, as early as 28-weeks (Cataldo et al., 2000). Typically however, Aβ accumulation begins in the teens and early-twenties; extracellular amyloid accumulation is seen in the brain parenchyma, beginning as diffuse plaques (made of non-fibrillary deposits), followed by the presence of dense-core plaques over the next decade (Ropper and Williams, 1980; Mann and Esiri, 1989; Lemere et al., 1996; Wegiel et al., 2022). An early site of extracellular Aβ accumulation in AD-DS is in the hippocampus (Leverenz and Raskind, 1998), while in LOAD the first plaques are reported to occur in the cortex (Thal et al., 2002). However, the earliest site of Aβ accumulation detected by positron emission tomography (PET) is in the striatum in AD-DS, consistent with reports for other genetic forms of EOAD (Handen et al., 2012; Annus et al., 2016; Hanseeuw et al., 2018; Zammit et al., 2020). In the brains of individuals who had LOAD, a higher density of Aβ plaques in the cortex is observed compared to AD-DS (Mann et al., 1987, 2018; Egensperger et al., 1999), however, the mean size of plaques in the brain of people who have AD-DS is larger than in LOAD (Armstrong, 2012). Cerebral amyloid angiopathy also occurs with higher frequency in AD-DS than in LOAD (Head et al., 2017; Mann et al., 2018), but microbleeds are thought to occur with the same frequency as in LOAD (Helman et al., 2019). Aβ42 deposition occurs earlier than Aβ40 in DS, with Aβ40 only identified in late-stage dense-core plaques (Lemere et al., 1996; Hirayama et al., 2003). These differences in Aβ pathology between people who had AD-DS and those who had LOAD may be caused by differences in the mechanisms triggering the accumulation of the peptide. In LOAD in the general population, Aβ accumulation is thought to be caused by impaired clearance of Aβ, whilst in AD-DS, increased Aβ production caused by three-copies of APP is the cause.
Like in EOAD and LOAD, NFT pathology occurs after Aβ deposition in AD-DS in a defined spatial-temporal pattern consistent with Braak tangle staging, starting in the entorhinal cortex, followed by the hippocampus and neocortex (Braak et al., 2006; Davidson et al., 2018; Wegiel et al., 2022). The density of NFTs in the DS brain greatly increases with age (Wisniewski et al., 1985; Davidson et al., 2018; Wegiel et al., 2022). The development of tau pathology is associated with clinical signs of cognitive impairment, as measured by tau PET (Rafii et al., 2017; Rafii, 2019; Lemoine et al., 2020). Other protein aggregates seen in neurodegeneration can also be found in the DS brain during AD-DS. TDP-43 pathology occurs with lower frequency in AD-DS compared to LOAD, but is comparable to that seen in EOAD, which may be caused by differences in the age of onset of disease in the subtypes of AD (Davidson et al., 2011, 2018; Wegiel et al., 2022). α-synuclein-positive Lewy bodies are found in the amygdala of people with DS at a slightly lower level than seen in EOAD (Gibb et al., 1989; Bodhireddy et al., 1994; Prasher et al., 2010; Wegiel et al., 2022), though the cause of this is currently unknown.
People with DS have a reduced number of neurons in childhood because of developmental differences, and accumulation of Aβ plaques and NFTs are associated with additional neuronal loss in AD-DS. Evidence suggests that neurofibrillary pathology strongly drives atrophy in many brain regions in DS (Wegiel et al., 2022). The pattern of neuronal loss in AD-DS is similar to that in AD, however, people with DS have increased selective loss of neurons from the nucleus basalis of Meynert (Mann et al., 1984; Casanova et al., 1985). Following AD-pathology onset, age and dementia status correlate with decreased neuronal volume, with huge neuronal loss seen in the entorhinal cortex and hippocampus from the 4th and 5th decades of life, followed by later loss of neurons from the amygdala and other regions (Wegiel et al., 2022). Decreased levels of neuronal PET imaging biomarkers, including N-acetylaspartate, are seen when people with DS develop AD, indicating that neuronal loss is coincident with cognitive decline (Lin et al., 2016; Montal et al., 2021).
Neuroinflammation is thought to make a significant contribution to the transition from pathology to disease in both LOAD and EOAD pathologies, with microgliosis and astrogliosis being identified in close proximity to plaques and tangles (Heneka et al., 2015; De Strooper and Karran, 2016), and gene-association studies identifying variation in a number of genes expressed in immune cells as predictive for disease (Kunkle et al., 2019). Microglial activation, with decreased branching, larger cell somas, and transcriptomic alterations, are seen in people with DS from a young age, with alterations being identified in post-mortem tissue from children as young as 1-year old (Xue and Streit, 2011; Flores-Aguilar et al., 2020; Palmer et al., 2021). As people who have DS age, ramified microglia (thought to have a homeostatic function) become less numerous, and there is an increase in hypertrophic and senescent cells (Xue and Streit, 2011; Martini et al., 2020). The Aβ and NFT load of the cases in these studies is unclear and this could contribute to the differences observed. Other studies have shown that AD-associated neuroinflammation differs in AD-DS compared to LOAD (Wilcock et al., 2015; Startin et al., 2019a; Martini et al., 2020). In the AD-DS posterior cingulate cortex, more ameboid and rod-shaped microglia have been identified compared to AD (Martini et al., 2020). mRNA levels of many pro-inflammatory markers, including IL-1β, TNFα, and IL-6, as well as proteins shown to be raised in activated microglia, CD64 and CD86, are higher in the temporal lobe of people with AD-DS compared to LOAD, despite having a younger age on average (Wilcock et al., 2015). Like in LOAD, plaques in AD-DS are surrounded by activated microglia and are positive for complement proteins, including C1q and C3 (Stoltzner et al., 2000), and activated microglia closely associate with C1q positive neurons (Head et al., 2001). The glial metabolite marker, myo-inositol, measured by proton magnetic resonance spectroscopy, is raised in the brain of people with DS and continues to increase in AD-DS (Lin et al., 2016; Montal et al., 2021). Many inflammatory cytokines, including IL-6, IL-10, TNFα, and IL-1β are raised in the serum of young adults with DS (Weber et al., 2020), and higher levels of IL-1β are seen in the plasma of people with DS compared to LOAD (Startin et al., 2019a). Furthermore, people with DS have higher CSF levels of YKL-40 (chitinase 3-like protein 1), a biomarker of neuroinflammation and astrogliosis, than carriers of EOAD causal mutations, both before and after a dementia diagnosis (Fagan et al., 2021).
People with DS have a perturbed peripheral immune response (Huggard et al., 2020), including increased susceptibility to infection (Hüls et al., 2021; Illouz et al., 2021), autoimmune disorders (Goldacre et al., 2004) and chronic inflammatory conditions (Ferreira et al., 2016). Peripheral or systemic infection can drive neuroinflammation and worsen cognitive decline and pathology in AD (Perry et al., 2007; Cunningham et al., 2009). Priming of microglia can occur in response to peripheral inflammation, with excessive cytokine production, including release of IL-1β and TNFα (Perry and Holmes, 2014). People with DS can experience chronic inflammatory conditions, such as periodontitis, and are susceptible to repeat infection; these systemic effects may be a driver of neuroinflammation and heightened microglial activation in AD-DS compared to LOAD (Kamer et al., 2016).
Due to pre-existing intellectual disability in people with DS, accurate diagnosis of AD-dementia has historically been challenging. Recent advances in cognitive assessments, as well as the use of biomarkers, have aided the diagnosis of dementia and mild-cognitive impairment in people with DS. As in EOAD and LOAD, the early signs of dementia in AD-DS include a decline in memory and language skills (Devenny et al., 2002; Krinsky-McHale et al., 2002; Startin et al., 2019b). Early studies in AD-DS suggested that changes to non-cognitive behaviors are more common in the early phase of dementia than in AD; these include apathy, behavioral impulsivity, decreased motivation and increased stubbornness (Nelson et al., 2001; Ball et al., 2008; Oliver et al., 2011); however more recent work has questioned this finding and indicates that memory loss is one of the earliest changes in people who have AD-DS, consistent with findings in the general population (Startin et al., 2019b).
Other clinical features of AD-DS include the onset of epileptic seizures and motor dysfunction (Hithersay et al., 2017). By contrast to LOAD, the majority of individuals with AD-DS develop seizures (Altuna et al., 2021), and onset of seizures in adults with DS is indicative of AD-onset and is associated with more severe AD progression (Lott et al., 2012). Decline in gait is also indicative of AD onset in people with DS (Anderson-Mooney et al., 2016) and symptoms such as dyspraxia, increased incontinence and grasping reflexes can also occur (Visser et al., 1997).
The average age of dementia diagnosis in people with DS is 55 years (Sinai et al., 2018), with death occurring about 4 years later in most individuals (Hithersay et al., 2019). By 60-years, 70% of people with DS will have a dementia diagnosis and by 80-years, virtually all people with DS will have a dementia diagnosis (McCarron et al., 2014, 2017), showing the age-dependant risk.
Unlike in LOAD, sex does not appear to significantly influence the incidence of AD in DS (Coppus et al., 2006; Lai et al., 2020), however, one study has shown an age-associated effect of a higher incidence of AD-DS in men over 60-years (Mhatre et al., 2021). The onset of menopause in women with DS correlates with the onset of AD (Schupf et al., 2003; Coppus et al., 2010), which is younger than in the general population (Cosgrave et al., 1999). As in LOAD, people with DS who carry an E4 allele of APOE have earlier changes to amyloid and clinical AD symptoms (Deb et al., 2000; Prasher et al., 2008; Fortea et al., 2020; Bejanin et al., 2021; Lemere et al., 2021).
Preclinical mouse models have been central to research of the genes and mechanisms that cause the early onset of AD in people who have DS. Several mouse models of DS have been created using a series of genome engineering and other state-of-the-art techniques (Table 1). Others have covered these in-depth (Herault et al., 2017), but those used in AD-DS preclinical research studies will be briefly introduced here.
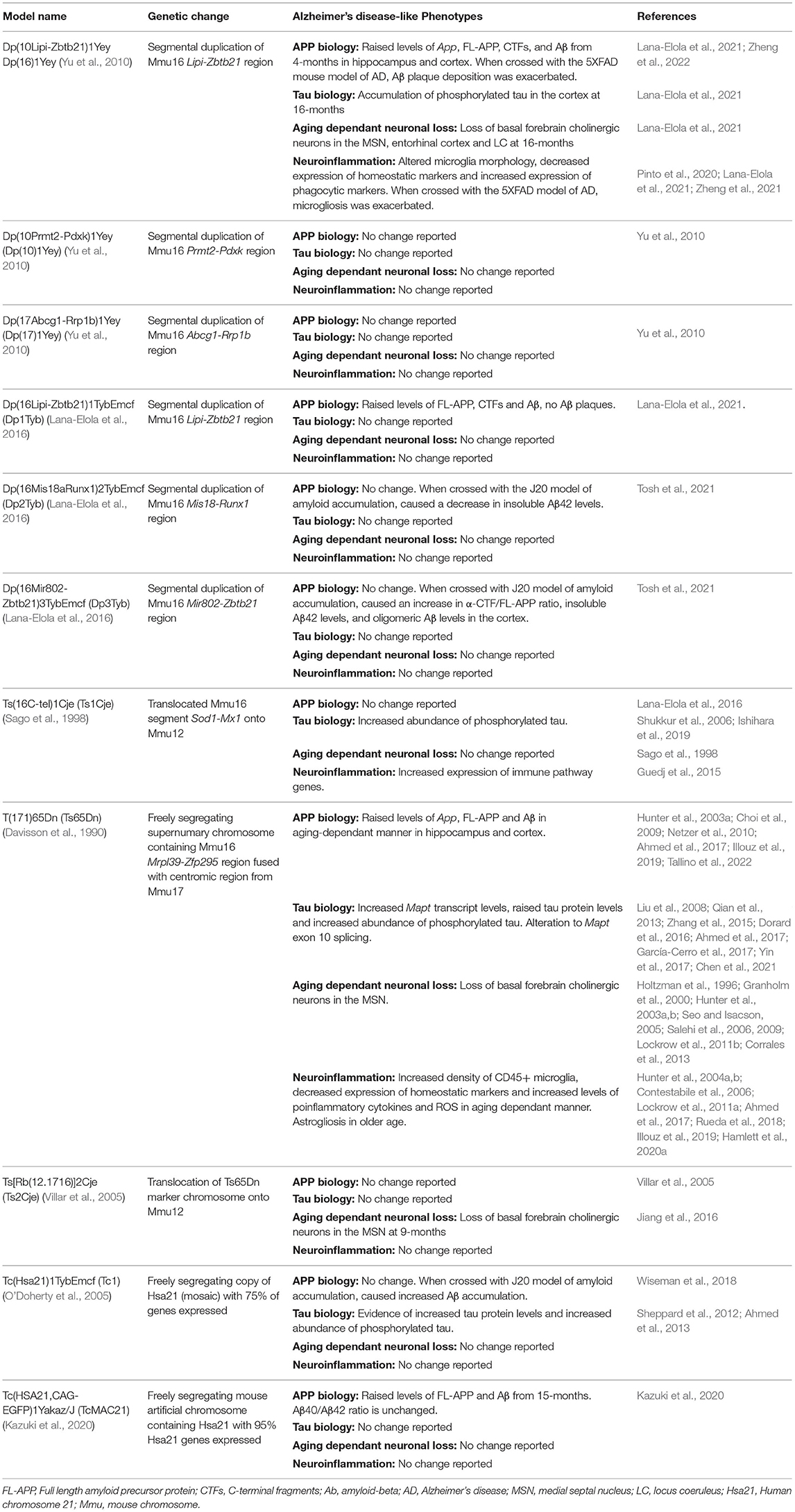
Table 1. Mouse models of Down syndrome and Alzheimer's disease phenotypes they exhibit.
The mouse orthologs of Hsa21 genes are spread across three regions of synteny on mouse chromosomes (Mmu) 10, 16, and 17 (Mouse Genome Informatics, Bult et al., 2019). Chromosomal engineering technology using Cre/LoxP recombination has enabled the creation of mouse models which have a duplication of the regions of the mouse chromosomes that are syntenic with Hsa21. These include several models that have been used in AD-DS research, which have different regions of Hsa21 orthologs on Mmu16 in three-copies; for example the Dp(16)1Yey (Yu et al., 2010) and Dp(16Lipi-Zbtb21)1TybEmcf (Dp1Tyb) (Lana-Elola et al., 2016) models with region Lipi-Zbtb21 in three-copies; Dp(16Mis18aRunx1)2TybEmcf (Dp2Tyb) with region Mis18-Runx1 in three-copies; and the Dp(16Mir802-Zbtb21)3TybEmcf (Dp3Tyb) with region Mir802-Zbtb21 in three-copies (Lana-Elola et al., 2016). Mouse models with the Mmu10 or Mmu17 syntenic Hsa21 regions in three-copies include the Dp (10Prmt2-Pdxk)1Yey (Dp10Yey) and Dp(17Abcg1-Rrp1b)1Yey (Dp17Yey) (Yu et al., 2010). The Ts(16C-tel)1Cje (Ts1Cje) mouse model (Sago et al., 1998), which originated from accidental translocation of Mmu16 segments onto Mmu12, has three-copies of the region Sod1-Mx1 on Mmu16 orthologous to Hsa21.
Using such models with different groups of Hsa21 orthologs in three-copies can be used to determine the combination of genes necessary to cause specific DS phenotypes and the identification of gene candidates. However, many DS phenotypes are multigenic, with many Hsa21 genes required to be in three-copies to result in a phenotype, and there may also be interactions between genes located far away from each other on the chromosome. Such complexities are not modeled when using the smaller segmental duplication models, thus it is important to keep these limitations in mind when interpreting experimental findings.
It is not clear which DS-associated phenotypes are caused by having an extra chromosome (aneuploidy) or by having three-copies of Hsa21 genes. The models discussed above have a duplication on one chromosome of a region of genes; while having three-copies of those genes, they remain euploid. Another approach to modeling DS is to also model the effect of aneuploidy. Models of aneuploidy have also been covered in-depth by others (Tosh et al., 2022), but those used in AD-DS preclinical research will be briefly discussed here. The T(171)65Dn (Ts65Dn) mouse model (Davisson et al., 1990) has a freely segregating, supernumerary chromosome containing the region Mrpl39-Zfp295 on Mmu16 orthologous to Hsa21 fused with the centromic region of ~10 Mb from Mmu17, resulting in a total of ~90 protein-coding Hsa21 orthologs in three-copies. However, this model also carries three-copies of an additional 35 protein-coding genes on Mmu17 that are not orthologous to Hsa21, thus, gene-dosage effects resulting from these are not relevant to the study of DS. Moreover, phenotypic drift has been recently reported in this model (Shaw et al., 2020), likely caused by its highly complex genetic background. Similarly, the Ts[Rb(12.1716)]2Cje (Ts2Cje) mouse model (Villar et al., 2005), created by Robertsonian translocation of the Ts65Dn's marker chromosome onto Mmu12, contains the same genetic region in three-copies as the Ts65Dn. Both models have been used for a number of AD-DS preclinical research studies.
The Tc(Hsa21)1TybEmcf (Tc1) mouse model (O'Doherty et al., 2005) contains a freely segregating copy of Hsa21. Tc1 mice are mosaic, with ~66% of brain cells carrying the Hsa21, though this percentage varies across tissues. Furthermore, the freely segregating copy of Hsa21 has some deletions and rearrangements, resulting in ~75% of Hsa21 genes being present and functional in the mouse (Gribble et al., 2013). Notably for the study of AD-DS, this model does not contain an additional functional copy of APP. Most recently, the Tc(HSA21,CAG-EGFP)1Yakaz/J (TcMAC21) model (Kazuki et al., 2020) was created which carries a mouse artificial chromosome (MAC) with a nearly complete copy of Hsa21; this model is not mosaic because of the use of mouse centromere to carry the human gene-content. While some deletions occurred during generation of the model where ~28% of Hsa21 is missing, only 14 protein-coding genes are within the deleted regions, leaving ~95% of the protein-coding Hsa21 genes intact. Thus, it is currently the most complete aneuploid model of DS and may be a highly useful tool for AD-DS preclinical research.
A range of mouse models exist to study the characteristic amyloid and tau pathology of AD (Götz et al., 2018), here we will briefly describe the amyloid models used in AD-DS preclinical research studies. Mouse Aβ does not readily form extracellular aggregates due to a difference in 3 amino acids between the mouse and human Aβ sequence (Lv et al., 2013). Thus, mouse models that express human Aβ and carry point mutations which cause familial AD in the general population have been used to model Aβ aggregation and accumulation in vivo. These include mutations to the APP gene, such as the Swedish mutations (Mullan et al., 1992) which are located near the β-secretase cleavage site of Aβ and increase APP processing by β-secretase, elevating Aβ production (Citron et al., 1992). The APP Indiana (Murrell et al., 1991) and Iberian (Guerreiro et al., 2010) mutations are located near the γ-secretase cleavage site of Aβ and increase the Aβ42 /Aβ40 ratio, thus enhancing Aβ aggregation and accelerating plaque formation. The Arctic mutation (Kamino et al., 1992), which changes the biophysical properties of the Aβ and cause the faster formation of protofibrils, has also been used in mouse models. There are also multiple AD-causal mutations to the PSEN1 gene which encodes presenilin-1, a subcomponent of the γ-secretase complex involved in APP processing into Aβ (Kelleher and Shen, 2017). PSEN1 mutations increase APP processing by γ-secretase, thus increasing the production of longer, more pathogenic Aβ peptides (Sherrington et al., 1995) and have mostly commonly been used in combination with mutant APP to model aspects of AD in the mouse.
Transgenic overexpression models overexpress human APP and/or PSEN1 carrying familial AD-causal mutations; in these models relatively high expression of the transgene is driven by an artificial promoter, resulting in time-dependent Aβ deposition and coinciding cognitive decline. The J20 model (Mucke et al., 2000) contains the Swedish and Indiana mutations in APP and presents robust Aβ plaque deposition from 5 to 7 months of age in the hippocampus and neocortex, with widespread plaques by 8–10 months, and the development of age-dependent deficits in spatial memory and learning (Cheng et al., 2007). It is worth noting that the J20 transgene integrated into the genome in an intron of Zbtb20, disrupting its expression (Tosh et al., 2017; Goodwin et al., 2019). ZBTB20 is important to hippocampal development (Mitchelmore et al., 2002), if and how disruption of Zbtb20 expression contributes to J20 phenotypes requires further study. The 5XFAD model contains three AD-causal mutations in APP and two AD-causal mutations in PSEN1; Aβ deposition occurs as early as 2-months old, significant Aβ accumulates with age, especially in subiculum and deep cortical layers, and the model also develops age-dependent spatial working memory deficits. Interestingly, the 5XFAD model is one of the few amyloid models to present neuronal loss, especially in brain regions with highest Aβ burden, starting at 6 months (Oakley et al., 2006).
Thus, transgenic overexpression models quickly and robustly present Aβ deposition and coinciding cognitive impairments. Yet, such models have many limitations, including: the highly elevated expression of other APP fragments (secreted APP, β-CTF, α-CTF), each with their own biological functions; the use of artificial promoters to drive transgene expression may result in non-physiological expression of the transgene; and the random insertion of the transgene into a gene locus of the host animal may disrupt endogenous genes (Jankowsky and Zheng, 2017). Furthermore, it has been largely unexplored how the presence of murine APP in these models affects the development of transgene-induced Aβ biology. Indeed, APP/PS1 mice with endogenous mouse App knocked-out had elevated levels of aggregated Aβ and higher speed of Aβ deposition than APP/PS1 mice with endogenous mouse App, indicating a reduction in the general aggregation propensity of human Aβ when co-existing with endogenous murine Aβ (Steffen et al., 2017).
To overcome some of the limitations of transgenic models, several “knock-in” models have been created which have various familial AD-causing mutations knocked into the murine App gene (Saito et al., 2014). In these models, the Aβ sequence is “humanized” (the 3 amino acids in the murine Aβ sequence are changed to match that of the human Aβ sequence), with expression driven by the endogenous murine App promoter. As a result, these knock-in models have elevated levels of pathogenic Aβ but physiological levels of FL-APP which is expressed in the correct spatial and temporal context (Saito et al., 2014). Furthermore, knock-in models of wild-type humanized Aβ APP containing no AD-causal point mutations have been created (Serneels et al., 2020; Baglietto-Vargas et al., 2021). Serneels et al. (2020) created both a rat and mouse humanized Aβ APP model and found that humanization increased the production of Aβ40 compared to the murine Aβ40, coinciding with increased β-CTF and decreased α-CTF levels. Furthermore, a genetically similar humanized App mouse model displayed age-dependent decreases in cognitive function, synaptic plasticity, and hippocampal volume (Baglietto-Vargas et al., 2021). Similar humanization of rodent App in DS preclinical models will provide the next generation of tools for AD-DS preclinical research.
Aβ accumulation is a key early event in the development of AD-DS, and initiates the pathological cascade of events that ultimately lead to brain atrophy and AD-dementia (Hardy and Higgins, 1992). Several key research studies in mouse model systems have been undertaken to understand how three-copies of APP cause this and how additional copies of other Hsa21 genes modulate this biology.
The Ts65Dn mouse model of DS exhibits raised App mRNA levels in the brain from 4-months (Holtzman et al., 1996), but raised levels of full-length APP (FL-APP) and Aβ have been primarily detected in the older Ts65Dn brain in an aging dependant manner, with increases in the hippocampus and cortex seen from 10-months of age (Hunter et al., 2003b; Choi et al., 2009; Illouz et al., 2019; Tallino et al., 2022). Furthermore, Aβ oligomers have been reported to form in the 12-month Ts65Dn hippocampus (Sansevero et al., 2016). Homeostatic processes in the young Ts65Dn brain may modulate APP protein level to compensate for the third copy of App, but with age, this process is less effective. Others have seen an increase in APP and Aβ from as young as 4-months (Netzer et al., 2010; Ahmed et al., 2017), but the level was less than the expected 50% increase, and the relative abundance increased with aging, consistent with aging-dependent breakdown of homeostasis contributing to raised protein levels in DS. Differences in experimental methods to quantify APP products may also account for the differing results between these experiments. Accumulation of insoluble Aβ in 15-month Ts65Dn mice has been reported, with raised abundance of thioflavin S+ inclusions compared to euploid controls reported (Illouz et al., 2019). Similarly, another study detected Aβ aggregates in the cerebellum in 12-month Ts65Dn mice (Lomoio et al., 2009). Notably, cerebellar Aβ deposition is not typically seen until late-stage disease in people with AD-DS and only after substantial accumulation is observed in other brain regions (Mann et al., 2018). Moreover, other studies of 20- and 21-month Ts65Dn mice have reported no detectable accumulation of Aβ aggregates (Reeves et al., 1995; Holtzman et al., 1996).
The Dp(16)1Yey mouse model of DS also exhibits raised levels of App mRNA, FL-APP, CTFs, and Aβ in the cortex and hippocampus from as young as 4-months, increasing further by 19-months (Lana-Elola et al., 2021). Importantly, the increase in abundance of CTF was greater than the 1.5-fold increase that would be predicted to occur because of an extra copy of App; the mechanism for this is not understood and may have considerable implications for the development of AD primary prevention therapy for people who have DS. The TcMAC21 model has increased levels of FL-APP and Aβ, but the Aβ40/Aβ42 ratio is unchanged compared to euploid mice (Kazuki et al., 2020). No plaque deposition was observed in this model at 24-months, but learning and memory deficits occur from as young as 3-months (Kazuki et al., 2020). Raised levels of APP or Aβ have not been seen in mouse models lacking three-copies of App, including the Tc1, Ts1Cje, Dp2Tyb, Dp3Tyb, Dp10Yey, and Dp17Yey models (Salehi et al., 2006; Wiseman et al., 2018; Tosh et al., 2021), consistent with human clinical-genetic data that indicates that three-copies of APP are necessary for the development of AD-pathology in people who have DS (Korbel et al., 2009; Doran et al., 2017).
Many mouse models of DS exhibit neurodevelopmental changes that cause deficits in cognition and changes to behavior (Herault et al., 2017). In addition to these alterations, aging dependent changes to cognition have also been reported in a number of mouse models of DS. Most notably, the Ts65Dn mouse model exhibits progressive cognitive decline (Hunter et al., 2003a; Seo and Isacson, 2005; Illouz et al., 2019), which has been linked to the increased Aβ in the brain. Aβ immunization using DNA or Aβ-fragment approaches successfully lowers soluble and insoluble Aβ toward the level seen in euploid mice, and this is associated with improvements in spatial learning, short-term memory and object recognition (Belichenko et al., 2016; Illouz et al., 2019). However, the response of Ts65Dn mice to the anti-Aβ was lower than euploid controls. Pharmacological reduction of Aβ using γ-secretase inhibition also rescued cognitive deficits in 4-month Ts65Dn mice (Netzer et al., 2010), suggesting that raised Aβ plays an important role in cognitive decline in AD-DS. However, use of another γ-secretase inhibitor in the same model was thought to improve cognition and neuron loss via an alternative mechanism by modulating the Sonic-hedgehog pathway, which is also altered in DS (Stagni et al., 2017). Furthermore, γ-secretase inhibition leads to increased production of CTFs (Netzer et al., 2010), which have been implicated in enlargement of endosomes in DS, contributing to perturbed neuronal transport (Filippone and Praticò, 2021). Given recent data indicating particularly highly elevated levels of this fragment in people with DS and preclinical DS mouse models (Lana-Elola et al., 2021), modulation of γ-secretase in people who have DS should be approached with caution.
Aβ is a key target for cognitive decline in AD-DS because it's role is proposed to occur upstream of other changes; other targets have been investigated in the Ts65Dn model, such as estrogen, which may have utility at later stages of disease progression (Hunter et al., 2004b). Notably, the use of bexarotene, a selective retinoid receptor agonist, to reduce Aβ in the hippocampus of the Ts65Dn mouse, worsened cognitive deficits; likely via an effect on thyroid function (Vidal et al., 2021), despite leading to Aβ clearance and improvements in cognitive decline in a model of Aβ accumulation (Cramer et al., 2012). Therapeutic targets for AD-DS must take into consideration the affect that trisomy has on AD-pathology and on wider physiology of individuals with DS, and such that effective drugs for AD might not always be suitable for the treatment of AD-DS, because of the specific physiology and co-morbidities of individuals with the condition. Importantly, DS mouse models recapitulate many DS-associated phenotypes and thus can be used to test the interaction of these co-morbidities with lead-candidates for AD primary prevention.
Notable differences occur between DS and Aβ accumulation mouse models. Firstly, levels of β-CTF are particularly raised in DS models; likely because of a combinatorial effect of an extra dose of App and other Hsa21 genes. Comparative studies using App segmental duplication models are needed to better understand this key biology. In addition, although Aβ levels are raised in DS mouse models, the peptide is not observed to sustainably aggregate within the brain. This is likely to occur because of differences in the processing and aggregation of mouse and human APP and Aβ (Lv et al., 2013); this limitation could be addressed by the humanization of the Aβ sequence in DS mouse models.
Although App is the primary gene-candidate for altered Aβ levels and associated cognitive decline in AD-DS, work in mouse preclinical systems has demonstrated that other genes on Hsa21, when in three-copies, can modulate Aβ pathology. DYRK1A [dual-specificity tyrosine-(Y)-phosphorylation regulated kinase 1A] is a protein kinase involved in multiple signaling pathways in the brain that are important for normal brain development and physiology. Normalizing the gene-dose of Dyrk1a to two-copies in the Ts65Dn mouse model reduced levels of FL-APP and Aβ in the hippocampus and cortex (García-Cerro et al., 2017), showing that this kinase may contribute to the development of AD-pathology in DS.
Further evidence of non-App Hsa21 genes modifying Aβ pathology in AD-DS has come from work crossing various DS mouse models with models of amyloid pathology. The Tc1 mouse model, which does not have an additional functional copy of APP (O'Doherty et al., 2005), was crossed with the J20 mouse model. This revealed that the extra copy of a gene(s) in the Tc1 model increased Aβ aggregation and Aβ plaque deposition and worsened plaque-associated cognitive deficits (Wiseman et al., 2018). The increased Aβ aggregation corresponded with an increase in the soluble Aβ42/Aβ40 ratio, independent of a change in γ-secretase activity or rate of extracellular Aβ clearance (Wiseman et al., 2018). To further investigate which gene or genes were modifying amyloid biology, several segmental duplication mouse models of DS were then crossed with the J20 mouse model, each containing smaller subregions of Hsa21 genes in three-copies; the Dp2Tyb, Dp3Tyb, Dp10Yey, and Dp17Yey (Tosh et al., 2021). Three-copies of the Dp3Tyb region, with ~37 genes in three-copies, increased the α-CTF/FL-APP ratio, insoluble Aβ42 levels, and oligomeric Aβ levels in the cortex (Tosh et al., 2021). Notably, duplication of the Dp3Tyb region alone was not sufficient to cause the increase in Aβ deposition as reported in the previous Tc1 study, which has many more genes in three-copies. This could be due to the multigenic nature of DS phenotypes. Alternatively, differences in the function of human and mouse genes may also have contributed to the differences observed. Interestingly, in the same study three-copies of the Dp2Tyb region was found to cause a significant decrease in insoluble Aβ42 levels, although interpretation of this data is confounded by the high mortality of this cross (Tosh et al., 2021).
Recently, the Dp(16)1Yey model crossed with the 5XFAD mouse model exhibited exacerbated Aβ plaque deposition (Zheng et al., 2022), consistent with the findings of Wiseman et al. (2018). The candidate causal gene for increased Aβ burden in 5XFAD mice was identified as USP25, which encodes a ubiquitin-specific protease (Zheng et al., 2022). Overexpressing USP25 in the 5XFAD mouse exacerbated Aβ deposition while conversely, genetic deficiency of Usp25 in the 5XFAD mouse reduced Aβ deposition. The proposed mechanism by which USP25 modifies Aβ deposition is via modification of APP processing; USP25 overexpression in 5XFAD mice increased APP and BACE1 levels while conversely Usp25 deficiency in 5XFAD mice reduced levels of FL-APP, α-CTFs and β-CTFs (Zheng et al., 2022). This overexpression paradigm is not exactly comparable to DS, when the expression level of genes in three-copies is only ~50% greater expression (Antonarakis et al., 2020). Notably, both the Tc1 and Dp(16)1Yey mouse model contain an additional copy of USP25 (Gribble et al., 2013) and the gene is significantly upregulated in the adult hippocampus of the Tc1 model (Granno et al., 2019). However, raised levels of full-length APP are not consistently observed in the Tc1 brain or elevated above 1.5-fold in the Dp(16)1Yey model (which also contains 3-copies of App), and raised levels of BACE1 are not observed in either model compared to euploid controls (Wiseman et al., 2018; Lana-Elola et al., 2021). Additional preclinical studies to understand the role of three-copies of USP25 are required to unravel this complexity.
Furthermore, both Wiseman et al. (2018) and Zheng et al. (2022) did not explore how the presence of murine App affects amyloid biology development in the 5XFAD and J20 models. As previously discussed, murine APP/Aβ may interfere with human Aβ pathology development (Steffen et al., 2017). Despite these caveats, studies such as these provide evidence that three-copies of non-App Hsa21 genes can modify amyloid biology, highlighting the complex nature of AD-DS development. Further research is required to identify the genes and mechanisms responsible for these differences.
In AD-DS, tau pathology occurs after the development of extensive Aβ within the brain, and in the general population Aβ accumulation is proposed to trigger tau misfolding, hyper-phosphorylation, mislocalization and accumulation (Braak et al., 2006; Davidson et al., 2018; Wegiel et al., 2022). Tau is a product of the Mapt gene and multiple isoforms of the protein occur; in particular, the relative abundance of the 3-repeat and 4-repeat forms are proposed to cause some genetic forms of frontotemporal dementia, independently of Aβ (Ghetti et al., 2015). Just as in the general population, the cellular and molecular mechanisms that cause tau pathology in AD-DS are not well understood. Research in DS mouse models has provided important insights into how the changes to transcriptional control, cell-signaling, and autophagy may impact on the development of tau pathology in people who have DS, but further research is needed in this important area.
In the Ts65Dn mouse model, increased levels of Mapt transcript occur in the brain because of changed transcript stability (Qian et al., 2013; Zhang et al., 2015). Consistent with this, raised tau protein levels have been reported in several regions of the Ts65Dn and Tc1 brain (Ahmed et al., 2013, 2017; García-Cerro et al., 2017), although this has not been observed in all studies (Sheppard et al., 2012; Chaves et al., 2020; Chen et al., 2021). In one report, the level of raised protein increased with age, consistent with observations for other proteins (Ahmed et al., 2017), suggesting that elevated levels of tau may contribute to development of pathology in older adults who have DS. How this relates to a recent report of alterations to the unfolded protein response, RNA translation and proteostasis in the Ts65Dn model (Lanzillotta et al., 2018; Alldred et al., 2021) is unknown and requires further research. A transitory alteration in Mapt exon 10 splicing also occurs in Ts65Dn mice at postnatal day 10 which results in a change to the 3-repeat/4-repeat isoform ratio of tau; notably use of a DYRK1A inhibitor corrected this perturbation to tau biology (Yin et al., 2017); whether such changes also occur during neurodegeneration is unknown. In addition, increased relative abundance of phosphorylated tau has been reported to occur in the Ts65Dn, Tc1 and Ts1Cje mouse models in numerous independent studies suggesting that trisomy of Hsa21 robustly enhances phosphorylation of the protein in the mouse (Shukkur et al., 2006; Liu et al., 2008; Sheppard et al., 2012; Dorard et al., 2016; Chen et al., 2021). However, the sites of phosphorylation vary between the studies, with reports of both extensive phosphorylation across the protein (Liu et al., 2008), and more limited phosphorylation (targeting Thr212) in the literature (Sheppard et al., 2012). Differences in the genetics of the mouse models, technical differences in detection and analysis of tau phosphorylation, and housing of the mice may contribute to this. Notably, great care during sample isolation and preparation is required to ensure the preservation of endogenous phosphorylation (Sheppard et al., 2012; Yin et al., 2017). Despite the reported robust increase in phospho-tau in DS mouse models, NFTs have not been detected in these models (Sheppard et al., 2012), consistent with reports for Aβ accumulation in these models (Hashimoto et al., 2019). Increased levels of tau-positive granular deposits have been reported in the Ts65Dn hippocampus (Kern et al., 2011).
A number of genes and mechanisms have been suggested to contribute to these changes in tau biology. Three recent mouse preclinical studies have suggested that raised abundance of APP/Aβ contributes to altered phosphorylation in the Ts65Dn model (Liu et al., 2008; Illouz et al., 2019; Chen et al., 2021). Notably, in the Tc1 mouse model, which does not carry an additional functional copy of APP, aberrant tau phosphorylation at position Thr212 has been reported (Sheppard et al., 2012), suggesting that genes on chromosome 21 other than APP may also contribute to the altered tau phosphorylation in people who have DS. DYRK1A, the Hsa21-encoded kinase, has been linked in multiple mouse studies to increased phosphorylation of tau particularly at position Thr212; and the abundance of the kinase has been found to correlate with phosphorylation at this site (Chaves et al., 2020). Thus, three-copies of this gene may contribute to the development of tau neuropathology in people with DS. Intranasal treatment of Ts65Dn mice with rapamycin, which targets the mTOR pathway, normalizes raised abundance of phosphorylated tau (Tramutola et al., 2018; Di Domenico et al., 2019), implicating mTOR signaling in altered tau biology in DS. The effect of rapamycin may occur via protein phosphatase 2A (PP2A) which has a key role in the dephosphorylation of tau. Indeed, another study has demonstrated that the PP2A inhibitor SET is increased in the Ts65Dn and that this may contribute to raised tau phosphorylation (Dorard et al., 2016). Lastly, a recent study in the Ts1Cje model has linked raised copper levels in the brain with enhanced phosphorylation of tau (Ishihara et al., 2019). The gene or mechanism underlying this intriguing biology has yet to be elucidated.
Differentiating between the effect of trisomy on the formation of tau pathology, from how it changes the response to the pathology, is highly challenging; two recent preclinical studies have started to address this complexity. The first study compared isolates from trisomy 21 and duplication of APP iPSC-derived neurons and measured how these altered hippocampal long-term potentiation (LTP) in wild-type mice (Hu et al., 2018). This demonstrated that the key protein responsible for changes to the electrophysiology of the mice differed between trisomy of Hsa21 and APP duplication. The second study demonstrated that tau in neuron-derived small extracellular vesicles isolated from adults with AD-DS can induce altered tau biology in the brain of wild-type mice (Ledreux et al., 2021). Further such preclinical studies are required to understand how trisomy of Hsa21 affects both the development of, and response to, tau pathology.
Neuronal loss occurs during the development of AD pathology in people with DS and this has been modeled in some mouse models of DS. An age-dependent loss of basal forebrain cholinergic neurons (BFCN) in the medial septal nucleus (MSN), as well as loss of norepinephrine neurons in the locus coeruleus, occur in the brain of the Ts65Dn mouse model (Holtzman et al., 1996; Granholm et al., 2000; Hunter et al., 2004a; Seo and Isacson, 2005; Salehi et al., 2006, 2009; Lockrow et al., 2011b; Corrales et al., 2013). Sex differences in the BFCNs of the Ts65Dn model have been identified, with females showing a larger decrease in BFCN number than age-matched males (Kelley et al., 2014). Loss of BFCNs was associated with a compensatory effect in the 12-month Ts65Dn brain, whereby remaining hippocampal neurons had increased ChAT activity compared to younger mice of the same genotype (Seo and Isacson, 2005). The Ts2Cje model exhibits a loss of BFCNs in the MSN at 9-months of age (Jiang et al., 2016) and the Dp(16)1Yey model exhibits loss of BFCNs in the MSN, entorhinal cortex and locus coeruleus in an age-dependent manner, with significant loss seen at 16-months, but not 4-months (Lana-Elola et al., 2021).
Interruptions in nerve growth factor (NGF) transport have been implicated to underlie the loss of BFCNs in DS preclinical models. Intracerebroventricular NGF supplementation to BFCN cell bodies of the Ts65Dn mouse reversed cholinergic degeneration (Cooper et al., 2001), and the loss of BFCNs in the Ts65Dn mouse is associated with a decreased expression of TrkA, a high-affinity NGF receptor (Granholm et al., 2000). NGF is a neurotrophic factor, which acts via its receptors, to modulate the maintenance of neuronal populations, including BFCNs. NGF is produced in the hippocampus, where it is endocytosed by MSN BFCNs and then undergoes retrograde transport toward the cell body (Sofroniew et al., 2001). Hippocampal NGF levels are decreased in the aged Ts65Dn brain and NGF retrograde transport is highly disrupted in degenerating BFCNs, correlating with cognitive decline (Cooper et al., 2001; Bimonte-Nelson et al., 2003; Hunter et al., 2003b; Salehi et al., 2006). NGF dysregulation is also seen in people with DS and is thought to be involved in the loss of BFCNs (Iulita et al., 2014).
BFCN degeneration and dysregulation of NGF transport in DS is driven by three-copies of App. Ts1Cje mice, which have only two-copies of App, have no alteration to BFCN number (Sago et al., 1998) and have only a moderate impairment of NGF transport compared to Ts65Dn mice (Salehi et al., 2006). Normalization of the gene dose of App in both the Ts65Dn and Dp(16)1Yey mice rescued the loss of BFCNs, as well as partial rescue of NGF transport in the Ts65Dn model (Salehi et al., 2006; Lana-Elola et al., 2021). Furthermore, drug treatment of Ts65Dn primary cortical neurons with posiphen, to lower the production of FL-APP, rescued deficits in axonal retrograde transport in BFCNs, indicating that it is raised levels of APP or it's cleavage products which are responsible for these deficits (Chen et al., 2021).
Endosomal enlargement, or an increase in the number of neuronal endosomes, has been shown to occur in DS and in mouse models of DS (Botté et al., 2020), and may contribute to BFCN degeneration in an APP-dependant manner. Increased levels of APP cause endosomal enlargement and disrupt NGF trafficking in Ts65Dn BFCNs, which is partially rescued through normalization of the App gene-dose or reduction of APP through siRNA targeting (Cataldo et al., 2003; Xu et al., 2016). Increased β-CTF levels correlate with deficits in NGF transport in the Ts65Dn brain (Salehi et al., 2006), and β-CTF is thought to be associated with enlargement of endosomes in DS. Reducing β-CTF levels through reduction of BACE1 activity in the Ts2Cje mouse model prevents endosomal enlargement and the BFCN neurodegeneration in the MSN, hippocampus and cortex (Jiang et al., 2016), showing similar findings as in DS fibroblasts (Jiang et al., 2010; Kim et al., 2016). A recent transcriptomic study of Ts65Dn BFCNs, isolated prior to extensive degeneration, highlighted that alterations to oxidation and phosphorylation, muscarinic M2 cholinergic receptor abundance, and downstream targets of CREB signaling and synaptogenesis signaling, may contribute to degeneration of these cells (Alldred et al., 2021).
Having three-copies of APP in DS is necessary for the development of AD-associated cognitive decline, but whether this is due to increased Aβ production, loss of cholinergic neurons, another mechanism, or these working in synergy, is unclear. In addition to the role of extra APP in BFCN degeneration and NGF transport disruptions, estrogen and melatonin supplementation have both been shown to improve cognitive deficits and cholinergic neuron loss in the Ts65Dn brain, despite the melatonin treatment not reducing Aβ or APP levels (Granholm et al., 2002; Corrales et al., 2013), showing that multiple pathways may contribute to this aspect of AD-DS pathology. Cholinergic neuronal loss in DS may also be driven by neuroinflammation, as treatment of Ts65Dn mice with the anti-inflammatory compound minocycline or anti-IL17 prevented cholinergic degeneration in the MSN and stopped the microglial activation or raised cytokine abundance, respectively, seen in untreated mice (Hunter et al., 2004a; Rueda et al., 2018).
As well as APP, the Hsa21 gene DYRK1A has been implicated to play a role in neurodegeneration in AD-DS. Normalizing the gene-dose of Dyrk1a in the Ts65Dn mouse model of DS rescued cholinergic neuron degeneration and modulated the density of senescent cells in the cingulate cortex and hippocampus back to euploid levels (García-Cerro et al., 2017), however, how this affected cognitive decline was not studied. Normalizing the gene-dose of Dyrk1a in glutamatergic neurons of the Dp(16)1Yey brain improved long-term explicit memory (Brault et al., 2021); this may have been the result of the gene's important role in neurodevelopment and further studies are thus needed to investigate how three-copies of DYRK1A may impact cognitive decline associated with AD-DS.
Microglial and astrocyte activation, and raised levels of proinflammatory cytokines occur in the brain of people with DS compared to euploid matched individuals (Flores-Aguilar et al., 2020; Palmer et al., 2021). These changes progressively worsen with age and the neuroinflammatory profile is different in people with AD-DS than in euploid individuals who have LOAD (Wilcock et al., 2015; Martini et al., 2020). Neuroinflammation occurs in the Dp(16)1Yey model from around postnatal day 22; hippocampal microglia have larger cell bodies, decreased branching, increased expression of CD68, LAMP1, and IBA1 and engulf greater numbers of synapses than their wild-type counterparts (Pinto et al., 2020; Zheng et al., 2021). This microglial phenotype is associated with worsened cognition, loss of neuronal spines and decreased synaptic potential; phenotypes which were rescued upon microglial depletion or anti-inflammatory drug treatment (Pinto et al., 2020). Gene-dose normalization of App partially rescued decreased microglial branching in the Dp(16)1Yey model (Lana-Elola et al., 2021). Another Hsa21 gene, USP25, has been implicated to contribute to the worsened AD-DS neuroinflammation in the Dp(16)1Yey mouse, whereby overexpression of this gene in the 5XFAD mouse model of AD caused increase microglial activation, loss of neuronal spines and worsened cognition (Zheng et al., 2021); as previously mentioned, this may occur indirectly via an effect on APP processing (Zheng et al., 2022).
The Ts65Dn mouse model exhibits neuroinflammation in an aging-dependant manner. There is an increased density of CD45+ microglia in the hippocampus and basal forebrain of the 10- and 18-month Ts65Dn model (Hunter et al., 2004a; Lockrow et al., 2011a; Hamlett et al., 2020a). IBA1 upregulation in the 12-month hippocampus occurs (Rueda et al., 2018), as well as decreased expression of the homeostatic microglial marker, P2RY12, in the 15-month Ts65Dn (Illouz et al., 2019). Increases in proinflammatory cytokine levels, including IL-1β, IFNγ, IL-17, and GM-CSF, occur in the 12-month hippocampus (Ahmed et al., 2017; Rueda et al., 2018), and serum-levels of proinflammatory markers IL-1, TNFα, and IL-6 are also increased in the 10-month Ts65Dn (Hamlett et al., 2020a). Ts65Dn mice have increased levels of reactive oxygen species in the cortex, which is further associated with BFCN degeneration and memory deficits (Lockrow et al., 2009). Upregulation in genes functionally associated with oxidative stress and immune pathways are elevated in the hippocampus of the Ts1Cje mouse (Guedj et al., 2015). Intriguingly, an exaggerated microglia response occurs in the Ts65Dn brain in which BFCN degeneration has been induced compared with euploid controls, consistent with the hypothesis that trisomy of Hsa21 perturbs neuroinflammation (Hamlett et al., 2020b). The use of anti-inflammatory or anti-oxidant treatments in the Ts65Dn model, including treatment with minocycline, vitamin E, resolvin E, granulocyte-macrophage colony-stimulating factor or anti-IL-17, rescues some of these neuroinflammatory phenotypes, neurodegeneration and cognition (Hunter et al., 2004a; Lockrow et al., 2009; Rueda et al., 2018; Hamlett et al., 2020a; Ahmed et al., 2022), showing the important role that neuroinflammation may have in AD-DS phenotypes.
Astrogliosis occurs in DS and there is thought to be a greater number of astrocytes in the DS brain due to a gliogenic shift that takes place during neurodevelopment (Lu et al., 2011; Ponroy Bally and Murai, 2021). Astrocytes become reactive in neurodegenerative disease, including in AD, and this has been shown to be potentially neurotoxic (Liddelow et al., 2017). Astrocytes in the 15–19-month Ts65Dn hippocampus have increased expression of GFAP, S100β and C3, indicating they have taken on a reactive phenotype (Holtzman et al., 1996; Contestabile et al., 2006; Illouz et al., 2019). Immunization with anti-Aβ reduced the expression of C3 in this model, indicating that this astrogliosis is likely in response to raised Aβ (Illouz et al., 2019). Interestingly, although the key astrocytic gene S100β is in three-copies in people with DS, the Ts65Dn model has only 2-copies of this gene (Reeves et al., 1995), and thus the increased expression is due to astrocyte reactivity, rather than an extra gene dose.
Neuroinflammation appears to differ between DS and Aβ accumulation mouse models. Neuroinflammatory responses have been reported in some mouse models of amyloid accumulation at the later stages of progression when substantial misfolded protein has accumulated within the brain, particularly in models that produce human Aβ carrying the Arctic mutation (Saito et al., 2014; Castillo et al., 2017). In contrast, in the Ts65Dn and Dp(16)1Yey model, mouse Aβ, although raised, does not accumulate or deposit within the brain; likely because of differences in the processing of mouse and human APP and different aggregation tendencies of Aβ in the two species (Lv et al., 2013) but an early and significant neuroinflammatory phenotype has been reported. Side-by-side studies are required to verify the observation that despite lower abundance of aggregated Aβ, DS mouse models exhibit elevated neuroinflammation; suggesting that genes on Hsa21 other than APP are likely to contribute to the perturbed neuroinflammatory phenotype seen in people who have AD-DS.
Like people with DS, mouse models of DS exhibit alterations in the peripheral immune response, and neuroinflammation may be mediated by peripheral or systemic inflammation. The Ts65Dn model exhibits an altered immune response, with decreased numbers of peripheral immune cells and increased production of ROS (Lorenzo et al., 2013), and the Dp(16)1Yey mouse exhibits peripheral interferon hypersensitivity and increased proinflammatory cytokine production (Tuttle et al., 2020). Ts65Dn and Dp1Tyb mice also develop otitis media (Han et al., 2009; Lana-Elola et al., 2021), or inner ear inflammation, as do many people with DS (Maris et al., 2014); this inflammation may contribute to neuroinflammatory priming in the brain of this model.
In current mouse models of DS, excluding the Tc1 due to its mosaicism, all cells and tissue are trisomic, making it difficult to understand the role of peripheral vs. central effects. The size of the genetic changes in some of these models means that it is not possible to use standard techniques to address this issue, and novel approaches such as iPSC chimera and blastocyst complementation are required to create novel models to address these questions (Chang et al., 2018; Real et al., 2018). Furthermore, it is difficult to study how peripheral infection may contribute to neuroinflammation in mouse models, as modern animal housing conditions often ensure that animals are free of a defined range of pathogens and commensal organisms. Artificial systemic inflammation can be employed to address this issue but may cause adverse welfare outcomes and mortality in DS model systems (Tuttle et al., 2020). This is a potential future route of investigation for understanding the complexities of neuroinflammation in DS.
Although mouse models of DS readily model many facets of the syndrome, some phenotypes are subtle and hard to interrogate in vivo. Rodent derived primary cell and tissue cultures have been established which allow single cell-types or whole pieces of brain tissue to be studied and manipulated, and these can give great insights into molecular and cellular processes in DS and AD-DS, allowing mechanisms to be explored independent of the confound of peripheral systematic inflammation, for example.
Primary brain cell cultures isolated from animal models are useful for determining the cellular and molecular mechanisms of both health and disease. Primary neuronal cultures are a commonly used tool to investigate neurological disease processes and have been extensively used in AD research. Primary cortical and hippocampal neurons from the Ts65Dn model exhibit aspects of AD and DS neurobiology, including raised levels of APP and CTF production (Chen et al., 2021), enlarged endosomes and disrupted axonal retrograde transport (Xu et al., 2016; Chen et al., 2021). Cortical neurons of the Ts2Cje mouse also exhibit raised APP levels and lysosomal dysfunction (Jiang et al., 2019). Alterations in GABA signaling have been observed in Ts65Dn primary neurons, as have been seen in vivo (Best et al., 2007; Lysenko et al., 2018). Neuroinflammation contributes hugely to AD and DS, however, unlike in AD, few studies to date have used primary-cultured astrocytes or microglia from DS mouse models to investigate how these cell types may differ due to trisomy. Although single-cell cultures are able to replicate in vivo phenotypes of those seen in people with AD-DS, recent studies have shown that single-cultured brain cells can have altered transcriptomic and proteomic profiles compared to neurons, astrocytes and microglia co-cultures, which express a more in vivo-like profile (Delbridge et al., 2020; Baxter et al., 2021). Therefore, although primary cultures can allow the simplified study of cellular mechanisms of brain cells, careful interpretation of results is necessary, as single-cultured cells do not represent cellular interactions normally seen in the brain. Despite their limitations, there are many benefits to primary cultures, and they can particularly be used in AD-DS research to understand how trisomy disrupts cell-autonomous processes. Primary cultures offer benefits according to the 3R's principals of replacement, reduction and refinement as well. Use of primary cultures can reduce the number of rodents required for an experiment compared to in vivo studies, can reduce variability due to animal-to-animal variation, and allow for treatments with noxious stimuli, which would be invasive in vivo, and which may cause adverse welfare outcomes.
Although single-cell cultures can give insight into cellular mechanisms, in the brain, neurons, astrocytes, microglia, oligodendrocytes and other cell types are present in the parenchyma. To try to model these cell-cell interactions, and work with cells in a more complex environment, organotypic brain slice cultures (OBSC) have begun to be used in research for AD, other neurodegenerative diseases, and stroke (Li et al., 2016). OBSCs are an “ex vivo” model system involving the maintenance of thinly sliced brain sections in culture (Humpel, 2015). OBSCs maintain the cytoarchitecture seen in vivo and contain all major cell types present in the brain, with cell populations representative of that in vivo (Staal et al., 2011). OBSCs are representative of in vivo cell-cell interactions, are anatomically like the brain region of interest, and have functioning synapses (Croft et al., 2019). OBSCs also offer 3Rs benefits. Multiple slices prepared from the same animal can be used for different treatments, reducing the number of animals required for experimentation and reducing inter-animal variability (Durrant, 2020). Furthermore, as with primary cultures, slices can be easily treated with noxious stimuli through addition into culture media, rendering challenges not possible in vivo in DS models because of welfare and mortality to be investigated.
OBSCs have been used to study neurodegenerative diseases, including AD. The pathogenesis of Aβ and tau proteins have been studied in slice cultures from transgenic mouse models, elucidating early mechanisms in protein accumulation (Harwell and Coleman, 2016; Croft et al., 2017; Miller et al., 2021). OBSCs have also been used to investigate neuroinflammation and seem to be a better model than single-cultured microglia, as OBSC-microglia have greater transcriptional and phenotypic similarity to acutely isolated and in vivo microglia (Delbridge et al., 2020). Stimulation of OBSCs with LPS resulted in the loss of synaptophysin, showing active microglial phagocytosis in slices (Sheppard et al., 2019), as well as release of proinflammatory cytokines (Hellstrom et al., 2005). Furthermore, NLRP3 inflammasome activity can be induced in OBSCs (Hoyle et al., 2020), showing that microglia can respond to stimuli similarly as seen in vivo and that OBSCs offer a useful model to study neuroinflammation in mouse models of neurological disease. OBSCs have been prepared from the brain of the Ts65Dn mouse model to examine synaptic connectivity of hippocampal CA3 neurons (Hanson et al., 2007). Alterations to excitatory and inhibitory synaptic function were identified, similar to what has been seen in vivo in this model (Siarey et al., 1997), and in people with DS (Contestabile et al., 2017). This study shows that OBSCs from DS models can recapitulate in vivo phenotypes and offer a model system for the future study of AD-DS mechanisms.
AD-DS has a unique and important place in AD research. The genetic cause of the increased risk of disease is fully established, a large number of individuals with the condition occur in the population, and their lifetime risk of AD is extremely high (Fortea et al., 2021). AD primary prevention trials in people who have DS are feasible, can be targeted at the earliest phase of disease, and thus, have high chances of success (Strydom et al., 2018). However, given the different biology of individuals who have DS, drug selection for these studies needs careful consideration to ensure both safety, and best chance of trial success. Current rodent models of DS have excellent construct validity and exhibit a range of AD relevant phenotypes. Notably, elegant work in preclinical models has already indicated that careful thought is required regarding the use of γ-secretase modulators in people who have DS (Lana-Elola et al., 2021), that bexarotene may clear Aβ effectively but also impairs cognitive performance in a DS preclinical model (Vidal et al., 2021), and that different vaccination protocols for anti-Aβ studies (Illouz et al., 2019) may be required in the context of trisomy of Hsa21.
Moreover, in recent years significant progress has been made to understand the differences and similarities that occur between AD-DS and other forms of AD and have highlighted that disease onset and progression is modulated by many processes. An impressive preclinical toolkit is available to unravel the complex multigenic-multi-cellular interactions that underpin this biology. Novel approaches including humanization of critical genes in DS rodent models, such as App, and iPSC-mouse chimeras are important to further our understanding of the development of neuropathology, the transition from pathology to disease and the efficacy and safety of potential treatments, in the context of an additional copy of chromosome 21. Future research using the next-generation of AD-DS rodent models will provide significant insight into which strategies have the best chance of preventing or delaying the development of AD in people who have DS.
All authors drafted sections of the manuscript, reviewed, edited the final draft, and read and approved the final manuscript.
FW and PM were supported by the UK Dementia Research Institute which receives its funding from DRI Ltd., funded by the UK Medical Research Council, Alzheimer's Society and Alzheimer's Research UK (UKDRI-1014), and FW also holds an Alzheimer's Research UK Senior Research Fellowship (ARUK-SRF2018A-001). CF was funded by an Alzheimer's Society PhD Studentship Awarded (AS-PhD-19a-007) to FW. FW also received funding that contributed to this paper from the MRC via CoEN award MR/S005145/1.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Ahmed, M. d. M., Block, A., Tong, S., Davisson, M. T., and Gardiner, K.J. (2017). Age exacerbates abnormal protein expression in a mouse model of Down syndrome. Neurobiol. Aging 57, 120–132. doi: 10.1016/j.neurobiolaging.2017.05.002
Ahmed, M. d. M., Dhanasekaran, A. R., Tong, S., Wiseman, F. K., Fisher, E. M. C., Tybulewicz, V. L. J., et al. (2013). Protein profiles in Tc1 mice implicate novel pathway perturbations in the Down syndrome brain. Hum. Mol. Genet. 22, 1709–1724. doi: 10.1093/hmg/ddt017
Ahmed, M. M., Wang, A. C.-J., Elos, M., Chial, H. J., Sillau, S., Solano, D. A., et al. (2022). The innate immune system stimulating cytokine GM-CSF improves learning/memory and interneuron and astrocyte brain pathology in Dp16 Down syndrome mice and improves learning/memory in wild-type mice. Neurobiol. Dis. 168, 105694. doi: 10.1016/j.nbd.2022.105694
Alldred, M. J., Penikalapati, S. C., Lee, S. H., Heguy, A., Roussos, P., and Ginsberg, S. D. (2021). Profiling basal forebrain cholinergic neurons reveals a molecular basis for vulnerability within the Ts65Dn model of down syndrome and Alzheimer's disease. Mol. Neurobiol. 58, 5141–5162. doi: 10.1007/s12035-021-02453-3
Altuna, M., Giménez, S., and Fortea, J. (2021). Epilepsy in down syndrome: a highly prevalent comorbidity. J. Clin. Med. 10, 2776. doi: 10.3390/jcm10132776
Anderson-Mooney, A. J., Schmitt, F. A., Head, E., Lott, I. T., and Heilman, K. M. (2016). Gait dyspraxia as a clinical marker of cognitive decline in down syndrome: a review of theory and proposed mechanisms. Brain Cogn. 104, 48–57. doi: 10.1016/j.bandc.2016.02.007
Annus, T., Wilson, L. R., Hong, Y. T., Acosta–Cabronero, J., Fryer, T. D., Cardenas–Blanco, A., et al. (2016). The pattern of amyloid accumulation in the brains of adults with Down syndrome. Alzheimers Dement. 12, 538–545. doi: 10.1016/j.jalz.2015.07.490
Antonarakis, S. E., Skotko, B. G., Rafii, M. S., Strydom, A., Pape, S. E., Bianchi, D. W., et al. (2020). Down syndrome. Nat. Rev. Dis. Primer 6, 9. doi: 10.1038/s41572-019-0143-7
Armstrong, R. A (2012). Size frequency distributions of β-amyloid (Aβ) deposits:a comparative study of four neurodegenerative disorders. Folia Neuropathol. 50, 240–249. doi: 10.5114/fn.2012.30524
Baglietto-Vargas, D., Forner, S., Cai, L., Martini, A. C., Trujillo-Estrada, L., Swarup, V., et al. (2021). Generation of a humanized Aβ expressing mouse demonstrating aspects of Alzheimer's disease-like pathology. Nat. Commun. 12, 2421. doi: 10.1038/s41467-021-22624-z
Ball, S. L., Holland, A. J., Treppner, P., Watson, P. C., and Huppert, F. A. (2008). Executive dysfunction and its association with personality and behaviour changes in the development of Alzheimer's disease in adults with Down syndrome and mild to moderate learning disabilities. Br. J. Clin. Psychol. 47, 1–29. doi: 10.1348/014466507X230967
Baxter, P. S., Dando, O., Emelianova, K., He, X., McKay, S., Hardingham, G. E., et al. (2021). Microglial identity and inflammatory responses are controlled by the combined effects of neurons and astrocytes. Cell Rep. 34, 108882. doi: 10.1016/j.celrep.2021.108882
Bejanin, A., Iulita, M. F., Vilaplana, E., Carmona-Iragui, M., Benejam, B., Videla, L., et al. (2021). Association of apolipoprotein E ε4 allele with clinical and multimodal biomarker changes of Alzheimer disease in adults with down syndrome. JAMA Neurol. 78, 937–947. doi: 10.1001/jamaneurol.2021.1893
Belichenko, P. V., Madani, R., Rey-Bellet, L., Pihlgren, M., Becker, A., Plassard, A., et al. (2016). An anti-β-amyloid vaccine for treating cognitive deficits in a mouse model of down syndrome. PLoS ONE 11, e0152471. doi: 10.1371/journal.pone.0152471
Best, T. K., Siarey, R. J., and Galdzicki, Z. (2007). Ts65Dn, a mouse model of down syndrome, exhibits increased GABAB-induced potassium current. J. Neurophysiol. 97, 892–900. doi: 10.1152/jn.00626.2006
Betthauser, T. J., Koscik, R. L., Jonaitis, E. M., Allison, S. L., Cody, K. A., Erickson, C. M., et al. (2020). Amyloid and tau imaging biomarkers explain cognitive decline from late middle-age. Brain 143, 320–335. doi: 10.1093/brain/awz378
Bimonte-Nelson, H. A., Hunter, C. L., Nelson, M. E., and Granholm, A.-C. E. (2003). Frontal cortex BDNF levels correlate with working memory in an animal model of Down syndrome. Behav. Brain Res. 139, 47–57. doi: 10.1016/S0166-4328(02)00082-7
Bodhireddy, S., Dickson, D. W., Mattiace, L., and Weidenheim, K. M. (1994). A case of Down's syndrome with diffuse Lewy body disease and Alzheimer's disease. Neurology 44, 159–159. doi: 10.1212/WNL.44.1.159
Botté, A., Lainé, J., Xicota, L., Heiligenstein, X., Fontaine, G., Kasri, A., et al. (2020). Ultrastructural and dynamic studies of the endosomal compartment in Down syndrome. Acta Neuropathol. Commun. 8, 89. doi: 10.1186/s40478-020-00956-z
Braak, H., Alafuzoff, I., Arzberger, T., Kretzschmar, H., and Del Tredici, K. (2006). Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol. (Berl.) 112, 389–404. doi: 10.1007/s00401-006-0127-z
Braak, H., and Braak, E. (1991). Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. (Berl.) 82, 239–259. doi: 10.1007/BF00308809
Brault, V., Nguyen, T. L., Flores-Gutiérrez, J., Iacono, G., Birling, M.-C., Lalanne, V., et al. (2021). Dyrk1a gene dosage in glutamatergic neurons has key effects in cognitive deficits observed in mouse models of MRD7 and Down syndrome. PLoS Genet. 17, e1009777. doi: 10.1371/journal.pgen.1009777
Bult C. J. Blake J. A. Smith C. L. Kadin J. A. Richardson J. E. The Mouse Genome Database Group (2019). Mouse genome database (MGD) 2019. Nucleic Acids Res. 47, D801–D806. doi: 10.1093/nar/gky1056
Casanova, M. F., Walker, L. C., Whitehouse, P. J., and Price, D. L. (1985). Abnormalities of the nucleus basalis in Down's syndrome. Ann. Neurol. 18, 310–313. doi: 10.1002/ana.410180306
Castillo, E., Leon, J., Mazzei, G., Abolhassani, N., Haruyama, N., Saito, T., et al. (2017). Comparative profiling of cortical gene expression in Alzheimer's disease patients and mouse models demonstrates a link between amyloidosis and neuroinflammation. Sci. Rep. 7, 17762. doi: 10.1038/s41598-017-17999-3
Cataldo, A. M., Petanceska, S., Peterhoff, C. M., Terio, N. B., Epstein, C. J., Villar, A., et al. (2003). App gene dosage modulates endosomal abnormalities of Alzheimer's disease in a segmental trisomy 16 mouse model of down syndrome. J. Neurosci. 23, 6788–6792. doi: 10.1523/JNEUROSCI.23-17-06788.2003
Cataldo, A. M., Peterhoff, C. M., Troncoso, J. C., Gomez-Isla, T., Hyman, B. T., and Nixon, R. A. (2000). Endocytic pathway abnormalities precede amyloid β deposition in sporadic Alzheimer's disease and Down syndrome: differential effects of APOE genotype and presenilin mutations. Am. J. Pathol. 157, 277–286. doi: 10.1016/s0002-9440(10)64538-5
Chang, A. N., Liang, Z., Dai, H.-Q., Chapdelaine-Williams, A. M., Andrews, N., Bronson, R. T., et al. (2018). Neural blastocyst complementation enables mouse forebrain organogenesis. Nature 563, 126–130. doi: 10.1038/s41586-018-0586-0
Chaves, J. C. S., Machado, F. T., Almeida, M. F., Bacovsky, T. B., and Ferrari, M. F. R. (2020). microRNAs expression correlates with levels of APP, DYRK1A, hyperphosphorylated Tau and BDNF in the hippocampus of a mouse model for Down syndrome during ageing. Neurosci. Lett. 714, 134541. doi: 10.1016/j.neulet.2019.134541
Chen, G., Xu, T., Yan, Y., Zhou, Y., Jiang, Y., Melcher, K., et al. (2017). Amyloid beta: structure, biology and structure-based therapeutic development. Acta Pharmacol. Sin. 38, 1205–1235. doi: 10.1038/aps.2017.28
Chen, X.-Q., Salehi, A., Pearn, M. L., Overk, C., Nguyen, P. D., Kleschevnikov, A. M., et al. (2021). Targeting increased levels of APP in Down syndrome: posiphen-mediated reductions in APP and its products reverse endosomal phenotypes in the Ts65Dn mouse model. Alzheimers Dement. 17, 271–292. doi: 10.1002/alz.12185
Cheng, I. H., Scearce-Levie, K., Legleiter, J., Palop, J. J., Gerstein, H., Bien-Ly, N., et al. (2007). Accelerating amyloid-beta fibrillization reduces oligomer levels and functional deficits in Alzheimer disease mouse models. J. Biol. Chem. 282, 23818–23828. doi: 10.1074/jbc.M701078200
Cheon, M. S., Dierssen, M., Kim, S. H., and Lubec, G. (2008). Protein expression of BACE1, BACE2 and APP in down syndrome brains. Amino Acids 35, 339–343. doi: 10.1007/s00726-007-0618-9
Choi, J. H. K., Berger, J. D., Mazzella, M. J., Morales-Corraliza, J., Cataldo, A. M., Nixon, R. A., et al. (2009). Age-dependent dysregulation of brain amyloid precursor protein in the Ts65Dn Down syndrome mouse model. J. Neurochem. 110, 1818–1827. doi: 10.1111/j.1471-4159.2009.06277.x
Citron, M., Oltersdorf, T., Haass, C., McConlogue, L., Hung, A. Y., Seubert, P., et al. (1992). Mutation of the beta-amyloid precursor protein in familial Alzheimer's disease increases beta-protein production. Nature 360, 672–674. doi: 10.1038/360672a0
Contestabile, A., Fila, T., Bartesaghi, R., Contestabile, A., and Ciani, E. (2006). Choline acetyltransferase activity at different ages in brain of Ts65Dn mice, an animal model for Down's syndrome and related neurodegenerative diseases. J. Neurochem. 97, 515–526. doi: 10.1111/j.1471-4159.2006.03769.x
Contestabile, A., Magara, S., and Cancedda, L. (2017). The GABAergic hypothesis for cognitive disabilities in down syndrome. Front. Cell. Neurosci. 11, 54. doi: 10.3389/fncel.2017.00054
Cooper, J. D., Salehi, A., Delcroix, J. D., Howe, C. L., Belichenko, P. V., Chua-Couzens, J., et al. (2001). Failed retrograde transport of NGF in a mouse model of Down's syndrome: reversal of cholinergic neurodegenerative phenotypes following NGF infusion. Proc. Natl. Acad. Sci. U. S. A. 98, 10439–10444. doi: 10.1073/pnas.181219298
Coppus, A., Evenhuis, H., Verberne, G.-J., Visser, F., van Gool, P., Eikelenboom, P., et al. (2006). Dementia and mortality in persons with Down's syndrome. J. Intellect. Disabil. Res. JIDR 50, 768–777. doi: 10.1111/j.1365-2788.2006.00842.x
Coppus, A. M. W., Evenhuis, H. M., Verberne, G.-J., Visser, F. E., Eikelenboom, P., van Gool, W. A., et al. (2010). Early age at menopause is associated with increased risk of dementia and mortality in women with Down syndrome. J. Alzheimers Dis. JAD 19, 545–550. doi: 10.3233/JAD-2010-1247
Corrales, A., Martínez, P., García, S., Vidal, V., García, E., Flórez, J., et al. (2013). Long-term oral administration of melatonin improves spatial learning and memory and protects against cholinergic degeneration in middle-aged Ts65Dn mice, a model of Down syndrome. J. Pineal Res. 54, 346–358. doi: 10.1111/jpi.12037
Cosgrave, M. P., Tyrrell, J., McCarron, M., Gill, M., and Lawlor, B. A. (1999). Age at onset of dementia and age of menopause in women with Down's syndrome. J. Intellect. Disabil. Res. JIDR 43, 461–465. doi: 10.1046/j.1365-2788.1999.00192.x
Cramer, P. E., Cirrito, J. R., Wesson, D. W., Lee, C. Y. D., Karlo, J. C., Zinn, A. E., et al. (2012). ApoE-directed therapeutics rapidly clear β-amyloid and reverse deficits in AD mouse models. Science 335, 1503–1506. doi: 10.1126/science.1217697
Croft, C. L., Futch, H. S., Moore, B. D., and Golde, T. E. (2019). Organotypic brain slice cultures to model neurodegenerative proteinopathies. Mol. Neurodegener. 14, 45. doi: 10.1186/s13024-019-0346-0
Croft, C. L., Wade, M. A., Kurbatskaya, K., Mastrandreas, P., Hughes, M. M., Phillips, E. C., et al. (2017). Membrane association and release of wild-type and pathological tau from organotypic brain slice cultures. Cell Death Dis. 8, e2671. doi: 10.1038/cddis.2017.97
Cunningham, C., Campion, S., Lunnon, K., Murray, C. L., Woods, J. F. C., Deacon, R. M. J., et al. (2009). Systemic inflammation induces acute behavioral and cognitive changes and accelerates neurodegenerative disease. Biol. Psychiatry 65, 304–312. doi: 10.1016/j.biopsych.2008.07.024
Das, U., Wang, L., Ganguly, A., Saikia, J. M., Wagner, S. L., Koo, E. H., et al. (2016). Visualization of APP and BACE-1 approximation in neurons: new insights into the amyloidogenic pathway. Nat. Neurosci. 19, 55–64. doi: 10.1038/nn.4188
Davidson, Y. S., Raby, S., Foulds, P. G., Robinson, A., Thompson, J. C., Sikkink, S., et al. (2011). TDP-43 pathological changes in early onset familial and sporadic Alzheimer's disease, late onset Alzheimer's disease and Down's Syndrome: association with age, hippocampal sclerosis and clinical phenotype. Acta Neuropathol. (Berl.) 122, 703–713. doi: 10.1007/s00401-011-0879-y
Davidson, Y. S., Robinson, A., Prasher, V. P., and Mann, D. M. A. (2018). The age of onset and evolution of Braak tangle stage and Thal amyloid pathology of Alzheimer's disease in individuals with Down syndrome. Acta Neuropathol. Commun. 6, 56. doi: 10.1186/s40478-018-0559-4
Davisson, M. T., Schmidt, C., and Akeson, E. C. (1990). Segmental trisomy of murine chromosome 16: a new model system for studying Down syndrome. Prog. Clin. Biol. Res. 360, 263–280.
de Graaf, G., Buckley, F., and Skotko, B. G. (2017). Estimation of the number of people with Down syndrome in the United States. Genet. Med. 19, 439–447. doi: 10.1038/gim.2016.127
De Strooper, B., and Karran, E. (2016). The cellular phase of Alzheimer's disease. Cell 164, 603–615. doi: 10.1016/j.cell.2015.12.056
Deb, S., Braganza, J., Norton, N., Williams, H., Kehoe, P. G., Williams, J., et al. (2000). APOE epsilon 4 influences the manifestation of Alzheimer's disease in adults with Down's syndrome. Br. J. Psychiatry J. Ment. Sci. 176, 468–472. doi: 10.1192/bjp.176.5.468
Delbridge, A. R. D., Huh, D., Brickelmaier, M., Burns, J. C., Roberts, C., Challa, R., et al. (2020). Organotypic brain slice culture microglia exhibit molecular similarity to acutely-isolated adult microglia and provide a platform to study neuroinflammation. Front. Cell. Neurosci. 14, 444. doi: 10.3389/fncel.2020.592005
Devenny, D. A., Zimmerli, E. J., Kittler, P., and Krinsky-McHale, S. J. (2002). Cued recall in early-stage dementia in adults with Down's syndrome. J. Intellect. Disabil. Res. JIDR 46, 472–483. doi: 10.1046/j.1365-2788.2002.00417.x
Di Domenico, F., Tramutola, A., Barone, E., Lanzillotta, C., Defever, O., Arena, A., et al. (2019). Restoration of aberrant mTOR signaling by intranasal rapamycin reduces oxidative damage: focus on HNE-modified proteins in a mouse model of down syndrome. Redox Biol. 23, 101162. doi: 10.1016/j.redox.2019.101162
Doran, E., Keator, D., Head, E., Phelan, M. J., Kim, R., Totoiu, M., et al. (2017). Down syndrome, partial trisomy 21, and absence of Alzheimer's disease: the role of APP. J. Alzheimers Dis. 56, 459–470. doi: 10.3233/JAD-160836
Dorard, E., Gorisse-Hussonnois, L., Guihenneuc-Jouyaux, C., Albac, C., Potier, M.-C., and Allinquant, B. (2016). Increases of SET level and translocation are correlated with tau hyperphosphorylation at ser202/thr205 in CA1 of Ts65Dn mice. Neurobiol. Aging 46, 43–48. doi: 10.1016/j.neurobiolaging.2016.06.010
Durrant, C. S (2020). “Preparation of organotypic hippocampal slice cultures for the study of CNS disease and damage, in: Axon Degeneration: Methods and Protocols, Methods in Molecular Biology, ed Babetto, E. (New York, NY: Springer US), 133–144.
Egensperger, R., Weggen, S., Ida, N., Multhaup, G., Schnabel, R., Beyreuther, K., et al. (1999). Reverse relationship between β-amyloid precursor protein and β-amyloid peptide plaques in Down's syndrome versus sporadic/familial Alzheimer's disease. Acta Neuropathol. (Berl.) 97, 113–118. doi: 10.1007/s004010050963
Fagan, A. M., Henson, R. L., Li, Y., Boerwinkle, A. H., Xiong, C., Bateman, R. J., et al. (2021). Comparison of CSF biomarkers in Down syndrome and autosomal dominant Alzheimer's disease: a cross-sectional study. Lancet Neurol. 20, 615–626. doi: 10.1016/S1474-4422(21)00139-3
Ferreira, R., Michel, R. C., Greghi, S. L. A., Resende, M. L. R., de Sant'Ana, A. C. P., Damante, C. A., et al. (2016). Prevention and periodontal treatment in down syndrome patients: a systematic review. PLoS ONE 11, e0158339. doi: 10.1371/journal.pone.0158339
Filippone, A., and Praticò, D. (2021). Endosome dysregulation in down syndrome: a potential contributor to Alzheimer disease pathology. Ann. Neurol. 90, 4–14. doi: 10.1002/ana.26042
Flores-Aguilar, L., Iulita, M. F., Kovecses, O., Torres, M. D., Levi, S. M., Zhang, Y., et al. (2020). Evolution of neuroinflammation across the lifespan of individuals with Down syndrome. Brain 143, 3653–3671. doi: 10.1093/brain/awaa326
Fortea, J., Vilaplana, E., Carmona-Iragui, M., Benejam, B., Videla, L., Barroeta, I., et al. (2020). Clinical and biomarker changes of Alzheimer's disease in adults with Down syndrome: a cross-sectional study. Lancet Lond. Engl. 395, 1988–1997. doi: 10.1016/S0140-6736(20)30689-9
Fortea, J., Zaman, S. H., Hartley, S., Rafii, M. S., Head, E., and Carmona-Iragui, M. (2021). Alzheimer's disease associated with Down syndrome: a genetic form of dementia. Lancet Neurol. 20, 930–942. doi: 10.1016/S1474-4422(21)00245-3
García-Cerro, S., Rueda, N., Vidal, V., Lantigua, S., and Martínez-Cué, C. (2017). Normalizing the gene dosage of Dyrk1A in a mouse model of Down syndrome rescues several Alzheimer's disease phenotypes. Neurobiol. Dis. 106, 76–88. doi: 10.1016/j.nbd.2017.06.010
Ghetti, B., Oblak, A. L., Boeve, B. F., Johnson, K. A., Dickerson, B. C., and Goedert, M. (2015). Invited review: frontotemporal dementia caused by microtubule-associated protein tau gene (MAPT) mutations: a chameleon for neuropathology and neuroimaging. Neuropathol. Appl. Neurobiol. 41, 24–46. doi: 10.1111/nan.12213
Gibb, W. R., Mountjoy, C. Q., Mann, D. M., and Lees, A. J. (1989). A pathological study of the association between Lewy body disease and Alzheimer's disease. J. Neurol. Neurosurg. Psychiatry 52, 701–708. doi: 10.1136/jnnp.52.6.701
Glasson, E. J., Jacques, A., Wong, K., Bourke, J., and Leonard, H. (2016). Improved survival in down syndrome over the last 60 years and the impact of perinatal factors in recent decades. J. Pediatr. 169, 214–220.e1. doi: 10.1016/j.jpeds.2015.10.083
Goldacre, M. J., Wotton, C. J., Seagroatt, V., and Yeates, D. (2004). Cancers and immune related diseases associated with Down's syndrome: a record linkage study. Arch. Dis. Child. 89, 1014–1017. doi: 10.1136/adc.2003.046219
Goodwin, L. O., Splinter, E., Davis, T. L., Urban, R., He, H., Braun, R. E., et al. (2019). Large-scale discovery of mouse transgenic integration sites reveals frequent structural variation and insertional mutagenesis. Genome Res. 29, 494–505. doi: 10.1101/gr.233866.117
Götz, J., Bodea, L.-G., and Goedert, M. (2018). Rodent models for Alzheimer disease. Nat. Rev. Neurosci. 19, 583–598. doi: 10.1038/s41583-018-0054-8
Granholm, A.-C. E., Ford, K. A., Hyde, L. A., Bimonte, H. A., Hunter, C. L., Nelson, M., et al. (2002). Estrogen restores cognition and cholinergic phenotype in an animal model of Down syndrome. Physiol. Behav. 77, 371–385. doi: 10.1016/S0031-9384(02)00884-3
Granholm, A.-C. E., Sanders, L. A., and Crnic, L. S. (2000). Loss of cholinergic phenotype in basal forebrain coincides with cognitive decline in a mouse model of down's syndrome. Exp. Neurol. 161, 647–663. doi: 10.1006/exnr.1999.7289
Granno, S., Nixon-Abell, J., Berwick, D. C., Tosh, J., Heaton, G., Almudimeegh, S., et al. (2019). Downregulated Wnt/β-catenin signalling in the Down syndrome hippocampus. Sci. Rep. 9, 7322. doi: 10.1038/s41598-019-43820-4
Gribble, S. M., Wiseman, F. K., Clayton, S., Prigmore, E., Langley, E., Yang, F., et al. (2013). Massively parallel sequencing reveals the complex structure of an irradiated human chromosome on a mouse background in the Tc1 model of down syndrome. PLoS ONE 8, e60482. doi: 10.1371/journal.pone.0060482
Guedj, F., Pennings, J. L. A., Wick, H. C., and Bianchi, D. W. (2015). Analysis of adult cerebral cortex and hippocampus transcriptomes reveals unique molecular changes in the Ts1Cje mouse model of down syndrome. Brain Pathol. 25, 11–23. doi: 10.1111/bpa.12151
Guerreiro, R. J., Baquero, M., Blesa, R., Boada, M., Brás, J. M., Bullido, M. J., et al. (2010). Genetic screening of Alzheimer's disease genes in Iberian and African samples yields novel mutations in presenilins and APP. Neurobiol. Aging 31, 725–731. doi: 10.1016/j.neurobiolaging.2008.06.012
Gyure, K. A., Durham, R., Stewart, W. F., Smialek, J. E., and Troncoso, J. C. (2001). Intraneuronal Aβ-amyloid precedes development of amyloid plaques in down syndrome. Arch. Pathol. Lab. Med. 125, 489–492. doi: 10.5858/2001-125-0489-IAAPDO
Hamlett, E. D., Hjorth, E., Ledreux, A., Gilmore, A., Schultzberg, M., and Granholm, A. C. (2020a). RvE1 treatment prevents memory loss and neuroinflammation in the Ts65Dn mouse model of Down syndrome. Glia 68, 1347–1360. doi: 10.1002/glia.23779
Hamlett, E. D., Ledreux, A., Gilmore, A., Vazey, E. M., Aston-Jones, G., Boger, H. A., et al. (2020b). Inhibitory designer receptors aggravate memory loss in a mouse model of down syndrome. Neurobiol. Dis. 134, 104616. doi: 10.1016/j.nbd.2019.104616
Han, F., Yu, H., Zhang, J., Tian, C., Schmidt, C., Nava, C., et al. (2009). Otitis media in a mouse model for Down syndrome. Int. J. Exp. Pathol. 90, 480–488. doi: 10.1111/j.1365-2613.2009.00677.x
Handen, B. L., Cohen, A. D., Channamalappa, U., Bulova, P., Cannon, S. A., Cohen, W. I., et al. (2012). Imaging brain amyloid in nondemented young adults with Down syndrome using Pittsburgh compound B. Alzheimers dement. J. Alzheimers Assoc. 8, 496–501. doi: 10.1016/j.jalz.2011.09.229
Hanseeuw, B. J., Betensky, R. A., Mormino, E. C., Schultz, A. P., Sepulcre, J., Becker, J. A., et al. (2018). PET staging of amyloidosis using striatum. Alzheimers dement. J. Alzheimers Assoc. 14, 1281–1292. doi: 10.1016/j.jalz.2018.04.011
Hanson, J. E., Blank, M., Valenzuela, R. A., Garner, C. C., and Madison, D. V. (2007). The functional nature of synaptic circuitry is altered in area CA3 of the hippocampus in a mouse model of Down's syndrome. J. Physiol. 579, 53–67. doi: 10.1113/jphysiol.2006.114868
Hardy, J. A., and Higgins, G. A. (1992). Alzheimer's disease: the amyloid cascade hypothesis. Science 256, 184–185. doi: 10.1126/science.1566067
Harwell, C. S., and Coleman, M. P. (2016). Synaptophysin depletion and intraneuronal Aβ in organotypic hippocampal slice cultures from huAPP transgenic mice. Mol. Neurodegener. 11, 44. doi: 10.1186/s13024-016-0110-7
Hashimoto, S., Matsuba, Y., Kamano, N., Mihira, N., Sahara, N., Takano, J., et al. (2019). Tau binding protein CAPON induces tau aggregation and neurodegeneration. Nat. Commun. 10, 2394. doi: 10.1038/s41467-019-10278-x
Head, E., Azizeh, B. Y., Lott, I. T., Tenner, A. J., Cotman, C. W., and Cribbs, D. H. (2001). Complement association with neurons and β-amyloid deposition in the brains of aged individuals with down syndrome. Neurobiol. Dis. 8, 252–265. doi: 10.1006/nbdi.2000.0380
Head, E., Phelan, M. J., Doran, E., Kim, R. C., Poon, W. W., Schmitt, F. A., et al. (2017). Cerebrovascular pathology in Down syndrome and Alzheimer disease. Acta Neuropathol. Commun. 5, 93. doi: 10.1186/s40478-017-0499-4
Hellstrom, I. C., Danik, M., Luheshi, G. N., and Williams, S. (2005). Chronic LPS exposure produces changes in intrinsic membrane properties and a sustained IL-β-dependent increase in GABAergic inhibition in hippocampal CA1 pyramidal neurons. Hippocampus 15, 656–664. doi: 10.1002/hipo.20086
Helman, A. M., Siever, M., McCarty, K. L., Lott, I. T., Doran, E., Abner, E. L., et al. (2019). Microbleeds and cerebral amyloid angiopathy in the brains of people with down syndrome with Alzheimer's disease. J. Alzheimers Dis. 67, 103–112. doi: 10.3233/JAD-180589
Heneka, M. T., Carson, M. J., El Khoury, J., Landreth, G. E., Brosseron, F., Feinstein, D. L., et al. (2015). Neuroinflammation in Alzheimer's disease. Lancet Neurol. 14, 388–405. doi: 10.1016/S1474-4422(15)70016-5
Herault, Y., Delabar, J. M., Fisher, E. M. C., Tybulewicz, V. L. J., Yu, E., and Brault, V. (2017). Rodent models in Down syndrome research: impact and future opportunities. Dis. Model. Mech. 10, 1165–1186. doi: 10.1242/dmm.029728
Hirayama, A., Horikoshi, Y., Maeda, M., Ito, M., and Takashima, S. (2003). Characteristic developmental expression of amyloid β40, 42 and 43 in patients with Down syndrome. Brain Dev. 25, 180–185. doi: 10.1016/S0387-7604(02)00209-7
Hithersay, R., Hamburg, S., Knight, B., and Strydom, A. (2017). Cognitive decline and dementia in Down syndrome. Curr. Opin. Psychiatry 30, 102–107. doi: 10.1097/YCO.0000000000000307
Hithersay, R., Startin, C. M., Hamburg, S., Mok, K. Y., Hardy, J., Fisher, E. M. C., et al. (2019). Association of dementia with mortality among adults with down syndrome older than 35 years. JAMA Neurol. 76, 152–160. doi: 10.1001/jamaneurol.2018.3616
Holtzman, D. M., Santucci, D., Kilbridge, J., Chua-Couzens, J., Fontana, D. J., Daniels, S. E., et al. (1996). Developmental abnormalities and age-related neurodegeneration in a mouse model of Down syndrome. Proc. Natl. Acad. Sci. 93, 13333–13338. doi: 10.1073/pnas.93.23.13333
Hong, S., Beja-Glasser, V. F., Nfonoyim, B. M., Frouin, A., Li, S., Ramakrishnan, S., et al. (2016). Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 352, 712–716. doi: 10.1126/science.aad8373
Hooli, B. V., Mohapatra, G., Mattheisen, M., Parrado, A. R., Roehr, J. T., Shen, Y., et al. (2012). Role of common and rare APP DNA sequence variants in Alzheimer disease. Neurology 78, 1250–1257. doi: 10.1212/WNL.0b013e3182515972
Hoyle, C., Redondo-Castro, E., Cook, J., Tzeng, T.-C., Allan, S. M., Brough, D., et al. (2020). Hallmarks of NLRP3 inflammasome activation are observed in organotypic hippocampal slice culture. Immunology 161, 39–52. doi: 10.1111/imm.13221
Hu, N.-W., Corbett, G. T., Moore, S., Klyubin, I., O'Malley, T. T., Walsh, D. M., et al. (2018). Extracellular forms of Aβ and tau from iPSC models of Alzheimer's disease disrupt synaptic plasticity. Cell Rep. 23, 1932–1938. doi: 10.1016/j.celrep.2018.04.040
Huggard, D., Doherty, D. G., and Molloy, E. J. (2020). Immune dysregulation in children with down syndrome. Front. Pediatr. 8, 73. doi: 10.3389/fped.2020.00073
Hüls, A., Costa, A. C. S., Dierssen, M., Baksh, R. A., Bargagna, S., Baumer, N. T., et al. (2021). Medical vulnerability of individuals with Down syndrome to severe COVID-19–data from the Trisomy 21 Research Society and the UK ISARIC4C survey. EClinicalMedicine 33, 100769. doi: 10.1016/j.eclinm.2021.100769
Humpel, C (2015). Organotypic brain slice cultures: a review. Neuroscience 305, 86–98. doi: 10.1016/j.neuroscience.2015.07.086
Hunter, C. L., Bachman, D., and Granholm, A.-C. (2004a). Minocycline prevents cholinergic loss in a mouse model of Down's syndrome. Ann. Neurol. 56, 675–688. doi: 10.1002/ana.20250
Hunter, C. L., Bimonte, H. A., and Granholm, A.-C. E. (2003a). Behavioral comparison of 4 and 6 month-old Ts65Dn mice: age-related impairments in working and reference memory. Behav. Brain Res. 138, 121–131. doi: 10.1016/S0166-4328(02)00275-9
Hunter, C. L., Bimonte-Nelson, H. A., Nelson, M., Eckman, C. B., and Granholm, A.-C. (2004b). Behavioral and neurobiological markers of Alzheimer's disease in Ts65Dn mice: effects of estrogen. Neurobiol. Aging 25, 873–884. doi: 10.1016/j.neurobiolaging.2003.10.010
Hunter, C. L., Isacson, O., Nelson, M., Bimonte-Nelson, H., Seo, H., et al. (2003b). Regional alterations in amyloid precursor protein and nerve growth factor across age in a mouse model of Down's syndrome. Neurosci. Res. 45, 437–445. doi: 10.1016/s0168-0102(03)00005-1
Illouz, T., Biragyn, A., Iulita, M. F., Flores-Aguilar, L., Dierssen, M., De Toma, I., et al. (2021). Immune dysregulation and the increased risk of complications and mortality following respiratory tract infections in adults with Down syndrome. Front. Immunol. 12, 621440. doi: 10.3389/fimmu.2021.621440
Illouz, T., Madar, R., Biragyn, A., and Okun, E. (2019). Restoring microglial and astroglial homeostasis using DNA immunization in a Down syndrome mouse model. Brain. Behav. Immun. 75, 163–180. doi: 10.1016/j.bbi.2018.10.004
Ishihara, K., Kawashita, E., Shimizu, R., Nagasawa, K., Yasui, H., Sago, H., et al. (2019). Copper accumulation in the brain causes the elevation of oxidative stress and less anxious behavior in Ts1Cje mice, a model of Down syndrome. Free Radic. Biol. Med. 134, 248–259. doi: 10.1016/j.freeradbiomed.2019.01.015
Iulita, M. F., Do Carmo, S., Ower, A. K., Fortress, A. M., Flores Aguilar, L., Hanna, M., et al. (2014). Nerve growth factor metabolic dysfunction in Down's syndrome brains. Brain J. Neurol. 137, 860–872. doi: 10.1093/brain/awt372
Jankowsky, J. L., and Zheng, H. (2017). Practical considerations for choosing a mouse model of Alzheimer's disease. Mol. Neurodegener. 12, 89. doi: 10.1186/s13024-017-0231-7
Jiang, Y., Mullaney, K. A., Peterhoff, C. M., Che, S., Schmidt, S. D., Boyer-Boiteau, A., et al. (2010). Alzheimer's-related endosome dysfunction in Down syndrome is Aβ-independent but requires APP and is reversed by BACE-1 inhibition. Proc. Natl. Acad. Sci. 107, 1630–1635. doi: 10.1073/pnas.0908953107
Jiang, Y., Rigoglioso, A., Peterhoff, C. M., Pawlik, M., Sato, Y., Bleiwas, C., et al. (2016). Partial BACE1 reduction in a Down syndrome mouse model blocks Alzheimer-related endosomal anomalies and cholinergic neurodegeneration: role of APP-CTF. Neurobiol. Aging 39, 90–98. doi: 10.1016/j.neurobiolaging.2015.11.013
Jiang, Y., Sato, Y., Im, E., Berg, M., Bordi, M., Darji, S., et al. (2019). Lysosomal dysfunction in down syndrome is APP-dependent and mediated by APP-βCTF (C99). J. Neurosci. 39, 5255–5268. doi: 10.1523/JNEUROSCI.0578-19.2019
Kamer, A. R., Fortea, J. O., Videla, S., Mayoral, A., Janal, M., Carmona-Iragui, M., et al. (2016). Periodontal disease's contribution to Alzheimer's disease progression in Down syndrome. Alzheimers Dement. Diagn. Assess. Dis. Monit. 2, 49–57. doi: 10.1016/j.dadm.2016.01.001
Kamino, K., Orr, H. T., Payami, H., Wijsman, E. M., Alonso, M. E., Pulst, S. M., et al. (1992). Linkage and mutational analysis of familial Alzheimer disease kindreds for the APP gene region. Am. J. Hum. Genet. 51, 998–1014.
Kazuki, Y., Gao, F. J., Li, Y., Moyer, A. J., Devenney, B., Hiramatsu, K., et al. (2020). A non-mosaic transchromosomic mouse model of Down syndrome carrying the long arm of human chromosome 21. eLife 9, e56223. doi: 10.7554/eLife.56223
Kelleher, R. J., and Shen, J. (2017). Presenilin-1 mutations and Alzheimer's disease. Proc. Natl. Acad. Sci. 114, 629–631. doi: 10.1073/pnas.1619574114
Kelley, C. M., Powers, B. E., Velazquez, R., Ash, J. A., Ginsberg, S. D., Strupp, B. J., et al. (2014). Sex differences in the cholinergic basal forebrain in the Ts65Dn mouse model of Down syndrome and Alzheimer's disease. Brain Pathol. Zurich Switz. 24, 33–44. doi: 10.1111/bpa.12073
Kern, D. S., Maclean, K. N., Jiang, H., Synder, E. Y., Sladek, J. R., and Bjugstad, K. B. (2011). Neural stem cells reduce hippocampal tau and reelin accumulation in aged Ts65Dn Down syndrome mice. Cell Transplant. 20, 371–379. doi: 10.3727/096368910X528085
Kim, S., Sato, Y., Mohan, P. S., Peterhoff, C., Pensalfini, A., Rigoglioso, A., et al. (2016). Evidence that the rab5 effector APPL1 mediates APP-βCTF-induced dysfunction of endosomes in Down syndrome and Alzheimer's disease. Mol. Psychiatry 21, 707–716. doi: 10.1038/mp.2015.97
Korbel, J. O., Tirosh-Wagner, T., Urban, A. E., Chen, X.-N., Kasowski, M., Dai, L., et al. (2009). The genetic architecture of Down syndrome phenotypes revealed by high-resolution analysis of human segmental trisomies. Proc. Natl. Acad. Sci. U. S. A. 106, 12031–12036. doi: 10.1073/pnas.0813248106
Krinsky-McHale, S. J., Devenny, D. A., and Silverman, W. P. (2002). Changes in explicit memory associated with early dementia in adults with Down's syndrome. J. Intellect. Disabil. Res. JIDR 46, 198–208. doi: 10.1046/j.1365-2788.2002.00365.x
Kunkle, B. W., Grenier-Boley, B., Sims, R., Bis, J. C., Damotte, V., Naj, A. C., et al. (2019). Genetic meta-analysis of diagnosed Alzheimer's disease identifies new risk loci and implicates Aβ, tau, immunity and lipid processing. Nat. Genet. 51, 414–430. doi: 10.1038/s41588-019-0358-2
Lai, F., Mhatre, P. G., Yang, Y., Wang, M.-C., Schupf, N., and Rosas, H. D. (2020). Sex differences in risk of Alzheimer's disease in adults with Down syndrome. Alzheimers Dement. Amst. Neth. 12, e12084. doi: 10.1002/dad2.12084
Lana-Elola, E., Cater, H., Watson-Scales, S., Greenaway, S., Müller-Winkler, J., Gibbins, D., et al. (2021). Comprehensive phenotypic analysis of the Dp1Tyb mouse strain reveals a broad range of Down syndrome-related phenotypes. Dis. Model. Mech. 14:dmm049157. doi: 10.1242/dmm.049157
Lana-Elola, E., Watson-Scales, S., Slender, A., Gibbins, D., Martineau, A., Douglas, C., et al. (2016). Genetic dissection of Down syndrome-associated congenital heart defects using a new mouse mapping panel. eLife 5, 11614. doi: 10.7554/eLife.11614
Lanzillotta, C., Tramutola, A., Meier, S., Schmitt, F., Barone, E., Perluigi, M., et al. (2018). Early and selective activation and subsequent alterations to the unfolded protein response in Down Syndrome mouse models. J. Alzheimers Dis. JAD 62, 347–359. doi: 10.3233/JAD-170617
Ledreux, A., Thomas, S., Hamlett, E. D., Trautman, C., Gilmore, A., Rickman Hager, E., et al. (2021). Small neuron-derived extracellular vesicles from individuals with Down syndrome propagate tau pathology in the wildtype mouse brain. J. Clin. Med. 10, 3931. doi: 10.3390/jcm10173931
Lemere, C. A., Blusztajn, J. K., Yamaguchi, H., Wisniewski, T., Saido, T. C., and Selkoe, D. J. (1996). Sequence of deposition of heterogeneous amyloid β-peptides and APO E in Down syndrome: implications for initial events in amyloid plaque formation. Neurobiol. Dis. 3, 16–32. doi: 10.1006/nbdi.1996.0003
Lemere, C. A., Head, E., and Holtzman, D. M. (2021). APOE ε4 association with cognition and alzheimer disease biomarkers in down syndrome-implications for clinical trials and treatments for all. JAMA Neurol. 78, 913–915. doi: 10.1001/jamaneurol.2021.1649
Lemoine, L., Ledreux, A., Mufson, E. J., Perez, S. E., Simic, G., Doran, E., et al. (2020). Regional binding of tau and amyloid PET tracers in Down syndrome autopsy brain tissue. Mol. Neurodegener. 15, 68. doi: 10.1186/s13024-020-00414-3
Leng, F., and Edison, P. (2021). Neuroinflammation and microglial activation in Alzheimer disease: where do we go from here? Nat. Rev. Neurol. 17, 157–172. doi: 10.1038/s41582-020-00435-y
Leverenz, J. B., and Raskind, M. A. (1998). Early amyloid deposition in the medial temporal lobe of young Down syndrome patients: a regional quantitative analysis. Exp. Neurol. 150, 296–304. doi: 10.1006/exnr.1997.6777
Li, Q., Han, X., and Wang, J. (2016). Organotypic hippocampal slices as models for stroke and traumatic brain injury. Mol. Neurobiol. 53, 4226–4237. doi: 10.1007/s12035-015-9362-4
Liddelow, S. A., Guttenplan, K. A., Clarke, L. E., Bennett, F. C., Bohlen, C. J., Schirmer, L., et al. (2017). Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541, 481–487. doi: 10.1038/nature21029
Lin, A.-L., Powell, D., Caban-Holt, A., Jicha, G., Robertson, W., Gold, B. T., et al. (2016). 1H-MRS metabolites in adults with Down syndrome: effects of dementia. NeuroImage Clin. 11, 728–735. doi: 10.1016/j.nicl.2016.06.001
Liu, F., Liang, Z., Wegiel, J., Hwang, Y.-W., Iqbal, K., Grundke-Iqbal, I., et al. (2008). Overexpression of Dyrk1A contributes to neurofibrillary degeneration in Down syndrome. FASEB J. 22, 3224–3233. doi: 10.1096/fj.07-104539
Lockrow, J., Boger, H., Bimonte-Nelson, H., and Granholm, A.-C. (2011a). Effects of long-term memantine on memory and neuropathology in Ts65Dn mice, a model for Down syndrome. Behav. Brain Res. 221, 610–622. doi: 10.1016/j.bbr.2010.03.036
Lockrow, J., Boger, H., Gerhardt, G., Aston-Jones, G., Bachman, D., and Granholm, A.-C. (2011b). A noradrenergic lesion exacerbates neurodegeneration in a Down syndrome mouse model. J. Alzheimers Dis. JAD 23, 471–489. doi: 10.3233/JAD-2010-101218
Lockrow, J., Prakasam, A., Huang, P., Bimonte-Nelson, H., Sambamurti, K., and Granholm, A.-C. (2009). Cholinergic degeneration and memory loss delayed by vitamin E in a Down syndrome mouse model. Exp. Neurol. 216, 278–289. doi: 10.1016/j.expneurol.2008.11.021
Lomoio, S., Scherini, E., and Necchi, D. (2009). β-Amyloid overload does not directly correlate with SAPK/JNK activation and tau protein phosphorylation in the cerebellar cortex of Ts65Dn mice. Brain Res. 1297. 198–206. doi: 10.1016/j.brainres.2009.08.052
Lorenzo, L. P. E., Shatynski, K. E., Clark, S., Yarowsky, P. J., and Williams, M. S. (2013). Defective thymic progenitor development and mature T-cell responses in a mouse model for Down syndrome. Immunology 139, 447–458. doi: 10.1111/imm.12092
Lott, I. T., Doran, E., Nguyen, V. Q., Tournay, A., Movsesyan, N., and Gillen, D. L. (2012). Down syndrome and dementia: seizures and cognitive decline. J. Alzheimers Dis. JAD 29, 177–185. doi: 10.3233/JAD-2012-111613
Lu, J., Esposito, G., Scuderi, C., Steardo, L., Delli-Bovi, L. C., Hecht, J. L., et al. (2011). S100B and APP promote a gliocentric shift and impaired neurogenesis in Down syndrome neural progenitors. PLoS ONE 6, e22126. doi: 10.1371/journal.pone.0022126
Lučiunaite, A., McManus, R. M., Jankunec, M., Rácz, I., Dansokho, C., Dalgediene, I., et al. (2020). Soluble Aβ oligomers and protofibrils induce NLRP3 inflammasome activation in microglia. J. Neurochem. 155, 650–661. doi: 10.1111/jnc.14945
Lv, X., Li, W., Luo, Y., Wang, D., Zhu, C., Huang, Z.-X., et al. (2013). Exploring the differences between mouse mAβ1–42 and human hAβ1–42 for Alzheimer's disease related properties and neuronal cytotoxicity. Chem. Commun. 49, 5865–5867. doi: 10.1039/C3CC40779A
Lysenko, L. V., Kim, J., Madamba, F., Tyrtyshnaia, A. A., Ruparelia, A., and Kleschevnikov, A. M. (2018). Developmental excitatory-to-inhibitory GABA polarity switch is delayed in Ts65Dn mice, a genetic model of Down syndrome. Neurobiol. Dis. 115, 1–8. doi: 10.1016/j.nbd.2018.03.005
Mann, D. M. A., Davidson, Y. S., Robinson, A. C., Allen, N., Hashimoto, T., Richardson, A., et al. (2018). Patterns and severity of vascular amyloid in Alzheimer's disease associated with duplications and missense mutations in APP gene, Down syndrome and sporadic Alzheimer's disease. Acta Neuropathol. (Berl.) 136, 569–587. doi: 10.1007/s00401-018-1866-3
Mann, D. M. A., and Esiri, M. M. (1989). The pattern of acquisition of plaques and tangles in the brains of patients under 50 years of age with Down's syndrome. J. Neurol. Sci. 89, 169–179. doi: 10.1016/0022-510X(89)90019-1
Mann, D. M. A., Yates, P. O., and Marcyniuk, B. (1984). Alzheimer's presenile dementia, senile dementia of Alzheimer type and Down's syndrome in middle age form an age related continuum of pathological changes. Neuropathol. Appl. Neurobiol. 10, 185–207. doi: 10.1111/j.1365-2990.1984.tb00351.x
Mann, D. M. A., Yates, P. O., Marcyniuk, B., and Ravindra, C. R. (1987). Loss of neurones from cortical and subcortical areas in Down's syndrome patients at middle age: quantitative comparisons with younger Down's patients and patients with Alzheimer's disease. J. Neurol. Sci. 80, 79–89. doi: 10.1016/0022-510X(87)90223-1
Maris, M., Wojciechowski, M., Van de Heyning, P., and Boudewyns, A. (2014). A cross-sectional analysis of otitis media with effusion in children with Down syndrome. Eur. J. Pediatr. 173, 1319–1325. doi: 10.1007/s00431-014-2323-5
Martini, A. C., Helman, A. M., McCarty, K. L., Lott, I. T., Doran, E., Schmitt, F. A., et al. (2020). Distribution of microglial phenotypes as a function of age and Alzheimer's disease neuropathology in the brains of people with Down syndrome. Alzheimers Dement. Diagn. Assess. Dis. Monit. 12, e12113. doi: 10.1002/dad2.12113
McCarron, M., McCallion, P., Reilly, E., Dunne, P., Carroll, R., and Mulryan, N. (2017). A prospective 20-year longitudinal follow-up of dementia in persons with Down syndrome: 20-year longitudinal follow-up of dementia. J. Intellect. Disabil. Res. 61, 843–852. doi: 10.1111/jir.12390
McCarron, M., McCallion, P., Reilly, E., and Mulryan, N. (2014). A prospective 14-year longitudinal follow-up of dementia in persons with Down syndrome. J. Intellect. Disabil. Res. JIDR 58, 61–70. doi: 10.1111/jir.12074
McNaughton, D., Knight, W., Guerreiro, R., Ryan, N., Lowe, J., Poulter, M., et al. (2012). Duplication of amyloid precursor protein (APP), but not prion protein (PRNP) gene is a significant cause of early onset dementia in a large UK series. Neurobiol. Aging 33, 426.e13–426.e21. doi: 10.1016/j.neurobiolaging.2010.10.010
Mendez, M. F (2017). Early-onset Alzheimer's disease. Neurol. Clin. 35, 263–281. doi: 10.1016/j.ncl.2017.01.005
Mhatre, P. G., Lee, J. H., Pang, D., Zigman, W. B., Tycko, B., Krinsky-McHale, S. J., et al. (2021). The association between sex and risk of Alzheimer's disease in adults with Down syndrome. J. Clin. Med. 10, 2966. doi: 10.3390/jcm10132966
Miller, L. V. C., Mukadam, A. S., Durrant, C. S., Vaysburd, M. J., Katsinelos, T., Tuck, B. J., et al. (2021). Tau assemblies do not behave like independently acting prion-like particles in mouse neural tissue. Acta Neuropathol. Commun. 9, 41. doi: 10.1186/s40478-021-01141-6
Mitchelmore, C., Kjaerulff, K. M., Pedersen, H. C., Nielsen, J. V., Rasmussen, T. E., Fisker, M. F., et al. (2002). Characterization of two novel nuclear BTB/POZ domain zinc finger isoforms. Association with differentiation of hippocampal neurons, cerebellar granule cells, and macroglia. J. Biol. Chem. 277, 7598–7609. doi: 10.1074/jbc.M110023200
Montal, V., Barroeta, I., Bejanin, A., Pegueroles, J., Carmona-Iragui, M., Altuna, M., et al. (2021). Metabolite signature of Alzheimer's disease in adults with Down syndrome. Ann. Neurol. 90, 407–416. doi: 10.1002/ana.26178
Mori, C., Spooner, E. T., Wisniewski, K. E., Wisniewski, T. M., Yamaguchi, H., Saido, T. C., et al. (2002). Intraneuronal Aβ42 accumulation in Down syndrome brain. Amyloid 9, 88–102. doi: 10.3109/13506120208995241
Mucke, L., Masliah, E., Yu, G. Q., Mallory, M., Rockenstein, E. M., Tatsuno, G., et al. (2000). High-level neuronal expression of abeta 1-42 in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J. Neurosci. Off. J. Soc. Neurosci. 20, 4050–4058. doi: 10.1523/JNEUROSCI.20-11-04050.2000
Mullan, M., Crawford, F., Axelman, K., Houlden, H., Lilius, L., Winblad, B., et al. (1992). A pathogenic mutation for probable Alzheimer's disease in the APP gene at the N-terminus of beta-amyloid. Nat. Genet. 1, 345–347. doi: 10.1038/ng0892-345
Murrell, J., Farlow, M., Ghetti, B., and Benson, M. D. (1991). A mutation in the amyloid precursor protein associated with hereditary Alzheimer's disease. Science 254, 97–99. doi: 10.1126/science.1925564
Naert, G., Ferré, V., Keller, E., Slender, A., Gibbins, D., Fisher, E. M., et al. (2018). In vivo and ex vivo analyses of amyloid toxicity in the Tc1 mouse model of Down syndrome. J. Psychopharmacol. Oxf. Engl. 32, 174–190. doi: 10.1177/0269881117743484
Nelson, L. D., Orme, D., Osann, K., and Lott, I. T. (2001). Neurological changes and emotional functioning in adults with Down Syndrome. J. Intellect. Disabil. Res. JIDR 45, 450–456. doi: 10.1046/j.1365-2788.2001.00379.x
Netzer, W. J., Powell, C., Nong, Y., Blundell, J., Wong, L., Duff, K., et al. (2010). Lowering β-amyloid levels rescues learning and memory in a Down syndrome mouse model. PLoS ONE 5, e10943. doi: 10.1371/journal.pone.0010943
Oakley, H., Cole, S. L., Logan, S., Maus, E., Shao, P., Craft, J., et al. (2006). Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer's disease mutations: potential factors in amyloid plaque formation. J. Neurosci. Off. J. Soc. Neurosci. 26, 10129–10140. doi: 10.1523/JNEUROSCI.1202-06.2006
O'Doherty, A., Ruf, S., Mulligan, C., Hildreth, V., Errington, M. L., Cooke, S., et al. (2005). An aneuploid mouse strain carrying human chromosome 21 with Down syndrome phenotypes. Science 309, 2033–2037. doi: 10.1126/science.1114535
Oliver, C., Kalsy, S., McQuillan, S., and Hall, S. (2011). Behavioural Excesses and deficits associated with dementia in adults who have Down syndrome. J. Appl. Res. Intellect. Disabil. 24, 208–216. doi: 10.1111/j.1468-3148.2010.00604.x
Palmer, C. R., Liu, C. S., Romanow, W. J., Lee, M.-H., and Chun, J. (2021). Altered cell and RNA isoform diversity in aging Down syndrome brains. Proc. Natl. Acad. Sci. 118, e2114326118. doi: 10.1073/pnas.2114326118
Parnetti, L., Chiasserini, D., Eusebi, P., Giannandrea, D., Bellomo, G., De Carlo, C., et al. (2012). Performance of aβ1-40, aβ1-42, total tau, and phosphorylated tau as predictors of dementia in a cohort of patients with mild cognitive impairment. J. Alzheimers Dis. JAD 29, 229–238. doi: 10.3233/JAD-2011-111349
Perry, V. H., Cunningham, C., and Holmes, C. (2007). Systemic infections and inflammation affect chronic neurodegeneration. Nat. Rev. Immunol. 7, 161–167. doi: 10.1038/nri2015
Perry, V. H., and Holmes, C. (2014). Microglial priming in neurodegenerative disease. Nat. Rev. Neurol. 10, 217–224. doi: 10.1038/nrneurol.2014.38
Pinto, B., Morelli, G., Rastogi, M., Savardi, A., Fumagalli, A., Petretto, A., et al. (2020). Rescuing over-activated microglia restores cognitive performance in juvenile animals of the Dp(16) mouse model of Down syndrome. Neuron 108, 887–904.e12. doi: 10.1016/j.neuron.2020.09.010
Ponroy Bally, B., and Murai, K. K. (2021). Astrocytes in Down syndrome across the lifespan. Front. Cell. Neurosci. 15, 702685. doi: 10.3389/fncel.2021.702685
Prasher, V. P., Airuehia, E., and Carey, M. (2010). The first confirmed case of Down syndrome with dementia with lewy bodies. J. Appl. Res. Intellect. Disabil. 23, 296–300. doi: 10.1111/j.1468-3148.2009.00526.x
Prasher, V. P., Farrer, M. J., Kessling, A. M., Fisher, E. M. C., West, R. J., Barber, P. C., et al. (1998). Molecular mapping of alzheimer-type dementia in Down's syndrome. Ann. Neurol. 43, 380–383. doi: 10.1002/ana.410430316
Prasher, V. P., Sajith, S. G., Rees, S. D., Patel, A., Tewari, S., Schupf, N., et al. (2008). Significant effect of APOE epsilon 4 genotype on the risk of dementia in Alzheimer's disease and mortality in persons with Down syndrome. Int. J. Geriatr. Psychiatry 23, 1134–1140. doi: 10.1002/gps.2039
Qian, W., Jin, N., Shi, J., Yin, X., Jin, X., Wang, S., et al. (2013). Dual-specificity tyrosine phosphorylation-regulated kinase 1A (Dyrk1A) enhances tau expression. J. Alzheimers Dis. JAD 37, 529–538. doi: 10.3233/JAD-130824
Rafii, M. S (2019). Tau PET imaging for staging of Alzheimer's disease in Down syndrome. Dev. Neurobiol. 79, 711–715. doi: 10.1002/dneu.22658
Rafii, M. S., Lukic, A. S., Andrews, R. D., Brewer, J., Rissman, R. A., Strother, S. C., et al. (2017). PET imaging of tau pathology and relationship to amyloid, longitudinal MRI, and cognitive change in Down syndrome: results from the Down syndrome biomarker initiative (DSBI). J. Alzheimers Dis. 60, 439–450. doi: 10.3233/JAD-170390
Real, R., Peter, M., Trabalza, A., Khan, S., Smith, M. A., Dopp, J., et al. (2018). In vivo modeling of human neuron dynamics and Down syndrome. Science 362, eaau1810. doi: 10.1126/science.aau1810
Reeves, R. H., Irving, N. G., Moran, T. H., Wohn, A., Kitt, C., Sisodia, S. S., et al. (1995). A mouse model for Down syndrome exhibits learning and behaviour deficits. Nat. Genet. 11, 177–184. doi: 10.1038/ng1095-177
Ropper, A. H., and Williams, R. S. (1980). Relationship between plaques, tangles, and dementia in Down syndrome. Neurology 30, 639–644. doi: 10.1212/wnl.30.6.639
Rovelet-Lecrux, A., Frebourg, T., Tuominen, H., Majamaa, K., Campion, D., and Remes, A. M. (2007). APP locus duplication in a Finnish family with dementia and intracerebral haemorrhage. J. Neurol. Neurosurg. Psychiatry 78, 1158–1159. doi: 10.1136/jnnp.2006.113514
Rovelet-Lecrux, A., Hannequin, D., Raux, G., Le Meur, N., Laquerrière, A., Vital, A., et al. (2006). APP locus duplication causes autosomal dominant early-onset Alzheimer disease with cerebral amyloid angiopathy. Nat. Genet. 38, 24–26. doi: 10.1038/ng1718
Rueda, N., Vidal, V., García-Cerro, S., Narcís, J. O., Llorens-Martín, M., Corrales, A., et al. (2018). Anti-IL17 treatment ameliorates Down syndrome phenotypes in mice. Brain. Behav. Immun. 73, 235–251. doi: 10.1016/j.bbi.2018.05.008
Sago, H., Carlson, E. J., Smith, D. J., Kilbridge, J., Rubin, E. M., Mobley, W. C., et al. (1998). Ts1Cje, a partial trisomy 16 mouse model for Down syndrome, exhibits learning and behavioral abnormalities. Proc. Natl. Acad. Sci. 95, 6256–6261. doi: 10.1073/pnas.95.11.6256
Saito, T., Matsuba, Y., Mihira, N., Takano, J., Nilsson, P., Itohara, S., et al. (2014). Single App knock-in mouse models of Alzheimer's disease. Nat. Neurosci. 17, 661–663. doi: 10.1038/nn.3697
Salehi, A., Delcroix, J.-D., Belichenko, P. V., Zhan, K., Wu, C., Valletta, J. S., et al. (2006). Increased app expression in a mouse model of Down's syndrome disrupts NGF transport and causes cholinergic neuron degeneration. Neuron 51, 29–42. doi: 10.1016/j.neuron.2006.05.022
Salehi, A., Faizi, M., Colas, D., Valletta, J., Laguna, J., Takimoto-Kimura, R., et al. (2009). Restoration of norepinephrine-modulated contextual memory in a mouse model of down syndrome. Sci. Transl. Med. 1, 7ra17. doi: 10.1126/scitranslmed.3000258
Sansevero, G., Begenisic, T., Mainardi, M., and Sale, A. (2016). Experience-dependent reduction of soluble β-amyloid oligomers and rescue of cognitive abilities in middle-age Ts65Dn mice, a model of Down syndrome. Exp. Neurol. 283, 49–56. doi: 10.1016/j.expneurol.2016.06.006
Schupf, N., Pang, D., Patel, B. N., Silverman, W., Schubert, R., Lai, F., et al. (2003). Onset of dementia is associated with age at menopause in women with Down's syndrome. Ann. Neurol. 54, 433–438. doi: 10.1002/ana.10677
Seo, H., and Isacson, O. (2005). Abnormal APP, cholinergic and cognitive function in Ts65Dn Down's model mice. Exp. Neurol. 193, 469–480. doi: 10.1016/j.expneurol.2004.11.017
Serneels, L., T'Syen, D., Perez-Benito, L., Theys, T., Holt, M. G., and De Strooper, B. (2020). Modeling the β-secretase cleavage site and humanizing amyloid-beta precursor protein in rat and mouse to study Alzheimer's disease. Mol. Neurodegener. 15, 60. doi: 10.1186/s13024-020-00399-z
Shaw, P. R., Klein, J. A., Aziz, N. M., and Haydar, T. F. (2020). Longitudinal neuroanatomical and behavioral analyses show phenotypic drift and variability in the Ts65Dn mouse model of Down syndrome. Dis. Model. Mech. 13, dmm046243. doi: 10.1242/dmm.046243
Sheppard, O., Coleman, M. P., and Durrant, C. S. (2019). Lipopolysaccharide-induced neuroinflammation induces presynaptic disruption through a direct action on brain tissue involving microglia-derived interleukin 1 beta. J. Neuroinflammation 16, 106. doi: 10.1186/s12974-019-1490-8
Sheppard, O., Plattner, F., Rubin, A., Slender, A., Linehan, J. M., Brandner, S., et al. (2012). Altered regulation of tau phosphorylation in a mouse model of down syndrome aging. Neurobiol. Aging 33, 828.e31–44. doi: 10.1016/j.neurobiolaging.2011.06.025
Sherrington, R., Rogaev, E. I., Liang, Y., Rogaeva, E. A., Levesque, G., Ikeda, M., et al. (1995). Cloning of a gene bearing missense mutations in early-onset familial Alzheimer's disease. Nature 375, 754–760. doi: 10.1038/375754a0
Shukkur, E. A., Shimohata, A., Akagi, T., Yu, W., Yamaguchi, M., Murayama, M., et al. (2006). Mitochondrial dysfunction and tau hyperphosphorylation in Ts1Cje, a mouse model for Down syndrome. Hum. Mol. Genet. 15, 2752–2762. doi: 10.1093/hmg/ddl211
Siarey, R. J., Stoll, J., Rapoport, S. I., and Galdzicki, Z. (1997). Altered long-term potentiation in the young and old Ts65Dn mouse, a model for Down Syndrome. Neuropharmacology 36, 1549–1554. doi: 10.1016/s0028-3908(97)00157-3
Sideris, D. I., Danial, J. S. H., Emin, D., Ruggeri, F. S., Xia, Z., Zhang, Y. P., et al. (2021). Soluble amyloid beta-containing aggregates are present throughout the brain at early stages of Alzheimer's disease. Brain Commun. 3, fcab147. doi: 10.1093/braincomms/fcab147
Sinai, A., Mokrysz, C., Bernal, J., Bohnen, I., Bonell, S., Courtenay, K., et al. (2018). Predictors of age of diagnosis and survival of Alzheimer's disease in Down syndrome. J. Alzheimers Dis. JAD 61, 717–728. doi: 10.3233/JAD-170624
Sleegers, K., Brouwers, N., Gijselinck, I., Theuns, J., Goossens, D., Wauters, J., et al. (2006). APP duplication is sufficient to cause early onset Alzheimer's dementia with cerebral amyloid angiopathy. Brain J. Neurol. 129, 2977–2983. doi: 10.1093/brain/awl203
Sofroniew, M. V., Howe, C. L., and Mobley, W. C. (2001). Nerve Growth Factor Signaling, Neuroprotection, and Neural Repair. Annu. Rev. Neurosci. 24, 1217–1281. doi: 10.1146/annurev.neuro.24.1.1217
Staal, J. A., Alexander, S. R., Liu, Y., Dickson, T. D., and Vickers, J. C. (2011). Characterization of cortical neuronal and glial alterations during culture of organotypic whole brain slices from neonatal and mature mice. PLoS ONE 6, e22040. doi: 10.1371/journal.pone.0022040
Stagni, F., Raspanti, A., Giacomini, A., Guidi, S., Emili, M., Ciani, E., et al. (2017). Long-term effect of neonatal inhibition of APP gamma-secretase on hippocampal development in the Ts65Dn mouse model of Down syndrome. Neurobiol. Dis. 103, 11–23. doi: 10.1016/j.nbd.2017.03.012
Startin, C. M., Ashton, N. J., Hamburg, S., Hithersay, R., Wiseman, F. K., Mok, K. Y., et al. (2019a). Plasma biomarkers for amyloid, tau, and cytokines in Down syndrome and sporadic Alzheimer's disease. Alzheimers Res. Ther. 11, 26. doi: 10.1186/s13195-019-0477-0
Startin, C. M., Hamburg, S., Hithersay, R., Al-Janabi, T., Mok, K. Y., Hardy, J., et al. (2019b). Cognitive markers of preclinical and prodromal Alzheimer's disease in Down syndrome. Alzheimers Dement. J. Alzheimers Assoc. 15, 245–257. doi: 10.1016/j.jalz.2018.08.009
Steffen, J., Krohn, M., Schwitlick, C., Brüning, T., Paarmann, K., Pietrzik, C. U., et al. (2017). Expression of endogenous mouse APP modulates β-amyloid deposition in hAPP-transgenic mice. Acta Neuropathol. Commun. 5, 49. doi: 10.1186/s40478-017-0448-2
Stoltzner, S. E., Grenfell, T. J., Mori, C., Wisniewski, K. E., Wisniewski, T. M., Selkoe, D. J., et al. (2000). Temporal accrual of complement proteins in amyloid plaques in Down's syndrome with Alzheimer's disease. Am. J. Pathol. 156, 489–499. doi: 10.1016/S0002-9440(10)64753-0
Strydom, A., Coppus, A., Blesa, R., Danek, A., Fortea, J., Hardy, J., et al. (2018). Alzheimer's disease in Down syndrome: AN overlooked population for prevention trials. Alzheimers Dement. N. Y. N 4, 703–713. doi: 10.1016/j.trci.2018.10.006
Tallino, S., Winslow, W., Bartholomew, S. K., and Velazquez, R. (2022). Temporal and brain region-specific elevations of soluble Amyloid-β40-42 in the Ts65Dn mouse model of Down syndrome and Alzheimer's disease. Aging Cell 21, e13590. doi: 10.1111/acel.13590
Thal, D. R., Rüb, U., Orantes, M., and Braak, H. (2002). Phases of A beta-deposition in the human brain and its relevance for the development of AD. Neurology 58, 1791–1800. doi: 10.1212/wnl.58.12.1791
Tosh, J., Tybulewicz, V., and Fisher, E. M. C. (2022). Mouse models of aneuploidy to understand chromosome disorders. Mamm. Genome 33, 157–168. doi: 10.1007/s00335-021-09930-z
Tosh, J. L., Rhymes, E. R., Mumford, P., Whittaker, H. T., Pulford, L. J., Noy, S. J., et al. (2021). Genetic dissection of down syndrome-associated alterations in APP/amyloid-β biology using mouse models. Sci. Rep. 11, 5736. doi: 10.1038/s41598-021-85062-3
Tosh, J. L., Rickman, M., Rhymes, E., Norona, F. E., Clayton, E., Mucke, L., et al. (2017). The integration site of the APP transgene in the J20 mouse model of Alzheimer's disease. Wellcome Open Res. 2:84. doi: 10.12688/wellcomeopenres.12237.2
Tramutola, A., Lanzillotta, C., Barone, E., Arena, A., Zuliani, I., Mosca, L., et al. (2018). Intranasal rapamycin ameliorates Alzheimer-like cognitive decline in a mouse model of Down syndrome. Transl. Neurodegener. 7, 28. doi: 10.1186/s40035-018-0133-9
Tuttle, K. D., Waugh, K. A., Araya, P., Minter, R., Orlicky, D. J., Ludwig, M., et al. (2020). JAK1 inhibition blocks lethal immune hypersensitivity in a mouse model of Down syndrome. Cell Rep. 33, 108407. doi: 10.1016/j.celrep.2020.108407
Vidal, V., Puente, A., García-Cerro, S., García Unzueta, M. T., Rueda, N., Riancho, J., et al. (2021). Bexarotene impairs cognition and produces hypothyroidism in a mouse model of Down syndrome and Alzheimer's disease. Front. Pharmacol. 12, 613211. doi: 10.3389/fphar.2021.613211
Villar, A. J., Belichenko, P. V., Gillespie, A. M., Kozy, H. M., Mobley, W. C., and Epstein, C. J. (2005). Identification and characterization of a new Down syndrome model, Ts[Rb(12.1716)]2Cje, resulting from a spontaneous Robertsonian fusion between T(1716)65Dn and mouseChromosome 12. Mamm. Genome 16, 79–90. doi: 10.1007/s00335-004-2428-7
Visser, F. E., Aldenkamp, A. P., van Huffelen, A. C., and Kuilman, M. (1997). Prospective study of the prevalence of Alzheimer-type dementia in institutionalized individuals with Down syndrome. Am. J. Ment. Retard. 101, 400–412.
Weber, G. E., Koenig, K. A., Khrestian, M., Shao, Y., Tuason, E. D., Gramm, M., et al. (2020). An altered relationship between soluble TREM2 and inflammatory markers in young adults with Down syndrome: a preliminary report. J. Immunol. 204, 1111–1118. doi: 10.4049/jimmunol.1901166
Wegiel, J., Flory, M., Kuchna, I., Nowicki, K., Wegiel, J., Ma, S. Y., et al. (2022). Developmental deficits and staging of dynamics of age associated Alzheimer's disease neurodegeneration and neuronal loss in subjects with Down syndrome. Acta Neuropathol. Commun. 10, 2. doi: 10.1186/s40478-021-01300-9
Wilcock, D. M., Hurban, J., Helman, A. M., Sudduth, T. L., McCarty, K. L., Beckett, T. L., et al. (2015). Down syndrome individuals with Alzheimer's disease have a distinct neuroinflammatory phenotype compared to sporadic Alzheimer's disease. Neurobiol. Aging 36, 2468–2474. doi: 10.1016/j.neurobiolaging.2015.05.016
Wiseman, F. K., Alford, K. A., Tybulewicz, V. L. J., and Fisher, E. M. C. (2009). Down syndrome–recent progress and future prospects. Hum. Mol. Genet. 18, R75–R83. doi: 10.1093/hmg/ddp010
Wiseman, F. K., Al-Janabi, T., Hardy, J., Karmiloff-Smith, A., Nizetic, D., Tybulewicz, V. L. J., et al. (2015). A genetic cause of Alzheimer disease: mechanistic insights from Down syndrome. Nat. Rev. Neurosci. 16, 564–574. doi: 10.1038/nrn3983
Wiseman, F. K., Pulford, L. J., Barkus, C., Liao, F., Portelius, E., Webb, R., et al. (2018). Trisomy of human chromosome 21 enhances amyloid-β deposition independently of an extra copy of APP. Brain 141, 2457–2474. doi: 10.1093/brain/awy159
Wisniewski, K. E., Wisniewski, H. M., and Wen, G. Y. (1985). Occurrence of neuropathological changes and dementia of Alzheimer's disease in Down's syndrome. Ann. Neurol. 17, 278–282. doi: 10.1002/ana.410170310
Wu, J., and Morris, J. K. (2013). The population prevalence of Down's syndrome in England and Wales in 2011. Eur. J. Hum. Genet. EJHG 21, 1016–1019. doi: 10.1038/ejhg.2012.294
Xu, W., Weissmiller, A. M., White, J. A., Fang, F., Wang, X., Wu, Y., et al. (2016). Amyloid precursor protein–mediated endocytic pathway disruption induces axonal dysfunction and neurodegeneration. J. Clin. Invest. 126, 1815–1833. doi: 10.1172/JCI82409
Xue, Q.-S., and Streit, W. J. (2011). Microglial pathology in Down syndrome. Acta Neuropathol. (Berl.) 122, 455–466. doi: 10.1007/s00401-011-0864-5
Yang, Y., Arseni, D., Zhang, W., Huang, M., Lövestam, S., Schweighauser, M., et al. (2022). Cryo-EM structures of amyloid-β 42 filaments from human brains. Science 375, 167–172. doi: 10.1126/science.abm7285
Yin, X., Jin, N., Shi, J., Zhang, Y., Wu, Y., Gong, C.-X., et al. (2017). Dyrk1A overexpression leads to increase of 3R-tau expression and cognitive deficits in Ts65Dn Down syndrome mice. Sci. Rep. 7, 619. doi: 10.1038/s41598-017-00682-y
Yu, T., Li, Z., Jia, Z., Clapcote, S. J., Liu, C., Li, S., et al. (2010). A mouse model of Down syndrome trisomic for all human chromosome 21 syntenic regions. Hum. Mol. Genet. 19, 2780–2791. doi: 10.1093/hmg/ddq179
Zammit, M. D., Laymon, C. M., Betthauser, T. J., Cody, K. A., Tudorascu, D. L., Minhas, D. S., et al. (2020). Amyloid accumulation in Down syndrome measured with amyloid load. Alzheimers Dement. Diagn. Assess. Dis. Monit. 12, e12020. doi: 10.1002/dad2.12020
Zhang, B., Wang, Q., Miao, T., Yu, B., Yuan, P., Kong, J., et al. (2015). Whether Alzheimer's diseases related genes also differently express in the hippocampus of Ts65Dn mice? Int. J. Clin. Exp. Pathol. 8, 4120–4125.
Zheng, Q., Li, G., Wang, S., Zhou, Y., Liu, K., Gao, Y., et al. (2021). Trisomy 21–induced dysregulation of microglial homeostasis in Alzheimer's brains is mediated by USP25. Sci. Adv. 7, eabe1340. doi: 10.1126/sciadv.abe1340
Keywords: Down syndrome, Alzheimer's disease, Amyloid-beta, tau, mouse model, neuronal loss, neuroinflammation
Citation: Farrell C, Mumford P and Wiseman FK (2022) Rodent Modeling of Alzheimer's Disease in Down Syndrome: In vivo and ex vivo Approaches. Front. Neurosci. 16:909669. doi: 10.3389/fnins.2022.909669
Received: 31 March 2022; Accepted: 16 May 2022;
Published: 07 June 2022.
Edited by:
Yann Herault, Centre National de la Recherche Scientifique (CNRS), FranceReviewed by:
Eric Daniel Hamlett, Medical University of South Carolina, United StatesCopyright © 2022 Farrell, Mumford and Wiseman. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Frances K. Wiseman, Zi53aXNlbWFuQHVjbC5hYy51aw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.