 Reed T. Bjork
Reed T. Bjork Nicholas P. Mortimore
Nicholas P. Mortimore Suvithanandhini Loganathan
Suvithanandhini Loganathan Daniela C. Zarnescu
Daniela C. Zarnescu- 1Department of Molecular and Cellular Biology, University of Arizona, Tucson, AZ, United States
- 2Neuroscience Graduate Interdisciplinary Program, University of Arizona, Tucson, AZ, United States
Local control of gene expression provides critical mechanisms for regulating development, maintenance and plasticity in the nervous system. Among the strategies known to govern gene expression locally, mRNA transport and translation have emerged as essential for a neuron’s ability to navigate developmental cues, and to establish, strengthen and remove synaptic connections throughout lifespan. Substantiating the role of RNA processing in the nervous system, several RNA binding proteins have been implicated in both developmental and age dependent neurodegenerative disorders. Of these, TDP-43 is an RNA binding protein that has emerged as a common denominator in amyotrophic lateral sclerosis (ALS), frontotemporal dementia (FTD) and related disorders due to the identification of causative mutations altering its function and its accumulation in cytoplasmic aggregates observed in a significant fraction of ALS/FTD cases, regardless of etiology. TDP-43 is involved in multiple aspects of RNA processing including splicing, transport and translation. Given that one of the early events in disease pathogenesis is mislocalization from the nucleus to the cytoplasm, several studies have focused on elucidating the pathogenic role of TDP-43 in cytoplasmic translation. Here we review recent findings describing TDP-43 translational targets and potential mechanisms of translation dysregulation in TDP-43 proteinopathies across multiple experimental models including cultured cells, flies, mice and patient derived neurons.
Introduction
One of the most unique features of a neuron is its dramatic morphology. Aside from the requisite cell body, which contains the nucleus and as little as 1% of the cytoplasm, neurons typically wield a robust network of processes including dendrites and a single axon (reviewed in Holt et al., 2019). In humans, the axon can extend beyond a meter in length and forge tens of thousands of synaptic connections (reviewed in Holt et al., 2019). Dendrites are highly branched processes and serve as the postsynaptic component of the synapse. This level of compartmentalization poses unique challenges and opportunities for spatiotemporally sensitive processes, especially for critical neuronal functions such as neural and synaptic plasticity. More specifically, neurite outgrowth, maintenance, branching, axonal turning, synaptogenesis, and synapse maintenance require carefully orchestrated, spatially and temporally controlled protein synthesis, which is achieved in part through mRNA localization and local translation (reviewed in Sutton and Schuman, 2006; Batista and Hengst, 2016; Costa and Willis, 2018; Holt et al., 2019). A growing body of work over the last two decades has revealed significant dysregulation of translation in several neurodevelopmental and neurodegenerative diseases such as Fragile X Syndrome (FXS), autism spectrum disorders (ASD), amyotrophic lateral sclerosis (ALS), spinal muscular atrophy (SMA), and Alzheimer’s Disease (AD) (reviewed in Bear et al., 2008; Akins et al., 2009; Liu-Yesucevitz et al., 2011; Baleriola et al., 2014; Ferro et al., 2018). Therefore, local translation has emerged as a critical mechanism underlying the pathogenesis of neurological disorders across lifespan.
Regulated primarily by RNA binding proteins, proper maintenance of RNA homeostasis is necessary for healthy function of the neuron. Aberrant function of several RNA binding proteins including TAR DNA-binding protein 43 (TDP-43), Fused in Sarcoma (FUS) and Senataxin (SETX) has been linked to neurodegenerative disease pathogenesis. TDP-43 is of particular interest as it participates in numerous RNA processing steps and has been identified as a major component of pathological cytoplasmic inclusions in ALS and frontotemporal dementia (FTD) (Andersen and Al-Chalabi, 2011). These cytoplasmic accumulations, collectively referred to as TDP-43 proteinopathy, are a hallmark of neuron degeneration in multiple disorders including ALS (97% of cases), FTD (45% of cases) (Ling et al., 2013), Alzheimer’s disease (AD, 57% of cases), and Lewy Body Dementia (McAleese et al., 2017). Together with the identification of disease causative mutations in 2–4% of ALS patients, these findings establish TDP-43 as a common denominator across multiple neurodegenerative diseases.
Recent reports have identified TDP-43 dependent alterations in the translatome (MacNair et al., 2016; Neelagandan et al., 2019; Marques et al., 2020; Lehmkuhl et al., 2021). Whether this occurs directly, via TDP-43 association with the translation machinery or indirectly, by disturbing cellular homeostasis, recently reported translation targets of TDP-43 proteinopathy have uncovered a plethora of cellular pathways that are providing insights into potential therapeutic strategies for ALS. Here we highlight recent discoveries and mechanistic insights into TDP-43 dependent translation dysregulation in ALS/FTD.
Translation Dysregulation in TDP-43 Proteinopathy
TDP-43 Structural Features—Implications for Translation
TDP-43 is an evolutionarily conserved DNA/RNA binding protein comprising four primary domains: an N-terminal domain (NTD, aa: 1–103) with a nuclear localization signal (NLS, aa: 82–98), two RNA-recognition motifs (RRMs, aa: 104–200, 191–262), and an intrinsically disordered C-terminal low-complexity domain (LCD, aa: 274–413) (see Figure 1). Insights from key structural features and disease causative mutations in TDP-43 highlight important mechanisms that drive TDP-43 pathogenesis. The vast majority of ALS/FTD causing mutations reside within its LCD and were shown to increase the propensity of TDP-43 to aggregate (Johnson et al., 2009; Buratti, 2015). In cases of familial ALS the A315 residue is mutated to E or T (A315E and A315T), which can lead to the assembly of tightly packing steric zippers that form stable fibrils (Nelson et al., 2005). The A315E mutation and phosphorylation of A315T also introduce electrostatic interactions into the amyloidogenic core regions of TDP-43, stabilizing larger fibril conformations that can seed irreversible aggregates (Cao et al., 2019). These TDP-43 aggregates could cause translation inhibition of sequestered mRNAs or affect translation by association with factors that regulate protein synthesis (Ramaswami et al., 2013). Interestingly, oligomerization of TDP-43 through the N-terminal domain via salt bridges between D22 and R55, E3 and R52, as well as E17 and R52 has been shown to antagonize TDP-43 proteinopathy by spatially separating LCDs that may otherwise aggregate (Afroz et al., 2017). Furthermore, the tandem RRMs of TDP-43 are of central importance to the specificity and activity of the wild-type protein, contributing to most of its RNA binding activity. The two RRMs have distinct nucleotide specificities, but mainly recognize (UG/TG)n repeat RNA via the organization of key aromatic and charged residues (Lukavsky et al., 2013). Recent findings suggest that the RNA binding activity of TDP-43 may be of particular interest in disease, as association with long sequences of UG rich RNA has been shown to dissolve optogenetically induced TDP-43 aggregates in vitro and reduce neurotoxicity (Mann et al., 2019; Grese et al., 2021). Another interesting feature of the RRM domains is the presence of a salt bridge, R151/D247, that is not required for RNA binding per se, but regulates affinity and specificity (Flores et al., 2019). Disrupting the R151/D247 salt bridge affects RNA binding and destabilizes the protein, which mitigates toxicity.
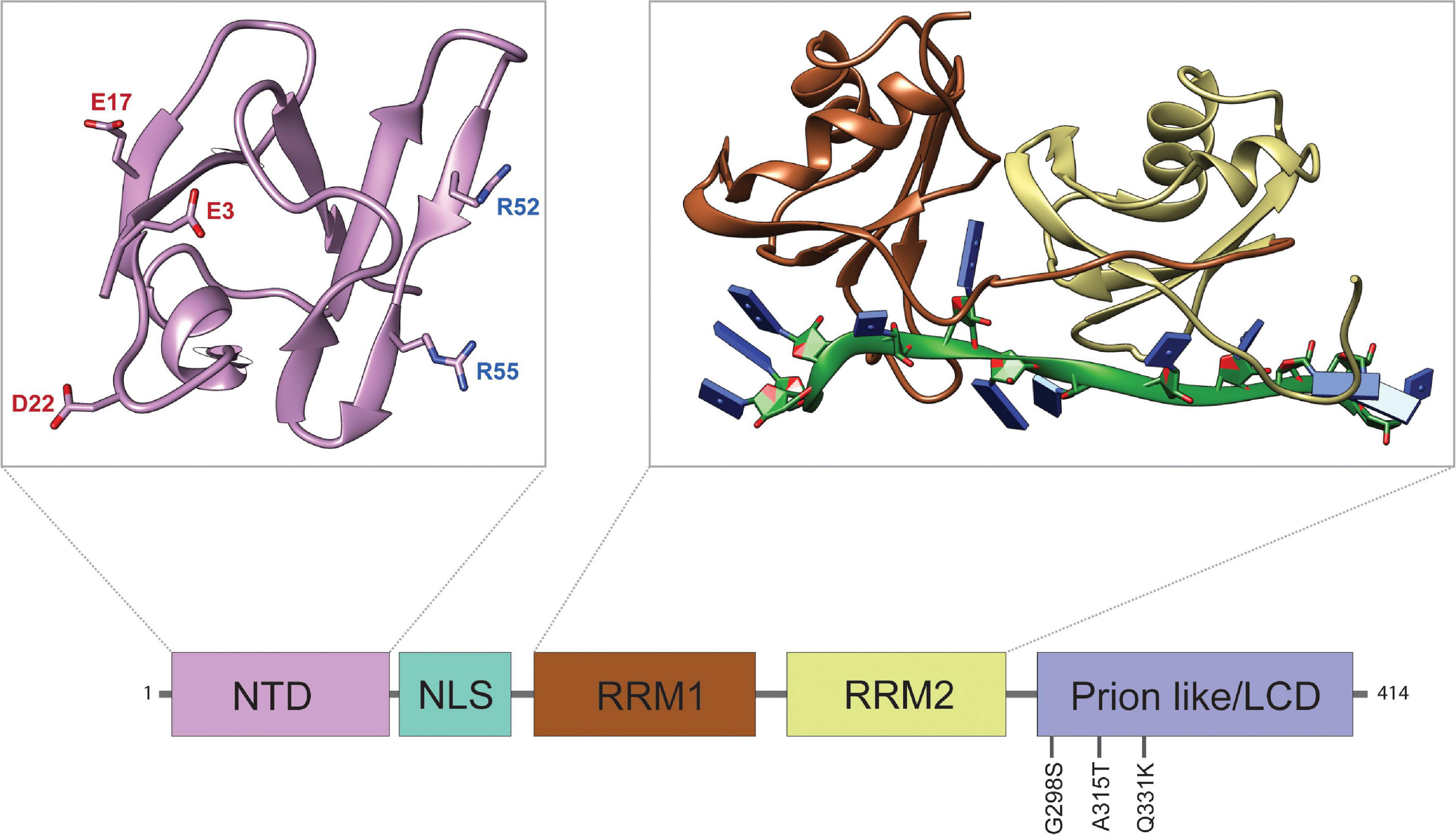
Figure 1. Structure and Domain Features of TDP-43. Domain map of TDP-43 depicting the approximate sizes of functional domains. The disease-associated mutations discussed in this review are highlighted. From left to right: N-terminal domain (NTD), nuclear localization signal (NLS), RNA-recognition motif 1 (RRM1), RNA-recognition motif 2 (RRM2), and low complexity domain (LCD). Top left panel: structure of the NTD (PDB ID: 5MDI) with aggregation antagonizing amino acid residues E3, E17, D22, R52, and R55, as indicated (Afroz et al., 2017). The negatively charged NTD residues (red) form salt bridges with positively charged residues (blue) of other TDP-43 NTDs to form oligomers. Top right panel: structure of the tandem RRMs bound to UG-rich RNA (PDB ID: 4BS2) (Lukavsky et al., 2013). The RNA-structure is shown in green with nucleobases represented by blue rectangles.
Recently, a short TDP-43 (sTDP-43) isoform upregulated by neuronal hyperactivity has been identified (Weskamp et al., 2020). It results from alternative splicing that removes most of the C terminus unstructured domain and introduces a functional nuclear export signal (NES) that causes it to localize to the cytoplasm where it can drive the aggregation of endogenous, full length TDP-43 (Weskamp et al., 2020). TDP-43 has also been shown to undergo liquid-liquid phase separation (LLPS) via its LCD, which may facilitate aggregation (Babinchak et al., 2019; Conicella et al., 2020; Pakravan et al., 2021). A recent study characterized a novel mouse model expressing LLPS-deficient TDP-43 (TDPΔCR) caused by deletion of residues 321–340 that represent the conserved region (CR) required for LLPS from within the LCD (Gao et al., 2021). Structure function studies defined residues 99–105 as the TDP-43 translation factor binding motif due to its ability to bind several translation factors and its requirement for enhanced protein synthesis observed in the TDPΔCR mouse model (Gao et al., 2021). Altogether, these findings highlight the importance of RNA binding, RNA sequestration as well as TDP-43 protein aggregation in disease pathology, and outline potential avenues for TDP-43 involvement in translational control.
TDP-43 Mislocalization to the Cytoplasm Is a Critical Event in Disease Pathogenesis
Under physiological conditions, TDP-43 resides predominantly in the nucleus where it modulates transcription, splicing and miRNA biogenesis (Casafont et al., 2009). Due to its scarce presence outside the nucleus, the extent to which full length TDP-43 performs physiological operations in the cytoplasm remains unclear, considering that the predicted NES does not function as such (Ederle et al., 2018). In numerous neurodegenerative diseases, however, TDP-43 is depleted from the nucleus and mislocalizes to the cytoplasm forming pathological inclusions (Neumann et al., 2006; McAleese et al., 2017). Substantiated by these observations, the mechanism of TDP-43 pathogenesis is postulated as a loss of nuclear function and/or cytoplasmic gain-of-function (Vanden Broeck et al., 2014; Ederle and Dormann, 2017). To test the nuclear depletion hypothesis, early studies using gene knockout methods revealed that loss of TDP-43 is lethal, both in embryonic development and postnatally when inactivated using Cre-inducible gene excision (Kraemer et al., 2010; Sephton et al., 2010; Wu et al., 2012). Other studies focused their attention on recapitulating the cytoplasmic accumulation of TDP-43, demonstrating that overexpression of either the full length wild type protein (Johnson et al., 2008; Li et al., 2010; Liachko et al., 2010; Tsai et al., 2010; Wils et al., 2010; Igaz et al., 2011) or a disease-associated variant (Kabashi et al., 2009; Wegorzewska et al., 2009; Liachko et al., 2010; Zhou et al., 2010; Estes et al., 2011; Huang et al., 2012), is sufficient to cause ALS phenotypes in various animal models. Using other mutant forms of TDP-43 to manipulate its localization and function provided additional support for cytoplasmic toxicity as a likely disease mechanism. For instance, variants of TDP-43 harboring RRM mutations that affect RNA binding do not result in cytoplasmic mislocalization or induce toxicity (Elden et al., 2010; Voigt et al., 2010; Ihara et al., 2013; Coyne et al., 2015). Conversely, deletion of the NLS causes TDP-43 (ΔNLS-TDP-43) to accumulate in the cytoplasm inducing toxicity in primary cortical neurons of rats and neurodegeneration in mice, with rare cytoplasmic inclusions in motor neurons (Barmada et al., 2010; Igaz et al., 2011; Walker et al., 2015; Spiller et al., 2016). Collectively, these animal models support both loss and gain of function mechanisms and highlight the relationship between cytoplasmic accumulation of TDP-43 and neurodegeneration.
TDP-43, Stress Granules, and Translation
As an RNA binding protein, TDP-43 has a broad influence on gene expression, however, its impact on translation in particular is not well understood. Aside from direct alterations to the translation machinery, disturbances in granule dynamics and mRNA localization may contribute considerably to changes in the translatome. As new transcripts are generated in canonical cap-dependent translation (reviewed in Jackson et al., 2010), the growing pre-mRNA strands are available to associate with RNA binding proteins and form highly dynamic ribonucleoprotein (RNP) complexes (Moore and Proudfoot, 2009). The composition of an RNP complex changes with respect to its location and function in RNA metabolism, providing a mechanism for controlling the translational fate of the mRNAs it associates with (Singh et al., 2015). In the cytoplasm, translationally repressed RNP complexes can remodel into higher order structures such as stress granules, neuronal RNA transport granules, and P-bodies (Bowden and Dormann, 2016). Stress granules are a well-studied subset of RNP complexes that can rapidly assemble in response to stress or when translation initiation is inhibited, resulting in sequestration of mRNAs and translation initiation factors (reviewed in Anderson and Kedersha, 2009; Buchan and Parker, 2009). The abundance of highly disordered, prion-like domains in several RNA binding proteins predisposes stress granules to pathogenicity (King et al., 2012; Ramaswami et al., 2013). As proposed in the ribostasis hypothesis, stochastic assembly of RNA binding proteins within stress granules into self-propagating amyloid structures creates aberrant RNP aggregates that disrupt RNA homeostasis eventually leading to cell death (Ramaswami et al., 2013). Disease associated TDP-43 variants (A315T, M337V, Q343R, R361S) have been shown to disrupt stress granule dynamics (McDonald et al., 2011; Liu-Yesucevitz et al., 2014; Gordon et al., 2019). Although recent reports do not support the involvement of stress granules in TDP-43 proteinopathy, or suggest that TDP-43 association with stress granules may in fact be protective (McGurk et al., 2018; Gasset-Rosa et al., 2019; Mann et al., 2019), loss of function studies for the stress granule assembly factor G3BP1 revealed that stress granules facilitate but are not required for TDP-43 aggregation (Fernandes et al., 2020).
Translation can also occur independent of the 5′ cap, driven by the presence of Internal Ribosome Entry Sites (IRESs), initially discovered within viral RNAs and subsequently identified within mRNAs with certain structural features (Pelletier and Sonenberg, 1988). Cap independent translation mechanisms remain less understood compared to canonical translation and rely on RNA binding proteins known as IRES Trans-Activating Factors (ITAFs), which also play a variety of other roles in the cell (Hellen and Sarnow, 2001; Komar and Hatzoglou, 2011). Cap independent translation is prevalent during cellular stress and may play a role in neurological disorders such as ALS/FTD where stress granule dynamics appears to be altered (reviewed in Pelletier and Sonenberg, 1988; Hellen and Sarnow, 2001; Spriggs et al., 2008; Komar and Hatzoglou, 2011; Yang and Wang, 2019).
TDP-43—Ribosome Association and Effects on Translation
The ribostasis hypothesis posits that cytoplasmic accumulation of TDP-43 leads to mRNA sequestration and translation inhibition. In support of this hypothesis, various reports indicate that TDP-43 may act as a global repressor of translation. Indeed, TDP-43 depletion in HEK cells by siRNA causes a global increase in translational yield (Freibaum et al., 2010; Fiesel et al., 2012). Corroborating these findings, recent studies show that overexpression of cytoplasmically restricted TDP-43 (ΔNLS-TDP-43) in HEK or SHSY5Y cells causes a global reduction in protein synthesis, as demonstrated by Surface Sensing of Translation (SUnSET) assays that quantify puromycilation of nascent proteins (Russo et al., 2017; Charif et al., 2020). Further substantiating these findings in vivo, overexpression of cytoplasmic TDP-43 in the forebrains of transgenic mice (ΔNLS-TDP-43) results in decreased global translation as evidenced by both SUnSET and polysome profiling (Charif et al., 2020). Interestingly, puromycin incorporation experiments in TDPΔCR mice and primary neurons revealed an increase in global protein synthesis. Since ribosomal protein levels were unaltered, the authors concluded that ribosome assembly was increased (Gao et al., 2021).
TDP-43-induced changes in translation may occur through TDP-43 association with components of the protein synthesis machinery (Freibaum et al., 2010). For instance, induction of short-term oxidative stress in HeLa cells causes TDP-43 to shift from non-ribosomal fractions to monosomal fractions where it associates with stalled ribosomes via mRNA binding (Higashi et al., 2013). Following longer recovery time after arsenite induced stress, TDP-43 associates temporarily with polyribosomes indicating its ability to act both as a negative and as a positive regulator of translation (Higashi et al., 2013). Subsequent reports that TDP-43 co-migrates with non-ribosomal fractions and polyribosomes in Drosophila models of TDP-43 proteinopathy lend support to the notion that TDP-43 also associates with the translation machinery in vivo and may play a pivotal role in the dysregulation of translation in disease (Coyne et al., 2015). Consistent with these findings, polysome profiling in a motor neuron-like cell line or primary cortical neurons transfected with human TDP-43 (hTDP-43) shows that in the absence of stress, TDP-43 fractionates with polyribosomes and shifts to lighter RNP fractions upon polysome disruption with EDTA treatment (Neelagandan et al., 2019). Further analyses using ribosome profiling confirmed that TDP-43 associates with polyribosomes in actively translating cells (Neelagandan et al., 2019). Furthermore, ribosome footprinting in primary cortical neurons identified specific mRNA translational targets of TDP-43 (Neelagandan et al., 2019; see more in “Specific Candidate Targets,” below and in Table 1).
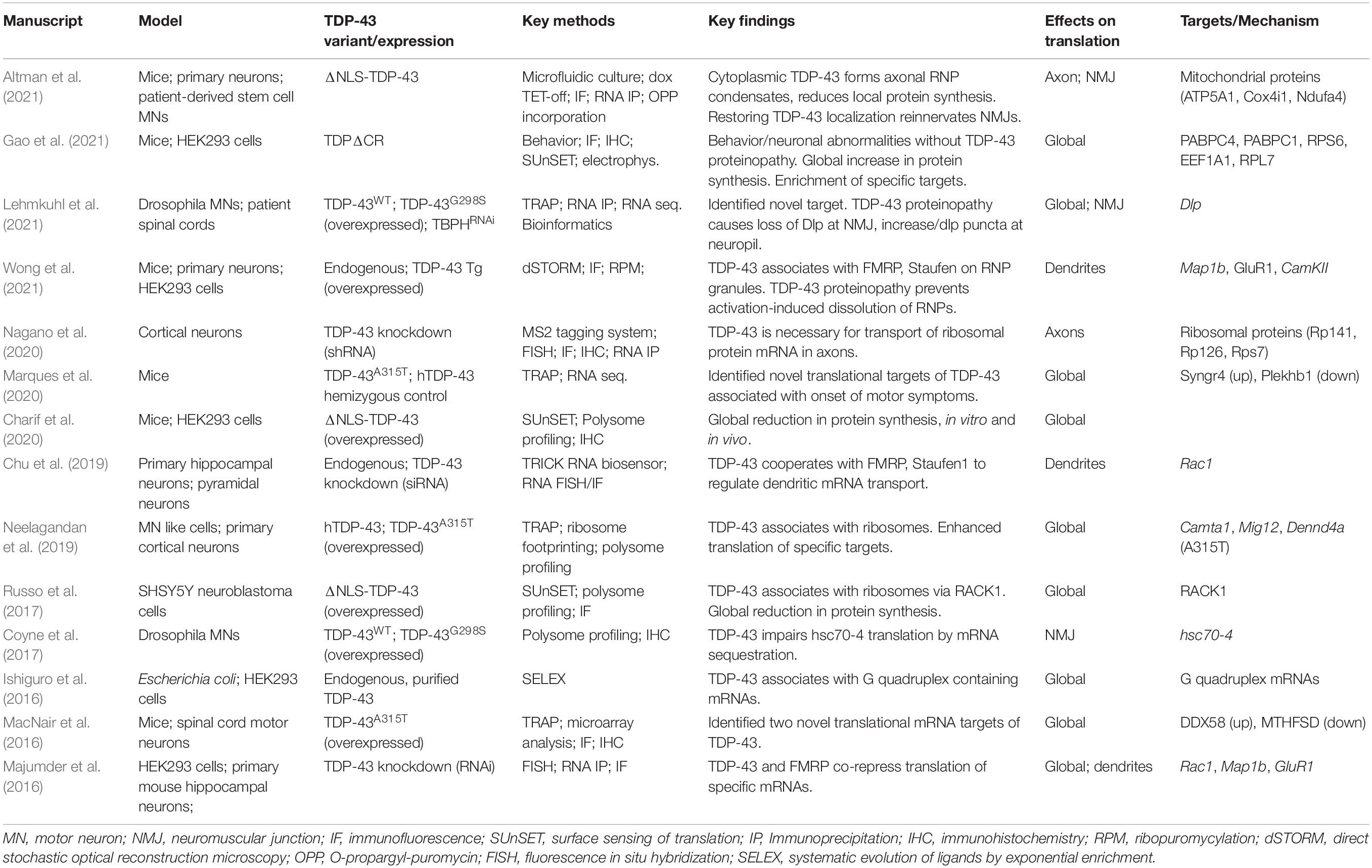
Table 1. Summary of key findings on the role and mechanism of TDP-43 in regulating translation.
These studies suggest that TDP-43 may play a direct role in translational control, perhaps by direct association with ribosomes. In SHSY5Y neuroblastoma cells, TDP-43 associates with ribosomes via RACK1, a WD40 scaffold protein that binds the 40S ribosomal subunit near the mRNA exit channel and functions as a docking site for several translation machinery proteins (Gallo and Manfrini, 2015; Russo et al., 2017). Immunostaining of mouse hippocampal neurons indicates that TDP-43 puncta strongly colocalize with RACK1 containing granules that correspond to ribosomes/polyribosomes within neurites. Furthermore, RACK1 colocalizes with TDP-43 in pathological inclusions in ALS patient spinal cords, further substantiating the notion that TDP-43’s association with ribosomes may promote TDP-43 aggregation and neurodegeneration (Russo et al., 2017). Several reports highlight TDP-43 dependent alterations in translation that may be indirect, resulting from impaired cellular homeostasis. When used in mouse or Drosophila models of TDP-43 proteinopathy, Translating Ribosomes Affinity Purification (TRAP) revealed complex changes in several targets and pathways within the in vivo motor neuron translatome, including RNA metabolism and cytoplasmic translation itself (MacNair et al., 2016; Lehmkuhl et al., 2021). Additionally, TDP-43 overexpression in Drosophila has been shown to cause the phosphorylation of eukaryotic initiation factor-2alpha (eIF2alpha), an indicator of stress granule formation and consequently, translational repression (Kim et al., 2014). Taken together, these findings support a role for TDP-43 in translation, mediated at least in part through direct association with ribosomes via RACK1 protein.
Local Dysregulation of Translation: Axons and Dendrites
RNP Granule Dynamics and Transport—A Role for TDP-43 in mRNA Localization
Neural plasticity during development and synaptic remodeling relies on the local synthesis of proteins in axons and dendrites (Holt et al., 2019). Within neurites, mRNAs are transported within RNP granules, which consist of mRNAs, ribosomes, translation factors and RNA binding proteins (Buffington et al., 2014; Glock et al., 2017). While mRNAs remain translationally inactive during transport within dendrites, they can be released from RNP granules in response to local activation and made available for translation at synaptic sites (reviewed in Akbalik and Schuman, 2014). Thus, RNA binding proteins play a critical role in RNP granule dynamics and, consequently, mRNA localization and translation (Kiebler and Bassell, 2006; Ayloo et al., 2017; Lim et al., 2017). Previous work has shown that TDP-43 is actively transported in motor neuron axons and mediates mRNA delivery to distal neuronal compartments, a mechanism that is impaired by ALS-linked mutations in TDP-43 (Fallini et al., 2012; Alami et al., 2014). Several reports indicate that TDP-43 colocalizes with the RNA binding proteins FMRP and Staufen1 in cultured neurons or in vivo, in Drosophila neurons, and cooperates with FMRP, an established translational regulator, to repress the translation of common mRNA targets (Wang et al., 2008; Majumder et al., 2012, 2016; Yu et al., 2012; Coyne et al., 2015).
The physical association between TDP-43, FMRP and Staufen in post-synaptic RNP granules was recently confirmed in primary mouse cortical neurons using super-resolution fluorescence microscopy (dSTORM) (Wong et al., 2021). To mechanistically investigate the potential co-regulation of RNP granule transport in dendrites via TDP-43, Staufen1, and FMRP, molecular beacons were used to characterize the dynamics of Rac1 RNP granules in primary hippocampal neurons (Chu et al., 2019). These elegant experiments indicate that TDP-43 cooperates with FMRP and Staufen1 for the transport of a specific subset of dendritic mRNAs. Upon depletion of TDP-43, Rac1 mRNA containing RNP granules remain stationary or exhibit brief bi-directional movement, indicating that TDP-43 is required for dendritic transport of Rac1 transcripts (Chu et al., 2019). Taken together, these findings support a role for TDP-43 in efficient mRNA transport and localization in neurons under basal conditions and in response to activity.
Evidence for TDP-43 Mediated Local Translation
In addition to its role in mRNA localization, TDP-43 was shown to be necessary for inhibition of translation within actively transporting RNP granules (Chu et al., 2019; see Figure 2). Indeed, depletion of TDP-43 by siRNA caused reduced entry of Rac1 mRNA into dendritic spines and premature translation as evidenced by an elegant TRICK RNA reporter that allows real time visualization of mRNA transport and translation (Chu et al., 2019). Substantiating this conclusion are findings that neuronal activation in wild type neurons but not in neurons with TDP-43 proteinopathy induces the disassembly of TDP-43-containing RNP granules, which in turn causes the release of TDP-43 target mRNAs for subsequent local translation at the synapse, as evidenced by puromycin labeling and immunofluorescence of target mRNAs (Wong et al., 2021). Taken together, these results suggest that TDP-43 proteinopathy impairs postsynaptic local translation and RNA metabolism through perturbation of activity-dependent RNP granule dynamics (Wang et al., 2008; Yu et al., 2012; Wong et al., 2021).
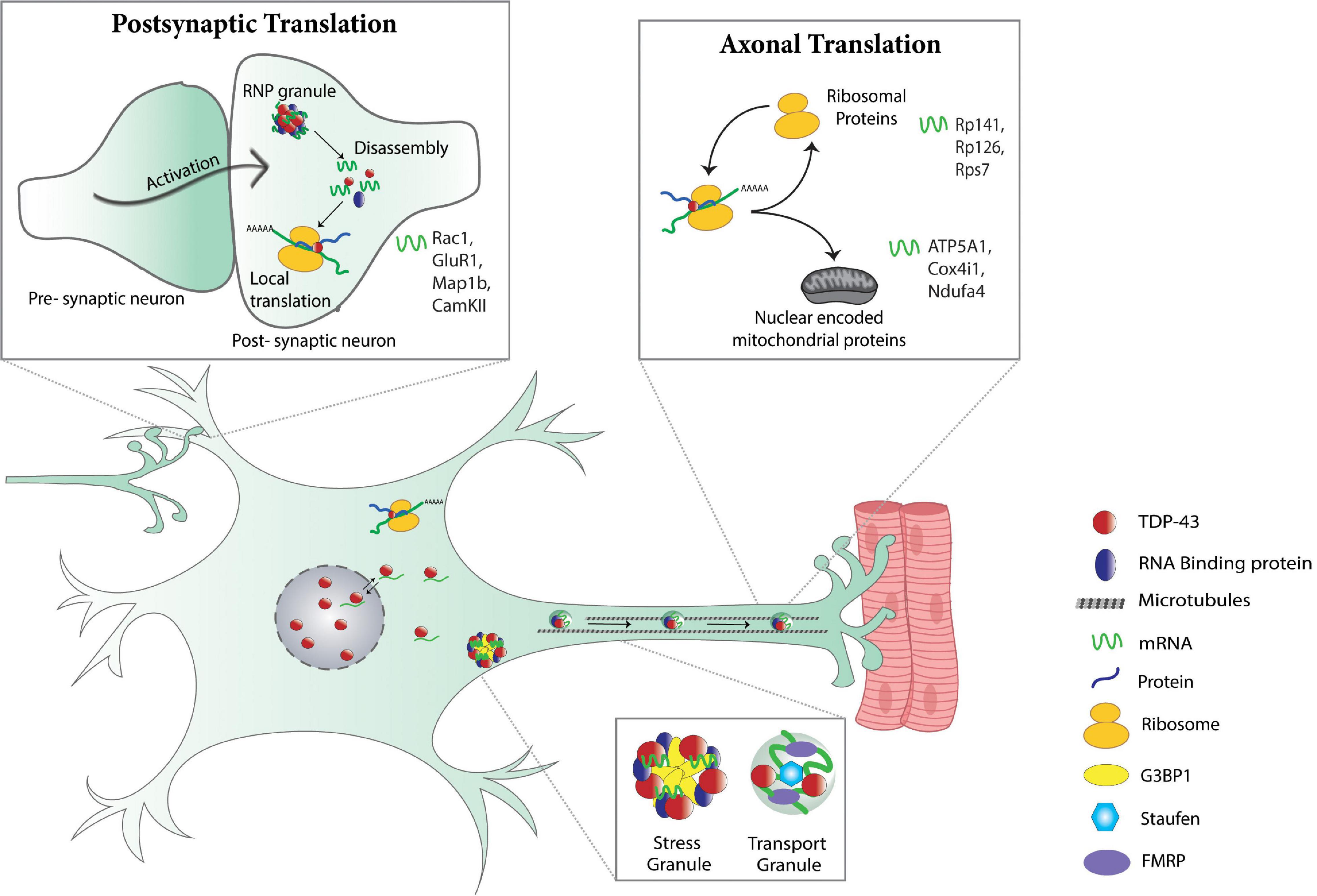
Figure 2. Physiological control of local translation by TDP-43. Post-synaptic translation (top left panel): in dendrites, activation of the post-synaptic neuron triggers the dissolution of dendritic RNP granules that in turn, causes the release of translationally silenced mRNAs making them available for translation. TDP-43 is a component of dendritic RNP granules and regulates dendritic translation of various mRNAs (Rac1, GluR1, Map1b, CamKII) (Wong et al., 2021). Stress and transport granules (bottom panel): TDP-43 associates with stress and RNA transport granules and is necessary for dendritic (Chu et al., 2019) and axonal transport (Alami et al., 2014) of mRNA-containing RNP granules. Axonal translation (top right panel): in axons, TDP-43 regulates local translation of mRNAs for ribosomal proteins (Rp141, Rp126, Rps7) and nuclear encoded mitochondrial proteins (ATP5A1, Cox4i1, Ndufa4) (Nagano et al., 2020; Altman et al., 2021).
Proper analysis of the spatiotemporal regulation of local translation in axons and at synaptic terminals requires the ability to experimentally manipulate them, independent from the soma. A recent report describes a novel platform for studying motor neuron axons and neuromuscular junctions by co-culturing primary motor neurons and muscles in microfluidic chambers, enabling fluidic separation and experimental manipulation of the motor neuron cell-bodies and axons, respectively (Altman et al., 2021). This setup allowed for compartment specific measurements of protein synthesis based on selective labeling with O-propargyl-puromycin (OPP) of nascent proteins in the motor neuron cell body, axon, or at the neuromuscular junction. Using the doxycycline (dox) TET-off system, transient accumulation of cytoplasmic TDP-43 (ΔNLS-TDP-43) was induced in axons of motor neurons, which resulted in the assembly of G3BP1-nucleated RNP condensates containing TDP-43. Formation of RNP condensates correlated with a reduction in local protein synthesis in axons and presynaptically at the NMJs, consistent with in vivo findings in ΔNLS-TDP-43 transgenic mice (Altman et al., 2021; Figure 2). Importantly, local protein synthesis recovered after dissolution of TDP-43 RNP condensates by a G3BP1 peptide (TAT-fused to residues 190–208 of G3BP1), suggesting that the reduction in local protein synthesis is a function of mRNA sequestration by TDP-43 RNP condensates. Clearance of TDP-43 from the cytoplasm by re-introducing doxycycline resulted in similar restoration of local protein synthesis and, remarkably, reinnervation of neuromuscular junctions, as evidenced by the co-expression of pre- and post-synaptic markers (Altman et al., 2021).
Additional evidence connecting mRNA transport and translation deficits was obtained from live imaging and microarray analyses of mouse cortical neurons depleted of TDP-43 (Nagano et al., 2020). RNAi knock-down of TDP-43 caused downregulation of a particular subset of ribosomal protein (RP) mRNAs in neurites but not in the cell-body, as shown by microarray analysis. To track the transport of RP mRNA by TDP-43 in living cells, fluorescence microscopy was conducted on cultured cortical neurons simultaneously expressing mCherry TDP-43 and mRNAs labeled using the MS2 tagging system (Bertrand et al., 1998). Loss of TDP-43 results in decreased axonal transport and subsequent local translation of RP mRNAs, along with diminished translational activity in axons. Further investigation by in situ hybridization of RP mRNAs, both with and without 5′UTRs, and TDP-43 immunostaining revealed that the 5′UTR is necessary for TDP-43-mediated axonal transport of RP mRNAs (Nagano et al., 2020). Taken together, these results show that TDP-43 proteinopathy impairs axonal translation with direct consequences on neuromuscular junction integrity and ribosomal protein expression, which in turn can cause more general alterations in local translation (Nagano et al., 2020; Altman et al., 2021).
Specific Candidate Targets as Mediators of TDP-43 Toxicity
Although TDP-43 appears to play a critical role in modulating RNP granule dynamics and mRNA localization, identifying specific translational targets of TDP-43 remains paramount in the development of novel therapeutics. Early reports identified Rac1 and futsch/Map1b, key regulators of neural plasticity, as specific mRNA targets of TDP-43 (Majumder et al., 2012; Coyne et al., 2014). Rac1 is a known positive regulator of AMPAR-mediated spinogenesis and is shown to be upregulated upon depletion of TDP-43 in mouse hippocampal neurons (Majumder et al., 2012). Consistent with these findings, TDP-43 has been shown to cooperate with FMRP to co-repress the translation initiation of Rac1, GluR1, and Map1b in dendrites (Majumder et al., 2016). In the context of TDP-43 proteinopathy modeled by TDP-43 overexpression, neuronal activation-induced dissolution of post-synaptic RNPs containing Map1b, GluR1, and CamkII is impaired and accompanied by reduced translation as evidenced by ribopuromycylation assays (Wong et al., 2021). In Drosophila, overexpression of TDP-43 (wild type or the disease associated variant G298S) in motor neurons represses the translation of futsch, as evidenced by a reduction in Futsch protein expression at the NMJ and a shift in futsch mRNA from actively translating polysome fractions to non-translating RNPs (Coyne et al., 2014). Dysregulation of futsch mRNA reduces microtubule stability at the neuromuscular junction, which is mitigated by restoring futsch levels using overexpression in the context of TDP-43 proteinopathy (Coyne et al., 2014). Another mRNA target with functional consequences on synaptic physiology is hsc70-4 mRNA (Coyne et al., 2017). More specifically, TDP-43 impairs the translation of hsc70-4 mRNA due to its sequestration by mutant TDP-43G298S, as evidenced by soluble vs. insoluble and polysome fractionations (Coyne et al., 2017). Upon restoration of hsc70-4 levels via overexpression, synaptic vesicle endocytosis measured using FM1-43 dye uptake is rescued, suggesting that dysregulation of hsc70-4 translation mediates TDP-43 toxicity at the neuromuscular synapse (Coyne et al., 2017). To identify specific mRNAs transported by TDP-43, in vitro systematic evolution of ligands by exponential enrichment (SELEX) demonstrated that RNAs bound by TDP-43 contained G-quadruplexes and were transported to distal neurites along with TDP-43 for local translation, consistent with G-quadruplexes being novel structural motifs within RNAs associated with TDP-43 (Ishiguro et al., 2016).
Additional translational targets of TDP-43 have recently been reported. In aged mice expressing mutant TDP-43A315T, TRAP and microarray analyses revealed seven genes that are differentially expressed. Of these, four candidates (Ddx58, Ccl4, Prickle4, and Mthfsd) were confirmed by immunofluorescence in mouse spinal cord motor neurons. Immunohistochemistry experiments in patient derived spinal cords confirmed that DDX58 and MTHFSD, both of which are RNA binding proteins, are differentially expressed in ALS compared to controls (MacNair et al., 2016). Another study characterized changes to the motor neuron translatome in TDP-43A315T transgenic mice, specifically at the onset of motor symptoms (Marques et al., 2020). By comparing transgenic mice with wild type littermates and asymptomatic mice hemizygous for the wild type hTDP-43 transgene (Chat bacTRAP; hTDP-43WT), translational changes specifically associated with disease could be distinguished. Candidate mRNAs Syngr4 and Plekhb1 were up- and downregulated, respectively, at the transition from asymptomatic to early symptomatic motor dysfunction, as identified by TRAP and RNA sequencing approaches in spinal cord motor neurons (Marques et al., 2020). Validation experiments confirmed that SYNGR4 and PLEKHB1 protein levels were dysregulated as predicted. Interestingly, these targets were altered in two different mouse models of TDP-43 proteinopathy (TDP-43A315T and TDP-43Q331K), suggesting that they may play critical role in the progression of mutant TDP-43 driven ALS (Marques et al., 2020).
Moreover, overexpression of TDP-43 (wild type and A315T) in motor neuron-like cells and in primary cortical neurons enhanced the translation of Camta1 and Mig12 mRNAs through 5′UTR binding. Conversely, translation of Dennd4a was enhanced specifically by mutant TDP-43A315T through the 3′UTR (Neelagandan et al., 2019). Importantly, independent studies in cultured ALS motor neurons and an in vivo murine model of Parkinson’s disease identified CAMTA1 and DENND4A as “master regulators” of transcriptional programs in neurodegenerative disease (Brichta et al., 2015; Ikiz et al., 2015).
A more recent study using RNA immunoprecipitation assays, TRAP, and bioinformatics analyses, reported the glypican Dally like protein (Dlp) as a novel TDP-43 candidate target, based on the enrichment of dlp mRNA in TDP-43 complexes and depletion from ribosomes in the context of TDP-43 proteinopathy (Lehmkuhl et al., 2021). This study further shows that surprisingly, while Dlp expression in synaptic terminals at the neuromuscular junction is significantly reduced, Dlp protein accumulates in puncta within the ventral cord neuropil suggesting that a combination of translation and transport defects are at play, and that these effects may be compartment specific. Interestingly, TDP-43 knock-down by RNAi (TBPHRNAi) was sufficient to deplete Dlp from the NMJ, but not induce significant ventral cord neuropil puncta, highlighting that TDP-43 nuclear depletion and cytoplasmic accumulation have distinct contributions to disease pathomechanism. Restoring Dlp by overexpression, in motor neurons specifically, restores Dlp expression at the synaptic terminal and mitigates TDP-43-dependent locomotor deficits. Importantly, these findings align with ALS patient data indicating that GPC6 protein, a human homolog of Dlp, accumulates in puncta within the spinal cord, mimicking the Dlp accumulations in the Drosophila ventral cord neuropil (Lehmkuhl et al., 2021). Additional targets identified in Drosophila using TRAP include metabolic pathways (e.g., pentose phosphate, nuclear encoded components electron transport chain components, oxidative stress). Of note, overexpressing glucose 6 phosphate dehydrogenase (G6PD), which is predicted to be downtranslated in the context of TDP-43G298S, mitigates locomotor deficits in Drosophila models and highlights a role for metabolic rewiring in degenerating motor neurons (Lehmkuhl et al., 2021).
Interestingly, several recent studies have identified mRNA targets of TDP-43 that encode distinct populations of proteins, such as ribosomal proteins (RPs), mitochondrial proteins, and translation factors (Nagano et al., 2020; Altman et al., 2021; Gao et al., 2021; Lehmkuhl et al., 2021). For instance, cytoplasmic TDP-43 caused a specific decrease in nuclear-encoded mitochondrial proteins, particularly ATP5A1, Cox4i1, and Ndufa4 (Altman et al., 2021). Notably, mRNA levels of these three proteins were unchanged and even moderately increased in TDP-43ΔNLS samples, suggesting TDP-43 accumulation in axons directly impairs local translation of nuclear encoded mitochondrial proteins (Altman et al., 2021). TDP-43 depletion has also been shown to reduce the number and functionality of mitochondria in MN axons, and subsequent treatment with nicotinamide (NAM), a precursor for nicotinamide adenine dinucleotide (NAD +), rescues axon growth defects and capacity for protein synthesis (Briese et al., 2020). In agreement with these findings, depletion of TDP-43 in cortical neurons causes a specific downregulation of ribosomal protein mRNAs in neurites that coincides with axonal extension defects, which are rescued upon overexpression of RP mRNAs Rp141, Rp126, or Rps7 (Nagano et al., 2020). In addition to translational changes in mitochondrial and ribosomal protein mRNA, several translation factors, namely PABPC4, PABPC1, RPS6, EEF1A1, and RPL7, have been shown to co-precipitate with wild type TDP-43 and are enriched in TDPΔCR mutant mice (Gao et al., 2021). Taken together, these findings highlight a role for TDP-43 in regulating various cellular pathways including synaptic metabolism, cellular energetics, and cytoplasmic translation (Marques et al., 2020; Nagano et al., 2020; Altman et al., 2021; Lehmkuhl et al., 2021).
Discussion
Growing evidence of alterations to the translatome in TDP-43 proteinopathies supports a multifaceted role for TDP-43 involvement in translation, highlighting both physiological and pathogenic functions. TDP-43 knock-down studies demonstrate that TDP-43 is necessary for the RNP-mediated transport of mRNAs into axons (Alami et al., 2014; Nagano et al., 2020) and dendrites (Chu et al., 2019), and that loss of TDP-43-mediated transport leads to a reduction in local translation. Underscoring the importance of mRNA transport in fueling local protein synthesis, these findings suggest that TDP-43 might dysregulate translation, in part, through indirect mechanisms involving the supply of mRNA to distal neurites. Indeed, additional studies have recently shown that cytoplasmic accumulation of TDP-43 disrupts RNP granule dynamics, impairing local translation in dendrites (Wong et al., 2021) and axons (Altman et al., 2021). Consistent with the ribostasis hypothesis (Ramaswami et al., 2013), cytoplasmic TDP-43 appears to inhibit translation by sequestering mRNAs in aberrant RNP granules, suggesting that TDP-43 intimately attenuates mRNA accessibility in neurites and thus alterations in TDP-43 structure or localization could destabilize the chain of events preceding local translation.
Although TDP-43 dependent alterations in the global translatome are well documented (Fiesel et al., 2012; MacNair et al., 2016; Russo et al., 2017; Neelagandan et al., 2019; Charif et al., 2020; Lehmkuhl et al., 2021), and TDP-43 co-fractionates with actively translating ribosomes (Coyne et al., 2015; Neelagandan et al., 2019), evidence for its direct association with ribosomes has only recently been uncovered (Russo et al., 2017) and requires further investigation, perhaps using more powerful structural techniques such as cryo-EM. Complicating the understanding of TDP-43’s role in translation is its global effect on cytoplasmic translation itself (Lehmkuhl et al., 2021). Mechanistically, this could occur via direct transport and translation regulation of ribosome protein mRNAs, specifically via the 5′UTR, as recently demonstrated (Nagano et al., 2020).
Regardless of mechanism, it is clear that TDP-43 accumulation in the cytoplasm is driving translation dysregulation (see Figure 3). The identification of specific targets is critical for understanding the mechanisms underlying TDP-43 dependent proteinopathies and has already uncovered common alterations across models, such as cellular metabolism, synaptic function and cytoplasmic translation (Coyne et al., 2014, 2017; Majumder et al., 2016; Chu et al., 2019; Nagano et al., 2020; Altman et al., 2021; Lehmkuhl et al., 2021). However, developing therapeutic strategies that mitigate neurodegeneration one target at a time does not seem like a practical approach as prioritizing targets is difficult and would require a combination strategy that simultaneously affects multiple pathways, one for each target. A possible solution to this challenge is the restoration of TDP-43 localization to the nucleus, which was shown to cause the reinnervation of neuromuscular junctions (Altman et al., 2021). That said, strategies to enhance translation locally at synapses could also offer mitigating approaches to TDP-43 dependent neurodegeneration.
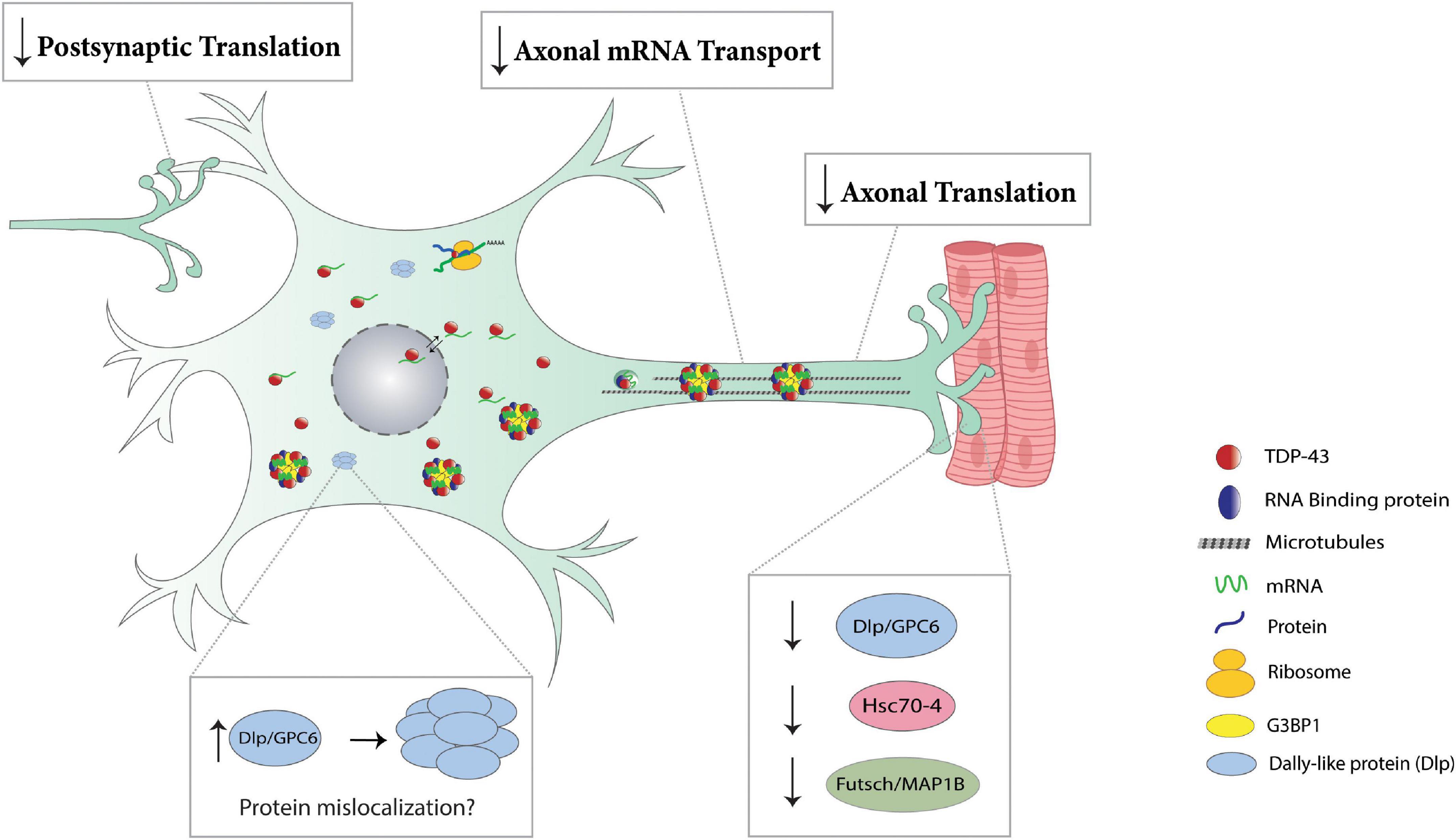
Figure 3. Translation impairments in TDP-43 proteinopathies. Post-synaptically, TDP-43 proteinopathy prevents the dissolution of dendritic RNP granules (Wong et al., 2021), thereby repressing the local translation of key mediators of synaptic plasticity (Rac1, GluR1, Map1b, CamKII). Cytoplasmic accumulation of TDP-43 in axons results in the formation of RNP condensates and causes a reduction in local translation of various mRNAs, including ribosomal proteins (Rp141, Rp126, Rps7) and nuclear encoded mitochondrial proteins (ATP5A1, Cox4i1, Ndufa4) (Nagano et al., 2020; Altman et al., 2021). TDP-43 proteinopathy reduces axonal mRNA transport as evidenced by a reduction in axonal levels of futsch mRNA at the Drosophila NMJ (Coyne et al., 2015). In Drosophila, TDP-43 proteinopathy represses the translation of dlp, futsch/Map1b, and hsc70-4 causing a reduction in respective protein expression at the NMJ (bottom right panel) (Coyne et al., 2015, 2017; Lehmkuhl et al., 2021). TDP-43 proteinopathy causes Dlp/GPC6 protein to accumulate in puncta at the ventral cord neuropil in Drosophila and ALS spinal cords, respectively (bottom left panel) (Lehmkuhl et al., 2021).
Questions remain on the potential connections between TDP-43’s role in transcription, splicing, RNA stability and translation that could help identify additional strategies for restoring more downstream translation deficits. A significant caveat is the limited knowledge regarding the physiological role of TDP-43 in RNA processing within the cytoplasm. The discovery of the cytoplasmically localized sTDP-43 (Weskamp et al., 2020), the increased availability of endogenously tagged TDP-43 models, and higher resolution imaging technologies, may help overcome this barrier and lead to a better understanding of the mechanistic differences between healthy and degenerating neurons that could in turn uncover novel therapeutic strategies for ALS and related neurodegenerative disorders.
Author Contributions
RTB, NPM, and DCZ wrote the manuscript. SL, RTB, and NPM made the figures. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Afroz, T., Hock, E. M., Ernst, P., Foglieni, C., Jambeau, M., Gilhespy, L. A. B., et al. (2017). Functional and dynamic polymerization of the ALS-linked protein TDP-43 antagonizes its pathologic aggregation. Nat. Commun. 8:45. doi: 10.1038/s41467-017-00062-0
Akbalik, G., and Schuman, E. M. (2014). Molecular biology. mRNA, live and unmasked. Science 343, 375–376. doi: 10.1126/science.1249623
Akins, M. R., Berk-Rauch, H. E., and Fallon, J. R. (2009). Presynaptic translation: stepping out of the postsynaptic shadow. Front. Neural Circuits 3:17. doi: 10.3389/neuro.04.017.2009
Alami, N. H., Smith, R. B., Carrasco, M. A., Williams, L. A., Winborn, C. S., Han, S. S., et al. (2014). Axonal transport of TDP-43 mRNA granules is impaired by ALS-causing mutations. Neuron 81, 536–543. doi: 10.1016/j.neuron.2013.12.018
Altman, T., Ionescu, A., Ibraheem, A., Priesmann, D., Gradus-Pery, T., Farberov, L., et al. (2021). Axonal TDP-43 condensates drive neuromuscular junction disruption through inhibition of local synthesis of nuclear encoded mitochondrial proteins. Nat. Commun. 12:6914. doi: 10.1038/s41467-021-27221-8
Andersen, P. M., and Al-Chalabi, A. (2011). Clinical genetics of amyotrophic lateral sclerosis: what do we really know? Nat. Rev. Neurol. 7, 603–615. doi: 10.1038/nrneurol.2011.150
Ayloo, S., Guedes-Dias, P., Ghiretti, A. E., and Holzbaur, E. L. F. (2017). Dynein efficiently navigates the dendritic cytoskeleton to drive the retrograde trafficking of BDNF/TrkB signaling endosomes. Mol. Biol. Cell 28, 2543–2554. doi: 10.1091/mbc.E17-01-0068
Babinchak, W. M., Haider, R., Dumm, B. K., Sarkar, P., Surewicz, K., Choi, J. K., et al. (2019). The role of liquid-liquid phase separation in aggregation of the TDP-43 low-complexity domain. J. Biol. Chem. 294, 6306–6317. doi: 10.1074/jbc.RA118.007222
Baleriola, J., Walker, C. A., Jean, Y. Y., Crary, J. F., Troy, C. M., Nagy, P. L., et al. (2014). Axonally synthesized ATF4 transmits a neurodegenerative signal across brain regions. Cell 158, 1159–1172. doi: 10.1016/j.cell.2014.07.001
Barmada, S. J., Skibinski, G., Korb, E., Rao, E. J., Wu, J. Y., and Finkbeiner, S. (2010). Cytoplasmic mislocalization of TDP-43 is toxic to neurons and enhanced by a mutation associated with familial amyotrophic lateral sclerosis. J. Neurosci. 30, 639–649. doi: 10.1523/JNEUROSCI.4988-09.2010
Batista, A. F., and Hengst, U. (2016). Intra-axonal protein synthesis in development and beyond. Int. J. Dev. Neurosci. 55, 140–149. doi: 10.1016/j.ijdevneu.2016.03.004
Bear, M. F., Dolen, G., Osterweil, E., and Nagarajan, N. (2008). Fragile X: translation in action. Neuropsychopharmacology 33, 84–87. doi: 10.1038/sj.npp.1301610
Bertrand, E., Chartrand, P., Schaefer, M., Shenoy, S. M., Singer, R. H., and Long, R. M. (1998). Localization of ASH1 mRNA particles in living yeast. Mol. Cell 2, 437–445. doi: 10.1016/s1097-2765(00)80143-4
Bowden, H. A., and Dormann, D. (2016). Altered mRNP granule dynamics in FTLD pathogenesis. J. Neurochem. 138(Suppl. 1), 112–133. doi: 10.1111/jnc.13601
Brichta, L., Shin, W., Jackson-Lewis, V., Blesa, J., Yap, E. L., Walker, Z., et al. (2015). Identification of neurodegenerative factors using translatome-regulatory network analysis. Nat. Neurosci. 18, 1325–1333. doi: 10.1038/nn.4070
Briese, M., Saal-Bauernschubert, L., Luningschror, P., Moradi, M., Dombert, B., Surrey, V., et al. (2020). Loss of Tdp-43 disrupts the axonal transcriptome of motoneurons accompanied by impaired axonal translation and mitochondria function. Acta Neuropathol. Commun. 8:116. doi: 10.1186/s40478-020-00987-6
Buchan, J. R., and Parker, R. (2009). Eukaryotic stress granules: the ins and outs of translation. Mol. Cell 36, 932–941. doi: 10.1016/j.molcel.2009.11.020
Buffington, S. A., Huang, W., and Costa-Mattioli, M. (2014). Translational control in synaptic plasticity and cognitive dysfunction. Annu. Rev. Neurosci. 37, 17–38. doi: 10.1146/annurev-neuro-071013-014100
Buratti, E. (2015). Functional significance of TDP-43 mutations in disease. Adv. Genet. 91, 1–53. doi: 10.1016/bs.adgen.2015.07.001
Cao, Q., Boyer, D. R., Sawaya, M. R., Ge, P., and Eisenberg, D. S. (2019). Cryo-EM structures of four polymorphic TDP-43 amyloid cores. Nat. Struct. Mol. Biol. 26, 619–627. doi: 10.1038/s41594-019-0248-4
Casafont, I., Bengoechea, R., Tapia, O., Berciano, M. T., and Lafarga, M. (2009). TDP-43 localizes in mRNA transcription and processing sites in mammalian neurons. J. Struct. Biol. 167, 235–241. doi: 10.1016/j.jsb.2009.06.006
Charif, S. E., Luchelli, L., Vila, A., Blaustein, M., and Igaz, L. M. (2020). Cytoplasmic expression of the ALS/FTD-related protein TDP-43 decreases global translation both in vitro and in vivo. Front. Cell Neurosci. 14:594561. doi: 10.3389/fncel.2020.594561
Chu, J. F., Majumder, P., Chatterjee, B., Huang, S. L., and Shen, C. J. (2019). TDP-43 regulates coupled dendritic mRNA transport-translation processes in co-operation with FMRP and Staufen1. Cell Rep. 29, 3118.e3116–3133.e3116. doi: 10.1016/j.celrep.2019.10.061
Conicella, A. E., Dignon, G. L., Zerze, G. H., Schmidt, H. B., D’ordine, A. M., Kim, Y. C., et al. (2020). TDP-43 alpha-helical structure tunes liquid-liquid phase separation and function. Proc. Natl. Acad. Sci. U.S.A. 117, 5883–5894. doi: 10.1073/pnas.1912055117
Costa, C. J., and Willis, D. E. (2018). To the end of the line: axonal mRNA transport and local translation in health and neurodegenerative disease. Dev. Neurobiol. 78, 209–220. doi: 10.1002/dneu.22555
Coyne, A. N., Lorenzini, I., Chou, C. C., Torvund, M., Rogers, R. S., Starr, A., et al. (2017). Post-transcriptional inhibition of Hsc70-4/HSPA8 expression leads to synaptic vesicle cycling defects in multiple models of ALS. Cell Rep. 21, 110–125.
Coyne, A. N., Siddegowda, B. B., Estes, P. S., Johannesmeyer, J., Kovalik, T., Daniel, S. G., et al. (2014). Futsch/MAP1B mRNA is a translational target of TDP-43 and is neuroprotective in a drosophila model of amyotrophic lateral sclerosis. J. Neurosci. 34, 15962–15974. doi: 10.1523/JNEUROSCI.2526-14.2014
Coyne, A. N., Yamada, S. B., Siddegowda, B. B., Estes, P. S., Zaepfel, B. L., Johannesmeyer, J. S., et al. (2015). Fragile X protein mitigates TDP-43 toxicity by remodeling RNA granules and restoring translation. Hum. Mol. Genet. 24, 6886–6898. doi: 10.1093/hmg/ddv389
Ederle, H., and Dormann, D. (2017). TDP-43 and FUS en route from the nucleus to the cytoplasm. FEBS Lett. 591, 1489–1507. doi: 10.1002/1873-3468.12646
Ederle, H., Funk, C., Abou-Ajram, C., Hutten, S., Funk, E. B. E., Kehlenbach, R. H., et al. (2018). Nuclear egress of TDP-43 and FUS occurs independently of Exportin-1/CRM1. Sci. Rep. 8:7084. doi: 10.1038/s41598-018-25007-5
Elden, A. C., Kim, H. J., Hart, M. P., Chen-Plotkin, A. S., Johnson, B. S., Fang, X., et al. (2010). Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature 466, 1069–1075. doi: 10.1038/nature09320
Estes, P. S., Boehringer, A., Zwick, R., Tang, J. E., Grigsby, B., and Zarnescu, D. C. (2011). Wild-type and A315T mutant TDP-43 exert differential neurotoxicity in a Drosophila model of ALS. Hum. Mol. Genet. 20, 2308–2321. doi: 10.1093/hmg/ddr124
Fallini, C., Bassell, G. J., and Rossoll, W. (2012). The ALS disease protein TDP-43 is actively transported in motor neuron axons and regulates axon outgrowth. Hum. Mol. Genet. 21, 3703–3718. doi: 10.1093/hmg/dds205
Fernandes, N., Nero, L., Lyons, S. M., Ivanov, P., Mittelmeier, T. M., Bolger, T. A., et al. (2020). Stress granule assembly can facilitate but is not required for TDP-43 cytoplasmic aggregation. Biomolecules 10:1367.
Ferro, D., Yao, S., and Zarnescu, D. C. (2018). Dynamic duo - FMRP and TDP-43: regulating common targets, causing different diseases. Brain Res. 1693(Pt A), 37–42. doi: 10.1016/j.brainres.2018.04.034
Fiesel, F. C., Weber, S. S., Supper, J., Zell, A., and Kahle, P. J. (2012). TDP-43 regulates global translational yield by splicing of exon junction complex component SKAR. Nucleic Acids Res. 40, 2668–2682. doi: 10.1093/nar/gkr1082
Flores, B. N., Li, X., Malik, A. M., Martinez, J., Beg, A. A., and Barmada, S. J. (2019). An intramolecular salt bridge linking TDP43 RNA binding, protein stability, and TDP43-dependent neurodegeneration. Cell Rep. 27, 1133.e1138–1150.e1138. doi: 10.1016/j.celrep.2019.03.093
Freibaum, B. D., Chitta, R. K., High, A. A., and Taylor, J. P. (2010). Global analysis of TDP-43 interacting proteins reveals strong association with RNA splicing and translation machinery. J. Proteome Res. 9, 1104–1120. doi: 10.1021/pr901076y
Gallo, S., and Manfrini, N. (2015). Working hard at the nexus between cell signaling and the ribosomal machinery: an insight into the roles of RACK1 in translational regulation. Translation (Austin) 3:e1120382. doi: 10.1080/21690731.2015.1120382
Gao, J., Wang, L., Ren, X., Dunn, J. R., Peters, A., Miyagi, M., et al. (2021). Translational regulation in the brain by TDP-43 phase separation. J. Cell Biol. 220:e202101019. doi: 10.1083/jcb.202101019
Gasset-Rosa, F., Lu, S., Yu, H., Chen, C., Melamed, Z., Guo, L., et al. (2019). Cytoplasmic TDP-43 de-mixing independent of stress granules drives inhibition of nuclear import, loss of nuclear TDP-43, and cell death. Neuron 102, 339–357e337. doi: 10.1016/j.neuron.2019.02.038
Glock, C., Heumuller, M., and Schuman, E. M. (2017). mRNA transport & local translation in neurons. Curr. Opin. Neurobiol. 45, 169–177.
Gordon, D., Dafinca, R., Scaber, J., Alegre-Abarrategui, J., Farrimond, L., Scott, C., et al. (2019). Single-copy expression of an amyotrophic lateral sclerosis-linked TDP-43 mutation (M337V) in BAC transgenic mice leads to altered stress granule dynamics and progressive motor dysfunction. Neurobiol. Dis. 121, 148–162. doi: 10.1016/j.nbd.2018.09.024
Grese, Z. R., Bastos, A. C., Mamede, L. D., French, R. L., Miller, T. M., and Ayala, Y. M. (2021). Specific RNA interactions promote TDP-43 multivalent phase separation and maintain liquid properties. EMBO Rep. 22:e53632. doi: 10.15252/embr.202153632
Hellen, C. U., and Sarnow, P. (2001). Internal ribosome entry sites in eukaryotic mRNA molecules. Genes Dev. 15, 1593–1612. doi: 10.1101/gad.891101
Higashi, S., Kabuta, T., Nagai, Y., Tsuchiya, Y., Akiyama, H., and Wada, K. (2013). TDP-43 associates with stalled ribosomes and contributes to cell survival during cellular stress. J. Neurochem. 126, 288–300. doi: 10.1111/jnc.12194
Holt, C. E., Martin, K. C., and Schuman, E. M. (2019). Local translation in neurons: visualization and function. Nat. Struct. Mol. Biol. 26, 557–566. doi: 10.1038/s41594-019-0263-5
Huang, C., Tong, J., Bi, F., Zhou, H., and Xia, X. G. (2012). Mutant TDP-43 in motor neurons promotes the onset and progression of ALS in rats. J. Clin. Investigat. 122, 107–118. doi: 10.1172/JCI59130
Igaz, L. M., Kwong, L. K., Lee, E. B., Chen-Plotkin, A., Swanson, E., Unger, T., et al. (2011). Dysregulation of the ALS-associated gene TDP-43 leads to neuronal death and degeneration in mice. J. Clin. Invest. 121, 726–738. doi: 10.1172/JCI44867
Ihara, R., Matsukawa, K., Nagata, Y., Kunugi, H., Tsuji, S., Chihara, T., et al. (2013). RNA binding mediates neurotoxicity in the transgenic Drosophila model of TDP-43 proteinopathy. Hum. Mol. Genet. 22, 4474–4484. doi: 10.1093/hmg/ddt296
Ikiz, B., Alvarez, M. J., Re, D. B., Le Verche, V., Politi, K., Lotti, F., et al. (2015). The regulatory machinery of neurodegeneration in in vitro models of amyotrophic lateral sclerosis. Cell Rep. 12, 335–345. doi: 10.1016/j.celrep.2015.06.019
Ishiguro, A., Kimura, N., Watanabe, Y., Watanabe, S., and Ishihama, A. (2016). TDP-43 binds and transports G-quadruplex-containing mRNAs into neurites for local translation. Genes Cells 21, 466–481. doi: 10.1111/gtc.12352
Jackson, R. J., Hellen, C. U., and Pestova, T. V. (2010). The mechanism of eukaryotic translation initiation and principles of its regulation. Nat. Rev. Mol. Cell Biol. 11, 113–127. doi: 10.1038/nrm2838
Johnson, B. S., Mccaffery, J. M., Lindquist, S., and Gitler, A. D. (2008). A yeast TDP-43 proteinopathy model: exploring the molecular determinants of TDP-43 aggregation and cellular toxicity. Proc. Natl. Acad. Sci. U.S.A. 105, 6439–6444. doi: 10.1073/pnas.0802082105
Johnson, B. S., Snead, D., Lee, J. J., Mccaffery, J. M., Shorter, J., and Gitler, A. D. (2009). TDP-43 is intrinsically aggregation-prone, and amyotrophic lateral sclerosis-linked mutations accelerate aggregation and increase toxicity. J. Biol. Chem. 284, 20329–20339. doi: 10.1074/jbc.M109.010264
Kabashi, E., Lin, L., Tradewell, M. L., Dion, P. A., Bercier, V., Bourgouin, P., et al. (2009). Gain and loss of function of ALS-related mutations of TARDBP (TDP-43) cause motor deficits in vivo. Hum. Mol. Genet. 19, 671–683. doi: 10.1093/hmg/ddp534
Kiebler, M. A., and Bassell, G. J. (2006). Neuronal RNA granules: movers and makers. Neuron 51, 685–690. doi: 10.1016/j.neuron.2006.08.021
Kim, H. J., Raphael, A. R., Ladow, E. S., Mcgurk, L., Weber, R. A., Trojanowski, J. Q., et al. (2014). Therapeutic modulation of eIF2alpha phosphorylation rescues TDP-43 toxicity in amyotrophic lateral sclerosis disease models. Nat. Genet. 46, 152–160. doi: 10.1038/ng.2853
King, O. D., Gitler, A. D., and Shorter, J. (2012). The tip of the iceberg: RNA-binding proteins with prion-like domains in neurodegenerative disease. Brain Res. 1462, 61–80. doi: 10.1016/j.brainres.2012.01.016
Komar, A. A., and Hatzoglou, M. (2011). Cellular IRES-mediated translation: the war of ITAFs in pathophysiological states. Cell Cycle 10, 229–240. doi: 10.4161/cc.10.2.14472
Kraemer, B. C., Schuck, T., Wheeler, J. M., Robinson, L. C., Trojanowski, J. Q., Lee, V. M., et al. (2010). Loss of murine TDP-43 disrupts motor function and plays an essential role in embryogenesis. Acta Neuropathol. 119, 409–419. doi: 10.1007/s00401-010-0659-0
Lehmkuhl, E. M., Loganathan, S., Alsop, E., Blythe, A. D., Kovalik, T., Mortimore, N. P., et al. (2021). TDP-43 proteinopathy alters the ribosome association of multiple mRNAs including the glypican Dally-like protein (Dlp)/GPC6. Acta Neuropathol. Commun. 9:52. doi: 10.1186/s40478-021-01148-z
Li, Y., Ray, P., Rao, E. J., Shi, C., Guo, W., Chen, X., et al. (2010). A Drosophila model for TDP-43 proteinopathy. Proc. Natl. Acad. Sci. U.S.A. 107, 3169–3174.
Liachko, N. F., Guthrie, C. R., and Kraemer, B. C. (2010). Phosphorylation promotes neurotoxicity in a Caenorhabditis elegans model of TDP-43 proteinopathy. J. Neurosci. 30, 16208–16219. doi: 10.1523/JNEUROSCI.2911-10.2010
Lim, A., Rechtsteiner, A., and Saxton, W. M. (2017). Two kinesins drive anterograde neuropeptide transport. Mol. Biol. Cell 28, 3542–3553. doi: 10.1091/mbc.E16-12-0820
Ling, S. C., Polymenidou, M., and Cleveland, D. W. (2013). Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron 79, 416–438. doi: 10.1016/j.neuron.2013.07.033
Liu-Yesucevitz, L., Bassell, G. J., Gitler, A. D., Hart, A. C., Klann, E., Richter, J. D., et al. (2011). Local RNA translation at the synapse and in disease. J. Neurosci. : Off. J. Soc. Neurosci. 31, 16086–16093. doi: 10.1523/JNEUROSCI.4105-11.2011
Liu-Yesucevitz, L., Lin, A. Y., Ebata, A., Boon, J. Y., Reid, W., Xu, Y. F., et al. (2014). ALS-linked mutations enlarge TDP-43-enriched neuronal RNA granules in the dendritic arbor. J. Neurosci. 34, 4167–4174. doi: 10.1523/JNEUROSCI.2350-13.2014
Lukavsky, P. J., Daujotyte, D., Tollervey, J. R., Ule, J., Stuani, C., Buratti, E., et al. (2013). Molecular basis of UG-rich RNA recognition by the human splicing factor TDP-43. Nat. Struct. Mol. Biol. 20, 1443–1449. doi: 10.1038/nsmb.2698
MacNair, L., Xiao, S., Miletic, D., Ghani, M., Julien, J. P., Keith, J., et al. (2016). MTHFSD and DDX58 are novel RNA-binding proteins abnormally regulated in amyotrophic lateral sclerosis. Brain 139, 86–100. doi: 10.1093/brain/awv308
Majumder, P., Chen, Y. T., Bose, J. K., Wu, C. C., Cheng, W. C., Cheng, S. J., et al. (2012). TDP-43 regulates the mammalian spinogenesis through translational repression of Rac1. Acta Neuropathol. 124, 231–245. doi: 10.1007/s00401-012-1006-4
Majumder, P., Chu, J. F., Chatterjee, B., Swamy, K. B., and Shen, C. J. (2016). Co-regulation of mRNA translation by TDP-43 and Fragile X Syndrome protein FMRP. Acta Neuropathol. 132, 721–738. doi: 10.1007/s00401-016-1603-8
Mann, J. R., Gleixner, A. M., Mauna, J. C., Gomes, E., Dechellis-Marks, M. R., Needham, P. G., et al. (2019). RNA binding antagonizes neurotoxic phase transitions of TDP-43. Neuron 102, 321.e328–338.e328. doi: 10.1016/j.neuron.2019.01.048
Marques, R. F., Engler, J. B., Kuchler, K., Jones, R. A., Lingner, T., Salinas, G., et al. (2020). Motor neuron translatome reveals deregulation of SYNGR4 and PLEKHB1 in mutant TDP-43 amyotrophic lateral sclerosis models. Hum. Mol. Genet. 29, 2647–2661. doi: 10.1093/hmg/ddaa140
McAleese, K. E., Walker, L., Erskine, D., Thomas, A. J., Mckeith, I. G., and Attems, J. (2017). TDP-43 pathology in Alzheimer’s disease, dementia with Lewy bodies and ageing. Brain Pathol. 27, 472–479. doi: 10.1111/bpa.12424
McDonald, K. K., Aulas, A., Destroismaisons, L., Pickles, S., Beleac, E., Camu, W., et al. (2011). TAR DNA-binding protein 43 (TDP-43) regulates stress granule dynamics via differential regulation of G3BP and TIA-1. Hum. Mol. Genet. 20, 1400–1410. doi: 10.1093/hmg/ddr021
McGurk, L., Gomes, E., Guo, L., Mojsilovic-Petrovic, J., Tran, V., Kalb, R. G., et al. (2018). Poly(ADP-Ribose) prevents pathological phase separation of TDP-43 by promoting liquid demixing and stress granule localization. Mol. Cell 71, 703.e709–717.e709. doi: 10.1016/j.molcel.2018.07.002
Moore, M. J., and Proudfoot, N. J. (2009). Pre-mRNA processing reaches back to transcription and ahead to translation. Cell 136, 688–700. doi: 10.1016/j.cell.2009.02.001
Nagano, S., Jinno, J., Abdelhamid, R. F., Jin, Y., Shibata, M., Watanabe, S., et al. (2020). TDP-43 transports ribosomal protein mRNA to regulate axonal local translation in neuronal axons. Acta Neuropathol. 140, 695–713. doi: 10.1007/s00401-020-02205-y
Neelagandan, N., Gonnella, G., Dang, S., Janiesch, P. C., Miller, K. K., Kuchler, K., et al. (2019). TDP-43 enhances translation of specific mRNAs linked to neurodegenerative disease. Nucleic Acids Res. 47, 341–361. doi: 10.1093/nar/gky972
Nelson, R., Sawaya, M. R., Balbirnie, M., Madsen, A. O., Riekel, C., Grothe, R., et al. (2005). Structure of the cross-beta spine of amyloid-like fibrils. Nature 435, 773–778. doi: 10.1038/nature03680
Neumann, M., Sampathu, D. M., Kwong, L. K., Truax, A. C., Micsenyi, M. C., Chou, T. T., et al. (2006). Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314, 130–133.
Pakravan, D., Michiels, E., Bratek-Skicki, A., De Decker, M., Van Lindt, J., Alsteens, D., et al. (2021). Liquid-Liquid phase separation enhances TDP-43 LCD aggregation but delays seeded aggregation. Biomolecules 11:548. doi: 10.3390/biom11040548
Pelletier, J., and Sonenberg, N. (1988). Internal initiation of translation of eukaryotic mRNA directed by a sequence derived from poliovirus RNA. Nature 334, 320–325. doi: 10.1038/334320a0
Ramaswami, M., Taylor, J. P., and Parker, R. (2013). Altered ribostasis: RNA-protein granules in degenerative disorders. Cell 154, 727–736. doi: 10.1016/j.cell.2013.07.038
Russo, A., Scardigli, R., La Regina, F., Murray, M. E., Romano, N., Dickson, D. W., et al. (2017). Increased cytoplasmic TDP-43 reduces global protein synthesis by interacting with RACK1 on polyribosomes. Hum. Mol. Genet. 26, 1407–1418. doi: 10.1093/hmg/ddx035
Sephton, C. F., Good, S. K., Atkin, S., Dewey, C. M., Mayer, P. 3rd., Herz, J., et al. (2010). TDP-43 is a developmentally regulated protein essential for early embryonic development. J. Biol. Chem. 285, 6826–6834. doi: 10.1074/jbc.M109.061846
Singh, G., Pratt, G., Yeo, G. W., and Moore, M. J. (2015). The clothes make the mRNA: past and present trends in mRNP fashion. Annu. Rev. Biochem. 84, 325–354. doi: 10.1146/annurev-biochem-080111-092106
Spiller, K. J., Cheung, C. J., Restrepo, C. R., Kwong, L. K., Stieber, A. M., Trojanowski, J. Q., et al. (2016). Selective motor neuron resistance and recovery in a new inducible mouse model of TDP-43 proteinopathy. J. Neurosci. 36, 7707–7717. doi: 10.1523/JNEUROSCI.1457-16.2016
Spriggs, K. A., Stoneley, M., Bushell, M., and Willis, A. E. (2008). Re-programming of translation following cell stress allows IRES-mediated translation to predominate. Biol. Cell 100, 27–38. doi: 10.1042/BC20070098
Sutton, M. A., and Schuman, E. M. (2006). Dendritic protein synthesis, synaptic plasticity, and memory. Cell 127, 49–58. doi: 10.1016/j.cell.2006.09.014
Tsai, K. J., Yang, C. H., Fang, Y. H., Cho, K. H., Chien, W. L., Wang, W. T., et al. (2010). Elevated expression of TDP-43 in the forebrain of mice is sufficient to cause neurological and pathological phenotypes mimicking FTLD-U. J. Exp. Med. 207, 1661–1673. doi: 10.1084/jem.20092164
Vanden Broeck, L., Callaerts, P., and Dermaut, B. (2014). TDP-43-mediated neurodegeneration: towards a loss-of-function hypothesis? Trends Mol. Med. 20, 66–71. doi: 10.1016/j.molmed.2013.11.003
Voigt, A., Herholz, D., Fiesel, F. C., Kaur, K., Muller, D., Karsten, P., et al. (2010). TDP-43-mediated neuron loss in vivo requires RNA-Binding activity. PLoS One 5:e12247. doi: 10.1371/journal.pone.0012247
Walker, A. K., Spiller, K. J., Ge, G., Zheng, A., Xu, Y., Zhou, M., et al. (2015). Functional recovery in new mouse models of ALS/FTLD after clearance of pathological cytoplasmic TDP-43. Acta Neuropathol. 130, 643–660. doi: 10.1007/s00401-015-1460-x
Wang, I. F., Wu, L. S., Chang, H. Y., and Shen, C. K. (2008). TDP-43, the signature protein of FTLD-U, is a neuronal activity-responsive factor. J. Neurochem. 105, 797–806. doi: 10.1111/j.1471-4159.2007.05190.x
Wegorzewska, I., Bell, S., Cairns, N. J., Miller, T. M., and Baloh, R. H. (2009). TDP-43 mutant transgenic mice develop features of ALS and frontotemporal lobar degeneration. Proc. Natl. Acad. Sci. U.S.A. 106, 18809–18814. doi: 10.1073/pnas.0908767106
Weskamp, K., Tank, E. M., Miguez, R., Mcbride, J. P., Gomez, N. B., White, M., et al. (2020). Shortened TDP43 isoforms upregulated by neuronal hyperactivity drive TDP43 pathology in ALS. J. Clin. Invest. 130, 1139–1155. doi: 10.1172/JCI130988
Wils, H., Kleinberger, G., Janssens, J., Pereson, S., Joris, G., Cuijt, I., et al. (2010). TDP-43 transgenic mice develop spastic paralysis and neuronal inclusions characteristic of ALS and frontotemporal lobar degeneration. Proc. Natl. Acad. Sci. U.S.A. 107, 3858–3863. doi: 10.1073/pnas.0912417107
Wong, C. E., Jin, L. W., Chu, Y. P., Wei, W. Y., Ho, P. C., and Tsai, K. J. (2021). TDP-43 proteinopathy impairs mRNP granule mediated postsynaptic translation and mRNA metabolism. Theranostics 11, 330–345. doi: 10.7150/thno.51004
Wu, L. S., Cheng, W. C., and Shen, C. K. (2012). Targeted depletion of TDP-43 expression in the spinal cord motor neurons leads to the development of amyotrophic lateral sclerosis-like phenotypes in mice. J. Biol. Chem. 287, 27335–27344. doi: 10.1074/jbc.M112.359000
Yang, Y., and Wang, Z. (2019). IRES-mediated cap-independent translation, a path leading to hidden proteome. J. Mol. Cell Biol. 11, 911–919. doi: 10.1093/jmcb/mjz091
Yu, Z., Fan, D., Gui, B., Shi, L., Xuan, C., Shan, L., et al. (2012). Neurodegeneration-associated TDP-43 Interacts with Fragile X mental retardation protein (FMRP)/Staufen (STAU1) and regulates SIRT1 expression in neuronal cells. J. Biol. Chem. 287, 22560–22572. doi: 10.1074/jbc.M112.357582
Keywords: TDP-43, ALS, FTD, translation, axon, dendrite, synapse, neurodegeneration
Citation: Bjork RT, Mortimore NP, Loganathan S and Zarnescu DC (2022) Dysregulation of Translation in TDP-43 Proteinopathies: Deficits in the RNA Supply Chain and Local Protein Production. Front. Neurosci. 16:840357. doi: 10.3389/fnins.2022.840357
Received: 21 December 2021; Accepted: 09 February 2022;
Published: 07 March 2022.
Edited by:
Lin Guo, Thomas Jefferson University, United StatesReviewed by:
Martin Lothar Duennwald, Western University, CanadaChiara F. Valori, German Center for Neurodegeneratives, Helmholtz Association of German Research Centers (HZ), Germany
Copyright © 2022 Bjork, Mortimore, Loganathan and Zarnescu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Daniela C. Zarnescu, emFybmVzY3VAYXJpem9uYS5lZHU=