 Can Zhang
Can Zhang- 1Department of Biology, Drexel University, Philadelphia, PA, United States
- 2Division of Biological Infrastructure, National Science Foundation, Alexandria, VA, United States
Alzheimer’s disease (AD) belongs to a class of diseases characterized by progressive accumulation and aggregation of pathogenic proteins, particularly Aβ proteins. Genetic analysis has identified UBQLN1 as an AD candidate gene. Ubiquilin-1 levels reduce with AD progression, suggesting a potential loss-of-function mechanism. The ubiquilin-1 protein is involved in protein quality control (PQC), which plays essential roles in cellular growth and normal cell function. Ubiquilin-1 regulates γ-secretase by increasing endoproteolysis of PS1, a key γ-secretase component. Presently, the effects of ubiquilin-1 on cellular physiology as well as Aβ-related events require further investigation. Here, we investigated the effects of ubiquilin-1 on cellular growth and viability in association with APP (amyloid-β protein precursor), APP processing-related β-secretase (BACE1, BACE) and γ-secretase using cell and animal-based models. We showed that loss-of-function in Drosophila ubqn suppresses human APP and human BACE phenotypes in wing veins and altered cell number and tissue compartment size in the wing. Additionally, we performed cell-based studies and showed that silencing UBQLN1 reduced cell viability and increased caspase-3 activity. Overexpression of UBQLN1 significantly reduced Aβ levels. Furthermore, pharmacological inhibition of γ-secretase increased ubiquilin-1 protein levels, suggesting a mechanism that regulates ubiquilin-1 levels which may associate with reduced Aβ reduction by inhibiting γ-secretase. Collectively, our results support not only a loss-of-function mechanism of ubiquilin-1 in association with AD, but also support the significance of targeting ubiquilin-1-mediated PQC as a potential therapeutic strategy for AD.
Introduction
Alzheimer’s disease (AD) is pathologically characterized by accumulation of neuronal amyloid plaques and fibrillary tangles, which ultimately lead to neuronal death and dementia (Hardy and Selkoe, 2002; Choi et al., 2014). The main components of the amyloid plaques are beta-amyloid peptides (or Aβ), which are primarily 37–43 amino acids long and generated by the proteolytic processing of a type-I transmembrane glycoprotein, amyloid-β protein precursor (APP) (Tanzi et al., 1987; Tanzi and Bertram, 2001). APP is present on almost every membrane containing intracellular compartment. After translation, APP is transported onto the membrane surface through intracellular trafficking. Aβ generation, also known as amyloidogenesis, requires two proteolytic processing events of APP mediated by β-secretase (BACE) and γ-secretase. Like APP, BACE is also a type-I transmembrane protein, while γ-secretase is a membrane protein complex which contains presenilin1 (PS1), nicastrin (NCSTN), Aph-1, and Pen-2. γ-Secretase cleavage can yield different length of Aβ species, among which Aβ40 and Aβ42 are the two main components in AD brains. Aβ42 is more aggregation prone than Aβ40 and increased Aβ42/Aβ40 ratios are common in early onset familial AD (FAD) cases. APP can also be cleaved by α-secretase, which cleaves within the Aβ sequence and precludes the process of amyloidogenesis (Zhang and Saunders, 2007).
In the past decade, there has been increasing interest in identifying and characterizing novel candidate genes in association with the progression of AD. One such gene of interest implicated in AD is UBQLN1, which encodes ubiquilin-1, a ubiquitin-like protein. Polymorphism of UBQLN1, has been shown to increase the risk of AD in family-based and large case-control samples (Bertram et al., 2005; Bensemain et al., 2006; Kamboh et al., 2006). Biologically, ubiquilin-1 is an intracellular protein quality control (PQC) protein and regulates related protein functions at various organelles or systems, particularly the ubiquitin-proteasome system (UPS) (Funakoshi et al., 2002; Ko et al., 2002; Medicherla et al., 2004; El Ayadi et al., 2013). Pathologically, ubiquilin-1 has been implicated to be involved in regulation of molecular process of AD (Hiltunen et al., 2006; Li et al., 2007; Zhang et al., 2007; Ganguly et al., 2008; Gross et al., 2008; Adegoke et al., 2017). Furthermore, ubiquilin-1 is associated with non-AD disorders and conditions, including Parkinson’s disease and a variety of cancers (Mah et al., 2000; Wang et al., 2006; Chen et al., 2007). These findings suggest a broad and important role of ubiquilin-1 in human pathophysiology. As a PQC protein, ubiquilin-1 levels reduce with AD progression, suggesting a potential link to AD pathology by a loss-of-function mechanism (Stieren et al., 2011). Related to its function in PQC, ubiquilin-1 has also been shown as an APP chaperon protein, reduction of which may lead to overproduction of pathogenic APP cleavage fragments and neuronal death (Stieren et al., 2011). Furthermore, ubiquilin-1 regulates γ-secretase by affecting the endoproteolysis of γ-secretase-related PS1, which harbors proteolytic cleavage activity of γ-secretase (Mah et al., 2000; Hiltunen et al., 2006; Zhang et al., 2007). Studies have also shown that ubiquilin-1 directly affects BACE trafficking and localization to recycling endosomal compartments of neuronal cells, where Aβ generation is most abundant (Takalo et al., 2017). Currently, the effects of ubiquilin-1 on AD-related events as well as the interaction of BACE and APP-related mechanistic phenotypes require further investigation.
Here, we investigated the effects of ubiquilin-1 on PQC and association with APP processing-related β-secretase and γ-secretase using cell and animal-based models. We analyzed the function of the Drosophila homolog of ubiquilin-1 (CG14224, ubqn) in the developing wing. Drosophila serves as a suitable in vivo system to model AD-associated protein function, as the fruit fly contains functional homologs of all the necessary components of the γ-secretase complex (Guo et al., 2003; Ganguly et al., 2008; Chakraborty et al., 2011), and can process expressed human APP (Fossgreen et al., 1998), displaying adult phenotypes that are easily identifiable and have broad phenotypic ranges for genetic studies. Additionally, human β-secretase can be expressed in fly tissues, along with human APP (Greeve et al., 2004) to generate adult phenotypes. Following animal studies, we also characterized the effects of silencing UBQLN1 on cell viability in vitro using the human neuroblastoma SH-SY5Y cells, as well as the effects of increasing UBQLN1 in AD-related Aβ levels. Thus, the results of our cell and animal-based studies may collectively be useful to further elucidate the underlying mechanism of ubiquilin-1 as a PQC protein in association with AD.
Materials and Methods
Drosophila Culture and Genetics
All flies were raised on standard cornmeal/molasses/agar media at 25°C. Experimental genotypes were generated using the Gal4/UAS system for expressing specific proteins in a temporal and tissue specific manner, as previously described (Brand and Perrimon, 1993). All crosses were performed using engrailed:Gal4 (Brower, 1986), which expresses the Gal4 protein specifically within the posterior compartment of Drosophila wings. The anterior compartment served as an internal control for each experiment. Transgenic Drosophila genotypes used were: UAS:APP (Fossgreen et al., 1998), UAS:APP and UAS:BACE (Greeve et al., 2004), UAS:p35 (Bloomington Stock Center # 5072), UAS:ubiquilin RNAi (Vienna Drosophila RNAi Center Transformant ID#s 47448 and 47449). UAS:GFP (Bloomington Stock Center # 4775) was used as the wild type control.
Fly Wing Analysis
All wings were dehydrated in ethanol and mounted in DPX (Zeiss). Digital photographs were taken under a Leica mZ 12.5 stereomicroscope using an attached Leica digital camera. Cell count and surface area measurements were performed as previously described (Marenda et al., 2003), using NIH Image J for analysis.
Chemical, Antibodies, and Plasmids
L-685,458 and puromycin were purchased from Sigma. β-Actin antibody (1:10,000) was purchased from Sigma. The APP C-terminal antibody (A8717; 1:1,000) was purchased from Sigma. The ubiquilin-1 antibody (1:160) was purchased from Zymed. The HRP-conjugated secondary antibodies (anti-mouse and anti-rabbit) (1:10,000) were purchased from GE. The plasmids APP-Gal4, Gal4-UAS-luciferase (encoding firefly luciferase) were kindly provided from Dr. Thomas Südof, and have been described (Cao and Sudhof, 2001). The previously reported ubiquilin-1 expressing plasmid was constructed from the pCMV vector and was kindly provided by Dr. Mervyn J. Monteiro (Mah et al., 2000).
Cell Culture
SH-SY5Y and HEK-293 cells were purchased from the American Type Culture Collection (ATCC). These cell lines were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum, 2 mM L-glutamine, 100 units/ml penicillin, and 100 μg/ml streptomycin. SH-SY5Y cells that stably express APP-Gal4 and the Gal4-UAS-luciferase reporter construct (named as “SY5Y-APP-Gal4 cells“) were described elsewhere (Zhang et al., 2007). These cells were maintained in media containing 200 μg/ml G418. SY5Y-APP-Gal4 cells were transfected with non-silencing or control shRNA construct (not targeting any known genes) or shUbqln1 construct (targeting ubiquilin-1 mRNA or “shUBQLN1”) (Zhang et al., 2007). The multiple clonal cells containing puromycin-resistant clones were selected and continued to grow in 1 μg/ml puromycin and 200 μg/ml G418-containing media. When cells reached 15 splitting times, they were replaced with fresh batches.
3-(4,5-Dimethylthiazol-2-yl)-2,5-Diphenyl-2H-Tetrazolium Bromide Assay
The 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) assay was carried out using the MTT cell proliferation assay kit (ATCC Bioproducts™) according to the manufacturer’s instructions. Briefly, 5,000 cells were plated per well in the 96-well plates. Cells were challenged with 500 μM H2O2 for 1 h and 10 μl MTT reagent was added to the cells. After 3 h of incubation at 37°C, 100 μl detergent reagent was added to the cells. After additional 2 h of incubation in darkness at 37°C, absorbance was read at 570 nm using a spectrophotometer (Multiskan spectrum, Thermo Labsystems).
Caspase-3 Activity Assay
The caspase-3 activity assay was following the manufacturer’s instructions (Molecular Probes). The EnzChek§ caspase-3 assay kit #2 was utilized. In brief, 2 × 106 cells were seeded on the 6-well plates. After the treatment using 500 μM H2O2 for 1 h, cells were washed with phosphate-buffered saline (PBS) and lyzed using the lysis buffer provided by the manufacturer, followed by centrifugation. Supernatant was transferred to a new white color 96-well plate and added with working solution containing caspase-3 substrate. After covering the plate in dark for 30 min, fluorescence was read with the excitation/emission of ∼496/520 nm on a microplate fluorometer (Fluoscan Ascent FL, Thermo Labsystems).
Western Blotting Analysis
Western Blotting (WB) analysis was carried out by the method described previously (Hiltunen et al., 2006; Zhang et al., 2007; Choi et al., 2014). Briefly cells were lysed in the radioimmunoprecipitation (RIPA) cell lysis buffer (50 mM Tris-HCL pH 7.4, 150 mM NaCl, 1 mM EDTA, 1% NP-40, 1 mM PMSF and 1 μg/ml aprotinin, 1 μg/ml leupeptin, and 1 μg/ml pepstatin). After centrifugation and protein concentration measurement, equal amount of protein was applied to the electrophoresis followed by membrane transfer, antibody incubation and signal development.
Aβ Measurement
Aβ measurement was following the method described previously (Hiltunen et al., 2006). In brief, Aβ40 and Aβ42 levels (pg/ml) were quantified using the sandwich enzyme-linked immunosorbent (ELISA) assay. Each experiment was carried out at least in triplicate.
Statistical Analysis
Values in the text and figures were demonstrated as means ± standard errors from at least three independent experiments. Equal variance two-sample student’s t-test was utilized to compare two groups followed by Bonferroni’s test if multiple comparisons were examined within a single experiment.
Results
Loss-of-Function in Drosophila ubqn Suppresses Human Amyloid-β Protein Precursor and Human BACE Phenotypes in Wing Veins
To analyze the function of ubqn in vivo, we used a publicly available ubqn RNAi construct (Ub-RNAi) under the transcriptional control of the Gal4/UAS system (Brand and Perrimon, 1993). Previous studies have shown that RNAi knockdown of ubqn in the entire Drosophila wing leads to a strong loss of vein phenotype (Li et al., 2007; Ganguly et al., 2008). To extend these findings, we expressed the well-established and previously reported engrailed-Gal4 which is restricted to the posterior wing compartment (Brower, 1986; Marenda et al., 2006; Vrailas-Mortimer et al., 2007). We analyzed the posterior domain of developing wings (below the dotted line in Figure 1A), and found a strong loss of vein phenotypes, including loss of wing vein L5 in all wings (arrowhead Figure 1D and Table 1), and loss of the posterior cross vein in 91% of wings (arrow in Figure 1D and Table 1). Our findings were consistent with expressing RNAi knockdown of ubqn in the entire Drosophila wing (Li et al., 2007; Ganguly et al., 2008). These results suggested that our Ub-RNAi construct was silencing ubqn function through RNAi knockdown.
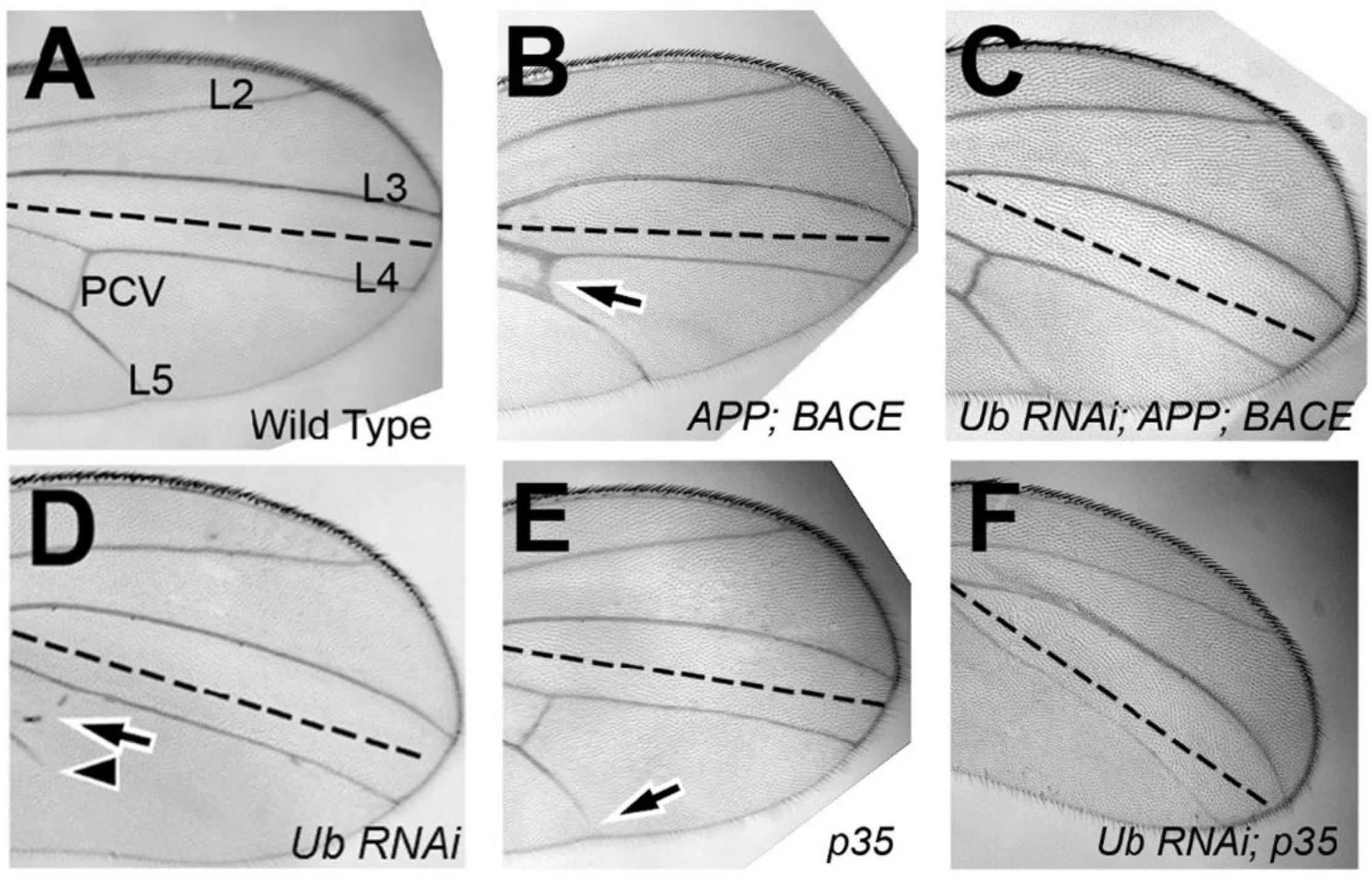
Figure 1. Drosophila ubqn genetically interacts with human APP and BACE in the wing. All wings are anterior up, distal right, same magnification. All wings express UAS constructs in the posterior compartment only (as driven by engrailed:Gal4). UAS constructs are listed in the bottom right of each panel. Posterior compartments are depicted below the dotted line in each panel. (A) Representation of a wild type wing where UAS:GFP is expressed. Longitudinal wing veins L2-L5 are depicted, as is the posterior crossvein (PCV). (B) Representation of a wing where human APP and human BACE are co-expressed. Arrow shows extra vein and thickening of the PCV. (C) Representation of a wing where ubiquilin-1 RNAi (shUbqln1) is expressed with human APP and human BACE. Note the suppression of the extra/thick PCV. (D) Representation of a wing where ubiquilin-1 RNAi is expressed. Arrowhead shows loss of vein L5 material. Arrow shows loss of PCV. (E) Representation of a wing where UAS: p35 is expressed. Arrow shows loss of vein L5. (F) Representation of a wing where both ubiquilin-1 RNAi and p35 are co-expressed. Note the enhancement of vein loss in both the PCV and L5 tissue. Ub, Ubiquilin-1.
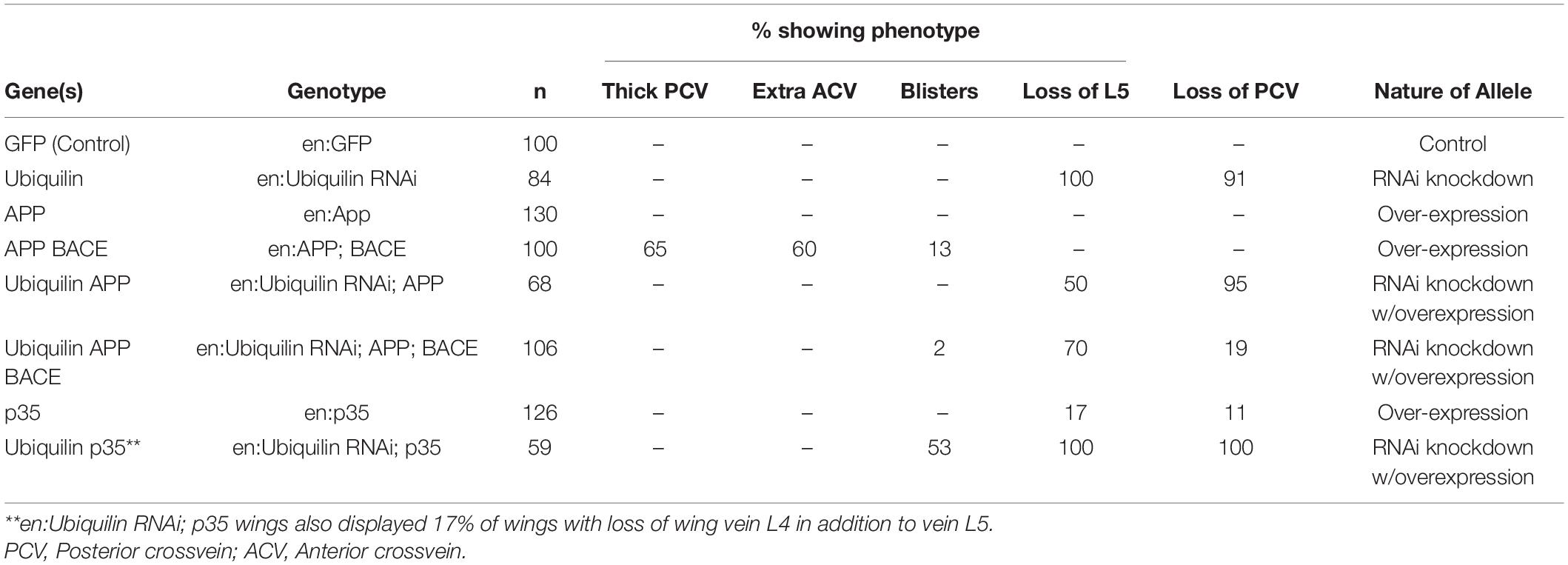
Table 1. Genetic Interaction between Ubiquilin, APP, BACE, and p35.
While expression of human APP in the posterior compartment of wings had no effect on adult wing vein structures (Table 1), expression of human APP with human BACE led to extra vein phenotypes (Figure 1B), with thicker posterior crossveins, extra anterior crossveins, and blistering (Table 1). This is consistent with what has been previously reported by Greeve et al. (2004). Specifically, Greeve et al. showed that expression of both BACE and Presenilin had no extra vein phenotype in wings, nor did expression of APP alone (Fossgreen et al., 1998). Taken together with our results, we suggest that this indicates the extra vein phenotypes observed are due to human β-secretase cleavage of expressed human APP protein. It is also important to note that previous analysis has shown that replacing human APP with Drosophila APPL protein, which does not contain an Aβ domain, suppressed extra vein formation, indicating the mechanism of extra vein formation is related to the presence of Aβ (Greeve et al., 2004; Li et al., 2007).
When both human APP, human BACE, and Ub-RNAi were expressed simultaneously, a strong suppression of both phenotypes associated with ubqn knockdown, and phenotypes associated with human APP and human BACE over-expression were observed (Figure 1C and Table 1). Further, suppression of the loss of L5 vein tissue associated with ubqn knockdown was also observed upon expression of human APP alone (Table 1). These data indicate that Drosophila ubqn facilitates human APP and human BACE in this vein phenotype in the wing, as these phenotypes are suppressed when ubqn is knocked down. Ganguly et al. have shown that Drosophila ubqn antagonizes presenilin function in the wing (Ganguly et al., 2008). Combined with our data, this suggests that ubqn function differs between components of the γ-secretase pathway and the β-secretase pathway during differentiation within this tissue.
Drosophila ubqn Genetically Interacts With p35 in Wing Vein Phenotypes
Drosophila ubqn has been previously suggested to be pro-apoptotic when over-expressed in the eye (Ganguly et al., 2008). To determine if the phenotypes we observe from ubqn knockdown in the Drosophila wing were also partially apoptotic in nature, we simultaneously expressed Ub-RNAi and the baculovirus pan-caspase inhibitor p35. Expression of p35 alone (UAS:p35) showed weak loss of vein structures (Figure 1E and Table 1), in agreement with previous findings (Marenda et al., 2006). Simultaneous expression of both Ub-RNAi and p35, showed a strong enhancement of loss of veins structures (Figure 1F and Table 1), in addition to wing blistering. These data indicate that ubqn may play a role in wing vein differentiation as a regulator of apoptosis in the Drosophila wing.
Drosophila ubqn Alters Cell Number and Tissue Compartment Size in the Wing
Apoptosis is part of the normal development of many epithelial tissues, including the Drosophila wing, and is a significant factor in both the pathology of cancer and neurodegenerative diseases. Increased ubiquilin-1 protein, and decreased phosphorylated ubiquilin-1 have been found to be associated with lung adenocarcinoma (Chen et al., 2007), linking ubiquilin-1 function to cancer. Consistent with our data above, we suggest that in the Drosophila wing, ubqn appears to function in a pro-apoptotic fashionf. Thus, to test whether the phenotypes we observed in the wing were due to increased cell number or due to a loss of apoptosis upon ubqn knockdown, we performed cell count and wing surface area measurements of these wings.
Each wing cell secretes one wing hair (Meyer et al., 2000), which we used as a proxy for cell number. We expressed our UAS constructs in the posterior domain of wings, and analyzed the difference in cell number between the posterior (P; experimental) and anterior (A; control) compartments of each genotype. To test specific expression of the constructs in the posterior compartments, we found that anterior compartments of Drosophila wing cell number displayed no significant differences comparing animals expressing different constructs in their posterior domain of wings, as expected (Supplementary Figure 1). Because there was no significant difference in cell number between the genotypes within the anterior compartment of wings, we next studied if there were significant differences between posterior compartments. We normalized each posterior cell count first to the anterior cell count of the same genotype, and then to the control cell count where UAS:GFP was expressed. We found that upon ubqn knockdown, the number of cells within the posterior compartment significantly increased compared to control wings (Figure 2A). Taken together, this suggests that ubqn functions to restrict cell number in the developing wing. Expression of human APP had no significant effect on cell number (Figure 2A), but co-expression of both human APP and human BACE increased cell number significantly (Figure 2A). Expression of Ub-RNAi with human APP and human BACE slightly increased cell number, indicating that ubqn antagonizes human APP and human BACE function in cell number of the wing.
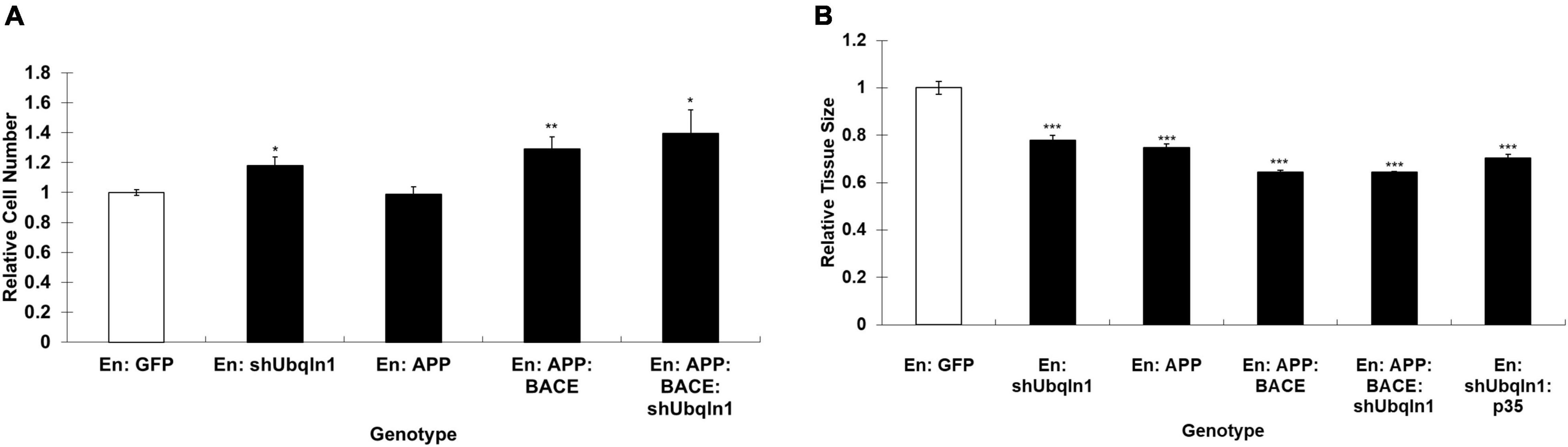
Figure 2. Effects of Drosophila ubqn on cell number (A) and growth (B) in the wing. The UAS constructs were expressed in the posterior domain of wings, and analyzed for the difference in cell number between the posterior (P; experimental) and anterior (A; control) compartments. For each genotype, five independent wing compartments were assayed (A). Total cell number for each genotype included: En: GFP (A: 300, 318, 327, 293, 363; P: 480, 566, 542, 499, 569); En: shUbqln1 (A: 275, 342, 318, 351, 317; P: 624, 651, 647, 572, 624); En: APP (A: 380, 355, 305, 270, 335; P: 550, 545, 500, 537, 532); En: APP:BACE (A: 256, 310, 318, 247, 325; P: 647, 615, 570, 601, 651); and En: APP: BACE: shUbqln1 (A: 344, 320, 238, 372, 350; P: 771, 638, 815, 712, 708). *p < 0.05, **p < 0.01; ***p < 0.001; compared to En:GFP. En, engrailed; P, posterior; A, Anterior.
To analyze cell growth within the wing, we expressed our UAS constructs as above, and measured the surface area of both the anterior compartment (control) and posterior compartment (experimental). As with the wing cell counts, we did not observe a significant difference in the surface area of anterior compartments between different genotypes (data not shown). We then normalized each posterior compartment measure as above. We found that upon ubqn knockdown, the size of the posterior compartment of adult wings significantly decreased compared to control wings (Figure 2B). When taken together with our cell count data, this suggests that though there are more cells within the posterior compartment of these wings, these cells are significantly smaller in size than normal. We suggest that this is due to a loss of normal apoptosis due to ubqn knockdown, resulting in an abnormally larger number of cells within the posterior compartment of these wings, and abnormal tissue morphology.
Interestingly, though expression of human APP in these wings had no effect on cell number, it did decrease overall tissue size (Figure 2B). This effect was enhanced when both human APP and human BACE were simultaneously expressed in these wings (Figure 2B). However, unlike with our cell number results, simultaneous expression of Ubiquilin RNAi with human APP and human BACE did not further enhance this effect (Figure 2B). Simultaneous expression of p35 with Ub-RNAi did show an enhancement of the tissue size defect from Ub-RNAi expression alone. These data were consistent with what we observed between these two genotypes in the differentiation of veins in the wing, and strengthen our conclusion that ubqn is normally pro-apoptotic in the developing Drosophila wing.
Silencing Ubiquilin-1 Alters Cells Viability and Caspase-3 Activity in Human Cells
Because ubiquilin-1 is indicated to be involved in PQC and we showed ubiquilin-1-related abnormality and apoptosis in Drosophila, we expect that silencing ubiquilin-1 may change cell viability and lead cells go through apoptosis. Therefore, we next set out to analyze the effects of silencing ubiquilin-1 on cell viability using human cell models. We utilized our previously reported cell lines with ubiquilin-1 stably knocked down using plasmid-based shRNA constructs which contained a puromycin resistance marker (Zhang et al., 2007). Cells stably expressing shRNA control or ubiquilin-1 were generated by selecting for puromycin resistant clones. The clonal cells with puromycin resistant constructs were selected and maintained in 1 μg/ml puromycin and 200 μg/ml G418 containing medium. The clonal cells were analyzed for ubiquilin-1 knock-down by Western blotting analysis with β-actin used as a loading control (Supplementary Figure 2).
Our control and shUbqln1 stably knockdown SY5Y-APP-Gal4 cells were challenged with 500 μM H2O2 for 1 h and applied to MTT cell viability analysis. We showed that ubiquilin-1 knockdown cells significantly decreased cell viability compared with control cells (p < 0.05).
Because increased caspase-3 activity has been shown to associate with cell viability and Aβ metabolism in AD (Xie et al., 2008), we also analyzed the caspase-3 activity in our cells. We showed that ubiquilin-1 knockdown cells displayed a significant elevation in the caspase-3 activity compared to control cells under the H2O2 treatment (p < 0.05) (Figure 3). These findings support the previous report showing overexpression of UBQLN1 in association with reduced caspase-3 activity (Liu et al., 2014), as well as a study showing reduced ubiquilin-1 expression in a brain injury mouse model (Luo et al., 2019). Collectively, our cell-based results extend the findings of Drosophila, suggesting a loss-of-function mechanism of ubiquilin-1 in AD.
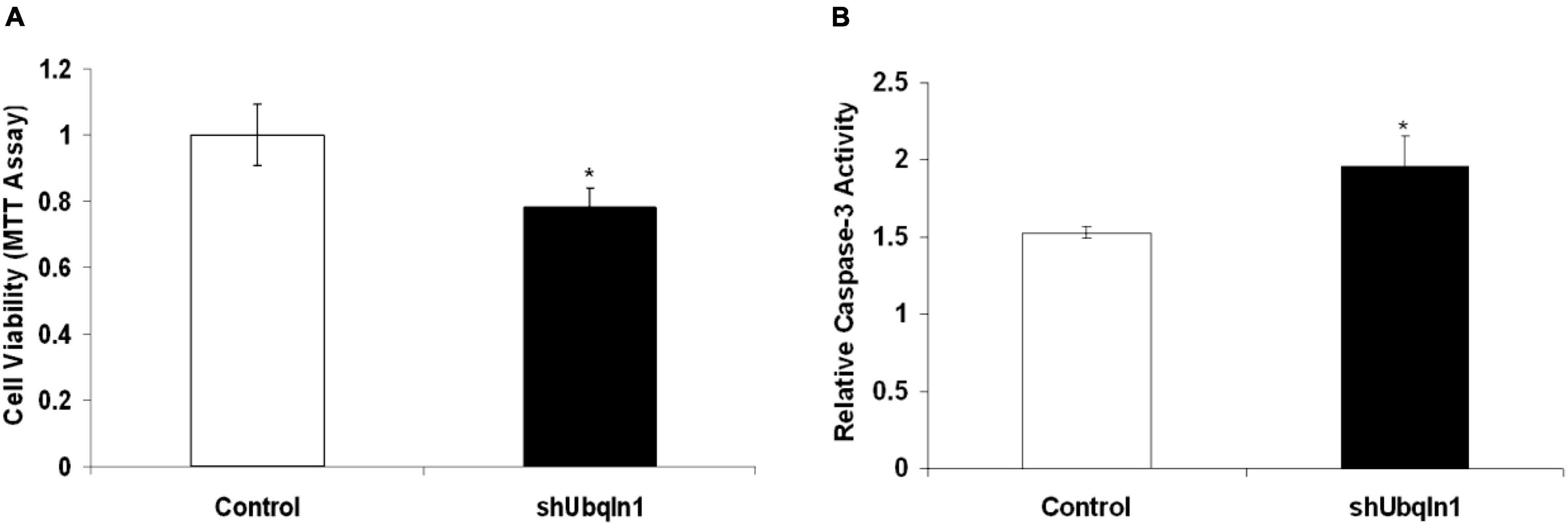
Figure 3. Down-regulation of ubiquilin-1 decreases cell viability and elevates caspase-3 activity. (A) Ubiquilin-1 knock-down significantly decreased cell viability in SY5Y-APP-Gal4 cells utilizing the MTT assay (p < 0.05). SY5Y-APP-Gal4 cells stably expressing the control (a non-specific shRNA not targeting any known genes) and shUbqln1 were incubated with 500 μM H2O2 for 1 h, and then subjected to MTT assay to measure cell viability. (B) Ubiquilin-1 knock-down significantly elevates caspase-3 activity. SY5Y-APP-Gal4 cells stably expressing the control and shUbqln1 were incubated with 500 μM H2O2 for 1 h, and then utilized in a caspase-3 activity assay (*p < 0.05). The caspase-3 activity of shUbqln1 cells was compared to control cells.
Ubiquilin-1 Over-Expression Decreases Aβ Levels
Evidence showed that UBQLN1 knockdown increases levels of Aβ40 and Aβ42 levels in cell-based models (Hiltunen et al., 2006). However, it hasn’t been demonstrated that increasing ubiquilin-1 expression alone leads to decreased Aβ levels in cell models. To test the effects of increasing ubiquilin-1 on Aβ levels, naïve HEK-293 cells were utilized and transiently transfected with previously reported control or UBQLN1 plasmid for 48 hrs. Conditioned media was prepared and utilized for the Aβ analysis by ELISA. Overexpression of ubiquilin-1 decreased Aβ40 levels by 40% compared to control (p < 0.05) and lowered Aβ42 levels by 60% (p = 0.09) (Figure 4A). Increasing ubiquilin-1 reduced Aβ(42:40) ratios by 40%, which however did not reach the statistical significance level (Figure 4B). Collectively, increasing UBQLN1 reduces Aβ levels, which is associated with the effects of UBQLN1 deficiency on increasing Aβ levels through a mechanism related to regulation of APP trafficking and metabolism (Hiltunen et al., 2006; Gross et al., 2008). Because reducing Aβ level is a therapeutic strategy for AD treatment, our results as well as others suggest that ubiquilin-1 is an AD therapeutic target and that overexpressing UBQLN1 may be considered as a potential therapeutic strategy.
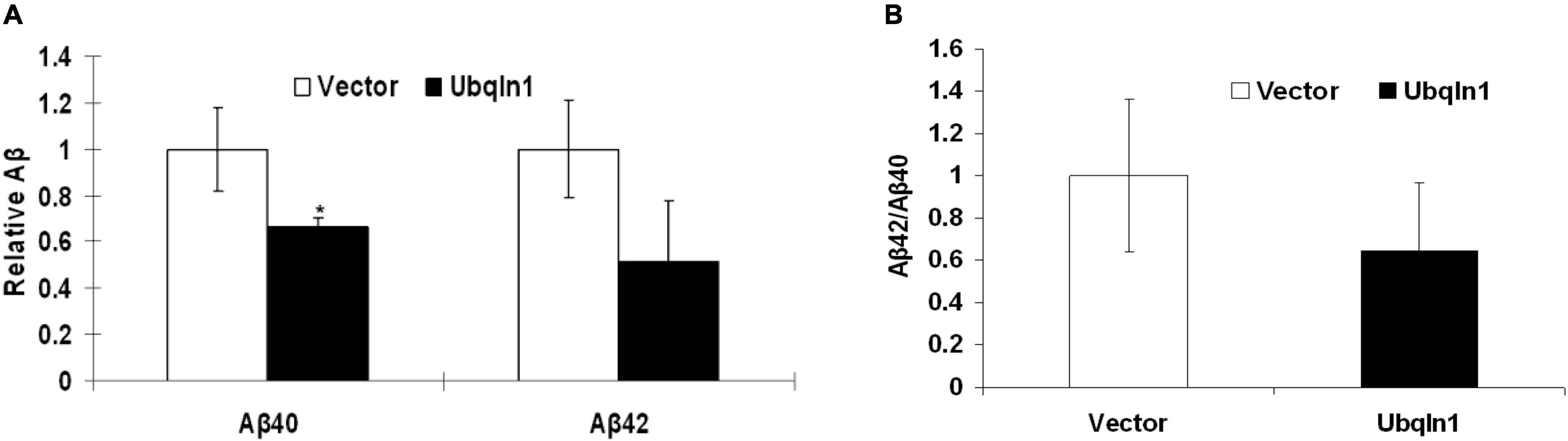
Figure 4. Ubiquilin-1 overexpression decreases Aβ levels. The effects of ubiquilin-1 over-expression on Aβ levels were evaluated using naïve HEK293 cells which were transiently transfected with the ubiquilin-1 or empty vector plasmids. Conditioned media was prepared 48 h post transfection and utilized for Aβ assay by ELISA. (A) Ubiquilin-1 over-expression significantly decreased Aβ40 (*p < 0.05) and showed a trend toward decreasing Aβ42 levels and Aβ(42:40) ratios (p > 0.05). (B) Ubiquilin-1 did not significantly change Aβ(42:40) ratios (p > 0.05).
γ-Secretase Inhibition Increases Ubiquilin-1 Protein Levels
Previous findings from our group and other groups showed that ubiquilin-1 modulates PS1 proteolysis and γ-secretase activity (Mah et al., 2000; Zhang et al., 2007). We next investigated if regulating γ-secretase activity may change ubiquilin-1 levels. First, we utilized L685,458, a common γ-secretase transition state inhibitor and contained in DMSO (Zhang et al., 2007), on naïve SH-SY5Y cells and cell lysates were then prepared and analyzed by WB. We showed that 5 μM L685,458 robustly increased ubiquilin-1 protein levels up to 5-fold comparing to the mock (DMSO) group (p < 0.05) in naïve SH-SY5Y cells (Figures 5A,B). We next attempted to generalize this finding using a different cell model. We utilized our naïve HEK-293 cells and found that L685,458 increased ubiquilin-1 levels up to 4-fold comparing to the mock group (p < 0.01) (Figures 5C,D). Thus, our results using pharmacological inhibition of γ-secretase resulted in a strong increase in ubiquilin-1 levels. These findings extends the previous studies showing the protein interactions of ubiquilin-1 and PS1 (Mah et al., 2000) as well as those showing protein-level changes of ubiquilin-1 in various pathophysiological conditions, including AD (Stieren et al., 2011; Luo et al., 2019).
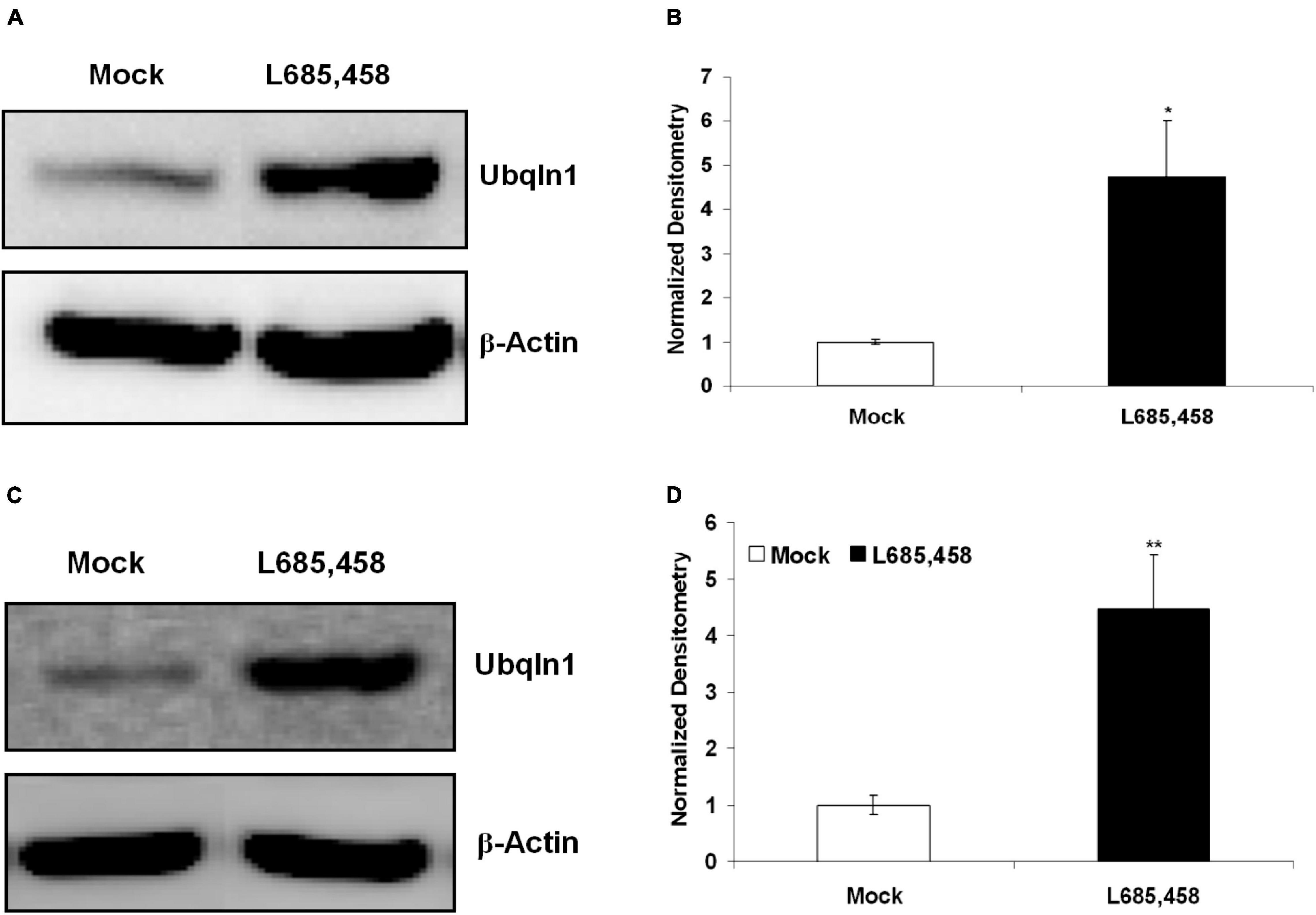
Figure 5. Inhibition of γ-secretase increases ubiquilin-1 protein levels. (A) L685,458 increased ubiquilin-1 protein levels in SH-SY5Y cells. Naïve SH-SY5Y cells were treated with the mock solution (DMSO) or 5 μM L685,458 (in DMSO) for 1 h. Cell lysates were prepared and applied to WB analysis. β-Actin served as the loading control. (B) Normalized densitometry for (A). *p < 0.05. (C) L685,458 increases ubiquilin-1 protein levels in HEK-293 cells. Similar to (A), naïve HEK293 cells were treated with the mock solution or 5μM L685,458 for 1 h. Cell lysates were prepared and applied to WB analysis. β-Actin served as the loading control. (D) Normalized densitometry for (C). **p < 0.01.
Discussion
In this study, we investigated the association of ubiquilin-1 with AD by analyzing the effects of ubiquilin-1 in cell and animal models. Particularly, we utilized both cell and animal-based models to analyze the consequences of changing ubiquilin-1 levels on PQC-related events as well as in the interaction of BACE and γ-secretase. We showed that loss of Drosophila ubiquilin-1 (ubqn) function in the wing leads to wing vein phenotypes, altered cell number, and altered tissue compartment size. Our results in the fly suggested that ubqn is normally pro-apoptotic in the developing wing, and also functionally interacts with human β-secretase and APP. These findings agreed with AD-related reduction of ubiquilin-1 and decreased quality control of APP (Stieren et al., 2011).
A number of previous studies showed that ubiquilin-1 is a PQC protein and can alter cellular toxicity and apoptosis (Ko et al., 2002; Wang et al., 2006; Zhang et al., 2020). Particularly, apoptosis, or programmed cell death, plays a critical role in development as well as in AD and other neurodegenerative diseases (Kim et al., 1997). Ubiquilin-1 could alleviate hypoxia-induced apoptosis in SH-SY5Y cells (Ko et al., 2002). Furthermore, ubiquilin-1 is a protein involved in the PQC system, which is a complex cellular mechanism that consists of the endoplasmic reticulum (ER) and UPS and other organelles (Ellgaard and Helenius, 2003; Kostova and Wolf, 2003). The ER is a eukaryotic organelle where protein folding and degradation can occur. The UPS is essential in regulating many cellular processes, including cell cycle, gene expression, and responses to oxidative stress. A recent study showed that overexpression of UBQLN1 reduces neuropathology in a mouse model of amyotrophic lateral sclerosis with frontotemporal dementia (ALS/FTD) that contains a mutation in UBQLN2, a gene that encode an ubiquilin-1 homolog protein (Wang et al., 2020). These collective data suggest that ubiquilin-1 is a potential player underlying AD which requires further study.
To analyze the association of ubiquilin-1 with AD, we investigated the effects of ubiquilin-1 on apoptosis and PQC in cell and animal models. We showed that ubiquilin-1 is involved in regulating cell viability and apoptosis activity both in vitro and in vivo. We identified genetic interactions between human APP, BACE, and ubqn in Drosophila, which may be due to the effects of these proteins on apoptosis. Drosophila ubqn facilitated human APP and human BACE in vein phenotype in the wing when these phenotypes were suppressed with ubqn deficiency. Our results of Drosophila ubqn in the wing agreed with previous results showing that Drosophila ubqn displays pro-apoptotic functions during over-expression in the eye (Ganguly et al., 2008). To gain insight into the regulatory effects of ubiquilin-1 on cell viability, we found that UBQLN1 knock-down decreased cell viability with elevated caspase-3 activity in the presence of H2O2 in SH-SY5Y cells. Our finding of ubiquilin-1 in association with apoptosis was supported by previous studies that analyzed ubiquilin-1 in apoptosis (Ko et al., 2002; Liu et al., 2014; Luo et al., 2019; Zhang et al., 2020). We note that apoptosis plays complex roles in both development and neurodegenerative conditions and ubiquilin-1 may modulate apoptosis in different conditions of related tissues (Ko et al., 2002; Ganguly et al., 2008).
Our results support a working model (Figure 6), which demonstrates two main mechanisms by which ubiquilin-1 may associate with AD. First our model showed APP and BACE overexpression-related wing veins and altered cell number and tissue compartment size in the Drosophila wing, which can be rescued by ubqn RNAi (Figure 6A, left column). This mechanism is likely due to Aβ, which agreed with previous findings of ubqn in Drosophila (Greeve et al., 2004; Li et al., 2007). The relationship of ubiquilin-1 and Aβ was further supported by our cell-based study overexpressing ubiquilin-1 in human cell models in this study. Next, we expressed the baculovirus pan-caspase inhibitor p35 in the Drosophila wing and found weak loss of vein structures, which can be enhanced by ubqn RNAi (Figure 6B; right column). This suggested an apoptosis-related (amyloid-independent) mechanism of Drosophila ubqn, consistent with a previous study (Ganguly et al., 2008). Additionally, our findings in Drosophila were further supported by our results showing an increase of caspase-3 activation caused by H2O2 in vitro. Collectively, our findings further extended previous results, and support an APP/BACE-related (amyloid-dependent) mechanism (Figure 6A) and an apoptosis-related (amyloid-independent) mechanism (Figure 6B) of ubiquilin-1.
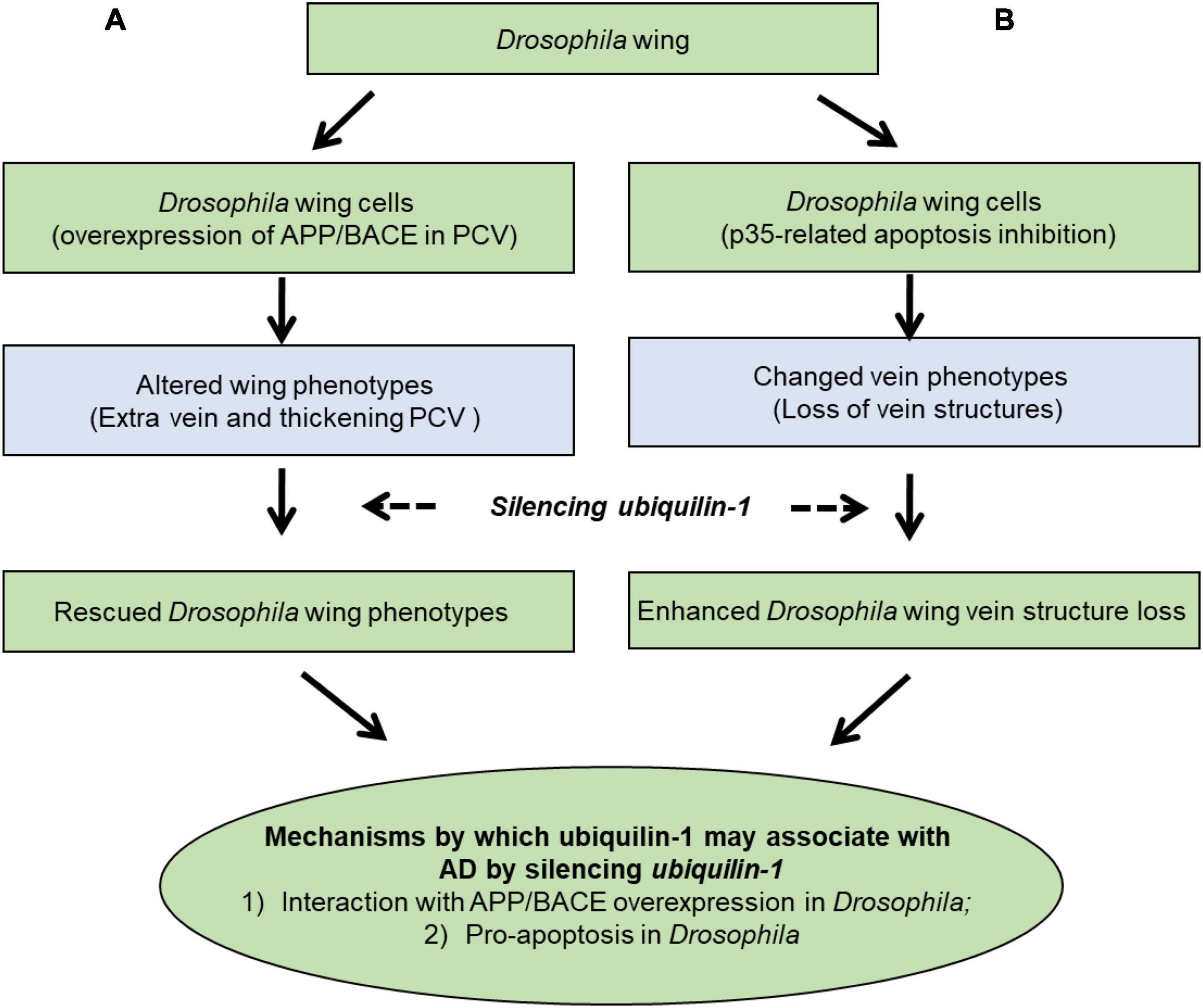
Figure 6. A working model showing mechanisms by which ubiquilin-1 may associate with AD. We characterized the mechanisms by which ubiquilin-1 may associate with AD using Drosophila and cell-based models. Our results extended previous reports and suggested two main mechanisms: (A) an APP/BACE-related (amyloid-dependent) mechanism, in which APP and BACE overexpression-related altered cell number and tissue compartment size in the Drosophila wing posterior crossvein (PCV) is observed and can be rescued by ubqn RNAi (left column). (B) An apoptosis-related (amyloid-independent) mechanism, in which expression of the baculovirus pan-caspase inhibitor p35 in the Drosophila wing resulted in weak loss of vein structures and can be enhanced by ubqn RNAi (right column).
Moreover, a number of proteins and pathways have been discovered to be pivotal mediators in apoptosis, e.g., members of the interleukin-1β-converting enzyme (ICE)/Ced-3 proteases (caspases) family of protease (Schlegel et al., 1995, 1996). A hallmark of apoptosis is caspase-mediated cleavage of specific Asp-Glu-Val-Asp (DEVD) amino acid sequence-containing proteins, including poly (ADP-ribose) polymerase (PARP), which is essentially involved in DNA repair in response to environmental and intracellular stress. PARP is a key player for AD pathogenesis and intervention (Salech et al., 2020). Thus, our findings may suggest future studies on ubiquilin-1-related changes in PARP metabolism as well as other AD-related apoptosis and other neuropathology.
Furthermore, our findings support that ubiquilin-1 may be a therapeutic target for the intervention of AD. On the pathophysiological level, ubiquilin-1 is a molecule that regulates PQC and the protein level of ubiquilin-1 reduces in association of AD progression (Stieren et al., 2011). On the therapeutic effects in preclinical models, we showed that overexpression of UBQLN1 leads to lowered Aβ levels in cells. Furthermore, increasing UBQLN1 reduces neuropathology in a mouse model of ALS/FTD conferred by the loss of a protein homologous to ubiquilin-1 (Wang et al., 2020).
Interestingly, our current study discovers a new mechanism in which ubiquilin-1 levels are positively regulated by the inhibition of γ-secretase in our cell models. Our results of the effects of L685, 458 may provide insights on the interaction of ubiquilin-1 and PS1 as well as targeting PS1 to influence ubiquilin-1 levels therapeutically. First, the mechanism that inhibition of γ-secretase increases ubiquilin-1 levels may associate with previous studies reporting ubiquilin-1 as a protein that binds PS1 and regulates PS1 endoproteolysis (Mah et al., 2000). Our results suggest a regulatory circuit which functionally coordinates the levels and activities of γ-secretase and ubiquilin-1. Furthermore, because it is important to maintain levels of ubiquilin-1 that is involved in PQC in responses of apoptotic signal and other stresses, our study implicates that targeting γ-secretase may regulate ubiquilin-1 levels in AD. Different from GSI, γ-secretase modulators (GSMs) have been developed that specifically modulate γ-secretase-mediate APP processing and preferentially reduce aggregation-prone Aβ42 other than Aβ40 (Raven et al., 2017; Ward et al., 2017; Rynearson et al., 2021). Future studies will be required to elucidate the mechanisms related to regulation of γ-secretase and changes of ubiquilin-1 levels.
Finally, we note that although our current study has not investigated specific variants of UBQLN1 and functional domains of ubiquilin-1, it is important that future studies may be performed to identify genetic variants of UBQLN1 and analyze their effects on PQC and in AD. Particularly, UBQLN1 is located on chromosome 9q22, and it can encode two isoforms of a protein that contains 589 amino acids (isoform 1) or 561 amino acid (isoform 2). Ubiquilin-1 contains a UBL (ubiquitin-like) domain and a UBA (ubiquitin-associated) domain. Utilizing these functional domains, ubiquilin-1 interacts with intracellular or surface proteins and modulates their stability and/or steady state levels (Mah et al., 2000; Bedford et al., 2001; Ko et al., 2002, 2004; Wu et al., 2002; N’Diaye and Brown, 2003; Ficklin et al., 2005; Regan-Klapisz et al., 2005; Heir et al., 2006). Ubiquilin-1 appears to be an adaptor protein, which binds polyubiquitinated proteins with its UBL or UBA domain and transport them to the proteasome for degradation, thus preventing aggregates of misfolded protein in the cytoplasm (Kleijnen et al., 2000; Funakoshi et al., 2002; Medicherla et al., 2004). Our current study, in combination with these previous findings, collectively warrant further studies to analyze the impacts of genetic variants and functional domains of ubiquilin-1 on cell viability and AD pathology.
In summary, we showed that silencing UBQLN1 reduces cell viability and increases caspase-3 activity and overexpression of UBQLN1 lowers Aβ levels, supporting a loss of function mechanism of UBQLN1 in AD. Furthermore, pharmacological inhibition of γ-secretase increases ubiquilin-1 protein levels, suggesting a mechanism that regulates the levels of ubiquilin-1. Collectively, our results suggest not only a loss-of-function mechanism of ubiquilin-1 in association with AD, but also support the significance of targeting ubiquilin-1 and PQC as potential therapeutic targets for AD.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author/s.
Author Contributions
AS and DM performed experimental design and data interpretation. CZ and SI performed the experiments and analyzed the results. CZ, SS, and DM performed literature review and prepared the draft of the manuscript. All authors revised and completed the manuscript.
Funding
This work was funded by the NIH NINDS R21NS048227 (AS), Commonwealth of Pennsylvania and Drexel University.
Author Disclaimer
The views expressed in this manuscript do not necessarily reflect those of the National Science Foundation or the United States Government.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank the members of the laboratories from AS and DM for helpful discussions and technical support on the research. This manuscript is based on work done in part by DM while serving at the U.S. National Science Foundation.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnins.2022.821059/full#supplementary-material
References
Adegoke, O. O., Qiao, F., Liu, Y., Longley, K., Feng, S., and Wang, H. (2017). Overexpression of ubiquilin-1 alleviates alzheimer’s disease-caused cognitive and motor deficits and reduces amyloid-beta accumulation in mice. J. Alzheimers Dis. 59, 575–590. doi: 10.3233/JAD-170173
Bedford, F. K., Kittler, J. T., Muller, E., Thomas, P., Uren, J. M., Merlo, D., et al. (2001). GABA(A) receptor cell surface number and subunit stability are regulated by the ubiquitin-like protein Plic-1. Nat. Neurosci. 4, 908–916. doi: 10.1038/nn0901-908
Bensemain, F., Chapuis, J., Tian, J., Shi, J., Thaker, U., Lendon, C., et al. (2006). Association study of the Ubiquilin gene with Alzheimer’s disease. Neurobiol. Dis. 22, 691–693. doi: 10.1016/j.nbd.2006.01.007
Bertram, L., Hiltunen, M., Parkinson, M., Ingelsson, M., Lange, C., Ramasamy, K., et al. (2005). Family-based association between Alzheimer’s disease and variants in UBQLN1. N. Engl. J. Med. 352, 884–894. doi: 10.1056/NEJMoa042765
Brand, A. H., and Perrimon, N. (1993). Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development 118, 401–415. doi: 10.1242/dev.118.2.401
Cao, X., and Sudhof, T. C. (2001). A transcriptionally [correction of transcriptively] active complex of APP with Fe65 and histone acetyltransferase Tip60. Science 293, 115–120. doi: 10.1126/science.1058783
Chakraborty, R., Vepuri, V., Mhatre, S. D., Paddock, B. E., Miller, S., Michelson, S. J., et al. (2011). Characterization of a Drosophila Alzheimer’s disease model: pharmacological rescue of cognitive defects. PLoS One 6:e20799. doi: 10.1371/journal.pone.0020799
Chen, G., Wang, X., Yu, J., Varambally, S., Thomas, D. G., Lin, M. Y., et al. (2007). Autoantibody profiles reveal ubiquilin 1 as a humoral immune response target in lung adenocarcinoma. Cancer Res. 67, 3461–3467. doi: 10.1158/0008-5472.CAN-06-4475
Choi, S. H., Kim, Y. H., Hebisch, M., Sliwinski, C., Lee, S., D’Avanzo, C., et al. (2014). A three-dimensional human neural cell culture model of Alzheimer’s disease. Nature 515, 274–278.
El Ayadi, A., Stieren, E. S., Barral, J. M., and Boehning, D. (2013). Ubiquilin-1 and protein quality control in Alzheimer disease. Prion 7, 164–169. doi: 10.4161/pri.23711
Ellgaard, L., and Helenius, A. (2003). Quality control in the endoplasmic reticulum. Nat. Rev. Mol. Cell Biol. 4, 181–191.
Ficklin, M. B., Zhao, S., and Feng, G. (2005). Ubiquilin-1 regulates nicotine-induced up-regulation of neuronal nicotinic acetylcholine receptors. J. Biol. Chem. 280, 34088–34095. doi: 10.1074/jbc.M506781200
Fossgreen, A., Bruckner, B., Czech, C., Masters, C. L., Beyreuther, K., and Paro, R. (1998). Transgenic Drosophila expressing human amyloid precursor protein show gamma-secretase activity and a blistered-wing phenotype. Proc. Natl. Acad. Sci. USA 95, 13703–13708. doi: 10.1073/pnas.95.23.13703
Funakoshi, M., Sasaki, T., Nishimoto, T., and Kobayashi, H. (2002). Budding yeast Dsk2p is a polyubiquitin-binding protein that can interact with the proteasome. Proc. Natl. Acad. Sci. USA 99, 745–750. doi: 10.1073/pnas.012585199
Ganguly, A., Feldman, R. M., and Guo, M. (2008). ubiquilin antagonizes presenilin and promotes neurodegeneration in Drosophila. Hum. Mol. Genet. 17, 293–302. doi: 10.1093/hmg/ddm305
Greeve, I., Kretzschmar, D., Tschape, J. A., Beyn, A., Brellinger, C., Schweizer, M., et al. (2004). Age-dependent neurodegeneration and Alzheimer-amyloid plaque formation in transgenic Drosophila. J. Neurosci. 24, 3899–3906. doi: 10.1523/JNEUROSCI.0283-04.2004
Gross, G. G., Feldman, R. M., Ganguly, A., Wang, J., Yu, H., and Guo, M. (2008). Role of X11 and ubiquilin as in vivo regulators of the amyloid precursor protein in Drosophila. PLoS One 3:e2495. doi: 10.1371/journal.pone.0002495
Guo, M., Hong, E. J., Fernandes, J., Zipursky, S. L., and Hay, B. A. (2003). A reporter for amyloid precursor protein gamma-secretase activity in Drosophila. Hum. Mol. Genet. 12, 2669–2678. doi: 10.1093/hmg/ddg292
Hardy, J., and Selkoe, D. J. (2002). The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science 297, 353–356. doi: 10.1126/science.1072994
Heir, R., Ablasou, C., Dumontier, E., Elliott, M., Fagotto-Kaufmann, C., and Bedford, F. K. (2006). The UBL domain of PLIC-1 regulates aggresome formation. EMBO Rep. 7, 1252–1258. doi: 10.1038/sj.embor.7400823
Hiltunen, M., Lu, A., Thomas, A. V., Romano, D. M., Kim, M., Jones, P. B., et al. (2006). Ubiquilin 1 modulates amyloid precursor protein trafficking and Abeta secretion. J. Biol. Chem. 281, 32240–32253. doi: 10.1074/jbc.M603106200
Kamboh, M. I., Minster, R. L., Feingold, E., and DeKosky, S. T. (2006). Genetic association of ubiquilin with Alzheimer’s disease and related quantitative measures. Mol. Psychiatry 11, 273–279. doi: 10.1038/sj.mp.4001775
Kim, T. W., Pettingell, W. H., Jung, Y. K., Kovacs, D. M., and Tanzi, R. E. (1997). Alternative cleavage of Alzheimer-associated presenilins during apoptosis by a caspase-3 family protease. Science 277, 373–376. doi: 10.1126/science.277.5324.373
Kleijnen, M. F., Shih, A. H., Zhou, P., Kumar, S., Soccio, R. E., Kedersha, N. L., et al. (2000). The hPLIC proteins may provide a link between the ubiquitination machinery and the proteasome. Mol. Cell 6, 409–419. doi: 10.1016/s1097-2765(00)00040-x
Ko, H. S., Uehara, T., and Nomura, Y. (2002). Role of ubiquilin associated with protein-disulfide isomerase in the endoplasmic reticulum in stress-induced apoptotic cell death. J. Biol. Chem. 277, 35386–35392. doi: 10.1074/jbc.M203412200
Ko, H. S., Uehara, T., Tsuruma, K., and Nomura, Y. (2004). Ubiquilin interacts with ubiquitylated proteins and proteasome through its ubiquitin-associated and ubiquitin-like domains. FEBS Lett. 566, 110–114. doi: 10.1016/j.febslet.2004.04.031
Kostova, Z., and Wolf, D. H. (2003). For whom the bell tolls: protein quality control of the endoplasmic reticulum and the ubiquitin-proteasome connection. EMBO J. 22, 2309–2317. doi: 10.1093/emboj/cdg227
Li, A., Xie, Z., Dong, Y., McKay, K. M., McKee, M. L., and Tanzi, R. E. (2007). Isolation and characterization of the Drosophila ubiquilin ortholog dUbqln: in vivo interaction with early-onset Alzheimer disease genes. Hum. Mol. Genet. 16, 2626–2639. doi: 10.1093/hmg/ddm219
Liu, Y., Lu, L., Hettinger, C. L., Dong, G., Zhang, D., Rezvani, K., et al. (2014). Ubiquilin-1 protects cells from oxidative stress and ischemic stroke caused tissue injury in mice. J. Neurosci. 34, 2813–2821. doi: 10.1523/JNEUROSCI.3541-13.2014
Luo, L., Liu, Y., Tu, X., Ren, X., Zhao, W., Liu, J., et al. (2019). Decreased expression of ubiquilin1 following neonatal hypoxiaischemic brain injury in mice. Mol. Med. Rep. 19, 4597–4602. doi: 10.3892/mmr.2019.10168
Mah, A. L., Perry, G., Smith, M. A., and Monteiro, M. J. (2000). Identification of ubiquilin, a novel presenilin interactor that increases presenilin protein accumulation. J. Cell Biol. 151, 847–862. doi: 10.1083/jcb.151.4.847
Marenda, D. R., Vrailas, A. D., Rodrigues, A. B., Cook, S., Powers, M. A., Lorenzen, J. A., et al. (2006). MAP kinase subcellular localization controls both pattern and proliferation in the developing Drosophila wing. Development 133, 43–51. doi: 10.1242/dev.02168
Marenda, D. R., Zraly, C. B., Feng, Y., Egan, S., and Dingwall, A. K. (2003). The Drosophila SNR1 (SNF5/INI1) subunit directs essential developmental functions of the Brahma chromatin remodeling complex. Mol. Cell Biol. 23, 289–305. doi: 10.1128/MCB.23.1.289-305.2003
Medicherla, B., Kostova, Z., Schaefer, A., and Wolf, D. H. (2004). A genomic screen identifies Dsk2p and Rad23p as essential components of ER-associated degradation. EMBO Rep. 5, 692–697. doi: 10.1038/sj.embor.7400164
Meyer, C. A., Jacobs, H. W., Datar, S. A., Du, W., Edgar, B. A., and Lehner, C. F. (2000). Drosophila Cdk4 is required for normal growth and is dispensable for cell cycle progression. EMBO J. 19, 4533–4542. doi: 10.1093/emboj/19.17.4533
N’Diaye, E. N., and Brown, E. J. (2003). The ubiquitin-related protein PLIC-1 regulates heterotrimeric G protein function through association with Gbetagamma. J. Cell Biol. 163, 1157–1165. doi: 10.1083/jcb.200307155
Raven, F., Ward, J. F., Zoltowska, K. M., Wan, Y., Bylykbashi, E., Miller, S. J., et al. (2017). Soluble gamma-secretase modulators attenuate alzheimer’s beta-amyloid pathology and induce conformational changes in presenilin 1. EBioMedicine 24, 93–101. doi: 10.1016/j.ebiom.2017.08.028
Regan-Klapisz, E., Sorokina, I., Voortman, J., de Keizer, P., Roovers, R. C., Verheesen, P., et al. (2005). Ubiquilin recruits Eps15 into ubiquitin-rich cytoplasmic aggregates via a UIM-UBL interaction. J. Cell Sci. 118, 4437–4450. doi: 10.1242/jcs.02571
Rynearson, K. D., Ponnusamy, M., Prikhodko, O., Xie, Y., Zhang, C., Nguyen, P., et al. (2021). Preclinical validation of a potent gamma-secretase modulator for Alzheimer’s disease prevention. J. Exp. Med. 218:2560. doi: 10.1084/jem.20202560
Salech, F., Ponce, D. P., Paula-Lima, A. C., SanMartin, C. D., and Behrens, M. I. (2020). Nicotinamide, a Poly [ADP-Ribose] Polymerase 1 (PARP-1) inhibitor, as an adjunctive therapy for the treatment of alzheimer’s disease. Front. Aging Neurosci. 12:255. doi: 10.3389/fnagi.2020.00255
Schlegel, J., Peters, I., and Orrenius, S. (1995). Isolation and partial characterization of a protease involved in Fas-induced apoptosis. FEBS Lett. 364, 139–142. doi: 10.1016/0014-5793(95)00374-i
Schlegel, J., Peters, I., Orrenius, S., Miller, D. K., Thornberry, N. A., Yamin, T. T., et al. (1996). CPP32/apopain is a key interleukin 1 beta converting enzyme-like protease involved in Fas-mediated apoptosis. J. Biol. Chem. 271, 1841–1844. doi: 10.1074/jbc.271.4.1841
Stieren, E. S., El Ayadi, A., Xiao, Y., Siller, E., Landsverk, M. L., Oberhauser, A. F., et al. (2011). Ubiquilin-1 is a molecular chaperone for the amyloid precursor protein. J. Biol. Chem. 286, 35689–35698. doi: 10.1074/jbc.M111.243147
Takalo, M., Natunen, T., Leskelä, S., Paldanius, K. M. A., and Soininen, H. (2017). New Implications for the role for ubiquilin-1 in molecular mechanisms of alzheime’s disease: interrelationship with BACE1. J. Alzheimer’s Dis. Parkins. 7:365.
Tanzi, R. E., and Bertram, L. (2001). New frontiers in Alzheimer’s disease genetics. Neuron 32, 181–184. doi: 10.1016/s0896-6273(01)00476-7
Tanzi, R. E., Gusella, J. F., Watkins, P. C., Bruns, G. A., GeorgeHyslop, P. St, Keuren, M.L., Van, et al. (1987). Amyloid beta protein gene: cDNA, mRNA distribution, and genetic linkage near the Alzheimer locus. Science 235, 880–884. doi: 10.1126/science.2949367
Vrailas-Mortimer, A. D., Majumdar, N., Middleton, G., Cooke, E. M., and Marenda, D. R. (2007). Delta and Egfr expression are regulated by Importin-7/Moleskin in Drosophila wing development. Dev. Biol. 308, 534–546. doi: 10.1016/j.ydbio.2007.06.011
Wang, H., Lim, P. J., Yin, C., Rieckher, M., Vogel, B. E., and Monteiro, M. J. (2006). Suppression of polyglutamine-induced toxicity in cell and animal models of Huntington’s disease by ubiquilin. Hum. Mol. Genet. 15, 1025–1041. doi: 10.1093/hmg/ddl017
Wang, S., Tatman, M., and Monteiro, M. J. (2020). Overexpression of UBQLN1 reduces neuropathology in the P497S UBQLN2 mouse model of ALS/FTD. Acta Neuropathol. Commun. 8:164. doi: 10.1186/s40478-020-01039-9
Ward, J., Wang, H., Saunders, A. J., Tanzi, R. E., and Zhang, C. (2017). Mechanisms that synergistically regulate eta-secretase processing of APP and Aeta-alpha protein levels: relevance to pathogenesis and treatment of Alzheimer’s disease. Discov. Med. 23, 121–128.
Wu, S., Mikhailov, A., Kallo-Hosein, H., Hara, K., Yonezawa, K., and Avruch, J. (2002). Characterization of ubiquilin 1, an mTOR-interacting protein. Biochim. Biophys. Acta 1542, 41–56. doi: 10.1016/s0167-4889(01)00164-1
Xie, Z., Culley, D. J., Dong, Y., Zhang, G., Zhang, B., Moir, R. D., et al. (2008). The common inhalation anesthetic isoflurane induces caspase activation and increases amyloid beta-protein level in vivo. Ann. Neurol. 64, 618–627. doi: 10.1002/ana.21548
Zhang, C., Khandelwal, P. J., Chakraborty, R., Cuellar, T. L., Sarangi, S., Patel, S. A., et al. (2007). An AICD-based functional screen to identify APP metabolism regulators. Mol. Neurodeg. 2:15. doi: 10.1186/1750-1326-2-15
Zhang, C., and Saunders, A. J. (2007). Therapeutic targeting of the alpha-secretase pathway to treat Alzheimer’s disease. Discov. Med. 7, 113–117.
Keywords: Alzheimer’s disease, ubiquilin 1 (UBQLN1), Drosophila, APP – amyloid precursor protein, gamma secretase (γ-secretase)
Citation: Zhang C, Inamdar SM, Swaminathan S, Marenda DR and Saunders AJ (2022) Association of the Protein-Quality-Control Protein Ubiquilin-1 With Alzheimer’s Disease Both in vitro and in vivo. Front. Neurosci. 16:821059. doi: 10.3389/fnins.2022.821059
Received: 23 November 2021; Accepted: 24 February 2022;
Published: 17 March 2022.
Edited by:
Peng Lei, Sichuan University, ChinaReviewed by:
Deng-Feng Zhang, Kunming Institute of Zoology (CAS), ChinaZhentao Zhang, Renmin Hospital of Wuhan University, China
Copyright © 2022 Zhang, Inamdar, Swaminathan, Marenda and Saunders. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Aleister J. Saunders, ajs56@drexel.edu
†Present addresses: Can Zhang, Genetics and Aging Research Unit, McCance Center for Brain Health, Mass General Institute for Neurodegenerative Diseases (MIND), Department of Neurology, Massachusetts General Hospital and Harvard Medical School, Boston, MA, United States; Shivangi M. Inamdar, Department of Anatomy and Cell Biology, University of Iowa, Iowa City, IA, United States