 Alessandra Rufini
Alessandra Rufini Florence Malisan
Florence Malisan Ivano Condò
Ivano Condò Roberto Testi
Roberto Testi
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Neurosci. , 09 February 2022
Sec. Neurodegeneration
Volume 16 - 2022 | https://doi.org/10.3389/fnins.2022.814445
This article is part of the Research Topic Basic and Clinical Mechanisms in Friedreich Ataxia View all 9 articles
Friedreich ataxia is a rare neurodegenerative disorder caused by insufficient levels of the essential mitochondrial protein frataxin. It is a severely debilitating disease that significantly impacts the quality of life of affected patients and reduces their life expectancy, however, an adequate cure is not yet available for patients. Frataxin function, although not thoroughly elucidated, is associated with assembly of iron-sulfur cluster and iron metabolism, therefore insufficient frataxin levels lead to reduced activity of many mitochondrial enzymes involved in the electron transport chain, impaired mitochondrial metabolism, reduced ATP production and inefficient anti-oxidant response. As a consequence, neurons progressively die and patients progressively lose their ability to coordinate movement and perform daily activities. Therapeutic strategies aim at restoring sufficient frataxin levels or at correcting some of the downstream consequences of frataxin deficiency. However, the classical pathways of drug discovery are challenging, require a significant amount of resources and time to reach the final approval, and present a high failure rate. Drug repositioning represents a viable alternative to boost the identification of a therapy, particularly for rare diseases where resources are often limited. In this review we will describe recent efforts aimed at the identification of a therapy for Friedreich ataxia through drug repositioning, and discuss the limitation of such strategies.
Friedreich ataxia (FRDA, OMIM#229300) is a rare genetic disorder caused by insufficient levels of the mitochondrial protein frataxin. It is inherited as an autosomal recessive disease with a prevalence of 1:50.000 individuals, which makes it the most common form of inherited ataxia. Typically, onset occurs during puberty, although late and early onset cases have been described. Patients initially present with gait and limb ataxia and progressively lose their ability to walk and accomplish daily activities. Clinical manifestations also include scoliosis and skeletal deformities, dysarthria, hearing and visual impairment. Neurological manifestations are associated with degeneration of sensory neurons in the dorsal root ganglia, in the dentate nuclei of the cerebellum and posterior column of the spinal cord (Koeppen and Mazurkiewicz, 2013; Koeppen et al., 2017). Although mostly considered a disorder of the nervous system, patients also develop a hypertrophic cardiomyopathy that often leads to premature death (Tsou et al., 2011; Tiano et al., 2020). Diabetes is also present in a minority of patients (Delatycki and Corben, 2012; Cnop et al., 2013).
The underlying mutation consists of a GAA triplet repeat expansion, within the first intron of the gene coding for frataxin. The repeat expansion is homozygous in >96% of cases, while a minority of patients presents the repeat expansion on one allele and a point mutation on the other allele of the frataxin gene (FXN) (Galea et al., 2016). The GAA repeat expansion alters the chromatin structure and leads to epigenetic modifications that reduce the accessibility of the DNA and impair transcription of the frataxin gene, resulting in insufficient production of the corresponding protein. Longer expansions of the GAA repeats correlate with lower levels of residual frataxin, earlier age at onset and more severe disease progression (Filla et al., 1996; Montermini et al., 1997; Chutake et al., 2014; Reetz et al., 2015; Patel et al., 2016), establishing a clear link between frataxin deficiency and the pathogenesis of the disease and highlighting the therapeutic relevance of restoring frataxin levels in patients.
Frataxin is an essential mitochondrial protein (Campuzano et al., 1997; Condò et al., 2007) involved in iron metabolism and in the biosynthesis of iron-sulfur clusters (ISC; Shan et al., 2007; Martelli and Puccio, 2014; Fox et al., 2019). Its deficiency results in impaired synthesis of ISC and reduced activity of many ISC-containing mitochondrial enzymes (Rotig et al., 1997; Poburski et al., 2016), causing inefficient function of the electron transport chain, reduced ATP production and consequently overall impaired mitochondrial metabolism and defective anti-oxidant response. The inability to cope with increased oxidative stress is considered one of the most important consequences of frataxin deficiency (Gomes and Santos, 2013). These alterations at the cellular level ultimately leads to cell death, the most susceptible cells being those with high mitochondrial metabolism such as neurons, cardiomyocytes and pancreatic beta-cells (Igoillo-Esteve et al., 2015). Therefore, frataxin levels are not only crucial for cell survival but also for handling different stress responses (Condò et al., 2006, 2010; Paupe et al., 2009; Guccini et al., 2011).
Currently, there is no approved therapy to treat Friedreich ataxia. Only palliative cares are available for patients, mostly consisting of antioxidants or iron chelators, all of which, however, do not impact significantly on the neurological dysfunctions associated with the disease. Since lack of frataxin is considered the direct cause of the disease, the most straightforward therapeutic approaches are aimed at increasing frataxin protein levels. Considering that the mutation in the frataxin gene does not affect the coding sequence and the residual frataxin protein that is produced in patients is functional, it is reasonable to invest in pharmacological approaches aimed to increase the production of endogenous frataxin. Strategies to elevate endogenous frataxin levels focus on either promoting its gene expression at the transcriptional (Herman et al., 2006; Li et al., 2016; Erwin et al., 2017; Shen et al., 2020) or translational (Bon et al., 2019) level, or preventing its degradation (Rufini et al., 2011, 2015; Benini et al., 2017). Alternatively, gene therapy (Perdomini et al., 2014; Piguet et al., 2018) or protein replacement approaches (Vyas et al., 2012; Britti et al., 2018; Castro et al., 2018) are being considered by various groups, reporting encouraging results (Delatycki and Bidichandani, 2019). Pharmacological treatments under development also focus on reducing the downstream consequences of frataxin deficiency, such as mitochondrial dysfunction and oxidative stress (Zesiewicz et al., 2018a,b; Lynch and Johnson, 2021). Indeed, the inability to mount an efficient anti-oxidant defense to the increased oxidative stress observed in patients’ cells is a major cause of cellular stress and death and is considered a relevant pathogenic mechanism for Friedreich ataxia. The inefficient anti-oxidant response has been linked to a defective activation of the nuclear factor erythroid 2-related factor 2 (Nrf2) pathway (Paupe et al., 2009) and its downstream target genes, both in cells derived from FRDA patients and in FRDA animal models. One of such models, the YG8R mice exhibit reduced expression of anti-oxidant genes, such as glutaredoxin, peroxiredoxin and glutathione S-transferase (Shan et al., 2013). In line with this evidence, strategies aimed at restoring an efficient Nrf2 activation are considered promising therapeutic approaches (Petrillo et al., 2019) and several drugs have been proposed which restore appropriate Nrf2 response in FRDA cellular and animal models.
Given the urgency to identify a therapy for such a devastating disease, many research efforts have focused on drug repositioning approaches. Drug repositioning or repurposing, which consists of finding a new therapeutic indication for an already existing and approved drug, has gained considerable attention over the last decades. Conventional drug discovery process involves significant high-risk investments by pharmaceutical industry, in terms of financial resources and time to reach market approval. The cost of bringing a new drug to the market was estimated to be approximately $2 billions (DiMasi, 2018) and it may take as long as 10–12 years for a new drug to be available for patients (Matthews et al., 2016). On the other side, drug repurposing approaches can take advantage of well-established safety and toxicity profiles for already approved drugs and therefore cut down the costs associated with the development of a new drug and significantly reduce the time to the final approval, allowing patients to benefit from the availability of new therapeutics (Schcolnik-Cabrera et al., 2021). This is particularly relevant in the context of rare diseases and orphan drugs, where the reduced business opportunity makes it often not convenient for pharma companies to invest in the development of new therapies from scratch (Sardana et al., 2011).
The research for a therapy for Friedreich ataxia has indeed recently pursued repositioning strategies to accelerate the pace toward the approval of an effective therapy for patients in need. In this review we will summarize the repurposing efforts made toward the identification of approved and market-available drugs for a successful treatment of Friedreich ataxia. Compounds identified and described below can be classified as chemical drugs, biological drugs and natural products, as summarized in Table 1, and are listed following the order of their initial studies in FRDA.
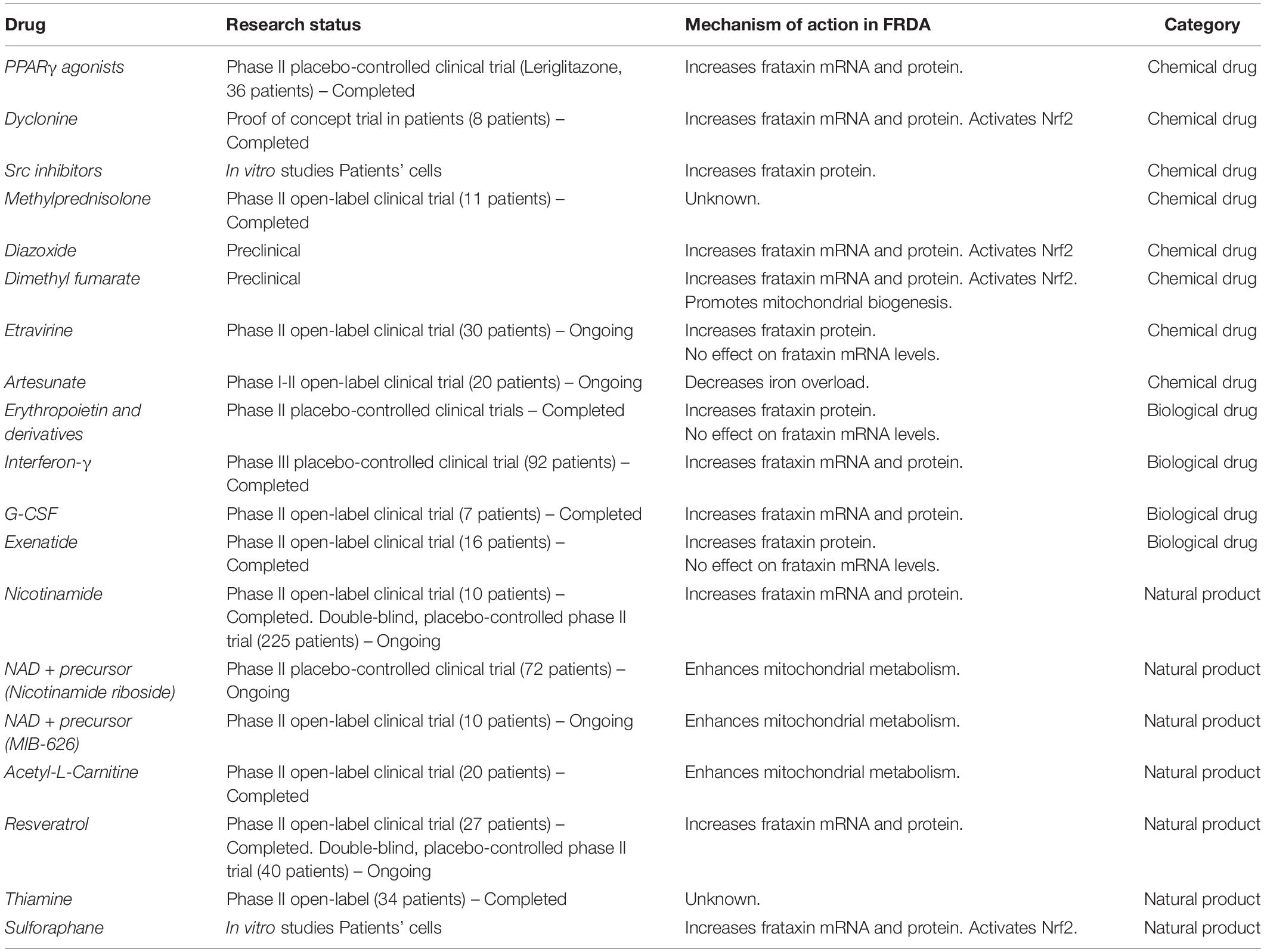
Table 1. List of repurposed drugs in FRDA.
PPARγ agonists are a class of drugs commonly used in the treatment of type II diabetes. They were initially proposed as a therapeutic for FRDA for their ability to activate PGC1α. The master transcriptional regulator PGC1α, which controls mitochondrial biogenesis and anti-oxidant response (Coppola et al., 2009), was indeed found to be reduced in primary fibroblasts form FRDA patients and in the KIKO mouse model (Lin et al., 2017). Initial observations on PPARγ agonists showed that the Azelaoyl-PAF increases frataxin mRNA and protein levels in fibroblasts derived from patients (Marmolino et al., 2009) and pioglitazone is able to restore anti-oxidant defense inducing the expression of SOD2 in FRDA fibroblasts and in the cerebellum of KIKO mice, through the activation of PGC1α (Coppola et al., 2009; Marmolino et al., 2010). More recently, the PPARγ agonist Leriglitazone (MIN-102, developed by Minoryx Therapeutics) was evaluated for its effect in cellular and murine models of FRDA. Leriglitazone, a metabolite a pioglitazone, currently under investigation for the treatment of X-linked adrenoleukodystrophy (Rodriguez-Pascau et al., 2021b), is an orally bioavailable drug that has been shown to have a good safety profile and, importantly, is able to cross the brain-blood barrier. Rodriguez-Pascau et al. (2021a) showed that leriglitazone promotes a 50% increase in frataxin levels in frataxin-deficient rat DRG (Rodriguez-Pascau et al., 2021a), rescues neurite morphology, mitochondrial membrane potential and prevents apoptosis. While no effects on frataxin levels were observed on frataxin-deficient rat cardiomyocytes, leriglitazone prevents lipid droplets accumulation in these cells, likely thanks to a positive effect on fatty acid beta-oxidation. The effects on the motor performances of YG8sR mice administered for 7 months with leriglitazone are, however, partial, with improvement observed only in the balance beam test (Rodriguez-Pascau et al., 2021a). A phase II, placebo-controlled, clinical trial in FRDA patient to evaluate the efficacy of this drug has been recently completed, however, the data have not been published yet1.
Dyclonine was proposed as a candidate therapeutic for Friedreich ataxia for its ability to increase frataxin levels and rescue downstream consequences of frataxin deficiency. In an effort to identify drugs able to restore anti-oxidant defense in FRDA cells, Sahdeo et al. (2014) used the thioredoxin oxidant diamide to induce oxidative stress and cell death in FRDA cells. Through a high-throughput screening of an FDA-approved drugs library, they identified dyclonine as a drug able to confer protection against diamide-induced oxidative stress. Dyclonine is a local oral anesthetic that has been used for more than 50 years and functions through inhibition of the sodium channels (Shelmire et al., 1955; Gargiulo et al., 1992). Dyclonine induces a significant increase in frataxin transcript and consequent increase in frataxin protein in FRDA lymphoblasts and in the cerebellum of YG8 and KIKO mouse models. Moreover, dyclonine activates the Nrf2 pathway and drives the expression of Antioxidant Responsive Elements (ARE)-containing genes. The identification of ARE elements in the frataxin locus suggests an interesting mechanistic link between the activation of Nrf2 and the upregulation of frataxin protein. However, not all Nrf2 inducers identified have been shown to upregulate frataxin (Abeti et al., 2018; Petrillo et al., 2019). Dyclonine rescues the activity of the iron-sulfur cluster (ISC)-containing enzymes aconitase and succinate dehydrogenase in FRDA lymphoblasts and YG8 mice and might provide an improvement in motor coordination in the KIKO mice. However, the dyclonine dose that induces frataxin upregulation is ∼30-fold higher than the dose required for protection against diamide, indicating that additional protective mechanisms may play a major role in protection from diamide-induced oxidative stress by dyclonine. Nevertheless, 8 FRDA patients were dosed with dyclonine oral rinse for 1 week. Six out of eight patients showed an induction of frataxin protein in cells from buccal swabs, with a trend showing a positive correlation between disease severity and responsiveness to treatment (Sahdeo et al., 2014).
Frataxin protein levels are controlled by the proteasome upon ubiquitination of target residue, K147, on frataxin (Rufini et al., 2011). This lysine represents a crucial site for frataxin stability since a frataxin mutant lacking K147 cannot be ubiquitinated and is more stable. Therefore, preventing ubiquitination on K147 is expected to grant frataxin an increased stability and a prolonged half-life (Rufini et al., 2011). Ubiquitination and phosphorylation are post-translational modifications that are important regulators of mitochondrial functions (Hebert-Chatelain, 2013). We have reported that frataxin is phosphorylated on Y118 by Src kinase (Cherubini et al., 2015). Interestingly, non-phosphorylatable frataxin-Y118F mutant is less ubiquitinated suggesting that phosphorylation on Y118 is required for ubiquitination. Moreover, blocking Src activity with specific inhibitors such as PP2, SU6656, Saracatinib, Bosutinib and Dasatinib increases frataxin levels. Accordingly, Src inhibitors are ineffective in human cells in which a frataxin-Y118F mutant was expressed. All the Src inhibitors can promote frataxin accumulation in a dose dependent manner, although with different efficacy. Among the Src inhibitors tested, Dasatinib appeared to be the most efficient in promoting frataxin accumulation, being still active in the nanomolar range of concentrations. Interestingly, frataxin accumulation was observed for all the different processing forms such as precursor, intermediate and mature forms. All the Src inhibitors tested could increase frataxin levels in cells overexpressing wild type frataxin, but not in cells overexpressing the non-phosphorylatable Y118F frataxin mutant, suggesting that they indeed act by inhibiting phosphorylation of Y118. In addition, these inhibitors are also effective on the levels of endogenous frataxin as shown in HEK293 cells (Cherubini et al., 2015). Dasatinib could upregulate frataxin in primary fibroblasts derived from a FRDA patient in a dose dependent manner. Furthermore, aconitase activity was also restored in FRDA lymphoblasts exposed to Dasatinib. Therefore, Src inhibitors induce frataxin expression and rescue the aconitase defect in cells derived from FRDA patients, supporting their potential therapeutic application (Cherubini et al., 2015). Src inhibitors Dasatinib and Bosutinib, reported to cross the blood-brain barrier (Porkka et al., 2008; Liang et al., 2009), have been approved for therapeutic use in humans (Hochhaus and Kantarjian, 2013; Hill et al., 2014) for the treatment of Philadelphia chromosome positive (Ph+) chronic myelogenous leukemia (CML). Further investigation should thus clarify the therapeutic potential of Src inhibitors in FRDA patients.
The rationale for considering corticosteroid treatment for FRDA derives from several lines of evidence. Comprehensive transcriptome analysis of PBMC derived from patients identified a number of upregulated genes associated with the innate immune response (Haugen et al., 2010; Nachun et al., 2018). Moreover, frataxin deficiency leads to steroid deficiency in flies and ovarian cells (Palandri et al., 2015). Whether inflammation contributes to the pathogenesis of FRDA or it is merely a consequence of prolonged and chronic cellular stress, has yet to be established. The observation that a FRDA patient treated with steroid for a concomitant nephrotic syndrome showed some improvement in the neurological symptoms, suggested to investigate the effects of steroid in other FRDA patients (Shinnick et al., 2016). However, an open label study on 11 FRDA patients treated with oral methylprednisolone showed that, although well tolerated, the drug induced non-significant improvement in neurological abilities except for the 1-min walk test. No changes in frataxin levels were detected in buccal cells (Patel et al., 2019).
Diazoxide, a well-known drug used to treat acute hypertension (Hutcheon and Barthalmus, 1962), was shown to increase frataxin protein levels in FRDA lymphoblastoid cell lines (Santoro et al., 2018). Diazoxide functions as a potassium channel opener that acts on the mitochondrial ATP-dependent potassium channel, resulting in vasodilation. Increase in frataxin mRNA and protein levels was observed also in the brain and the heart of YG8sR mice (Anjomani Virmouni et al., 2015). Moreover, treatment with diazoxide resulted in increase in aconitase activity and reduced protein oxidation in the brain in this mouse model (Santoro et al., 2018). The activation of the mTOR pathway, already described as an effect of diazoxide (Kwon et al., 2006), could in turn be responsible for the activation of Nrf2 (Lim et al., 2014) observed upon treatment with diazoxide in FRDA cells and could promote the anti-oxidant response. Interestingly, the mTOR pathway was also directly linked to the increase in frataxin levels following IGF-1 stimulation (Franco et al., 2012). However, results on neurological parameters in YG8sR were variable, contrasting and difficult to interpret, with some improvement on the beam walk test but overall reduced locomotor function (Santoro et al., 2018).
Through the screening of a library of market-available drugs, Jasoliya et al. (2019) identified dimethyl fumarate (DMF) as a drug able to protect FRDA cells from cell death. DMF was previously shown to promote the survival of motor neurons and neuronal stem cells from oxidative stress-induced cell death by activating Nrf2 pathway (Wang et al., 2015; Petrillo et al., 2017). DMF is a fumaric acid derivative currently used for the treatment of multiple sclerosis and psoriasis (Venci and Gandhi, 2013) and recently proposed for the treatment of Parkinson’s disease (Lastres-Becker et al., 2016). In multiple sclerosis patients, DMF was shown to promote mitochondrial biogenesis, through the activation of Nrf2 pathway.
Treatment with DMF promotes an increase in frataxin levels in lymphoblastoid cell lines from FRDA patients and in the cerebellum of YG8 and KIKO mice (Jasoliya et al., 2019). Interestingly, an increase in frataxin mRNA had been previously observed in peripheral blood mononuclear cells (PBMC) from multiple sclerosis patients treated with DMF for 3 months. The proposed mechanism of action implies an increase in the transcription initiation at the frataxin gene and a reduction in the R-loops content. As it is also known that DMF triggers the activation of the Nrf2 pathway (Linker et al., 2011), binding of Nrf2 to putative ARE in the promoter region of frataxin could account for the increased frataxin transcription. The increased transcriptional activation of the frataxin gene could in turn reduce the formation of R-loops that is favored during transcriptional pausing of the RNA polymerase, further increasing frataxin mRNA elongation. Accordingly, cells treated with DMF present reduced R-loops frequency, which could further contribute to the resulting enhanced frataxin transcription (Jasoliya et al., 2019). DMF was also shown to promote mitochondrial biogenesis in fibroblasts derived from FRDA patients (Hayashi et al., 2017), possibly by activating the PGC-1α and Nrf1 dependent mitochondrial biogenesis pathway (Wu et al., 1999; Piantadosi et al., 2008).
More recently, the activity of DMF was tested in a FXN knock down model of FRDA (Hui et al., 2021). The FXN knock down mice represents a recently developed FRDA model, in which frataxin expression is silenced by a doxycycline-inducible frataxin siRNA. This system allows a progressive down regulation of frataxin in all the tissues, which induces a progressive and more physiological phenotype. Defects in mitochondrial biogenesis were observed in these mice following frataxin silencing (Chandran et al., 2017). Of relevance, DMF treatment rescues mitochondrial deficit caused by frataxin knock down by inducing an increase in mitochondrial copy number and restoring mitochondrial enzymes activity. Conversely, DMF orally administered to wild type mice for 2 weeks increases frataxin and cytochrome c oxidase-4 expression and aconitase activity in the brain and muscle, without affecting mitochondrial biogenesis, indicating that DMF promotes mitochondrial biogenesis only when there is a deficit caused by low frataxin expression (Hui et al., 2021).
Importantly the maximal effective dose observed in mice is equivalent or below the maximal approved dose in humans for the treatment of MS and psoriasis. Future investigations are required to assess the effect of DMF on clinical parameters in Friedreich ataxia and clinical trials in FRDA patients are currently being planned.
Using a frataxin reporter system in a cell-based assay, our group performed a screening of an FDA-approved and commercially available drugs library to search for compounds able to increase frataxin amount in cells. Among a few potentially interesting candidates, our attention was focused on etravirine, given its highly favorable safety profile and its potential to be used as a chronic treatment (Croxtall, 2012; Havens et al., 2020). Etravirine (Intelence) is a non-nucleoside reverse transcriptase inhibitor (NNRTI) of the HIV enzyme (Namasivayam et al., 2019; Zhuang et al., 2020). it is currently used an anti-HIV therapy in life-long treatment regimens (Lazzarin et al., 2007; Katlama et al., 2020). Its use has been approved by FDA in patients starting from 2 years of age and above. Etravirine promotes a significant increase in frataxin levels in lymphoblasts and fibroblasts derived from FRDA patients (Alfedi et al., 2019). However, etravirine does not appear to elicit an increase in frataxin mRNA levels nor does it affect its stability. Data suggest that etravirine treatment results in an enhancement of the translation rate of frataxin, through a mechanism involving the redistribution of frataxin mRNA to the actively translating heavy polysome fraction. Interestingly, frataxin accumulation in treated patients’ cells is comparable to frataxin levels in cells from unaffected carrier sibling, suggesting a potential beneficial role of etravirine in a Friedreich ataxia therapy. Indeed, etravirine can correct some defects associated with frataxin deficiency. In lymphoblasts from FRDA patients, etravirine restores the activity of aconitase and confers protection to oxidative stress (Alfedi et al., 2019). Further studies will be required to assess whether a functional improvement can be achieved in tissues relevant for FRDA pathology, such as the heart and the nervous system. In this regard, etravirine was shown to pass into the cerebrospinal fluid of treated HIV patients, although it was found to be highly protein-bound (Nguyen et al., 2013; Havens et al., 2020).
Etravirine functions by binding to HIV reverse transcriptase (RT) in the NNRTI binding pocket and allosterically regulating its catalytic activity (Zhuang et al., 2020). However, HIV RT does not have any human counterpart and etravirine binding mode on RT is not suggestive of any obvious human target that could account for the effect on frataxin. Efforts are now focused on the identification of the relevant molecular target through which etravirine elicits frataxin accumulation. This could in turn unveil new pathways relevant for the regulation of frataxin synthesis.
Considering the excellent safety profile of etravirine and its availability as on oral medication, a phase II clinical trial to test the efficacy of etravirine in FRDA patients is currently ongoing2.
Defects in the metabolic pathway for Transferrin receptor 1 (TfR1) palmitoylation could lead to increased cell surface expression of TfR1 (Alvarez et al., 1990). Lack of palmitoylation is also associated with increased endocytosis of transferrin-TfR1 complex and defective recycling of transferrin, leading to intracellular iron accumulation (Drecourt et al., 2018). Petit et al. (2021) have shown that fibroblasts derived from FRDA patients develop iron overload and fail to negatively regulate iron transport in response to ferric ammonium citrate. Interestingly, an increased expression of TfR1 at the plasma membrane was observed in these cells, associated with a defect in the palmitoylation pathway. Defects in the assembly of iron-sulfur cluster, observed in frataxin deficient cells, and consequent defects in the activity of ISC-containing enzymes involved in palmitate synthesis, could be responsible for the reduced palmitoylation of TfR1. This could in turn account for the increase in intracellular iron observed in FRDA patients. Artesunate is a derivative of artemisinin extracted from the artemisia plant, used for the treatment of malaria, and proposed as anti-cancer treatment for its anti-proliferative effects (Yang et al., 2021). Artesunate was shown to promote palmitoylation of TfR1 (Ba et al., 2012; Drecourt et al., 2018). Indeed, treatment of FRDA fibroblasts with artesunate restored palmitoylation levels and reduced iron overload (Petit et al., 2021). Similar effects were observed in PBMC from FRDA patients. The possibility to target TfR1 palmitoylation therapeutically in FRDA using artesunate will be explored in a clinical trial in FRDA patients3.
Erythropoietin (EPO) is a cytokine produced by the kidney that plays a major role in the regulation of erythropoiesis (Jelkmann, 1992). The initial attention toward considering EPO for FRDA came from the observation that recombinant human erythropoietin (rhuEpo) can promote frataxin increase in neuronal and cardiac cells and in PBMCs (Sturm et al., 2005) and fibroblast derived from FRDA patients, through a mechanism that does not involve an increase in frataxin mRNA transcription (Acquaviva et al., 2008). Moreover, erythropoietin exerts effects on iron metabolism, by inducing heme synthesis (Camaschella et al., 2016), and promotes mitochondrial biogenesis and oxidative respiration (Carraway et al., 2010; Plenge et al., 2012), further favoring it as a candidate therapy for FRDA. Erythropoietin is able to cross the blood-brain barrier (Brines et al., 2000), and was shown to have neuroprotective functions (Sakanaka et al., 1998; Celik et al., 2002; Bianchi et al., 2006), making it potentially attractive as a therapeutic for neurodegenerative diseases. However, a minimal portion of intravenously administered EPO can be found in the cerebrospinal fluid, suggesting that elevated doses are required to achieve significant concentration in the nervous system (Xenocostas et al., 2005).
Several clinical trials have been conducted in FRDA patients, giving different and sometimes inconsistent results. A pilot open label, 8 weeks study in 10 FRDA patients, reported increased frataxin levels, reduced expression of oxidative stress markers and improvement in neurological performances, as measured by the Scale for the Assessment and Rating of Ataxia (SARA) score (Boesch et al., 2007). Similar results were observed in a 6-months follow up study (Boesch et al., 2008) and in a dose escalation study (Nachbauer et al., 2011a), without showing, however, a clear dose-dependent correlation in frataxin accumulation. These studies highlighted also a significant drop in ferritin levels and an increase in hematocrit that required phlebotomy. Interestingly, increase in frataxin amount could be observed also in skeletal muscle biopsies (Nachbauer et al., 2011a,b, 2012). In another open-label trial, the effect of the administration of two single doses of erythropoietin at 3 months interval was assessed. This study showed long term increase in frataxin levels without altering hematocrit, but failed to show clinical benefit (Sacca et al., 2011). Another small dose-escalating placebo-controlled trial, however, failed to show significative increase in frataxin levels and had no effect on SARA scale (Mariotti et al., 2012). Similarly, inconsistent results were obtained in a larger placebo-controlled randomized trial, which highlighted, however, potential benefit in the upper limb performance (Sacca et al., 2016). Recent discoveries of an extra-mitochondrial frataxin in erythrocytes raise doubts about the possibility that the increase in frataxin promoted by erythropoietin could be ascribed to red blood cells (Guo et al., 2018).
To avoid undesired side effects due to stimulation of erythropoiesis promoted by erythropoietin, the possibility of using erythropoietin mimetics has been considered (Leist et al., 2004). Although an Epo derivative, such as carbamylated erythropoietin (CEPO), exerts similar effects on frataxin upregulation in vitro (Sturm et al., 2010), it does not show evidence of function in patients (Boesch et al., 2014). More recently, attention was drawn on two EPO mimetics, STS-E412 and STS-E424, which are agonists of the tissue protective EPO receptor. They were shown to promote increase in frataxin mRNA levels in human neuronal cells in vitro and in FRDA patients derived PBMC. Moreover, STS-E412 and STS-E424 stimulate frataxin expression in the brain of KIKO mice without exerting effect on hematopoiesis. Their ability to cross the blood brain barrier, together with their favorable safety profile and lack of hematopoiesis stimulation, encourage further studies on these compounds (Miller et al., 2017; Boesch and Indelicato, 2019).
Interferon gamma (IFN-γ) is a pleiotropic cytokine able to drive innate and adaptive immune responses against a variety of pathogens. A major immunological function of IFN-γ appears to involve the regulation of iron metabolism, as iron represents a key strategic asset for both bacteria and immune cells (Nairz et al., 2014). In order to prevent iron access to pathogens, in fact, IFN-γ upregulates the expression of multiple genes involved in iron storage, trafficking and redistribution between the cellular and extracellular environment (Abreu et al., 2020; Nairz and Weiss, 2020). Moreover, IFN-γ shows effects on various non-immune cell types, with direct modulation of metabolic pathways, tissue remodeling and neural cell survival (Ivashkiv, 2018). Notably, among the non-immune cells that might be affected by IFN-γ are the DRG sensory neurons, that are both capable of releasing and responding to IFN-γ, in an autocrine fashion (Neumann et al., 1997). Perhaps not surprisingly, therefore, this cytokine has been identified as potential therapeutic for the treatment of FRDA because of its ability to upregulate frataxin expression in vitro and in vivo. The treatment with IFN-γ has been shown to increase frataxin mRNA and protein levels in primary cells derived from FRDA patients (Tomassini et al., 2012). Same results were observed for frataxin protein in dorsal root ganglia (DRG) neurons from the YG8R FRDA mouse model. Importantly, FRDA mice treated with IFN-γ demonstrated enhancements of locomotor activity, improvements in motor coordination and prevention of DRG degeneration (Tomassini et al., 2012). Since IFN-γ is a drug already approved for use in other rare pediatric disorders, i.e., chronic granulomatous disease and severe malignant osteopetrosis, two different open-label pilot trials were rapidly performed. Both short-term clinical studies showed that IFN-γ was well tolerated in FRDA patients and without serious adverse effects (Seyer et al., 2015; Marcotulli et al., 2016). The first study reported a significant benefit in neurologic measures, with an improvement in the Friedreich Ataxia Rating Scale (FARS) score over 3 months of treatment (Seyer et al., 2015). Subsequently, a larger placebo-controlled phase III trial to assess the efficacy of IFN-γ in FRDA has been conducted. Unfortunately, because of underestimation of experimental variability and systematic wrong timing in clinical measure assessment, this part of the study did not allow to appreciate significant differences between the treated and placebo groups, within the 6 months of treatment. However, the 12 months open-label extension of the same study showed a potential benefit toward expected disease progression in treated patients (Lynch et al., 2019). More recently, an open-label phase II trial measuring the SARA score and using instrumental evaluations including electro/echocardiography (Vavla et al., 2020b) and Diffusion Tensor Magnetic Resonance Imaging of the brain (Vavla et al., 2020a), was completed over an observation time of 18 months. The progression of SARA was completely halted during IFN-γ treatment and slightly resumed after termination of the treatment. Significant improvements in some cardiac parameters were also observed, with a clear rebound effect after discontinuation of IFN-γ treatment. Interestingly, similar results from a case report showed reduction of cardiomyocyte damage and improvement of diastolic function after 8 months IFN-γ therapy in a patient with severe FRDA cardiomyopathy (Wyller et al., 2016). Overall, four separate clinical studies reported no critical involvement of immune responses, a satisfactory safety profile and possible disease-modifying effects of IFN-γ in FRDA patients. Frataxin measurement in blood and/or buccal cells showed heterogeneous response among patients and did not provide significant data. The results suggesting that IFN-γ treatment may slow the natural rate of FRDA progression are encouraging. However, to definitively establish the benefits of IFN-γ therapy, future clinical studies and concomitant design optimization are required.
The granulocyte-colony stimulating factor (G-CSF) is a hematopoietic cytokine promoting the proliferation and differentiation of myeloid precursors into mature granulocytes. The pharmacologic administration of this growth factor is widely used in healthy (Anderlini and Champlin, 2008) and diseased individuals (Hubel and Engert, 2003), with numerous clinical applications including allogeneic peripheral blood progenitor cell transplantation, reproductive medicine, treatment of hematopoietic and solid cancers, acute infections and myocardial infarction. The experimental administration of G-CSF, alone or in combination with stem cell factor (SCF), showed benefits toward neurological disorders in animal models (Chang et al., 2011; Song et al., 2011; Kemp et al., 2017) and has been investigated in the YG8R FRDA mice. The treatment with G-CSF or combined G-CSF/SCF revealed a transcriptional frataxin upregulation in spinal cord and cerebellum of YG8R animals, with the possible involvement of HIF-2a transcription factor (Kemp et al., 2017). Moreover, rescuing effects on aconitase activity and expression levels of Nrf2, SOD1, SOD2, catalase and glutathione peroxidase were observed. Finally, cytokine administration led to reduced neuroinflammation, reduced DRG vacuolization and significant improvements in motor coordination and locomotor activities (Kemp et al., 2017). A first open-label phase II trial to determine the effects of G-CSF on frataxin expression was recently conducted (EudraCT 2017-003084-34). The public report of this small clinical study4 describes interesting efficacy results concerning increased frataxin expression in PBMC and platelets of FRDA patients. G-CSF therapy showed known, mild side effects and no serious adverse events. Full results have not yet been published.
In FRDA patients, frataxin deficiency causes mitochondrial dysfunction and increased oxidative stress, which lead to the apoptotic cell death of neurons and pancreatic beta cells. Loss of beta cells is the main cause of the increased prevalence of diabetes observed in FRDA patients (Cnop et al., 2012). Incretin mimetics, such as GLP-1 analogues, are currently used for the treatment and management of type 2 diabetes (Hinnen, 2017; Chia and Egan, 2020). Recently, GLP-1 analogues were shown to protect FRDA patients beta cells from apoptosis and promote survival of neurons by reducing oxidative stress (Igoillo-Esteve et al., 2020). These effects could be the consequence of restored anti-oxidant response due to the activation of Nrf2 and its downstream targets, which has been associated with GLP-1 treatment (Igoillo-Esteve et al., 2015; Fernandez-Millan et al., 2016). In addition to promoting beta cells function and improving glucose tolerance, a further study has shown that the GLP-1 analog exenatide promotes an increase in frataxin protein levels in the brain of KIKO mice. A similar effect was also observed in beta cells and neurons derived from FRDA patients iPSCs (Igoillo-Esteve et al., 2020). Along with this modest increase in frataxin amount, an increased activity of ISC-containing enzymes was also noticed, together with an overall improved mitochondrial function. Considering also its potential to cross the blood-brain barrier (Kastin and Akerstrom, 2003; Christensen et al., 2015), the protective effects of exenatide were further investigated in an open label pilot clinical trial on FRDA patients. A mild increase in frataxin levels was observed in platelets derived from patients treated for 5 weeks with exenatide. However, no improvement on neurological functions as assessed by the SARA score or Activities of Daily Living scales, was detectable. Whether this is because the period of treatment was not long enough or whether other parameters might influence the response in patients should be assessed in further studies.
The GAA repeat expansion in the frataxin intron 1 is known to cause epigenetic modification leading to condensed chromatin structure and reduced accessibility of the gene for transcription, resulting in reduced amount of frataxin protein. Hence, the use of HDAC inhibitors has been widely investigated for their potential ability to reverse epigenetic silencing of the frataxin gene (Herman et al., 2006; Soragni and Gottesfeld, 2016). In line with these studies, Festenstein and colleagues explored the possibility to use the class III HDAC sirtuin inhibitor nicotinamide for the treatment of Friedreich ataxia. Nicotinamide (vitamin B3) is a class III HDAC inhibitor (Avalos et al., 2005; Vaquero et al., 2007) that has been widely used as a food additive over the last years for a number of indications, without presenting significant adverse effects. Importantly, nicotinamide has the ability to cross the blood-brain barrier (Stratford and Dennis, 1994). Initial studies reported an upregulation of frataxin mRNA and protein in lymphoblastoid cell lines derived from FRDA patients, in patients PBMC and in the YG8 mouse model (Chan et al., 2013). This effect is concomitant with a reduced DNA methylation, a reduction in histone H3K9 and H3K27 methylation and an increase in histone H3 and H4 acetylation in the FXN gene. Importantly, nicotinamide can also restore the expression of a number of genes known to be dysregulated in frataxin deficient cells. However, other studies argue against the involvement of sirtuin in the regulation of FXN gene expression (Xu et al., 2009), therefore the mechanism of nicotinamide action in this context needs further evaluation. A later study, indeed, reported that while levels of H3K9 acetylation were increased in patients iPSC-derived neurons treated with nicotinamide, no significant alteration of frataxin mRNA and protein levels could be detected (Georges et al., 2019).
In an open-label, dose escalation trial in FRDA patients a significant and sustained increase in frataxin mRNA and protein amount was observed, together with a reduction in chromatin condensation markers (Libri et al., 2014). However, no effect on neurological parameters was observed in patients, possibly due to the short duration of the trial. Although nicotinamide was used at doses 200 times higher than the daily recommended allowance for its use as a vitamin supplement, it did not cause significant side effects beside nausea and vomiting. These effects could, however, hamper its use over longer regimen. Nevertheless, to assess nicotinamide long-term effects on FRDA progression through the SARA score, a multicenter, placebo-controlled, 24 months trial, using the individually determined highest tolerated doses, is about to start in Europe (Reetz et al., 2019)5.
Nicotinamide riboside (NR) is a NAD + precursor that presents numerous potential benefits for health and its use as a dietary supplement has been proposed to help in the treatment of cardiovascular, neurodegenerative, metabolic and aging-related disorders. Indeed, a natural decline in NAD + content is associated with aging (Mehmel et al., 2020). NAD + is an important cofactor in many oxidation-reduction reactions in the cell and plays a crucial role in glycolysis and oxidative phosphorylation. Accordingly, NR was shown to enhance mitochondrial function and promote oxidative metabolism (Canto et al., 2012). Moreover, the activity of the NAD + -dependent mitochondrial protein deacetylase SIRT3 plays an important role in cardiac function and stress response, and heart defects in muscle/cardiac FXN-KO mouse model can be rescued by inducing SIRT3 activity through NAD + precursor supplementation (Martin et al., 2017). Given the important function of NAD + in modulating mitochondrial metabolism and heart bioenergetic, supplementation with the NAD + precursor MIB-626 (Metro International Biotech, LLC) or with NR is currently being tested in FRDA patients6,7.
Acetyl-L-Carnitine (ALCAR) is a naturally occurring compound that facilitates energy production in the mitochondria and exerts neurotrophic effects. It is proposed as beneficial in a number of conditions such as cardiovascular and neurodegenerative disorders and is currently marketed as a food supplement for the treatment of neuropathies (Maldonado et al., 2020; Pennisi et al., 2020). L-carnitine has been proposed as a therapeutic for FRDA and its effects have been tested in a placebo-controlled trial in 16 FRDA patients, showing mild but encouraging improvements in energy metabolism (Schols et al., 2005). More recently, an open-label trial with ALCAR has been performed on 20 FRDA patients, but results have not been published yet8.
Resveratrol is a natural product extracted from the grape, berries and nuts that has been proposed to have anti-oxidant, anti-tumor and neuroprotective properties. Although not approved for any specific indication, it is commonly used as a dietary supplement (Salehi et al., 2018). It is also characterized as an activator of the sirtuin class of HDAC (Pyo et al., 2020). Starting from the screening of a collection of FDA-approved drugs and natural compounds, Li and colleagues identified resveratrol, among others, as a compound able to elicit an increase in frataxin levels (Li et al., 2013) in cells expressing a FXN-EGFP genomic reporter containing a normal copy of the frataxin, as well as in lymphoblasts and fibroblasts derived from FRDA patients, both at the mRNA and protein level. Moreover, frataxin protein levels were found elevated in the brain of YG8R transgenic mice, administered with resveratrol for 3 days, suggesting the in vivo efficacy of this compound and indicating its ability to cross the blood-brain barrier. Nevertheless, when administered in vitro to neurons derived from patients iPSCs, resveratrol failed to elicit a significant increase in frataxin mRNA and protein levels (Georges et al., 2019). Similarly, a successive 3-months open label study on FRDA patients failed to detect an increase in frataxin levels. However, improvements in neurological parameters, as measured by the FARS score, and reduction in oxidative stress, as measured by reduction in the F2-isoprostanes marker, were observed in patients treated with the highest dose of 5 g/day (Yiu et al., 2015). A double-blind, placebo-controlled trial with micronized resveratrol is currently ongoing to further assess the impact of this treatment in FRDA patients9.
Thiamine (Vitamin B1) is an important co-factor of many enzymes involved in energy metabolism within the cells, such as pyruvate dehydrogenase and alpha-keto-glutarate dehydrogenase. It also plays a role in neurotransmission, in the maintenance of mitochondrial membrane potential, glucose metabolism, oxidative stress and inflammation (Jhala et al., 2014; Xu et al., 2021). It is proposed to have a neuroprotective function. Indeed, thiamine deficiency is associated with cerebellar dysfunction (Mulholland, 2006) and neurodegenerative diseases (Jhala and Hazell, 2011). Non-coenzyme functions for thiamine have also been reported which may play a role in neuroprotection (Aleshin et al., 2019). Earlier studies reported a thiamine deficiency in the cerebrospinal fluid of FRDA patients (Pedraza and Botez, 1992; Botez and Young, 2001) and an impaired activity of pyruvate dehydrogenase (Barbeau et al., 1976; Dijkstra et al., 1984). However, the relevance of this deficiency is controversial (Bettendorff et al., 1996).
A 3-months study in two FRDA patients reported significant improvement in motor coordination and reduction in fatigue after administration of high doses of thiamine (Costantini et al., 2013). A longer open-label study in a cohort of 34 FRDA patients, which lasted between 90 and 930 days, reported a significant improvement in the SARA score and a decrease in the thickness of interventricular septum. Some patients presented an improvement in tendon reflexes and in swallowing. The effects on frataxin levels in PBMC were variable and not consistent. The treatment was well tolerated and did not present adverse effects. However, the lack of a placebo control and the fact that patients did not present a deficit in thiamine before the start of the trial, warrant careful evaluation of the effect of this vitamin in FRDA.
Sulforaphane is an isothiocyanate derived from cruciferous plants that has recently gained considerable attention thanks to its potential anti-oxidant (Ruhee and Suzuki, 2020), antitumor (Iahtisham Ul et al., 2021), anti-inflammatory and neuroprotective effects (Calabrese and Kozumbo, 2021; Mangla et al., 2021), through the activation of the Nrf2 pathway (Petrillo et al., 2019; Uddin et al., 2020). It is currently sold as a nutritional supplement. Its activity was tested in a motor neuron cell line in which frataxin gene had been silenced (Petrillo et al., 2017). These cells present mitochondrial dysfunction, impaired anti-oxidant defense with an increased GSSG/GSH ratio and decreased Nrf2 response, and axonal degeneration (D’Oria et al., 2013; Carletti et al., 2014). Sulforaphane treatment promotes Nrf2 response and the expression of its downstream target genes, restores glutathione homeostasis and promotes axonal regeneration. Moreover, in accordance with the hypothesis linking the activation of the Nrf2 pathway with frataxin expression, an increase in frataxin levels was also observed upon treatment with sulforaphane in frataxin-silenced motor neurons. Similar results were also observed in FRDA patients derived fibroblasts (Petrillo et al., 2017). These data lend further support to the idea of targeting the Nrf2 pathway as therapeutic strategy for FRDA. Additional studies should clarify the therapeutic potential of sulforaphane in FRDA patients.
Currently, there is no approved treatment to cure Friedreich ataxia or halt the progression of the disease. A number of efforts have been put into the development of an effective therapy, focusing on targeting mitochondrial dysfunction, oxidative stress and augmenting frataxin levels (Clay et al., 2019; Delatycki and Bidichandani, 2019; Gottesfeld, 2019; Zesiewicz et al., 2020). Nonetheless, drug discovery for FRDA has been challenging, for at least two reasons. First, the classical process of drug discovery implies high investments form pharmaceuticals companies and requires a very long process of development that includes medicinal chemistry-driven optimization and experimental validation before moving to the clinical phases. Second, designing clinical trials for rare neurodegenerative diseases, such as FRDA, has often proved to be tricky because of the paucity of patients and difficulties in enrolling a sufficiently large and stratified sample, and because of the heterogeneous nature of the disease with a slow and variable progression rate, which makes it challenging to identify reliable outcome measures to monitor the progression of the disease (Patel et al., 2016; Reetz et al., 2021; Rodden and Lynch, 2021; Savelieff and Feldman, 2021).
Drug repurposing is a viable alternative approach to classical drug discovery that can potentially bypass some of these hurdles (Pushpakom et al., 2019; Talevi and Bellera, 2020) and boost the discovery of a therapy for FRDA. Some of the repurposed drugs identified for FRDA could actually be turned into an available therapy for patients in the next years. By taking advantage of already established safety profiles, toxicology and pharmacokinetic studies, repurposed drugs can indeed more rapidly enter clinical trials to directly evaluate their efficacy in patients.
A repurposed drug could prove effective as it is, or could undergo some additional development and reformulation and be approved for use in a different dosage, using a different route of administration or even used in combination with other drugs. Indeed, drugs identified through repositioning screening can sometimes require optimization to increase their efficacy and achieve a therapeutically relevant effect. However, one of the most important limitation in drug repurposing approaches is that most of the times, even though the drug has been identified as able to rescue Friedreich ataxia phenotype, its relevant molecular targets in FRDA and its specific mechanism of action remain unknown. This poses a significant drawback, as lack of this knowledge significantly hinders the optimization process of the candidate therapeutic compound and the development of more efficient derivatives. On the other side, although implying a considerable research effort, target identification of potentially active compounds, identified through drug repositioning screening for FRDA, holds the possibility to unveil new molecular targets and pathways that could be relevant for understanding FRDA pathophysiology. This could add to the research field and open the way for the development of target-specific therapies for FRDA.
By integrating these efforts there is great hope that patients will benefit from a therapeutic opportunity in the near future.
AR and RT contributed to conception and designed of the study. AR wrote the first draft of the manuscript. FM and IC wrote sections of the manuscript and made substantial, direct and intellectual contribution to the work. AR, FM, IC, and RT contributed to manuscript revision. All authors read and approved the submitted version.
This work was supported by Ataxia UK (grant to AR), Telethon-AFM (grant 22974 to FM), and Association Française Ataxie de Friedreich (AFAF, grant to FM).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abeti, R., Baccaro, A., Esteras, N., and Giunti, P. (2018). Novel Nrf2-inducer prevents mitochondrial defects and oxidative stress in friedreich’s ataxia models. Front. Cell Neurosci. 12:188. doi: 10.3389/fncel.2018.00188
Abreu, R., Essler, L., Giri, P., and Quinn, F. (2020). Interferon-gamma promotes iron export in human macrophages to limit intracellular bacterial replication. PLoS One 15:e0240949. doi: 10.1371/journal.pone.0240949
Acquaviva, F., Castaldo, I., Filla, A., Giacchetti, M., Marmolino, D., Monticelli, A., et al. (2008). Recombinant human erythropoietin increases frataxin protein expression without increasing mRNA expression. Cerebellum 7, 360–365. doi: 10.1007/s12311-008-0036-x
Aleshin, V. A., Mkrtchyan, G. V., and Bunik, V. I. (2019). Mechanisms of non-coenzyme action of thiamine: protein targets and medical significance. Biochemistry (Mosc) 84, 829–850. doi: 10.1134/S0006297919080017
Alfedi, G., Luffarelli, R., Condo, I., Pedini, G., Mannucci, L., Massaro, D. S., et al. (2019). Drug repositioning screening identifies etravirine as a potential therapeutic for friedreich’s ataxia. Mov. Disord. 34, 323–334. doi: 10.1002/mds.27604
Alvarez, E., Girones, N., and Davis, R. J. (1990). Inhibition of the receptor-mediated endocytosis of diferric transferrin is associated with the covalent modification of the transferrin receptor with palmitic acid. J. Biol. Chem. 265, 16644–16655. doi: 10.1016/s0021-9258(17)46270-3
Anderlini, P., and Champlin, R. E. (2008). Biologic and molecular effects of granulocyte colony-stimulating factor in healthy individuals: recent findings and current challenges. Blood 111, 1767–1772. doi: 10.1182/blood-2007-07-097543
Anjomani Virmouni, S., Ezzatizadeh, V., Sandi, C., Sandi, M., Al-Mahdawi, S., Chutake, Y., et al. (2015). A novel GAA-repeat-expansion-based mouse model of Friedreich’s ataxia. Dis. Model Mech. 8, 225–235. doi: 10.1242/dmm.018952
Avalos, J. L., Bever, K. M., and Wolberger, C. (2005). Mechanism of sirtuin inhibition by nicotinamide: altering the NAD(+) cosubstrate specificity of a Sir2 enzyme. Mol. Cell 17, 855–868. doi: 10.1016/j.molcel.2005.02.022
Ba, Q., Zhou, N., Duan, J., Chen, T., Hao, M., Yang, X., et al. (2012). Dihydroartemisinin exerts its anticancer activity through depleting cellular iron via transferrin receptor-1. PLoS One 7:e42703. doi: 10.1371/journal.pone.0042703
Barbeau, A., Butterworth, R. F., Ngo, T., Breton, G., Melancon, S., Shapcott, D., et al. (1976). Pyruvate metabolism in Friedreich’s ataxia. Can. J. Neurol. Sci. 3, 379–388.
Benini, M., Fortuni, S., Condo, I., Alfedi, G., Malisan, F., Toschi, N., et al. (2017). E3 Ligase RNF126 directly ubiquitinates frataxin, promoting its degradation: identification of a potential therapeutic target for friedreich ataxia. Cell Rep. 18, 2007–2017. doi: 10.1016/j.celrep.2017.01.079
Bettendorff, L., Mastrogiacomo, F., Lamarche, J., Dozic, S., and Kish, S. J. (1996). Brain levels of thiamine and its phosphate esters in Friedreich’s ataxia and spinocerebellar ataxia type 1. Mov. Disord. 11, 437–439. doi: 10.1002/mds.870110415
Bianchi, R., Brines, M., Lauria, G., Savino, C., Gilardini, A., Nicolini, G., et al. (2006). Protective effect of erythropoietin and its carbamylated derivative in experimental Cisplatin peripheral neurotoxicity. Clin. Cancer Res. 12, 2607–2612. doi: 10.1158/1078-0432.CCR-05-2177
Boesch, S., and Indelicato, E. (2019). Erythropoietin and friedreich ataxia: time for a reappraisal? Front. Neurosci. 13:386. doi: 10.3389/fnins.2019.00386
Boesch, S., Nachbauer, W., Mariotti, C., Sacca, F., Filla, A., Klockgether, T., et al. (2014). Safety and tolerability of carbamylated erythropoietin in Friedreich’s ataxia. Mov. Disord. 29, 935–939. doi: 10.1002/mds.25836
Boesch, S., Sturm, B., Hering, S., Goldenberg, H., Poewe, W., and Scheiber-Mojdehkar, B. (2007). Friedreich’s ataxia: clinical pilot trial with recombinant human erythropoietin. Ann. Neurol. 62, 521–524. doi: 10.1002/ana.21177
Boesch, S., Sturm, B., Hering, S., Scheiber-Mojdehkar, B., Steinkellner, H., Goldenberg, H., et al. (2008). Neurological effects of recombinant human erythropoietin in Friedreich’s ataxia: a clinical pilot trial. Mov. Disord. 23, 1940–1944. doi: 10.1002/mds.22294
Bon, C., Luffarelli, R., Russo, R., Fortuni, S., Pierattini, B., Santulli, C., et al. (2019). SINEUP non-coding RNAs rescue defective frataxin expression and activity in a cellular model of Friedreich’s Ataxia. Nucleic Acids Res. 47, 10728–10743. doi: 10.1093/nar/gkz798
Botez, M. I., and Young, S. N. (2001). Biogenic amine metabolites and thiamine in cerebrospinal fluid in heredo-degenerative ataxias. Can. J. Neurol. Sci. 28, 134–140. doi: 10.1017/s0317167100052811
Brines, M. L., Ghezzi, P., Keenan, S., Agnello, D., De Lanerolle, N. C., Cerami, C., et al. (2000). Erythropoietin crosses the blood-brain barrier to protect against experimental brain injury. Proc. Natl. Acad. Sci. U. S. A. 97, 10526–10531. doi: 10.1073/pnas.97.19.10526
Britti, E., Delaspre, F., Feldman, A., Osborne, M., Greif, H., Tamarit, J., et al. (2018). Frataxin-deficient neurons and mice models of Friedreich ataxia are improved by TAT-MTScs-FXN treatment. J. Cell Mol. Med. 22, 834–848. doi: 10.1111/jcmm.13365
Calabrese, E. J., and Kozumbo, W. J. (2021). The phytoprotective agent sulforaphane prevents inflammatory degenerative diseases and age-related pathologies via Nrf2-mediated hormesis. Pharmacol. Res. 163:105283. doi: 10.1016/j.phrs.2020.105283
Camaschella, C., Pagani, A., Nai, A., and Silvestri, L. (2016). The mutual control of iron and erythropoiesis. Int. J. Lab. Hematol. 38, 20–26. doi: 10.1111/ijlh.12505
Campuzano, V., Montermini, L., Lutz, Y., Cova, L., Hindelang, C., Jiralerspong, S., et al. (1997). Frataxin is reduced in Friedreich ataxia patients and is associated with mitochondrial membranes. Hum. Mol. Genet. 6, 1771–1780. doi: 10.1093/hmg/6.11.1771
Canto, C., Houtkooper, R. H., Pirinen, E., Youn, D. Y., Oosterveer, M. H., Cen, Y., et al. (2012). The NAD(+) precursor nicotinamide riboside enhances oxidative metabolism and protects against high-fat diet-induced obesity. Cell Metab. 15, 838–847. doi: 10.1016/j.cmet.2012.04.022
Carletti, B., Piermarini, E., Tozzi, G., Travaglini, L., Torraco, A., Pastore, A., et al. (2014). Frataxin silencing inactivates mitochondrial Complex I in NSC34 motoneuronal cells and alters glutathione homeostasis. Int. J. Mol. Sci. 15, 5789–5806. doi: 10.3390/ijms15045789
Carraway, M. S., Suliman, H. B., Jones, W. S., Chen, C. W., Babiker, A., and Piantadosi, C. A. (2010). Erythropoietin activates mitochondrial biogenesis and couples red cell mass to mitochondrial mass in the heart. Circ. Res. 106, 1722–1730. doi: 10.1161/CIRCRESAHA.109.214353
Castro, I. H., Ferrari, A., Herrera, M. G., Noguera, M. E., Maso, L., Benini, M., et al. (2018). Biophysical characterisation of the recombinant human frataxin precursor. FEBS Open Bio 8, 390–405. doi: 10.1002/2211-5463.12376
Celik, M., Gokmen, N., Erbayraktar, S., Akhisaroglu, M., Konakc, S., Ulukus, C., et al. (2002). Erythropoietin prevents motor neuron apoptosis and neurologic disability in experimental spinal cord ischemic injury. Proc. Natl. Acad. Sci. U. S. A. 99, 2258–2263. doi: 10.1073/pnas.042693799
Chan, P. K., Torres, R., Yandim, C., Law, P. P., Khadayate, S., Mauri, M., et al. (2013). Heterochromatinization induced by GAA-repeat hyperexpansion in Friedreich’s ataxia can be reduced upon HDAC inhibition by vitamin B3. Hum. Mol. Genet. 22, 2662–2675. doi: 10.1093/hmg/ddt115
Chandran, V., Gao, K., Swarup, V., Versano, R., Dong, H., Jordan, M. C., et al. (2017). Inducible and reversible phenotypes in a novel mouse model of Friedreich’s Ataxia. Elife 6:e30054. doi: 10.7554/eLife.30054
Chang, Y. C., Lin, C. Y., Hsu, C. M., Lin, H. C., Chen, Y. H., Lee-Chen, G. J., et al. (2011). Neuroprotective effects of granulocyte-colony stimulating factor in a novel transgenic mouse model of SCA17. J. Neurochem. 118, 288–303. doi: 10.1111/j.1471-4159.2011.07304.x
Cherubini, F., Serio, D., Guccini, I., Fortuni, S., Arcuri, G., Condo, I., et al. (2015). Src inhibitors modulate frataxin protein levels. Hum. Mol. Genet. 24, 4296–4305. doi: 10.1093/hmg/ddv162
Chia, C. W., and Egan, J. M. (2020). Incretins in obesity and diabetes. Ann. N. Y. Acad. Sci. 1461, 104–126.
Christensen, M., Sparre-Ulrich, A. H., Hartmann, B., Grevstad, U., Rosenkilde, M. M., Holst, J. J., et al. (2015). Transfer of liraglutide from blood to cerebrospinal fluid is minimal in patients with type 2 diabetes. Int. J. Obes. (Lond) 39, 1651–1654. doi: 10.1038/ijo.2015.136
Chutake, Y. K., Lam, C., Costello, W. N., Anderson, M., and Bidichandani, S. I. (2014). Epigenetic promoter silencing in Friedreich ataxia is dependent on repeat length. Ann. Neurol. 76, 522–528. doi: 10.1002/ana.24249
Clay, A., Hearle, P., Schadt, K., and Lynch, D. R. (2019). New developments in pharmacotherapy for Friedreich ataxia. Expert. Opin. Pharmacother. 20, 1855–1867. doi: 10.1080/14656566.2019.1639671
Cnop, M., Igoillo-Esteve, M., Rai, M., Begu, A., Serroukh, Y., Depondt, C., et al. (2012). Central role and mechanisms of beta-cell dysfunction and death in friedreich ataxia-associated diabetes. Ann. Neurol. 72, 971–982. doi: 10.1002/ana.23698
Cnop, M., Mulder, H., and Igoillo-Esteve, M. (2013). Diabetes in Friedreich ataxia. J. Neurochem. 126, 94–102. doi: 10.1111/jnc.12216
Condò, I., Malisan, F., Guccini, I., Serio, D., Rufini, A., and Testi, R. (2010). Molecular control of the cytosolic aconitase/IRP1 switch by extramitochondrial frataxin. Hum. Mol. Genet. 19, 1221–9. doi: 10.1093/hmg/ddp592
Condò, I., Ventura, N., Malisan, F., Rufini, A., Tomassini, B., and Testi, R. (2007). In vivo maturation of human frataxin. Hum. Mol. Genet. 16, 1534–1540.
Condò, I., Ventura, N., Malisan, F., Tomassini, B., and Testi, R. (2006). A pool of extramitochondrial frataxin that promotes cell survival. J. Biol. Chem. 281, 16750–16756. doi: 10.1074/jbc.M511960200
Coppola, G., Marmolino, D., Lu, D., Wang, Q., Cnop, M., Rai, M., et al. (2009). Functional genomic analysis of frataxin deficiency reveals tissue-specific alterations and identifies the PPARgamma pathway as a therapeutic target in Friedreich’s ataxia. Hum. Mol. Genet. 18, 2452–2461. doi: 10.1093/hmg/ddp183
Costantini, A., Giorgi, R., D’agostino, S., and Pala, M. I. (2013). High-dose thiamine improves the symptoms of Friedreich’s ataxia. BMJ Case Rep. 2013:bcr2013009424. doi: 10.1136/bcr-2013-009424
Croxtall, J. D. (2012). Etravirine: a review of its use in the management of treatment-experienced patients with HIV-1 infection. Drugs 72, 847–869. doi: 10.2165/11209110-000000000-00000
Delatycki, M. B., and Bidichandani, S. I. (2019). Friedreich ataxia- pathogenesis and implications for therapies. Neurobiol. Dis. 132:104606. doi: 10.1016/j.nbd.2019.104606
Delatycki, M. B., and Corben, L. A. (2012). Clinical features of Friedreich ataxia. J. Child. Neurol. 27, 1133–1137.
Dijkstra, U., Gabreels, F., Joosten, E., Wevers, R., Lamers, K., Doesburg, W., et al. (1984). Friedreich’s ataxia: intravenous pyruvate load to demonstrate a defect in pyruvate metabolism. Neurology 34, 1493–1497. doi: 10.1212/wnl.34.11.1493
DiMasi, J. A. (2018). Assessing pharmaceutical research and development costs. JAMA Intern. Med. 178:587. doi: 10.1001/jamainternmed.2017.8706
D’Oria, V., Petrini, S., Travaglini, L., Priori, C., Piermarini, E., Petrillo, S., et al. (2013). Frataxin deficiency leads to reduced expression and impaired translocation of NF-E2-related factor (Nrf2) in cultured motor neurons. Int. J. Mol. Sci. 14, 7853–7865. doi: 10.3390/ijms14047853
Drecourt, A., Babdor, J., Dussiot, M., Petit, F., Goudin, N., Garfa-Traore, M., et al. (2018). Impaired transferrin receptor palmitoylation and recycling in neurodegeneration with brain iron accumulation. Am. J. Hum. Genet. 102, 266–277. doi: 10.1016/j.ajhg.2018.01.003
Erwin, G. S., Grieshop, M. P., Ali, A., Qi, J., Lawlor, M., Kumar, D., et al. (2017). Synthetic transcription elongation factors license transcription across repressive chromatin. Science 358, 1617–1622. doi: 10.1126/science.aan6414
Fernandez-Millan, E., Martin, M. A., Goya, L., Lizarraga-Mollinedo, E., Escriva, F., Ramos, S., et al. (2016). Glucagon-like peptide-1 improves beta-cell antioxidant capacity via extracellular regulated kinases pathway and Nrf2 translocation. Free Radic. Biol. Med. 95, 16–26. doi: 10.1016/j.freeradbiomed.2016.03.002
Filla, A., De Michele, G., Cavalcanti, F., Pianese, L., Monticelli, A., Campanella, G., et al. (1996). The relationship between trinucleotide (GAA) repeat length and clinical features in Friedreich ataxia. Am. J. Hum. Genet. 59, 554–560.
Fox, N. G., Yu, X., Feng, X., Bailey, H. J., Martelli, A., Nabhan, J. F., et al. (2019). Structure of the human frataxin-bound iron-sulfur cluster assembly complex provides insight into its activation mechanism. Nat. Commun. 10:2210. doi: 10.1038/s41467-019-09989-y
Franco, C., Fernandez, S., and Torres-Aleman, I. (2012). Frataxin deficiency unveils cell-context dependent actions of insulin-like growth factor I on neurons. Mol. Neurodegener. 7:51. doi: 10.1186/1750-1326-7-51
Galea, C. A., Huq, A., Lockhart, P. J., Tai, G., Corben, L. A., Yiu, E. M., et al. (2016). Compound heterozygous FXN mutations and clinical outcome in friedreich ataxia. Ann. Neurol. 79, 485–495. doi: 10.1002/ana.24595
Gargiulo, A. V., Burns, G. M., and Huck, C. P. (1992). Dyclonine hydrochloride–a topical agent for managing pain. Ill. Dent. J. 61, 303–304.
Georges, P., Boza-Moran, M. G., Gide, J., Peche, G. A., Foret, B., Bayot, A., et al. (2019). Induced pluripotent stem cells-derived neurons from patients with Friedreich ataxia exhibit differential sensitivity to resveratrol and nicotinamide. Sci. Rep. 9:14568. doi: 10.1038/s41598-019-49870-y
Gomes, C. M., and Santos, R. (2013). Neurodegeneration in Friedreich’s ataxia: from defective frataxin to oxidative stress. Oxid. Med. Cell Longev. 2013:487534. doi: 10.1155/2013/487534
Gottesfeld, J. M. (2019). Molecular mechanisms and therapeutics for the GAA.TTC expansion disease friedreich ataxia. Neurotherapeutics 16, 1032–1049. doi: 10.1074/jbc.M110.194035
Guccini, I., Serio, D., Condo, I., Rufini, A., Tomassini, B., Mangiola, A., et al. (2011). Frataxin participates to the hypoxia-induced response in tumors. Cell Death Dis. 2:e123. doi: 10.1038/cddis.2011.5
Guo, L., Wang, Q., Weng, L., Hauser, L. A., Strawser, C. J., Mesaros, C., et al. (2018). Characterization of a new N-terminally acetylated extra-mitochondrial isoform of frataxin in human erythrocytes. Sci. Rep. 8:17043. doi: 10.1038/s41598-018-35346-y
Haugen, A. C., Di Prospero, N. A., Parker, J. S., Fannin, R. D., Chou, J., Meyer, J. N., et al. (2010). Altered gene expression and DNA damage in peripheral blood cells from Friedreich’s ataxia patients: cellular model of pathology. PLoS Genet. 6:e1000812. doi: 10.1371/journal.pgen.1000812
Havens, J. P., Podany, A. T., Scarsi, K. K., and Fletcher, C. V. (2020). Clinical pharmacokinetics and pharmacodynamics of etravirine: an updated review. Clin. Pharmacokinet. 59, 137–154. doi: 10.1007/s40262-019-00830-9
Hayashi, G., Jasoliya, M., Sahdeo, S., Sacca, F., Pane, C., Filla, A., et al. (2017). Dimethyl fumarate mediates Nrf2-dependent mitochondrial biogenesis in mice and humans. Hum. Mol. Genet. 26, 2864–2873. doi: 10.1093/hmg/ddx167
Hebert-Chatelain, E. (2013). Src kinases are important regulators of mitochondrial functions. Int. J. Biochem. Cell Biol. 45, 90–98. doi: 10.1016/j.biocel.2012.08.014
Herman, D., Jenssen, K., Burnett, R., Soragni, E., Perlman, S. L., and Gottesfeld, J. M. (2006). Histone deacetylase inhibitors reverse gene silencing in Friedreich’s ataxia. Nat. Chem. Biol. 2, 551–558. doi: 10.1038/nchembio815
Hill, B. G., Kota, V. K., and Khoury, H. J. (2014). Bosutinib: a third generation tyrosine kinase inhibitor for the treatment of chronic myeloid leukemia. Expert Rev. Anticancer Ther. 14, 765–770. doi: 10.1586/14737140.2014.924400
Hinnen, D. (2017). Glucagon-like peptide 1 receptor agonists for type 2 diabetes. Diab. Spectr. 30, 202–210.
Hochhaus, A., and Kantarjian, H. (2013). The development of dasatinib as a treatment for chronic myeloid leukemia (CML): from initial studies to application in newly diagnosed patients. J. Cancer Res. Clin. Oncol. 139, 1971–1984. doi: 10.1007/s00432-013-1488-z
Hubel, K., and Engert, A. (2003). Clinical applications of granulocyte colony-stimulating factor: an update and summary. Ann. Hematol. 82, 207–213. doi: 10.1007/s00277-003-0628-y
Hui, C. K., Dedkova, E. N., Montgomery, C., and Cortopassi, G. (2021). Dimethyl fumarate dose-dependently increases mitochondrial gene expression and function in muscle and brain of Friedreich’s ataxia model mice. Hum. Mol. Genet. 29, 3954–3965. doi: 10.1093/hmg/ddaa282
Hutcheon, D. E., and Barthalmus, K. S. (1962). Antihypertensive action of diazoxide. A new benzothiazine with antidiuretic properties. Br. Med. J. 2, 159–161. doi: 10.1136/bmj.2.5298.159
Iahtisham Ul, H., Khan, S., Awan, K. A., and Iqbal, M. J. (2021). Sulforaphane as a potential remedy against cancer: comprehensive mechanistic review. J. Food Biochem. doi: 10.1111/jfbc.13886 [Epub Online ahead of Print].
Igoillo-Esteve, M., Gurgul-Convey, E., Hu, A., Romagueira Bichara Dos Santos, L., Abdulkarim, B., Chintawar, S., et al. (2015). Unveiling a common mechanism of apoptosis in beta-cells and neurons in Friedreich’s ataxia. Hum. Mol. Genet. 24, 2274–2286. doi: 10.1093/hmg/ddu745
Igoillo-Esteve, M., Oliveira, A. F., Cosentino, C., Fantuzzi, F., Demarez, C., Toivonen, S., et al. (2020). Exenatide induces frataxin expression and improves mitochondrial function in Friedreich ataxia. JCI Insight 5:e134221. doi: 10.1172/jci.insight.134221
Ivashkiv, L. B. (2018). IFNgamma: signalling, epigenetics and roles in immunity, metabolism, disease and cancer immunotherapy. Nat. Rev. Immunol. 18, 545–558. doi: 10.1038/s41577-018-0029-z
Jasoliya, M., Sacca, F., Sahdeo, S., Chedin, F., Pane, C., Brescia Morra, V., et al. (2019). Dimethyl fumarate dosing in humans increases frataxin expression: a potential therapy for Friedreich’s Ataxia. PLoS One 14:e0217776. doi: 10.1371/journal.pone.0217776
Jelkmann, W. (1992). Erythropoietin: structure, control of production, and function. Physiol. Rev. 72, 449–489. doi: 10.1152/physrev.1992.72.2.449
Jhala, S. S., and Hazell, A. S. (2011). Modeling neurodegenerative disease pathophysiology in thiamine deficiency: consequences of impaired oxidative metabolism. Neurochem. Int. 58, 248–260. doi: 10.1016/j.neuint.2010.11.019
Jhala, S. S., Wang, D., and Hazell, A. S. (2014). Thiamine deficiency results in release of soluble factors that disrupt mitochondrial membrane potential and downregulate the glutamate transporter splice-variant GLT-1b in cultured astrocytes. Biochem. Biophys. Res. Commun. 448, 335–341. doi: 10.1016/j.bbrc.2014.04.017
Kastin, A. J., and Akerstrom, V. (2003). Entry of exendin-4 into brain is rapid but may be limited at high doses. Int. J. Obes. Relat. Metab. Disord. 27, 313–318. doi: 10.1038/sj.ijo.0802206
Katlama, C., Assoumou, L., Valantin, M. A., Soulie, C., Martinez, E., Beniguel, L., et al. (2020). Dual therapy combining raltegravir with etravirine maintains a high level of viral suppression over 96 weeks in long-term experienced HIV-infected individuals over 45 years on a PI-based regimen: results from the Phase II ANRS 163 ETRAL study-authors’ response. J. Antimicrob. Chemother. 75, 3699–3700. doi: 10.1093/jac/dkaa341
Kemp, K. C., Cerminara, N., Hares, K., Redondo, J., Cook, A. J., Haynes, H. R., et al. (2017). Cytokine therapy-mediated neuroprotection in a Friedreich’s ataxia mouse model. Ann. Neurol. 81, 212–226. doi: 10.1002/ana.24846
Koeppen, A. H., Becker, A. B., Qian, J., and Feustel, P. J. (2017). Friedreich ataxia: hypoplasia of spinal cord and dorsal root ganglia. J. Neuropathol. Exp. Neurol. 76, 101–108. doi: 10.1093/jnen/nlw111
Koeppen, A. H., and Mazurkiewicz, J. E. (2013). Friedreich ataxia: neuropathology revised. J. Neuropathol. Exp. Neurol. 72, 78–90. doi: 10.1097/NEN.0b013e31827e5762
Kwon, G., Marshall, C. A., Liu, H., Pappan, K. L., Remedi, M. S., and Mcdaniel, M. L. (2006). Glucose-stimulated DNA synthesis through mammalian target of rapamycin (mTOR) is regulated by KATP channels: effects on cell cycle progression in rodent islets. J. Biol. Chem. 281, 3261–3267. doi: 10.1074/jbc.M508821200
Lastres-Becker, I., Garcia-Yague, A. J., Scannevin, R. H., Casarejos, M. J., Kugler, S., Rabano, A., et al. (2016). Repurposing the NRF2 activator dimethyl fumarate as therapy against synucleinopathy in parkinson’s disease. Antioxid. Redox Signal. 25, 61–77. doi: 10.1089/ars.2015.6549
Lazzarin, A., Campbell, T., Clotet, B., Johnson, M., Katlama, C., Moll, A., et al. (2007). Efficacy and safety of TMC125 (etravirine) in treatment-experienced HIV-1-infected patients in DUET-2: 24-week results from a randomised, double-blind, placebo-controlled trial. Lancet 370, 39–48. doi: 10.1016/S0140-6736(07)61048-4
Leist, M., Ghezzi, P., Grasso, G., Bianchi, R., Villa, P., Fratelli, M., et al. (2004). Derivatives of erythropoietin that are tissue protective but not erythropoietic. Science 305, 239–242. doi: 10.1126/science.1098313
Li, L., Matsui, M., and Corey, D. R. (2016). Activating frataxin expression by repeat-targeted nucleic acids. Nat. Commun. 7:10606. doi: 10.1038/ncomms10606
Li, L., Voullaire, L., Sandi, C., Pook, M. A., Ioannou, P. A., Delatycki, M. B., et al. (2013). Pharmacological screening using an FXN-EGFP cellular genomic reporter assay for the therapy of Friedreich ataxia. PLoS One 8:e55940. doi: 10.1371/journal.pone.0055940
Liang, S., Pong, K., Gonzales, C., Chen, Y., Ling, H. P., Mark, R. J., et al. (2009). Neuroprotective profile of novel SRC kinase inhibitors in rodent models of cerebral ischemia. J. Pharmacol. Exp. Ther. 331, 827–835. doi: 10.1124/jpet.109.156562
Libri, V., Yandim, C., Athanasopoulos, S., Loyse, N., Natisvili, T., Law, P. P., et al. (2014). Epigenetic and neurological effects and safety of high-dose nicotinamide in patients with Friedreich’s ataxia: an exploratory, open-label, dose-escalation study. Lancet 384, 504–513. doi: 10.1016/S0140-6736(14)60382-2
Lim, J. L., Wilhelmus, M. M., De Vries, H. E., Drukarch, B., Hoozemans, J. J., and Van Horssen, J. (2014). Antioxidative defense mechanisms controlled by Nrf2: state-of-the-art and clinical perspectives in neurodegenerative diseases. Arch. Toxicol. 88, 1773–1786. doi: 10.1007/s00204-014-1338-z
Lin, H., Magrane, J., Rattelle, A., Stepanova, A., Galkin, A., Clark, E. M., et al. (2017). Early cerebellar deficits in mitochondrial biogenesis and respiratory chain complexes in the KIKO mouse model of Friedreich ataxia. Dis. Model. Mech. 10, 1343–1352. doi: 10.1242/dmm.030502
Linker, R. A., Lee, D. H., Ryan, S., Van Dam, A. M., Conrad, R., Bista, P., et al. (2011). Fumaric acid esters exert neuroprotective effects in neuroinflammation via activation of the Nrf2 antioxidant pathway. Brain 134, 678–692. doi: 10.1093/brain/awq386
Lynch, D. R., Hauser, L., Mccormick, A., Wells, M., Dong, Y. N., Mccormack, S., et al. (2019). Randomized, double-blind, placebo-controlled study of interferon-gamma 1b in Friedreich Ataxia. Ann. Clin. Transl. Neurol. 6, 546–553. doi: 10.1002/acn3.731
Lynch, D. R., and Johnson, J. (2021). Omaveloxolone: potential new agent for Friedreich ataxia. Neurodegener. Dis. Manag. 11, 91–98. doi: 10.2217/nmt-2020-0057
Maldonado, C., Vazquez, M., and Fagiolino, P. (2020). Potential therapeutic role of carnitine and acetylcarnitine in neurological disorders. Curr. Pharm. Des. 26, 1277–1285. doi: 10.2174/1381612826666200212114038
Mangla, B., Javed, S., Sultan, M. H., Kumar, P., Kohli, K., Najmi, A., et al. (2021). Sulforaphane: a review of its therapeutic potentials, advances in its nanodelivery, recent patents, and clinical trials. Phytother. Res. 35, 5440–5458. doi: 10.1002/ptr.7176
Marcotulli, C., Fortuni, S., Arcuri, G., Tomassini, B., Leonardi, L., Pierelli, F., et al. (2016). GIFT-1, a phase IIa clinical trial to test the safety and efficacy of IFNgamma administration in FRDA patients. Neurol. Sci. 37, 361–364. doi: 10.1007/s10072-015-2427-3
Mariotti, C., Fancellu, R., Caldarazzo, S., Nanetti, L., Di Bella, D., Plumari, M., et al. (2012). Erythropoietin in Friedreich ataxia: no effect on frataxin in a randomized controlled trial. Mov. Disord. 27, 446–449. doi: 10.1002/mds.24066
Marmolino, D., Acquaviva, F., Pinelli, M., Monticelli, A., Castaldo, I., Filla, A., et al. (2009). PPAR-gamma agonist Azelaoyl PAF increases frataxin protein and mRNA expression: new implications for the Friedreich’s ataxia therapy. Cerebellum 8, 98–103. doi: 10.1007/s12311-008-0087-z
Marmolino, D., Manto, M., Acquaviva, F., Vergara, P., Ravella, A., Monticelli, A., et al. (2010). PGC-1alpha down-regulation affects the antioxidant response in Friedreich’s ataxia. PLoS One 5:e10025. doi: 10.1371/journal.pone.0010025
Martelli, A., and Puccio, H. (2014). Dysregulation of cellular iron metabolism in Friedreich ataxia: from primary iron-sulfur cluster deficit to mitochondrial iron accumulation. Front. Pharmacol. 5:130. doi: 10.3389/fphar.2014.00130
Martin, A. S., Abraham, D. M., Hershberger, K. A., Bhatt, D. P., Mao, L., Cui, H., et al. (2017). Nicotinamide mononucleotide requires SIRT3 to improve cardiac function and bioenergetics in a Friedreich’s ataxia cardiomyopathy model. JCI Insight 2:e93885. doi: 10.1172/jci.insight.93885
Matthews, H., Hanison, J., and Nirmalan, N. (2016). “Omics”-informed drug and biomarker discovery: opportunities, challenges and future perspectives. Proteomes 4:28. doi: 10.3390/proteomes4030028
Mehmel, M., Jovanovic, N., and Spitz, U. (2020). Nicotinamide riboside-the current state of research and therapeutic uses. Nutrients 12:1616. doi: 10.3390/nu12061616
Miller, J. L., Rai, M., Frigon, N. L., Pandolfo, M., Punnonen, J., and Spencer, J. R. (2017). Erythropoietin and small molecule agonists of the tissue-protective erythropoietin receptor increase FXN expression in neuronal cells in vitro and in Fxn-deficient KIKO mice in vivo. Neuropharmacology 123, 34–45. doi: 10.1016/j.neuropharm.2017.05.011
Montermini, L., Richter, A., Morgan, K., Justice, C. M., Julien, D., Castellotti, B., et al. (1997). Phenotypic variability in Friedreich ataxia: role of the associated GAA triplet repeat expansion. Ann. Neurol. 41, 675–682. doi: 10.1002/ana.410410518
Mulholland, P. J. (2006). Susceptibility of the cerebellum to thiamine deficiency. Cerebellum 5, 55–63. doi: 10.1080/14734220600551707
Nachbauer, W., Boesch, S., Reindl, M., Eigentler, A., Hufler, K., Poewe, W., et al. (2012). Skeletal muscle involvement in friedreich ataxia and potential effects of recombinant human erythropoietin administration on muscle regeneration and neovascularization. J. Neuropathol. Exp. Neurol. 71, 708–715. doi: 10.1097/NEN.0b013e31825fed76
Nachbauer, W., Hering, S., Seifert, M., Steinkellner, H., Sturm, B., Scheiber-Mojdehkar, B., et al. (2011a). Effects of erythropoietin on frataxin levels and mitochondrial function in Friedreich ataxia–a dose-response trial. Cerebellum 10, 763–769. doi: 10.1007/s12311-011-0287-9
Nachbauer, W., Wanschitz, J., Steinkellner, H., Eigentler, A., Sturm, B., Hufler, K., et al. (2011b). Correlation of frataxin content in blood and skeletal muscle endorses frataxin as a biomarker in Friedreich ataxia. Mov. Disord. 26, 1935–1938. doi: 10.1002/mds.23789
Nachun, D., Gao, F., Isaacs, C., Strawser, C., Yang, Z., Dokuru, D., et al. (2018). Peripheral blood gene expression reveals an inflammatory transcriptomic signature in Friedreich’s ataxia patients. Hum. Mol. Genet. 27, 2965–2977. doi: 10.1093/hmg/ddy198
Nairz, M., Haschka, D., Demetz, E., and Weiss, G. (2014). Iron at the interface of immunity and infection. Front. Pharmacol. 5:152. doi: 10.3389/fphar.2014.00152
Namasivayam, V., Vanangamudi, M., Kramer, V. G., Kurup, S., Zhan, P., Liu, X., et al. (2019). The journey of HIV-1 Non-Nucleoside Reverse Transcriptase Inhibitors (NNRTIs) from lab to clinic. J. Med. Chem. 62, 4851–4883. doi: 10.1021/acs.jmedchem.8b00843
Neumann, H., Schmidt, H., Wilharm, E., Behrens, L., and Wekerle, H. (1997). Interferon gamma gene expression in sensory neurons: evidence for autocrine gene regulation. J. Exp. Med. 186, 2023–2031. doi: 10.1084/jem.186.12.2023
Nguyen, A., Rossi, S., Croteau, D., Best, B. M., Clifford, D., Collier, A. C., et al. (2013). Etravirine in CSF is highly protein bound. J. Antimicrob. Chemother. 68, 1161–1168. doi: 10.1093/jac/dks517
Palandri, A., L’hote, D., Cohen-Tannoudji, J., Tricoire, H., and Monnier, V. (2015). Frataxin inactivation leads to steroid deficiency in flies and human ovarian cells. Hum. Mol. Genet. 24, 2615–2626. doi: 10.1093/hmg/ddv024
Patel, M., Isaacs, C. J., Seyer, L., Brigatti, K., Gelbard, S., Strawser, C., et al. (2016). Progression of Friedreich ataxia: quantitative characterization over 5 years. Ann. Clin. Transl. Neurol. 3, 684–694. doi: 10.1002/acn3.332
Patel, M., Schadt, K., Mccormick, A., Isaacs, C., Dong, Y. N., and Lynch, D. R. (2019). Open-label pilot study of oral methylprednisolone for the treatment of patients with friedreich ataxia. Muscle Nerve 60, 571–575. doi: 10.1002/mus.26610
Paupe, V., Dassa, E. P., Goncalves, S., Auchere, F., Lonn, M., Holmgren, A., et al. (2009). Impaired nuclear Nrf2 translocation undermines the oxidative stress response in Friedreich ataxia. PLoS One 4:e4253. doi: 10.1371/journal.pone.0004253
Pedraza, O. L., and Botez, M. I. (1992). Thiamine status in inherited degenerative ataxias. J. Neurol. Neurosurg. Psychiatry 55, 136–137. doi: 10.1136/jnnp.55.2.136
Pennisi, M., Lanza, G., Cantone, M., D’amico, E., Fisicaro, F., Puglisi, V., et al. (2020). Acetyl-L-carnitine in dementia and other cognitive disorders: a critical update. Nutrients 12:1389. doi: 10.3390/nu12051389
Perdomini, M., Belbellaa, B., Monassier, L., Reutenauer, L., Messaddeq, N., Cartier, N., et al. (2014). Prevention and reversal of severe mitochondrial cardiomyopathy by gene therapy in a mouse model of Friedreich’s ataxia. Nat. Med. 20, 542–547. doi: 10.1038/nm.3510
Petit, F., Drecourt, A., Dussiot, M., Zangarelli, C., Hermine, O., Munnich, A., et al. (2021). Defective palmitoylation of transferrin receptor triggers iron overload in Friedreich ataxia fibroblasts. Blood 137, 2090–2102. doi: 10.1182/blood.2020006987
Petrillo, S., D’amico, J., La Rosa, P., Bertini, E. S., and Piemonte, F. (2019). Targeting NRF2 for the treatment of friedreich’s ataxia: a comparison among drugs. Int. J. Mol. Sci. 20:5211. doi: 10.3390/ijms20205211
Petrillo, S., Piermarini, E., Pastore, A., Vasco, G., Schirinzi, T., Carrozzo, R., et al. (2017). Nrf2-inducers counteract neurodegeneration in frataxin-silenced motor neurons: disclosing new therapeutic targets for friedreich’s ataxia. Int. J. Mol. Sci. 18:2173. doi: 10.3390/ijms18102173
Piantadosi, C. A., Carraway, M. S., Babiker, A., and Suliman, H. B. (2008). Heme oxygenase-1 regulates cardiac mitochondrial biogenesis via Nrf2-mediated transcriptional control of nuclear respiratory factor-1. Circ. Res. 103, 1232–1240. doi: 10.1161/01.RES.0000338597.71702.ad
Piguet, F., De Montigny, C., Vaucamps, N., Reutenauer, L., Eisenmann, A., and Puccio, H. (2018). Rapid and complete reversal of sensory ataxia by gene therapy in a novel model of friedreich ataxia. Mol. Ther. 26, 1940–1952. doi: 10.1016/j.ymthe.2018.05.006
Plenge, U., Belhage, B., Guadalupe-Grau, A., Andersen, P. R., Lundby, C., Dela, F., et al. (2012). Erythropoietin treatment enhances muscle mitochondrial capacity in humans. Front. Physiol. 3:50. doi: 10.3389/fphys.2012.00050
Poburski, D., Boerner, J. B., Koenig, M., Ristow, M., and Thierbach, R. (2016). Time-resolved functional analysis of acute impairment of frataxin expression in an inducible cell model of Friedreich ataxia. Biol. Open 5, 654–661. doi: 10.1242/bio.017004
Porkka, K., Koskenvesa, P., Lundan, T., Rimpilainen, J., Mustjoki, S., Smykla, R., et al. (2008). Dasatinib crosses the blood-brain barrier and is an efficient therapy for central nervous system Philadelphia chromosome-positive leukemia. Blood 112, 1005–1012. doi: 10.1182/blood-2008-02-140665
Pushpakom, S., Iorio, F., Eyers, P. A., Escott, K. J., Hopper, S., Wells, A., et al. (2019). Drug repurposing: progress, challenges and recommendations. Nat. Rev. Drug Discov. 18, 41–58. doi: 10.1038/nrd.2018.168
Pyo, I. S., Yun, S., Yoon, Y. E., Choi, J. W., and Lee, S. J. (2020). Mechanisms of aging and the preventive effects of resveratrol on age-related diseases. Molecules 25:4649. doi: 10.3390/molecules25204649
Reetz, K., Dogan, I., Costa, A. S., Dafotakis, M., Fedosov, K., Giunti, P., et al. (2015). Biological and clinical characteristics of the European Friedreich’s Ataxia Consortium for Translational Studies (EFACTS) cohort: a cross-sectional analysis of baseline data. Lancet Neurol. 14, 174–182. doi: 10.1016/S1474-4422(14)70321-7
Reetz, K., Dogan, I., Hilgers, R. D., Giunti, P., Parkinson, M. H., Mariotti, C., et al. (2021). Progression characteristics of the European Friedreich’s Ataxia Consortium for Translational Studies (EFACTS): a 4-year cohort study. Lancet Neurol. 20, 362–372. doi: 10.1016/S1474-4422(21)00027-2
Reetz, K., Hilgers, R. D., Isfort, S., Dohmen, M., Didszun, C., Fedosov, K., et al. (2019). Protocol of a randomized, double-blind, placebo-controlled, parallel-group, multicentre study of the efficacy and safety of nicotinamide in patients with Friedreich ataxia (NICOFA). Neurol. Res. Pract. 1:33. doi: 10.1186/s42466-019-0038-9
Rodden, L. N., and Lynch, D. R. (2021). Designing phase II clinical trials in Friedreich ataxia. Expert Opin. Emerg. Drugs 26, 415–423. doi: 10.1080/14728214.2021.1998452
Rodriguez-Pascau, L., Vilalta, A., Cerrada, M., Traver, E., Forss-Petter, S., Weinhofer, I., et al. (2021b). The brain penetrant PPARgamma agonist leriglitazone restores multiple altered pathways in models of X-linked adrenoleukodystrophy. Sci. Transl. Med. 13:eabc0555. doi: 10.1126/scitranslmed.abc0555
Rodriguez-Pascau, L., Britti, E., Calap-Quintana, P., Dong, Y. N., Vergara, C., Delaspre, F., et al. (2021a). PPAR gamma agonist leriglitazone improves frataxin-loss impairments in cellular and animal models of Friedreich Ataxia. Neurobiol. Dis. 148:105162. doi: 10.1016/j.nbd.2020.105162
Rotig, A., De Lonlay, P., Chretien, D., Foury, F., Koenig, M., Sidi, D., et al. (1997). Aconitase and mitochondrial iron-sulphur protein deficiency in Friedreich ataxia. Nat. Genet. 17, 215–217. doi: 10.1038/ng1097-215
Rufini, A., Cavallo, F., Condo, I., Fortuni, S., De Martino, G., Incani, O., et al. (2015). Highly specific ubiquitin-competing molecules effectively promote frataxin accumulation and partially rescue the aconitase defect in Friedreich ataxia cells. Neurobiol. Dis. 75, 91–99. doi: 10.1016/j.nbd.2014.12.011
Rufini, A., Fortuni, S., Arcuri, G., Condo, I., Serio, D., Incani, O., et al. (2011). Preventing the ubiquitin-proteasome-dependent degradation of frataxin, the protein defective in Friedreich’s ataxia. Hum. Mol. Genet. 20, 1253–1261. doi: 10.1093/hmg/ddq566
Ruhee, R. T., and Suzuki, K. (2020). The integrative role of sulforaphane in preventing inflammation, oxidative stress and fatigue: a review of a potential protective phytochemical. Antioxidants (Basel) 9:521. doi: 10.3390/antiox9060521
Sacca, F., Piro, R., De Michele, G., Acquaviva, F., Antenora, A., Carlomagno, G., et al. (2011). Epoetin alfa increases frataxin production in Friedreich’s ataxia without affecting hematocrit. Mov. Disord. 26, 739–742. doi: 10.1002/mds.23435
Sacca, F., Puorro, G., Marsili, A., Antenora, A., Pane, C., Casali, C., et al. (2016). Long-term effect of epoetin alfa on clinical and biochemical markers in friedreich ataxia. Mov. Disord. 31, 734–741. doi: 10.1002/mds.26552
Sahdeo, S., Scott, B. D., Mcmackin, M. Z., Jasoliya, M., Brown, B., Wulff, H., et al. (2014). Dyclonine rescues frataxin deficiency in animal models and buccal cells of patients with Friedreich’s ataxia. Hum. Mol. Genet. 23, 6848–6862. doi: 10.1093/hmg/ddu408
Sakanaka, M., Wen, T. C., Matsuda, S., Masuda, S., Morishita, E., Nagao, M., et al. (1998). In vivo evidence that erythropoietin protects neurons from ischemic damage. Proc. Natl. Acad. Sci. U. S. A. 95, 4635–4640. doi: 10.1073/pnas.95.8.4635
Salehi, B., Mishra, A. P., Nigam, M., Sener, B., Kilic, M., Sharifi-Rad, M., et al. (2018). Resveratrol: a double-edged sword in health benefits. Biomedicines 6:91. doi: 10.3390/biomedicines6030091
Santoro, A., Anjomani Virmouni, S., Paradies, E., Villalobos Coa, V. L., Al-Mahdawi, S., Khoo, M., et al. (2018). Effect of diazoxide on Friedreich ataxia models. Hum. Mol. Genet. 27, 992–1001. doi: 10.1093/hmg/ddy016
Sardana, D., Zhu, C., Zhang, M., Gudivada, R. C., Yang, L., and Jegga, A. G. (2011). Drug repositioning for orphan diseases. Brief Bioinform. 12, 346–356. doi: 10.1093/bib/bbr021
Savelieff, M. G., and Feldman, E. L. (2021). Lessons for clinical trial design in Friedreich’s ataxia. Lancet Neurol. 20, 330–332. doi: 10.1016/S1474-4422(21)00064-8
Schcolnik-Cabrera, A., Juarez-Lopez, D., and Duenas-Gonzalez, A. (2021). Perspectives on Drug Repurposing. Curr. Med. Chem. 28, 2085–2099. doi: 10.2174/0929867327666200831141337
Schols, L., Zange, J., Abele, M., Schillings, M., Skipka, G., Kuntz-Hehner, S., et al. (2005). L-carnitine and creatine in Friedreich’s ataxia. A randomized, placebo-controlled crossover trial. J. Neural. Transm. (Vienna) 112, 789–796. doi: 10.1007/s00702-004-0216-x
Seyer, L., Greeley, N., Foerster, D., Strawser, C., Gelbard, S., Dong, Y., et al. (2015). Open-label pilot study of interferon gamma-1b in Friedreich ataxia. Acta Neurol. Scand. 132, 7–15. doi: 10.1111/ane.12337
Shan, Y., Napoli, E., and Cortopassi, G. (2007). Mitochondrial frataxin interacts with ISD11 of the NFS1/ISCU complex and multiple mitochondrial chaperones. Hum. Mol. Genet. 16, 929–941. doi: 10.1093/hmg/ddm038
Shan, Y., Schoenfeld, R. A., Hayashi, G., Napoli, E., Akiyama, T., Iodi Carstens, M., et al. (2013). Frataxin deficiency leads to defects in expression of antioxidants and Nrf2 expression in dorsal root ganglia of the Friedreich’s ataxia YG8R mouse model. Antioxid. Redox Signal. 19, 1481–1493. doi: 10.1089/ars.2012.4537
Shelmire, B., Gastineau, F. M., and Shields, T. L. (1955). Evaluation of a new topical anesthetic, dyclonine hydrochloride. AMA Arch. Derm. 71, 728–730. doi: 10.1001/archderm.1955.01540300050013
Shen, X., Wong, J., Prakash, T. P., Rigo, F., Li, Y., Napierala, M., et al. (2020). Progress towards drug discovery for Friedreich’s Ataxia: identifying synthetic oligonucleotides that more potently activate expression of human frataxin protein. Bioorg. Med. Chem. 28:115472. doi: 10.1016/j.bmc.2020.115472
Shinnick, J. E., Isaacs, C. J., Vivaldi, S., Schadt, K., and Lynch, D. R. (2016). Friedreich Ataxia and nephrotic syndrome: a series of two patients. BMC Neurol. 16:3. doi: 10.1186/s12883-016-0526-2
Song, S., Sava, V., Rowe, A., Li, K., Cao, C., Mori, T., et al. (2011). Granulocyte-colony stimulating factor (G-CSF) enhances recovery in mouse model of Parkinson’s disease. Neurosci. Lett. 487, 153–157. doi: 10.1016/j.neulet.2010.10.012
Soragni, E., and Gottesfeld, J. M. (2016). Translating HDAC inhibitors in Friedreich’s ataxia. Expert Opin. Orphan Drugs 4, 961–970. doi: 10.1080/21678707.2016.1215910
Stratford, M. R., and Dennis, M. F. (1994). Pharmacokinetics and biochemistry studies on nicotinamide in the mouse. Cancer Chemother. Pharmacol. 34, 399–404. doi: 10.1007/BF00685564
Sturm, B., Helminger, M., Steinkellner, H., Heidari, M. M., Goldenberg, H., and Scheiber-Mojdehkar, B. (2010). Carbamylated erythropoietin increases frataxin independent from the erythropoietin receptor. Eur. J. Clin. Invest. 40, 561–565. doi: 10.1111/j.1365-2362.2010.02292.x
Sturm, B., Stupphann, D., Kaun, C., Boesch, S., Schranzhofer, M., Wojta, J., et al. (2005). Recombinant human erythropoietin: effects on frataxin expression in vitro. Eur. J. Clin. Invest. 35, 711–717. doi: 10.1111/j.1365-2362.2005.01568.x
Talevi, A., and Bellera, C. L. (2020). Challenges and opportunities with drug repurposing: finding strategies to find alternative uses of therapeutics. Expert Opin. Drug Discov. 15, 397–401. doi: 10.1080/17460441.2020.1704729
Tiano, F., Amati, F., Cherubini, F., Morini, E., Vancheri, C., Maletta, S., et al. (2020). Frataxin deficiency in Friedreich’s ataxia is associated with reduced levels of HAX-1, a regulator of cardiomyocyte death and survival. Hum. Mol. Genet. 29, 471–482. doi: 10.1093/hmg/ddz306
Tomassini, B., Arcuri, G., Fortuni, S., Sandi, C., Ezzatizadeh, V., Casali, C., et al. (2012). Interferon gamma upregulates frataxin and corrects the functional deficits in a Friedreich ataxia model. Hum. Mol. Genet. 21, 2855–2861. doi: 10.1093/hmg/dds110
Tsou, A. Y., Paulsen, E. K., Lagedrost, S. J., Perlman, S. L., Mathews, K. D., Wilmot, G. R., et al. (2011). Mortality in Friedreich ataxia. J. Neurol. Sci. 307, 46–49. doi: 10.1016/j.jns.2011.05.023
Uddin, M. S., Mamun, A. A., Jakaria, M., Thangapandiyan, S., Ahmad, J., Rahman, M. A., et al. (2020). Emerging promise of sulforaphane-mediated Nrf2 signaling cascade against neurological disorders. Sci. Total Environ. 707:135624. doi: 10.1016/j.scitotenv.2019.135624
Vaquero, A., Scher, M., Erdjument-Bromage, H., Tempst, P., Serrano, L., and Reinberg, D. (2007). SIRT1 regulates the histone methyl-transferase SUV39H1 during heterochromatin formation. Nature 450, 440–444. doi: 10.1038/nature06268
Vavla, M., D’angelo, M. G., Arrigoni, F., Toschi, N., Peruzzo, D., Gandossini, S., et al. (2020b). Safety and efficacy of interferon gamma in friedreich’s ataxia. Mov. Disord. 35, 370–371. doi: 10.1002/mds.27979
Vavla, M., Arrigoni, F., Toschi, N., Peruzzo, D., D’angelo, M. G., Gandossini, S., et al. (2020a). Sensitivity of neuroimaging indicators in monitoring the effects of interferon gamma treatment in friedreich’s ataxia. Front. Neurosci. 14:872. doi: 10.3389/fnins.2020.00872
Venci, J. V., and Gandhi, M. A. (2013). Dimethyl fumarate (Tecfidera): a new oral agent for multiple sclerosis. Ann. Pharmacother. 47, 1697–1702. doi: 10.1177/1060028013509232
Vyas, P. M., Tomamichel, W. J., Pride, P. M., Babbey, C. M., Wang, Q., Mercier, J., et al. (2012). A TAT-frataxin fusion protein increases lifespan and cardiac function in a conditional Friedreich’s ataxia mouse model. Hum. Mol. Genet. 21, 1230–1247. doi: 10.1093/hmg/ddr554
Wang, Q., Chuikov, S., Taitano, S., Wu, Q., Rastogi, A., Tuck, S. J., et al. (2015). Dimethyl fumarate protects neural stem/progenitor cells and neurons from oxidative damage through Nrf2-ERK1/2 MAPK Pathway. Int. J. Mol. Sci. 16, 13885–13907. doi: 10.3390/ijms160613885
Wu, Z., Puigserver, P., Andersson, U., Zhang, C., Adelmant, G., Mootha, V., et al. (1999). Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell 98, 115–124. doi: 10.1016/S0092-8674(00)80611-X
Wyller, V. B., Jacobsen, K., Dahl, M. B., Nilsen, H., Proske, S., Horter, T., et al. (2016). Interferon gamma may improve cardiac function in Friedreich’s ataxia cardiomyopathy. Int. J. Cardiol. 221, 376–378. doi: 10.1016/j.ijcard.2016.06.288
Xenocostas, A., Cheung, W. K., Farrell, F., Zakszewski, C., Kelley, M., Lutynski, A., et al. (2005). The pharmacokinetics of erythropoietin in the cerebrospinal fluid after intravenous administration of recombinant human erythropoietin. Eur. J. Clin. Pharmacol. 61, 189–195. doi: 10.1007/s00228-005-0896-7
Xu, C., Soragni, E., Chou, C. J., Herman, D., Plasterer, H. L., Rusche, J. R., et al. (2009). Chemical probes identify a role for histone deacetylase 3 in Friedreich’s ataxia gene silencing. Chem. Biol. 16, 980–989. doi: 10.1016/j.chembiol.2009.07.010
Xu, Y., Zhao, L., Qiu, H., Qian, T., Sang, S., and Zhong, C. (2021). The impact of thiamine deficiency and benfotiamine treatment on Nod-like receptor protein-3 inflammasome in microglia. Neuroreport 32, 1041–1048. doi: 10.1097/WNR.0000000000001691
Yang, X., Zheng, Y., Liu, L., Huang, J., Wang, F., and Zhang, J. (2021). Progress on the study of the anticancer effects of artesunate. Oncol. Lett. 22:750. doi: 10.3892/ol.2021.13011
Yiu, E. M., Tai, G., Peverill, R. E., Lee, K. J., Croft, K. D., Mori, T. A., et al. (2015). An open-label trial in Friedreich ataxia suggests clinical benefit with high-dose resveratrol, without effect on frataxin levels. J. Neurol. 262, 1344–1353. doi: 10.1007/s00415-015-7719-2
Zesiewicz, T., Heerinckx, F., De Jager, R., Omidvar, O., Kilpatrick, M., Shaw, J., et al. (2018a). Randomized, clinical trial of RT001: early signals of efficacy in Friedreich’s ataxia. Mov. Disord. 33, 1000–1005. doi: 10.1002/mds.27353
Zesiewicz, T., Salemi, J. L., Perlman, S., Sullivan, K. L., Shaw, J. D., Huang, Y., et al. (2018b). Double-blind, randomized and controlled trial of EPI-743 in Friedreich’s ataxia. Neurodegener. Dis. Manag. 8, 233–242. doi: 10.2217/nmt-2018-0013
Zesiewicz, T. A., Hancock, J., Ghanekar, S. D., Kuo, S. H., Dohse, C. A., and Vega, J. (2020). Emerging therapies in Friedreich’s Ataxia. Expert Rev. Neurother. 20, 1215–1228.
Keywords: friedreich ataxia, drug development, frataxin, drug repositioning, therapeutics
Citation: Rufini A, Malisan F, Condò I and Testi R (2022) Drug Repositioning in Friedreich Ataxia. Front. Neurosci. 16:814445. doi: 10.3389/fnins.2022.814445
Received: 13 November 2021; Accepted: 07 January 2022;
Published: 09 February 2022.
Edited by:
David Lynch, University of Pennsylvania, United StatesReviewed by:
M. Grazia Cotticelli, Children’s Hospital of Philadelphia, United StatesCopyright © 2022 Rufini, Malisan, Condò and Testi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alessandra Rufini, cnVmaW5pQG1lZC51bmlyb21hMi5pdA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.