
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Neurosci. , 24 February 2023
Sec. Translational Neuroscience
Volume 16 - 2022 | https://doi.org/10.3389/fnins.2022.1100256
 Viviam Sanabria1
Viviam Sanabria1 Simone Romariz1
Simone Romariz1 Matheus Braga1
Matheus Braga1 Maira Licia Foresti1,2
Maira Licia Foresti1,2 Maria da Graça Naffah-Mazzacoratti1
Maria da Graça Naffah-Mazzacoratti1 Luiz Eugênio Mello1,2
Luiz Eugênio Mello1,2 Beatriz M. Longo1*
Beatriz M. Longo1*Interest in the use of anticholinergics to prevent the development of epilepsy after traumatic brain injury (TBI) has grown since recent basic studies have shown their effectiveness in modifying the epileptogenic process. These studies demonstrated that treatment with anticholinergics, in the acute phase after brain injury, decreases seizure frequency, and severity, and the number of spontaneous recurrent seizures (SRS). Therefore, anticholinergics may reduce the risk of developing posttraumatic epilepsy (PTE). In this brief review, we summarize the role of the cholinergic system in epilepsy and the key findings from using anticholinergic drugs to prevent PTE in animal models and new clinical trial protocols. Furthermore, we discuss why treatment with anticholinergics is more likely to prevent PTE than treatment for other epilepsies.
Traumatic brain injury (TBI) is an interruption in normal brain function when a sudden force (i.e., rapid acceleration or deceleration of the brain, direct impact or injury due to explosions, or penetrating head injury) damages the brain (Carteri and da Silva, 2021). TBI has several causes, including traffic accidents, sports injuries, and falls (de Almeida et al., 2016). TBI is associated with long-term disability, neurological disorders, and early mortality, constituting a public health problem with socioeconomic burden in low- and middle-income countries (Arciniegas et al., 2002; de Almeida et al., 2016; Maas et al., 2017).
One of the main consequences of TBI is posttraumatic epilepsy (PTE), which refers to the development of epilepsy after brain injury. According to the International League Against Epilepsy, epilepsy is considered in the following conditions: one unprovoked seizure and probability of recurrent seizure risk over 60%, a diagnosis of epilepsy syndrome, and two or more seizures occurring more than 24 h apart (Fisher et al., 2014).
Epileptogenesis in PTE is a continuous process in which a normal brain shows hypersynchronous excitability after a head injury (for review, see Golub and Reddy, 2022). After brain injury, seizures are categorized as immediate (≤24 h post-TBI), early (≤7 days), or late (>7 days) (Jennett, 1969). Trauma severity is correlated with the risk of epileptogenesis, with a 2.1% risk in mild, 4.2% in moderate, and 16.7% in severe cases of TBI (Annegers et al., 1998). The risk of developing epilepsy in patients with TBI is 29-fold higher than in the general population (Lowenstein, 2009), with TBI causing 10–20% of all symptomatic epilepsies (Annegers et al., 1998). Moreover, follow-up studies have indicated that PTE is diagnosed in approximately 80% of TBI cases within 2 years (Haltiner et al., 1997). Critical risk factors for PTE include intracranial hematoma, unconsciousness at hospital admission, chronic alcoholism, age ≥ 65 years, injury severity, and skull fracture (Annegers et al., 1998).
Unfortunately, the cascade of changes that transforms the non-epileptic brain into one that generates spontaneous recurrent seizures (SRS) is not completely understood due to its complexity (for review, see Löscher et al., 2015); In fact, Löscher et al. (2015) proposed that there is no latent period, and that epileptogenesis can start immediately after brain injury, suggesting that antiepileptogenic therapies should be administered as soon as possible. Thus, PTE is not yet preventable. Although brain alterations are correlated with brain injury severity, they are not necessarily involved in epileptogenesis. Moreover, not all individuals develop epilepsy after an acute brain injury, probably due to the endogenous repair response, absence of risk factors, or low insult severity (Pignataro et al., 2008).
One significant research benchmark from the U.S. National Institute of Neurological Disorders and Stroke (NINDS) has been focusing on preventing the occurrence of epilepsy and its progression (National Institute of Neurological Disorders and Stroke, 2022). Therefore, developing novel therapies for preventing the development and progression of epilepsy is vital in at-risk patients.
A prospective window of opportunity to prevent, intervene, and stop the development of PTE is the latent period between brain injury and SRS onset; a period that can vary from months to years in humans (French et al., 1993) and from days to weeks in rodent models (Mello et al., 1993; Cavalheiro, 1995). Neurochemical interventions during this period may modify focal development (Cavalheiro et al., 1994). This concept has led to various experimental studies, and some clinical trials evaluated whether prolonged administration of anti-seizure drugs after a brain injury can prevent or modify epilepsy development (for review, see Löscher et al., 2015). However, no compound test has been advanced into clinical practice (Saletti et al., 2019).
To date, the crucial roles of the glutamatergic and GABAergic systems in the expression and suppression of seizures and the development of epileptic conditions have been widely investigated in preclinical and clinical studies due to the classical mechanism of imbalance between excitation and inhibition. Nonetheless, studies focusing on the involvement of the cholinergic system in epileptic conditions have raised some interest, with recent studies showing the effectiveness of anticholinergics in modifying the epileptogenic process (Bittencourt et al., 2017; Benassi et al., 2021; Meller et al., 2021).
In this review, we discuss the role of the cholinergic system in epilepsy and the use of anticholinergic drugs to treat PTE in animal models and clinical trials. Additionally, we discuss why PTE is more likely to be prevented than other types of epilepsy. Further, we considered that administering a selective cholinergic antagonist immediately after a brain injury attenuates excitotoxicity and consequently inhibits the development of PTE by interfering with the epileptogenic process at a critical moment. This review aims to provide new insights based on what has been proposed regarding PTE treatment using an anticholinergic strategy.
Acetylcholine (ACh) is an endogenous neurotransmitter synthesized from choline and acetyl-CoA in the cytoplasm of nerve terminals via the acetyltransferase enzyme and subsequently transported into vesicles (for review, see Zaagsma and Meurs, 2006). After release, ACh binds to two types of membrane proteins: metabotropic muscarinic receptors, which signal through either Gq proteins (M1, M3, and M5 subtypes) that activate phospholipase C, Gi/o proteins that are negatively related to adenylate cyclase (M2 and M4 subtypes), and ionotropic nicotinic receptors, which function as non-selective, excitatory channels. Within the synapse, ACh is converted back into choline and acetic acid by the acetylcholinesterase enzyme. Finally, choline is reuptaken by choline transporters (Pavel et al., 2006).
In the brain, ACh is broadly distributed; all regions of the forebrain, midbrain, and brainstem contain cholinergic neurons. Projections originating in the medial septum and diagonal band of Broca’s canal via the pre-commissural branch of the fimbria-fornix pathway constitute the main cholinergic inputs of the hippocampus, providing direct information to both principal neurons and interneurons (for a review, see Drever et al., 2011). In the substantia innominata of the basal forebrain, the nucleus basalis of Meynert has neurons projecting throughout the cortex and amygdala (Mesulam et al., 1983). Moreover, all the peripheral and central nervous systems cholinergic neurons use ACh as their neurotransmitter (pre- and post-ganglionic parasympathetic neurons and all pre-ganglionic sympathetic neurons) (for review, see Wang et al., 2021).
Cholinergic neurons are implicated in several crucial physiological processes, such as attention, learning, memory, and stress response (Wang et al., 2021). Additionally, ACh functions as a neuromodulator by coordinating the firing of neuronal clusters, increasing or decreasing synaptic dynamics, neuroplasticity, and spine density, increasing the release of growth factors, and hippocampal neurogenesis. Cognitive processes improve through an increase in cholinergic synapses, and the degeneration of central cholinergic neurons impairs memory (for review, see Maurer and Williams, 2017).
Due to its critical role in cognitive processes, ACh is highlighted as an essential factor in several diseases, such as depression (Saricicek et al., 2012), Alzheimer’s (Wilcock et al., 1982), Parkinson’s (Bohnen and Albin, 2011), and Huntington’s diseases (Ferrante et al., 1987) which show ACh denervation and imbalance.
Considering these findings, anticholinergic drugs that block and decrease the activity of ACh synapses in the central nervous systems have been prescribed for Parkinson’s, amyotrophic lateral sclerosis, and depression (Hori et al., 2015; McGeachan et al., 2017; López-Álvarez et al., 2019). Anticholinergic medications achieve the intended therapeutic effect by competing one-for-one with ACh, mostly at muscarinic receptors, modulating the receptor’s affinity, and reducing the physiological response by avoiding neuronal injury and subsequent inflammation (for review, see Scarr, 2012). However, various adverse effects on the central and peripheral nervous systems have been described with anticholinergics, including cognitive impairment and blurred vision (for review, see López-Álvarez et al., 2019).
In PTE, the hippocampus presents a persistent reactive gliosis and a consistent neuronal loss in the hilar, CA1, and CA2 regions (Diaz-Arrastia et al., 2000). Although this structure commonly presents hippocampal sclerosis, other structures also undergo neurodegeneration; the brain stem, cerebellum, basal forebrain, and selected thalamic nuclei are also damaged (Maxwell et al., 2004). However, no studies have investigated neuropathological differences between the hippocampus and other structures in patients with TBI who did not develop PTE and those who developed PTE (Pitkänen and McIntosh, 2006). Most patients with PTE have seizure foci in the temporal lobe (56% of the cases), followed by the frontal lobe (36%), parietal lobe (5%), and occipital lobe (3%) (Hudak et al., 2004). This high percentage of patients with PTE with temporal lobe lesions reflects the relevance of the entorhinal cortex-hippocampus complex as these regions are frequently the site of origin of seizure activity, and both areas are enriched with cholinergic innervation (for review, see Friedman et al., 2007).
Early after TBI, the cholinergic system activates a large number of muscarinic receptors leading to an immediate massive release of ACh (increasing 49%), which can lead to status epilepticus (SE) (Turski et al., 1983; Saija et al., 1988; Shin and Dixon, 2015). However, in the chronic periods after TBI, cholinergic hypoactivity occurs. It has been speculated that cholinergic activity may be reduced due to cholinergic neuron dysfunction caused by functional deficits or neuronal loss (Östberg et al., 2011) (see Figure 1).
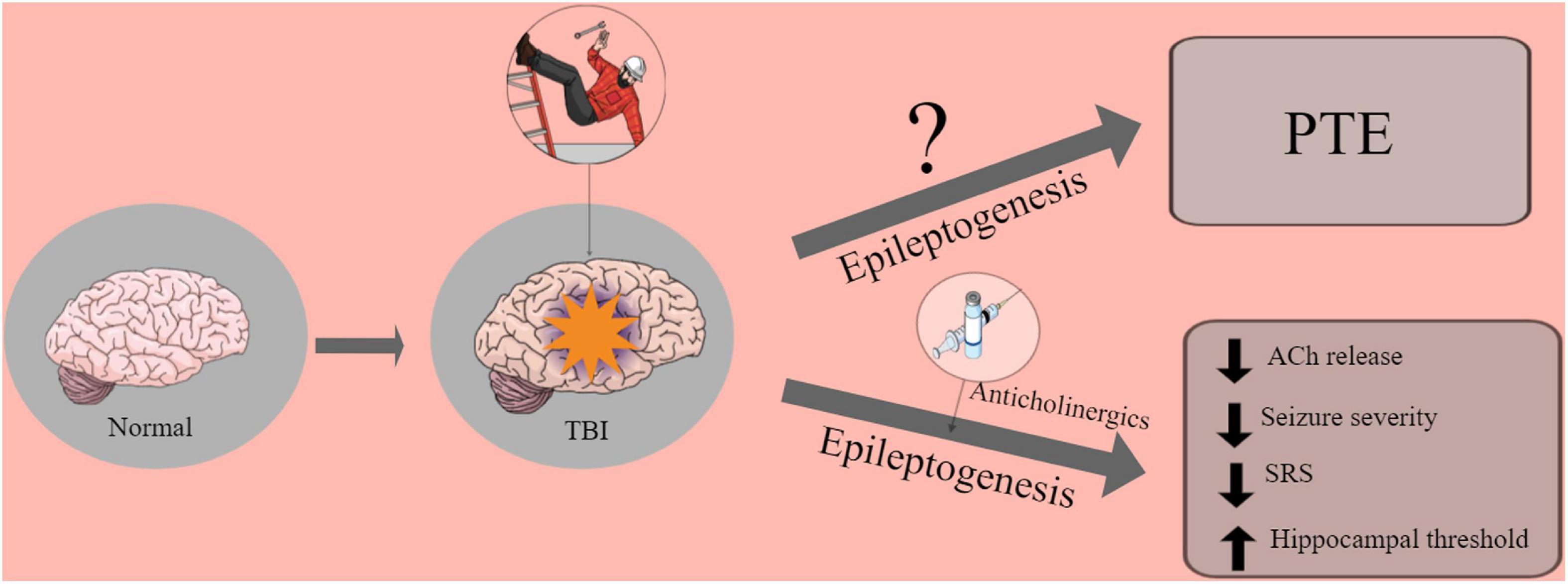
Figure 1. Graphic abstract of the role of anticholinergics drugs in the brain.
The mechanism underlying ACh hyperexcitability has been demonstrated by a stretch injury system applied to cerebral axons in vitro, inducing abnormal sodium influx through mechanically sensitive Na+ channels leading to increased intra-axonal calcium via the opening of voltage-gated calcium channels and reversal of the Na+-Ca2+ exchanger (Wolf et al., 2001; Arciniegas, 2003). Consequently, deformed axons were depolarized, and action potentials were propagated, leading to an excessive release of ACh or glutamate, excitotoxic to presynaptic and postsynaptic targets. Glutamate excitotoxicity may lead to excessive ACh and neuronal injury secondary release (Palmer et al., 1993). Moreover, TBI injury is related to blood-brain barrier damage, glial cell activation, and the release of pro- and anti-inflammatory cytokines (Korn et al., 2005; Pan et al., 2012). These pathological processes lead to excessive excitation of neurons and ultimately drive PTE.
In laboratory rats exposed to head trauma, the levels of ACh increased by 33% after TBI (Saija et al., 1988). Additionally, a study by Hillert et al. (2014) assessed changes in ACh release in the hippocampus before, during, and after SE via microdialysis in rats exposed to the lithium-pilocarpine model. Administration of a subconvulsant dose of pilocarpine increased hippocampal ACh release by six-fold, mainly in the striatum and hippocampus (Hillert et al., 2014).
Another study by Benassi et al. (2021) tested the relevance of the cholinergic system in epileptogenesis through a sophisticated experiment in which 192 IgG-saporin (a toxin that selectively kills cholinergic basal forebrain neurons) was injected into the right lateral ventricle, followed by induction of SE with subconvulsant doses of either pilocarpine or kainic acid. The results showed that 192 IgG-saporin-treated animals with a complete lesion of the cholinergic basal forebrain, did not exhibit SRS over 10 weeks after SE. Thus, this study showed that basal forebrain ACh release was relevant in epileptogenesis as a modulator of hippocampal transmission (Benassi et al., 2021).
These studies indicate that cholinergic innervation is essential for the onset and propagation of epileptic seizures, seizure-induced brain damage, and epileptogenesis, irrespective of the mechanism of seizure induction, at least in rodent models of epilepsy (for review, see Friedman et al., 2007).
Reliable animal models are essential to understand how the progression of trauma contributes to epileptogenesis in PTE and to help develop antiepileptogenic treatment. Fluid percussion injury (FPI) and controlled cortical impact (CCI) are the most extensively used PTE models. In the FPI model, a precise pressure pulse is distinctly delivered in the animal dura mater using a closed hydraulic system (Pitkänen and McIntosh, 2006; Alder et al., 2011), followed by a chemoconvulsant as a second hit. In contrast, the CCI model produces brain injury using a pneumatic or electromagnetic impact to compress the exposed brain, causing brain injury (Scheff et al., 1997).
Along with the PTE models, conventional models, such as pilocarpine, kindling, pentylenetetrazol, and genetics models, have been extensively used to study epilepsy and investigate the prevention of this disease (Löscher, 2011; Meller et al., 2021). These animal models also play a crucial role in identifying cellular and epileptogenic mechanisms that are key to the discovery of novel treatments for PTE.
Many anticholinergics have been investigated and proposed as potential treatments for PTE using different animal models of epilepsy. Scopolamine and biperiden are the most well-known of these drugs (Eşkazan et al., 1999; Pereira et al., 2005; Bittencourt et al., 2017). Till now, no studies have evaluated the effects of cholinergic drugs in animal PTE models. Therefore, this section revisits the principal findings highlighting anticholinergics as a promising strategy to prevent PTE (see Table 1).
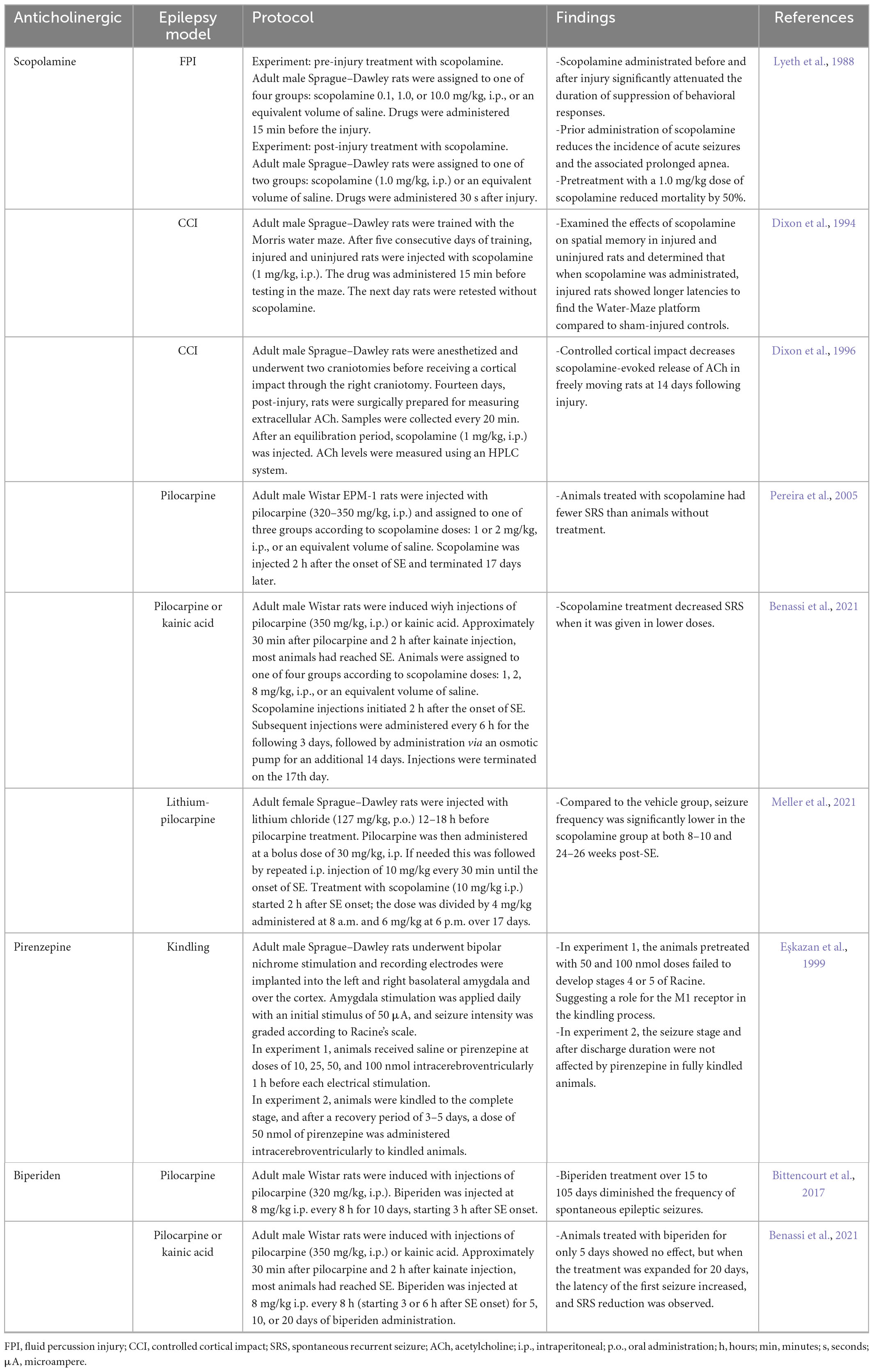
Table 1. Summary of laboratory studies using anticholinergics with animal models for posttraumatic epilepsy (PTE).
In Lyeth et al. (1988) performed different experiments with scopolamine (a high-affinity muscarinic antagonist) to attenuate transient behavioral suppression and physiological responses in rats that had experienced brain injury through the FPI model. They observed that transient behavioral suppression produced by moderate brain injury could be mitigated by prior administration of scopolamine (Lyeth et al., 1988).
Accordingly, Dixon et al. (1994) examined the effects of scopolamine on spatial memory by the Morris water maze test in uninjured and injured rats using the CCI model (see Table 1). Interestingly, when scopolamine was administered, the injured rats demonstrated longer latencies to find the hidden platform than the controls (which may be due to the adverse effects of the anticholinergic drug). This result led them to speculate that changes in cholinergic neurotransmission by receptor blockade rendered this system more vulnerable to the impact (Dixon et al., 1994). Similarly, 14 days post-brain injury, scopolamine evokes less ACh in neocortex and hippocampus of injured animals, which may propose a posttraumatic cognitive deficit (Dixon et al., 1996). However, in vivo study did not demonstrate that the site of action of scopolamine is presynaptic. Thus, muscarinic antagonists may block receptors located on the cell bodies of inhibitory interneurons, leading to increased brain ACh release at late TBI phase (Dixon et al., 1996).
Furthermore, a study by Pereira et al. (2005) evaluated the use of scopolamine after the onset of pilocarpine-induced SE. Although they did not observe any change in the behavioral characteristics of the ongoing SE, scopolamine was found to reduce SRS frequency by 50%, although it did not affect mossy fiber sprouting in the dentate gyrus. Therefore, scopolamine interferes with the epileptogenic process (Pereira et al., 2005). More importantly, this is the first report to show the effect of anticholinergic agents on the epileptogenic process, in which the tested compound was injected hours after the insult (not before).
Besides acting on acute provoked seizures, scopolamine has also shown encouraging results as an antiepileptogenic in two animal models of epilepsy: pilocarpine and kainic acid. More recently, the effects of scopolamine were also reported by Benassi et al. (2021), who validated different animal models of epilepsy and searched for the optimal dose for treatment (Benassi et al., 2021). Intriguingly, the results indicated that the lowest dose of scopolamine presented the best results in both models, considering the SRS occurrence. In contrast, higher-dose animals were indistinguishable from those that did not receive any scopolamine treatment in the SE group suggesting an inverted dose-effect curve (Benassi et al., 2021).
Another 2021 study by Meller et al. (2021) treated laboratory rats with scopolamine and compared it with vehicle control treatment (see Table 1). Scopolamine treatment during the latent period following lithium-pilocarpine-induced SE did not significantly reduce the number of rats with SRS 2 months post-SE. However, at 6 months post-SE, the number of rats with SRS was reduced considerably (>60%) (Meller et al., 2021), suggesting that prolonged treatment with scopolamine after lithium-pilocarpine-induced SE blocks muscarinic receptors during epileptogenesis and prevents hypersensitivity to ACh release (Meller et al., 2021).
Pirenzepine, a competitive M1 muscarinic, was also analyzed, and its administration was evaluated pre- and post-completely kindle laboratory rats. Anticholinergic effect was not observed in completely kindled animals but was involved in the epileptogenesis of the kindling model (Eşkazan et al., 1999).
Biperiden, another muscarinic antagonist widely prescribed for Parkinson’s disease, have also been investigated (Benassi et al., 2021). Bittencourt et al. (2017) performed an experiment in which rats were injected with pilocarpine and then administered biperiden (Table 1; Bittencourt et al., 2017). The authors concluded that biperiden treatment reduced the severity and number of SRS by elevating the threshold of hippocampal excitability (Bittencourt et al., 2017).
The duration of the blockade of cholinergic transmission necessary to suppress epileptogenesis was tested by Mello’s group. Biperiden was administered for 5, 10, or 20 days to determine the optimal treatment duration (Benassi et al., 2021). No effect was found in the group that received biperiden for only 5 days. However, the groups that received treatment for 10 and 20 days showed a significant increase in the latency for the first seizure and a substantial reduction in SRS frequency (Benassi et al., 2021). Additionally, animals treated with biperiden performed better in learning and memory tests after 3 months than those without treatment (Benassi et al., 2021). The authors proposed that biperiden decreased SRS incidence and may effectively reduce epilepsy after an insult.
Overall, essential differences between scopolamine and biperiden have been identified for their therapeutic applications in epileptic conditions. Scopolamine acts as a non-selective muscarinic antagonist that produces both peripheral and central antimuscarinic effects, such as sedative, antiemetic, and amnestic effects (Renner et al., 2005).
In contrast, biperiden is a relatively specific Ml antimuscarinic antagonist with weak peripheral anticholinergic action and with pronounced effects on the central nervous system, resulting in relevant properties for therapeutic application (DRUGBANK, 2022). Furthermore, biperiden is on the World Health Organization’s List of Essential Medicine (World Health Organization, 2021), making it an attractive potential antiepileptogenic treatment for PTE.
To date, no favorable results for avoiding epileptogenic processes have been obtained using anti-seizure drugs such as phenytoin, carbamazepine, or valproate. Their use is frequently recommended only during the first-week post-injury to suppress immediate and early seizures (Carney et al., 2017).
According to the ClinicalTrials.gov database, only six out of 12 active studies related to PTE are performing drug interventions. These include studies on levetiracetam (ClinicalTrials.gov Identifier: NCT01463033, a completed phase 2 study), [18F] DPA-714 (NCT03999164, ongoing only in phase 1), allopregnanolone (NCT01673828, completed phase 2), valproate sodium (NCT00004817, completed phase 3), and biperiden (NCT01048138, recruiting in phase 3; NCT04945213, not yet recruiting in phase 3).
Among these, biperiden is the only anticholinergic drug that has been investigated in a PTE clinical trial to evaluates its safety and efficacy. Although presenting dose- and concentration-dependent temporary declines in cognitive functioning (Bakker et al., 2021), biperiden was proved safe in a phase 2 study that tested its use after TBI, showing the protocol feasibility and possible prospective efficacy (Benassi et al., 2021; Foresti et al., 2022).
As mentioned earlier, PTE is likely to develop after a TBI of great severity; however, PTE does not necessarily occur in all cases (Löscher, 2020). Furthermore, epilepsy after TBI is potentially preventable because posttraumatic seizures that follow this injury have a high incidence of presenting only after several years (Christensen, 2015). This period between the trauma event and the putative appearance of seizures, offers an open window; if treated correctly, it might stop the epileptogenesis process.
Therefore, considering the predicted risk of epileptogenesis after TBI (Pitkänen et al., 2016), prevention of primary brain injury is key to preventing PTE. This aspect differentiates PTE from other epilepsies in which the causes and development of the disease remain unknown until the first seizure. Furthermore, prevention is vital, as patients with PTE are frequently pharmacoresistant and usually unsuitable surgical candidates (Garga and Lowenstein, 2006).
Posttraumatic epilepsy is a frequent consequence of TBI, depending on the trauma severity and internal and external risk factors. Interest in the cholinergic pathway for preventing PTE has recently grown because of the effectiveness of anticholinergics in modifying the epileptogenic process. Consequently, different anticholinergics have been studied in various laboratories. Scopolamine and biperiden have demonstrated promising results by reducing seizure severity and frequency, decreasing SRS, and increasing hippocampal threshold in different animal models. In particular, biperiden has drawn researchers’ attention and shown exciting results. To date, biperiden is the only anticholinergic drug studied in clinical trials and passed all safety and efficacy tests, raising expectations as the first antiepileptogenic drug. Additionally, considering the need to find effective strategies to prevent epileptic conditions, PTE seems potentially preventable compared to other types of epilepsies, since TBI has a latent period for interventions, thereby increasing the chance of a positive result. Future studies should clarify the anti-epileptogenic properties and overcome the challenge of preventing epileptic conditions caused by TBI.
VS wrote the first draft of the manuscript. VS, BL, and MF contributed to the article’s literature search, analysis, and conception. SR, MB, MN-M, and LM contributed to the revision of the manuscript. All authors approved and read the submitted version.
This work was supported by the Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP grant 2018/24561-5), Coordenação de Aperfeiçoamento de Pessoal de Nível Superior, Brazil (CAPES; Finance Code 001), and Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq; grant 311619/2019-3). VS (2022/10520-0) and SR (2017/05242-3) receive post-doctoral fellowships from São Paulo Research Foundation (FAPESP).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Alder, J., Fujioka, W., Lifshitz, J., Crockett, D., and Thakker-Varia, S. (2011). Lateral fluid percussion: Model of traumatic brain injury in mice. J. Vis. Exp. 54:3063. doi: 10.3791/3063
Annegers, J. F., Hauser, W. A., Coan, S. P., and Rocca, W. A. (1998). A population-based study of seizures after traumatic brain injuries. N. Engl. J. Med. 338, 8–12.
Arciniegas, D. B. (2003). The cholinergic hypothesis of cognitive impairment caused by traumatic brain injury. Curr. Psychiatry Rep. 5, 391–399. doi: 10.1007/s11920-003-0074-5
Arciniegas, D. B., Held, K., and Wagner, P. (2002). Cognitive impairment following TBI. Curr. Treat. Options Neurol. 4, 43–57. doi: 10.1007/s11940-002-0004-6
Bakker, C., Esdonk, M. J., Stuurman, R. F., Borghans, L. G., Kam, M. L., Gerven, J. M., et al. (2021). Biperiden challenge model in healthy elderly as proof-of-pharmacology tool: A randomized, placebo-controlled trial. J. Clin. Pharmacol. 61, 1466–1478. doi: 10.1002/jcph.1913
Benassi, S. K., Alves, J. G., Guidoreni, C. G., Massant, C. G., Queiroz, C. M., Garrido-Sanabria, E., et al. (2021). Two decades of research towards a potential first anti-epileptic drug. Seizure 90, 99–109. doi: 10.1016/j.seizure.2021.02.031
Bittencourt, A. S., Ferrazoli, E., Valente, M., Romariz, S., Janisset, N., Macedo, C., et al. (2017). Modification of the natural progression of epileptogenesis by means of biperiden in the pilocarpine model of epilepsy. Epilepsy Res. 138, 88–97. doi: 10.1016/j.eplepsyres.2017.10.019
Bohnen, N. I., and Albin, R. L. (2011). The cholinergic system in Parkinson’s disease. Behav. Brain Res. 221, 564–573. doi: 10.1016/j.bbr.2009.12.048.The
Carney, N., Totten, A. M., O’Reilly, C., Ullman, J. S., Hawryluk, G. W., Bell, M. J., et al. (2017). Guidelines for the management of severe traumatic brain injury, fourth edition. Neurosurgery 80, 6–15. doi: 10.1227/NEU.0000000000001432
Carteri, R. B. K., and da Silva, R. A. (2021). Traumatic brain injury hospital incidence in Brazil: An analysis of the past 10 years. Rev. Bras. Ter. Intensiva 33, 282–289. doi: 10.5935/0103-507X.20210036
Cavalheiro, E. A., Fernandes, M. J., Turski, L., and Naffah-Mazzacoratti, M. G. (1994). Spontaneous recurrent seizures in rats: Amino acid and monoamine determination in the hippocampus. Epilepsia 35, 1–11. doi: 10.1111/j.1528-1157.1994.tb02905.x
Christensen, J. (2015). The epidemiology of posttraumatic epilepsy. Semin. Neurol. 35, 218–222. doi: 10.1055/s-0035-1552923
de Almeida, C. E., Filho, J. L., Dourado, J. C., Gontijo, P. A., Dellaretti, M. A., and Costa, B. S. (2016). Traumatic brain injury epidemiology in Brazil. World Neurosurg. 87, 540–547. doi: 10.1016/j.wneu.2015.10.020
Diaz-Arrastia, R., Agostini, M. A., Frol, A. B., Mickey, B., Fleckenstein, J., Bigio, E., et al. (2000). Neurophysiologic and neuroradiologic features of intractable epilepsy after traumatic brain injury in adults. Arch. Neurol. 57, 1611–1616. doi: 10.1001/archneur.57.11.1611
Dixon, C. E., Bao, J., Long, D. A., and Hayes, R. L. (1996). Reduced evoked release of acetylcholine in the rodent hippocampus following traumatic brain injury. Pharmacol. Biochem. Behav. 53, 679–686. doi: 10.1016/0091-3057(95)02069-1
Dixon, C. E., Hamm, R. J., Taft, W. C., and Hayes, R. L. (1994). Increased anticholinergic sensitivity following closed skull impact and controlled cortical impact traumatic brain injury in the rat. J. Neurotrauma 11, 275–287. doi: 10.1089/neu.1994.11.275
Drever, B. D., Riedel, G., and Platt, B. (2011). The cholinergic system and hippocampal plasticity. Behav. Brain Res. 221, 505–514. doi: 10.1016/j.bbr.2010.11.037
Eşkazan, E., Aker, R., Onat, F., Köseoǧlu, S., Gören, M. Z., and Hasanoǧlu, A. (1999). Effect of pirenzepine, a muscarinic M1 receptor antagonist, on amygdala kindling in rat. Epilepsy Res. 37, 133–140. doi: 10.1016/S0920-1211(99)00040-6
Ferrante, R. J., Beal, M. F., Kowall, N. W., Jr, E. P., and Martin, J. B. (1987). Sparing of acetylcholinesterase-containing striatal neurons in Huntington’s disease. Brain Res. 411, 162–166. doi: 10.1016/0006-8993(87)90694-9
Fisher, R. S., Acevedo, C., Arzimanoglou, A., Bogacz, A., Cross, J. H., Elger, C. E., et al. (2014). ILAE official report: A practical clinical definition of epilepsy. Epilepsia 55, 475–482. doi: 10.1111/epi.12550
Foresti, M. L., Garzon, E., Pinheiro, C., Pacheco, R., Riera, R., Mello, L., et al. (2022). Biperiden for prevention of post-traumatic epilepsy: A protocol of a double-blinded placebo-controlled randomized clinical trial (biperiden trial). PLoS One 17:e0273584. doi: 10.1371/journal.pone.0273584
French, J. A., Williamson, P. D., Thadani, V. M., Darcey, T. M., Mattson, R. H., Spencer, S. S., et al. (1993). Characteristics of medial temporal lobe epilepsy: I. results of history and physical examination. Ann. Neurol. 34, 774–780. doi: 10.1002/ana.410340604
Friedman, A., Behrens, C. J., and Heinemann, U. (2007). Cholinergic dysfunction in temporal lobe epilepsy. Epilepsia 48(Suppl. 5) 126–130. doi: 10.1111/j.1528-1167.2007.01300.x
Garga, N., and Lowenstein, D. H. (2006). Posttraumatic epilepsy: A major problem in desperate need of major advances. Epilepsy Curr. 6, 1–5. doi: 10.1111/j.1535-7511.2005.00083.x
Golub, V. M., and Reddy, D. S. (2022). Post-traumatic epilepsy and comorbidities: Advanced models, molecular mechanisms, biomarkers, and novel therapeutic interventions. Pharmacol. Rev. 74, 387–438. doi: 10.1124/pharmrev.121.000375
Haltiner, A. M., Temkin, N. R., and Dikmen, S. S. (1997). Risk of seizure recurrence after the first late posttraumatic seizure. Arch. Phys. Med. Rehabil. 78, 835–840. doi: 10.1016/S0003-9993(97)90196-9
Hillert, M. H., Imran, I., Zimmermann, M., Lau, H., Weinfurter, S., and Klein, J. (2014). Dynamics of hippocampal acetylcholine release during lithium-pilocarpine-induced status epilepticus in rats. J. Neurochem. 131, 42–52. doi: 10.1111/jnc.12787
Hori, K., Konishi, K., Hanashi, T., Tani, M., Tomioka, H., Kitajima, Y., et al. (2015). Demonstrating the role of anticholinergic activity in a mood disorder. Neurodegener. Dis. 15, 175–181. doi: 10.1159/000381525
Hudak, A. M., Trivedi, K., Harper, C. R., Booker, K., Caesar, R. R., Agostini, M., et al. (2004). Evaluation of seizure-like episodes in survivors of moderate and severe traumatic brain injury. J. Head Trauma Rehabil. 19, 290–295. doi: 10.1097/00001199-200407000-00003
Jennett, W. B. (1969). Early traumatic epilepsy. Lancet 293, 1023–1025. doi: 10.1016/s0140-6736(69)91822-4
Korn, A., Golan, H., Melamed, I., Pascual-Marqui, R., and Friedman, A. (2005). Focal cortical dysfunction and blood-brain barrier disruption in patients with postconcussion syndrome. J. Clin. Neurophysiol. 22, 1–9. doi: 10.1097/01.wnp.0000150973.24324.a7
López-Álvarez, J., Sevilla-Llewellyn-Jones, J., and Agüera-Ortiz, L. (2019). Anticholinergic drugs in geriatric psychopharmacology. Front. Neurosci. 13:1309. doi: 10.3389/fnins.2019.01309
Löscher, W. (2011). Critical review of current animal models of seizures and epilepsy used in the discovery and development of new antiepileptic drugs. Seizure 20, 359–368.
Löscher, W. (2020). The holy grail of epilepsy prevention: Preclinical approaches to antiepileptogenic treatments. Neuropharmacology 167:107605. doi: 10.1016/j.neuropharm.2019.04.011
Löscher, W., Hirsch, L. J., and Schmidt, D. (2015). The enigma of the latent period in the development of symptomatic acquired epilepsy–traditional view versus new concepts. Epilepsy Behav. 52, 78–92. doi: 10.1016/j.yebeh.2015.08.037
Lowenstein, D. H. (2009). Epilepsy after head injury: An overview. Epilepsia 50(Suppl. 2) 4–9. doi: 10.1111/j.1528-1167.2008.02004.x
Lyeth, B. G., Dixon, C. E., Hamm, R. J., Jenkins, L. W., Young, H. F., Stonnington, H. H., et al. (1988). Effects of anticholinergic treatment on transient behavioral suppression and physiological responses following concussive brain injury to the rat. Brain Res. 448, 88–97. doi: 10.1016/0006-8993(88)91104-3
Maas, A. I. R., Menon, D., Adelson, P., Andelic, N., Bell, M., Belli, A., et al. (2017). Traumatic brain injury: Integrated approaches to improve prevention, clinical care, and research. Lancet Neurol. 16, 987–1048. doi: 10.1016/S1474-4422(17)30371-X
Maurer, S. V., and Williams, C. L. (2017). The cholinergic system modulates memory and hippocampal plasticity via its interactions with non-neuronal cells. Front. Immunol. 8:1489. doi: 10.3389/fimmu.2017.01489
Maxwell, W. L., Pennington, K., MacKinnon, M. A., Smith, D. H., McIntosh, T. K., Wilson, J. T., et al. (2004). Differential responses in three thalamic nuclei in moderately disabled, severely disabled and vegetative patients after blunt head injury. Brain 127, 2470–2478. doi: 10.1093/brain/awh294
McGeachan, A. J., Hobson, E. V., Al-Chalabi, A., Stephenson, J., Chandran, S., Crawley, F., et al. (2017). A multicentre evaluation of oropharyngeal secretion management practices in amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Frontotemporal Degener. 18, 1–9. doi: 10.1080/21678421.2016.1221433
Meller, S., Käufer, C., Gailus, B., Brandt, C., and Löscher, W. (2021). Scopolamine prevents aberrant mossy fiber sprouting and facilitates remission of epilepsy after brain injury. Neurobiol. Dis. 158:105446. doi: 10.1016/j.nbd.2021.105446
Mello, L. E., Cavalheiro, E. A., Tan, A. M., Kupfer, W. R., Pretorius, J. K., Babb, T. L., et al. (1993). Circuit mechanisms of seizures in the pilorcarpine model of chronic epilepsy: Cell loss and mossy fiber sprouting. Epilepsia 34, 985–995. doi: 10.1111/j.1528-1157.1993.tb02123.x
Mesulam, M. M., Mufson, E., Levey, A., and Wainer, B. (1983). Cholinergic innervation of cortex by the basal forebrain: Cytochemistry and cortical connections of the septal area, diagonal band nuclei, nucleus basalis (Substantia innominata), and hypothalamus in the rhesus monkey. J. Comp. Neurol. 214, 170–197. doi: 10.1002/cne.902140206
National Institute of Neurological Disorders and Stroke (2022). 2021 AES/NINDS epilepsy research benchmarks. Bethesda, ML: National Institute of Neurological Disorders and Stroke.
Östberg, A., Virta, J., Rinne, J. O., Oikonen, V., Luoto, P., Någren, K., et al. (2011). Cholinergic dysfunction after traumatic brain injury: Preliminary findings from a PET study. Neurology 76, 1046–1050. doi: 10.1212/WNL.0b013e318211c1c4
Palmer, A. M., Marion, D. W., Botscheller, M. L., Swedlow, P. E., Styren, S. D., and DeKosky, S. T. (1993). Traumatic brain injury-induced excitotoxicity assessed in a controlled cortical impact model. J. Neurochem. 61, 2015–2024. doi: 10.1111/j.1471-4159.1993.tb07437.x
Pan, H., Wang, H., Wang, X., Zhu, L., and Mao, L. (2012). The absence of Nrf2 enhances NF-κB-dependent inflammation following scratch injury in mouse primary cultured astrocytes. Mediators Inflamm. 2012:217580. doi: 10.1155/2012/217580
Pavel, O., William, W., and Rolf, S. (2006). “Molecular mechanisms of synaptic function in the hippocampus: Neurotransmitter exocytosis and glutamatergic, GABAergic, and cholinergic transmission,” in The hippocampus book, eds P. Andersen et al. (Oxford University Press: Oxford), 243–296. doi: 10.1093/acprof:oso/9780195100273.003.0007
Pereira, H. A. A., Benassi, S. K., and Mello, L. E. (2005). Plastic changes and disease-modifying effects of scopolamine in the pilocarpine model of epilepsy in rats. Epilepsia 46(Suppl. 5) 118–124. doi: 10.1111/j.1528-1167.2005.01017.x
Pignataro, G., Maysami, S., Studer, F. E., Wilz, A., Simon, R. P., and Boison, D. (2008). Downregulation of hippocampal adenosine kinase after focal ischemia as potential endogenous neuroprotective mechanism. J. Cereb. Blood Flow Metab. 28, 17–23. doi: 10.1038/sj.jcbfm.9600499
Pitkänen, A., and McIntosh, T. K. (2006). Animal models of post-traumatic epilepsy. J. Neurotrauma 23, 241–261.
Pitkänen, A., Roivainen, R., and Lukasiuk, K. (2016). Development of epilepsy after ischaemic stroke. Lancet Neurol. 15, 185–197.
Renner, U. D., Oertel, R., and Kirch, W. (2005). Pharmacokinetics and pharmacodynamics in clinical use of scopolamine. Ther. Drug Monit. 27, 655–665. doi: 10.1097/01.ftd.0000168293.48226.57
Saija, A., Robinson, S. E., Lyeth, B. G., Dixon, C. E., Yamamoto, T., Clifton, G. L., et al. (1988). The effects of scopolamine and traumatic brain injury on central cholinergic neurons. J. Neurotrauma 5, 161–170. doi: 10.1089/neu.1988.5.161
Saletti, P. G., Ali, I., Casillas-Espinosa, P. M., Semple, B. D., Lisgaras, C. P., Mosh, S. L., et al. (2019). In search of antiepileptogenic treatments for post-traumatic epilepsy. Neurobiol. Dis. 123, 86–99. doi: 10.1016/j.nbd.2018.06.017
Saricicek, A., Esterlis, I., Maloney, K. H., Mineur, Y. S., Ruf, B. M., Muralidharan, A., et al. (2012). Persistent β2*-nicotinic acetylcholinergic receptor dysfunction in major depressive disorder. Am. J. Psychiatry 169, 851–859. doi: 10.1176/appi.ajp.2012.11101546
Scarr, E. (2012). Muscarinic receptors: Their roles in disorders of the central nervous system and potential as therapeutic targets. CNS Neurosci. Ther. 18, 369–379. doi: 10.1111/j.1755-5949.2011.00249.x
Scheff, S. W., Baldwin, S. A., Brown, R. W., and Kraemer, P. J. (1997). Morris water maze deficits in rats following traumatic brain injury: Lateral controlled cortical impact. J. Neurotrauma 14, 615–627. doi: 10.1089/neu.1997.14.615
Shin, S. S., and Dixon, C. E. (2015). Alterations in cholinergic pathways and therapeutic strategies targeting cholinergic system after traumatic brain injury. J. Neurotrauma 32, 1429–1440. doi: 10.1089/neu.2014.3445
Turski, W. A., Cavalheiro, E. A., Schwarz, M., Czuczwar, S. J., Kleinrok, Z., and Turski, L. (1983). Limbic seizures produced by pilocarpine in rats: Behavioural, electroencephalographic and neuropathological study. Behav. Brain Res. 9, 315–335.
Wang, Y., Tan, B., Wang, Y., and Chen, Z. (2021). Cholinergic signaling, neural excitability, and epilepsy. Molecules 26, 1–14. doi: 10.3390/molecules26082258
Wilcock, G. K., Esiri, M. M., Bowen, D. M., and Smith, C. C. (1982). Alzheimer’s disease. Correlation of cortical choline acetyltransferase activity with the severity of dementia and histological abnormalities. J. Neurol. Sci. 57, 407–417. doi: 10.1016/0022-510X(82)90045-4
Wolf, J. A., Stys, P. K., Lusardi, T., Meaney, D., and Smith, D. H. (2001). Traumatic axonal injury induces calcium influx modulated by tetrodotoxin-sensitive sodium channels. J. Neurosci. 21, 1923–1930. doi: 10.1523/jneurosci.21-06-01923.2001
World Health Organization (2021). WHO model list of essential medicines–22nd list, 2021. Geneva: World Health Organization.
Keywords: traumatic brain injury, epileptogenesis, prevention, pharmacological treatment, anticholinergics
Citation: Sanabria V, Romariz S, Braga M, Foresti ML, Naffah-Mazzacoratti MG, Mello LE and Longo BM (2023) Anticholinergics: A potential option for preventing posttraumatic epilepsy. Front. Neurosci. 16:1100256. doi: 10.3389/fnins.2022.1100256
Received: 16 November 2022; Accepted: 30 December 2022;
Published: 24 February 2023.
Edited by:
Christos Panagiots Lisgaras, New York University, United StatesReviewed by:
Anna Maslarova, Langone Medical Center, New York University, United StatesCopyright © 2023 Sanabria, Romariz, Braga, Foresti, Naffah-Mazzacoratti, Mello and Longo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Beatriz M. Longo,  YmlhbW9udGVpcm9uZXVyb0BnbWFpbC5jb20=, YmVhdHJpei5sb25nb0B1bmlmZXNwLmJy
YmlhbW9udGVpcm9uZXVyb0BnbWFpbC5jb20=, YmVhdHJpei5sb25nb0B1bmlmZXNwLmJy
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.