 Xurui Shen
Xurui Shen Peixin Sun
Peixin Sun Hao Zhang
Hao Zhang Hanting Yang
Hanting Yang- Institute for Translational Brain Research, State Key Laboratory of Medical Neurobiology, MOE Frontiers Center for Brain Science, Department of Neurology, Zhongshan Hospital, Fudan University, Shanghai, China
The human brain has high energetic expenses and consumes over 20% of total oxygen metabolism. Abnormal brain energy homeostasis leads to various brain diseases. Among multiple factors that contribute to these diseases, mitochondrial dysfunction is one of the most common causes. Maintenance of mitochondrial integrity and functionality is of pivotal importance to brain energy generation. Mitochondrial quality control (MQC), employing the coordination of multiple mechanisms, is evolved to overcome many mitochondrial defects. Thus, not surprisingly, aberrant mitochondrial quality control results in a wide range of brain disorders. Targeting MQC to preserve and restore mitochondrial function has emerged as a promising therapeutic strategy for the prevention and treatment of brain diseases. Here, we set out to summarize the current understanding of mitochondrial quality control in brain homeostasis. We also evaluate potential pharmaceutically and clinically relevant targets in MQC-associated brain disorders.
Introduction
The brain is the most important and intricate component of the central nervous system (CNS). In addition to controlling how the body moves, it regulates higher neural activities including spirit, language, learning, memory, and consciousness. Brain damage causes reduced body function, such as memory loss, cognitive impairment, sensory deficits, and behavioral abnormalities. Brain disorders, from neurodegenerative to psychiatric illnesses, have drawn increasing attention in recent years (VanItallie, 2019; Xie et al., 2019; Oh et al., 2021; Wang et al., 2021).
In humans, the brain accounts for approximately 2% of the body weight, while it consumes over 20% of the body’s energy needs (Ambekar et al., 2021). It is well-known that mitochondria are the center of energy metabolism. They are essential for brain metabolism, development, and function. Although a variety of factors contribute to brain disorders, evidence postulates that mitochondrial dysfunction is one of the leading causes (Guntuku et al., 2016; Lan et al., 2022). Unhealthy and aged brains often show aberrant mitochondrial structures and excessive reactive oxygen species (ROS), which is related to many adult-onset brain diseases, ranging from injuries and infections to brain tumors and dementia (Figure 1; Sultana et al., 2011; Chouchani et al., 2014; Zorov et al., 2014; Cheng et al., 2020; Iranmanesh et al., 2021). Accordingly, the maintenance of mitochondrial homeostasis is crucial for brain function. Cell employed numerous strategies to coordinate protein and organellar quality control, including mechanisms to monitor the mitochondria. In this review, we discuss the pathways of mitochondrial quality control (MQC) and its role in the progression of brain diseases, and briefly summarized the known MQC-related potential drug targets.
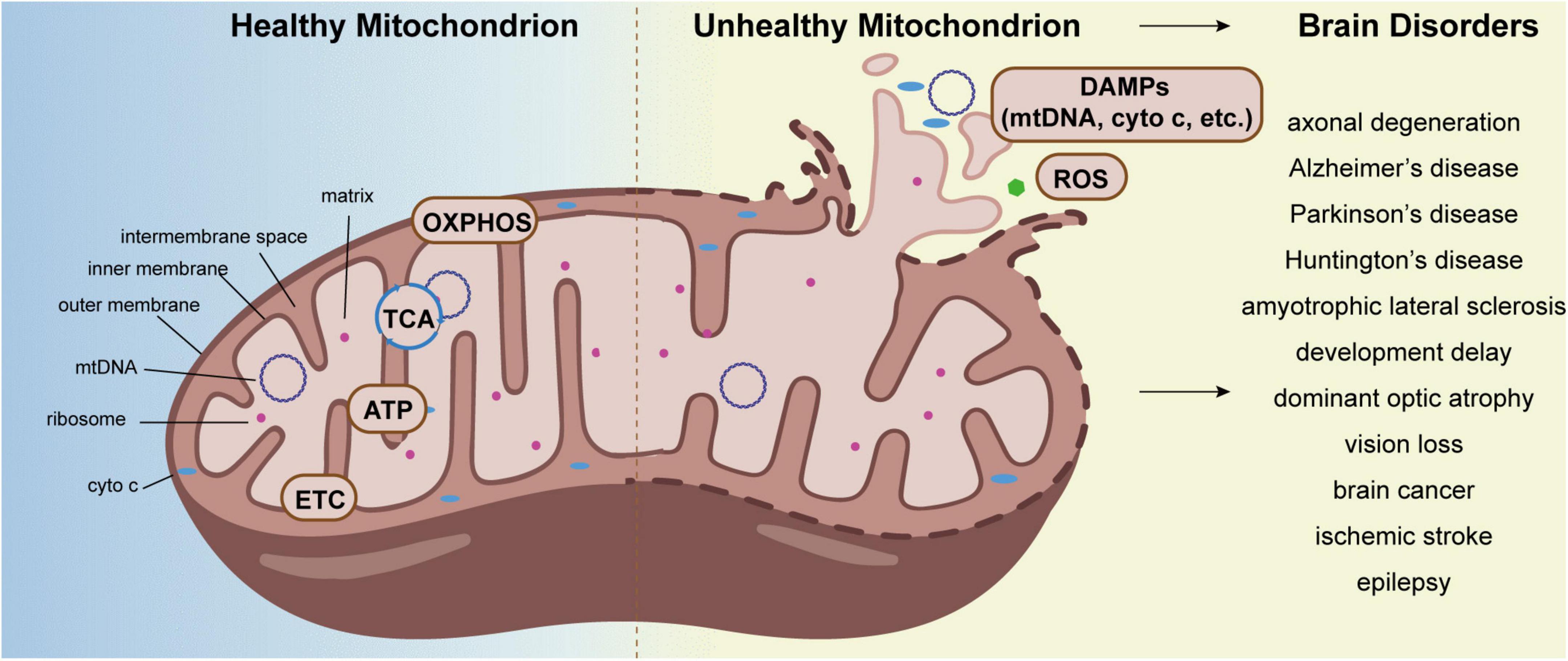
Figure 1. Aberrant mitochondrion causes brain disorders. The tricarboxylic acid cycle (TCA cycle), oxidative phosphorylation (OXPHOS), electron transport chain (ETC), and ATP synthesis all take place primarily in mitochondria. Under stress, mitochondria produce DAMPs and excessive ROS, which cause a variety of brain disorders, including axonal degeneration, neurodegeneration diseases, dominant optic atrophy, epilepsy, and so on.
Physiological functions of mitochondria in the brain
ATP production, metabolism, and oxidative phosphorylation
Mitochondria participate in energy and free radicals production, cell metabolism, cell death, and inflammation in the brain (Martin, 2010; Yin et al., 2016; Stefanatos and Sanz, 2018; Bader and Winklhofer, 2020). Mitochondria are the primary sites of ATP production as well as catabolic biochemical processes such as glycolysis, tricarboxylic acid (TCA) cycle, and oxidative phosphorylation (OXPHOS). At synapses, neurons in the brain exchange chemical and electrical signals with one another. Maintaining electrochemical gradients, liberating and recycling synaptic vesicles, and other very energy-intensive procedures rely on mitochondrial ATP synthesis (Devine and Kittler, 2018). It has been predicted that axonal terminals consume 4.5 × 108 ATP during an action potential (and downstream synaptic events), compared to 3 × 106 ATP used by resting potentials and housekeeping (Harris et al., 2012). The ATP-dependent membrane pumps, such as Na+/K+ ATPase and Ca2+ ATPase, are powered by about 55% of the total ATP produced by neurons in order to maintain the resting potential by resetting ionic gradients (Harris and Attwell, 2012). In addition, synaptic vesicle recycling also consumes a significant amount of energy. Each glutamate synaptic vesicle recycling event requires more than 2 × 104 ATP molecules, and in order to restore ionic gradients at a steady state, 1 × 106 ATP molecules must be restored within each individual neuron terminal (Rangaraju et al., 2014). Besides that, the process of cargo transportation along axons, which is carried out by motors, kinesins, and cytoplasmic dynein, is also ATP-dependent (Gibbs et al., 2015). Therefore, mitochondrial ATP synthesis is essential to keep the brain functioning normally.
Glucose serves as the main source of energy in neurons. Initially, glucose catabolism generates pyruvate, which is then transferred to mitochondria for TCA and OXPHOS (Mergenthaler et al., 2013). By combining electron transport with the phosphorylation of ADP on the inner mitochondrial membrane (IMM), OXPHOS produces ATP. NADH CoQ reductase (complex I), succinate dehydrogenase (complex II), ubiquinol-cytochrome c reductase (complex III), cytochrome c oxidase (complex IV), and ATP synthase (complex V) are all involved in the process (Sousa et al., 2018; Vercellino and Sazanov, 2022).
Free radicals
Free radicals, particularly ROS, are generated by mitochondria. ROS acts a significant role in the regulation of multiple neuronal cell life processes, including nucleic acid oxidation, immune response, and NF-κB pathway. The major source of free radicals, also known as “mitochondrial ROS,” is the electron transport chain (ETC). There is a strong correlation between the rate of ROS production, mitochondrial membrane potential (MMP), and the activity of the ETC complexes (Islam, 2017; Kalpage et al., 2019).
Cell death
To maintain organ size and function, mitochondria are required for cell death processes such as apoptosis, necroptosis, pyroptosis, and ferroptosis (Bock and Tait, 2020). The most widely understood mitochondria-related mechanism among them is apoptosis. Mitochondrial apoptosis also referred to as intrinsic apoptosis, is dependent on mitochondrial outer membrane permeabilization (MOMP). The procedure enables the release of proteins from the mitochondrial intermembrane space into the cytoplasm, which causes cell death. The establishment of functional circuitry, upkeep of healthy cell bodies and axons, promotion of myelination, and effective synaptic contact with target muscle are all facilitated by mitochondrial apoptosis in the brain (Buss et al., 2006; Fricker et al., 2018).
Neuroinflammation
Under certain stress conditions, the outer and inner membrane of mitochondria are damaged, and mitochondrial components such as mitochondrial DNA (mtDNA), formyl peptides, cytochrome c (cyto c) and cardiolipin are released into the cytoplasm, which is regarded as danger-associated molecular patterns (DAMPs), inducing the assembly and activation of the inflammasome, the release of cytokines and the elicitation of innate immune responses (Bader and Winklhofer, 2020). DAMPs released by mitochondria activate microglia in the brain, which represent the primary form of immune defense. In addition to oxidative stress, metabolism, and OXPHOS regulation, mitochondria play an important role in neuroinflammation (Regen et al., 2017; Gu et al., 2021; Zhao et al., 2021).
Mitochondrial quality control in the brain
Mitochondrial dysfunction causes various diseases in the brain. Mitochondria are semi-autonomous organelles, and their proteome includes about 1,500 human proteins, which are derived from the nuclear genome and mitochondrial genome (Morgenstern et al., 2017). Among them, only 13 proteins are encoded by the mitochondrial genome. About 99% of mitochondrial proteins are synthesized by cytosolic ribosomes, followed by sorted and imported to mitochondria. Mitochondria are the central sites for the development of the TCA and energy production, while also participates in cell metabolism, cell growth, cell death, inflammation, and cell homeostasis. Therefore, MQC mechanisms are essential to ensure proper protein folding and maintain a normal mitochondrial environment. The processes of MQC include mitochondrial morphology control (fission and fusion), macromitophagy (mitophagy), micromitophagy, and the mitochondrial protease system (Figure 2).
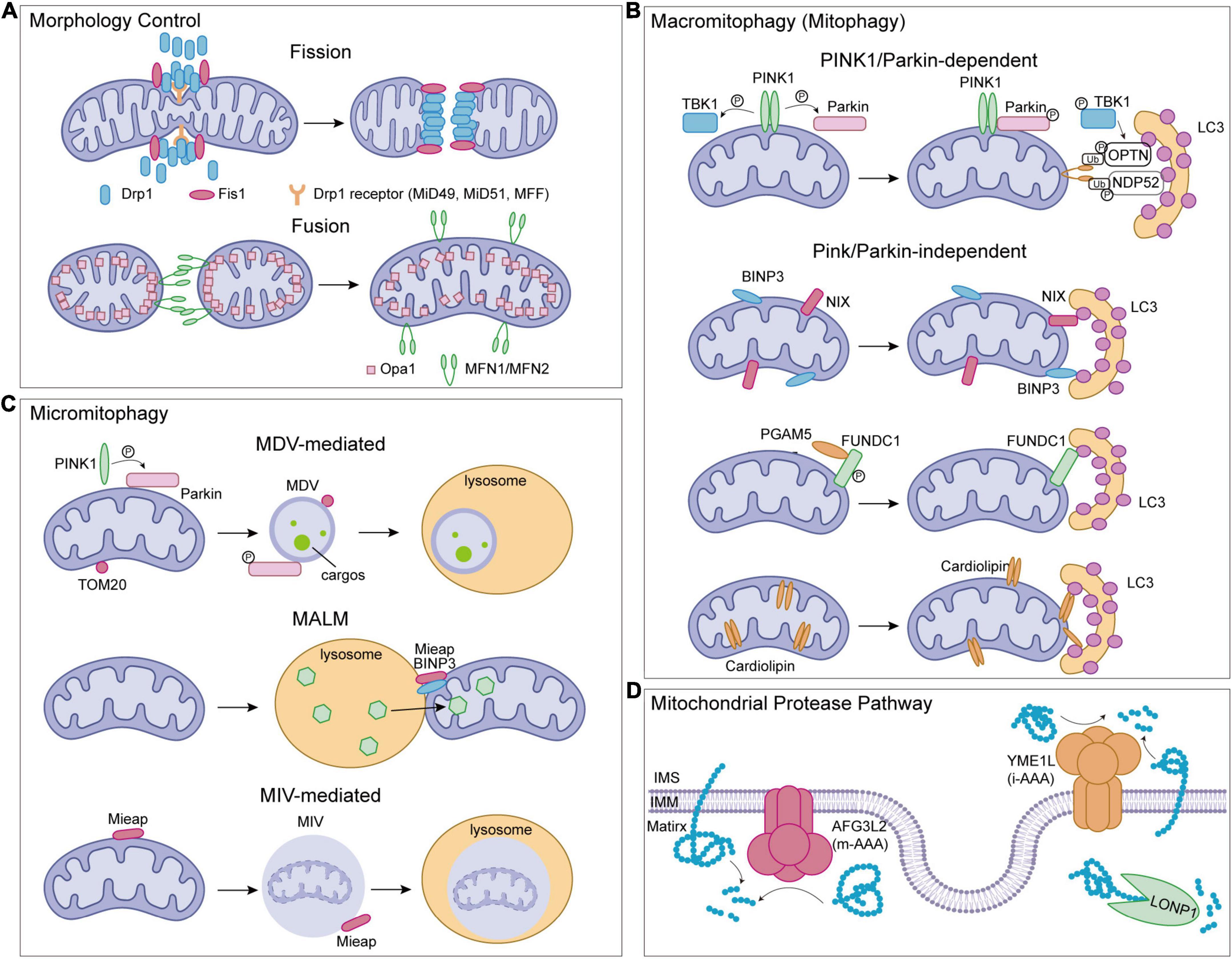
Figure 2. Mitochondrial quality control (MQC) pathways in human. (A) Mitochondrial morphology control. (B) Mitophagy pathways. (C) Micromitophagy pathways. (D) Mitochondrial protease pathways. Drp1, dynamic-related protein 1; Fis1, fission 1; MFF, mitochondrial fission factor; MiD49/MiD51, mitochondrial dynamics proteins 49/51; OPA1, optic atrophy 1; MFN1/MFN2, mitofusin1/mitofusin2; PINK1, serine/threonine-protein kinase PINK1; Parkin, E3 ubiquitin-protein ligase parkin; LC3, microtubule-associated protein light chain 3; TBK1, TANK binding kinase 1; NIX, NIP3-like protein X; BNIP3, Bcl-2/adenovirus E1B 19 kDa interacting protein 3; FUNDC1, FUN14 domain containing 1; PGAM5, phosphoglycerate mutase family member 5 phosphatase; Mieap, spermatogenesis-associated protein 18; TOM20, translocase of outer mitochondrial membrane 20; MDV, mitochondrial-derived vesicles; MALM, Mieap-induced accumulation of lysosome-like organelles within mitochondria; MIV, Mieap-induced vacuoles.
Mitochondrial morphology control
Mitochondria, as highly dynamic organelles, participate in calcium networks and apoptosis, which are coupled to molecular patterns signaling, amino acid and lipid metabolism, and cell death. Thus, the maintenance of mitochondrial integrity and homeostasis is critical, which is accomplished through continuous fusion and fission. The process by which two mitochondria fuse into one is known as mitochondrial fusion. Because of the double membranes, mitochondrial fusion includes both outer and inner membrane fusion. Three large dynamin-related GTP-hydrolyzing enzymes, mitofusin 1 (MFN1), mitofusin 2 (MFN2), and optic atrophy 1 (OPA1) are involved in the fusion process (Bertholet et al., 2016; Chan, 2020). In particular, MFN1 and MFN2 are localized on the outer mitochondrial membrane (OMM) and are required for outer membrane fusion (Santel and Fuller, 2001; Rojo et al., 2002; Koshiba et al., 2004). Trans interactions between mitofusin are commonly accepted to mediate the tethering of mitochondria during the fusion process because they are present on opposing mitochondrial membranes and form homo-oligomeric and heterooligomeric complexes for fusion. Models of outer membrane fusion have been proposed. The crystal structures of the MFN1 suggest that an intermolecular interface of the globular GTPase domains modulates membrane tethering (Cao et al., 2017; Yan et al., 2018), whereas another model indicates that the C-terminal domain is also needed (Koshiba et al., 2004; Franco et al., 2016). It has recently been suggested that mitochondrial fusion tethers outer membranes through nucleotide-dependent dimerization (Qi et al., 2016; Cao et al., 2017). Following outer membrane fusion, OPA1 mediates mitochondrial inner membrane fusion. OPA1 is found in two topologically distinct isoforms in different tissues due to alternative splicing and proteolytic processing by mitochondrial proteases OMA1 and YME1L (Xiao et al., 2014; Wai et al., 2015; Anderson et al., 2020). Long-form OPA1 (L-OPA1) and cardiolipin are sufficient to facilitate membrane fusion, and loss of OMA1 delays neurodegeneration by preventing stress-induced OPA1 cleavage processing in mitochondria (Korwitz et al., 2016; Ban et al., 2017). In the OPA1-null cells, mitochondria could only show mitochondrial outer membrane fusion but never progress to inner membrane fusion. In this case, mitochondria appear to fission (Song et al., 2009; Mishra et al., 2014).
Fission is undeniably important for mitochondrial division and quality control, and dynamic-related protein 1 (Drp1) plays a key role in this process. Three Drp1 receptors, mitochondrial fission factor (MFF), mitochondrial dynamics proteins 49 (MiD49), and mitochondrial dynamics proteins 51 (MiD51), are all involved in recruiting Drp1 from the cytoplasm to the OMM. A fission defect similar to Drp1 depletion is generated by the loss of any of the receptors, which causes the mitochondria to elongate noticeably (Losón et al., 2013; Osellame et al., 2016; Otera et al., 2016). Fission 1 (Fis1), another OMM-located protein, has also been shown to recruit Drp1. Overexpression of Fis1 in cells promotes mitochondrial fragmentation, however, deletion of the FIS1 gene has no effect on mitochondrial morphology or Drp1 recruitment to mitochondria (Yoon et al., 2003; Stojanovski et al., 2004; Otera et al., 2010). Drp1 undergoes structural changes after being recruited to mitochondria, constricting the mitochondrial tubule and inducing mitochondrial fission. The cryo-EM studies indicate that cardiolipin, a lipid enriched in mitochondrial membranes, can modulate the Drp1 structure and thus activate the fission process (Figure 2A; Francy et al., 2017).
Macromitophagy (Mitophagy)
Autophagy is an important quality control system in the nervous system. In mammals, three different types of autophagy processes have been described: macroautophagy, microautophagy, and chaperone-mediated autophagy (CMA). The primary mechanism of MQC in cells is the macroautophagic degradation of mitochondria, or mitophagy, which is necessary for basal mitochondrial turnover.
In the process of mitophagy, dysfunctional mitochondria are first detected, then separated from the mitochondrial network, and recruited by the mitophagosome. The mitophagosome structures are formed in the absence of the ATG8 family proteins, which are classified as microtubule-associated protein light chain 3 (LC3, including LC3A, LC3B, and LC3C) and GABARAP (GABARAP, GABARAP-L1, and GABARAP-L2) subfamilies. Fusion of mitophagosome with lysosomes for degradation is necessary for the last stage of the elimination of damaged mitochondria (Tsuboyama et al., 2016; Onishi et al., 2021). Four major mitophagy pathways include serine/threonine-protein kinase PINK1 (PINK1)/E3 ubiquitin-protein ligase parkin (Parkin)-mediated mitophagy, Bcl-2/adenovirus E1B 19 kDa interacting protein 3 (BNIP3)/NIP3-like protein X (NIX)-regulated mitophagy, FUN14 domain containing 1 (FUNDC1)-mediated mitophagy, and lipid-related pathways (Figure 2B). To trigger the degradation process, members of the ATG8 family should interact with all four pathways.
PINK1/Parkin-mediated mitophagy is the most well-known pathway. PINK1 is found on the IMM of normal mitochondria and is rapidly degraded by the mitochondrial membrane peptidase and presenilin-associated rhomboid-like protease (PARL) (Mokranjac and Neupert, 2007; Meissner et al., 2011). As a result, PINK1 remains at a low level under healthy conditions. However, when the inner membrane potential is depolarized, PINK1 moves to the OMM instead of IMM to form a dimer and is auto-phosphorylated at Ser228 and Ser402 residues (Okatsu et al., 2012). After being phosphorylated at Ser65 in its ubiquitin-like (Ubl) domain and activated by PINK1, Parkin, one of the E3 ubiquitin ligases, ubiquitinates its substrates like mitofusin (Kondapalli et al., 2012; Shiba-Fukushima et al., 2012; Koyano et al., 2014; Onishi et al., 2021). The autophagy adaptors, such as OPTN and NDP52, are phosphorylated by TANK-binding kinase 1 (TBK1), which recognizes these poly-ubiquitin chains, and binds with autophagy-related ATG8 family proteins via LIR motif, leading to mitophagy (Narendra et al., 2010; Kane et al., 2014; Kazlauskaite et al., 2014; Heo et al., 2015; Okatsu et al., 2015; Ordureau et al., 2015).
Unlike PINK1/Parkin, the BNIP3/NIX pathway is activated independently of changes in mitochondrial membrane potential (Rikka et al., 2011). In the normal state, BNIP3 is typically expressed as an inactive monomer in the cytoplasm, but under the hypoxia condition, BNIP3 is up-regulated, homodimerized, and anchored to the OMM by its C-terminal domain, while simultaneously exposing its N-terminal domain to the cytoplasm (Ray et al., 2000; Kubli et al., 2008; Hanna et al., 2012). At the N-terminal domain of BNIP3, the LC3-interacting region (LIR) motif recognizes and binds LC3, and mutations in the LIR motif prevent the contact with LC3, resulting in mitophagy abnormalities. Besides, phosphorylation at Ser17 and Ser24 near the LIR motif of BLIP3 is also important for BNIP3-LC3 interactions (Zhu et al., 2013). NIX is homology to BNIP3 and contains an LIR motif binding to ATG8 family members LC3A, LC3B, GABARAP, GABARAP-L1, and GABARAP-L2 among others (Hamacher-Brady et al., 2007; Sandoval et al., 2008; Novak et al., 2010). Ser34 and Ser35 of NIX, two serine residues close to the LIR motif, are phosphorylated similarly to BNIP3 in order to stabilize NIX-LC3 interactions and induce mitophagy (Rogov et al., 2017).
Additionally, the FUN14 domain containing 1 (FUNDC1) is also an OMM protein that has an LIR motif. It contains a characteristic LIR motif close to the N-terminus and three transmembrane domains (Liu et al., 2012). Phosphorylation and dephosphorylation on residues Ser13 and Tyr18 near the LIR motif of FUNDC1 regulate the process of mitophagy. Under hypoxia conditions, FUNDC1 interacts with LC3 via phosphoglycerate mutase family member 5 phosphatase (PGAM5) dephosphorylation at Ser13, and FUNDC1 phosphorylated by CK2 can reverse the effect of PGAM5 on mitophagy activation (Chen G. et al., 2014). In addition, SRC tyrosine kinase mediates the phosphorylation of Tyr18 to negatively regulate the FUNDC1-LC3 interactions field (Chen et al., 2016). Moreover, the phosphorylation of Ser17 in FUNDC1 by ULK1 enhanced the interaction between FUNDC1 and LC3, which could promote the mitophagy process (Wu et al., 2014).
Likewise, lipids including cardiolipin, cholesterol, and fatty acids have a role in the regulation of mitophagy. Among them, cardiolipin is a mitochondria-specific phospholipid located in IMM and is involved in receptor-mediated mitophagy in cells (Chu et al., 2013). When oxidized, cardiolipin is redistributed and translocated from IMM to OMM in damaged mitochondria and recognized by LC3. This process is coordinated by a hexameric intermembrane space protein, NDPK-D. The knockdown of endogenous NDPK-D decreases cardiolipin externalization and mitochondrial degradation (Kagan et al., 2016). Meanwhile, fatty acids could support the stability of PINK1 and translocation of the Parkin protein, participating in the regulation of mitophagy in the presence of PINK1. In the meantime, it has been shown that cholesterol has a dual role in PINK1/Parkin-mediated mitophagy (Roca-Agujetas et al., 2021).
Micromitophagy
Micromitophagy, which is defined as mitochondrial degradation independent of mitophagosomes, is another MQC mechanism that ensures mitochondrial homeostasis (Figure 2C; Wang et al., 2022). Under oxidative stress, mitochondria can generate mitochondrial-derived vesicles (MDVs) to proceed micromitophagy. In mammalian cells, MDVs formation is required OMM protein TOM20, or PINK1, Parkin, and soluble N-ethylmaleimide-sensitive factor attachment protein receptors (SNAREs). Recognition and initiation of micromitophagy are different from the mechanism mediated by canonical autophagy regulators such as ATG5 or LC3 (Soubannier et al., 2012; McLelland et al., 2014, 2016; König et al., 2021). Internalization of MDVs into the lysosomal lumen for degradation occurs following the formation of MDVs (Soubannier et al., 2012; Wang et al., 2022). An alternative micromitophagy mechanism known as spermatogenesis-associated protein 18 (SPATA18; also called Mieap)-induced accumulation of lysosome-like organelles within mitochondria (MALM) is characterized by the transfer of lysosomal proteins into mitochondria to destroy the oxidized mitochondrial protein. This process varies from MDVs uptaking microautophagic payloads into lysosomes (Kitamura et al., 2011; Nakamura et al., 2012). Under mitochondrial stress, Mieap is up-regulated and works with BNIP3 to promote MALM (Nakamura et al., 2012). When mitochondria are severely damaged or MALM is inhibited, Mieap-induced vacuoles (MIVs) uptake the entire damaged mitochondria into the lysosome for decay (Kitamura et al., 2011; Miyamoto et al., 2011). Although the micromitophagy processes have been characterized in yeast and some mammalian cells such as hepatocytes, its role in brain and brain-related diseases need to be further investigated (Kissová et al., 2007; Bhatia-Kiššová and Camougrand, 2010; Lemasters and Zhong, 2018).
Mitochondrial protease system
Mitochondrial proteome is encoded by both nuclear- and mitochondrial-genome. The fidelity and synchronization of these two protein synthesis systems are essential for ATP production and mitochondrial function. The nuclear-encoded proteins are synthesized by cytosolic ribosomes and imported into mitochondria by elaborate machinery embedded in the outer and inner mitochondrial membrane. During this coordination, the production of mitochondrial encoded proteins is regulated by the levels of imported proteins, preventing the redundant unassembled subunits (Ott et al., 2016; Priesnitz and Becker, 2018). The mitochondrial proteases could remove the unassembled proteins of the IMM. Furthermore, upon being imported into mitochondria, proteins synthesized by cytosolic ribosomes are monitored by mitochondrial proteases. Mitochondria have various mitoproteases, which can be divided into processing peptidases, ATP-dependent peptidases, and other mitochondrial peptidases (Deshwal et al., 2020). Among them, mitochondrial processing peptidases remove sorting signals from newly imported nuclear-encoded proteins, which is required for the maturation of many mitochondrial proteins (Mossmann et al., 2012; Poveda-Huertes et al., 2017; Deshwal et al., 2020). The mitochondrial processing protease MPP, for example, cleaves off mitochondrial targeting sequences (MTSs) in the matrix (Mach et al., 2013). Meanwhile, the inner membrane protease (IMMP) or ATP23 promotes the maturation of some proteins into the intermembrane space (IMS) (Weckbecker et al., 2012).
Another type of MQC-related protease is ATP-dependent proteases, which are the core consistency of the mitochondrial proteolytic system, activating in all mitochondrial compartments. The LONP1, caseinolytic mitochondrial matrix peptidase (CLPXP), m-AAA protease, and i-AAA protease are the four ATP-dependent proteases. LONP1 regulates mitochondrial oxidative phosphorylation (OXPHOS) via degrading damaged aconitase, and enzyme of the Krebs cycle in the mitochondrial matrix (Figure 2D; Bota and Davies, 2016). Besides, LONP1 modulates mitochondrial gene expression and some protein maturation (Lagouge et al., 2015; Zurita Rendón and Shoubridge, 2018). LONP1 also facilitates degrade COX4-1 to promote the assembly of the terminal electron transport chain (ETC) enzyme cytochrome c oxidase (Sepuri et al., 2017). Because of the critical role of LONP1 in mitochondrial functions, abnormal LONP1 causes a variety of diseases in humans. CLPXP, other than LONP1, is also reported to participate in the degradation of damaged OXPHOS complex I and II subunits (Seo et al., 2016). Meanwhile, CLPXP regulates gene expression by controlling the mitochondrial RNA (mtRNA) stability (Matsushima et al., 2017).
The m-AAA and i-AAA proteases are mitochondrial membrane-localized proteases (Figure 2D). They are necessary for the proteolysis of misfolded or damaged proteins and some IMM proteins, which helps to maintain the stability of mitochondria. In mammalian mitochondria, the m-AAA protease is composed of either an AFG3L2 homohexamer or an AFG3L2 and SPG7 heterohexamer. The hexameric AAA protease p97 (VCP) has a critical role in the degradation of outer mitochondrial membrane proteins, such as MFN1 (Xu et al., 2011), and the i-AAA protease YME1L removes translocase of the inner membrane 17A protein (TIM17A) to reduce protein import into mitochondria under stress and also regulates mitochondrial lipid composition by degradation of some lipid transfer proteins that shuttle phospholipids across the intermembrane space between the OMM and the IMM (Rainbolt et al., 2013; Saita et al., 2018). Additionally, the IMM proteases also regulate mitochondrial morphology by cleaving OPA1. Deletion of YME1L in the nervous system causes spinal cord axon degeneration in mice. Ablation of metalloproteinase OMA1, which is located on IMM, prevents neurodegeneration in YME1L-mutant mice, demonstrating the role of proteolytic processing in regulating mitochondrial function and physiology in the brain (Sprenger et al., 2019). In addition, the OMM-located ATPase family AAA domain-containing 1 (ATAD1) may promote the extraction and degradation of mislocalized tail-anchored (TA) proteins to preserve mitochondrial integrity, performing a crucial role in the regulation of synaptic activities in neurons (Chen Y. C. et al., 2014; Han et al., 2020).
Aberrant mitochondrial quality control and brain disorders
Mitochondrial dysfunction impairs mitochondrial respiration, energy generation, mitochondrial oxidative stress, and cell death (Martin, 2010; Yin et al., 2016; Stefanatos and Sanz, 2018; Bader and Winklhofer, 2020). Prior studies have largely focused on how abnormal MQC contributes to various neurological diseases. As an essential part of the nervous system, brain abnormality has always been linked to mitochondrial dysfunction. Here, we briefly outline some MQC disruption-related brain disorders and set out to summarize identified proteins in pathological pathways.
Neurodegenerative diseases
Neurodegenerative diseases, including Alzheimer’s disease (AD), Parkinson’s disease (PD), Huntington’s disease (HD), and amyotrophic lateral sclerosis (ALS), are characterized by the loss of selective neuron subtypes in the CNS. It has been demonstrated that the aberrant MQC plays a significant role in the progression of these diseases. If we take AD as an example, the mitochondrial fission and fusion proteins are disrupted in the hippocampus in various AD animal models and AD patients. The fission protein Fis1 is up-regulated, while fusion proteins MFN1, MFN2, and OPA1 are down-regulated (Wang et al., 2009). Moreover, Drp1 phosphorylation at Ser616 is higher in the brains of AD patients (Wang et al., 2009). Even though Parkin-mediated mitophagy was initially characterized in PD, the Parkin protein level has been shown higher and Parkin-mediated mitophagy is ineffective in the brains of AD patients (Ye et al., 2015). In addition, abnormal MQC and dysfunctional mitochondria are associated with chronic inflammation. It is well-known that, activated microglia function as innate immune cells in the CNS. In AD mice and patients, as observed, microglia stimulate phagocytosis, clearance, and degradation to minimize the accumulation of Aβ (Graeber et al., 2011). However, chronic microglia activation leads to the secretion of inflammatory cytokines and further neuronal dysfunction (Jiang et al., 2012). These findings suggest that aberrant MQC contributes to the development of neurodegenerative diseases.
Ischemic stroke
Ischemic stroke is one of the major diseases that cause death and disability of the nervous system. Although revascularization via reperfusion has led to a reduction in the mortality rate of ischemic stroke, the reperfusion itself also causes additional damage to the brain tissue, which is called ischemia-reperfusion (I/R) injury. It has been suggested that several MQC-related processes contribute to I/R damage. The removal of damaged mitochondria and mitochondrial apoptosis of neuron cells is aided by mitochondrial fission and mitophagy during cerebral I/R injury (Kumar et al., 2016; Zhao et al., 2018b). Overexpression of Sirtuin 3 (Sirt3) can inhibit mitochondrial fission and trigger pro-survival signals in neurons subjected to I/R injury (Zhao et al., 2018a). Furthermore, excessive mitochondrial fission stimulates energy imbalance and mtDNA damage, which worsens brain damage (Yue et al., 2015).
Epilepsy
Epilepsy is typified by recurrent unprovoked seizures of neurons. Mitochondrial stress and MQC are involved in the pathogenesis of epilepsy. The levels of MQC-related proteins OPA1, MFN2, MFF, and Drp1 are elevated in the mice models of acute maximal electroshock and 6 Hz 44 mA seizure (Cho et al., 2022). MQC-related serine peptidase LONP1 is up-regulated in the mitochondria during status epilepticus (SE), and LONP1 knockdown enhances SE-induced mitochondrial apoptosis in neuron (Kim et al., 2021).
Mitochondrial quality control-related gene mutation or abnormal expression
Mitochondrial quality control-related gene mutation or abnormal expression is related to brain dysplasia (Table 1). In cultured neurons, over-expression of MFN2 mutation disrupted axonal mitochondrial positioning and promoted axon degeneration (Detmer and Chan, 2007; Misko et al., 2012). Moreover, neuron-specific knockout mice show that MFN2 is required for dendritic outgrowth, axonal projection, and survival (Chen et al., 2003, 2007; Lee et al., 2012; Pham et al., 2012). It has been improved that OPA1 can protect neurons from excitotoxicity (Jahani-Asl et al., 2011; Nguyen et al., 2011; Kushnareva et al., 2013). Some research shows heterozygous OPA1 mutations both on the GTPase or GED domains lead to a decrease of protein quantity and mice of Opa1 heterozygous mutations have become autosomal dominant optic atrophy (DOA) models. These DOA mice perform retinal ganglionic cell (RGC) loss or dysfunction and optic nerve dysfunction including axonal degeneration and demyelination (Williams et al., 2011). Moreover, mutations of mitophagy-associated gene PARK6 (encoded PINK1) or PARK2 (encoded Parkin) contribute to autosomal recessive juvenile parkinsonism (Lücking et al., 1998; Valente et al., 2001, 2002, 2004).
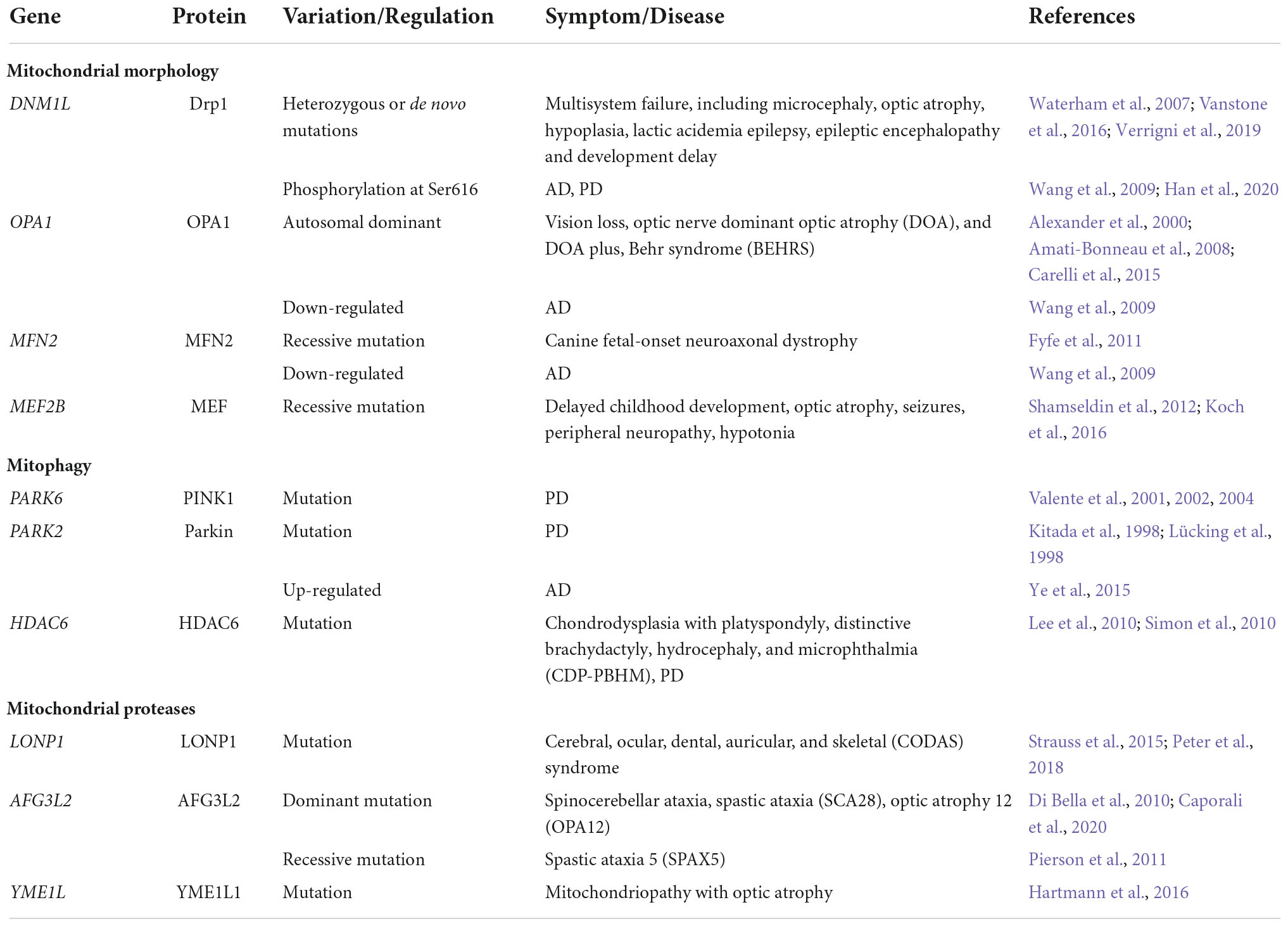
Table 1. Mitochondrial quality control gene abnormality-related diseases.
Potential of mitochondrial quality control-related targets for brain disorders
Mitochondrial quality control-related protein inhibitors and agonists have been recently shown to suppress pathological processes by regulating mitochondrial functions, including nervous system diseases, cardiovascular diseases, metabolic diseases, and cancer. In light of this, researchers are looking into whether MQC-related proteins can be drug targets for these diseases. For instance, a small molecule SIRT1 activator, SRT-1720, markedly improves renal tubular pathology and overall renal function in adult mice following I/R via regulating mitophagy (Fan et al., 2013). Mdivi-1, the inhibitor of Drp1, can induce apoptosis of hepatocellular carcinoma cells, suggesting a new approach of targeting MQC in cancer treatment (Akita et al., 2014; Wang et al., 2015; Lin et al., 2020). Thus, MQC-related proteins may become possible targets for disease treatment. Likewise, MQC is crucial for preserving healthy mitochondria and preventing the pathological effects of dysfunctional mitochondria in the brain. MQC-related targets, accordingly, represent potential future therapeutic strategies for brain diseases.
The mitochondrial morphology-associated protein Drp1 is highly expressed in the brain, implying that it is an important component in the brain. It has been demonstrated to modulate both the death of the neuronal cell and the survival of post-mitotic neurons (Cribbs and Strack, 2007; Kageyama et al., 2012). The inhibitor of Drp1, Mdvi-1, provides neuroprotection in vitro and in vivo (Cassidy-Stone et al., 2008; Park et al., 2011; Grohm et al., 2012). Pre- and post-treated AD or PD cells with Drp1 inhibitor Mdivi-1 show decreasing interaction between Drp1 and phosphorylated tau, reducing Aβ or α-syn aggregates, suppressing mitochondrial dysfunction, and maintaining cell viability, mitochondrial dynamics, mitochondrial biogenesis, and synaptic activity, indicating neuroprotection effects (Manczak and Reddy, 2012; Reddy et al., 2017; Wang et al., 2017). Meanwhile, inhibition of mitochondrial fission also suppresses the progress of ALS. The SOD1G93A mouse model is used for preclinical testing of treatments for ALS (Gurney et al., 1994). P100, which inhibits the interaction of Drp1 and Fis1, improves the mitochondrial structure and function by reducing oxidative stress in this model. Besides, P110 treatment also suppresses mitochondrial dysfunction in motor neurons and patient-derived fibroblasts, suggesting that Drp1 may be a drug target in ALS therapy strategy (Joshi et al., 2018). Furthermore, treatment with mitochondrial-targeted donor AP39 can transfer mitochondrial fission to fusion by increasing OPA1 and MFN1 levels and decreasing Fis1 levels in early-onset AD model APP/PS1 neurons and transgenic mice (Cassidy-Stone et al., 2008; Park et al., 2011; Grohm et al., 2012).
Mitophagy-associated proteins and mitochondrial proteases are also potential drug targets for brain diseases. Compound BC1464, which disrupts the FBXO7/PINK1 interaction, can rescue mitophagy and provides neuroprotection in PD models (Liu et al., 2020). The ATPase inhibitor, KUS121, improves the average readable letter counts, visual field scores, and retinal sensitivities of all nine patients with acute central retinal artery occlusion (CRAO) in phase I/II clinical trial (Hanako Ohashi et al., 2020). In the meanwhile, KUS121 shows the effect of preventing retinal ganglion cell death in animal models of glaucoma (Nakano et al., 2016).
As can be seen, these studies indicate that MQC-associated proteins are suitable as therapeutic pharmaceutical targets for brain disorders (Table 2).
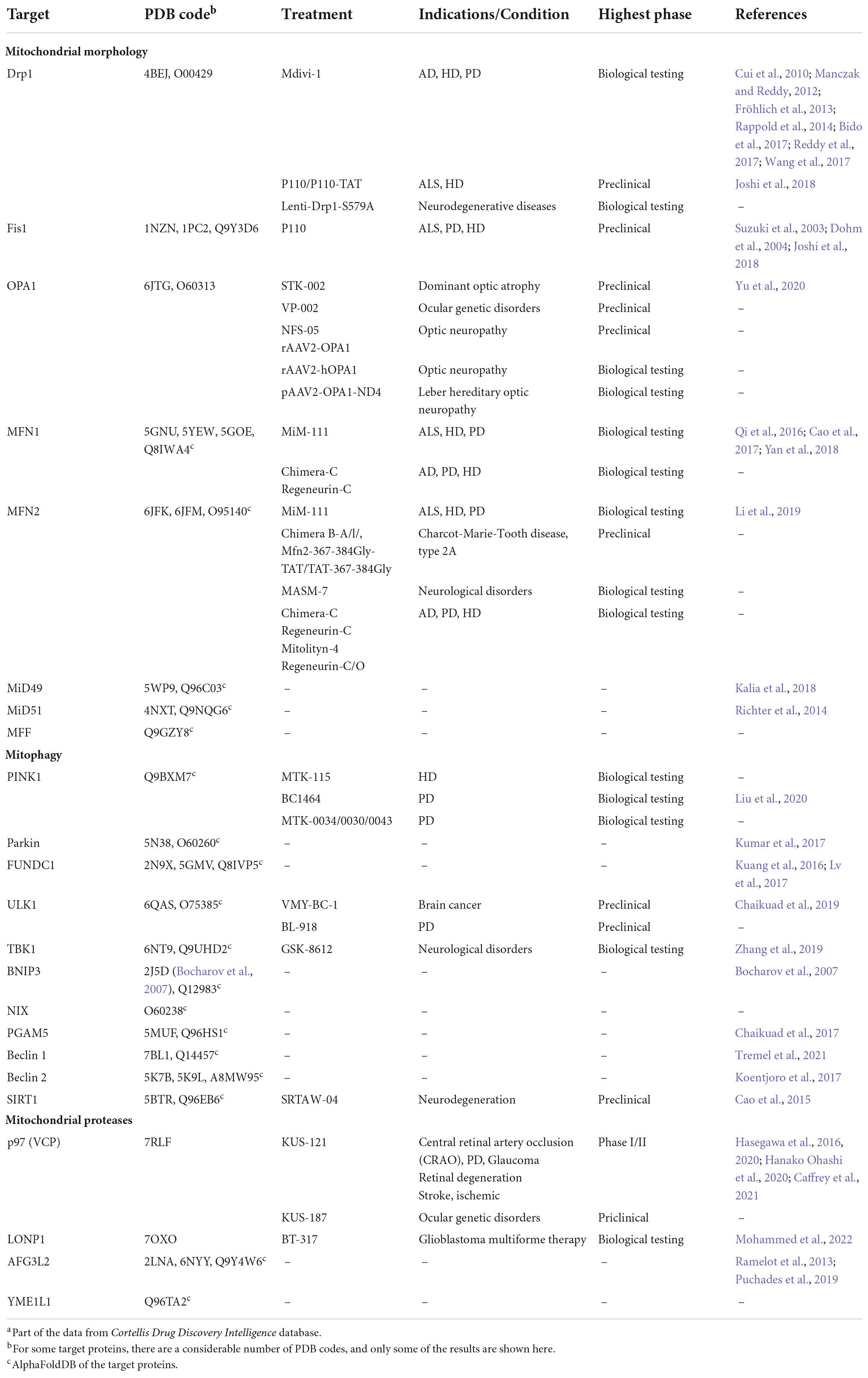
Table 2. Mitochondrial quality control (MQC)-associated proteins and therapeutic agents in brain diseases.a
Conclusion and perspective
In this review, we discuss the physiological roles of mitochondria and the MQC mechanism in the brain. MQC not only plays a vital role in maintaining mitochondrial morphology and functioning, but also participates in the pathological progression of a range of brain illnesses. Regulation of MQC through the pharmacological intervention of mitochondrial morphology, mitophagy, or the activity of mitochondrial proteases is emerging as a strategy for the treatment of mitochondrial-associated brain disorders.
Although MQC regulation can improve the process of brain disease, only a few regulators of MQC-related proteins have been identified as novel therapeutic targets or used in preclinical research (Table 2). Thus, there is still a lack of effective regulators, and developing targeted drugs is incredibly challenging. Because of the massive data sets available for drug candidates, computer-aided drug design (CADD) offers new approaches to efficacy and safety evaluations of drug candidates based on big data modeling, artificial intelligence modeling, and molecular docking (Zhu, 2020). This targeted drug development is dependent on the protein structure of the target (Yang et al., 2021). Some MQC-related essential proteins’ structures have been analyzed or predicted as structural biology and structure prediction methods have advanced, but PDB structures of full-length proteins under different conditions, as well as functional complexes, require further investigation (Table 2).
Finally, as we reviewed, although it is of bright prospects to develop MQC-related proteins as novel drug targets for brain disorders, the treatment of which still has a long way to go. Meanwhile, when some MQC-regulated drugs are in clinical trials, larger-scale clinical studies will be required to verify the safety and effectiveness of the drugs. Hence, in the future, more in-depth understanding of MQC would give rise to the development in the treatment of neurological related diseases, upon which more innovative therapeutic options will come to fruit.
Author contributions
HY and XS conceived the topic for this review. XS, PS, and HZ prepared the figures and tables. All authors listed wrote the manuscript and approved the submitted version.
Funding
This work was supported by grants from the National Natural Science Foundation of China (No. 32171216) and the Ministry of Science and Technology China Brain Initiative Project (No. 2022ZD0212600).
Acknowledgments
We thank all members of the Yang laboratory for helpful discussions.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Akita, M., Suzuki-Karasaki, M., Fujiwara, K., Nakagawa, C., Soma, M., Yoshida, Y., et al. (2014). Mitochondrial division inhibitor-1 induces mitochondrial hyperfusion and sensitizes human cancer cells to TRAIL-induced apoptosis. Int. J. Oncol. 45, 1901–1912. doi: 10.3892/ijo.2014.2608
Alexander, C., Votruba, M., Pesch, U. E., Thiselton, D. L., Mayer, S., Moore, A., et al. (2000). OPA1, encoding a dynamin-related GTPase, is mutated in autosomal dominant optic atrophy linked to chromosome 3q28. Nat. Genet. 26, 211–215. doi: 10.1038/79944
Amati-Bonneau, P., Valentino, M. L., Reynier, P., Gallardo, M. E., Bornstein, B., Boissière, A., et al. (2008). OPA1 mutations induce mitochondrial DNA instability and optic atrophy ‘plus’ phenotypes. Brain 131, 338–351. doi: 10.1093/brain/awm298
Ambekar, T., Pawar, J., Rathod, R., Patel, M., Fernandes, V., Kumar, R., et al. (2021). Mitochondrial quality control: Epigenetic signatures and therapeutic strategies. Neurochem. Int. 148:105095. doi: 10.1016/j.neuint.2021.105095
Anderson, C. J., Kahl, A., Fruitman, H., Qian, L., Zhou, P., Manfredi, G., et al. (2020). Prohibitin levels regulate OMA1 activity and turnover in neurons. Cell Death Differ. 27, 1896–1906. doi: 10.1038/s41418-019-0469-4
Bader, V., and Winklhofer, K. F. (2020). Mitochondria at the interface between neurodegeneration and neuroinflammation. Semin. Cell Dev. Biol. 99, 163–171. doi: 10.1016/j.semcdb.2019.05.028
Ban, T., Ishihara, T., Kohno, H., Saita, S., Ichimura, A., Maenaka, K., et al. (2017). Molecular basis of selective mitochondrial fusion by heterotypic action between OPA1 and cardiolipin. Nat. Cell Biol. 19, 856–863. doi: 10.1038/ncb3560
Bertholet, A. M., Delerue, T., Millet, A. M., Moulis, M. F., David, C., Daloyau, M., et al. (2016). Mitochondrial fusion/fission dynamics in neurodegeneration and neuronal plasticity. Neurobiol. Dis. 90, 3–19. doi: 10.1016/j.nbd.2015.10.011
Bhatia-Kiššová, I., and Camougrand, N. (2010). Mitophagy in yeast: Actors and physiological roles. FEMS Yeast Res. 10, 1023–1034. doi: 10.1111/j.1567-1364.2010.00659.x
Bido, S., Soria, F. N., Fan, R. Z., Bezard, E., and Tieu, K. (2017). Mitochondrial division inhibitor-1 is neuroprotective in the A53T-α-synuclein rat model of Parkinson’s disease. Sci. Rep. 7:7495. doi: 10.1038/s41598-017-07181-0
Bocharov, E. V., Pustovalova, Y. E., Pavlov, K. V., Volynsky, P. E., Goncharuk, M. V., Ermolyuk, Y. S., et al. (2007). Unique dimeric structure of BNip3 transmembrane domain suggests membrane permeabilization as a cell death trigger. J. Biol. Chem. 282, 16256–16266. doi: 10.1074/jbc.M701745200
Bock, F. J., and Tait, S. W. G. (2020). Mitochondria as multifaceted regulators of cell death. Nat. Rev. Mol. Cell Biol. 21, 85–100. doi: 10.1038/s41580-019-0173-8
Bota, D. A., and Davies, K. J. (2016). Mitochondrial Lon protease in human disease and aging: Including an etiologic classification of Lon-related diseases and disorders. Free Radic. Biol. Med. 100, 188–198. doi: 10.1016/j.freeradbiomed.2016.06.031
Buss, R. R., Gould, T. W., Ma, J., Vinsant, S., Prevette, D., Winseck, A., et al. (2006). Neuromuscular development in the absence of programmed cell death: Phenotypic alteration of motoneurons and muscle. J. Neurosci. 26, 13413–13427. doi: 10.1523/jneurosci.3528-06.2006
Caffrey, B., Zhu, X., Berezuk, A., Tuttle, K., Chittori, S., and Subramaniam, S. (2021). AAA+ ATPase p97/VCP mutants and inhibitor binding disrupt inter-domain coupling and subsequent allosteric activation. J. Biol. Chem. 297:101187. doi: 10.1016/j.jbc.2021.101187
Cao, D., Wang, M., Qiu, X., Liu, D., Jiang, H., Yang, N., et al. (2015). Structural basis for allosteric, substrate-dependent stimulation of SIRT1 activity by resveratrol. Genes Dev. 29, 1316–1325. doi: 10.1101/gad.265462.115
Cao, Y. L., Meng, S., Chen, Y., Feng, J. X., Gu, D. D., Yu, B., et al. (2017). MFN1 structures reveal nucleotide-triggered dimerization critical for mitochondrial fusion. Nature 542, 372–376. doi: 10.1038/nature21077
Caporali, L., Magri, S., Legati, A., Del Dotto, V., Tagliavini, F., Balistreri, F., et al. (2020). ATPase domain AFG3L2 mutations alter OPA1 processing and cause optic neuropathy. Ann. Neurol. 88, 18–32. doi: 10.1002/ana.25723
Carelli, V., Sabatelli, M., Carrozzo, R., Rizza, T., Schimpf, S., Wissinger, B., et al. (2015). ‘Behr syndrome’ with OPA1 compound heterozygote mutations. Brain 138:e321. doi: 10.1093/brain/awu234
Cassidy-Stone, A., Chipuk, J. E., Ingerman, E., Song, C., Yoo, C., Kuwana, T., et al. (2008). Chemical inhibition of the mitochondrial division dynamin reveals its role in Bax/Bak-dependent mitochondrial outer membrane permeabilization. Dev. Cell 14, 193–204. doi: 10.1016/j.devcel.2007.11.019
Chaikuad, A., Filippakopoulos, P., Marcsisin, S. R., Picaud, S., Schröder, M., Sekine, S., et al. (2017). Structures of PGAM5 provide insight into active site plasticity and multimeric assembly. Structure 25, 1089–1099.e3. doi: 10.1016/j.str.2017.05.020
Chaikuad, A., Koschade, S. E., Stolz, A., Zivkovic, K., Pohl, C., Shaid, S., et al. (2019). Conservation of structure, function and inhibitor binding in UNC-51-like kinase 1 and 2 (ULK1/2). Biochem. J. 476, 875–887. doi: 10.1042/bcj20190038
Chan, D. C. (2020). Mitochondrial dynamics and its involvement in disease. Annu. Rev. Pathol. 15, 235–259. doi: 10.1146/annurev-pathmechdis-012419-032711
Chen, G., Han, Z., Feng, D., Chen, Y., Chen, L., Wu, H., et al. (2014). A regulatory signaling loop comprising the PGAM5 phosphatase and CK2 controls receptor-mediated mitophagy. Mol. Cell 54, 362–377. doi: 10.1016/j.molcel.2014.02.034
Chen, H., Detmer, S. A., Ewald, A. J., Griffin, E. E., Fraser, S. E., and Chan, D. C. (2003). Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J. Cell Biol. 160, 189–200. doi: 10.1083/jcb.200211046
Chen, H., McCaffery, J. M., and Chan, D. C. (2007). Mitochondrial fusion protects against neurodegeneration in the cerebellum. Cell 130, 548–562. doi: 10.1016/j.cell.2007.06.026
Chen, M., Chen, Z., Wang, Y., Tan, Z., Zhu, C., Li, Y., et al. (2016). Mitophagy receptor FUNDC1 regulates mitochondrial dynamics and mitophagy. Autophagy 12, 689–702. doi: 10.1080/15548627.2016.1151580
Chen, Y. C., Umanah, G. K., Dephoure, N., Andrabi, S. A., Gygi, S. P., Dawson, T. M., et al. (2014). Msp1/ATAD1 maintains mitochondrial function by facilitating the degradation of mislocalized tail-anchored proteins. EMBO J. 33, 1548–1564. doi: 10.15252/embj.201487943
Cheng, X., Geng, F., Pan, M., Wu, X., Zhong, Y., Wang, C., et al. (2020). Targeting DGAT1 ameliorates glioblastoma by increasing fat catabolism and oxidative stress. Cell Metab. 32, 229–242.e8. doi: 10.1016/j.cmet.2020.06.002
Cho, C., Zeigler, M., Mizuno, S., Morrison, R. S., Totah, R. A., and Barker-Haliski, M. (2022). Reductions in hydrogen sulfide and changes in mitochondrial quality control proteins are evident in the early phases of the corneally kindled mouse model of epilepsy. Int. J. Mol. Sci. 23:1434. doi: 10.3390/ijms23031434
Chouchani, E. T., Pell, V. R., Gaude, E., Aksentijević, D., Sundier, S. Y., Robb, E. L., et al. (2014). Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 515, 431–435. doi: 10.1038/nature13909
Chu, C. T., Ji, J., Dagda, R. K., Jiang, J. F., Tyurina, Y. Y., Kapralov, A. A., et al. (2013). Cardiolipin externalization to the outer mitochondrial membrane acts as an elimination signal for mitophagy in neuronal cells. Nat. Cell Biol. 15, 1197–1205. doi: 10.1038/ncb2837
Cribbs, J. T., and Strack, S. (2007). Reversible phosphorylation of Drp1 by cyclic AMP-dependent protein kinase and calcineurin regulates mitochondrial fission and cell death. EMBO Rep. 8, 939–944. doi: 10.1038/sj.embor.7401062
Cui, M., Tang, X., Christian, W. V., Yoon, Y., and Tieu, K. (2010). Perturbations in mitochondrial dynamics induced by human mutant PINK1 can be rescued by the mitochondrial division inhibitor mdivi-1. J. Biol. Chem. 285, 11740–11752. doi: 10.1074/jbc.M109.066662
Deshwal, S., Fiedler, K. U., and Langer, T. (2020). Mitochondrial proteases: Multifaceted regulators of mitochondrial plasticity. Annu. Rev. Biochem. 89, 501–528. doi: 10.1146/annurev-biochem-062917-012739
Detmer, S. A., and Chan, D. C. (2007). Complementation between mouse Mfn1 and Mfn2 protects mitochondrial fusion defects caused by CMT2A disease mutations. J. Cell Biol. 176, 405–414. doi: 10.1083/jcb.200611080
Devine, M. J., and Kittler, J. T. (2018). Mitochondria at the neuronal presynapse in health and disease. Nat. Rev. Neurosci. 19, 63–80. doi: 10.1038/nrn.2017.170
Di Bella, D., Lazzaro, F., Brusco, A., Plumari, M., Battaglia, G., Pastore, A., et al. (2010). Mutations in the mitochondrial protease gene AFG3L2 cause dominant hereditary ataxia SCA28. Nat. Genet. 42, 313–321. doi: 10.1038/ng.544
Dohm, J. A., Lee, S. J., Hardwick, J. M., Hill, R. B., and Gittis, A. G. (2004). Cytosolic domain of the human mitochondrial fission protein fis1 adopts a TPR fold. Proteins 54, 153–156. doi: 10.1002/prot.10524
Fan, H., Yang, H. C., You, L., Wang, Y. Y., He, W. J., and Hao, C. M. (2013). The histone deacetylase, SIRT1, contributes to the resistance of young mice to ischemia/reperfusion-induced acute kidney injury. Kidney Int. 83, 404–413. doi: 10.1038/ki.2012.394
Franco, A., Kitsis, R. N., Fleischer, J. A., Gavathiotis, E., Kornfeld, O. S., Gong, G., et al. (2016). Correcting mitochondrial fusion by manipulating mitofusin conformations. Nature 540, 74–79. doi: 10.1038/nature20156
Francy, C. A., Clinton, R. W., Fröhlich, C., Murphy, C., and Mears, J. A. (2017). Cryo-EM studies of Drp1 reveal cardiolipin interactions that activate the helical oligomer. Sci. Rep. 7:10744. doi: 10.1038/s41598-017-11008-3
Fricker, M., Tolkovsky, A. M., Borutaite, V., Coleman, M., and Brown, G. C. (2018). Neuronal cell death. Physiol. Rev. 98, 813–880. doi: 10.1152/physrev.00011.2017
Fröhlich, C., Grabiger, S., Schwefel, D., Faelber, K., Rosenbaum, E., Mears, J., et al. (2013). Structural insights into oligomerization and mitochondrial remodelling of dynamin 1-like protein. EMBO J. 32, 1280–1292. doi: 10.1038/emboj.2013.74
Fyfe, J. C., Al-Tamimi, R. A., Liu, J., Schäffer, A. A., Agarwala, R., and Henthorn, P. S. (2011). A novel mitofusin 2 mutation causes canine fetal-onset neuroaxonal dystrophy. Neurogenetics 12, 223–232. doi: 10.1007/s10048-011-0285-6
Gibbs, K. L., Greensmith, L., and Schiavo, G. (2015). Regulation of axonal transport by protein kinases. Trends Biochem. Sci. 40, 597–610. doi: 10.1016/j.tibs.2015.08.003
Graeber, M. B., Li, W., and Rodriguez, M. L. (2011). Role of microglia in CNS inflammation. FEBS Lett. 585, 3798–3805. doi: 10.1016/j.febslet.2011.08.033
Grohm, J., Kim, S. W., Mamrak, U., Tobaben, S., Cassidy-Stone, A., Nunnari, J., et al. (2012). Inhibition of Drp1 provides neuroprotection in vitro and in vivo. Cell Death Differ. 19, 1446–1458. doi: 10.1038/cdd.2012.18
Gu, C., Wang, F., Zhang, Y. T., Wei, S. Z., Liu, J. Y., Sun, H. Y., et al. (2021). Microglial MT1 activation inhibits LPS-induced neuroinflammation via regulation of metabolic reprogramming. Aging Cell 20:e13375. doi: 10.1111/acel.13375
Guntuku, L., Naidu, V. G., and Yerra, V. G. (2016). Mitochondrial dysfunction in gliomas: Pharmacotherapeutic potential of natural compounds. Curr. Neuropharmacol. 14, 567–583. doi: 10.2174/1570159x14666160121115641
Gurney, M. E., Pu, H., Chiu, A. Y., Dal Canto, M. C., Polchow, C. Y., Alexander, D. D., et al. (1994). Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science 264, 1772–1775. doi: 10.1126/science.8209258
Hamacher-Brady, A., Brady, N. R., Logue, S. E., Sayen, M. R., Jinno, M., Kirshenbaum, L. A., et al. (2007). Response to myocardial ischemia/reperfusion injury involves Bnip3 and autophagy. Cell Death Differ. 14, 146–157. doi: 10.1038/sj.cdd.4401936
Han, H., Tan, J., Wang, R., Wan, H., He, Y., Yan, X., et al. (2020). PINK1 phosphorylates Drp1(S616) to regulate mitophagy-independent mitochondrial dynamics. EMBO Rep. 21:e48686. doi: 10.15252/embr.201948686
Hanako Ohashi, I., Muraoka, Y., Hata, M., Sumi, E., Ikeda, T., Nakagawa, T., et al. (2020). Safety and effectiveness of a novel neuroprotectant, KUS121, in patients with non-arteritic central retinal artery occlusion: An open-label, non-randomized, first-in-humans, phase 1/2 trial. PLoS One 15:e0229068. doi: 10.1371/journal.pone.0229068
Hanna, R. A., Quinsay, M. N., Orogo, A. M., Giang, K., Rikka, S., and Gustafsson, Å. B. (2012). Microtubule-associated protein 1 light chain 3 (LC3) interacts with Bnip3 protein to selectively remove endoplasmic reticulum and mitochondria via autophagy. J. Biol. Chem. 287, 19094–19104. doi: 10.1074/jbc.M111.322933
Harris, J. J., and Attwell, D. (2012). The energetics of CNS white matter. J. Neurosci. 32, 356–371. doi: 10.1523/JNEUROSCI.3430-11.2012
Harris, J. J., Jolivet, R., and Attwell, D. (2012). Synaptic energy use and supply. Neuron 75, 762–777. doi: 10.1016/j.neuron.2012.08.019
Hartmann, B., Wai, T., Hu, H., MacVicar, T., Musante, L., Fischer-Zirnsak, B., et al. (2016). Homozygous YME1L1 mutation causes mitochondriopathy with optic atrophy and mitochondrial network fragmentation. eLife 5:e16078. doi: 10.7554/eLife.16078
Hasegawa, T., Ikeda, H. O., Gotoh, N., Iida, K., Iwai, S., Nakano, N., et al. (2020). Effect of VCP modulators on gene expression profiles of retinal ganglion cells in an acute injury mouse model. Sci. Rep. 10:4251. doi: 10.1038/s41598-020-61160-6
Hasegawa, T., Muraoka, Y., Ikeda, H. O., Tsuruyama, T., Kondo, M., Terasaki, H., et al. (2016). Neuoroprotective efficacies by KUS121, a VCP modulator, on animal models of retinal degeneration. Sci. Rep. 6:31184. doi: 10.1038/srep31184
Heo, J. M., Ordureau, A., Paulo, J. A., Rinehart, J., and Harper, J. W. (2015). The PINK1-PARKIN mitochondrial ubiquitylation pathway drives a program of OPTN/NDP52 recruitment and TBK1 activation to promote mitophagy. Mol. Cell 60, 7–20. doi: 10.1016/j.molcel.2015.08.016
Iranmanesh, Y., Jiang, B., Favour, O. C., Dou, Z., Wu, J., Li, J., et al. (2021). Mitochondria’s role in the maintenance of cancer stem cells in glioblastoma. Front. Oncol. 11:582694. doi: 10.3389/fonc.2021.582694
Islam, M. T. (2017). Oxidative stress and mitochondrial dysfunction-linked neurodegenerative disorders. Neurol. Res. 39, 73–82. doi: 10.1080/01616412.2016.1251711
Jahani-Asl, A., Pilon-Larose, K., Xu, W., MacLaurin, J. G., Park, D. S., McBride, H. M., et al. (2011). The mitochondrial inner membrane GTPase, optic atrophy 1 (Opa1), restores mitochondrial morphology and promotes neuronal survival following excitotoxicity. J. Biol. Chem. 286, 4772–4782. doi: 10.1074/jbc.M110.167155
Jiang, T., Yu, J. T., and Tan, L. (2012). Novel disease-modifying therapies for Alzheimer’s disease. J. Alzheimers Dis. 31, 475–492. doi: 10.3233/jad-2012-120640
Joshi, A. U., Saw, N. L., Vogel, H., Cunnigham, A. D., Shamloo, M., and Mochly-Rosen, D. (2018). Inhibition of Drp1/Fis1 interaction slows progression of amyotrophic lateral sclerosis. EMBO Mol. Med. 10:e8166. doi: 10.15252/emmm.201708166
Kagan, V. E., Jiang, J., Huang, Z., Tyurina, Y. Y., Desbourdes, C., Cottet-Rousselle, C., et al. (2016). NDPK-D (NM23-H4)-mediated externalization of cardiolipin enables elimination of depolarized mitochondria by mitophagy. Cell Death Differ. 23, 1140–1151. doi: 10.1038/cdd.2015.160
Kageyama, Y., Zhang, Z., Roda, R., Fukaya, M., Wakabayashi, J., Wakabayashi, N., et al. (2012). Mitochondrial division ensures the survival of postmitotic neurons by suppressing oxidative damage. J. Cell Biol. 197, 535–551. doi: 10.1083/jcb.201110034
Kalia, R., Wang, R. Y., Yusuf, A., Thomas, P. V., Agard, D. A., Shaw, J. M., et al. (2018). Structural basis of mitochondrial receptor binding and constriction by DRP1. Nature 558, 401–405. doi: 10.1038/s41586-018-0211-2
Kalpage, H. A., Bazylianska, V., Recanati, M. A., Fite, A., Liu, J., Wan, J., et al. (2019). Tissue-specific regulation of cytochrome c by post-translational modifications: Respiration, the mitochondrial membrane potential, ROS, and apoptosis. FASEB J. 33, 1540–1553. doi: 10.1096/fj.201801417R
Kane, L. A., Lazarou, M., Fogel, A. I., Li, Y., Yamano, K., Sarraf, S. A., et al. (2014). PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J. Cell Biol. 205, 143–153. doi: 10.1083/jcb.201402104
Kazlauskaite, A., Kondapalli, C., Gourlay, R., Campbell, D. G., Ritorto, M. S., Hofmann, K., et al. (2014). Parkin is activated by PINK1-dependent phosphorylation of ubiquitin at Ser65. Biochem. J. 460, 127–139. doi: 10.1042/bj20140334
Kim, J. E., Park, H., Kim, T. H., and Kang, T. C. (2021). LONP1 regulates mitochondrial accumulations of HMGB1 and caspase-3 in CA1 and PV neurons following status epilepticus. Int. J. Mol. Sci. 22:2275. doi: 10.3390/ijms22052275
Kissová, I., Salin, B., Schaeffer, J., Bhatia, S., Manon, S., and Camougrand, N. (2007). Selective and non-selective autophagic degradation of mitochondria in yeast. Autophagy 3, 329–336. doi: 10.4161/auto.4034
Kitada, T., Asakawa, S., Hattori, N., Matsumine, H., Yamamura, Y., Minoshima, S., et al. (1998). Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392, 605–608. doi: 10.1038/33416
Kitamura, N., Nakamura, Y., Miyamoto, Y., Miyamoto, T., Kabu, K., Yoshida, M., et al. (2011). Mieap, a p53-inducible protein, controls mitochondrial quality by repairing or eliminating unhealthy mitochondria. PLoS One 6:e16060. doi: 10.1371/journal.pone.0016060
Koch, J., Feichtinger, R. G., Freisinger, P., Pies, M., Schrödl, F., Iuso, A., et al. (2016). Disturbed mitochondrial and peroxisomal dynamics due to loss of MFF causes Leigh-like encephalopathy, optic atrophy and peripheral neuropathy. J. Med. Genet. 53, 270–278. doi: 10.1136/jmedgenet-2015-103500
Koentjoro, B., Park, J. S., and Sue, C. M. (2017). Nix restores mitophagy and mitochondrial function to protect against PINK1/Parkin-related Parkinson’s disease. Sci. Rep. 7:44373. doi: 10.1038/srep44373
Kondapalli, C., Kazlauskaite, A., Zhang, N., Woodroof, H. I., Campbell, D. G., Gourlay, R., et al. (2012). PINK1 is activated by mitochondrial membrane potential depolarization and stimulates Parkin E3 ligase activity by phosphorylating Serine 65. Open Biol. 2:120080. doi: 10.1098/rsob.120080
König, T., Nolte, H., Aaltonen, M. J., Tatsuta, T., Krols, M., Stroh, T., et al. (2021). MIROs and DRP1 drive mitochondrial-derived vesicle biogenesis and promote quality control. Nat. Cell Biol. 23, 1271–1286. doi: 10.1038/s41556-021-00798-4
Korwitz, A., Merkwirth, C., Richter-Dennerlein, R., Tröder, S. E., Sprenger, H. G., Quirós, P. M., et al. (2016). Loss of OMA1 delays neurodegeneration by preventing stress-induced OPA1 processing in mitochondria. J. Cell Biol. 212, 157–166. doi: 10.1083/jcb.201507022
Koshiba, T., Detmer, S. A., Kaiser, J. T., Chen, H., McCaffery, J. M., and Chan, D. C. (2004). Structural basis of mitochondrial tethering by mitofusin complexes. Science 305, 858–862. doi: 10.1126/science.1099793
Koyano, F., Okatsu, K., Kosako, H., Tamura, Y., Go, E., Kimura, M., et al. (2014). Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature 510, 162–166. doi: 10.1038/nature13392
Kuang, Y., Ma, K., Zhou, C., Ding, P., Zhu, Y., Chen, Q., et al. (2016). Structural basis for the phosphorylation of FUNDC1 LIR as a molecular switch of mitophagy. Autophagy 12, 2363–2373. doi: 10.1080/15548627.2016.1238552
Kubli, D. A., Quinsay, M. N., Huang, C., Lee, Y., and Gustafsson, A. B. (2008). Bnip3 functions as a mitochondrial sensor of oxidative stress during myocardial ischemia and reperfusion. Am. J. Physiol. Heart Circ. Physiol. 295, H2025–H2031. doi: 10.1152/ajpheart.00552.2008
Kumar, A., Chaugule, V. K., Condos, T. E. C., Barber, K. R., Johnson, C., Toth, R., et al. (2017). Parkin-phosphoubiquitin complex reveals cryptic ubiquitin-binding site required for RBR ligase activity. Nat. Struct. Mol. Biol. 24, 475–483. doi: 10.1038/nsmb.3400
Kumar, R., Bukowski, M. J., Wider, J. M., Reynolds, C. A., Calo, L., Lepore, B., et al. (2016). Mitochondrial dynamics following global cerebral ischemia. Mol. Cell. Neurosci. 76, 68–75. doi: 10.1016/j.mcn.2016.08.010
Kushnareva, Y. E., Gerencser, A. A., Bossy, B., Ju, W. K., White, A. D., Waggoner, J., et al. (2013). Loss of OPA1 disturbs cellular calcium homeostasis and sensitizes for excitotoxicity. Cell Death Differ. 20, 353–365. doi: 10.1038/cdd.2012.128
Lagouge, M., Mourier, A., Lee, H. J., Spåhr, H., Wai, T., Kukat, C., et al. (2015). SLIRP regulates the rate of mitochondrial protein synthesis and protects LRPPRC from degradation. PLoS Genet. 11:e1005423. doi: 10.1371/journal.pgen.1005423
Lan, B., Zhao, H., He, Y., Zhao, Z., Wang, N., and Gao, Y. (2022). Inhibition of human peptide deformylase by actinonin sensitizes glioblastoma cells to temozolomide chemotherapy. Exp. Cell Res. 420:113358. doi: 10.1016/j.yexcr.2022.113358
Lee, J. Y., Nagano, Y., Taylor, J. P., Lim, K. L., and Yao, T. P. (2010). Disease-causing mutations in parkin impair mitochondrial ubiquitination, aggregation, and HDAC6-dependent mitophagy. J. Cell Biol. 189, 671–679. doi: 10.1083/jcb.201001039
Lee, S., Sterky, F. H., Mourier, A., Terzioglu, M., Cullheim, S., Olson, L., et al. (2012). Mitofusin 2 is necessary for striatal axonal projections of midbrain dopamine neurons. Hum. Mol. Genet. 21, 4827–4835. doi: 10.1093/hmg/dds352
Lemasters, J. J., and Zhong, Z. (2018). Mitophagy in hepatocytes: Types, initiators and role in adaptive ethanol metabolism ✩. Liver Res. 2, 125–132. doi: 10.1016/j.livres.2018.09.005
Li, Y. J., Cao, Y. L., Feng, J. X., Qi, Y., Meng, S., Yang, J. F., et al. (2019). Structural insights of human mitofusin-2 into mitochondrial fusion and CMT2A onset. Nat. Commun. 10:4914. doi: 10.1038/s41467-019-12912-0
Lin, X. H., Qiu, B. Q., Ma, M., Zhang, R., Hsu, S. J., Liu, H. H., et al. (2020). Suppressing DRP1-mediated mitochondrial fission and mitophagy increases mitochondrial apoptosis of hepatocellular carcinoma cells in the setting of hypoxia. Oncogenesis 9:67. doi: 10.1038/s41389-020-00251-5
Liu, L., Feng, D., Chen, G., Chen, M., Zheng, Q., Song, P., et al. (2012). Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat. Cell Biol. 14, 177–185. doi: 10.1038/ncb2422
Liu, Y., Lear, T. B., Verma, M., Wang, K. Z., Otero, P. A., McKelvey, A. C., et al. (2020). Chemical inhibition of FBXO7 reduces inflammation and confers neuroprotection by stabilizing the mitochondrial kinase PINK1. JCI Insight 5:e131834. doi: 10.1172/jci.insight.131834
Losón, O. C., Song, Z., Chen, H., and Chan, D. C. (2013). Fis1, Mff, MiD49, and MiD51 mediate Drp1 recruitment in mitochondrial fission. Mol. Biol. Cell 24, 659–667. doi: 10.1091/mbc.E12-10-0721
Lücking, C. B., Abbas, N., Dürr, A., Bonifati, V., Bonnet, A. M., de Broucker, T., et al. (1998). Homozygous deletions in parkin gene in European and North African families with autosomal recessive juvenile parkinsonism. The European consortium on genetic susceptibility in Parkinson’s disease and the French Parkinson’s disease genetics study group. Lancet 352, 1355–1356. doi: 10.1016/s0140-6736(05)60746-5
Lv, M., Wang, C., Li, F., Peng, J., Wen, B., Gong, Q., et al. (2017). Structural insights into the recognition of phosphorylated FUNDC1 by LC3B in mitophagy. Protein Cell 8, 25–38. doi: 10.1007/s13238-016-0328-8
Mach, J., Poliak, P., Matusková, A., Zárský, V., Janata, J., Lukes, J., et al. (2013). An advanced system of the mitochondrial processing peptidase and core protein family in Trypanosoma brucei and multiple origins of the core I subunit in eukaryotes. Genome Biol. Evol. 5, 860–875. doi: 10.1093/gbe/evt056
Manczak, M., and Reddy, P. H. (2012). Abnormal interaction between the mitochondrial fission protein Drp1 and hyperphosphorylated tau in Alzheimer’s disease neurons: Implications for mitochondrial dysfunction and neuronal damage. Hum. Mol. Genet. 21, 2538–2547. doi: 10.1093/hmg/dds072
Martin, L. J. (2010). Mitochondrial and cell death mechanisms in neurodegenerative diseases. Pharmaceuticals 3, 839–915. doi: 10.3390/ph3040839
Matsushima, Y., Hirofuji, Y., Aihara, M., Yue, S., Uchiumi, T., Kaguni, L. S., et al. (2017). Drosophila protease ClpXP specifically degrades DmLRPPRC1 controlling mitochondrial mRNA and translation. Sci. Rep. 7:8315. doi: 10.1038/s41598-017-08088-6
McLelland, G. L., Lee, S. A., McBride, H. M., and Fon, E. A. (2016). Syntaxin-17 delivers PINK1/parkin-dependent mitochondrial vesicles to the endolysosomal system. J. Cell Biol. 214, 275–291. doi: 10.1083/jcb.201603105
McLelland, G. L., Soubannier, V., Chen, C. X., McBride, H. M., and Fon, E. A. (2014). Parkin and PINK1 function in a vesicular trafficking pathway regulating mitochondrial quality control. EMBO J. 33, 282–295. doi: 10.1002/embj.201385902
Meissner, C., Lorenz, H., Weihofen, A., Selkoe, D. J., and Lemberg, M. K. (2011). The mitochondrial intramembrane protease PARL cleaves human Pink1 to regulate Pink1 trafficking. J. Neurochem. 117, 856–867. doi: 10.1111/j.1471-4159.2011.07253.x
Mergenthaler, P., Lindauer, U., Dienel, G. A., and Meisel, A. (2013). Sugar for the brain: The role of glucose in physiological and pathological brain function. Trends Neurosci. 36, 587–597. doi: 10.1016/j.tins.2013.07.001
Mishra, P., Carelli, V., Manfredi, G., and Chan, D. C. (2014). Proteolytic cleavage of Opa1 stimulates mitochondrial inner membrane fusion and couples fusion to oxidative phosphorylation. Cell Metab. 19, 630–641. doi: 10.1016/j.cmet.2014.03.011
Misko, A. L., Sasaki, Y., Tuck, E., Milbrandt, J., and Baloh, R. H. (2012). Mitofusin2 mutations disrupt axonal mitochondrial positioning and promote axon degeneration. J. Neurosci. 32, 4145–4155. doi: 10.1523/jneurosci.6338-11.2012
Miyamoto, Y., Kitamura, N., Nakamura, Y., Futamura, M., Miyamoto, T., Yoshida, M., et al. (2011). Possible existence of lysosome-like organella within mitochondria and its role in mitochondrial quality control. PLoS One 6:e16054. doi: 10.1371/journal.pone.0016054
Mohammed, I., Schmitz, K. A., Schenck, N., Balasopoulos, D., Topitsch, A., Maier, T., et al. (2022). Catalytic cycling of human mitochondrial Lon protease. Structure 30, 1254–1268.e7. doi: 10.1016/j.str.2022.06.006
Mokranjac, D., and Neupert, W. (2007). Protein import into isolated mitochondria. Methods Mol. Biol. 372, 277–286. doi: 10.1007/978-1-59745-365-3_20
Morgenstern, M., Stiller, S. B., Lübbert, P., Peikert, C. D., Dannenmaier, S., Drepper, F., et al. (2017). Definition of a high-confidence mitochondrial proteome at quantitative scale. Cell Rep. 19, 2836–2852. doi: 10.1016/j.celrep.2017.06.014
Mossmann, D., Meisinger, C., and Vögtle, F. N. (2012). Processing of mitochondrial presequences. Biochim. Biophys. Acta 1819, 1098–1106. doi: 10.1016/j.bbagrm.2011.11.007
Nakamura, Y., Kitamura, N., Shinogi, D., Yoshida, M., Goda, O., Murai, R., et al. (2012). BNIP3 and NIX mediate Mieap-induced accumulation of lysosomal proteins within mitochondria. PLoS One 7:e30767. doi: 10.1371/journal.pone.0030767
Nakano, N., Ikeda, H. O., Hasegawa, T., Muraoka, Y., Iwai, S., Tsuruyama, T., et al. (2016). Neuroprotective effects of VCP modulators in mouse models of glaucoma. Heliyon 2:e00096. doi: 10.1016/j.heliyon.2016.e00096
Narendra, D. P., Jin, S. M., Tanaka, A., Suen, D. F., Gautier, C. A., Shen, J., et al. (2010). PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 8:e1000298. doi: 10.1371/journal.pbio.1000298
Nguyen, D., Alavi, M. V., Kim, K. Y., Kang, T., Scott, R. T., Noh, Y. H., et al. (2011). A new vicious cycle involving glutamate excitotoxicity, oxidative stress and mitochondrial dynamics. Cell Death Dis. 2:e240. doi: 10.1038/cddis.2011.117
Novak, I., Kirkin, V., McEwan, D. G., Zhang, J., Wild, P., Rozenknop, A., et al. (2010). Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep. 11, 45–51. doi: 10.1038/embor.2009.256
Oh, H., Prevot, T. D., Newton, D., and Sibille, E. (2021). From serendipity to rational drug design in brain disorders: In silico, in vitro, and in vivo approaches. Curr. Opin. Pharmacol. 60, 177–182. doi: 10.1016/j.coph.2021.07.012
Okatsu, K., Koyano, F., Kimura, M., Kosako, H., Saeki, Y., Tanaka, K., et al. (2015). Phosphorylated ubiquitin chain is the genuine Parkin receptor. J. Cell Biol. 209, 111–128. doi: 10.1083/jcb.201410050
Okatsu, K., Oka, T., Iguchi, M., Imamura, K., Kosako, H., Tani, N., et al. (2012). PINK1 autophosphorylation upon membrane potential dissipation is essential for Parkin recruitment to damaged mitochondria. Nat. Commun. 3:1016. doi: 10.1038/ncomms2016
Onishi, M., Yamano, K., Sato, M., Matsuda, N., and Okamoto, K. (2021). Molecular mechanisms and physiological functions of mitophagy. EMBO J. 40:e104705. doi: 10.15252/embj.2020104705
Ordureau, A., Heo, J. M., Duda, D. M., Paulo, J. A., Olszewski, J. L., Yanishevski, D., et al. (2015). Defining roles of PARKIN and ubiquitin phosphorylation by PINK1 in mitochondrial quality control using a ubiquitin replacement strategy. Proc. Natl. Acad. Sci. U.S.A. 112, 6637–6642. doi: 10.1073/pnas.1506593112
Osellame, L. D., Singh, A. P., Stroud, D. A., Palmer, C. S., Stojanovski, D., Ramachandran, R., et al. (2016). Cooperative and independent roles of the Drp1 adaptors Mff, MiD49 and MiD51 in mitochondrial fission. J. Cell Sci. 129, 2170–2181. doi: 10.1242/jcs.185165
Otera, H., Miyata, N., Kuge, O., and Mihara, K. (2016). Drp1-dependent mitochondrial fission via MiD49/51 is essential for apoptotic cristae remodeling. J. Cell Biol. 212, 531–544. doi: 10.1083/jcb.201508099
Otera, H., Wang, C., Cleland, M. M., Setoguchi, K., Yokota, S., Youle, R. J., et al. (2010). Mff is an essential factor for mitochondrial recruitment of Drp1 during mitochondrial fission in mammalian cells. J. Cell Biol. 191, 1141–1158. doi: 10.1083/jcb.201007152
Ott, M., Amunts, A., and Brown, A. (2016). Organization and regulation of mitochondrial protein synthesis. Annu. Rev. Biochem. 85, 77–101. doi: 10.1146/annurev-biochem-060815-014334
Park, S. W., Kim, K. Y., Lindsey, J. D., Dai, Y., Heo, H., Nguyen, D. H., et al. (2011). A selective inhibitor of drp1, mdivi-1, increases retinal ganglion cell survival in acute ischemic mouse retina. Invest. Ophthalmol. Vis. Sci. 52, 2837–2843. doi: 10.1167/iovs.09-5010
Peter, B., Waddington, C. L., Oláhová, M., Sommerville, E. W., Hopton, S., Pyle, A., et al. (2018). Defective mitochondrial protease LonP1 can cause classical mitochondrial disease. Hum. Mol. Genet. 27, 1743–1753. doi: 10.1093/hmg/ddy080
Pham, A. H., Meng, S., Chu, Q. N., and Chan, D. C. (2012). Loss of Mfn2 results in progressive, retrograde degeneration of dopaminergic neurons in the nigrostriatal circuit. Hum. Mol. Genet. 21, 4817–4826. doi: 10.1093/hmg/dds311
Pierson, T. M., Adams, D., Bonn, F., Martinelli, P., Cherukuri, P. F., Teer, J. K., et al. (2011). Whole-exome sequencing identifies homozygous AFG3L2 mutations in a spastic ataxia-neuropathy syndrome linked to mitochondrial m-AAA proteases. PLoS Genet. 7:e1002325. doi: 10.1371/journal.pgen.1002325
Poveda-Huertes, D., Mulica, P., and Vögtle, F. N. (2017). The versatility of the mitochondrial presequence processing machinery: Cleavage, quality control and turnover. Cell Tissue Res. 367, 73–81. doi: 10.1007/s00441-016-2492-9
Priesnitz, C., and Becker, T. (2018). Pathways to balance mitochondrial translation and protein import. Genes Dev. 32, 1285–1296. doi: 10.1101/gad.316547.118
Puchades, C., Ding, B., Song, A., Wiseman, R. L., Lander, G. C., and Glynn, S. E. (2019). Unique structural features of the mitochondrial AAA+ protease AFG3L2 reveal the molecular basis for activity in health and disease. Mol. Cell 75, 1073–1085.e6. doi: 10.1016/j.molcel.2019.06.016
Qi, Y., Yan, L., Yu, C., Guo, X., Zhou, X., Hu, X., et al. (2016). Structures of human mitofusin 1 provide insight into mitochondrial tethering. J. Cell Biol. 215, 621–629. doi: 10.1083/jcb.201609019
Rainbolt, T. K., Atanassova, N., Genereux, J. C., and Wiseman, R. L. (2013). Stress-regulated translational attenuation adapts mitochondrial protein import through Tim17A degradation. Cell Metab. 18, 908–919. doi: 10.1016/j.cmet.2013.11.006
Ramelot, T. A., Yang, Y., Sahu, I. D., Lee, H. W., Xiao, R., Lorigan, G. A., et al. (2013). NMR structure and MD simulations of the AAA protease intermembrane space domain indicates peripheral membrane localization within the hexaoligomer. FEBS Lett. 587, 3522–3528. doi: 10.1016/j.febslet.2013.09.009
Rangaraju, V., Calloway, N., and Ryan, T. A. (2014). Activity-driven local ATP synthesis is required for synaptic function. Cell 156, 825–835. doi: 10.1016/j.cell.2013.12.042
Rappold, P. M., Cui, M., Grima, J. C., Fan, R. Z., de Mesy-Bentley, K. L., Chen, L., et al. (2014). Drp1 inhibition attenuates neurotoxicity and dopamine release deficits in vivo. Nat. Commun. 5:5244. doi: 10.1038/ncomms6244
Ray, R., Chen, G., Vande Velde, C., Cizeau, J., Park, J. H., Reed, J. C., et al. (2000). BNIP3 heterodimerizes with Bcl-2/Bcl-X(L) and induces cell death independent of a Bcl-2 homology 3 (BH3) domain at both mitochondrial and nonmitochondrial sites. J. Biol. Chem. 275, 1439–1448. doi: 10.1074/jbc.275.2.1439
Reddy, P. H., Manczak, M., and Yin, X. (2017). Mitochondria-division inhibitor 1 protects against amyloid-β induced mitochondrial fragmentation and synaptic damage in Alzheimer’s disease. J. Alzheimers Dis. 58, 147–162. doi: 10.3233/jad-170051
Regen, F., Hellmann-Regen, J., Costantini, E., and Reale, M. (2017). Neuroinflammation and Alzheimer’s disease: Implications for microglial activation. Curr. Alzheimer Res. 14, 1140–1148.
Richter, V., Palmer, C. S., Osellame, L. D., Singh, A. P., Elgass, K., Stroud, D. A., et al. (2014). Structural and functional analysis of MiD51, a dynamin receptor required for mitochondrial fission. J. Cell Biol. 204, 477–486. doi: 10.1083/jcb.201311014
Rikka, S., Quinsay, M. N., Thomas, R. L., Kubli, D. A., Zhang, X., Murphy, A. N., et al. (2011). Bnip3 impairs mitochondrial bioenergetics and stimulates mitochondrial turnover. Cell Death Differ. 18, 721–731. doi: 10.1038/cdd.2010.146
Roca-Agujetas, V., Barbero-Camps, E., de Dios, C., Podlesniy, P., Abadin, X., Morales, A., et al. (2021). Cholesterol alters mitophagy by impairing optineurin recruitment and lysosomal clearance in Alzheimer’s disease. Mol. Neurodegener. 16:15. doi: 10.1186/s13024-021-00435-6
Rogov, V. V., Suzuki, H., Marinković, M., Lang, V., Kato, R., Kawasaki, M., et al. (2017). Phosphorylation of the mitochondrial autophagy receptor Nix enhances its interaction with LC3 proteins. Sci. Rep. 7:1131. doi: 10.1038/s41598-017-01258-6
Rojo, M., Legros, F., Chateau, D., and Lombès, A. (2002). Membrane topology and mitochondrial targeting of mitofusins, ubiquitous mammalian homologs of the transmembrane GTPase Fzo. J. Cell Sci. 115, 1663–1674. doi: 10.1242/jcs.115.8.1663
Saita, S., Tatsuta, T., Lampe, P. A., König, T., Ohba, Y., and Langer, T. (2018). PARL partitions the lipid transfer protein STARD7 between the cytosol and mitochondria. EMBO J. 37:e97909. doi: 10.15252/embj.201797909
Sandoval, H., Thiagarajan, P., Dasgupta, S. K., Schumacher, A., Prchal, J. T., Chen, M., et al. (2008). Essential role for Nix in autophagic maturation of erythroid cells. Nature 454, 232–235. doi: 10.1038/nature07006
Santel, A., and Fuller, M. T. (2001). Control of mitochondrial morphology by a human mitofusin. J. Cell Sci. 114, 867–874. doi: 10.1242/jcs.114.5.867
Seo, J. H., Rivadeneira, D. B., Caino, M. C., Chae, Y. C., Speicher, D. W., Tang, H. Y., et al. (2016). The mitochondrial unfoldase-peptidase complex ClpXP controls bioenergetics stress and metastasis. PLoS Biol. 14:e1002507. doi: 10.1371/journal.pbio.1002507
Sepuri, N. B. V., Angireddy, R., Srinivasan, S., Guha, M., Spear, J., Lu, B., et al. (2017). Mitochondrial LON protease-dependent degradation of cytochrome c oxidase subunits under hypoxia and myocardial ischemia. Biochim. Biophys. Acta Bioenerget. 1858, 519–528. doi: 10.1016/j.bbabio.2017.04.003
Shamseldin, H. E., Alshammari, M., Al-Sheddi, T., Salih, M. A., Alkhalidi, H., Kentab, A., et al. (2012). Genomic analysis of mitochondrial diseases in a consanguineous population reveals novel candidate disease genes. J. Med. Genet. 49, 234–241. doi: 10.1136/jmedgenet-2012-100836
Shiba-Fukushima, K., Imai, Y., Yoshida, S., Ishihama, Y., Kanao, T., Sato, S., et al. (2012). PINK1-mediated phosphorylation of the Parkin ubiquitin-like domain primes mitochondrial translocation of Parkin and regulates mitophagy. Sci. Rep. 2:1002. doi: 10.1038/srep01002
Simon, D., Laloo, B., Barillot, M., Barnetche, T., Blanchard, C., Rooryck, C., et al. (2010). A mutation in the 3’-UTR of the HDAC6 gene abolishing the post-transcriptional regulation mediated by hsa-miR-433 is linked to a new form of dominant X-linked chondrodysplasia. Hum. Mol. Genet. 19, 2015–2027. doi: 10.1093/hmg/ddq083
Song, Z., Ghochani, M., McCaffery, J. M., Frey, T. G., and Chan, D. C. (2009). Mitofusins and OPA1 mediate sequential steps in mitochondrial membrane fusion. Mol. Biol. Cell 20, 3525–3532. doi: 10.1091/mbc.e09-03-0252
Soubannier, V., McLelland, G. L., Zunino, R., Braschi, E., Rippstein, P., Fon, E. A., et al. (2012). A vesicular transport pathway shuttles cargo from mitochondria to lysosomes. Curr. Biol. 22, 135–141. doi: 10.1016/j.cub.2011.11.057
Sousa, J. S., D’Imprima, E., and Vonck, J. (2018). Mitochondrial respiratory chain complexes. Subcell. Biochem. 87, 167–227. doi: 10.1007/978-981-10-7757-9_7
Sprenger, H. G., Wani, G., Hesseling, A., König, T., Patron, M., MacVicar, T., et al. (2019). Loss of the mitochondrial i-AAA protease YME1L leads to ocular dysfunction and spinal axonopathy. EMBO Mol. Med. 11:e9288. doi: 10.15252/emmm.201809288
Stefanatos, R., and Sanz, A. (2018). The role of mitochondrial ROS in the aging brain. FEBS Lett. 592, 743–758. doi: 10.1002/1873-3468.12902
Stojanovski, D., Koutsopoulos, O. S., Okamoto, K., and Ryan, M. T. (2004). Levels of human Fis1 at the mitochondrial outer membrane regulate mitochondrial morphology. J. Cell Sci. 117, 1201–1210. doi: 10.1242/jcs.01058
Strauss, K. A., Jinks, R. N., Puffenberger, E. G., Venkatesh, S., Singh, K., Cheng, I., et al. (2015). CODAS syndrome is associated with mutations of LONP1, encoding mitochondrial AAA+ Lon protease. Am. J. Hum. Genet. 96, 121–135. doi: 10.1016/j.ajhg.2014.12.003
Sultana, R., Mecocci, P., Mangialasche, F., Cecchetti, R., Baglioni, M., and Butterfield, D. A. (2011). Increased protein and lipid oxidative damage in mitochondria isolated from lymphocytes from patients with Alzheimer’s disease: Insights into the role of oxidative stress in Alzheimer’s disease and initial investigations into a potential biomarker for this dementing disorder. J. Alzheimers Dis. 24, 77–84. doi: 10.3233/jad-2011-101425
Suzuki, M., Jeong, S. Y., Karbowski, M., Youle, R. J., and Tjandra, N. (2003). The solution structure of human mitochondria fission protein Fis1 reveals a novel TPR-like helix bundle. J. Mol. Biol. 334, 445–458. doi: 10.1016/j.jmb.2003.09.064
Tremel, S., Ohashi, Y., Morado, D. R., Bertram, J., Perisic, O., Brandt, L. T. L., et al. (2021). Structural basis for VPS34 kinase activation by Rab1 and Rab5 on membranes. Nat. Commun. 12:1564. doi: 10.1038/s41467-021-21695-2
Tsuboyama, K., Koyama-Honda, I., Sakamaki, Y., Koike, M., Morishita, H., and Mizushima, N. (2016). The ATG conjugation systems are important for degradation of the inner autophagosomal membrane. Science 354, 1036–1041. doi: 10.1126/science.aaf6136
Valente, E. M., Abou-Sleiman, P. M., Caputo, V., Muqit, M. M., Harvey, K., Gispert, S., et al. (2004). Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science 304, 1158–1160. doi: 10.1126/science.1096284
Valente, E. M., Bentivoglio, A. R., Dixon, P. H., Ferraris, A., Ialongo, T., Frontali, M., et al. (2001). Localization of a novel locus for autosomal recessive early-onset parkinsonism, PARK6, on human chromosome 1p35-p36. Am. J. Hum. Genet. 68, 895–900. doi: 10.1086/319522
Valente, E. M., Brancati, F., Ferraris, A., Graham, E. A., Davis, M. B., Breteler, M. M., et al. (2002). PARK6-linked parkinsonism occurs in several European families. Ann. Neurol. 51, 14–18.
VanItallie, T. B. (2019). Traumatic brain injury (TBI) in collision sports: Possible mechanisms of transformation into chronic traumatic encephalopathy (CTE). Metabolism 100S:153943. doi: 10.1016/j.metabol.2019.07.007
Vanstone, J. R., Smith, A. M., McBride, S., Naas, T., Holcik, M., Antoun, G., et al. (2016). DNM1L-related mitochondrial fission defect presenting as refractory epilepsy. Eur. J. Hum. Genet. 24, 1084–1088. doi: 10.1038/ejhg.2015.243
Vercellino, I., and Sazanov, L. A. (2022). The assembly, regulation and function of the mitochondrial respiratory chain. Nat. Rev. Mol. Cell Biol. 23, 141–161. doi: 10.1038/s41580-021-00415-0
Verrigni, D., Di Nottia, M., Ardissone, A., Baruffini, E., Nasca, A., Legati, A., et al. (2019). Clinical-genetic features and peculiar muscle histopathology in infantile DNM1L-related mitochondrial epileptic encephalopathy. Hum. Mutat. 40, 601–618. doi: 10.1002/humu.23729
Wai, T., García-Prieto, J., Baker, M. J., Merkwirth, C., Benit, P., Rustin, P., et al. (2015). Imbalanced OPA1 processing and mitochondrial fragmentation cause heart failure in mice. Science 350:aad0116. doi: 10.1126/science.aad0116
Wang, J., Hansen, K., Edwards, R., Van Houten, B., and Qian, W. (2015). Mitochondrial division inhibitor 1 (mdivi-1) enhances death receptor-mediated apoptosis in human ovarian cancer cells. Biochem. Biophys. Res. Commun. 456, 7–12. doi: 10.1016/j.bbrc.2014.11.010
Wang, L., Klionsky, D. J., and Shen, H. M. (2022). The emerging mechanisms and functions of microautophagy. Nat. Rev. Mol. Cell Biol. [Epub ahead of print]. doi: 10.1038/s41580-022-00529-z
Wang, W., Yin, J., Ma, X., Zhao, F., Siedlak, S. L., Wang, Z., et al. (2017). Inhibition of mitochondrial fragmentation protects against Alzheimer’s disease in rodent model. Hum. Mol. Genet. 26, 4118–4131. doi: 10.1093/hmg/ddx299
Wang, X., Su, B., Lee, H. G., Li, X., Perry, G., Smith, M. A., et al. (2009). Impaired balance of mitochondrial fission and fusion in Alzheimer’s disease. J. Neurosci. 29, 9090–9103. doi: 10.1523/jneurosci.1357-09.2009
Wang, Y., Zhan, G., Cai, Z., Jiao, B., Zhao, Y., Li, S., et al. (2021). Vagus nerve stimulation in brain diseases: Therapeutic applications and biological mechanisms. Neurosci. Biobehav. Rev. 127, 37–53. doi: 10.1016/j.neubiorev.2021.04.018
Waterham, H. R., Koster, J., van Roermund, C. W., Mooyer, P. A., Wanders, R. J., and Leonard, J. V. (2007). A lethal defect of mitochondrial and peroxisomal fission. N. Engl. J. Med. 356, 1736–1741. doi: 10.1056/NEJMoa064436
Weckbecker, D., Longen, S., Riemer, J., and Herrmann, J. M. (2012). Atp23 biogenesis reveals a chaperone-like folding activity of Mia40 in the IMS of mitochondria. EMBO J. 31, 4348–4358. doi: 10.1038/emboj.2012.263
Williams, P. A., Morgan, J. E., and Votruba, M. (2011). Mouse models of dominant optic atrophy: What do they tell us about the pathophysiology of visual loss? Vision Res. 51, 229–234. doi: 10.1016/j.visres.2010.08.031
Wu, W., Tian, W., Hu, Z., Chen, G., Huang, L., Li, W., et al. (2014). ULK1 translocates to mitochondria and phosphorylates FUNDC1 to regulate mitophagy. EMBO Rep. 15, 566–575. doi: 10.1002/embr.201438501
Xiao, X., Hu, Y., Quirós, P. M., Wei, Q., López-Otín, C., and Dong, Z. (2014). OMA1 mediates OPA1 proteolysis and mitochondrial fragmentation in experimental models of ischemic kidney injury. Am. J. Physiol. Renal Physiol. 306, F1318–F1326. doi: 10.1152/ajprenal.00036.2014
Xie, J., Shen, Z., Anraku, Y., Kataoka, K., and Chen, X. (2019). Nanomaterial-based blood-brain-barrier (BBB) crossing strategies. Biomaterials 224:119491. doi: 10.1016/j.biomaterials.2019.119491
Xu, S., Peng, G., Wang, Y., Fang, S., and Karbowski, M. (2011). The AAA-ATPase p97 is essential for outer mitochondrial membrane protein turnover. Mol. Biol. Cell 22, 291–300. doi: 10.1091/mbc.E10-09-0748
Yan, L., Qi, Y., Huang, X., Yu, C., Lan, L., Guo, X., et al. (2018). Structural basis for GTP hydrolysis and conformational change of MFN1 in mediating membrane fusion. Nat. Struct. Mol. Biol. 25, 233–243. doi: 10.1038/s41594-018-0034-8
Yang, D., Zhou, Q., Labroska, V., Qin, S., Darbalaei, S., Wu, Y., et al. (2021). G protein-coupled receptors: Structure- and function-based drug discovery. Signal Transduct. Target. Ther. 6:7. doi: 10.1038/s41392-020-00435-w
Ye, X., Sun, X., Starovoytov, V., and Cai, Q. (2015). Parkin-mediated mitophagy in mutant hAPP neurons and Alzheimer’s disease patient brains. Hum. Mol. Genet. 24, 2938–2951. doi: 10.1093/hmg/ddv056
Yin, F., Sancheti, H., Patil, I., and Cadenas, E. (2016). Energy metabolism and inflammation in brain aging and Alzheimer’s disease. Free Radic. Biol. Med. 100, 108–122. doi: 10.1016/j.freeradbiomed.2016.04.200
Yoon, Y., Krueger, E. W., Oswald, B. J., and McNiven, M. A. (2003). The mitochondrial protein hFis1 regulates mitochondrial fission in mammalian cells through an interaction with the dynamin-like protein DLP1. Mol. Cell. Biol. 23, 5409–5420. doi: 10.1128/mcb.23.15.5409-5420.2003
Yu, C., Zhao, J., Yan, L., Qi, Y., Guo, X., Lou, Z., et al. (2020). Structural insights into G domain dimerization and pathogenic mutation of OPA1. J. Cell Biol. 219:e201907098. doi: 10.1083/jcb.201907098
Yue, Z. Y., Dong, H., Wang, Y. F., Liu, Y., Song, C. Y., Yang, W. C., et al. (2015). Propofol prevents neuronal mtDNA deletion and cerebral damage due to ischemia/reperfusion injury in rats. Brain Res. 1594, 108–114. doi: 10.1016/j.brainres.2014.10.016
Zhang, C., Shang, G., Gui, X., Zhang, X., Bai, X. C., and Chen, Z. J. (2019). Structural basis of STING binding with and phosphorylation by TBK1. Nature 567, 394–398. doi: 10.1038/s41586-019-1000-2
Zhao, H., Luo, Y., Chen, L., Zhang, Z., Shen, C., Li, Y., et al. (2018a). Sirt3 inhibits cerebral ischemia-reperfusion injury through normalizing Wnt/β-catenin pathway and blocking mitochondrial fission. Cell Stress Chaperones 23, 1079–1092. doi: 10.1007/s12192-018-0917-y
Zhao, H., Pan, W., Chen, L., Luo, Y., and Xu, R. (2018b). Nur77 promotes cerebral ischemia-reperfusion injury via activating INF2-mediated mitochondrial fragmentation. J. Mol. Histol. 49, 599–613. doi: 10.1007/s10735-018-9798-8
Zhao, Y., Zhang, J., Zheng, Y., Zhang, Y., Zhang, X. J., Wang, H., et al. (2021). NAD(+) improves cognitive function and reduces neuroinflammation by ameliorating mitochondrial damage and decreasing ROS production in chronic cerebral hypoperfusion models through Sirt1/PGC-1α pathway. J. Neuroinflammation 18:207. doi: 10.1186/s12974-021-02250-8
Zhu, H. (2020). Big data and artificial intelligence modeling for drug discovery. Annu. Rev. Pharmacol. Toxicol. 60, 573–589. doi: 10.1146/annurev-pharmtox-010919-023324
Zhu, Y., Massen, S., Terenzio, M., Lang, V., Chen-Lindner, S., Eils, R., et al. (2013). Modulation of serines 17 and 24 in the LC3-interacting region of Bnip3 determines pro-survival mitophagy versus apoptosis. J. Biol. Chem. 288, 1099–1113. doi: 10.1074/jbc.M112.399345
Zorov, D. B., Juhaszova, M., and Sollott, S. J. (2014). Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 94, 909–950. doi: 10.1152/physrev.00026.2013
Keywords: mitochondrial quality control, mitochondrial dysfunction, brain disorders, mitochondrial homeostasis, therapeutic target
Citation: Shen X, Sun P, Zhang H and Yang H (2022) Mitochondrial quality control in the brain: The physiological and pathological roles. Front. Neurosci. 16:1075141. doi: 10.3389/fnins.2022.1075141
Received: 20 October 2022; Accepted: 24 November 2022;
Published: 12 December 2022.
Edited by:
Jun Zhao, United States Food and Drug Administration, United StatesReviewed by:
Liming Wang, Hunan University, ChinaKaige Yan, Southern University of Science and Technology, China
Copyright © 2022 Shen, Sun, Zhang and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hanting Yang, eWFuZ2h0QGZ1ZGFuLmVkdS5jbg==; Hao Zhang, aGFvX3poYW5nQGZ1ZGFuLmVkdS5jbg==