
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Neurosci. , 09 December 2022
Sec. Neurodegeneration
Volume 16 - 2022 | https://doi.org/10.3389/fnins.2022.1064566
This article is part of the Research Topic Experimental and Innovative Approaches to Multi-Target Treatment of Parkinson’s and Alzheimer’s Diseases - Volume II View all 7 articles
 Lili Gao1*
Lili Gao1* Chunlan Shi2Qing Lin2Yujing Wu2Liqi Hu2Mingwang Wang2Jianhua Guan1Sheng Lin1Yuansheng Liao1Chenghan Wu1
Chunlan Shi2Qing Lin2Yujing Wu2Liqi Hu2Mingwang Wang2Jianhua Guan1Sheng Lin1Yuansheng Liao1Chenghan Wu1Background: Early onset Parkinson's disease (EOPD) is a neurodegenerative disease associated with the action ofto genetic factors. A mutated phospholipase A2 type VI gene (PLA2G6) is considered to be one of pathogenic genes involved in EOPD development. Although EOPD caused by a mutated PLA2G6 has been recorded in major databases, not all mutant genotypes have been reported. Here, we report a case of PLA2G6-related EOPD caused by a novel compound heterozygous mutation.
Case presentation: The case was an of 26-year-old young male with a 2-year course of disease. The onset of the disease was insidious and developed gradually. The patient presented with unsteady walking, bradykinesia, unresponsiveness, and decreased facial expression. Auxiliary examination showed a compound heterozygous mutation of the PLA2G6gene with c.991G > T and c.1427 + 1G > A. Mild atrophy of the cerebrum and cerebellum was detected on brain MRI. The patient was diagnosed with EOPD. We administered treatment with Madopar, which was effective. After a two-year disease course, we observed progression to stage 5 according to the Hoehn-Yahr Scale (without medicine in the off-stage). An MDS-UPDRS III score of 62 was obtained, with characteristics of severe disease and rapid progress. The diagnosis was an EOPD phenotype caused by a combination of mutations at the c.991G > T and c.1427 + 1G > A sites of the PLA2G6gene.
Conclusion: After active treatment, the disease was set under control, with no significant progression during the three-month follow-up period. Dyskinesia did not recur after reducing the Madopar dose. The freezing sign was slightly decreased and the wearing-off was delayed to 2 h.
Parkinson's disease (PD) is a multifactorial disease, and its pathogenesis may be related to factors, such as oxidative stress, mitochondrial dysfunction, and abnormal protein expression. However, from 10 to 15% of PD patients have a genetic predisposition (Zhu et al., 2019). Early-onset PD (EOPD) refers to an earlier age onset (50 or younger) of this disease. EOPD is related mainly to more than 20 gene variants such as ATP13A2, PLA2G6, and PRKN (Zhao et al., 2020). A disease caused by mutation of PLA2G6 is highly genetically and clinically heterogeneous. The clinical manifestations of PLA2G6-related EOPD include mental disorders and/or cognitive decline, non-motor symptoms such as dystonia, and motor symptoms such as resting tremor, muscle rigidity, and bradykinesia (Chan et al., 2008). Although the incidence of EOPD caused by a PLA2G6 mutation is not high, it has a very high disability and mortality rate, and the average age of onset is 22 years (Oliveira et al., 2021). These disease characteristics bring a heavy burden to patients and families with no curative treatment currently available. Therefore, PLA2G6-related EOPD needs more extensive in-depth research. Here, we report a case of EOPD associated with a heterozygous mutation of PLA2G6.
A 26-year-old male patient was admitted to the Second People's Hospital Affiliated to Fujian University of Traditional Chinese Medicine (Fuzhou, Fujian, China) in May 2022 due to unsteady walking for more than 2 years with progressive aggravation, and hand tremor for more than 1 year. We reviewed his medical history and established that disease symptoms, such as unstable walking, slow movement, and decreased facial expression, gradually developed in early 2020. In April 2020, he attended a local hospital, where he was diagnosed with Parkinson's syndrome. Then, he received treatment with Madopar at 0.125 g tid, which improved the unstable walking and bradykinesia, but not the facial expression. At the end of 2020, the unstable walking and the bradykinesia significantly progressed, and symptoms such as trunk leaning back and tremor of hands (mainly postural maintenance tremor) occurred. The patient increased the amount of Madopar by himself (details unknown). However, involuntary convulsions, outstretched tongue, and other abnormal movement symptoms appeared. These symptoms gradually progressed along with the appearance of freezing symptoms such as inability to move and turn over voluntarily. Thus, the patient gradually increased further the amount of Madopar by himself, up to 1.25 g/day. In April 2022, the patient's off-stage condition progressed to stage 5 with retarded speech, resting tremors in both hands, and the gradual appearance of the “masked face” syndrome of PD. Moreover, he also had by Madopar-induced dyskinesia, freezing symptoms, and wearing off. He was married with two sons and denied family history of genetic diseases and similar diseases. The two sons did not suffer from similar symptoms.
Blood pressure in lying and standing position was normal. The off-stage nervous system examination showed clear consciousness, unresponsiveness, dysfluent speech, reduced facial expression, normal higher cortical function by rough test, normal bilateral eye movements, grade V of limb strength, increased muscle tone of the right upper and lower limbs (1 score), normal left limb muscle tone, tendon reflexes of the limbs (-), and bilateral pathological signs (-). Besides, the results of the bilateral rotation, the finger-finger test, and the foot tapping were normal. The patient could not cooperate to complete the bilateral finger-nose and heel-knee-shin tests with Romberg's sign (+) and back pull test (+). Support was required during walking. The pace of the case was reduced with lower limbs dragging.
The thyroid function was assessed with the following results: free thyroid hormone level was 36.940 pmol/L and anti-thyroid peroxidase antibody > 1300.0 IU/mL. Blood, urine, stool routine, complete biochemical items, homocysteine, ceruloplasmin, hepatitis, syphilis, HIV, coagulation, and rheumatism indexes were normal. Brain MRI showed atrophy of the cerebrum and cerebellum (Figure 1). EEG indicated moderate abnormality. Electromyography revealed no peripheral neurogenic damage. SEP of the right lower extremity was generally normal. Ophthalmological examination showed no obvious abnormality. The following examination scores were received: AD8, 3; Mini-Mental State Examination (MMSE) 24; Montreal Cognitive Assay (MoCA), 20; HAMD, 49 scores; and HAMA, 23. The Hoehn-Yahr Scale showed stage 5 (without medicine in the off-stage). The following MDS-UPDRS I scores were obtained: MDS-UPDRS I, 8; MDS-UPDRS II, 38; MDS-UPDRS III, 62, and MDS-UPDRS IV, 7. MRI showed atrophy of the cerebrum and cerebellum.
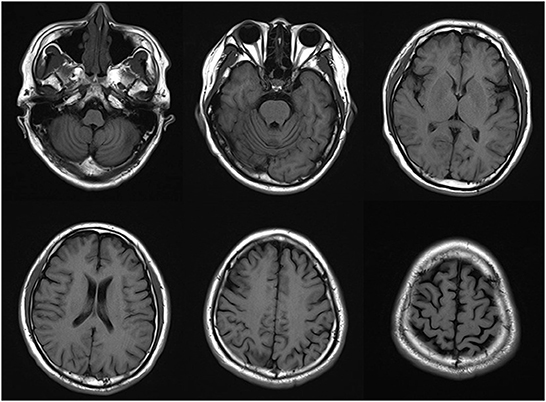
Figure 1. Brain MRI showed atrophy of the cerebrum and cerebellum.
Two heterozygous mutations were found in the exon and a splice site region of PLA2G6 including c.991G > T (guanine > thymine), which led to p.D331Y (aspartic acid > tyrosine) and c.1427 + 1G > A (guanine > adenine) (Figure 2).
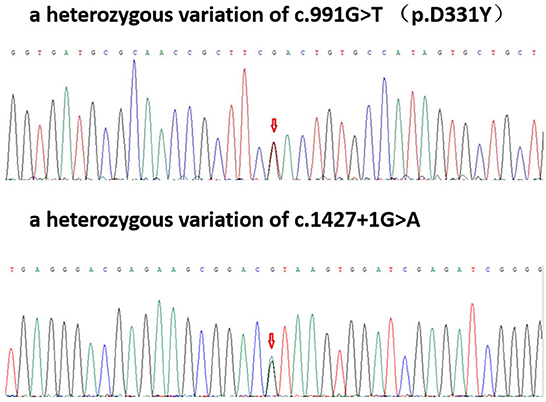
Figure 2. Patient gene map: A heterozygous variation of c.991G > T (p.D331Y); a heterozygous variation of c.1427 + 1G>A.
Based on the patient's symptoms, including bradykinesia, unsteady walking, tremor of hands, and decreased facial expression, the disease was located in the extrapyramidal system. The qualitative diagnosis findings were as follows: factors, such as poisoning, trauma, and infection were denied. According to the characteristics of insidious onset in adolescence and the gradual progression, combined with the genetic test results, the disorder was considered hereditary; the final conclusion was genetic Parkinson's disease type 14 (PARK14). Based on the PD diagnostic criteria of International Parkinson and Movement Disorder Society (MDS) in 2015 (Postuma et al., 2015), the case was diagnosed as EOPD. The following treatment was administered: Madopar 0.25 g tid combined with ropinirole hydrochloride tablets 0.25 g tid. The patient's dyskinesia episodes were significantly less than at the onset, and the motor symptoms were slightly improved. The significant effect of Madopar was achieved within 1 h after its administration. During the three-month follow-up period, the patient's walking was unsteady. However, the bradykinesia did not progress, and dyskinesia did not recur. The wearing-off was prolonged from 1 h to approximately 2 h, and the overall mental state was better than before the treatment.
As mentioned earlier, the patient, whose case has been described in this paper, was a young male. The disease course lasted for more than 2 years, with an onset of unstable walking and bradykinesia, and gradually increasing resting tremor. The symptoms progressed rapidly. Brain MRI showed mild atrophic changes in the whole brain. The treatment with Madopar was effective. The genetic test results showed two mutation sites of PLA2G6, including c.991G > T and c.1427 + 1G > A. On the basis of the present findings and the existing evidence base, this is the first reported EOPD case with mutations in these two sites. The overall prognosis of this disease is poor and with a very high disability and mortality rate. Thus, early identification and treatment are of critical significance to patient prognosis.
Abnormalities in the PLA2G6 gene are often manifested in various neurodegenerative diseases (Ji et al., 2019), such as infantile neuroaxonal dystrophy (INAD), atypical infantile neuroaxonal dystrophy (ANAD), dystonia parkinsonism (DP), and autosomal recessive EOPD (Cheng et al., 2019). The mechanism of action may be associated with the D331Y and T572I mutations in the PLA2G6 domain, leading to the reduction of the formation of dopamine neurons and the manifestation of the neuromotor symptom phenotype (Yeh et al., 2021). Several studies have also suggested that potential enzyme activity, DNA methylation, synergistic genetics, and environmental factors may be involved in PLA2G6 gene-related neurological diseases (Guo et al., 2018), which, therefore, have diverse phenotypes. Among these diseases, EOPD related to the PLA2G6 gene is more common in young people. The PLA2G6 gene has been characterized as a locus for parkinsonism with early onset and associated neurological features, particularly dystonia. Many identified mutations have been detected in PLA2G6 gene-related EOPD (Table 1). However, no other compound with a heterozygous mutation of the PLA2G6 gene with c.991G > T and c.1427 + 1G > A has been reported. The c.991G > T site of the PLA2G6 gene is a missense mutation, which may lead to the loss of function of the expressed protein and various motor symptoms (Cheng et al., 2022). In the ClinVar database, the pathogenicity of this variant is classified as pathogenic, and the corresponding disease is EOPD (Shen et al., 2019). Another variant site, c.1427 + 1G > A, is reported to be related to the INAD phenotype (Wan et al., 2022), but the reports of the EOPD phenotype have been scarce. Previous investigations showed that the clinical symptoms of EOPD patients caused by only c.991G > T mutation were relatively mild with typical PD symptoms, no dystonia, and relatively slow disease progression (more than 10 years). In addition, most MRI showed no abnormal signs of cerebral iron deposition (Lu et al., 2012; Xie et al., 2015).
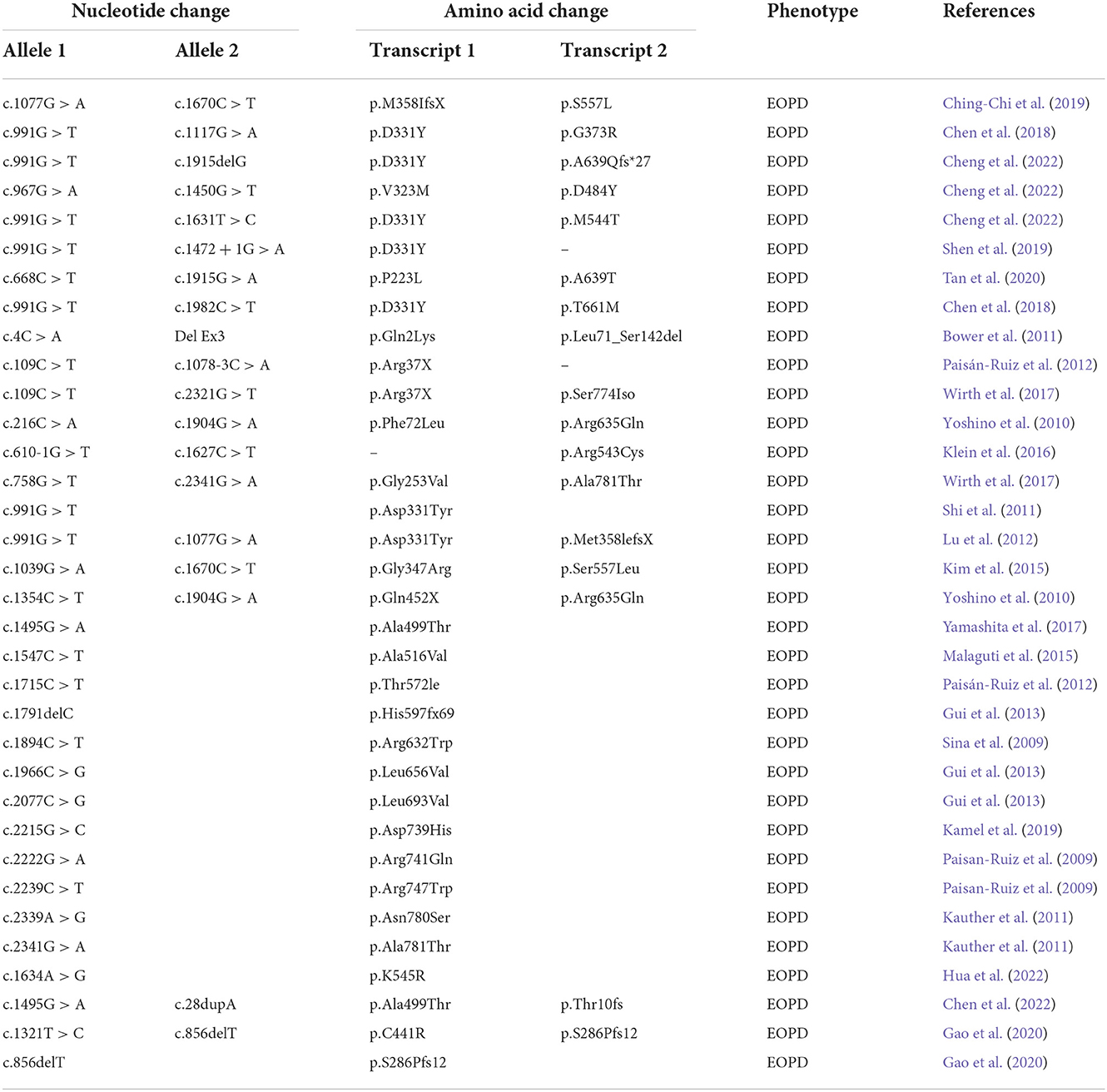
Table 1. Summary of all variant sites of EOPD caused by PLA2G6 mutation.
The c.991G > T of a PLA2G6 mutant is associated with approximately 30% of the residual protein activity (Shi et al., 2011), which may explain why EOPD patients with only c.991G > T mutation have generally mild disease symptoms and relatively slow disease progression. Although c.991G > T mutation was also present in this case, the condition of the patient was severe, with rapid disease progression. Two years later, the disease had developed into stage 5 according to the Hoehn-Yahr Scale (off-stage), and the case needed special care with his diet and daily life activities. Thus, the disease seriously affected the patients' quality of life, which is inconsistent with findings concerning patients with a single c.991G > T mutation. The patient also differed from other EOPD patients with PLA2G6 compound heterozygous mutations in terms of cognitive decline and response to madopar. Unlike most patients with PLA2G6 gene-related EOPD, which is associated with cognitive dysfunction, the patient did not experience cognitive decline. In addition, although the patient began to use madopar at the early stage of the disease, the disorder was not well controlled and progressed rapidly and gradually worsened. During the 2 years of disease course, the bradykinesia symptoms gradually progressed along with the appearance of freezing symptoms, such as inability to move and turn over voluntarily. Most patients have a good response to madopar 5–10 years before onset, which was in contrast to the response of this patient, whose side effects appeared early. The reported here results suggests that these characteristics are associated with the presence of a compound heterozygous mutation of the PLA2G6 gene with c.991G > T and c.1427 + 1G > A. Several studies have indicated that the rapid progression of the disease in EOPD patients may be related to the fluctuation of the hormone levels in the body (Magrinelli et al., 2022). However, this case had no obvious history of hormone fluctuations. Thus, it was considered that the severe disease with rapid progression was related to the combination of a c.991G > T mutation and a c.1427 + 1G > A mutation. The identification and research of this case laid the foundation for further understanding of PLA2G6 gene-related neurodegenerative diseases.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethical review and approval was not required for the study on human participants in accordance with the local legislation and institutional requirements. The patients/participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
LG, JG, SL, and YL carried out the studies and participated in collecting data. LG and CW performed the statistical analysis and participated in its design. CS, QL, YW, MW, and LH participated in acquisition and draft the manuscript. All authors read and approved the final manuscript.
This work was supported by the Fujian Provincial Health Commission (No. 2019-ZQN-78) and the Natural Science Foundation of the Fujian Province (No. 2020J01247).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Bower, M. A., Khalaf, B., Dempsey, M. A., Soma, D., and Tuite, P. J. (2011). Novel mutations in siblings with later-onset PLA2G6-associated neurodegeneration (PLAN). Mov. Disord. 26, 1768–1769. doi: 10.1002/mds.23617
Chan, D. K., Mok, V., Ng, P. W., Yeung, J., Kwok, J. B., Fang, Z. M., et al. (2008). PARK2 mutations and clinical features in a Chinese population with early-onset Parkinson's disease. J Neural. Transm. 115, 715–719. doi: 10.1007/s00702-007-0011-6
Chen, C., Lou, M.-M., Sun, Y-M., Luo, F., Liu, F.-T, Luo, S.-S., et al. (2022). Serum metabolomic characterization of PLA2G6-associated dystonia-parkinsonism: A case-control biomarker study. Front. Neurosci. 16, 879548. doi: 10.3389/fnins.2022.879548
Chen, Y.-J., Chen, Y.-C., Dong, H.-L., Li, L.-X., Ni, W., Li, H.-F., et al. (2018). Novel PLA2G6 mutations and clinical heterogeneity in Chinese cases with phospholipase A2-associated neurodegeneration. Parkinsonism Relat. Disord. 49, 88–94. doi: 10.1016/j.parkreldis.2018.02.010
Cheng, H.-L., Chen, Y.-J., Xue, Y.-Y., Wu, Z.-Y., Li, H.-F., Wang, N., et al. (2022). Clinical characterization and founder effect analysis in chinese patients with phospholipase a2-associated neurodegeneration. Brain Sci. 12:517. doi: 10.3390/brainsci12050517
Cheng, Y. C., Lin, H. I., Syu, S. H., Lu, H. E., Huang, C. Y., Lin, C. H., et al. (2019). Reprogramming of a human induced pluripotent stem cell (iPSC) line (IBMSi012-A) from an early-onset Parkinson's disease patient harboring a homozygous p.D331Y mutation in the PLA2G6 gene. Stem Cell Res. 37, 101432. doi: 10.1016/j.scr.2019.101432
Ching-Chi, C., Hung-Li, W., Yi-Hsin, W., Rou-Shayn, C., Chiung-Mei, C., Tu-Hsueh, Y., et al. (2019). Generation of induced pluripotent stem cells from a young-onset Parkinson's disease patient carrying the compound heterozygous PLA2G6 p.D331Y/p.M358IfsX mutations. Stem. Cell Res. 40, 101552. doi: 10.1016/j.scr.2019.101552
Gao, C., Huang, T., Chen, R., Yuan, Z., Tian, Y., and Zhang, Y. (2020). PRKNA han Chinese family with early-onset Parkinson's Disease carrying novel frameshift mutation and compound heterozygous mutation of appearing incompatible with MDS clinical diagnostic criteria. Front. Neurol. 11, 582323. doi: 10.3389/fneur.2020.582323
Gui, Y.-X., Xu, Z.-P., Wen-Lv, Liu, H.-M., Zhao, J.-J., and Hu, X.-Y. (2013). Four novel rare mutations of PLA2G6 in Chinese population with Parkinson's disease. Parkinsonism Relat. Disord. 19, 21–26. doi: 10.1016/j.parkreldis.2012.07.016
Guo, Y. P., Tang, B. S., and Guo, J. F. (2018). PLA2G6-associated neurodegeneration (PLAN): review of clinical phenotypes and genotypes. Front. Neurol. 9, 1100. doi: 10.3389/fneur.2018.01100
Hua, P., Zhao, Y., Zeng, Q., Li, L., Ren, J., Guo, J., et al. (2022). Genetic analysis of patients with early-onset Parkinson's Disease in Eastern China. Front. Aging Neurosci. 14, 849462. doi: 10.3389/fnagi.2022.849462
Ji, Y., Li, Y., Shi, C., Gao, Y., Yang, J., Liang, D., et al. (2019). Identification of a novel mutation in PLA2G6 gene and phenotypic heterogeneity analysis of PLA2G6-related neurodegeneration. Parkinsonism Relat. Disord. 65, 159–164. doi: 10.1016/j.parkreldis.2019.04.002
Kamel, W. A., Al-Hashel, J. Y., Abdulsalam, A. J., Damier, P., and Al-Mejalhem, A. Y. (2019). PLA2G6-related parkinsonism presenting as adolescent behavior. Acta. Neurol. Belg. 119, 621–622. doi: 10.1007/s13760-018-1003-z
Kauther, K. M., Höft, C., Rissling, I., Oertel, W. H., and Möller, J. C. (2011). The PLA2G6 gene in early-onset Parkinson's disease. Mov. Disord. 26, 2415–2417. doi: 10.1002/mds.23851
Kim, Y. J., Lyoo, C. H., Hong, S., Kim, N. Y., and Lee, M. S. (2015). Neuroimaging studies and whole exome sequencing of PLA2G6-associated neurodegeneration in a family with intrafamilial phenotypic heterogeneity. Parkinsonism Relat. Disord. 21, 402–406. doi: 10.1016/j.parkreldis.2015.01.010
Klein, C., Löchte, T., Delamonte, S. M., Braenne, I., Hicks, A. A., Zschiedrich-Jansen, K., et al. (2016). PLA2G6 mutations and Parkinsonism: Long-term follow-up of clinical features and neuropathology. Mov. Disord. 31, 1927–1929. doi: 10.1002/mds.26814
Lu, C. S., Lai, S. C., Wu, R. M., Weng, Y. H., Huang, C. L., Chen, R. S., et al. (2012). PLA2G6 mutations in PARK14-linked young-onset parkinsonism and sporadic Parkinson's disease. Am. J. Med. Genet. B. Neuropsychiatr. Genet. 159b, 183–191. doi: 10.1002/ajmg.b.32012
Magrinelli, F., Rajapaksha, I., Kobylecki, C., Latorre, A., Mulroy, E., Estevez-Fraga, C., et al. (2022). Reply to: juvenile PLA2G6-parkinsonism due to indian 'asian' p.R741Q mutation, and response to STN DBS. Mov. Disord. 37, 658–662. doi: 10.1002/mds.28955
Malaguti, M. C., Melzi, V., Di Giacopo, R., Monfrini, E., Di Biase, E., Franco, G., et al. (2015). A novel homozygous PLA2G6 mutation causes dystonia-parkinsonism. Parkinsonism Relat. Disord. 21, 337–339. doi: 10.1016/j.parkreldis.2015.01.001
Oliveira, D., Montanaro, P., Carvalho, V., Martins, D., Ferreira, B. A., and Cardoso, F. (2021). Severe early-onset parkinsonian syndrome caused by PLA2G6 heterozygous variants. Mov. Disord. Clin. Pract 8, 794–796. doi: 10.1002/mdc3.13230
Paisan-Ruiz, C., Bhatia, K. P., Li, A., Hernandez, D., Davis, M., Wood, N. W., et al. (2009). Characterization of PLA2G6 as a locus for dystonia-parkinsonism. Ann. Neurol. 65, 19–23. doi: 10.1002/ana.21415
Paisán-Ruiz, C., Li, A., Schneider, S. A., Holton, J. L., Robert, J., Desmond, K., et al. (2012). Widespread Lewy body and tau accumulation in childhood and adult onset dystonia-parkinsonism cases with PLA2G6 mutations. Neurobiol. Aging 33, 814–823. doi: 10.1016/j.neurobiolaging.2010.05.009
Postuma, R. B., Berg, D., Stern, M., Poewe, W., Olanow, C. W., Oertel, W., et al. (2015). MDS clinical diagnostic criteria for Parkinson's disease. Mov. Disord. 30, 1591–1601. doi: 10.1002/mds.26424
Shen, T., Hu, J., Jiang, Y., Zhao, S., Lin, C., Yin, X., et al. (2019). Early-onset parkinson's disease caused by PLA2G6 compound heterozygous mutation, a case report and literature review. Front. Neurol. 10, 915. doi: 10.3389/fneur.2019.00915
Shi, C. H., Tang, B. S., Wang, L., Lv, Z. Y., Wang, J., Luo, L. Z., et al. (2011). PLA2G6 gene mutation in autosomal recessive early-onset parkinsonism in a Chinese cohort. Neurology 77, 75–81. doi: 10.1212/WNL.0b013e318221acd3
Sina, F., Shojaee, S., Elahi, E., and Paisán-Ruiz, C. (2009). R632W mutation in PLA2G6 segregates with dystonia-parkinsonism in a consanguineous Iranian family. Eur. J. Neurol. 16, 101–104. doi: 10.1111/j.1468-1331.2008.02356.x
Tan, J., Yan, T., Chang, R., Yuan, D., Pan, L., and Cai, R. (2020). Analysis of PLA2G6 gene variant in a family affected with infantile neuroaxonal dystrophy. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 37, 21–24. doi: 10.3760/cma.j.issn.1003-9406.2020.01.006
Wan, Y., Jiang, Y., Xie, Z., Ling, C., Du, K., Li, R., et al. (2022). Novel PLA2G6 pathogenic variants in chinese patients with PLA2G6-associated neurodegeneration. Front. Neurol. 13, 922528. doi: 10.3389/fneur.2022.922528
Wirth, T., Weibel, S., Montaut, S., Bigaut, K., Rudolf, G., Chelly, J., et al. (2017). Severe early-onset impulsive compulsive behavior and psychosis in PLA2G6-related juvenile Parkinson's disease. Parkinsonism Relat. Disord. 41, 127–129. doi: 10.1016/j.parkreldis.2017.05.014
Xie, F., Cen, Z., Ouyang, Z., Wu, S., Xiao, J., Luo, W., et al. (2015). Homozygous p.D331Y mutation in PLA2G6 in two patients with pure autosomal-recessive early-onset parkinsonism: further evidence of a fourth phenotype of PLA2G6-associated neurodegeneration. Parkinsonism Relat. Disord. 21, 420–422. doi: 10.1016/j.parkreldis.2015.01.012
Yamashita, C., Funayama, M., Li, Y., Yoshino, H., Yamada, H., Seino, Y., et al. (2017). Mutation screening of PLA2G6 in Japanese patients with early onset dystonia-parkinsonism. J. Neural. Transm. (Vienna). 124, 431–435. doi: 10.1007/s00702-016-1658-7
Yeh, T. H., Liu, H. F., Chiu, C. C., Cheng, M. L., Huang, G. J., Huang, Y. C., et al. (2021). PLA2G6 mutations cause motor dysfunction phenotypes of young-onset dystonia-parkinsonism type 14 and can be relieved by DHA treatment in animal models. Exp. Neurol. 346, 113863. doi: 10.1016/j.expneurol.2021.113863
Yoshino, H., Tomiyama, H., Tachibana, N., Ogaki, K., Li, Y., Funayama, M., et al. (2010). Phenotypic spectrum of patients with PLA2G6 mutation and PARK14-linked parkinsonism. Neurology 75, 1356–1361. doi: 10.1212/WNL.0b013e3181f73649
Zhao, Y., Qin, L., Pan, H., Liu, Z., Jiang, L., He, Y., et al. (2020). The role of genetics in Parkinson's disease: a large cohort study in Chinese mainland population. Brain 143, 2220–2234. doi: 10.1093/brain/awaa167
Keywords: EOPD, PLA2G6, PARK14, PD, heterozygous mutation, case report
Citation: Gao L, Shi C, Lin Q, Wu Y, Hu L, Wang M, Guan J, Lin S, Liao Y and Wu C (2022) Case Report: A case of PLA2G6 gene-related early-onset Parkinson's disease and review of literature. Front. Neurosci. 16:1064566. doi: 10.3389/fnins.2022.1064566
Received: 08 October 2022; Accepted: 28 November 2022;
Published: 09 December 2022.
Edited by:
Sandeep Kumar Singh, Indian Scientific Education and Technology Foundation, IndiaReviewed by:
Saurabh Srivastav, Rice University, United StatesCopyright © 2022 Gao, Shi, Lin, Wu, Hu, Wang, Guan, Lin, Liao and Wu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lili Gao, Z2FvbGlsaWRleGluZ2Z1QDE2My5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.