 Genevieve Hayes
Genevieve Hayes Joana Pinto
Joana Pinto Sierra N. Sparks
Sierra N. Sparks Congxiyu Wang
Congxiyu Wang Sana Suri
Sana Suri Daniel P. Bulte
Daniel P. Bulte
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Neurosci., 10 November 2022
Sec. Neurodegeneration
Volume 16 - 2022 | https://doi.org/10.3389/fnins.2022.1010164
This article is part of the Research TopicVascular and Perivascular Contributions to NeurodegenerationView all 7 articles
Vascular smooth muscle cells (VSMCs) are the key moderators of cerebrovascular dynamics in response to the brain’s oxygen and nutrient demands. Crucially, VSMCs may provide a sensitive biomarker for neurodegenerative pathologies where vasculature is compromised. An increasing body of research suggests that VSMCs have remarkable plasticity and their pathophysiology may play a key role in the complex process of neurodegeneration. Furthermore, extrinsic risk factors, including environmental conditions and traumatic events can impact vascular function through changes in VSMC morphology. VSMC dysfunction can be characterised at the molecular level both preclinically, and clinically ex vivo. However the identification of VSMC dysfunction in living individuals is important to understand changes in vascular function at the onset and progression of neurological disorders such as dementia, Alzheimer’s disease, and Parkinson’s disease. A promising technique to identify changes in the state of cerebral smooth muscle is cerebrovascular reactivity (CVR) which reflects the intrinsic dynamic response of blood vessels in the brain to vasoactive stimuli in order to modulate regional cerebral blood flow (CBF). In this work, we review the role of VSMCs in the most common neurodegenerative disorders and identify physiological systems that may contribute to VSMC dysfunction. The evidence collected here identifies VSMC dysfunction as a strong candidate for novel therapeutics to combat the development and progression of neurodegeneration, and highlights the need for more research on the role of VSMCs and cerebrovascular dynamics in healthy and diseased states.
Cerebrovascular disease is among the leading disease-related causes of societal and economic burden in the developed world (Wang et al., 2016). The brain consumes 20% of the human body’s metabolic reserve, but it has a limited capability for energy storage. As a result, small changes in blood supply can lead to severe alterations in metabolic function, which can also lead to the secretion of neurotoxic and inflammatory factors, a reduction in cerebral blood flow (CBF), and hypoxia that might initiate and/or contribute to neuronal degradation (Zlokovic, 2011).
The aim of this review is to identify how vascular smooth muscle cells (VSMCs) participate in these vascular insults, and whether VSMC dysfunction may, in turn, contribute to neuronal degradation. Understanding the role of VSMC dysfunction in neurodegeneration is critical as it may inform on the underlying mechanisms behind cerebrovascular impairment that sometimes accompany and precede the onset of various neurological disorders.
In this review, we first summarise how VSMCs normally function to control cerebrovascular dynamics and blood flow, and key mechanisms by which VSMCs grow and communicate. Additionally, we discuss the best candidates for identifying the presence of cerebral VSMC dysfunction. We then present studies on VSMC phenotype, signalling pathways, and general contractility seen in a host of neurodegenerative disorders, presented in order of disorder prevalence. While it is not possible to encompass all neurodegenerative disorders, nor all the mechanisms at play, we review the role of VSMCs in Alzheimer’s disease (AD), Parkinson’s disease (PD), amyotrophic lateral sclerosis (ALS), multiple sclerosis (MS), Huntington’s disease (HD), cerebral small vessel disease (CSVD), post-stroke dementia (PSD), Down’s syndrome (DS), cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL), Lewy body dementia (LBD), and Moyamoya disease (MMD). In assessing VSMC dysfunction in these disorders, the studies have only considered correlation not causation. Finally, we review how extrinsic influences can affect VSMC function and touch on a few potential therapeutic opportunities in the context of the evidence we have collected on VSMC dysfunction in neurodegeneration.
The vascular tree supplies the brain with blood flow and spans from the circle of Willis, which is known to be highly variable, down to the capillary network. The circle of Willis interconnects the anterior and posterior cerebral circulation network and gives rise to pairs of anterior, middle, and posterior cerebral arteries. Each artery divides into progressively smaller arteries and arterioles which run along the brain’s surface until they penetrate into the brain tissue (Ovsenik et al., 2021).
Appropriate CBF is critical for brain function and survival. The regulation of CBF involves a coordinated interplay between different types of cells, including neurons, glia, and vascular cells. The neurons and glia generate signals which are translated into vascular changes by the collaboration of endothelial and mural cells. In particular, the mural cells, which include VSMCs and pericytes, surround the endothelial cell layer and regulate cerebrovascular resistance and blood flow to downstream capillary beds. Surrounding the smooth muscle is the adventitia layer that fuses to the basement membrane of astrocyte end feet, an important mediator of signals from neurons to blood vessels (MacVicar and Newman, 2015). In recent years, much attention has also been placed on the participation of glial cells, primarily astrocytes signalling pathways for cerebrovascular tone (Attwell et al., 2010; Filosa and Iddings, 2013). Together, these vascular and perivascular components make up the blood brain barrier (BBB), the selective interface between the blood and central nervous system (CNS). A simplified diagram of the anatomy of a cerebral artery is illustrated in Figure 1.
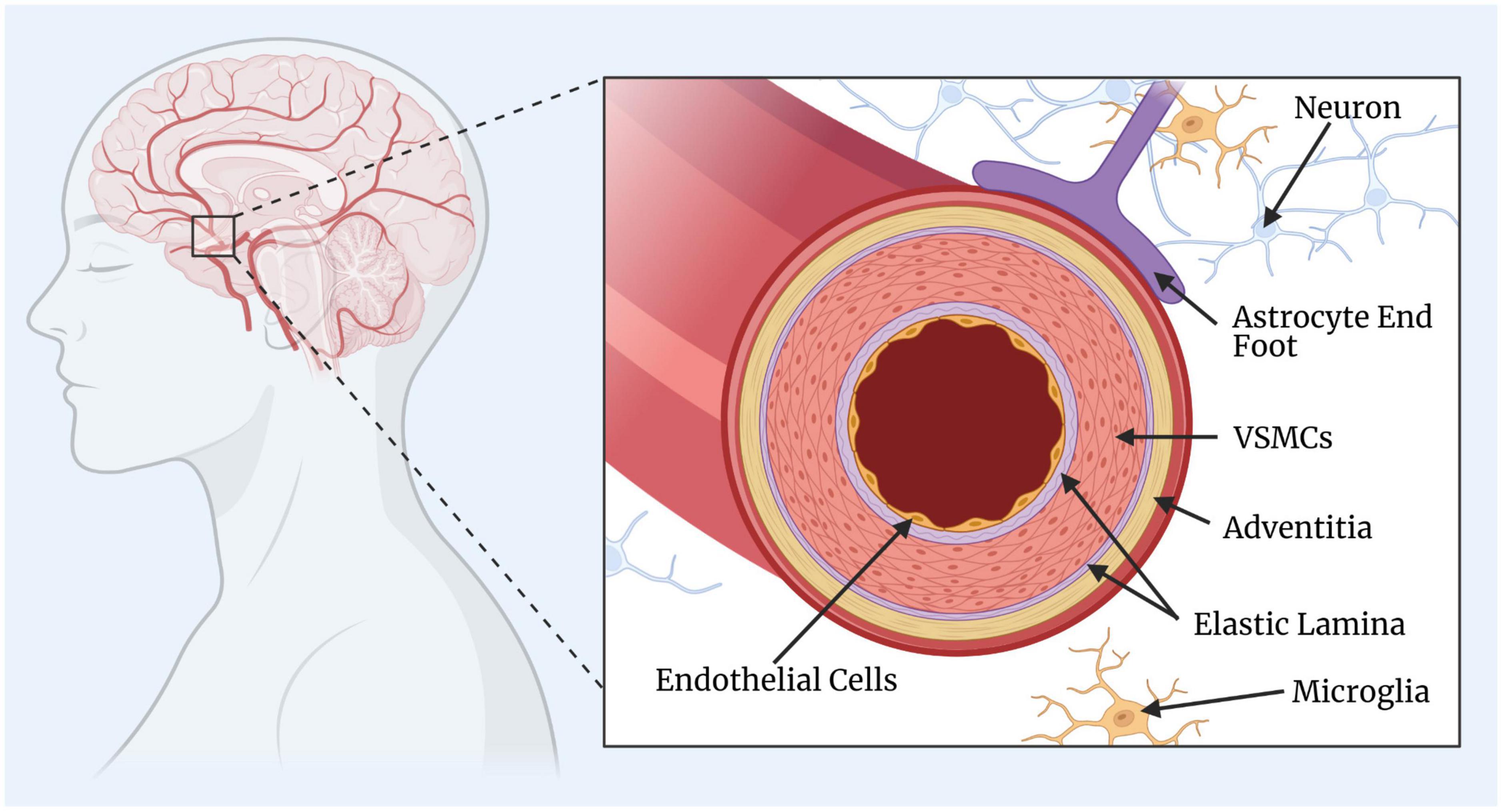
Figure 1. Simplified diagram of a cerebral artery. The key components of the cerebral artery include the endothelial cells, elastic lamina (one internal and one external), vascular smooth muscle cells (VSMCs), and the adventitia layer. The surrounding and supporting cells include the astrocytes, microglia, and neurons. This figure was created by GH rendering using BioRender.com.
Vascular smooth muscle cells are a particular contractile cell type characterised by their expression of contractile proteins such as smooth muscle actin, myosin heavy chain, and myosin light chain (Poittevin et al., 2014). Through contraction and relaxation, VSMCs alter blood vessel diameter and enable the maintenance of appropriate cerebral blood pressure and flow. A cascade of events occurs in response to changing oxygen and nutrient needs, as chemical signals are converted by VSMCs into mechanical constriction or relaxation by inducing changes in calcium ion (Ca2+) concentrations, activating potassium channels, and altering the contractile state of the light chain of myosin. In response to mechanical forces, such as high circumferential stress, VSMCs undergo mechanical signalling to influence gene expression, regulate cellular function, and alter vascular tone, which occurs even faster than the chemical signal cascade and is known as mechanotransduction (Na et al., 2008; Liu and Lin, 2022). VSMCs are coupled by gap junctions which mediate the intramural propagation of vascular signals between cells (Iadecola, 2004). The renin-angiotensin system (RAS) is a key mediator of VSMC constriction and the Notch signalling pathway is essential in the coordination of VSMC migration and adhesion (Griendling et al., 1997; Sorrentino et al., 2022). Additionally, the free radical, nitric oxide (NO) is a powerful vasodilator, produced most readily through endothelial NO synthesis and is a vital component of the signalling cascade (Zhao et al., 2015). Notably, changes in these pathways may play a role in pathological states and are discussed in more detail below.
Some studies have reported that capillary pericyte contractility affects CBF as well, though at a much slower time-scale than VSMCs due to their lack of smooth muscle actin (Yemisci et al., 2009; Hall et al., 2014; Hartmann et al., 2021). While there are still unresolved controversies regarding pericyte contractility, it remains that in arteries and arterioles in the brain, VSMCs are likely to be the primary regulators of cerebrovascular dynamics and blood flow changes in response to the brain’s oxygen and nutrient demands (Hartmann et al., 2022).
Vascular smooth muscle cells and endothelial cells exhibit a spectrum of genetically distinct phenotypes along the arteriovenous pathway, known as zonation (Vanlandewijck et al., 2018). Within arterial zones, VSMCs can also express different phenotypes that change their properties, assuring that each vessel can adapt to changes in the local conditions. VSMC phenotype modulation is affected by innate genetic programmes and environmental cues. Contractile and synthetic VSMCs represent the two ends of a spectrum of intermediate phenotypes that have clearly different morphologies. While many different phenotypes exist, the contractile and synthetic VSMCs are by far the most abundant and better researched than the others.
Contractile VSMCs are long, spindly, and contain lots of contractile filaments, while synthetic VSMCs are wider, with large extracellular matrices (Rensen et al., 2007). Contractile VSMCs are more abundant in healthy vessels than the synthetic VSMC phenotype. Furthermore, synthetic VSMCs tend to exhibit higher proliferative and migratory activity than contractile VSMCs, and increase local inflammation (Touyz et al., 2018). The VSMC phenotype and function can change in response to cell-signalling proteins, cell-to-cell contact, extracellular matrix interactions, injury stimuli, and mechanical forces. For example, immediately after an insult, VSMCs dedifferentiate, becoming more synthetic-like to promote the repair of the vessel. Then, once the injury is resolved, healthy VSMCs return to a non-proliferative, contractile phenotype (Davis-Dusenbery et al., 2011). In rare cases, VSMCs may also become hypercontractile due to excess contractile filaments which can increase the speed and amplitude of shortening (contraction) in response to stimulation and impair vasorelaxation (Ma et al., 1998).
These transitions between the different phenotypes can be transmitted and regulated at multiple levels, including gene transcription, epigenetic modification, signal generation, and transduction (Frismantiene et al., 2018). It has been suggested that the switch to and from differentiated/contractile and dedifferentiated/synthetic phenotypes is a key element of disease progression and imbalanced VSMC plasticity might lead to progression of VSMC-driven vascular disorders (Frismantiene et al., 2018). An illustration of contractile and synthetic phenotypes is presented in Figure 2 along with their key attributes.
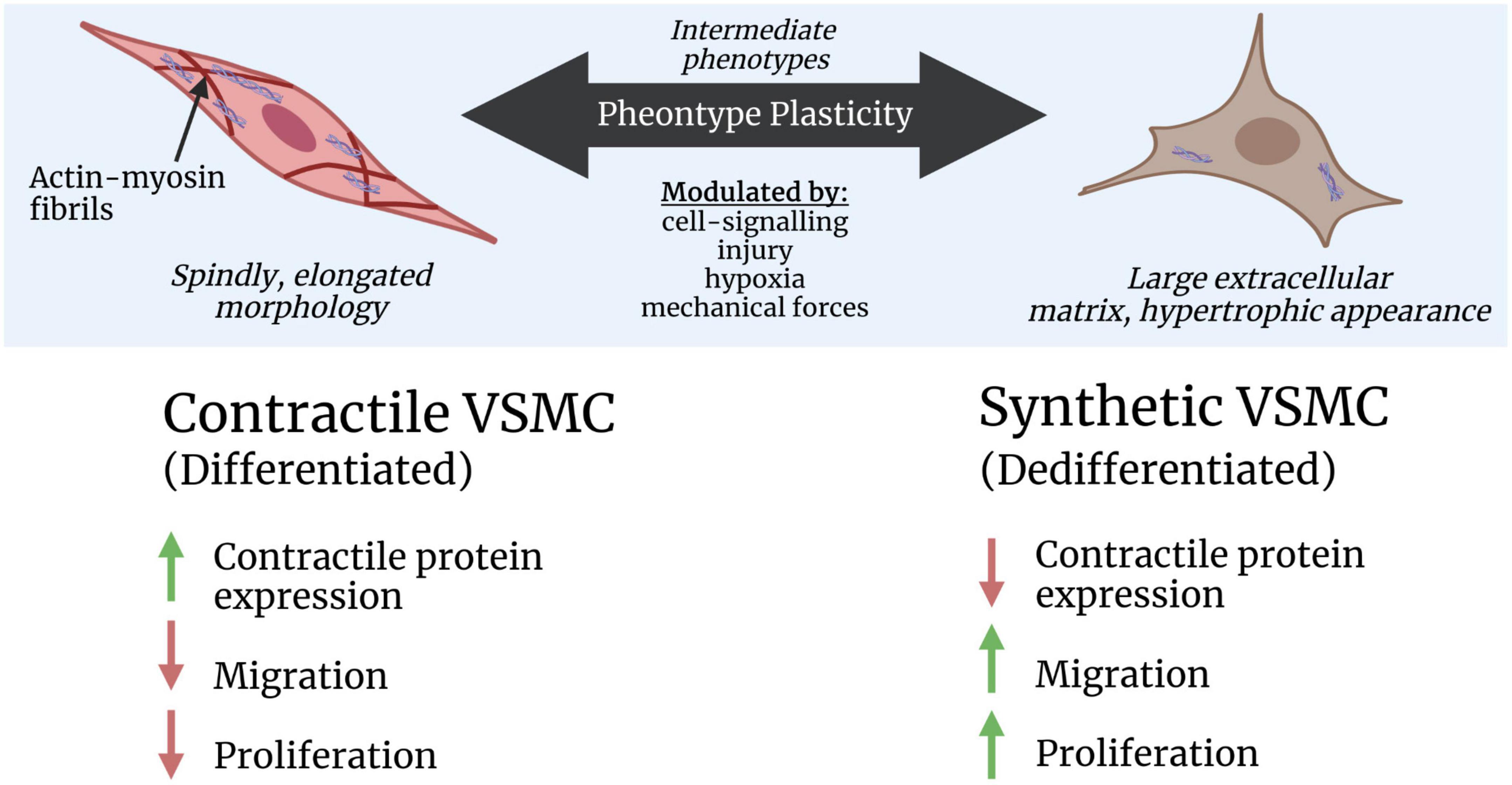
Figure 2. Simplified illustration of contractile and synthetic vascular smooth muscle cell (VSMC) phenotype plasticity. The key attributes for each phenotype are presented relative to each other, including their expression of contractile protein genes, migration, and proliferation. Higher levels are indicated by green, upward-facing arrows, and lower levels are indicated by red, downward-facing arrows. This figure was created by GH rendering using BioRender.com.
The RAS is an important mediator of VSMC contractility (Griendling et al., 1997). When RAS is stimulated, the level of renin in the blood increases and promotes the production of angiotensin II (Ang II). Ang II is essential for the function and proliferation of healthy VSMCs but also plays a role in diseased states due to its growth-promoting effects on dysfunctional cells. The two major isoforms of the Ang II receptors, type-1 (AT1) and type-2 (AT2), appear to have opposing effects. Most of the known effects of Ang II are attributed to the AT1 receptors, which have a hypertensive effect, while AT2 receptors tend to produce hypotension (St. Paul et al., 2020).
The response of VSMCs to Ang II is multiphasic, first involving the mobilisation of Ca2+ and phospholipase C (Wynne et al., 2009). Phospholipase C mediates two distinct pathways which commonly result in VSMC contraction by activating several protein kinases, including myosin light chain kinase and Rho-kinase (Hilgers and Webb, 2005). The removal of Ca2+ from the cytosol and the increase in myosin phosphatase initiate VSMC relaxation.
The prolonged effect of Ang II binded to AT1 receptors is the activation of NADH/NADPH oxidase which stimulates the intracellular formation of reactive oxygen species (ROS) such as superoxide anions (Griendling et al., 1997). While their precise mechanisms remain unexplored, ROS appear to play an important role in VSMC proliferation, DNA synthesis, and apoptosis (Rao and Berk, 1992; Tsai et al., 1996).
Notch receptors in VSMCs appear to promote the phenotypic transition toward a contractile, differentiated state and promote VSMC survival (Baeten et al., 2015; Baeten and Lilly, 2017). The Notch signalling pathway is one the prominent communication routes between vascular cells and has been found to be upregulated in VSMCs after vessel injury, particularly in VSMC regions close to endothelial cells in the regenerating endothelium (Lindner et al., 2001). Notch signalling is reiteratively used in VSMCs to positively regulate differentiation, arterial specification, and maturation. The connection between notch signalling and vasculature was first recognised when dominant mutations in the Notch3 receptor were found to be responsible for CADASIL (Joutel et al., 1996).
A pure representation of VSMC dysfunction can be identified at the molecular level both preclinically, and clinically ex vivo, however in vivo methods are necessary for the identification of VSMC dysfunction in patients when interventions may still be possible. Characterising VSMC dysfunction can be difficult in vivo due to the high number of physiological and environmental parameters affecting in vivo measurements. Here, we will discuss the most promising parameters and techniques for identifying the presence of cerebral VSMC dysfunction.
Vascular smooth muscle cells directly participate in the regulation of CBF and due to their contractile properties, they largely mediate vascular contraction and relaxation (Hill et al., 2015). Decreased baseline CBF correlates with the severity of cognitive symptoms of dementia, showing the potential contribution of CBF alteration to cognitive decline (Bracko et al., 2020).
Cerebrovascular reactivity (CVR) is an indicator of the intrinsic ability of brain vessels to contract or dilate in response to vasoactive stimuli and therefore characterises the brain’s ability to support neuronal function under stress. Changes in vessel calibre occur in response to a stimulus, such as alterations in the brain’s oxygen/nutrient requirements, or changes in the partial pressure of carbon dioxide (CO2). As a result, CVR might provide additional and more sensitive information on brain health compared with baseline CBF. For example, in a comparison of magnetic resonance imaging (MRI)-derived CVR and CBF using arterial spin labelling (ASL) in healthy adults and adults with mild cognitive impairment (MCI), whole-brain CVR in both grey and white matter was positively associated with cognitive performance in response to hypercapnia, even in the absence of a statistical relationship between baseline CBF and cognitive performance (Kim et al., 2021).
Furthermore, because the dilation of cerebral blood vessels, known as vasodilation, is induced during CVR measurements, CVR may also be more representative of VSMC function and morphology than baseline CBF.
Whole brain CBF and CVR mapping can be acquired using positron emission tomography (PET) and single-photon emission computed tomography (SPECT), however, they require the administration of radiotracers, limiting their ease of use and appropriateness in many studies. Non-invasively, CBF and CVR can be measured using MRI methodologies such as ASL and blood-oxygen-level-dependent (BOLD) imaging (Pinto et al., 2021). ASL allows direct and quantitative measurement of CBF, while the BOLD signal results from a combination of several physiological parameters (CBF, cerebral blood volume, and cerebral metabolic rate of oxygen). When using a challenge that increases the arterial blood partial pressure of CO2, it is assumed that the BOLD signal changes predominantly result from changes in CBF. It should also be noted that the BOLD signal has a better signal-to-noise ratio, higher temporal resolution, and is more widely available in conventional MRI scanners than ASL (Catchlove et al., 2018). Nevertheless, CVR and CBF quantification obtained using MRI techniques have been shown to have similar accuracy and precision to 15O H2O PET, which is considered to be the gold-standard measurement of CBF (Heijtel et al., 2014; Hauser et al., 2019). Hauser et al. (2019) found that CO2-triggerent BOLD MRI correlates strongly with 15O H2O PET acetazolamide challenge. Heijtel et al. (2014) compared ASL MRI and PET in both baseline and CO2-induced hypercapnia, and found intra- and inter-session measurements to be highly comparable with similar precision.
Transcranial doppler (TCD) ultrasound, near infrared spectroscopy (NIRS) can also be used as non-invasive and indirect measures of CBF regulation and CVR (Ainslie and Duffin, 2009; Amyot et al., 2022). TCD measures blood velocities within an artery, often the middle cerebral artery, as a proxy for CBF. Although it is inexpensive, portable, and relatively easy to use, TCD has low spatial resolution and cannot be used for regional analysis. NIRS provides a real-time assessment of fluctuations in cerebral blood haemoglobin differences in large blood vessels, which is highly correlated with CBF (Tsuji et al., 1998). It should be noted that localised pathologies may not manifest as global changes in cerebrovascular dynamics, and therefore CVR measured using TCD and NIRS might not show changes, while regional CVR measured using PET, SPECT, or MRI may. Other imaging techniques exist for the quantification of CBF in preclinical models including two-photon fluorescence microscopy and laser Doppler flowmetry which can both image the motion of red blood cells in microvasculature and at the subcellular level in the cortex in vivo (Dirnagl et al., 1989; Kleinfeld et al., 1998).
The most common vasoactive challenge used for CVR assessment involves inducting hypercapnia and/or hypocapnia. In hypercapnia, the arterial blood partial pressure of CO2 is increased leading to vasodilation and increased CBF. This is achieved by the direct effect of CO2 and decreased extracellular pH on VSMCs to open their potassium channels, resulting in hyperpolarization (Brayden, 1996). This decreases the activity of voltage-dependent Ca2+ channels, thus decreasing intracellular Ca2+ and leading to VSMC relaxation and vasodilation (Pfefferkorn et al., 2001). Hypocapnia represents the inverse, whereby the arterial blood partial pressure of CO2 is decreased leading to vasoconstriction. Cerebral vasodilation and vasoconstriction requires the orchestration of the whole neurovascular unit including endothelial cells, astrocytes, parenchymal neurons, and perivascular nerves, in addition to VSMCs (Lavi et al., 2006; Iadecola, 2013; Haidey et al., 2021). Therefore, VSMC dysfunction cannot be completely isolated under hypocapnic and hypercapnic conditions, however their functionality may still be well characterised under these conditions as they change and maintain vascular tone (Brian, 1998; Cipolla, 2009).
One well-known method of inducing hypercapnia in subjects is the inhalation of air with increased CO2 content. Different techniques have been developed in order to more precisely control CO2 concentrations, including those based on fixed inspired CO2 (Liu et al., 2017). Some more complex respiratory gas manipulation techniques allow for the precise targeting and maintenance of end-tidal carbon dioxide (PETCO2), a non-invasive surrogate for the corresponding arterial gas concentration (Spano et al., 2013). Since room air has very low concentrations of CO2, hypocapnia is usually achieved through hyperventilation to increase CO2 removal from the blood. PETCO2 is derived from air expelled from the lungs during hypocapnia or hypercapnia. PETCO2 and CVR exhibit a sigmoidal relationship which is best modelled using progressively applied levels of hypo-/hypercapnia (Bhogal et al., 2014).
Hypercapnic vasodilation can also be induced with exogenous chemicals such as acetazolamide, which can be done in a single dose with a high reproducibility. In most cases, the administration of CO2 is still preferred over intravenous acetazolamide because the final serum concentration for a given dose of acetazolamide and the cerebrovascular responses to that serum level can vary considerably between subjects. These variations are reduced at higher concentrations of acetazolamide, but higher concentrations also increase the severity and frequency of side-effects and subject discomfort (Grossmann and Koeberle, 2000).
Strategies based on breathing tasks have also been used, such as breath holding or paced deep breathing that can readily induce changes in arterial pressure of blood gases and consequently drive hypercapnia (vasodilation) and hypocapnia (vasoconstriction), respectively. Breathing tasks are often simple and inexpensive to implement, therefore they may offer an accessible option for CVR mapping. However due to the many factors that affect arterial blood CO2 other than the breathing rate, subject variability must be considered to make this a reliable stimulus to identify cerebrovascular pathology (Fierstra et al., 2013).
Many neurological disorders share a common pathological triad of vascular damage, neuronal degeneration, and neuroinflammation. This vasculo-neuronal-inflammatory triad was termed by Zlokovic and colleagues, and involved not just neurons, but brain endothelium, VSMCs, pericytes, astrocytes, and activated microglia (Zlokovic, 2011). Specifically, many brain disorders share VSMC dysfunction as a mechanism for the progression of neurodegenerative pathology. Although there may be overlap between many of these pathologies, here we discuss and summarise existing research on the role of VSMCs in prevalent neurodegenerative disorders.
Alzheimer’s disease is characterised by progressively impaired memory, cognition, and behavioural changes, and it is the most common cause of dementia. Of the three polymorphic alleles of the human apolipoprotein E (APOE) gene, ε2, ε3, and ε4, the presence of APOE ε4 has been identified as the most prominent genetic risk factor for late-onset AD (Bertram et al., 2007). Notably, increasing evidence indicates that APOE genotypes differentially modulate cerebrovascular function, and that APOE ε4 induces detrimental cerebrovascular effects including reduced CBF, increased BBB leakiness, and disrupted transport of nutrients and toxins (Alonzo et al., 1998; Zerbi et al., 2014; Halliday et al., 2016).
Decreased VSMC density was identified in the blood vessels of the arachnoid, grey matter, and white matter of patients with AD compared with age-matched, healthy controls (Ervin et al., 2004). Patients with AD expressing the APOE ε4 gene also have less smooth muscle actin immunoreactivity, associated with more severe AD, than patients with AD expressing APOE ε3 (Ervin et al., 2004). Furthermore, APOE ε4 is associated with impaired intramural periarterial drainage (IPAD) in mice (Hawkes et al., 2012).
Studies have reported that in patients with AD and mouse models of AD, the overexpression of molecules that control the rate of cell transcript in cerebral VSMCs generates a hypercontractile VSMC phenotype with reduced clearance receptors for the notorious amyloid-beta (Aβ) peptides (Chow et al., 2007; Bell et al., 2009). Autoimmune response mechanisms may also be involved in the degeneration of VSMCs during the development of AD. For example, an increase in anti-smooth muscle antibodies was found to be strongly correlated with brain atrophy in patients with early AD (Giulia et al., 2015).
Furthermore, specific genes have been associated with changes in VSMC in AD. An upregulation of synthetic-VSMC genes has been found in patients with AD, while the expression of VSMC contractile-genes is significantly reduced in AD cerebral pathology compared to healthy brains (Owens et al., 2004; Mack, 2011; Frismantiene et al., 2018). Notably, changes in the gene encoding for the sarco-endoplasmic reticulum calcium ATPase isoform 2 (SERCA2), a gatekeeper of normal VSMC function, have been associated with AD (Britzolaki et al., 2020). The overexpression of SERCA2b was shown to lead to elevated Aβ production, while the knockdown of SERCA2b caused a drastic decline in Aβ (Green et al., 2008). Gallego et al. (2018) further support the importance of maintaining SERCA activity and show that dysfunctional SERCA can have damaging effects on VSMC Ca2+ homeostasis causing AD-associated neuronal cell death.
Growing evidence shows that cerebrovascular contractility may be impaired in AD as well as in MCI, a stage of cognitive decline more severe than what is expected through normal ageing, but less serious than the decline seen in AD. People with MCI are significantly more likely to progress to AD and dementia than healthy adults and are therefore a group of interest for better understanding the development and progression of AD (Flanagan et al., 2016). Several studies have shown that neurocognitive decline in MCI and AD is paralleled by reduced CVR and deficits in vascular contractility, identified using hypercapnic challenge in CO2 BOLD MRI, TCD, and PET imaging (Vicenzini et al., 2007; Cantin et al., 2011; Yezhuvath et al., 2012; Richiardi et al., 2015). A recent investigation using ASL MRI showed that impaired CVR in grey matter regions and the whole brain white matter may be an early imaging biomarker revealing the relationship between cerebrovascular function and cognitive decline (Kim et al., 2021). However, other studies have shown that baseline CBF and global CVR are preserved in AD (Jagust et al., 1997; Rodell et al., 2012; Glodzik et al., 2013). Rodell et al. (2012) found that after factoring out the effects of CO2 on blood flow, people with AD no longer showed significant inter-subject global CBF variability, though reduced water and oxygen clearance was reduced in specific regions of the brain. While clinical studies are not yet conclusive, there is a substantial body of research that highlights that CVR may be a prospective means to detect early vascular dysfunction in subjects at risk (Glodzik et al., 2011). However, it is also clear that the way in which CVR is measured must be standardised to make it clinically useful.
A common pathological feature associated with AD is cerebral amyloid angiopathy (CAA). CAA results from the build-up of Aβ in the basement membrane of VSMCs in cerebral arteries and the basement membrane of cortical capillaries. Studies have demonstrated that VSMCs can undergo dramatic phenotypic transitions in CAA-like neuroinflammatory conditions, adopting synthetic and proliferative phenotypes (Aguilar-Pineda et al., 2021b). Additionally, these phenotypic transitions often occur in conjunction with tau protein hyperphosphorylation and accumulation in the brain (Aguilar-Pineda et al., 2021b).
Aldea et al. (2019) have studied the impact of vasomotion, a rhythmic relaxations and contractions of cerebral vasculature of ∼0.1 Hz, on the IPAD mechanism in CAA, and propose that the failure of the vasomotion-driven IPAD mechanisms are responsible for the progression of CAA (Rayshubskiy et al., 2014). Their mathematical models showed that the contractile VSMCs of cerebral arteries act as the drivers of IPAD in the brain by inducing basement membrane deformation in the direction of the vasomotion, contributing to the drainage of fluid and soluble metabolites from the brain. It has been suggested that the force of contractile VSMCs in arteries and arterioles contribute to drainage along the IPAD pathway all the way down to the capillary level, and that impaired VSMCs could result in the accumulation of Aβ in the walls of macro- and microvasculature, contributing to CAA pathology (Aldea et al., 2019). In CAA, Aβ peptides may also contribute to the transition of VSMCs from contractile to synthetic phenotypes. One study showed that VSMCs exposed to Aβ1-40-peptide, the main amyloid peptide found to accumulate in the vessel wall in sporadic forms of CAA before the addition of proinflammatory cytokines, was greatly intensified compared to those only exposed to the cytokines (Vromman et al., 2013). Additionally, changes in transcription factors were suggested as a possible path leading to VSMC phenotypic changes and vascular dysfunction in CAA.
Further research into VSMC function and signalling in AD and CAA pathologies is required to better understand the mechanisms underlying vascular and neuronal degradation. However, the cumulation of these findings support that VSMC dysfunction may be a potential biomarker for characterisation or treatment during the development and progression of disease.
Parkinson’s disease is characterised by the presence of intracellular Lewy bodies containing alpha-synuclein (aSyn) fibrils, which are thought to lead to the cell death of dopaminergic neurons in the substantia nigra, affecting motor control, tremors, and gait (Yasuda and Mochizuki, 2010). The mechanism by which aSyn causes neurodegeneration is unclear, however these fibrils are linked to mural cell function because VSMCs and pericytes appear to be able to take up, secrete, and transfer aSyn (Tamo et al., 2003; Fields et al., 2019). A loss in BBB integrity has been observed post-mortem and it has been suggested that aSyn may be responsible for the breakdown of mural and endothelial cells in the BBB (Dohgu et al., 2019).
The specific role of VSMC dysfunction is not yet clear in PD pathologies. One study showed that the global response of CBF and CVR did not differ significantly between patients with PD and healthy controls, though a correlation was identified between increased CBF in posterior regions of the brain and severity of cognitive impairment in the PD group (Al-Bachari et al., 2014). Another study also did not find any significant difference between the whole-brain CBF and CVR measurements between patients with PD and health controls (Pelizzari et al., 2019). However, similar to Al-Bachari et al. (2014), Pelizzari’s group found that regional increases in CBF correlated with the severity of PD motor symptoms, most significantly in the postcentral gyrus. Furthermore, significant negative correlations were found between CVR and PD severity, corrected for age, in specific regions of the brain, such as the corpus striatum, which is involved in the generation and inhibition of movement.
Genetics also play a role in PD pathogenesis, such as protein deglycase Dj-1 and NDUFV2 (Rui et al., 2018; Lin et al., 2021). Dj-1 is a gene accountable for the autosomal recessive early onset form of PD and is multifunctional (Bandopadhyay et al., 2004; Clements et al., 2006). Mutations in Dj-1 result in neurodegeneration, leading to an early onset familial form of PD (Bonifati et al., 2003). Dj-1 also responds to oxidative stress in VSMCs, inhibits hyperplasia, and maintains vasorelaxation by participating in endothelial NO synthesis, though the effects of Dj-1 in VSMC phenotype switching remain unclear (Won et al., 2013). The NDUFV2 gene, also involved in AD pathogenesis, contributes to VSMC communication and may play a role in phenotypic switching to synthetic, pro-inflammatory phenotypes (Gan et al., 2017). Furthermore, like with AD, changes in the gene encoding for SERCA2 have recently been associated with aSyn and PD pathology. The investigators proposed that abnormal accumulation of aSyn could lead to intracellular Ca2+ dyshomeostasis due to the downregulation of SERCA2 activity in preclinical models (Tuon et al., 2012).
Lewy body dementia is a type of dementia in which the same aSyn Lewy bodies found in PD clump in the cytoplasm of neurons, which disrupts the production of dopamine and causes cognitive decline often as well as motor symptoms such as muscle weakness and rigidity. The role of VSMCs in LBD has been very minimally studied and is largely based on evidence of changes to general cerebrovascular function. Reductions in CBF and microvessel density associated with deficient vascular endothelial growth factor have been described in the occipital cortex of LBD patients compared with controls, though it was unclear whether this was secondary to the accumulation of aSyn (Miners et al., 2014). The contribution of hypoxia and hemodynamic changes has been hypothesised based on the high levels of cortical microinfarcts in LBD which is also found in CAA and vascular dementia (Reuck et al., 2014). LBD patients appear to experience a significant inflammatory response and vascular abnormalities, which exacerbate Lewy body-induced neuropathology, though specific research on VSMCs in LBD has not yet been explored (Raz et al., 2015). Changes in CVR and CBF mechanisms have yet to be investigated in patients with LBD.
Amyotrophic lateral sclerosis is a progressive neurodegenerative disease of the motor system characterised by focal to generalised weakness leading to paralysis and respiratory failure (Peters and Brown, 2015).
Patients with ALS can exhibit more than a 50% reduction in pericyte numbers in the spinal cord and a significant increase in platelet-derived growth factor (PDGF) expression, a potent protein, which enhances the rate of cell division and is involved in VSMC recruitment, differentiation, and homeostasis (Winkler et al., 2013). PDGF also induces VSMC phenotypic switching from a contractile to proliferative and proinflammatory state, and it has been suggested that this plays an important role in atherosclerosis (Krettek et al., 1997; Zhao et al., 2011).
There have been very few studies that have investigated the role of VSMC dysfunction in ALS pathology, however it has been speculated that VSMCs could be involved in ALS due to upregulation of Ang II in the CNS (Sorrentino et al., 2022). This is accompanied by a significant reduction in Ang II found in cerebrospinal fluid (CSF) from patients with ALS, which has been associated with disease severity and progression rates (Kawajiri et al., 2009). This indicates that changes to RAS and Ang II levels may induce VSMC dysfunction and ALS pathology, but more research is required to better understand the mechanisms and integrity of VSMCs and cerebrovascular dynamics in ALS.
Multiple sclerosis is a complex inflammatory autoimmune disease, largely affecting the white matter of the CNS characterised by demyelination, axonal loss, and the formation of focal and diffuse lesions (Hernandez et al., 2014, p. 52).
Compared with other neurodegenerative diseases, the role of VSMCs in MS progression is not very well understood, though it’s clear that Ang II plays a role in the pathogenesis of MS. Enhanced levels of Ang II in the CNS are culprits of vascular disease and inflammation, and are also seen in MS along with decreased CSF Ang II and increased serum angiotensin-converting enzyme (Guzik et al., 2007; Matsushita et al., 2010). Increased CNS Ang II was also found in a mouse model of MS accompanied by an upregulation of AT1 receptors in MS brain lesions (Platten et al., 2009). In VSMCs, upregulated AT1 can promote hypertensive effects and increase ROS activity.
A significant decrease in grey matter CVR has been shown in patients with MS compared to healthy controls (Marshall et al., 2014). The same study found that impaired CVR was significantly correlated with increased lesion volume and grey matter atrophy, suggesting that impaired vasodilation may be an underlying cause of neurodegeneration in MS. More research is needed to properly understand the role and mechanisms of VSMC dysfunction and altered vascular dynamics in MS.
Huntington’s disease is an autosomal-dominant neurodegenerative disorder affecting the CNS, which can lead to involuntary choreiform movements, personality changes, and dementia (Parsons and Raymond, 2015).
The role of VSMCs in HD is not yet conclusive and is largely based on preclinical studies. Models using mice expressing the huntingtin gene indicate that vascular dysfunction might develop earlier in life in the HD mice than in wild-type mice (Rahman et al., 2013). The HD mice presented with reduced contractility in arteries of varying vascular beds, attributed to altered Ca2+ fluctuations in the arterial VSMCs. However, in a different HD mouse model, the response to potassium chloride was tested as a global measure of vascular smooth muscle structure, mass, and function, and no difference was found compared to wild-type mice (Kane et al., 2016).
Studies have also investigated the role of myosin phosphatase target subunit 1 and Rho kinase in HD (Narayanan et al., 2016). Myosin phosphatase target subunit 1 is essential for VSMC relaxation through the regulation of myosin light chain phosphatase, and Rho kinase contributes to Ca2+ sensitization in VSMC contraction. Both the myosin phosphatase target subunit 1 and Rho kinase were significantly increased in patients with HD and were found to lead to increased apoptotic cell death (Narayanan et al., 2016). More research is required to better understand the implications of VSMC dysfunction and their molecular pathways in HD.
Down’s syndrome is caused by complete or partial trisomy of chromosome 21, frequently causing intellectual disability and reduced lifespan (Parra et al., 2017). People with DS are at a high risk of developing AD at an early age, but are much less likely to develop cardiovascular disease than those without, despite DS being associated with premature ageing, the early onset of AD, and a higher prevalence of risk factors for cardiovascular disease such as obesity, dyslipidemia, and sedentarism (Murdoch et al., 1977; Sobey et al., 2015; Parra et al., 2017).
Structural MRI studies have shown notable increases in cerebrovascular pathology in people with DS, including lobar microbleeds, infarcts, and white matter hyperintensities (WMHs), not strongly associated with Aβ pathology (DiProspero et al., 2022). CBF is significantly lower among adults with DS with probable AD compared with controls and participants with DS without dementia when measured using pulsed ASL (Thalman et al., 2020). The implications of VSMCs on these changes have not yet been explored.
Two genes are found on chromosome 21 in DS that appear to play a role in vascular health, neural development, and the onset of AD: DYRK1A and RCAN1 (Roy-Vallejo et al., 2020). DYRK1A has recently been shown to decrease neprilysin enzyme levels in subjects with DS (Kawakubo et al., 2017). Neprilysin degrades natriuretic peptides and vasodilators, which are negatively correlated with VSMC migration and proliferation, and the inhibition of neprilysin is related to the accumulation of Aβ peptides and the development of AD in adults with DS (Garg and Hassid, 1989; Miners et al., 2011; Karoor et al., 2013). Interestingly, it has the opposite effect in cardiovascular disease where its inhibition has been shown as an effective treatment for heart failure and hypertension (von Lueder et al., 2014; Vodovar et al., 2015). The effect of RCAN1 on VSMC phenotype is unclear, though it appears to play a role in VSMC migration. The genetic ablation of RCAN1 in mice led to decreased VSMC migration and resistance to Ang II induced aneurysm and restenosis (Esteban et al., 2011). In contrast, endogenous RCAN1 knockout was found to increase migration in cancer cells in vitro, whereas the expression of exogenous RCAN1 reduced migration (Espinosa et al., 2009). RCAN1 thus appears to have different effects on cell migration depending on the setting.
Here we review the existing research on the role of VSMCs in pathologies that are well known to present a clear vascular component: CSVD, CADASIL, stroke, PSD, and MMD.
Cerebral small vessel disease is composed of several diseases affecting the small arteries, arterioles, capillaries, venules, and small veins, which may include genetic, idiopathic, infectious, and immune-related pathologies (Cannistraro et al., 2019). While the manifestation and progression of CSVD is broad reaching, VSMC migration and hypertrophy due to the upregulation of Ang II and leading to increased ROS may be a common link to vascular remodelling and dysfunction (Virdis et al., 2004). In preclinical and in vitro studies of Ang II-induced CSVD, ROS have been shown to alter blood pressure, vascular structure, and collagen deposition (Griendling et al., 1994; Virdis et al., 2004).
Beyond investigating the role of Ang II and ROS in VSMC function in CSVD, much of the research on VSMCs in CSVD has been based on CVR and CBF studies. It has recently been demonstrated that patients with advanced hypertensive CSVD have impaired CVR in the basal ganglia, temporal lobe, and frontal lobe, determined using ASL MRI with an intravenous vasoactive stimulus (Lee et al., 2021). The same study found that the reduced vasoconstriction was significantly associated with conventional MRI markers of CSVD, including cerebral microbleeds, WMHs, and deep lacunar infarcts. Furthermore, lower CVR determined using BOLD MRI under hypercapnic challenge was found to be indicative of WMHs and perivascular spaces associated with CSVD when controlling for patient characteristics (Blair et al., 2020). Another BOLD MRI study showed that areas of reduced CVR precede the progression of normal-appearing white matter to WMHs suggesting that impairment of vascular contractility may contribute to the pathogenesis and progression of CSVD (Sam et al., 2016). A systematic review of CSVD and ischaemia associations showed that baseline CBF was also negatively related to WMH severity linked to CSVD, and Shi et al. (2016) noted that investigating CVR in patients with CSVD may provide new insights.
Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy is a rare, hereditary CSVD characterised by VSMC degradation and the accumulation of Notch3 receptors on the VSMCs of small and middle-sized arteries (Joutel et al., 2000, 1997). A common pathological feature of CADASIL is the dramatic reduction of VSMCs in cerebral arterioles walls, which may be attributed to irregular mitochondrial proliferation and function in CADASIL VSMCs (Viitanen et al., 2013).
In a study comparing proteomic expression of cultured CADASIL and control VSMCs isolated from human umbilical cord, it was found that CADASIL is likely caused by misfolded Notch3 molecules (Ihalainen et al., 2007). This impairs the Notch3 signalling cascade, increases ROS, inhibits cell proliferation, and degrades VSMCs. Ihalainen et al. (2007) found that this also induces the overproduction of collagen type I, leading to fibrosis, vascular stenosis, and ischaemic lesions as seen in patients with CADASIL. Similarly, cultured CADASIL VSMCs have been shown to have a lower proliferation rate compared to control VSMCs and also showed an increased gene expression of transforming growth factor-beta that, when neutralised, corrected the proliferation rate (Panahi et al., 2018).
The CVR and mean CBF in the middle cerebral artery, measured using TCD and MRI, have been shown to be reduced in CADASIL compared with controls, attributed to VSMC dysfunction (Pfefferkorn et al., 2001; Liem et al., 2009; Moreton et al., 2018; Atwi et al., 2019). Pfefferkorn et al. (2001) also found reduced CO2 reactivity in patients with CADASIL before they became disabled, suggesting that CVR plays an early role in the evolution of the disease. In an MRI analysis using an intravenous acetazolamide vasoconstrictive challenge, CVR was significantly reduced in CADASIL and was identified as an important determinant of development of WMHs (Liem et al., 2009). Furthermore, an overall decrease in relative CBF and cerebral blood volume, before and after an acetazolamide challenge, were detected in patients with CADASIL when measured using MRI (Chabriat et al., 2000).
There have also been studies that found that CVR was not impaired in patients with CADASIL when measuring large arterial flow using MRI and TCD (van den Boom et al., 2003; Singhal and Markus, 2005). Both of these studies found that baseline CBF was lower in subjects with CADASIL than in controls. As a result, van den Boom et al. (2003) suggested that flow impairment may give rise to the development of WMHs and lacunar infarcts in patients with CADASIL.
More research specifically on the contribution of VSMCs to vascular changes in CSVD and CADASIL is essential to better understand the underlying mechanisms of these disorders.
Ischaemic strokes are caused by a sudden blockage of blood flow to an area of the CNS. Having a stroke doubles the risk of dementia and the prevalence of PSD has been predicted to increase in the future because of better survival after stroke and ageing of the population (Leys et al., 2005). VSMCs appear to play an important role in the early development of ischaemic stroke and PSD due to their role in stabilising BBB integrity and perfusion, and maintaining healthy vasculature (Meng et al., 2021).
Phenotypic switching of VSMCs to a synthetic state has been shown to induce BBB disruption after ischaemic stroke in mice (Meng et al., 2021). Meng et al. (2021) found that a deficiency in myosin phosphatase target subunit 1 stimulated the phenotypic switching to a synthetic, pro-inflammatory state in human brain VSMCs and after ischaemic stroke in mice. It was also found that modifications in VSMC phenotype after stroke in vivo can impact the incidence, severity, pattern, and outcome of ischaemic stroke (Poittevin et al., 2014).
Furthermore, stroke-induced hypoxia can lead to the overexpression of amyloid precursor protein (APP) in VSMCs, which could exacerbate or expedite the development and progression of CAA, worsen stroke outcome, and lead to PSD (Rensink et al., 2003). Notably, before the deposition of Aβ in the brain and blood vessels, mice with APP mutations show reduced hypercapnic CVR and altered neurovascular coupling (Niwa et al., 2000). Zlokovic proposed a model to explain how cerebrovascular diseases, such as stroke, can lead to AD and dementia (Zlokovic, 2008). He proposed that stroke-induced hypoxia compromises the BBB and the overexpression of APP alters the balance between Aβ production and clearance in favour of production, which induces neuroinflammation. As a result, this may lead to synaptic dysfunction, and the accelerated onset of CAA and PSD.
Atherosclerotic plaque stability, a leading cause of ischaemic stroke, also appears to be highly dependent on the VSMC phenotype, which may either undergo apoptosis or activate the production of inflammatory mediators that can trigger plaque rupture and thrombosis (Galis and Khatri, 2002; Herrington et al., 2016). In an experimental focal cerebral ischaemia model, simvastatin promoted arteriogenesis through increased Notch receptor expression in the ischaemic cerebral arteries and peri-infarct area and it was suggested that the regulation of VSMC function through Notch signalling may be a determinant of the outcome of cerebral ischaemic disease (Zacharek et al., 2009).
Further assessment of the role of VSMCs in ischaemic brain disease and PSD may offer a new avenue for understanding the development and severity of stroke, and its progression to AD and dementia.
Moyamoya disease is a rare cerebrovascular disease characterised by progressive stenosis or occlusion of the terminal portion of the internal carotid arteries (Kuroda and Houkin, 2008).
Vascular smooth muscle cells in patients with MMD have shown specific differentially expressed contractile protein genes such as increased expression of smooth muscle actin and myosin heavy chain, compared with healthy control volunteers (Kang H. -S. et al., 2014). Notably, these mutations are related to VSMC adhesion, cell migration, immune response, and vascular development. The increased expression of α-smooth muscle actin may be involved in the increased proliferation of VSMCs, contributing to MMD stenosis (Guo et al., 2009). Post-mortem analysis of patients with MMD has shown degeneration in the cerebral VSMCs, and migration and morphological changes of VSMCs in the thickened intima (Lin et al., 2012).
It has also been suggested that endothelial cell dysfunction contributes to the early development of MMD and that VSMCs may require additional factors to contribute to the pathophysiology of MMD. In the analysis of VSMCs from neural crest stem cells, MMD patients and healthy control subjects were observed to have similar VSMCs proliferation, migration and contractile abilities, in contrast to the endothelial cells, which displayed distinct transcriptome profiles (Tokairin et al., 2020).
Studies have shown a significant CVR impairment in patients with MMD, based on ASL MRI and PET measurements following the injection of vasodilator acetazolamide (Deckers et al., 2021; Zhao et al., 2022). These studies showed that the CBF of patients with MMD did not increase after the injection of acetazolamide, suggesting that the cerebral vessels were already at maximal vasodilation to compensate for CBF reduction. Heyn et al. (2010) also identified decreased regional CVR in patients with MMD using CO2 BOLD MRI. Interestingly, unilateral surgical revascularization, a widely used preventative treatment for MMD, was found to improve PET-derived CVR in MMD patients not only in the hemisphere ipsilateral to the flow restoration, but also in the non-intervened hemisphere (Sam et al., 2015; Deckers et al., 2021). It remains that more research is required to better understand how and at what stage VSMC dysfunction may contribute to MMD development and progression.
Extrinsic influences such as lifestyle choices, environmental effects, and traumatic events can impact the function, migration, and proliferation of VSMCs, and are likely to be factors in the onset and progression of neurodegenerative disorders. Many of these have been identified as risk factors for cerebrovascular and cardiovascular disorders as well as neurodegenerative disorders (Livingston et al., 2020). Understanding how extrinsic factors may influence VSMCs may inform novel interventions and therapies for preventing and arresting neurodegeneration.
Toxins such as those introduced to the body through cigarette smoking, alcohol or drug consumption, or air pollution are major risk factors for the early onset of neurodegenerative disorders (Mash et al., 2003; Cataldo et al., 2010; Sabia et al., 2018; Jankowska-Kieltyka et al., 2021). Exposure to toxins can also lead to profound remodelling and apoptosis of cerebral VSMCs.
Exposing cultured cerebral VSMCs to cigarette smoke extract was found to induce phenotypic switching to a pro-inflammatory, synthetic state, and suggested an avenue for the major risk that cigarette smoking causes for cerebral vascular injury such as atherosclerosis and stroke (Starke et al., 2013). In a preclinical study, nicotine exposure has been shown to stimulate abnormal growth of VSMCs and fibroblasts due to an enhanced Ang II type 1 receptor-mediated functional response (Li J. -M. et al., 2004). The same study showed that nicotine can also cause phenotypic changes of contractile VSMCs to synthetic VSMCs. The effects of nicotine on VSMCs in humans is not yet conclusive, but it has been proposed that nicotine can induce imbalances in RAS, which can cause VSMC dysfunction (Oakes et al., 2018).
High concentrations of ethanol have been shown to increase neuronal apoptosis due to increased ROS and induced apoptosis of cerebral VSMCs (Li W. et al., 2004). Similarly, exposure to even small doses of cocaine induces rapid apoptosis in cerebral VSMCs, contributing to the development of stroke and neurodegeneration (Su et al., 2003). Exposure to smaller concentrations of ethanol has been found to inhibit Notch signalling and, therefore, decrease proliferation and migration in VSMCs in vitro and during vessel remodelling in response to flow reduction in vivo in mice (Hendrickson et al., 1998; Morrow et al., 2010). In these studies, VSMC proliferation was found to increase when Notch pathways were artificially turned back on. Morrow et al. (2010) suggest that the inhibitory effect of moderate alcohol on VSMC growth may prevent vessel thickening associated with atherosclerosis and stroke. However, recent studies suggest that no level of alcohol consumption is safe in humans. These studies show that even moderate alcohol consumption has been associated with ROS generation, hypertension, stroke, vascular alterations in the peripheral and central nervous systems, cognitive decline, and more (Gill et al., 1986; Roerecke et al., 2017; Cho et al., 2022; Ohlrogge et al., 2022; Topiwala et al., 2022).
Exposure to air pollutants and ambient particulate matter has been shown to induce the transitioning of VSMCs from contractile to synthetic phenotypes, increasing cell migration and expression of proinflammatory cytokines (Ho et al., 2021). Furthermore, organic matter from ambient particulates and polycyclic aromatic hydrocarbons induce VSMC migration through ROS generation leading to vascular pathology (Ju et al., 2022). NO is a common free radical which is both present in the environment and can be produced in the body as a vital signalling molecule. In the blood vessels, NO decreases the intracellular concentration of Ca2+ and causes VSMC relaxation, and is thus a potent vasodilator (Zhao et al., 2015). In the environment, NO can react with other gases in the atmosphere to form nitrogen dioxide (NO2) and nitrous oxide (N2O). Notably, evidence is emerging that air pollution, including high ambient particulate matter, NO, and carbon monoxide, is also associated with increased risk of dementia and vascular damage (Cerza et al., 2019; Peters et al., 2019). More research on the effect of poor air quality on cerebral VSMCs and in neurodegenerative pathology is required to better understand the neurotoxic effect of air pollution.
Pulsatility is generated by oscillations in arterial blood flow and pressure that occur with each heartbeat. Normal haemodynamic pulsatility can be affected by age, hypertension, diabetes mellitus, hyperlipidaemia, and haematocrit levels (Jeong et al., 2018). Pulsatility factors such as blood flow-induced vessel wall shear stress, cyclic strain, and hydrostatic pressure can affect the function and phenotype of VSMCs, which has been attributed to vascular disorders (Shi and Tarbell, 2011). A recent review by Thorin-Trescases et al. (2018) suggests that age-related increased arterial stiffness may promote high pulse pressure in cerebrovasculature that leads to functional and structural alterations that could promote neuronal dysfunction and cognitive decline. Notably, a chronic increase in pulse pressure has been associated with cognitive decline and dementia (Scuteri et al., 2007; Pase et al., 2016).
Vascular smooth muscle cells have multiple sensing mechanisms to perceive mechanical stimuli generated from pulsatile stretch and transduce signals resulting in the modulation of gene expression, and cellular functions such as proliferation, apoptosis, migration, and remodelling (Haga et al., 2007; Qiu et al., 2014). A recent review found that high pulsatile shear stress induces VSMC apoptosis due to the increased release of NO from endothelial cells and low shear stress increases VSMC proliferation and migration through endothelial-released platelet-derived growth factor (Qiu et al., 2014). Other studies have shown that both the pressure amplitude and frequency of pulsatile flow can affect VSMC morphology and alterations in the cyclic oscillations modulated VSMC function and tissue stiffness (Neutel et al., 2021; Meki et al., 2022).
While more research is needed to understand the vascular changes that occur in response to pulsatile load, the effects of pulsatility on blood vessel contractility and VSMC function are notable and warrant further investigation in healthy and pathological states.
Traumatic brain injury (TBI) leads to an increased risk of progressive neurodegeneration and dementia (Graham and Sharp, 2019). While the mechanisms for this are not yet clearly understood, it has been shown that TBI results in a loss in cerebrovascular tone and changes in the function of VSMCs and the neurovascular unit, which may contribute to the increased risk of neurodegenerative disease.
Traumatic brain injury has been reported to increase concentrations of endothelial-released NO, severely blunting pressure-induced vasoconstriction (Villalba et al., 2014). This was found in conjunction with a decrease in VSMC Ca2+ levels, which collectively led to VSMC apoptosis and profound cerebral artery dilation after TBI.
In preclinical studies, it has been demonstrated that TBI reduces gap junction coupling in VSMCs through mechanisms related to increased ROS (Yu et al., 2014). Substantial increases in hydrogen peroxide were found in VSMCs of cerebral arteries after TBI leading to decreased pressure-induced constriction of cerebral arteries in rats (Szarka et al., 2018). The impaired VSMC contractility was restored after the VSMCs were treated with a mitochondria-targeted antioxidant that blocked specific ion signalling channels in VSMCs.
A study of patients with blast-induced TBI found an overexpression of mRNA markers in contractile VSMCs and suggested that TBI leads to phenotype switching mechanisms in VSMCs of cerebral arteries, as well as prolonged hypercontraction and vasospasm (Alford et al., 2011). Alford et al. (2011) proposed that trauma causing small perturbations in tissue tension results in contractile-dominant VSMC phenotypes, while larger perturbations shift VSMCs toward a synthetic phenotype, causing large-scale remodelling.
The key to developing novel therapeutic treatments for neurodegenerative disorders depends on the understanding and investigation of the underlying molecular mechanisms. Some of these are genetic, such as in HD, DS, and CADASIL, however others such as AD, PD, ALS, and MS, can occur sporadically, and their mechanisms have been harder to establish.
Despite their variability in symptoms and progression, VSMC dysfunction appears to be a common link between these disorders and could prove to be a promising target for intervention. Studies have shown that targeting the vascular tissue with drugs may be effective at modulating and/or preventing neurodegenerative disorders such as AD and PD (Speer and Ratan, 2009; Aguilar-Pineda et al., 2021a). It has also been suggested that intervening during the differentiation, proliferation, and/or migration mechanisms of VSMCs could be a therapeutic strategy to reduce pathological VSMC functions while maintaining their stabilising roles in CBF (Worssam and Jørgensen, 2021). Here, we will review recent insights on VSMC-based therapeutics in vascular pathology and discuss how VSMCs could be targeted to intervene in the development and progression of neurodegenerative disorders.
It has been proposed that targeting VSMC signalling mechanisms could be protective against the onset of neurodegeneration (Marsboom and Archer, 2008; Lin et al., 2021). Notably, increased VSMC proliferation and migration exhibited in AD, ALS, and MMD can be harmful to cerebrovascular function and may contribute to the accumulation of toxic plaques in the central nervous system (Krettek et al., 1997; Guo et al., 2009; Aguilar-Pineda et al., 2021b). In other pathologies, such as CADASIL, a dramatic reduction in VSMC proliferation can lead to vessel thinning and reduced CVR amplitude (Liem et al., 2009; Panahi et al., 2018). VSMCs have several signalling pathways to control cell function and proliferation, however a few players have been identified as key contributors in VSMC proliferation.
Vascular smooth muscle cell proliferation and migration can be manipulated by inhibiting the degradation of cyclin-dependent kinase (CDK) with pharmaceuticals like thiazolidinediones, which have been developed for treating type II diabetes (Wakino et al., 2000). These drugs are also peroxisome proliferator-activated receptor γ (PPARγ) agonists and the activation of PPARγ in VSMCs has been shown to inhibit synthetic VSMC proliferation and migration, which may have vascular-protective effects (Law et al., 2000; Yang et al., 2013). The CDK pathway is also involved in telomere activity, which stabilises chromosomes. Telomerase reverse transcriptase (TERT) phosphorylation, which is upregulated during and following hypoxia, has been shown to increase VSMC proliferation (Minamino et al., 2001). Some evidence has shown that inhibiting TERT in vivo, such as via the CDK pathway, may reduce vascular disease (Fuster and Andrés, 2006).
The transient receptor potential vanilloid (TRPV) family is expressed abundantly in cerebral artery VSMCs and has been shown to be an important mediator of vascular tone (Guibert et al., 2011). The overexpression of TRPV1 has been found to inhibit Ang II induced VSMC migration and phenotype transitions from a contractile to secretory state (Wang and Jia, 2020). Another study found that activated TRPV1 inhibited VSMC proliferation, migration, and ROS production by upregulating the expression of PPARα (Zhou et al., 2021). Additionally, TRPV4 channel activity has been found to increase vasoconstriction and elevate resting blood pressure in mice and patients with hypertension (Chen et al., 2022).
RhoA/Rho-kinase have been shown to modulate VSMC phenotype switching including affecting proliferation, dedifferentiation, apoptosis, and migration in VSMCs, which is discussed extensively in the review paper by Sawma et al. (2022). Notably, the inhibition of Rho signalling using fasudil hydrochloride is currently used clinically for the treatment of vasospasm (Montagnoli et al., 2021). It has been suggested that it may also reduce inflammation and lesion volume in MS (Aktas et al., 2010).
Interestingly, berberine has been increasingly found to have vascular- and neuro-protective effects against neurodegenerative pathology, which could be attributed to its effects on signalling pathways (Kumar et al., 2016; Ye C. et al., 2021). Berberine is an organic compound in classical natural medicine, proven by modern research to have various pharmacological effects, including antiviral, anti-inflammatory, and lipid regulation (Ye Y. et al., 2021). Preclinically, berberine was found to affect smooth muscle contractility by regulating the VSMC handling of intracellular Ca2+ (Ma et al., 2016). In vitro, berberine inhibited early signalling pathways induced by VSMC injury, reducing the contraction and proliferation of dysfunctional VSMCs (Liang et al., 2006). Other studies have shown that berberine inhibits induced VSMC proliferation via the PPARγ signalling pathway by stimulating a beneficial increase in NO (Calvier et al., 2017; Qiu et al., 2017).
Notch signalling has gained significant attention in the context of vascular biology and has been identified as a possible culprit for VSMC and pericyte reduction seen in some neurodegenerative disorders (Lin et al., 2021). As mentioned earlier, Notch3 is a gene involved in the pathogenesis of CADASIL, though it is currently not known if the mutations in Notch3 are causative (Ihalainen et al., 2007). One study showed that Notch3 is necessary for pericyte proliferation in zebrafish (Wang et al., 2014). In contrast, another study found that while Notch3 mutations disrupt and compromise the BBB, the mutations have no direct effect on mural cells (Henshall et al., 2015). While very minimal research has been conducted on Notch-based therapeutics in neurodegeneration, targeting Notch receptors in mice models of inflammatory disorders that share features with human MS has shown very minimal side effects while safely preventing and reversing inflammation (Minter et al., 2005; Sandy et al., 2013).
Renin-angiotensin system may also be a potential target for therapeutic intervention for VSMC dysfunction. Angiotensin-converting enzyme inhibitors and angiotensin receptor blockers have been shown as effective modulators of AD in preclinical models (Lee et al., 2020). In a mouse model, angiotensin receptor blockers alone were not found to restore Aβ-related cognitive and cerebrovascular deficits (Royea et al., 2020). However, captopril (another angiotensin converting enzyme inhibitor) alone was found to reduce Aβ deposition in vitro and in vivo, and improve cognition in animal models (AbdAlla et al., 2013; Asraf et al., 2018).
Another major signalling molecule, adenosine, is synthesised by VSMCs and endothelial cells via multiple pathways, and may be useful for controlling VSMC growth in vascular disease (Pearson et al., 1980). Studies have shown that adenosine inhibits VSMC growth and that a reduction in local adenosine levels may initiate VSMC growth and contribute to the vascular remodelling in hypertension and atherosclerosis (Dubey et al., 1996). Additionally, abnormal adenosine homeostasis has been observed in PD, HD, and ALS (Yoshida et al., 1999; Armentero et al., 2011; Lee and Chern, 2014). The review by Chang et al. (2021) summarises this topic more extensively and highlights the therapeutic opportunities for targeting adenosine homeostasis in neurodegenerative disorders.
Vascular smooth muscle cells are crucial in hypertension progression and the response of VSMCs to statins, most commonly used to treat and prevent hypertension, has therefore been more extensively researched, though only more recently in the context of neurodegeneration (Sarafidis et al., 2007). Statins can affect several components of RAS pathways in conjunction with lowering low-density lipoprotein cholesterol. Notably, statins can inhibit the harmful overexpression of Ang II and AT1 receptors, and support healthy VSMC contraction (Kiaie et al., 2021). It has been shown that statins can inhibit VSMC proliferation and reduce vascular inflammation, likely by decreasing the sensitivity of VSMCs to Ca2+ leading to structural remodelling, and reduced vascular constriction (Garg and Hassid, 1989; Kang S. et al., 2014). As a result, statins may have vasodilatory and antithrombotic properties, which could enhance collateral circulation and improve CVR, notably in cases where VSMCs are hypercontractile (Giannopoulos et al., 2012). It appears that statins may also reduce the accumulation of Aβ in the brain and have protective effects against cognitive decline, however it is unclear which mechanisms drive these outcomes (Schultz et al., 2018).
Here we have identified signalling pathways such as PPARγ, CDK, Notch, Rho, TRPV, adenosine, and RAS, which may be potential targets for intervening in VSMC dysfunction. Further research is critical to clearly understand the effect of these signalling pathways on VSMC physiology in both healthy and diseased states.
Another method of inhibiting VSMC phenotype transitions could be through the inhibition of ROS. In VSMCs, ROS are known to mediate many physiological processes such as phenotypic switching and signalling pathways, as well as pathophysiological processes, including migration, proliferation, apoptosis, and inflammatory cytokine secretion (Rao and Berk, 1992; Tsai et al., 1996; Griendling et al., 1997).
In mice, the inhibition of ROS via the oral administration of the antioxidant drug apocynin suppresses proinflammatory and proliferative synthetic VSMCs associated with vascular disorders (Pi et al., 2013). While the precise mechanisms underlying the effect of ROS remain unclear, the same study suggested that ROS induces VSMC phenotype switching through the upregulation of toll-like receptor 4 (TLR4) in endothelial cells, which can be modulated using drugs such as apocynin. Shimokawa (2013) showed that the growth factor cyclophilin A contributes to ROS effects in VSMC by promoting growth and, as a result, the effect of ROS on VSMC function may be possible by blocking the cyclophilin A signalling or binding pathways.
It has been demonstrated that ROS mediates vascular contraction and remodelling by activating Rho-signalling (Jin et al., 2004). For example, the long-term blockage of the Rho-kinase pathway was shown to inhibit induced vascular remodelling in mice by blocking the synthesis of ROS, such as NO (Ikegaki et al., 2001). As a result, the Rho pathways may be a target for inhibiting ROS.
These findings suggest that novel pharmacological strategies aimed at mediating the effect of VSMC phenotype switching through the inhibition of ROS may have therapeutic potentials in treating VSMC dysfunction.
Gene therapy is an emerging therapeutic tool by which functional genetic material is delivered to specific cells to correct a defective gene. Several clinical studies have tested gene therapy for treating neurodegenerative disorders, including PD and AD, though the efficacy of these approaches has yet to be determined (Bartus et al., 2013; Palfi et al., 2014; Tuszynski et al., 2015).
Among the most promising of potential gene therapy targets for VSMC dysfunction is SERCA2a, the SERCA isoform present in the nervous system, which has been approved as a human gene therapy target to treat heart failure (Jessup et al., 2011). This has been attributed to the normalisation of Ca2+ cycling in VSMCs and inhibiting Ca2+-dependent transcription factors (Merlet et al., 2013). This could be used to target VSMC dysfunction as forced expression of SERCA2a in contractile VSMCs has been shown to prevent injury-induced dedifferentiation toward a synthetic, inflammatory phenotype, whereas forced expression in synthetic VSMCs reduces their proliferation and migration, but has no effect on their phenotype (Bobe et al., 2011).
The review papers by Piguet et al. (2017), Martier and Konstantinova (2020) explore the past, current, and future perspectives of gene therapy for neurodegenerative disorders. Importantly, it has been noted that delivering therapies administered before the onset of neurodegeneration is essential to improve the efficacy of gene therapies (Martier and Konstantinova, 2020). Therefore, the discovery of biomarkers for early detection of neurodegenerative disorders and a better understanding of factors leading to these disorders is essential to improve clinical applications of gene therapy.
In this work, we reviewed the role of VSMCs in the most common neurodegenerative disorders and identified mechanisms by which VSMC dysfunction may contribute to neurodegeneration. In particular, changes in VSMC phenotype, signalling pathways, and interactions with ROS are seen in a host of neurodegenerative disorders and could serve as targets for novel therapeutic interventions. We identified CVR and CBF measurements as promising techniques by which VSMC dysfunction could be characterised in vivo, but further research is required to understand how to isolate VSMC contributions. Note that correlations between VSMC dysfunction and some of the disorders were more extensively studied than others, and methodologies varied greatly between them. Furthermore, inferences drawn from some of the studies analysed in this review should be taken with caution as not all of them were specifically assessing VSMC dysfunction in this context, and it remains unclear whether VSMC dysfunction is a cause or a consequence of these disorders. In conclusion, the evidence collected here highlights the critical role of VSMC dysfunction in the development and progression of neurodegeneration, and the need for more investigation into VSMCs and cerebrovascular dynamics in neurodegenerative disease.
GH, JP, and DB contributed to the conception and initial design of the manuscript. GH performed the search and selection of the relevant studies and wrote the manuscript. JP, SNS, CW, and SS contributed to the manuscript revision and editing. All authors read and approved the submitted manuscript.
This work was supported by Engineering and Physical Sciences Research Council UK through grant EP/S021507/1. GH was supported by Clarendon, SNS by the Rhodes Trust, and SS by the Alzheimer’s Society.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
VSMC, vascular smooth muscle cell; CBF, cerebral blood flow; BBB, blood brain barrier; CVR, cerebrovascular reactivity; MRI, magnetic resonance imaging; BOLD, blood-oxygen-level-dependent; ASL, arterial spin labelling; TCD, transcranial doppler; ROS, reactive oxygen species; Ang II, angiotensin II; A β, amyloid-beta; AT1, type-1 Ang II receptor; APP, amyloid precursor protein; AD, Alzheimer’s disease; APOE, apolipoprotein E; CAA, cerebral amyloid angiopathy; ALS, amyotrophic lateral sclerosis; MS, multiple sclerosis; HD, Huntington’s disease; CSVD, cerebral small vessel disease; PSD, post-stroke dementia; DS, Down’s syndrome; CADASIL, cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy; LBD, lewy body dementia; MMD, Moyamoya disease; WMH, white matter hyperintensities; SERCA2, sarco-endoplasmic reticulum calcium ATPase isoform 2; PPAR γ, peroxisome proliferator-activated receptor γ; CDK, cyclin-dependent kinase; TERT, telomerase reverse transcriptase.
AbdAlla, S., Langer, A., Fu, X., and Quitterer, U. (2013). ACE Inhibition with captopril retards the development of signs of neurodegeneration in an animal model of Alzheimer’s disease. Int. J. Mol. Sci. 14, 16917–16942. doi: 10.3390/ijms140816917
Aguilar-Pineda, J. A., Vera-Lopez, K. J., Shrivastava, P., Chávez-Fumagalli, M. A., Nieto-Montesinos, R., Alvarez-Fernandez, K. L., et al. (2021a). Vascular smooth muscle cell dysfunction contribute to neuroinflammation and Tau hyperphosphorylation in Alzheimer disease. iScience 24:102993. doi: 10.1016/j.isci.2021.102993
Aguilar-Pineda, J. A., Vera-Lopez, K. J., Shrivastava, P., Nieto-Montesinos, R., Chávez-Fumagalli, M. A., Del-Carpio, G. D., et al. (2021b). Dysfunctional vascular smooth muscle cells mediate early and late-stage neuroinflammation and Tau hyperphosphorylation. bioRxiv [Preprint] doi: 10.1101/2021.04.13.439741
Ainslie, P. N., and Duffin, J. (2009). Integration of cerebrovascular CO2 reactivity and chemoreflex control of breathing: Mechanisms of regulation, measurement, and interpretation. Am. J. Physiol. Regul. Integr. Comp. Physiol. 296, R1473–R1495. doi: 10.1152/ajpregu.91008.2008
Aktas, O., Küry, P., Kieseier, B., and Hartung, H.-P. (2010). Fingolimod is a potential novel therapy for multiple sclerosis. Nat. Rev. Neurol. 6, 373–382. doi: 10.1038/nrneurol.2010.76
Al-Bachari, S., Parkes, L. M., Vidyasagar, R., Hanby, M. F., Tharaken, V., Leroi, I., et al. (2014). Arterial spin labelling reveals prolonged arterial arrival time in idiopathic Parkinson’s disease. Neuroimage Clin. 6, 1–8. doi: 10.1016/j.nicl.2014.07.014
Aldea, R., Weller, R. O., Wilcock, D. M., Carare, R. O., and Richardson, G. (2019). Cerebrovascular smooth muscle cells as the drivers of intramural periarterial drainage of the brain. Front. Aging Neurosci. 11:1. doi: 10.3389/fnagi.2019.00001
Alford, P. W., Dabiri, B. E., Goss, J. A., Hemphill, M. A., Brigham, M. D., and Parker, K. K. (2011). Blast-induced phenotypic switching in cerebral vasospasm. Proc. Natl. Acad. Sci. U.S.A. 108, 12705–12710. doi: 10.1073/pnas.1105860108
Alonzo, N. C., Hyman, B. T., Rebeck, G. W., and Greenberg, S. M. (1998). Progression of cerebral amyloid angiopathy: Accumulation of amyloid-β40 in affected vessels. J. Neuropathol. Exp. Neurol. 57, 353–359. doi: 10.1097/00005072-199804000-00008
Amyot, F., Davis, C., Sangobowale, M., Moore, C., Silverman, E., Gandjbakhche, A., et al. (2022). Cerebrovascular reactivity measurement with functional near infrared spectroscopy. J. Vis. Exp. 2:184. doi: 10.3791/61284
Armentero, M. T., Pinna, A., Ferré, S., Lanciego, J. L., Müller, C. E., and Franco, R. (2011). Past, present and future of A2A adenosine receptor antagonists in the therapy of Parkinson’s disease. Pharmacol. Ther. 132, 280–299. doi: 10.1016/j.pharmthera.2011.07.004
Asraf, K., Torika, N., Apte, R. N., and Fleisher-Berkovich, S. (2018). Microglial activation is modulated by captopril: In vitro and in vivo studies. Front. Cell. Neurosci. 12:116. doi: 10.3389/fncel.2018.00116
Attwell, D., Buchan, A. M., Charpak, S., Lauritzen, M., MacVicar, B. A., and Newman, E. A. (2010). Glial and neuronal control of brain blood flow. Nature 468, 232–243. doi: 10.1038/nature09613
Atwi, S., Shao, H., Crane, D. E., da Costa, L., Aviv, R. I., Mikulis, D. J., et al. (2019). BOLD-based cerebrovascular reactivity vascular transfer function isolates amplitude and timing responses to better characterize cerebral small vessel disease. NMR Biomed. 32:e4064. doi: 10.1002/nbm.4064
Baeten, J. T., Jackson, A. R., McHugh, K. M., and Lilly, B. (2015). Loss of Notch2 and Notch3 in vascular smooth muscle causes patent ductus arteriosus. Genesis 53, 738–748. doi: 10.1002/dvg.22904
Baeten, J. T., and Lilly, B. (2017). Notch signaling in vascular smooth muscle cells. Adv. Pharmacol. San Diego Calif. 78, 351–382. doi: 10.1016/bs.apha.2016.07.002
Bandopadhyay, R., Kingsbury, A. E., Cookson, M. R., Reid, A. R., Evans, I. M., Hope, A. D., et al. (2004). The expression of DJ-1 (PARK7) in normal human CNS and idiopathic Parkinson’s disease. Brain J. Neurol. 127, 420–430. doi: 10.1093/brain/awh054
Bartus, R. T., Baumann, T. L., Siffert, J., Herzog, C. D., Alterman, R., Boulis, N., et al. (2013). Safety/feasibility of targeting the substantia nigra with AAV2-neurturin in Parkinson patients. Neurology 80, 1698–1701. doi: 10.1212/WNL.0b013e3182904faa
Bell, R. D., Deane, R., Chow, N., Long, X., Sagare, A., Singh, I., et al. (2009). SRF and myocardin regulate LRP-mediated amyloid-β clearance in brain vascular cells. Nat. Cell Biol. 11, 143–153. doi: 10.1038/ncb1819
Bertram, L., McQueen, M. B., Mullin, K., Blacker, D., and Tanzi, R. E. (2007). Systematic meta-analyses of Alzheimer disease genetic association studies: The AlzGene database. Nat. Genet. 39, 17–23. doi: 10.1038/ng1934
Bhogal, A. A., Siero, J. C. W., Fisher, J. A., Froeling, M., Luijten, P., Philippens, M., et al. (2014). Investigating the non-linearity of the BOLD cerebrovascular reactivity response to targeted hypo/hypercapnia at 7T. Neuroimage 98, 296–305. doi: 10.1016/j.neuroimage.2014.05.006
Blair, G. W., Thrippleton, M. J., Shi, Y., Hamilton, I., Stringer, M., Chappell, F., et al. (2020). Intracranial hemodynamic relationships in patients with cerebral small vessel disease. Neurology 94, e2258–e2269. doi: 10.1212/WNL.0000000000009483
Bobe, R., Hadri, L., Lopez, J. J., Sassi, Y., Atassi, F., Karakikes, I., et al. (2011). SERCA2a controls the mode of agonist-induced intracellular Ca2+ signal, transcription factor NFAT and proliferation in human vascular smooth muscle cells. J. Mol. Cell. Cardiol. 50, 621–633. doi: 10.1016/j.yjmcc.2010.12.016
Bonifati, V., Rizzu, P., van Baren, M. J., Schaap, O., Breedveld, G. J., Krieger, E., et al. (2003). Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science 299, 256–259. doi: 10.1126/science.1077209
Bracko, O., Njiru, B. N., Swallow, M., Ali, M., Haft-Javaherian, M., and Schaffer, C. B. (2020). Increasing cerebral blood flow improves cognition into late stages in Alzheimer’s disease mice. J. Cereb. Blood Flow Metab. 40, 1441–1452. doi: 10.1177/0271678X19873658
Brayden, J. E. (1996). Potassium channels in vascular smooth muscle. Clin. Exp. Pharmacol. Physiol. 23, 1069–1076. doi: 10.1111/j.1440-1681.1996.tb01172.x
Brian, J. E. (1998). Carbon dioxide and the cerebral circulation. Anesthesiology 88, 1365–1386. doi: 10.1097/00000542-199805000-00029
Britzolaki, A., Saurine, J., Klocke, B., and Pitychoutis, P. M. (2020). A role for SERCA pumps in the neurobiology of neuropsychiatric and neurodegenerative disorders. Adv. Exp. Med. Biol. 1131, 131–161. doi: 10.1007/978-3-030-12457-1_6
Calvier, L., Chouvarine, P., Legchenko, E., Hoffmann, N., Geldner, J., Borchert, P., et al. (2017). PPARγ links BMP2 and TGFβ1 pathways in vascular smooth muscle cells, regulating cell proliferation and glucose metabolism. Cell Metab. 25, 1118–1134.e7. doi: 10.1016/j.cmet.2017.03.011
Cannistraro, R. J., Badi, M., Eidelman, B. H., Dickson, D. W., Middlebrooks, E. H., and Meschia, J. F. (2019). CNS small vessel disease: A clinical review. Neurology 92, 1146–1156. doi: 10.1212/WNL.0000000000007654
Cantin, S., Villien, M., Moreaud, O., Tropres, I., Keignart, S., Chipon, E., et al. (2011). Impaired cerebral vasoreactivity to CO2 in Alzheimer’s disease using BOLD fMRI. Neuroimage 58, 579–587. doi: 10.1016/j.neuroimage.2011.06.070
Cataldo, J. K., Prochaska, J. J., and Glantz, S. A. (2010). Cigarette smoking is a risk factor for Alzheimer’s disease: An analysis controlling for tobacco industry affiliation. J. Alzheimers Dis. 19, 465–480. doi: 10.3233/jad-2010-1240
Catchlove, S. J., Pipingas, A., Hughes, M. E., and Macpherson, H. (2018). Magnetic resonance imaging for assessment of cerebrovascular reactivity and its relationship to cognition: A systematic review. BMC Neurosci. 19:21. doi: 10.1186/s12868-018-0421-4
Cerza, F., Renzi, M., Gariazzo, C., Davoli, M., Michelozzi, P., Forastiere, F., et al. (2019). Long-term exposure to air pollution and hospitalization for dementia in the Rome longitudinal study. Environ. Health 18:72. doi: 10.1186/s12940-019-0511-5
Chabriat, H., Pappata, S., Ostergaard, L., Clark, C. A., Pachot-Clouard, M., Vahedi, K., et al. (2000). Cerebral hemodynamics in CADASIL before and after acetazolamide challenge assessed with MRI bolus tracking. Stroke 31, 1904–1912. doi: 10.1161/01.STR.31.8.1904
Chang, C.-P., Wu, K.-C., Lin, C.-Y., and Chern, Y. (2021). Emerging roles of dysregulated adenosine homeostasis in brain disorders with a specific focus on neurodegenerative diseases. J. Biomed. Sci. 28:70. doi: 10.1186/s12929-021-00766-y
Chen, Y.-L., Daneva, Z., Kuppusamy, M., Ottolini, M., Baker, T. M., Klimentova, E., et al. (2022). Novel smooth muscle Ca2+-signaling nanodomains in blood pressure regulation. Circulation 146, 548–564. doi: 10.1161/CIRCULATIONAHA.121.058607
Cho, K.-H., Nam, H.-S., Kang, D.-J., Park, M.-H., and Kim, J.-H. (2022). Long-term alcohol consumption caused a significant decrease in serum high-density lipoprotein (HDL)-cholesterol and apolipoprotein A-I with the atherogenic changes of HDL in middle-aged Korean women. Int. J. Mol. Sci. 23:8623. doi: 10.3390/ijms23158623
Chow, N., Bell, R. D., Deane, R., Streb, J. W., Chen, J., Brooks, A., et al. (2007). Serum response factor and myocardin mediate arterial hypercontractility and cerebral blood flow dysregulation in Alzheimer’s phenotype. Proc. Natl. Acad. Sci. 104, 823–828. doi: 10.1073/pnas.0608251104
Cipolla, M. J. (2009). Control of cerebral blood flow. The cerebral circulation. San Rafael, CA: Morgan & Claypool Life Sciences.
Clements, C. M., McNally, R. S., Conti, B. J., Mak, T. W., and Ting, J. P.-Y. (2006). DJ-1, a cancer- and Parkinson’s disease-associated protein, stabilizes the antioxidant transcriptional master regulator Nrf2. Proc. Natl. Acad. Sci. U.S.A. 103, 15091–15096. doi: 10.1073/pnas.0607260103
Davis-Dusenbery, B. N., Wu, C., Hata, A., and Sessa, W. C. (2011). Micromanaging vascular smooth muscle cell differentiation and phenotypic modulation. Arterioscler. Thromb. Vasc. Biol. 31, 2370–2377. doi: 10.1161/ATVBAHA.111.226670
Deckers, P. T., van Hoek, W., Kronenburg, A., Yaqub, M., Siero, J. C. W., Bhogal, A. A., et al. (2021). Contralateral improvement of cerebrovascular reactivity and TIA frequency after unilateral revascularization surgery in moyamoya vasculopathy. Neuroimage Clin. 30:102684. doi: 10.1016/j.nicl.2021.102684
DiProspero, N. D., Kim, S., and Yassa, M. A. (2022). “Chapter 8 – Magnetic resonance imaging biomarkers for cognitive decline in Down syndrome,” in The neurobiology of aging and Alzheimer disease in down syndrome, eds E. Head and I. Lott (Cambridge, MA: Academic Press), 149–172. doi: 10.1016/B978-0-12-818845-3.00014-1
Dirnagl, U., Kaplan, B., Jacewicz, M., and Pulsinelli, W. (1989). Continuous measurement of cerebral cortical blood flow by laser—doppler flowmetry in a rat stroke model. J. Cereb. Blood Flow Metab. 9, 589–596. doi: 10.1038/jcbfm.1989.84
Dohgu, S., Takata, F., Matsumoto, J., Kimura, I., Yamauchi, A., and Kataoka, Y. (2019). Monomeric α-synuclein induces blood-brain barrier dysfunction through activated brain pericytes releasing inflammatory mediators in vitro. Microvasc. Res. 124, 61–66. doi: 10.1016/j.mvr.2019.03.005
Dubey, R. K., Gillespie, D. G., Osaka, K., Suzuki, F., and Jackson, E. K. (1996). Adenosine inhibits growth of rat aortic smooth muscle cells. Hypertension 27, 786–793. doi: 10.1161/01.HYP.27.3.786
Ervin, J. F., Pannell, C., Szymanski, M., Welsh-Bohmer, K., Schmechel, D. E., and Hulette, C. M. (2004). Vascular smooth muscle actin is reduced in Alzheimer disease brain: A quantitative analysis. J. Neuropathol. Exp. Neurol. 63, 735–741. doi: 10.1093/jnen/63.7.735
Espinosa, A. V., Shinohara, M., Porchia, L. M., Chung, Y. J., McCarty, S., Saji, M., et al. (2009). Regulator of calcineurin 1 modulates cancer cell migration in vitro. Clin. Exp. Metastasis 26, 517–526. doi: 10.1007/s10585-009-9251-1
Esteban, V., Méndez-Barbero, N., Jiménez-Borreguero, L. J., Roqué, M., Novensá, L., García-Redondo, A. B., et al. (2011). Regulator of calcineurin 1 mediates pathological vascular wall remodeling. J. Exp. Med. 208, 2125–2139. doi: 10.1084/jem.20110503
Fields, C. R., Bengoa-Vergniory, N., and Wade-Martins, R. (2019). Targeting alpha-synuclein as a therapy for Parkinson’s disease. Front. Mol. Neurosci. 12:299. doi: 10.3389/fnmol.2019.00299
Fierstra, J., Sobczyk, O., Battisti-Charbonney, A., Mandell, D. M., Poublanc, J., Crawley, A. P., et al. (2013). Measuring cerebrovascular reactivity: What stimulus to use? J. Physiol. 591, 5809–5821. doi: 10.1113/jphysiol.2013.259150
Filosa, J. A., and Iddings, J. A. (2013). Astrocyte regulation of cerebral vascular tone. Am. J. Physiol. Heart Circ. Physiol. 305, H609–H619. doi: 10.1152/ajpheart.00359.2013
Flanagan, M., Larson, E. B., Latimer, C. S., Cholerton, B., Crane, P. K., Montine, K. S., et al. (2016). Clinical-pathologic correlations in vascular cognitive impairment and dementia. Biochim. Biophys. Acta 1862, 945–951. doi: 10.1016/j.bbadis.2015.08.019
Frismantiene, A., Philippova, M., Erne, P., and Resink, T. J. (2018). Smooth muscle cell-driven vascular diseases and molecular mechanisms of VSMC plasticity. Cell. Signal. 52, 48–64. doi: 10.1016/j.cellsig.2018.08.019
Fuster, J. J., and Andrés, V. (2006). Telomere biology and cardiovascular disease. Circ. Res. 99, 1167–1180. doi: 10.1161/01.RES.0000251281.00845.18
Galis, Z. S., and Khatri, J. J. (2002). Matrix metalloproteinases in vascular remodeling and atherogenesis. Circ. Res. 90, 251–262. doi: 10.1161/res.90.3.251
Gallego, J., Pedraza, A., Lopez, S., Steiner, G., Gonzalez, L., Laurinavicius, A., et al. (2018). Glomerulus classification and detection based on convolutional neural networks. J. Imaging 4:20. doi: 10.3390/jimaging4010020
Gan, S., Qiu, S., Feng, Y., Zhang, Y., Qian, Q., Wan, Z., et al. (2017). Identification of genes associated with the effect of inflammation on the neurotransmission of vascular smooth muscle cell. Exp. Ther. Med. 13, 1303–1312. doi: 10.3892/etm.2017.4138
Garg, U. C., and Hassid, A. (1989). Nitric oxide-generating vasodilators and 8-bromo-cyclic guanosine monophosphate inhibit mitogenesis and proliferation of cultured rat vascular smooth muscle cells. J. Clin. Invest. 83, 1774–1777. doi: 10.1172/JCI114081
Giannopoulos, S., Katsanos, A. H., Tsivgoulis, G., and Marshall, R. S. (2012). Statins and cerebral hemodynamics. J. Cereb. Blood Flow Metab. 32, 1973–1976. doi: 10.1038/jcbfm.2012.122
Gill, J. S., Zezulka, A. V., Shipley, M. J., Gill, S. K., and Beevers, D. G. (1986). Stroke and alcohol consumption. N. Engl. J. Med. 315, 1041–1046. doi: 10.1056/NEJM198610233151701
Giulia, P., Michele, L., Andrea, F., Grazia, D., Filomena, C., Francesco, P., et al. (2015). Brain atrophy, anti-smooth muscle antibody and cognitive impairment: An association study. Aging Dis. 7, 318–325. doi: 10.14336/AD.2015.1124
Glodzik, L., Randall, C., Rusinek, H., and de Leon, M. J. (2013). Cerebrovascular reactivity to carbon dioxide in Alzheimer’s disease. J. Alzheimers Dis. 35, 427–440. doi: 10.3233/jad-122011
Glodzik, L., Rusinek, H., Brys, M., Tsui, W. H., Switalski, R., Mosconi, L., et al. (2011). Framingham cardiovascular risk profile correlates with impaired hippocampal and cortical vasoreactivity to hypercapnia. J. Cereb. Blood Flow Metab. 31, 671–679. doi: 10.1038/jcbfm.2010.145
Graham, N. S., and Sharp, D. J. (2019). Understanding neurodegeneration after traumatic brain injury: From mechanisms to clinical trials in dementia. J. Neurol. Neurosurg. Psychiatry 90, 1221–1233. doi: 10.1136/jnnp-2017-317557
Green, K. N., Demuro, A., Akbari, Y., Hitt, B. D., Smith, I. F., Parker, I., et al. (2008). SERCA pump activity is physiologically regulated by presenilin and regulates amyloid β production. J. Cell Biol. 181, 1107–1116. doi: 10.1083/jcb.200706171
Griendling, K. K., Minieri, C. A., Ollerenshaw, J. D., and Alexander, R. W. (1994). Angiotensin II stimulates NADH and NADPH oxidase activity in cultured vascular smooth muscle cells. Circ. Res. 74, 1141–1148. doi: 10.1161/01.res.74.6.1141
Griendling, K. K., Ushio-Fukai, M., Lassègue, B., and Alexander, R. W. (1997). Angiotensin II signaling in vascular smooth muscle. Hypertension 29, 366–370. doi: 10.1161/01.hyp.29.1.366
Grossmann, W. M., and Koeberle, B. (2000). The dose-response relationship of acetazolamide on the cerebral blood flow in normal subjects. Cerebrovasc. Dis. 10, 65–69. doi: 10.1159/000016027
Guibert, C., Ducret, T., and Savineau, J.-P. (2011). “Expression and physiological roles of TRP channels in smooth muscle cells,” in Transient receptor potential channels, advances in experimental medicine and biology, ed. M. S. Islam (Dordrecht: Springer), 687–706. doi: 10.1007/978-94-007-0265-3_36
Guo, D.-C., Papke, C. L., Tran-Fadulu, V., Regalado, E. S., Avidan, N., Johnson, R. J., et al. (2009). Mutations in smooth muscle alpha-actin (ACTA2) cause coronary artery disease, stroke, and Moyamoya disease, along with thoracic aortic disease. Am. J. Hum. Genet. 84, 617–627. doi: 10.1016/j.ajhg.2009.04.007
Guzik, T. J., Hoch, N. E., Brown, K. A., McCann, L. A., Rahman, A., Dikalov, S., et al. (2007). Role of the T cell in the genesis of angiotensin II induced hypertension and vascular dysfunction. J. Exp. Med. 204, 2449–2460. doi: 10.1084/jem.20070657
Haga, J. H., Li, Y.-S. J., and Chien, S. (2007). Molecular basis of the effects of mechanical stretch on vascular smooth muscle cells. J. Biomech. 40, 947–960. doi: 10.1016/j.jbiomech.2006.04.011
Haidey, J. N., Peringod, G., Institoris, A., Gorzo, K. A., Nicola, W., Vandal, M., et al. (2021). Astrocytes regulate ultra-slow arteriole oscillations via stretch-mediated TRPV4-COX-1 feedback. Cell Rep. 36:109405. doi: 10.1016/j.celrep.2021.109405
Hall, C. N., Reynell, C., Gesslein, B., Hamilton, N. B., Mishra, A., Sutherland, B. A., et al. (2014). Capillary pericytes regulate cerebral blood flow in health and disease. Nature 508, 55–60. doi: 10.1038/nature13165
Halliday, M. R., Rege, S. V., Ma, Q., Zhao, Z., Miller, C. A., Winkler, E. A., et al. (2016). Accelerated pericyte degeneration and blood–brain barrier breakdown in apolipoprotein E4 carriers with Alzheimer’s disease. J. Cereb. Blood Flow Metab. 36, 216–227. doi: 10.1038/jcbfm.2015.44
Hartmann, D. A., Berthiaume, A.-A., Grant, R. I., Harrill, S. A., Koski, T., Tieu, T., et al. (2021). Brain capillary pericytes exert a substantial but slow influence on blood flow. Nat. Neurosci. 24, 633–645. doi: 10.1038/s41593-020-00793-2
Hartmann, D. A., Coelho-Santos, V., and Shih, A. Y. (2022). Pericyte control of blood flow across microvascular zones in the central nervous system. Annu. Rev. Physiol. 84, 331–354. doi: 10.1146/annurev-physiol-061121-040127
Hauser, T.-K., Seeger, A., Bender, B., Klose, U., Thurow, J., Ernemann, U., et al. (2019). Hypercapnic BOLD MRI compared to H215O PET/CT for the hemodynamic evaluation of patients with Moyamoya disease. Neuroimage Clin. 22:101713. doi: 10.1016/j.nicl.2019.101713
Hawkes, C. A., Sullivan, P. M., Hands, S., Weller, R. O., Nicoll, J. A. R., and Carare, R. O. (2012). Disruption of arterial perivascular drainage of amyloid-β from the brains of mice expressing the human APOE ε4 allele. PLoS One 7:e41636. doi: 10.1371/journal.pone.0041636
Heijtel, D. F. R., Mutsaerts, H. J. M. M., Bakker, E., Schober, P., Stevens, M. F., Petersen, E. T., et al. (2014). Accuracy and precision of pseudo-continuous arterial spin labeling perfusion during baseline and hypercapnia: A head-to-head comparison with 15O H2O positron emission tomography. Neuroimage 92, 182–192. doi: 10.1016/j.neuroimage.2014.02.011
Hendrickson, R. J., Cahill, P. A., McKillop, I. H., Sitzmann, J. V., and Redmond, E. M. (1998). Ethanol inhibits mitogen activated protein kinase activity and growth of vascular smooth muscle cells in vitro. Eur. J. Pharmacol. 362, 251–259. doi: 10.1016/S0014-2999(98)00771-7
Henshall, T. L., Keller, A., He, L., Johansson, B. R., Wallgard, E., Raschperger, E., et al. (2015). Notch3 is necessary for blood vessel integrity in the central nervous system. Arterioscler. Thromb. Vasc. Biol. 35, 409–420. doi: 10.1161/ATVBAHA.114.304849
Hernandez, A. L., O’Connor, K. C., and Hafler, D. A. (2014). “Chapter 52 – multiple sclerosis,” in The autoimmune diseases, 5th Edn, eds N. R. Rose and I. R. Mackay (Boston, MA: Academic Press), 735–756. doi: 10.1016/B978-0-12-384929-8.00052-6
Herrington, W., Lacey, B., Sherliker, P., Armitage, J., and Lewington, S. (2016). Epidemiology of atherosclerosis and the potential to reduce the global burden of atherothrombotic disease. Circ. Res. 118, 535–546. doi: 10.1161/CIRCRESAHA.115.307611
Heyn, C., Poublanc, J., Crawley, A., Mandell, D., Han, J. S., Tymianski, M., et al. (2010). Quantification of cerebrovascular reactivity by blood oxygen level–dependent MR imaging and correlation with conventional angiography in patients with moyamoya disease. Am. J. Neuroradiol. 31, 862–867. doi: 10.3174/ajnr.A1922
Hilgers, R. H. P., and Webb, R. C. (2005). Molecular aspects of arterial smooth muscle contraction: Focus on Rho. Exp. Biol. Med. 230, 829–835. doi: 10.1177/153537020523001107
Hill, R. A., Tong, L., Yuan, P., Murikinati, S., Gupta, S., and Grutzendler, J. (2015). Regional blood flow in the normal and ischemic brain is controlled by arteriolar smooth muscle cell contractility and not by capillary pericytes. Neuron 87, 95–110. doi: 10.1016/j.neuron.2015.06.001
Ho, C.-C., Chen, Y.-C., Tsai, M.-H., Tsai, H.-T., Weng, C.-Y., Yet, S.-F., et al. (2021). Ambient particulate matter induces vascular smooth muscle cell phenotypic changes via NOX1/ROS/NF-κB dependent and independent pathways: Protective effects of polyphenols. Antioxidants 10:782. doi: 10.3390/antiox10050782
Iadecola, C. (2004). Neurovascular regulation in the normal brain and in Alzheimer’s disease. Nat. Rev. Neurosci. 5, 347–360. doi: 10.1038/nrn1387
Iadecola, C. (2013). The pathobiology of vascular dementia. Neuron 80, 844–866. doi: 10.1016/j.neuron.2013.10.008
Ihalainen, S., Soliymani, R., Iivanainen, E., Mykkänen, K., Sainio, A., Pöyhönen, M., et al. (2007). Proteome analysis of cultivated vascular smooth muscle cells from a CADASIL patient. Mol. Med. 13, 305–314. doi: 10.2119/2006-00069.ihalainen
Ikegaki, I., Hattori, T., Yamaguchi, T., Sasaki, Y., Satoh, S., Asano, T., et al. (2001). Involvement of Rho-kinase in vascular remodeling caused by long-term inhibition of nitric oxide synthesis in rats. Eur. J. Pharmacol. 427, 69–75. doi: 10.1016/S0014-2999(01)01181-5
Jagust, W. J., Eberling, J. L., Reed, B. R., Mathis, C. A., and Budinger, T. F. (1997). Clinical studies of cerebral blood flow in Alzheimer’s disease. Ann. N. Y. Acad. Sci. 826, 254–262. doi: 10.1111/j.1749-6632.1997.tb48477.x
Jankowska-Kieltyka, M., Roman, A., and Nalepa, I. (2021). The air we breathe: Air pollution as a prevalent proinflammatory stimulus contributing to neurodegeneration. Front. Cell. Neurosci. 15:647643. doi: 10.3389/fncel.2021.647643
Jeong, H. T., Kim, D. S., Kang, K. W., Nam, Y. T., Oh, J. E., and Cho, E. K. (2018). Factors affecting basilar artery pulsatility index on transcranial doppler. Korean J. Clin. Lab. Sci. 50, 477–483. doi: 10.15324/kjcls.2018.50.4.477
Jessup, M., Greenberg, B., Mancini, D., Cappola, T., Pauly, D. F., Jaski, B., et al. (2011). Calcium upregulation by percutaneous administration of gene therapy in cardiac disease (CUPID). Circulation 124, 304–313. doi: 10.1161/CIRCULATIONAHA.111.022889
Jin, L., Ying, Z., and Webb, R. C. (2004). Activation of Rho/Rho kinase signaling pathway by reactive oxygen species in rat aorta. Am. J. Physiol. Heart Circ. Physiol. 287, H1495–H1500. doi: 10.1152/ajpheart.01006.2003
Joutel, A., Andreux, F., Gaulis, S., Domenga, V., Cecillon, M., Battail, N., et al. (2000). The ectodomain of the Notch3 receptor accumulates within the cerebrovasculature of CADASIL patients. J. Clin. Invest. 105, 597–605. doi: 10.1172/JCI8047
Joutel, A., Corpechot, C., Ducros, A., Vahedi, K., Chabriat, H., Mouton, P., et al. (1996). Notch3 mutations in CADASIL, a hereditary adult-onset condition causing stroke and dementia. Nature 383, 707–710. doi: 10.1038/383707a0
Joutel, A., Vahedi, K., Corpechot, C., Troesch, A., Chabriat, H., Vayssière, C., et al. (1997). Strong clustering and stereotyped nature of Notch3 mutations in CADASIL patients. Lancet Lond. Engl. 350, 1511–1515. doi: 10.1016/S0140-6736(97)08083-5
Ju, S., Lim, L., Ki, Y.-J., Choi, D.-H., and Song, H. (2022). Oxidative stress generated by polycyclic aromatic hydrocarbons from ambient particulate matter enhance vascular smooth muscle cell migration through MMP upregulation and actin reorganization. Part. Fibre Toxicol. 19:29. doi: 10.1186/s12989-022-00472-z
Kane, A. D., Niu, Y., Herrera, E. A., Morton, A. J., and Giussani, D. A. (2016). Impaired nitric oxide mediated vasodilation in the peripheral circulation in the R6/2 mouse model of Huntington’s disease. Sci. Rep. 6:25979. doi: 10.1038/srep25979
Kang, H.-S., Moon, Y.-J., Kim, Y.-Y., Park, W.-Y., Park, A. K., Wang, K.-C., et al. (2014). Smooth-muscle progenitor cells isolated from patients with moyamoya disease: Novel experimental cell model: Laboratory investigation. J. Neurosurg. 120, 415–425. doi: 10.3171/2013.9.JNS131000
Kang, S., Woo, H.-H., Kim, K., Lim, K.-M., Noh, J.-Y., Lee, M.-Y., et al. (2014). Dysfunction of vascular smooth muscle and vascular remodeling by simvastatin. Toxicol. Sci. 138, 446–556. doi: 10.1093/toxsci/kfu011
Karoor, V., Oka, M., Walchak, S. J., Hersh, L. B., Miller, Y. E., and Dempsey, E. C. (2013). Neprilysin regulates pulmonary artery smooth muscle cell phenotype through a platelet-derived growth factor receptor–dependent mechanism.c Hypertension 61, 921–930. doi: 10.1161/HYPERTENSIONAHA.111.199588
Kawajiri, M., Mogi, M., Higaki, N., Tateishi, T., Ohyagi, Y., Horiuchi, M., et al. (2009). Reduced angiotensin II levels in the cerebrospinal fluid of patients with amyotrophic lateral sclerosis. Acta Neurol. Scand. 119, 341–344. doi: 10.1111/j.1600-0404.2008.01099.x
Kawakubo, T., Mori, R., Shirotani, K., Iwata, N., and Asai, M. (2017). Neprilysin is suppressed by dual-specificity tyrosine-phosphorylation regulated kinase 1A (DYRK1A) in down-syndrome-derived fibroblasts. Biol. Pharm. Bull. 40, 327–333. doi: 10.1248/bpb.b16-00825
Kiaie, N., Gorabi, A. M., Reiner, Ž, Jamialahmadi, T., Ruscica, M., and Sahebkar, A. (2021). Effects of statins on renin–angiotensin system. J. Cardiovasc. Dev. Dis. 8:80. doi: 10.3390/jcdd8070080
Kim, D., Hughes, T. M., Lipford, M. E., Craft, S., Baker, L. D., Lockhart, S. N., et al. (2021). Relationship between cerebrovascular reactivity and cognition among people with risk of cognitive decline. Front. Physiol. 12:645342. doi: 10.3389/fphys.2021.645342
Kleinfeld, D., Mitra, P. P., Helmchen, F., and Denk, W. (1998). Fluctuations and stimulus-induced changes in blood flow observed in individual capillaries in layers 2 through 4 of rat neocortex. Proc. Natl. Acad. Sci. U.S.A. 95, 15741–15746. doi: 10.1073/pnas.95.26.15741
Krettek, A., Fager, G., Jernberg, P., Östergren-Lundén, G., and Lustig, F. (1997). Quantitation of platelet-derived growth factor receptors in human arterial smooth muscle cells in vitro. Arterioscler. Thromb. Vasc. Biol. 17, 2395–2404. doi: 10.1161/01.ATV.17.11.2395
Kumar, A., Ekavali Mishra, J., Chopra, K., and Dhull, D. K. (2016). Possible role of P-glycoprotein in the neuroprotective mechanism of berberine in intracerebroventricular streptozotocin-induced cognitive dysfunction. Psychopharmacology 233, 137–152. doi: 10.1007/s00213-015-4095-7
Kuroda, S., and Houkin, K. (2008). Moyamoya disease: Current concepts and future perspectives. Lancet Neurol. 7, 1056–1066. doi: 10.1016/S1474-4422(08)70240-0
Lavi, S., Gaitini, D., Milloul, V., and Jacob, G. (2006). Impaired cerebral CO2 vasoreactivity: Association with endothelial dysfunction. Am. J. Physiol. Heart Circ. Physiol. 291, H1856–H1861. doi: 10.1152/ajpheart.00014.2006
Law, R. E., Goetze, S., Xi, X.-P., Jackson, S., Kawano, Y., Demer, L., et al. (2000). Expression and function of PPARγ in rat and human vascular smooth muscle cells. Circulation 101, 1311–1318. doi: 10.1161/01.CIR.101.11.1311
Lee, B.-C., Tsai, H.-H., Huang, A. P.-H., Lo, Y.-L., Tsai, L.-K., Chen, Y.-F., et al. (2021). Arterial spin labeling imaging assessment of cerebrovascular reactivity in hypertensive small vessel disease. Front. Neurol. 12:640069. doi: 10.3389/fneur.2021.640069
Lee, C., and Chern, Y. (2014). “Chapter ten – adenosine receptors and Huntington’s disease,” in International review of neurobiology, adenosine receptors in neurology and psychiatry, ed. A. Mori (Cambridge, MA: Academic Press), 195–232. doi: 10.1016/B978-0-12-801022-8.00010-6
Lee, S.-H., Gomes, S. M., Ghalayini, J., Iliadi, K. G., and Boulianne, G. L. (2020). Angiotensin converting enzyme inhibitors and angiotensin receptor blockers rescue memory defects in Drosophila-expressing Alzheimer’s disease-related transgenes independently of the canonical renin angiotensin system. eNeuro 7:ENEURO.0235-20.2020. doi: 10.1523/ENEURO.0235-20.2020
Leys, D., Hénon, H., Mackowiak-Cordoliani, M.-A., and Pasquier, F. (2005). Poststroke dementia. Lancet Neurol. 4, 752–759. doi: 10.1016/S1474-4422(05)70221-0
Li, J.-M., Cui, T.-X., Shiuchi, T., Liu, H.-W., Min, L.-J., Okumura, M., et al. (2004). Nicotine enhances angiotensin II-induced mitogenic response in vascular smooth muscle cells and fibroblasts. Arterioscler. Thromb. Vasc. Biol. 24, 80–84. doi: 10.1161/01.ATV.0000104007.17365.1c
Li, W., Li, J., Liu, W., Altura, B. T., and Altura, B. M. (2004). Alcohol-induced apoptosis of canine cerebral vascular smooth muscle cells: Role of extracellular and intracellular calcium ions. Neurosci. Lett. 354, 221–224. doi: 10.1016/j.neulet.2003.10.047
Liang, K.-W., Ting, C.-T., Yin, S.-C., Chen, Y.-T., Lin, S.-J., Liao, J. K., et al. (2006). Berberine suppresses MEK/ERK-dependent Egr-1 signaling pathway and inhibits vascular smooth muscle cell regrowth after in vitro mechanical injury. Biochem. Pharmacol. 71, 806–817. doi: 10.1016/j.bcp.2005.12.028
Liem, M. K., Oberstein, S. A. J. L., Haan, J., Boom, R. V. D., Ferrari, M. D., Buchem, M. A. V., et al. (2009). Cerebrovascular reactivity is a main determinant of white matter hyperintensity progression in CADASIL. Am. J. Neuroradiol. 30, 1244–1247. doi: 10.3174/ajnr.A1533
Lin, A., Peiris, N. J., Dhaliwal, H., Hakim, M., Li, W., Ganesh, S., et al. (2021). Mural cells: Potential therapeutic targets to bridge cardiovascular disease and neurodegeneration. Cells 10:593. doi: 10.3390/cells10030593
Lin, R., Xie, Z., Zhang, J., Xu, H., Su, H., Tan, X., et al. (2012). Clinical and immunopathological features of moyamoya disease. PLoS One 7:e36386. doi: 10.1371/journal.pone.0036386
Lindner, V., Booth, C., Prudovsky, I., Small, D., Maciag, T., and Liaw, L. (2001). Members of the jagged/notch gene families are expressed in injured arteries and regulate cell phenotype via alterations in cell matrix and cell-cell interaction. Am. J. Pathol. 159, 875–883. doi: 10.1016/S0002-9440(10)61763-4
Liu, P., Welch, B. G., Li, Y., Gu, H., King, D., Yang, Y., et al. (2017). Multiparametric imaging of brain hemodynamics and function using gas-inhalation MRI. Neuroimage 146, 715–723. doi: 10.1016/j.neuroimage.2016.09.063
Liu, S., and Lin, Z. (2022). Vascular smooth muscle cells mechanosensitive regulators and vascular remodeling. J. Vasc. Res. 59, 90–113. doi: 10.1159/000519845
Livingston, G., Huntley, J., Sommerlad, A., Ames, D., Ballard, C., Banerjee, S., et al. (2020). Dementia prevention, intervention, and care: 2020 report of the lancet commission. Lancet 396, 413–446. doi: 10.1016/s0140-6736(20)30367-6
Ma, X., Wang, Y., and Stephens, N. L. (1998). Serum deprivation induces a unique hypercontractile phenotype of cultured smooth muscle cells. Am. J. Physiol. Cell Physiol. 274, C1206–C1214. doi: 10.1152/ajpcell.1998.274.5.c1206
Ma, Y.-G., Zhang, Y.-B., Bai, Y.-G., Dai, Z.-J., Liang, L., Liu, M., et al. (2016). Berberine alleviates the cerebrovascular contractility in streptozotocin-induced diabetic rats through modulation of intracellular Ca2+ handling in smooth muscle cells. Cardiovasc. Diabetol. 15:63. doi: 10.1186/s12933-016-0382-9
Mack, C. P. (2011). Signaling mechanisms that regulate smooth muscle cell differentiation. Arterioscler. Thromb. Vasc. Biol. 31, 1495–1505. doi: 10.1161/ATVBAHA.110.221135
MacVicar, B. A., and Newman, E. A. (2015). Astrocyte regulation of blood flow in the brain. Cold Spring Harb. Perspect. Biol. 7:a020388. doi: 10.1101/cshperspect.a020388
Marsboom, G., and Archer, S. L. (2008). Pathways of proliferation: New targets to inhibit the growth of vascular smooth muscle cells. Circ. Res. 103, 1047–1049. doi: 10.1161/CIRCRESAHA.108.188003
Marshall, O., Lu, H., Brisset, J.-C., Xu, F., Liu, P., Herbert, J., et al. (2014). Impaired cerebrovascular reactivity in multiple sclerosis. JAMA Neurol. 71, 1275–1281. doi: 10.1001/jamaneurol.2014.1668
Martier, R., and Konstantinova, P. (2020). Gene therapy for neurodegenerative diseases: Slowing down the ticking clock. Front. Neurosci. 14:580179. doi: 10.3389/fnins.2020.580179
Mash, D. C., Ouyang, Q., Pablo, J., Basile, M., Izenwasser, S., Lieberman, A., et al. (2003). Cocaine abusers have an overexpression of α-synuclein in dopamine neurons. J. Neurosci. 23, 2564–2571. doi: 10.1523/JNEUROSCI.23-07-02564.2003
Matsushita, T., Isobe, N., Kawajiri, M., Mogi, M., Tsukuda, K., Horiuchi, M., et al. (2010). CSF angiotensin II and angiotensin-converting enzyme levels in anti-aquaporin-4 autoimmunity. J. Neurol. Sci. 295, 41–45. doi: 10.1016/j.jns.2010.05.014
Meki, M., El-Baz, A., Sethu, P., and Giridharan, G. (2022). Effects of pulsatility on arterial endothelial and smooth muscle cells. Cells Tissues Organs [Epub ahead of print]. doi: 10.1159/000524317
Meng, H., Fan, L., Zhang, C.-J., Zhu, L., Liu, P., Chen, J., et al. (2021). Synthetic VSMCs induce BBB disruption mediated by MYPT1 in ischemic stroke. iScience 24:103047. doi: 10.1016/j.isci.2021.103047
Merlet, E., Lipskaia, L., Marchand, A., Hadri, L., Mougenot, N., Atassi, F., et al. (2013). A calcium-sensitive promoter construct for gene therapy. Gene Ther. 20, 248–254. doi: 10.1038/gt.2012.30
Minamino, T., Mitsialis, S. A., and Kourembanas, S. (2001). Hypoxia extends the life span of vascular smooth muscle cells through telomerase activation. Mol. Cell. Biol. 21, 3336–3342. doi: 10.1128/MCB.21.10.3336-3342.2001
Miners, J. S., Morris, S., Love, S., and Kehoe, P. G. (2011). Accumulation of insoluble amyloid-β in Down’s syndrome is associated with increased BACE-1 and Neprilysin activities. J. Alzheimers Dis. 23, 101–108. doi: 10.3233/jad-2010-101395
Miners, S., Moulding, H., Silva, R., and Love, S. (2014). Reduced vascular endothelial growth factor and capillary density in the occipital cortex in dementia with lewy bodies. Brain Pathol. 24, 334–343. doi: 10.1111/bpa.12130
Minter, L. M., Turley, D. M., Das, P., Shin, H. M., Joshi, I., Lawlor, R. G., et al. (2005). Inhibitors of γ-secretase block in vivo and in vitro T helper type 1 polarization by preventing Notch upregulation of Tbx21. Nat. Immunol. 6, 680–688. doi: 10.1038/ni1209x
Montagnoli, T. L., da Silva, J. S., Sudo, S. Z., Santos, A. D., Gomide, G. F., de Sá, M. P. L., et al. (2021). ROCK inhibition as potential target for treatment of pulmonary hypertension. Cells 10:1648. doi: 10.3390/cells10071648
Moreton, F. C., Cullen, B., Delles, C., Santosh, C., Gonzalez, R. L., Dani, K., et al. (2018). Vasoreactivity in CADASIL: Comparison to structural MRI and neuropsychology. J. Cereb. Blood Flow Metab. 38, 1085–1095. doi: 10.1177/0271678X17710375
Morrow, D., Cullen, J. P., Liu, W., Cahill, P. A., and Redmond, E. M. (2010). Alcohol inhibits smooth muscle cell proliferation via regulation of the notch signaling pathway. Arterioscler. Thromb. Vasc. Biol. 30, 2597–2603. doi: 10.1161/ATVBAHA.110.215681
Murdoch, J. C., Rodger, J. C., Rao, S. S., Fletcher, C. D., and Dunnigan, M. G. (1977). Down’s syndrome: An atheroma-free model? Br. Med. J. 2, 226–228. doi: 10.1136/bmj.2.6081.226
Na, S., Collin, O., Chowdhury, F., Tay, B., Ouyang, M., Wang, Y., et al. (2008). Rapid signal transduction in living cells is a unique feature of mechanotransduction. Proc. Natl. Acad. Sci. U.S.A. 105, 6626–6631. doi: 10.1073/pnas.0711704105
Narayanan, K. L., Chopra, V., Rosas, H. D., Malarick, K., and Hersch, S. (2016). Rho kinase pathway alterations in the brain and leukocytes in Huntington’s disease. Mol. Neurobiol. 53, 2132–2140. doi: 10.1007/s12035-015-9147-9
Neutel, C. H. G., Corradin, G., Puylaert, P., De Meyer, G. R. Y., Martinet, W., and Guns, P.-J. (2021). High pulsatile load decreases arterial stiffness: An ex vivo study. Front. Physiol. 12:741346. doi: 10.3389/fphys.2021.741346
Niwa, K., Younkin, L., Ebeling, C., Turner, S. K., Westaway, D., Younkin, S., et al. (2000). Aβ1–40-related reduction in functional hyperemia in mouse neocortex during somatosensory activation. Proc. Natl. Acad. Sci. U.S.A. 97, 9735–9740. doi: 10.1073/pnas.97.17.9735
Oakes, J. M., Fuchs, R. M., Gardner, J. D., Lazartigues, E., and Yue, X. (2018). Nicotine and the renin-angiotensin system. Am. J. Physiol. Regul. Integr. Comp. Physiol. 315, R895–R906. doi: 10.1152/ajpregu.00099.2018
Ohlrogge, A. H., Frost, L., and Schnabel, R. B. (2022). Harmful impact of tobacco smoking and alcohol consumption on the atrial myocardium. Cells 11:2576. doi: 10.3390/cells11162576
Ovsenik, A., Podbregar, M., and Fabjan, A. (2021). Cerebral blood flow impairment and cognitive decline in heart failure. Brain Behav. 11:e02176. doi: 10.1002/brb3.2176
Owens, G. K., Kumar, M. S., and Wamhoff, B. R. (2004). Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol. Rev. 84, 767–801. doi: 10.1152/physrev.00041.2003
Palfi, S., Gurruchaga, J. M., Ralph, G. S., Lepetit, H., Lavisse, S., Buttery, P. C., et al. (2014). Long-term safety and tolerability of ProSavin, a lentiviral vector-based gene therapy for Parkinson’s disease: A dose escalation, open-label, phase 1/2 trial. Lancet 383, 1138–1146. doi: 10.1016/S0140-6736(13)61939-X
Panahi, M., Yousefi Mesri, N., Samuelsson, E., Coupland, K. G., Forsell, C., Graff, C., et al. (2018). Differences in proliferation rate between CADASIL and control vascular smooth muscle cells are related to increased TGFβ expression. J. Cell. Mol. Med. 22, 3016–3024. doi: 10.1111/jcmm.13534
Parra, P., Costa, R., de Asúa, D. R., Moldenhauer, F., and Suárez, C. (2017). Atherosclerotic surrogate markers in adults with down syndrome: A case-control study. J. Clin. Hypertens. 19, 205–211. doi: 10.1111/jch.12890
Parsons, M. P., and Raymond, L. A. (2015). “Chapter 20 – Huntington disease,” in Neurobiology of brain disorders, eds M. J. Zigmond, L. P. Rowland, and J. T. Coyle (San Diego, CA: Academic Press), 303–320. doi: 10.1016/B978-0-12-398270-4.00020-3
Pase, M. P., Beiser, A., Himali, J. J., Tsao, C., Satizabal, C. L., Vasan, R. S., et al. (2016). Aortic stiffness and the risk of incident mild cognitive impairment and dementia. Stroke 47, 2256–2261. doi: 10.1161/STROKEAHA.116.013508
Pearson, J. D., Carleton, J. S., and Gordon, J. L. (1980). Metabolism of adenine nucleotides by ectoenzymes of vascular endothelial and smooth-muscle cells in culture. Biochem. J. 190, 421–429. doi: 10.1042/bj1900421
Pelizzari, L., Laganà, M. M., Rossetto, F., Bergsland, N., Galli, M., Baselli, G., et al. (2019). Cerebral blood flow and cerebrovascular reactivity correlate with severity of motor symptoms in Parkinson’s disease. Ther. Adv. Neurol. Disord. 12:1756286419838354. doi: 10.1177/1756286419838354
Peters, O. M., and Brown, R. H. (2015). “Chapter 18 – Amyotrophic lateral sclerosis,” in Neurobiology of brain disorders, eds M. J. Zigmond, L. P. Rowland, and J. T. Coyle (San Diego, CA: Academic Press), 262–280. doi: 10.1016/B978-0-12-398270-4.00018-5
Peters, R., Ee, N., Peters, J., Booth, A., Mudway, I., and Anstey, K. J. (2019). Air pollution and dementia: A systematic review. J. Alzheimers Dis. 70, S145–S163. doi: 10.3233/JAD-180631
Pfefferkorn, T., von Stuckrad-Barre, S., Herzog, J., Gasser, T., Hamann, G. F., and Dichgans, M. (2001). Reduced cerebrovascular CO2 reactivity in CADASIL. Stroke 32, 17–21. doi: 10.1161/01.str.32.1.17
Pi, Y., Zhang, L., Li, B., Guo, L., Cao, X., Gao, C., et al. (2013). Inhibition of reactive oxygen species generation attenuates TLR4-mediated proinflammatory and proliferative phenotype of vascular smooth muscle cells. Lab. Invest. 93, 880–887. doi: 10.1038/labinvest.2013.79
Piguet, F., Alves, S., and Cartier, N. (2017). Clinical gene therapy for neurodegenerative diseases: Past, present, and future. Hum. Gene Ther. 28, 988–1003. doi: 10.1089/hum.2017.160
Pinto, J., Bright, M. G., Bulte, D. P., and Figueiredo, P. (2021). Cerebrovascular reactivity mapping without gas challenges: A methodological guide. Front. Physiol. 11:608475. doi: 10.3389/fphys.2020.608475
Platten, M., Youssef, S., Hur, E. M., Ho, P. P., Han, M. H., Lanz, T. V., et al. (2009). Blocking angiotensin-converting enzyme induces potent regulatory T cells and modulates TH1- and TH17-mediated autoimmunity. Proc. Natl. Acad. Sci. U.S.A. 106, 14948–14953. doi: 10.1073/pnas.0903958106
Poittevin, M., Lozeron, P., Hilal, R., Levy, B. I., Merkulova-Rainon, T., and Kubis, N. (2014). Smooth muscle cell phenotypic switching in stroke. Transl. Stroke Res. 5, 377–384. doi: 10.1007/s12975-013-0306-x
Qiu, H., Wu, Y., Wang, Q., Liu, C., Xue, L., Wang, H., et al. (2017). Effect of berberine on PPARα-NO signalling pathway in vascular smooth muscle cell proliferation induced by angiotensin IV. Pharm. Biol. 55, 227–232. doi: 10.1080/13880209.2016.1257642
Qiu, J., Zheng, Y., Hu, J., Liao, D., Gregersen, H., Deng, X., et al. (2014). Biomechanical regulation of vascular smooth muscle cell functions: From in vitro to in vivo understanding. J. R. Soc. Interface 11:20130852. doi: 10.1098/rsif.2013.0852
Rahman, A., Ekman, M., Shakirova, Y., Andersson, K. E., Mörgelin, M., Erjefält, J. S., et al. (2013). Late onset vascular dysfunction in the R6/1 model of Huntington’s disease. Eur. J. Pharmacol. 698, 345–353. doi: 10.1016/j.ejphar.2012.10.026
Rao, G. N., and Berk, B. C. (1992). Active oxygen species stimulate vascular smooth muscle cell growth and proto-oncogene expression. Circ. Res. 70, 593–599. doi: 10.1161/01.RES.70.3.593
Rayshubskiy, A., Wojtasiewicz, T. J., Mikell, C. B., Bouchard, M. B., Timerman, D., Youngerman, B. E., et al. (2014). Direct, intraoperative observation of ∼0.1 Hz hemodynamic oscillations in awake human cortex: Implications for fMRI. Neuroimage 87, 323–331. doi: 10.1016/j.neuroimage.2013.10.044
Raz, L., Knoefel, J., and Bhaskar, K. (2015). The neuropathology and cerebrovascular mechanisms of dementia. J. Cereb. Blood Flow Metab. 36, 172–186. doi: 10.1038/jcbfm.2015.164
Rensen, S. S. M., Doevendans, P. A. F. M., and van Eys, G. J. J. M. (2007). Regulation and characteristics of vascular smooth muscle cell phenotypic diversity. Neth. Heart J. 15, 100–108. doi: 10.1007/bf03085963
Rensink, A. A. M., de Waal, R. M. W., Kremer, B., and Verbeek, M. M. (2003). Pathogenesis of cerebral amyloid angiopathy. Brain Res. Rev. 43, 207–223. doi: 10.1016/j.brainresrev.2003.08.001
Reuck, J. D., Deramecourt, V., Auger, F., Durieux, N., Cordonnier, C., Devos, D., et al. (2014). Post-mortem 7.0-tesla magnetic resonance study of cortical microinfarcts in neurodegenerative diseases and vascular dementia with neuropathological correlates. J. Neurol. Sci. 346, 85–89. doi: 10.1016/j.jns.2014.07.061
Richiardi, J., Monsch, A. U., Haas, T., Barkhof, F., de Ville, D. V., Radü, E. W., et al. (2015). Altered cerebrovascular reactivity velocity in mild cognitive impairment and Alzheimer’s disease. Neurobiol. Aging 36, 33–41. doi: 10.1016/j.neurobiolaging.2014.07.020
Rodell, A., Aanerud, J., Braendgaard, H., and Gjedde, A. (2012). Low residual CBF variability in Alzheimer’s disease after correction for CO2 effect. Front. Neuroenergetics 4:8. doi: 10.3389/fnene.2012.00008
Roerecke, M., Kaczorowski, J., Tobe, S. W., Gmel, G., Hasan, O. S. M., and Rehm, J. (2017). The effect of a reduction in alcohol consumption on blood pressure: A systematic review and meta-analysis. Lancet Public Health 2, e108–e120. doi: 10.1016/S2468-2667(17)30003-8
Royea, J., Lacalle-Aurioles, M., Trigiani, L. J., Fermigier, A., and Hamel, E. (2020). AT2R’s (Angiotensin II type 2 receptor’s) role in cognitive and cerebrovascular deficits in a mouse model of Alzheimer disease. Hypertension 75, 1464–1474. doi: 10.1161/HYPERTENSIONAHA.119.14431
Roy-Vallejo, E., Galván-Román, J. M., Moldenhauer, F., and de Asúa, D. R. (2020). Adults with Down syndrome challenge another paradigm: When aging no longer entails arterial hypertension. J. Clin. Hypertens. 22, 1127–1133. doi: 10.1111/jch.13930
Rui, Q., Ni, H., Li, D., Gao, R., and Chen, G. (2018). The role of LRRK2 in neurodegeneration of Parkinson disease. Curr. Neuropharmacol. 16, 1348–1357. doi: 10.2174/1570159X16666180222165418
Sabia, S., Fayosse, A., Dumurgier, J., Dugravot, A., Akbaraly, T., Britton, A., et al. (2018). Alcohol consumption and risk of dementia: 23 year follow-up of Whitehall II cohort study. BMJ 362:k2927. doi: 10.1136/bmj.k2927
Sam, K., Crawley, A. P., Conklin, J., Poublanc, J., Sobczyk, O., Mandell, D. M., et al. (2016). Development of white matter hyperintensity is preceded by reduced cerebrovascular reactivity. Ann. Neurol. 80, 277–285. doi: 10.1002/ana.24712
Sam, K., Poublanc, J., Sobczyk, O., Han, J. S., Battisti-Charbonney, A., Mandell, D. M., et al. (2015). Assessing the effect of unilateral cerebral revascularisation on the vascular reactivity of the non-intervened hemisphere: A retrospective observational study. BMJ Open 5:e006014. doi: 10.1136/bmjopen-2014-006014
Sandy, A. R., Stoolman, J., Malott, K., Pongtornpipat, P., Segal, B. M., and Maillard, I. (2013). Notch signaling regulates T cell accumulation and function in the central nervous system during experimental autoimmune encephalomyelitis. J. Immunol. 191, 1606–1613. doi: 10.4049/jimmunol.1301116
Sarafidis, P. A., Kanaki, A. I., and Lasaridis, A. N. (2007). Statins and blood pressure: Is there an effect or not? J. Clin. Hypertens. 9, 460–467. doi: 10.1111/j.1524-6175.2007.06625.x
Sawma, T., Shaito, A., Najm, N., Sidani, M., Orekhov, A., El-Yazbi, A. F., et al. (2022). Role of RhoA and Rho-associated kinase in phenotypic switching of vascular smooth muscle cells: Implications for vascular function. Atherosclerosis 358, 12–28. doi: 10.1016/j.atherosclerosis.2022.08.012
Schultz, B. G., Patten, D. K., and Berlau, D. J. (2018). The role of statins in both cognitive impairment and protection against dementia: A tale of two mechanisms. Transl. Neurodegener. 7:5. doi: 10.1186/s40035-018-0110-3
Scuteri, A., Tesauro, M., Appolloni, S., Preziosi, F., Brancati, A. M., and Volpe, M. (2007). Arterial stiffness as an independent predictor of longitudinal changes in cognitive function in the older individual. J. Hypertens. 25, 1035–1040. doi: 10.1097/HJH.0b013e3280895b55
Shi, Y., Thrippleton, M. J., Makin, S. D., Marshall, I., Geerlings, M. I., de Craen, A. J., et al. (2016). Cerebral blood flow in small vessel disease: A systematic review and meta-analysis. J. Cereb. Blood Flow Metab. 36, 1653–1667. doi: 10.1177/0271678X16662891
Shi, Z.-D., and Tarbell, J. M. (2011). Fluid flow mechanotransduction in vascular smooth muscle cells and fibroblasts. Ann. Biomed. Eng. 39, 1608–1619. doi: 10.1007/s10439-011-0309-2
Shimokawa, H. (2013). Reactive oxygen species promote vascular smooth muscle cell proliferation. Circ. Res. 113, 1040–1042. doi: 10.1161/CIRCRESAHA.113.302049
Singhal, S., and Markus, H. S. (2005). Cerebrovascular reactivity and dynamic autoregulation in nondemented patients with CADASIL (cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy). J. Neurol. 252, 163–167. doi: 10.1007/s00415-005-0624-3
Sobey, C. G., Judkins, C. P., Sundararajan, V., Phan, T. G., Drummond, G. R., and Srikanth, V. K. (2015). Risk of major cardiovascular events in people with down syndrome. PLoS One 10:e0137093. doi: 10.1371/journal.pone.0137093
Sorrentino, S., Polini, A., Arima, V., Romano, A., Quattrini, A., Gigli, G., et al. (2022). Neurovascular signals in amyotrophic lateral sclerosis. Curr. Opin. Biotechnol. 74, 75–83. doi: 10.1016/j.copbio.2021.10.021
Spano, V. R., Mandell, D. M., Poublanc, J., Sam, K., Battisti-Charbonney, A., Pucci, O., et al. (2013). CO2 blood oxygen level–dependent MR mapping of cerebrovascular reserve in a clinical population: Safety, tolerability, and technical feasibility. Radiology 266, 592–598. doi: 10.1148/radiol.12112795
Speer, R. E., and Ratan, R. R. (2009). Drug treatment for neurodegenerative disorders among the elderly: Re-engaging homeostatic programs. Aging Health 5, 133–136. doi: 10.2217/ahe.09.11
St. Paul, A., Corbett, C. B., Okune, R., and Autieri, M. V. (2020). Angiotensin II, hypercholesterolemia, and vascular smooth muscle cells: A perfect trio for vascular pathology. Int. J. Mol. Sci. 21:4525. doi: 10.3390/ijms21124525
Starke, R. M., Ali, M. S., Jabbour, P. M., Tjoumakaris, S. I., Gonzalez, F., Hasan, D. M., et al. (2013). Cigarette smoke modulates vascular smooth muscle phenotype: Implications for carotid and cerebrovascular disease. PLoS One 8:e71954. doi: 10.1371/journal.pone.0071954
Su, J., Li, J., Li, W., Altura, B. T., and Altura, B. M. (2003). Cocaine induces apoptosis in cerebral vascular muscle cells: Potential roles in strokes and brain damage. Eur. J. Pharmacol. 482, 61–66. doi: 10.1016/j.ejphar.2003.09.056
Szarka, N., Pabbidi, M. R., Amrein, K., Czeiter, E., Berta, G., Pohoczky, K., et al. (2018). Traumatic brain injury impairs myogenic constriction of cerebral arteries: Role of mitochondria-derived H2O2 and TRPV4-dependent activation of BKca channels. J. Neurotrauma 35, 930–939. doi: 10.1089/neu.2017.5056
Tamo, W., Imaizumi, T., Tanji, K., Yoshida, H., Mori, F., Fukuda, I., et al. (2003). Expression of α-synuclein in vascular endothelial and smooth muscle cells. Int. Congr. Ser. 1251, 173–179. doi: 10.1016/S0531-5131(03)00120-1
Thalman, S., Van Pelt, K. L., Lin, A.-L., Johnson, N. F., Jicha, G., Caban-Holt, A., et al. (2020). A preliminary study of cerebral blood flow, aging and dementia in people with Down syndrome. J. Intellect. Disabil. Res. 64, 934–945. doi: 10.1111/jir.12784
Thorin-Trescases, N., de Montgolfier, O., Pinçon, A., Raignault, A., Caland, L., Labbé, P., et al. (2018). Impact of pulse pressure on cerebrovascular events leading to age-related cognitive decline. Am. J. Physiol. Heart Circ. Physiol. 314, H1214–H1224. doi: 10.1152/ajpheart.00637.2017
Tokairin, K., Hamauchi, S., Ito, M., Kazumata, K., Sugiyama, T., Nakayama, N., et al. (2020). Vascular smooth muscle cell derived from IPS cell of moyamoya disease – comparative characterization with endothelial cell transcriptome. J. Stroke Cerebrovasc. Dis. 29:105305. doi: 10.1016/j.jstrokecerebrovasdis.2020.105305
Topiwala, A., Wang, C., Ebmeier, K. P., Burgess, S., Bell, S., Levey, D. F., et al. (2022). Associations between moderate alcohol consumption, brain iron, and cognition in UK Biobank participants: Observational and mendelian randomization analyses. PLoS Med. 19:e1004039. doi: 10.1371/journal.pmed.1004039
Touyz, R. M., Alves-Lopes, R., Rios, F. J., Camargo, L. L., Anagnostopoulou, A., Arner, A., et al. (2018). Vascular smooth muscle contraction in hypertension. Cardiovasc. Res. 114, 529–539. doi: 10.1093/cvr/cvy023
Tsai, J.-C., Jain, M., Hsieh, C.-M., Lee, W.-S., Yoshizumi, M., Patterson, C., et al. (1996). Induction of apoptosis by pyrrolidinedithiocarbamate and n-acetylcysteine in vascular smooth muscle cells. J. Biol. Chem. 271, 3667–3670. doi: 10.1074/jbc.271.7.3667
Tsuji, M., Duplessis, A., Taylor, G., Crocker, R., and Volpe, J. J. (1998). Near infrared spectroscopy detects cerebral ischemia during hypotension in piglets. Pediatr. Res. 44, 591–595. doi: 10.1203/00006450-199810000-00020
Tuon, T., Valvassori, S. S., Lopes-Borges, J., Luciano, T., Trom, C. B., Silva, L. A., et al. (2012). Physical training exerts neuroprotective effects in the regulation of neurochemical factors in an animal model of Parkinson’s disease. Neuroscience 227, 305–312. doi: 10.1016/j.neuroscience.2012.09.063
Tuszynski, M. H., Yang, J. H., Barba, D., U, H.-S., Bakay, R. A. E., Pay, M. M., et al. (2015). Nerve growth factor gene therapy: Activation of neuronal responses in Alzheimer disease. JAMA Neurol. 72, 1139–1147. doi: 10.1001/jamaneurol.2015.1807
van den Boom, R., Oberstein, S. A. L., Spilt, A., Behloul, F., Ferrari, M. D., Haan, J., et al. (2003). Cerebral hemodynamics and white matter hyperintensities in CADASIL. J. Cereb. Blood Flow Metab. 23, 599–604. doi: 10.1097/01.WCB.0000062341.61367.D3
Vanlandewijck, M., He, L., Mäe, M. A., Andrae, J., Ando, K., Del Gaudio, F., et al. (2018). A molecular atlas of cell types and zonation in the brain vasculature. Nature 554, 475–480. doi: 10.1038/nature25739
Vicenzini, E., Ricciardi, M. C., Altieri, M., Puccinelli, F., Bonaffini, N., Piero, V. D., et al. (2007). Cerebrovascular reactivity in degenerative and vascular dementia: A transcranial doppler study. Eur. Neurol. 58, 84–89. doi: 10.1159/000103642
Viitanen, M., Sundström, E., Baumann, M., Poyhonen, M., Tikka, S., and Behbahani, H. (2013). Experimental studies of mitochondrial function in CADASIL vascular smooth muscle cells. Exp. Cell Res. 319, 134–143. doi: 10.1016/j.yexcr.2012.09.015
Villalba, N., Sonkusare, S. K., Longden, T. A., Tran, T. L., Sackheim, A. M., Nelson, M. T., et al. (2014). Traumatic brain injury disrupts cerebrovascular tone through endothelial inducible nitric oxide synthase expression and nitric oxide gain of function. J. Am. Heart Assoc. 3:e001474. doi: 10.1161/JAHA.114.001474
Virdis, A., Neves, M. F., Amiri, F., Touyz, R. M., and Schiffrin, E. L. (2004). Role of NAD(P)H oxidase on vascular alterations in angiotensin II-infused mice. J. Hypertens. 22, 535–542.
Vodovar, N., Paquet, C., Mebazaa, A., Launay, J.-M., Hugon, J., and Cohen-Solal, A. (2015). Neprilysin, cardiovascular, and Alzheimer’s diseases: The therapeutic split? Eur. Heart J. 36, 902–905. doi: 10.1093/eurheartj/ehv015
von Lueder, T. G., Atar, D., and Krum, H. (2014). Current role of neprilysin inhibitors in hypertension and heart failure. Pharmacol. Ther. 144, 41–49. doi: 10.1016/j.pharmthera.2014.05.002
Vromman, A., Trabelsi, N., Rouxel, C., Béréziat, G., Limon, I., and Blaise, R. (2013). β−amyloid context intensifies vascular smooth muscle cells induced inflammatory response and de-differentiation. Aging Cell 12, 358–369. doi: 10.1111/acel.12056
Wakino, S., Kintscher, U., Kim, S., Yin, F., Hsueh, W. A., and Law, R. E. (2000). Peroxisome proliferator-activated receptor γ ligands inhibit retinoblastoma phosphorylation and G1 → S transition in vascular smooth muscle cells. J. Biol. Chem. 275, 22435–22441. doi: 10.1074/jbc.M910452199
Wang, H., Naghavi, M., Allen, C., Barber, R. M., Bhutta, Z. A., Carter, A., et al. (2016). Global, regional, and national life expectancy, all-cause mortality, and cause-specific mortality for 249 causes of death, 1980–2015: A systematic analysis for the Global Burden of Disease Study 2015. Lancet 388, 1459–1544. doi: 10.1016/S0140-6736(16)31012-1
Wang, S., and Jia, C. (2020). TRPV1 inhibits smooth muscle cell phenotype switching in a mouse model of abdominal aortic aneurysm. Channels 14, 59–68. doi: 10.1080/19336950.2020.1730020
Wang, Y., Pan, L., Moens, C. B., and Appel, B. (2014). Notch3 establishes brain vascular integrity by regulating pericyte number. Development 141, 307–317. doi: 10.1242/dev.096107
Winkler, E. A., Sengillo, J. D., Sullivan, J. S., Henkel, J. S., Appel, S. H., and Zlokovic, B. V. (2013). Blood-spinal cord barrier breakdown and pericyte reductions in amyotrophic lateral sclerosis. Acta Neuropathol. 125, 111–120. doi: 10.1007/s00401-012-1039-8
Won, K. J., Jung, S. H., Lee, C.-K., Na, H. R., Lee, K. P., Lee, D.-Y., et al. (2013). DJ-1/park7 protects against neointimal formation via the inhibition of vascular smooth muscle cell growth. Cardiovasc. Res. 97, 553–561. doi: 10.1093/cvr/cvs363
Worssam, M. D., and Jørgensen, H. F. (2021). Mechanisms of vascular smooth muscle cell investment and phenotypic diversification in vascular diseases. Biochem. Soc. Trans. 49, 2101–2111. doi: 10.1042/BST20210138
Wynne, B. M., Chiao, C.-W., and Webb, R. C. (2009). Vascular smooth muscle cell signaling mechanisms for contraction to angiotensin II and endothelin-1. J. Am. Soc. Hypertens. 3, 84–95. doi: 10.1016/j.jash.2008.09.002
Yang, H.-M., Kim, B.-K., Kim, J.-Y., Kwon, Y.-W., Jin, S., Lee, J.-E., et al. (2013). PPARγ modulates vascular smooth muscle cell phenotype via a protein kinase G-dependent pathway and reduces neointimal hyperplasia after vascular injury. Exp. Mol. Med. 45:e65. doi: 10.1038/emm.2013.112
Yasuda, T., and Mochizuki, H. (2010). The regulatory role of α-synuclein and parkin in neuronal cell apoptosis; possible implications for the pathogenesis of Parkinson’s disease. Apoptosis 15, 1312–1321. doi: 10.1007/s10495-010-0486-8
Ye, C., Liang, Y., Chen, Y., Xiong, Y., She, Y., Zhong, X., et al. (2021). Berberine improves cognitive impairment by simultaneously impacting cerebral blood flow and β-amyloid accumulation in an APP/tau/PS1 mouse model of Alzheimer’s disease. Cells 10:1161. doi: 10.3390/cells10051161
Ye, Y., Liu, X., Wu, N., Han, Y., Wang, J., Yu, Y., et al. (2021). Efficacy and safety of berberine alone for several metabolic disorders: A systematic review and meta-analysis of randomized clinical trials. Front. Pharmacol. 12:653887. doi: 10.3389/fphar.2021.653887
Yemisci, M., Gursoy-Ozdemir, Y., Vural, A., Can, A., Topalkara, K., and Dalkara, T. (2009). Pericyte contraction induced by oxidative-nitrative stress impairs capillary reflow despite successful opening of an occluded cerebral artery. Nat. Med. 15, 1031–1037. doi: 10.1038/nm.2022
Yezhuvath, U. S., Uh, J., Cheng, Y., Martin-Cook, K., Weiner, M., Diaz-Arrastia, R., et al. (2012). Forebrain-dominant deficit in cerebrovascular reactivity in Alzheimer’s disease. Neurobiol. Aging 33, 75–82. doi: 10.1016/j.neurobiolaging.2010.02.005
Yoshida, Y., Une, F., Utatsu, Y., Nomoto, M., Furukawa, Y., Maruyama, Y., et al. (1999). Adenosine and neopterin levels in cerebrospinal fluid of patients with neurological disorders. Intern. Med. 38, 133–139. doi: 10.2169/internalmedicine.38.133
Yu, G.-X., Mueller, M., Hawkins, B. E., Mathew, B. P., Parsley, M. A., Vergara, L. A., et al. (2014). Traumatic brain injury in vivo and in vitro contributes to cerebral vascular dysfunction through impaired gap junction communication between vascular smooth muscle cells. J. Neurotrauma 31, 739–748. doi: 10.1089/neu.2013.3187
Zacharek, A., Chen, J., Cui, X., Yang, Y., and Chopp, M. (2009). Simvastatin increases notch signaling activity and promotes arteriogenesis after stroke. Stroke 40, 254–260. doi: 10.1161/STROKEAHA.108.524116
Zerbi, V., Wiesmann, M., Emmerzaal, T. L., Jansen, D., Beek, M. V., Mutsaers, M. P. C., et al. (2014). Resting-state functional connectivity changes in aging apoE4 and apoE-KO mice. J. Neurosci. 34, 13963–13975. doi: 10.1523/JNEUROSCI.0684-14.2014
Zhao, M. Y., Fan, A. P., Chen, D. Y.-T., Ishii, Y., Khalighi, M. M., Moseley, M., et al. (2022). Using arterial spin labeling to measure cerebrovascular reactivity in Moyamoya disease: Insights from simultaneous PET/MRI. J. Cereb. Blood Flow Metab. 42, 1493–1506. doi: 10.1177/0271678X221083471
Zhao, Y., Biswas, S. K., McNulty, P. H., Kozak, M., Jun, J. Y., and Segar, L. (2011). PDGF-induced vascular smooth muscle cell proliferation is associated with dysregulation of insulin receptor substrates. Am. J. Physiol. Cell Physiol. 300, C1375–C1385. doi: 10.1152/ajpcell.00670.2008
Zhao, Y., Vanhoutte, P. M., and Leung, S. W. S. (2015). Vascular nitric oxide: Beyond eNOS. J. Pharmacol. Sci. 129, 83–94. doi: 10.1016/j.jphs.2015.09.002
Zhou, Y., Wang, X., Guo, L., Chen, L., Zhang, M., Chen, X., et al. (2021). TRPV1 activation inhibits phenotypic switching and oxidative stress in vascular smooth muscle cells by upregulating PPARα. Biochem. Biophys. Res. Commun. 545, 157–163. doi: 10.1016/j.bbrc.2021.01.072
Zlokovic, B. V. (2008). New therapeutic targets in the neurovascular pathway in Alzheimer’s disease. Neurotherapeutics 5, 409–414. doi: 10.1016/j.nurt.2008.05.011
Keywords: vascular smooth muscle cells, neurodegeneration, cerebrovascular reactivity, cerebral blood flow, dementia, Alzheimer’s disease, MRI
Citation: Hayes G, Pinto J, Sparks SN, Wang C, Suri S and Bulte DP (2022) Vascular smooth muscle cell dysfunction in neurodegeneration. Front. Neurosci. 16:1010164. doi: 10.3389/fnins.2022.1010164
Received: 02 August 2022; Accepted: 24 October 2022;
Published: 10 November 2022.
Edited by:
Yuan Ma, Harvard University, United StatesReviewed by:
Jessica Lorena Presa, University of Buenos Aires, ArgentinaCopyright © 2022 Hayes, Pinto, Sparks, Wang, Suri and Bulte. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Genevieve Hayes, Z2VuZXZpZXZlLmhheWVzQGVuZy5veC5hYy51aw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.