 Ilaria Parenti1*
Ilaria Parenti1* Frank J. Kaiser1,2
Frank J. Kaiser1,2- 1Institut für Humangenetik, Universitätsklinikum Essen, Universität Duisburg-Essen, Essen, Germany
- 2Essener Zentrum für Seltene Erkrankungen (EZSE), Universitätsklinikum Essen, Essen, Germany
Chromatinopathies can be defined as a class of neurodevelopmental disorders caused by mutations affecting proteins responsible for chromatin remodeling and transcriptional regulation. The resulting dysregulation of gene expression favors the onset of a series of clinical features such as developmental delay, intellectual disability, facial dysmorphism, and behavioral disturbances. Cornelia de Lange syndrome (CdLS) is a prime example of a chromatinopathy. It is caused by mutations affecting subunits or regulators of the cohesin complex, a multisubunit protein complex involved in various molecular mechanisms such as sister chromatid cohesion, transcriptional regulation and formation of topologically associated domains. However, disease-causing variants in non-cohesin genes with overlapping functions have also been described in association with CdLS. Notably, the majority of these genes had been previously found responsible for distinct neurodevelopmental disorders that also fall within the category of chromatinopathies and are frequently considered as differential diagnosis for CdLS. In this review, we provide a systematic overview of the current literature to summarize all mutations in non-cohesin genes identified in association with CdLS phenotypes and discuss about the interconnection of proteins belonging to the chromatinopathies network.
Introduction
Cornelia de Lange syndrome (CdLS, OMIM # 122470, #300590, #610759, #614701, and #300882) is a multisystem developmental disorder named after the Dutch pediatrician Cornelia de Lange, who reported in 1933 two unrelated patients with comparable features. Nowadays, its prevalence is estimated between 1/10,000 and 1/30,000 live births (Kline et al., 2007). A distinct craniofacial appearance, pre- and post-natal growth retardation, intellectual disability, developmental delay, behavioral issues, and limb anomalies are the main clinical features of CdLS, albeit observed with variable expressivity (Kline et al., 2018). The first international consensus statement for CdLS has recently introduced a scoring system to classify the severity of the syndrome and help select the most appropriate pipeline for genetic testing. A score ≥11 confirms the clinical diagnosis of CdLS also in the absence of a molecular diagnosis (Kline et al., 2018).
The genetic etiology of CdLS is mainly attributable to variants affecting the function of the deeply conserved protein complex known as cohesin (Kline et al., 2018). Variants in the cohesin regulator NIPBL are the most frequent cause of CdLS and account for approximately 70% of cases. Other subunits or regulators of the complex (SMC1A, SMC3, RAD21, and HDAC8) are responsible altogether for 10–15% of cases (Kline et al., 2018). Variants in additional cohesin-associated proteins like MAU2, STAG1, and STAG2 have been associated with CdLS or phenotypes reminiscent of CdLS in few individuals (Lehalle et al., 2017; Mullegama et al., 2017; Soardi et al., 2017; Yuan et al., 2019; Parenti et al., 2020).
The cohesin complex performs numerous functions that are essential for cell survival, including sister chromatid cohesion, DNA repair, maintenance of genomic stability, transcriptional regulation, and chromatin regulation by mediating long-range interactions between distant genomic regions and contributing to the formation of topologically associating domains (Zhu and Wang, 2019). Sister chromatid cohesion is the best-characterized role of the complex. However, cell lines of individuals with CdLS do not display cohesion defects (Castronovo et al., 2009). A global dysregulation of gene expression is instead observed in these cells (Liu et al., 2009; Izumi et al., 2015; Yuan et al., 2015).
Hence, an altered functionality of the cohesin complex in the context of transcriptional regulation and chromatin remodeling rather than sister chromatid cohesion can be held accountable for the onset of the disease phenotype (Yuan et al., 2015). In line with these findings, several patients with CdLS were found to carry variants in regulators of gene expression and chromatin architecture other than cohesin. Notably, the majority of these genes have been previously associated with neurodevelopmental disorders sharing a partial phenotypical overlap with CdLS, such as Rubinstein-Taybi syndrome (RSTS, OMIM #180849), KBG syndrome (KBGS, OMIM #148050), Coffin-Siris syndrome (CSS, OMIM #135900), or Wiedemann-Steiner syndrome (WDSTS, OMIM #605130) (Petrif et al., 1995; Roelfsema et al., 2005; Sirmaci et al., 2011; Jones et al., 2012; Tsurusaki et al., 2012; Asadollahi et al., 2013; Grozeva et al., 2014; Hamdan et al., 2014; Hao et al., 2015; O’Rawe et al., 2015; Olley et al., 2018). Not surprisingly, the aforementioned disorders are often considered as a differential diagnosis for CdLS. On the other hand, variants in cohesin genes have been identified in individuals with neurodevelopmental disorders other than CdLS, such as CSS, WDSTS, Rett-like syndrome, or syndromic intellectual disability (Harakalova et al., 2012; Tzschach et al., 2015; Yuan et al., 2015; Yuen et al., 2015; Retterer et al., 2016; Huisman et al., 2017; Parenti et al., 2017; Saikusa et al., 2018; Xiao et al., 2018; Iwama et al., 2019; Kruszka et al., 2019; Downie et al., 2020; Goel and Parasivam, 2020).
Supported by these findings, a new class of disorders, named chromatinopathies, has started to emerge. Chromatinopathies are caused by variants in proteins responsible for chromatin remodeling and transcriptional regulation. The resulting global gene expression dysregulation favors the onset of a series of clinical features such as developmental delay, intellectual disability, and behavioral disturbances. CdLS, CSS, RSTS, WDSTS, and KBGS all fall within this growing family of disorders.
In this review, we aim to provide a systematic overview of the current literature to summarize all mutations in non-cohesin genes identified in association with CdLS phenotypes. For this purpose, we will discuss the functions of the affected genes, the type of variants, and the clinical features observed. By this, we will acknowledge the role of CdLS as paradigm of chromatinopathies.
Non-canonical Cornelia de Lange Syndrome-Causing Variants
Numerous CdLS patients have been reported to carry mutations in chromatin remodelers and transcriptional regulators other than cohesin. Table 1 summarizes the described variants and provides information on the coordinates, origin and zygosity of the variants as well as gender and phenotypic CdLS scores of the individuals. Scores in parenthesis were calculated based on the published clinical features. A detailed list of the clinical features of each individual is available in Supplementary Table 1. For the purpose of this review, only individuals with a monogenic molecular diagnosis were considered. Individuals with multiple molecular diagnoses or gross deletions/insertions encompassing multiple genes were not included.
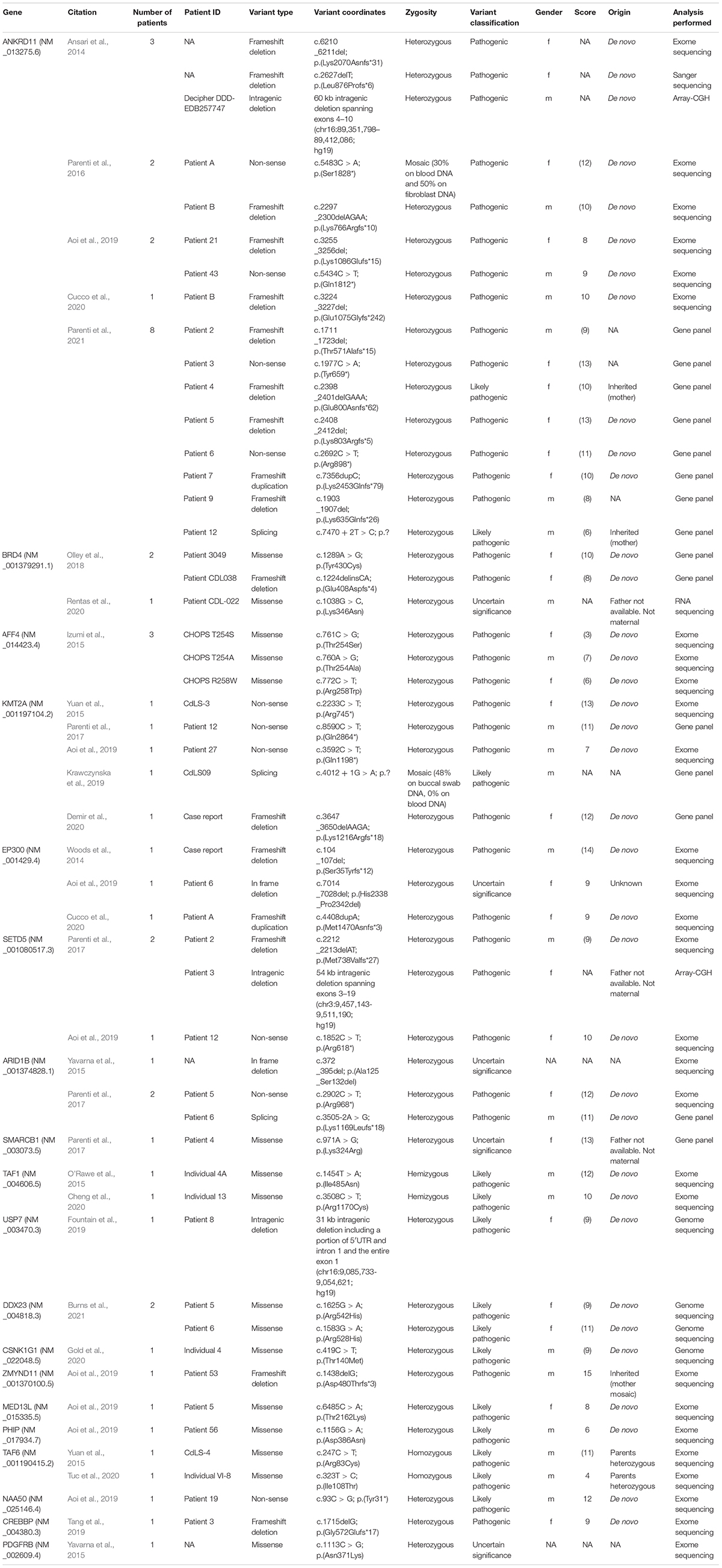
Table 1. Summary of variants in non-cohesin genes identified in CdLS-patients.
Many variants identified in CdLS individuals affect bona fide transcriptional regulators such as ANKRD11, AFF4, BRD4, SETD5, TAF1, TAF6, ZMYND11, PHIP, and MED13L.
ANKRD11 regulates gene expression through the interaction with histone-modifying proteins (Zhang et al., 2007; Li et al., 2008). Variants affecting the ANKRD11 gene were formerly associated with KBGS (Sirmaci et al., 2011). To date, 16 individuals who received a clinical diagnosis of CdLS during infancy were found to harbor loss-of-function variants in ANKRD11 (Ansari et al., 2014; Parenti et al., 2016, 2021; Aoi et al., 2019; Cucco et al., 2020). Clinical scores could be assessed for 13 of these 16 individuals. With an average score of 10, variants in ANKRD11 appear to be associated with non-classic CdLS phenotypes. The relatively high frequency of ANKRD11 variants in CdLS cohorts has motivated the inclusion of ANKRD11 among the CdLS-genes (Kline et al., 2018).
BRD4 binds to super-enhancers elements and promotes the release of the paused RNA polymerase II (Olley et al., 2018). Three CdLS individuals with two missense substitutions and a frameshift deletion-insertion affecting BRD4 were so far described (Olley et al., 2018). Clinical scores of 8 and 10 could be calculated for two of the three patients, thus indicating a partial overlap with CdLS.
Loss-of-function variants in SETD5 had been initially reported in patients with moderate-to-severe intellectual disability (OMIM, #615761) (Grozeva et al., 2014). Recently, SETD5 has been recognized as one of the most frequently mutated genes in the context of neurodevelopmental disorders (Deciphering Developmental Disorders Study, 2017; Kaplanis et al., 2020). The resulting protein carries out its function as transcriptional regulator upon interaction with two protein complexes, namely an HDAC3-containing chromatin remodeler known as Nuclear Receptor Co-Repressor (NCoR) and the RNA polymerase II-interacting complex known as Polymerase-Associated Factor 1 Complex (PAF1C) (Osipovich et al., 2016; Deliu et al., 2018). A total of three individuals carrying SETD5 variants were identified in two independent CdLS cohorts (Parenti et al., 2017; Aoi et al., 2019). The resulting clinical scores (9 and 10) suggest a non-classic form of CdLS.
TAF1 and TAF6 are both subunits of Transcription Factor II D (TFIID), a megadalton-sized protein complex that promotes transcriptional initiation (Bieniossek et al., 2013). Variants affecting TAF1 and TAF6 are, respectively, associated with X-linked recessive intellectual disability (OMIM #300966) and autosomal recessive Alazami-Yuan syndrome (OMIM #617126) (Alazami et al., 2015; O’Rawe et al., 2015). Hemizygous missense substitutions in TAF1 were identified in two individuals with CdLS (clinical scores 12 and 10), whereas two individuals were found to carry homozygous missense variants in TAF6 (clinical scores 11 and 4) (O’Rawe et al., 2015; Yuan et al., 2015; Cheng et al., 2020; Tuc et al., 2020).
ZMYND11, PHIP, and MED13L were each found mutated in a single CdLS individual (Aoi et al., 2019). ZMYND11 was the only non-cohesin-related gene altered in an individual with a clinical score of 15 and presenting with oligodactyly (Aoi et al., 2019). Prior to this discovery, ZMYND11 had been associated with intellectual disability and behavioral disturbances (OMIM #616083); furthermore, it appears to be a critical gene in the context of the 10p15.3 microdeletion syndrome (Coe et al., 2014). The resulting protein specifically binds to trimethylated lysine 36 of histone H3 to modulate elongation of RNA polymerase II (Wen et al., 2014). PHIP encodes for a DNA-binding protein that localizes at promoters and transcriptional cis-regulatory elements (Aoi et al., 2019). Variants in PHIP are responsible for the obesity-associated neurodevelopmental syndrome known as Chung-Jansen syndrome (OMIM #617991) (de Ligt et al., 2012; Jansen et al., 2018). Variants in MED13L, a subunit of the transcriptional regulator known as Mediator complex, are instead responsible for a form of intellectual disability with dysmorphic features (OMIM #616789). Missense substitutions in MED13L and PHIP were described in two patients with CdLS-like phenotypes (clinical scores 8 and 6, respectively) (Aoi et al., 2019).
In addition, missense substitutions in AFF4, a subunit of the super elongation complex which coordinates pausing of RNA polymerase II, were identified in individuals with CHOPS (cognitive impairment, coarse facies, heart defects, obesity, pulmonary involvement, short stature, and skeletal dysplasia; OMIM #616368), who were initially suspected of having CdLS (Izumi et al., 2015). The low clinical scores of these individuals (3, 7, and 6) suggest a limited phenotypical overlap with CdLS.
Proteins that have an impact on chromatin conformation are also occasionally altered in CdLS individuals. The list of chromatin remodelers associated with CdLS comprises KMT2A, ARID1B, SMARCB1, CREBBP, and EP300.
KMT2A is a histone methyltransferase whose mutations are responsible for the onset of WDSTS (Jones et al., 2012). Five loss-of-function variants affecting KMT2A were reported in CdLS individuals (Yuan et al., 2015; Parenti et al., 2017; Aoi et al., 2019; Krawczynska et al., 2019; Demir et al., 2020). Clinical scores could be assessed for four of the five individuals. A score equal to or higher than 11 was calculated for three of these individuals, suggesting that KMT2A might be contemplated in the future as additional CdLS-gene.
ARID1B and SMARCB1 are structural components of the multisubunit protein complex named SWItch/Sucrose Non-Fermentable complex (SWI/SNF), which is known for its role as ATP-dependent chromatin remodeler (Kassabov et al., 2003). Mutations in ARID1B, SMARCB1, and other subunits of the SWI/SNF remodeler cause CSS (Santen et al., 2012; Tsurusaki et al., 2012). To date, three CdLS individuals were found to carry loss-of-function variants in ARID1B and one individual carried a missense substitution in SMARCB1 (Yavarna et al., 2015; Parenti et al., 2017). Similar to KMT2A, the clinical scores of these patients fell within the range of classic manifestation of CdLS.
CREBBP and EP300 are part of a coactivator family characterized by intrinsic ability to acetylate histone as well as non-histone proteins and to interact with core transcription factors (Vo and Goodman, 2001; Jin et al., 2011). Mutations in CREBBP and EP300 result in distinct subtypes of RSTS (Petrif et al., 1995; Roelfsema et al., 2005). In CdLS cohorts, exome sequencing led to the identification of three loss-of-function mutations in EP300 and one out-of-frame deletion in CREBBP (Woods et al., 2014; Aoi et al., 2019; Tang et al., 2019; Cucco et al., 2020). With the exception of a single patient presenting with classic CdLS (Woods et al., 2014), the other individuals with variants in CREBBP and EP300 appear to be associated with a rather non-classic form of CdLS (average clinical score of 9) (Aoi et al., 2019; Tang et al., 2019; Cucco et al., 2020).
The remaining CdLS-associated proteins USP7, DDX23, CSNK1G1, NAA50, and PDGFRB act indirectly on nuclear processes through their interaction with several proteins involved in genomic stability, transcriptional regulation, and chromatin remodeling.
DDX23 is a RNA helicase with a role in RNA splicing and maintenance of genomic stability through suppression of incorrect R-loops formed during transcription (Mathew et al., 2008; Sridhara et al., 2017). Two out of the nine recently published individuals with DDX23-related neurodevelopmental disorders presented with clinical features suggestive of CdLS and clinical scores of 9 and 11 (Burns et al., 2021).
USP7 is a deubiquitinating proteolytic enzyme with a variety of targets, including DNMT1 and members of the Polycomb multiprotein complex. By preventing their ubiquitin-dependent degradation, it promotes DNA methylation and chromatin remodeling (Maertens et al., 2010; Felle et al., 2011). Variants in USP7 are responsible for a neurodevelopmental disorder with speech delay, altered behavior, and neurologic anomalies (Hao-Fountain syndrome, OMIM #616863) (Hao et al., 2015; Fountain et al., 2019). An individual with a CdLS score of 9 was found to carry an intragenic deletion affecting the 5′UTR and exon 1 of USP7 (Fountain et al., 2019).
A missense substitution in NAA50 was identified in an individual with classic CdLS (clinical score 12). NAA50 interacts with the highly conserved NatA complex composed of NAA10 and NAA15 to form the NatE complex (Deng et al., 2019; Armbruster et al., 2020). The main function of these proteins is to carry out N-terminal acetylation, a major post-translational modification to which 70–90% of proteins are subject in humans (Reddi et al., 2016; Gottlieb and Marmorstein, 2018; Deng et al., 2019). Strikingly, individuals with NAA10 variants often show phenotypes reminiscent of CdLS (Saunier et al., 2016).
CSNK1G1 and PDGFRB possess intrinsic kinase activity through which they regulate several cellular processes including signal transduction, cell migration, and proliferation (Mori et al., 1993; Li et al., 2015). The corresponding genes have been associated with two distinct forms of syndromic neurodevelopmental disorder (Foster et al., 2020; Gold et al., 2020). Missense substitutions of each gene were identified in single individuals with CdLS-overlapping phenotypes (Yavarna et al., 2015; Gold et al., 2020).
In view of the high CdLS scores reported, KMT2A and the subunits of the SWI/SNF complex can be included within the extended list of CdLS genes. Variants in ANKRD11, SETD5, EP300, CREBBP, BRD4, and TAF1 can similarly result in non-classic forms of CdLS. For this reason, these genes should be taken into account for the molecular diagnostic pipeline of CdLS. Individuals with AFF4 variants instead present with a distinct phenotype that is only minimally overlapping with CdLS. The contribution of the other genes presented in this review in the context of CdLS still remains to be assessed (USP7, TAF6, DDX23, CSNK1G1, ZMYND11, MED13L, PHIP, NAA50, and PDGFRB).
The Chromatinopathies Protein Network
Cohesin and non-cohesin proteins involved in the pathogenesis of CdLS and other neurodevelopmental disorders do not only share overlapping functions. These proteins are profoundly interconnected and give rise to a genuine chromatinopathies protein network. Figure 1 provides a schematic overview of the network; here, the chromatinopathies proteins are illustrated in light of their physical and functional interactions. Central nodes of the network such as HDAC3 or POLR2A, despite not being associated with CdLS so far, are depicted to allow a more comprehensive outlook of the network. It is apparent how the proteins involved act concertedly and regulate each other with the aim of controlling transcription. The tightly regulated interplay of components is in fact responsible for the coordinated expression of numerous genes. Given the major role of RNA polymerase II, mediator, and TFIID complexes in the context of gene expression regulation, it is not surprising that several chromatinopathies proteins either interact with or indirectly control the levels or activity of these three main effectors. For instance, the canonical CdLS-protein complex, i.e., cohesin, can directly influence the amount of RNA polymerase II available at the promoters of several genes (Schaaf et al., 2013). Furthermore, cohesin functionally and physically interacts with the mediator complex to connect enhancers and promoters of active genes (Kagey et al., 2010). The recruitment of RNA polymerase II is also dependent on HDAC3 (Wang et al., 2018), a histone deacetylase that equally appears to be one of the central nodes of the chromatinopathies network. The roles of HDAC3 within the network are in fact plentiful, as it was reported to interact directly with numerous players with the aim of “fine-tuning” transcription. The HDAC3-interacting proteins comprise SETD5, ANKRD11, EP300, CREBBP, and the cohesin loader NIPBL (Zhang et al., 2004; Jahnke et al., 2008; Sankar et al., 2008; Osipovich et al., 2016; Deliu et al., 2018). Remarkably, whereas mutations affecting RNA polymerase II have already been associated with a neurodevelopmental disorder that overlaps with chromatinopathies (OMIM, #618603) (Haijes et al., 2019), variants in HDAC3 have never been reported. Taking into account the central role of HDAC3 in the transcription process, a possible identification of disease-causing HDAC3 variants can be envisaged.
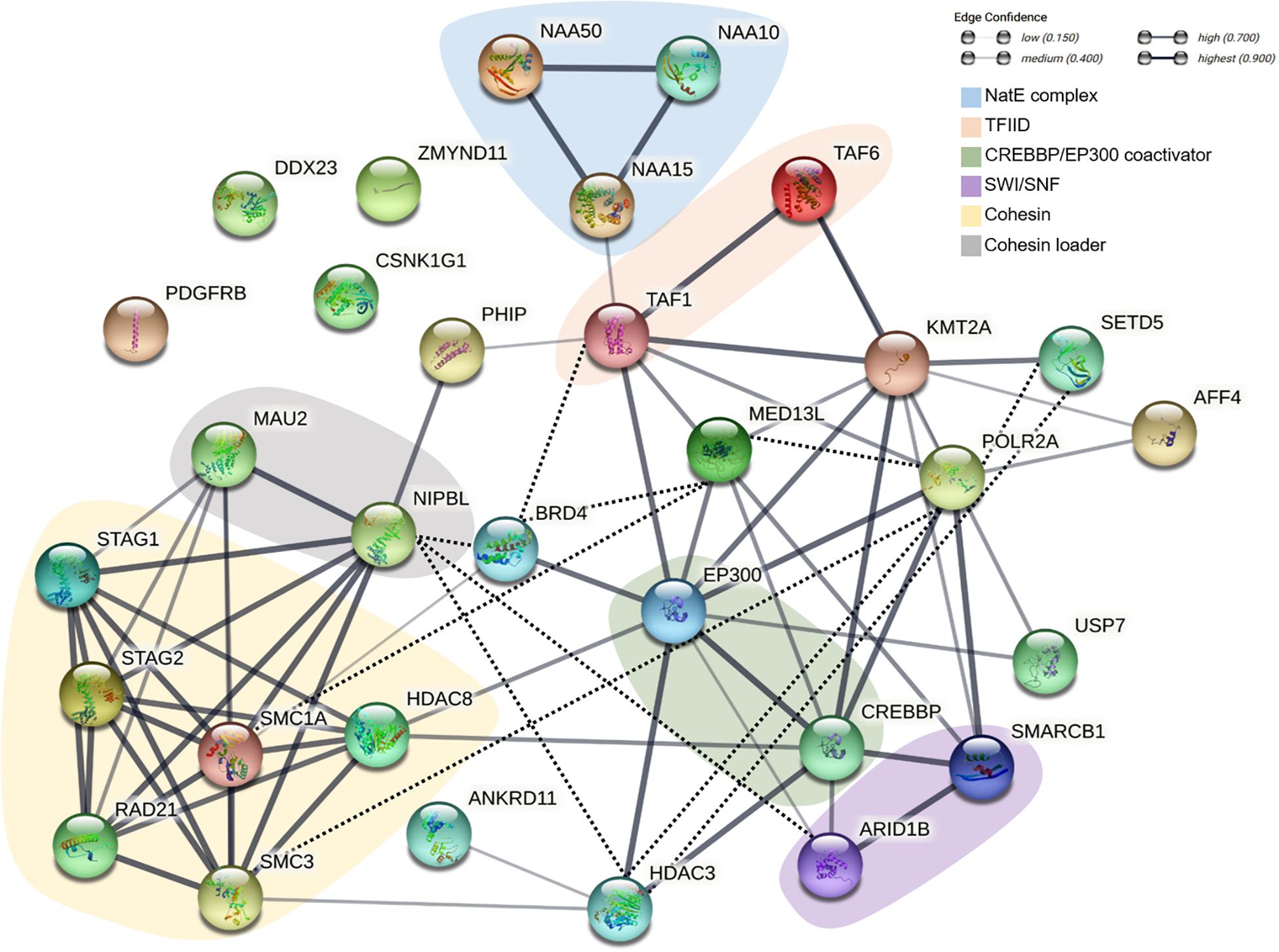
Figure 1. Schematic representation of the functional and physical interactions of the chromatinopathies protein network. The network was generated with the String Database (v. 11.5) (Szklarczyk et al., 2019). Empty nodes represent proteins of unknown 3D structure, while filled nodes indicate proteins with known or predicted protein structure. Line thickness indicates the strength of data support. Interactions were established based on co-expression or data from either curated databases or experimentally determined. The network was subsequently manually curated (dotted black line) based on more recent literature.
Following its recruitment to the DNA, the dynamics and activity of RNA polymerase II are further subject to regulation through proteins like SETD5 and BRD4 (Osipovich et al., 2016; Lee et al., 2017; Deliu et al., 2018). Specifically, BRD4 can control transcription by promoting the enrichment of RNA polymerase II, mediator and TFIID at target genes (Lee et al., 2017) and through its interaction with NIPBL and different cohesin subunits (Olley et al., 2018). In turn, the acetyltransferase EP300 and CREBBP seem to be responsible for BRD4 recruitment to enhancers (Lee et al., 2017). Additional data suggest that EP300 and CREBBP contribute to chromatin architecture along with the mediator complex (Zhang et al., 2020), the methyltransferase KMT2A (Goto et al., 2002), and the SWI/SNF complex (Alver et al., 2017). The latter is itself responsible for the recruitment of the cohesin loader to nucleosome-free regions (Lopez-Serra et al., 2014) and is as well able to interact with RNA polymerase II and the TFIID complex (Sharma et al., 2003).
This is certainly a simplistic view of the incredibly complex and perfectly orchestrated process that is transcription, but conveys the idea of how much interconnected the chromatinopathies protein network is. The level of synergy of the network is so high that variants of a single factor will inevitably result in an altered function of the other players.
Conclusion
Several proteins with interdependent roles belong to the chromatinopathies protein network. Disease-causing variants in the corresponding genes are accountable for the onset of distinct but overlapping neurodevelopmental disorders, of which CdLS is a paradigm. Whether or not the resulting transcriptional dysregulation converge on a common pathway or set of genes is an intriguing possibility that is worth exploring for therapeutic purposes.
Author Contributions
Both authors contributed to the manuscript drafting, read and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
This work has been generated within the European Reference Network on Rare Congenital Malformations and Rare Intellectual Disability (ERN-ITHACA) (EU Framework Partnership Agreement ID: 3HP-HP-FPA ERN-01-2016/739516).
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnins.2021.774950/full#supplementary-material
References
Alazami, A. M., Patel, N., Shamseldin, H. E., Anazi, S., Al-Dosari, M. S., Alzahrani, F., et al. (2015). Accelerating novel candidate gene discovery in neurogenetic disorders via whole-exome sequencing of prescreened multiplex consanguineous families. Cell Rep. 10, 148–161. doi: 10.1016/j.celrep.2014.12.015
Alver, B. H., Kim, K. H., Lu, P., Wang, X., Manchester, H. E., Wang, W., et al. (2017). The SWI/SNF chromatin remodelling complex is required for maintenance of lineage specific enhancers. Nat. Commun. 8:14648. doi: 10.1038/ncomms14648
Ansari, M., Poke, G., Ferry, Q., Williamson, K., Aldridge, R., Meynert, A. M., et al. (2014). Genetic heterogeneity in Cornelia de Lange syndrome (CdLS) and CdLS-like phenotypes with observed and predicted levels of mosaicism. J. Med. Genet. 51, 659–668. doi: 10.1136/jmedgenet-2014-102573
Aoi, H., Mizuguchi, T., Ceroni, J. R., Kim, V. E. H., Furquim, I., Honjo, R. S., et al. (2019). Comprehensive genetic analysis of 57 families with clinically suspected Cornelia de Lange syndrome. J. Hum. Genet. 64, 967–978. doi: 10.1038/s10038-019-0643-z
Armbruster, L., Linster, E., Boyer, J.-B., Brünje, A., Eirich, J., Stephan, I., et al. (2020). NAA50 is an enzymatically active N α -Acetyltransferase that is crucial for development and regulation of stress responses. Plant Physiol. 183, 1502–1516. doi: 10.1104/pp.20.00222
Asadollahi, R., Oneda, B., Sheth, F., Azzarello-Burri, S., Baldinger, R., Joset, P., et al. (2013). Dosage changes of MED13L further delineate its role in congenital heart defects and intellectual disability. Eur. J. Hum. Genet. 21, 1100–1104. doi: 10.1038/ejhg.2013.17
Bieniossek, C., Papai, G., Schaffitzel, C., Garzoni, F., Chaillet, M., Scheer, E., et al. (2013). The architecture of human general transcription factor TFIID core complex. Nature 493, 699–702. doi: 10.1038/nature11791
Burns, W., Bird, L. M., Heron, D., Keren, B., Ramachandra, D., Thiffault, I., et al. (2021). Syndromic neurodevelopmental disorder associated with de novo variants in DDX23. Am. J. Med. Genet. 185, 2863–2872. doi: 10.1002/ajmg.a.62359
Castronovo, P., Gervasini, C., Cereda, A., Masciadri, M., Milani, D., Russo, S., et al. (2009). Premature chromatid separation is not a useful diagnostic marker for Cornelia de Lange syndrome. Chromosome Res. 17, 763–771. doi: 10.1007/s10577-009-9066-6
Cheng, H., Capponi, S., Wakeling, E., Marchi, E., Li, Q., Zhao, M., et al. (2020). Missense variants in TAF1 and developmental phenotypes: challenges of determining pathogenicity. Hum. Mutat. 41, 449–464. doi: 10.1002/humu.23936
Coe, B. P., Witherspoon, K., Rosenfeld, J. A., van Bon, B. W. M., Vulto-van Silfhout, A. T., Bosco, P., et al. (2014). Refining analyses of copy number variation identifies specific genes associated with developmental delay. Nat. Genet. 46, 1063–1071. doi: 10.1038/ng.3092
Cucco, F., Sarogni, P., Rossato, S., Alpa, M., Patimo, A., Latorre, A., et al. (2020). Pathogenic variants in EP300 and ANKRD11 in patients with phenotypes overlapping Cornelia de Lange syndrome. Am. J. Med. Genet. 182, 1690–1696. doi: 10.1002/ajmg.a.61611
de Ligt, J., Willemsen, M. H., van Bon, B. W. M., Kleefstra, T., Yntema, H. G., Kroes, T., et al. (2012). diagnostic exome sequencing in persons with severe intellectual disability. N. Engl. J. Med. 367, 1921–1929. doi: 10.1056/NEJMoa1206524
Deciphering Developmental Disorders Study (2017). Prevalence and architecture of de novo mutations in developmental disorders. Nature 542, 433–438. doi: 10.1038/nature21062
Deliu, E., Arecco, N., Morandell, J., Dotter, C. P., Contreras, X., Girardot, C., et al. (2018). Haploinsufficiency of the intellectual disability gene SETD5 disturbs developmental gene expression and cognition. Nat. Neurosci. 21, 1717–1727. doi: 10.1038/s41593-018-0266-2
Demir, S., Gürkan, H., Öz, V., Yalçıntepe, S., Atlı, E. Ý, and Atlı, E. (2020). Wiedemann-steiner syndrome as a differential diagnosis of Cornelia de Lange syndrome using targeted next-generation sequencing: a case report. Mol. Syndromol. 12, 1–6. doi: 10.1159/000511971
Deng, S., Magin, R. S., Wei, X., Pan, B., Petersson, E. J., and Marmorstein, R. (2019). Structure and mechanism of acetylation by the N-Terminal dual enzyme NatA/Naa50 complex. Structure 27, 1057.e4–1070.e4. doi: 10.1016/j.str.2019.04.014
Downie, L., Halliday, J., Burt, R., Lunke, S., Lynch, E., Martyn, M., et al. (2020). Exome sequencing in infants with congenital hearing impairment: a population-based cohort study. Eur. J. Hum. Genet. 28, 587–596. doi: 10.1038/s41431-019-0553-8
Felle, M., Joppien, S., Németh, A., Diermeier, S., Thalhammer, V., Dobner, T., et al. (2011). The USP7/Dnmt1 complex stimulates the DNA methylation activity of Dnmt1 and regulates the stability of UHRF1. Nucleic Acids Res. 39, 8355–8365. doi: 10.1093/nar/gkr528
Foster, A., Chalot, B., Antoniadi, T., Schaefer, E., Keelagher, R., Ryan, G., et al. (2020). Kosaki overgrowth syndrome: a novel pathogenic variant in PDGFRB and expansion of the phenotype including cerebrovascular complications. Clin. Genet. 98, 19–31. doi: 10.1111/cge.13752
Fountain, M. D., Oleson, D. S., Rech, M. E., Segebrecht, L., Hunter, J. V., McCarthy, J. M., et al. (2019). Pathogenic variants in USP7 cause a neurodevelopmental disorder with speech delays, altered behavior, and neurologic anomalies. Genet. Med. 21, 1797–1807. doi: 10.1038/s41436-019-0433-1
Goel, H., and Parasivam, G. (2020). Another case of holoprosencephaly associated with RAD21 loss-of-function variant. Brain 143:e64. doi: 10.1093/brain/awaa173
Gold, N. B., Li, D., Chassevent, A., Kaiser, F. J., Parenti, I., Strom, T. M., et al. (2020). Heterozygous de novo variants in CSNK1G1 are associated with syndromic developmental delay and autism spectrum disorder. Clin. Genet. 98, 571–576. doi: 10.1111/cge.13851
Goto, N. K., Zor, T., Martinez-Yamout, M., Dyson, H. J., and Wright, P. E. (2002). Cooperativity in transcription factor binding to the coactivator CREB-binding Protein (CBP). J. Biol. Chem. 277, 43168–43174. doi: 10.1074/jbc.M207660200
Gottlieb, L., and Marmorstein, R. (2018). Structure of Human NatA and its regulation by the huntingtin interacting protein HYPK. Structure 26, 925.e8–935.e8. doi: 10.1016/j.str.2018.04.003
Grozeva, D., Carss, K., Spasic-Boskovic, O., Parker, M. J., Archer, H., Firth, H. V., et al. (2014). De novo loss-of-function mutations in SETD5, encoding a methyltransferase in a 3p25 microdeletion syndrome critical region, cause intellectual disability. Am. J. Hum. Genet. 94, 618–624. doi: 10.1016/j.ajhg.2014.03.006
Haijes, H. A., Koster, M. J. E., Rehmann, H., Li, D., Hakonarson, H., Cappuccio, G., et al. (2019). De novo heterozygous POLR2A variants cause a neurodevelopmental syndrome with profound infantile-onset hypotonia. Am. J. Hum. Genet. 105, 283–301. doi: 10.1016/j.ajhg.2019.06.016
Hamdan, F. F., Srour, M., Capo-Chichi, J.-M., Daoud, H., Nassif, C., Patry, L., et al. (2014). De novo mutations in moderate or severe intellectual disability. PLoS Genet. 10:e1004772. doi: 10.1371/journal.pgen.1004772
Hao, Y.-H., Fountain, M. D., Fon Tacer, K., Xia, F., Bi, W., Kang, S.-H. L., et al. (2015). USP7 acts as a molecular rheostat to promote WASH-dependent endosomal protein recycling and is mutated in a human neurodevelopmental disorder. Mol. Cell 59, 956–969. doi: 10.1016/j.molcel.2015.07.033
Harakalova, M., van den Boogaard, M.-J., Sinke, R., van Lieshout, S., van Tuil, M. C., Duran, K., et al. (2012). X-exome sequencing identifies a HDAC8 variant in a large pedigree with X-linked intellectual disability, truncal obesity, gynaecomastia, hypogonadism and unusual face. J. Med. Genet. 49, 539–543. doi: 10.1136/jmedgenet-2012-100921
Huisman, S., Mulder, P. A., Redeker, E., Bader, I., Bisgaard, A.-M., Brooks, A., et al. (2017). Phenotypes and genotypes in individuals with SMC1A variants. Am. J. Med. Genet. 173, 2108–2125. doi: 10.1002/ajmg.a.38279
Iwama, K., Mizuguchi, T., Takeshita, E., Nakagawa, E., Okazaki, T., Nomura, Y., et al. (2019). Genetic landscape of Rett syndrome-like phenotypes revealed by whole exome sequencing. J. Med. Genet. 56, 396–407. doi: 10.1136/jmedgenet-2018-105775
Izumi, K., Nakato, R., Zhang, Z., Edmondson, A. C., Noon, S., Dulik, M. C., et al. (2015). Germline gain-of-function mutations in AFF4 cause a developmental syndrome functionally linking the super elongation complex and cohesin. Nat. Genet. 47, 338–344. doi: 10.1038/ng.3229
Jahnke, P., Xu, W., Wulling, M., Albrecht, M., Gabriel, H., Gillessen-Kaesbach, G., et al. (2008). The Cohesin loading factor NIPBL recruits histone deacetylases to mediate local chromatin modifications. Nucleic Acids Res. 36, 6450–6458. doi: 10.1093/nar/gkn688
Jansen, S., Hoischen, A., Coe, B. P., Carvill, G. L., Van Esch, H., Bosch, D. G. M., et al. (2018). A genotype-first approach identifies an intellectual disability-overweight syndrome caused by PHIP haploinsufficiency. Eur. J. Hum. Genet. 26, 54–63. doi: 10.1038/s41431-017-0039-5
Jin, Q., Yu, L.-R., Wang, L., Zhang, Z., Kasper, L. H., Lee, J.-E., et al. (2011). Distinct roles of GCN5/PCAF-mediated H3K9ac and CBP/p300-mediated H3K18/27ac in nuclear receptor transactivation: histone acetylation and gene activation. EMBO J. 30, 249–262. doi: 10.1038/emboj.2010.318
Jones, W. D., Dafou, D., McEntagart, M., Woollard, W. J., Elmslie, F. V., Holder-Espinasse, M., et al. (2012). De novo mutations in MLL cause wiedemann-steiner syndrome. Am. J. Hum. Genet. 91, 358–364. doi: 10.1016/j.ajhg.2012.06.008
Kagey, M. H., Newman, J. J., Bilodeau, S., Zhan, Y., Orlando, D. A., van Berkum, N. L., et al. (2010). Mediator and cohesin connect gene expression and chromatin architecture. Nature 467, 430–435. doi: 10.1038/nature09380
Kaplanis, J., Samocha, K. E., Wiel, L., Zhang, Z., Arvai, K. J., Eberhardt, R. Y., et al. (2020). Evidence for 28 genetic disorders discovered by combining healthcare and research data. Nature 586, 757–762. doi: 10.1038/s41586-020-2832-5
Kassabov, S. R., Zhang, B., Persinger, J., and Bartholomew, B. (2003). SWI/SNF Unwraps. Slides, and rewraps the nucleosome. Mol. Cell 11, 391–403. doi: 10.1016/S1097-2765(03)00039-X
Kline, A. D., Krantz, I. D., Sommer, A., Kliewer, M., Jackson, L. G., FitzPatrick, D. R., et al. (2007). Cornelia de Lange syndrome: clinical review, diagnostic and scoring systems, and anticipatory guidance. Am. J. Med. Genet. 143A, 1287–1296. doi: 10.1002/ajmg.a.31757
Kline, A. D., Moss, J. F., Selicorni, A., Bisgaard, A.-M., Deardorff, M. A., Gillett, P. M., et al. (2018). Diagnosis and management of Cornelia de Lange syndrome: first international consensus statement. Nat. Rev. Genet. 19, 649–666. doi: 10.1038/s41576-018-0031-0
Krawczynska, N., Wierzba, J., and Wasag, B. (2019). Genetic mosaicism in a group of patients with Cornelia de Lange Syndrome. Front. Pediatr. 7:203. doi: 10.3389/fped.2019.00203
Kruszka, P., Berger, S. I., Casa, V., Dekker, M. R., Gaesser, J., Weiss, K., et al. (2019). Cohesin complex-associated holoprosencephaly. Brain 142, 2631–2643. doi: 10.1093/brain/awz210
Lee, J.-E., Park, Y.-K., Park, S., Jang, Y., Waring, N., Dey, A., et al. (2017). Brd4 binds to active enhancers to control cell identity gene induction in adipogenesis and myogenesis. Nat. Commun. 8:2217. doi: 10.1038/s41467-017-02403-5
Lehalle, D., Mosca-Boidron, A.-L., Begtrup, A., Boute-Benejean, O., Charles, P., Cho, M. T., et al. (2017). STAG1 mutations cause a novel cohesinopathy characterised by unspecific syndromic intellectual disability. J. Med. Genet. 54, 479–488. doi: 10.1136/jmedgenet-2016-104468
Li, C.-W., Dinh, G. K., Zhang, A., and Chen, J. D. (2008). Ankyrin repeats-containing cofactors interact with ADA3 and modulate its co-activator function. Biochem. J. 413, 349–357. doi: 10.1042/BJ20071484
Li, D.-P., Zhou, J.-J., and Pan, H.-L. (2015). Endogenous casein kinase-1 modulates NMDA receptor activity of hypothalamic presympathetic neurons and sympathetic outflow in hypertension: casein kinase-1 and synaptic plasticity in hypertension. J. Physiol. 593, 4439–4452. doi: 10.1113/JP270831
Liu, J., Zhang, Z., Bando, M., Itoh, T., Deardorff, M. A., Clark, D., et al. (2009). Transcriptional dysregulation in NIPBL and cohesin mutant human cells. PLoS Biol. 7:e1000119. doi: 10.1371/journal.pbio.1000119
Lopez-Serra, L., Kelly, G., Patel, H., Stewart, A., and Uhlmann, F. (2014). The Scc2–Scc4 complex acts in sister chromatid cohesion and transcriptional regulation by maintaining nucleosome-free regions. Nat. Genet. 46, 1147–1151. doi: 10.1038/ng.3080
Maertens, G. N., El Messaoudi-Aubert, S., Elderkin, S., Hiom, K., and Peters, G. (2010). Ubiquitin-specific proteases 7 and 11 modulate Polycomb regulation of the INK4a tumour suppressor. EMBO J. 29, 2553–2565. doi: 10.1038/emboj.2010.129
Mathew, R., Hartmuth, K., Möhlmann, S., Urlaub, H., Ficner, R., and Lührmann, R. (2008). Phosphorylation of human PRP28 by SRPK2 is required for integration of the U4/U6-U5 tri-snRNP into the spliceosome. Nat. Struct. Mol. Biol. 15, 435–443. doi: 10.1038/nsmb.1415
Mori, S., Rönnstrand, L., Yokote, K., Engström, A., Courtneidge, S. A., Claesson-Welsh, L., et al. (1993). Identification of two juxtamembrane autophosphorylation sites in the PDGF beta-receptor; involvement in the interaction with Src family tyrosine kinases. EMBO J. 12, 2257–2264. doi: 10.1002/j.1460-2075.1993.tb05879.x
Mullegama, S. V., Klein, S. D., Mulatinho, M. V., Senaratne, T. N., and Singh, K., Ucla Clinical Genomics Center, et al. (2017). De novo loss-of-function variants in STAG2 are associated with developmental delay, microcephaly, and congenital anomalies. Am. J. Med. Genet. 173, 1319–1327. doi: 10.1002/ajmg.a.38207
Olley, G., Ansari, M., Bengani, H., Grimes, G. R., Rhodes, J., von Kriegsheim, A., et al. (2018). BRD4 interacts with NIPBL and BRD4 is mutated in a Cornelia de Lange–like syndrome. Nat. Genet. 50, 329–332. doi: 10.1038/s41588-018-0042-y
O’Rawe, J. A., Wu, Y., Dörfel, M. J., Rope, A. F., Au, P. Y. B., Parboosingh, J. S., et al. (2015). TAF1 variants are associated with dysmorphic features, intellectual disability, and neurological manifestations. Am. J. Hum. Genet. 97, 922–932. doi: 10.1016/j.ajhg.2015.11.005
Osipovich, A. B., Gangula, R., Vianna, P. G., and Magnuson, M. A. (2016). Setd5 is essential for mammalian development and co-transcriptional regulation of histone acetylation. Development 143, 4595–4607. doi: 10.1242/dev.141465
Parenti, I., Diab, F., Gil, S. R., Mulugeta, E., Casa, V., Berutti, R., et al. (2020). MAU2 and NIPBL variants impair the heterodimerization of the cohesin loader subunits and cause Cornelia de Lange syndrome. Cell Rep. 31:107647. doi: 10.1016/j.celrep.2020.107647
Parenti, I., Gervasini, C., Pozojevic, J., Graul-Neumann, L., Azzollini, J., Braunholz, D., et al. (2016). Broadening of cohesinopathies: exome sequencing identifies mutations in ANKRD11 in two patients with Cornelia de Lange-overlapping phenotype: broadening of cohesinopathies. Clin. Genet. 89, 74–81. doi: 10.1111/cge.12564
Parenti, I., Mallozzi, M. B., Hüning, I., Gervasini, C., Kuechler, A., Agolini, E., et al. (2021). ANKRD11 variants: KBG syndrome and beyond. Clin. Genet. 100, 187–200. doi: 10.1111/cge.13977
Parenti, I., Teresa-Rodrigo, M. E., Pozojevic, J., Ruiz Gil, S., Bader, I., Braunholz, D., et al. (2017). Mutations in chromatin regulators functionally link Cornelia de Lange syndrome and clinically overlapping phenotypes. Hum. Genet. 136, 307–320. doi: 10.1007/s00439-017-1758-y
Petrif, F., Giles, R. H., Dauwerse, H. G., Saris, J. J., Hennekam, R. C. M., Masuno, M., et al. (1995). Rubinstein-Taybi syndrome caused by mutations in the transcriptional co-activator CBP. Nature 376, 348–351. doi: 10.1038/376348a0
Reddi, R., Saddanapu, V., Chinthapalli, D. K., Sankoju, P., Sripadi, P., and Addlagatta, A. (2016). Human Naa50 protein displays broad substrate specificity for amino-terminal acetylation. J. Biol. Chem. 291, 20530–20538. doi: 10.1074/jbc.M116.730432
Rentas, S., Rathi, K. S., Kaur, M., Raman, P., Krantz, I. D., Sarmady, M., et al. (2020). Diagnosing Cornelia de Lange syndrome and related neurodevelopmental disorders using RNA sequencing. Genet. Med. 22, 927–936. doi: 10.1038/s41436-019-0741-5
Retterer, K., Juusola, J., Cho, M. T., Vitazka, P., Millan, F., Gibellini, F., et al. (2016). Clinical application of whole-exome sequencing across clinical indications. Genet. Med. 18, 696–704. doi: 10.1038/gim.2015.148
Roelfsema, J. H., White, S. J., Ariyürek, Y., Bartholdi, D., Niedrist, D., Papadia, F., et al. (2005). Genetic heterogeneity in rubinstein-taybi syndrome: mutations in both the CBP and EP300 genes cause disease. Am. J. Hum. Genet. 76, 572–580. doi: 10.1086/429130
Saikusa, T., Hara, M., Iwama, K., Yuge, K., Ohba, C., Okada, J., et al. (2018). De novo HDAC8 mutation causes Rett-related disorder with distinctive facial features and multiple congenital anomalies. Brain Dev. 40, 406–409. doi: 10.1016/j.braindev.2017.12.013
Sankar, N., Baluchamy, S., Kadeppagari, R.-K., Singhal, G., Weitzman, S., and Thimmapaya, B. (2008). p300 provides a corepressor function by cooperating with YY1 and HDAC3 to repress c-Myc. Oncogene 27, 5717–5728. doi: 10.1038/onc.2008.181
Santen, G. W. E., Kriek, M., and van Attikum, H. (2012). SWI/SNF complex in disorder: SWItching from malignancies to intellectual disability. Epigenetics 7, 1219–1224. doi: 10.4161/epi.22299
Saunier, C., Støve, S. I., Popp, B., Gérard, B., Blenski, M., AhMew, N., et al. (2016). Expanding the phenotype associated with NAA10-Related N-Terminal acetylation deficiency. Hum. Mutat. 37, 755–764. doi: 10.1002/humu.23001
Schaaf, C. A., Misulovin, Z., Gause, M., Koenig, A., and Dorsett, D. (2013). The Drosophila Enhancer of split gene complex: architecture and coordinate regulation by notch. Cohesin, and Polycomb Group Proteins. G3 Genes Genomes Genet. 3, 1785–1794. doi: 10.1534/g3.113.007534
Sharma, V. M., Li, B., and Reese, J. C. (2003). SWI/SNF-dependent chromatin remodeling of RNR3 requires TAFIIs and the general transcription machinery. Genes Dev. 17, 502–515. doi: 10.1101/gad.1039503
Sirmaci, A., Spiliopoulos, M., Brancati, F., Powell, E., Duman, D., Abrams, A., et al. (2011). Mutations in ANKRD11 cause KBG Syndrome, characterized by intellectual disability, skeletal malformations, and macrodontia. Am. J. Hum. Genet. 89, 289–294. doi: 10.1016/j.ajhg.2011.06.007
Soardi, F. C., Machado-Silva, A., Linhares, N. D., Zheng, G., Qu, Q., Pena, H. B., et al. (2017). Familial STAG2 germline mutation defines a new human cohesinopathy. npj Genomic Med. 2:7. doi: 10.1038/s41525-017-0009-4
Sridhara, S. C., Carvalho, S., Grosso, A. R., Gallego-Paez, L. M., Carmo-Fonseca, M., and de Almeida, S. F. (2017). Transcription dynamics prevent RNA-mediated genomic instability through SRPK2-Dependent DDX23 phosphorylation. Cell Rep. 18, 334–343. doi: 10.1016/j.celrep.2016.12.050
Szklarczyk, D., Gable, A. L., Lyon, D., Junge, A., Wyder, S., Huerta-Cepas, J., et al. (2019). STRING v11: protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 47, D607–D613. doi: 10.1093/nar/gky1131
Tang, H., Guo, J., Linpeng, S., and Wu, L. (2019). Next generation sequencing identified two novel mutations in NIPBL and a frame shift mutation in CREBBP in three Chinese children. Orphanet J. Rare Dis. 14:45. doi: 10.1186/s13023-019-1022-8
Tsurusaki, Y., Okamoto, N., Ohashi, H., Kosho, T., Imai, Y., Hibi-Ko, Y., et al. (2012). Mutations affecting components of the SWI/SNF complex cause Coffin-Siris syndrome. Nat. Genet. 44, 376–378. doi: 10.1038/ng.2219
Tuc, E., Bengur, F. B., Aykut, A., Sahin, O., and Alanay, Y. (2020). The third family with TAF6-related phenotype: alazami-Yuan syndrome. Clin. Genet. 97, 795–796. doi: 10.1111/cge.13711
Tzschach, A., Grasshoff, U., Beck-Woedl, S., Dufke, C., Bauer, C., Kehrer, M., et al. (2015). Next-generation sequencing in X-linked intellectual disability. Eur. J. Hum. Genet. 23, 1513–1518. doi: 10.1038/ejhg.2015.5
Vo, N., and Goodman, R. H. (2001). CREB-binding Protein and p300 in transcriptional regulation. J. Biol. Chem. 276, 13505–13508. doi: 10.1074/jbc.R000025200
Wang, J., Zhao, Y., Zhou, X., Hiebert, S. W., Liu, Q., and Shyr, Y. (2018). Nascent RNA sequencing analysis provides insights into enhancer-mediated gene regulation. BMC Genomics 19:633. doi: 10.1186/s12864-018-5016-z
Wen, H., Li, Y., Xi, Y., Jiang, S., Stratton, S., Peng, D., et al. (2014). ZMYND11 links histone H3.3K36me3 to transcription elongation and tumour suppression. Nature 508, 263–268. doi: 10.1038/nature13045
Woods, S. A., Robinson, H. B., Kohler, L. J., Agamanolis, D., Sterbenz, G., and Khalifa, M. (2014). Exome sequencing identifies a novel EP300 frame shift mutation in a patient with features that overlap cornelia de lange syndrome. Am. J. Med. Genet. 164, 251–258. doi: 10.1002/ajmg.a.36237
Xiao, B., Qiu, W., Ji, X., Liu, X., Huang, Z., Liu, H., et al. (2018). Marked yield of re-evaluating phenotype and exome/target sequencing data in 33 individuals with intellectual disabilities. Am. J. Med. Genet. 176, 107–115. doi: 10.1002/ajmg.a.38542
Yavarna, T., Al-Dewik, N., Al-Mureikhi, M., Ali, R., Al-Mesaifri, F., Mahmoud, L., et al. (2015). High diagnostic yield of clinical exome sequencing in Middle Eastern patients with Mendelian disorders. Hum. Genet. 134, 967–980. doi: 10.1007/s00439-015-1575-0
Yuan, B., Neira, J., Pehlivan, D., Santiago-Sim, T., Song, X., Rosenfeld, J., et al. (2019). Clinical exome sequencing reveals locus heterogeneity and phenotypic variability of cohesinopathies. Genet. Med. 21, 663–675. doi: 10.1038/s41436-018-0085-6
Yuan, B., Pehlivan, D., Karaca, E., Patel, N., Charng, W.-L., Gambin, T., et al. (2015). Global transcriptional disturbances underlie Cornelia de Lange syndrome and related phenotypes. J. Clin. Invest. 125, 636–651. doi: 10.1172/JCI77435
Yuen, R. K. C., Thiruvahindrapuram, B., Merico, D., Walker, S., Tammimies, K., Hoang, N., et al. (2015). Whole-genome sequencing of quartet families with autism spectrum disorder. Nat. Med. 21, 185–191. doi: 10.1038/nm.3792
Zhang, A., Li, C.-W., and Chen, J. D. (2007). Characterization of transcriptional regulatory domains of ankyrin repeat cofactor-1. Biochem. Biophys. Res. Commun. 358, 1034–1040. doi: 10.1016/j.bbrc.2007.05.017
Zhang, A., Yeung, P. L., Li, C.-W., Tsai, S.-C., Dinh, G. K., Wu, X., et al. (2004). Identification of a novel family of ankyrin repeats containing cofactors for p160 nuclear receptor coactivators. J. Biol. Chem. 279, 33799–33805. doi: 10.1074/jbc.M403997200
Zhang, N., Song, Y., Xu, Y., Liu, J., Shen, Y., Zhou, L., et al. (2020). MED13L integrates Mediator-regulated epigenetic control into lung cancer radiosensitivity. Theranostics 10, 9378–9394. doi: 10.7150/thno.48247
Keywords: Cornelia de Lange syndrome (CdLS), chromatinopathies, transcriptional regulators, chromatin remodelers, cohesin
Citation: Parenti I and Kaiser FJ (2021) Cornelia de Lange Syndrome as Paradigm of Chromatinopathies. Front. Neurosci. 15:774950. doi: 10.3389/fnins.2021.774950
Received: 13 September 2021; Accepted: 18 October 2021;
Published: 05 November 2021.
Edited by:
Debbie L. C. van den Berg, Erasmus Medical Center, NetherlandsReviewed by:
Xiaoli Chen, Capital Institute of Pediatrics, ChinaLeda Torres, National Institute of Pediatrics, Mexico
Copyright © 2021 Parenti and Kaiser. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ilaria Parenti, aWxhcmlhLnBhcmVudGlAdWstZXNzZW4uZGU=