 Katsuya Uchida
Katsuya Uchida Mao Suzuki
Mao Suzuki- 1Laboratory of Information Biology, Graduate School of Information Sciences, Tohoku University, Sendai, Japan
- 2Laboratory of Biomodeling, Graduate School of Information Sciences, Tohoku University, Sendai, Japan
Thyroid hormones play an important role in brain development, and thyroid hormone insufficiency during the perinatal period results in severe developmental delays. Perinatal thyroid hormone deficiency is clinically known as congenital hypothyroidism, which is caused by dysgenesis of the thyroid gland or low iodine intake. If the disorder is not diagnosed or not treated early, the neuronal architecture is perturbed by thyroid hormone insufficiency, and neuropathological findings, such as abnormal synapse formation, defects in neuronal migration, and impairment of myelination, are observed in the brains of such patients. Furthermore, the expression of psychiatric disorder-related molecules, especially parvalbumin, is significantly decreased by thyroid hormone insufficiency during the perinatal period. Animal experiments using hypothyroidism models display decreased parvalbumin expression and abnormal brain architecture, and these experimental results show reproducibility and stability. These basic studies reinforce the results of epidemiological studies, suggesting the relevance of thyroid dysfunction in psychiatric disorders. In this review, we discuss the disruption of brain function associated with congenital hypothyroidism from the perspective of basic and clinical research.
Introduction
Thyroid hormones are synthesized in and released by the thyroid gland, with thyroxine (T4) comprising the highest concentration of these hormones. T4 is released from the thyroid gland and converted to triiodothyronine (T3) by deiodinase; T3 is highly biologically active as a transcription factor that plays important roles in brain development. This includes its roles in glial myelination, neuronal migration, cortical layer formation, synaptogenesis, and neurogenesis (Nicholson and Altman, 1972; Oppenheimer and Schwartz, 1997; Koibuchi and Chin, 2000; Uchida et al., 2005). Therefore, thyroid hormones during the perinatal period are important for normal development of the brain, and congenital hypothyroidism causes serious developmental delay if proper treatment is not implemented immediately after birth in such patients (Morreale de Escobar et al., 1987; Rastogi and LaFranchi, 2010). Thyroid dysfunction can be clinically detected by mass screening immediately after birth, and developmental disorders can be avoided by treatment with levothyroxine (T4).
Therapy for thyroid dysfunction has been established; however, neuroscientific research using rodent models is still ongoing because thyroid hormones are involved in diverse aspects of neurodevelopment, and thyroid hormone research in turn has the potential to provide new insights. A typical neuropathological finding caused by thyroid hormone insufficiency is a decrease in the parvalbumin of GABAergic neurons. This phenomenon is of interest to many researchers because it is observed not only in thyroid hormone insufficiency, but also in the dysfunction of thyroid hormone receptors and iodothyronine deiodinase (Berbel et al., 1996; Gilbert et al., 2007; Wallis et al., 2008; Barez-Lopez et al., 2019). In other words, the decrease in parvalbumin in GABAergic neurons is closely linked to the thyroid system. In addition, in recent years, a decrease in parvalbumin has been observed in the postmortem brains of patients with autism and schizophrenia, reaffirming the importance of thyroid hormone research in these conditions (Hashimoto et al., 2003; Lawrence et al., 2010; Soghomonian et al., 2017). Furthermore, parvalbumin neurons are lost in the cerebral cortex of mice lacking methyl CpG binding protein 2 (MeCP2), the gene responsible for Rett syndrome (RTT) (Fukuda et al., 2005). Recently, abnormalities in thyroid function in RTT patients have also been reported (Stagi et al., 2015). Herein, we present the neuropathological findings observed in experimental hypothyroidism (animal study) and congenital hypothyroidism, discuss the relationship between some psychiatric disorders and hypothyroidism, and review the importance of thyroid hormones in brain development.
Role of Thyroid Hormone on the Brain Architecture
The role of thyroid hormones in brain development has long been studied, and numerous studies have been published to date. Congenital hypothyroidism that is not diagnosed or not treated early causes developmental delay, which, as mentioned above, is triggered by abnormalities in neural architecture during brain development. Notably, maternal hypothyroidism during pregnancy has long-lasting effects on the cortical morphology of their offspring, with specific effects reflecting both the severity and timing of maternal thyroid hormone insufficiency (Lischinsky et al., 2016). Furthermore, Cooper et al. (2019) reported that individuals with severe congenital hypothyroidism are at risk of developing white matter microstructural abnormalities, despite early detection and treatment. In congenital hypothyroidism, neuropathological features such as abnormalities in neuritogenesis of Purkinje cells in the cerebellum, myelin sheath hypoplasia in myelinated nerves, and dysgenesis of dendrite spine formation are observed in the developmental and mature brain. However, the effects of thyroid hormone deficiency are not only observed in local cellular structures, but also in neural architecture and signal transmission between the cerebral hemisphere via the corpus callosum (Berbel et al., 1993; Samadi et al., 2015). Although cell positioning during corticogenesis follows an inside-out pattern, radial neurogenic gradients are more diffuse than in normal animals. This difference in radial migration may be attributed to reduced reelin mRNA and protein in Cajal–Retzius cells observed in hypothyroid animals during the perinatal period. Since the administration of T3 to hypothyroid rats restores reelin mRNA expression both in vitro and in vivo (Alvarez-Dolado et al., 1999), the formation of radial neurogenic gradients may be highly dependent on thyroid hormones. Regarding the commissural fibers, the commissural neurons that form cortical layer II/III in the cortex are connected to the contralateral hemisphere via the corpus callosum. In general, retrograde neural tracers administered to the primary auditory cortex are widely distributed in the cortical layers of the contralateral side; however, in hypothyroid animals, tracer signals converge in cortical layers IV–V of the contralateral side of the primary auditory cortex (Berbel et al., 1993). Furthermore, Goodman and Gilbert (2007) reported that thyroid hormone insufficiency induced cellular malformation in the corpus callosum. Abnormalities in neural connections between the cerebral hemispheres, hence, would have a very strong impact on integrated brain functions (Table 1). In fact, abnormalities associated with commissural fibers have been observed not only in hypothyroidism but also in autism spectrum disorder (ASD) and attention deficit hyperactivity disorder (Casanova et al., 2011; Qiu et al., 2011; Ameis et al., 2016), indicating that defects in brain structures involved in functional integration affect behavioral expression. Thus, thyroid hormones participate in various aspects of the developing brain.
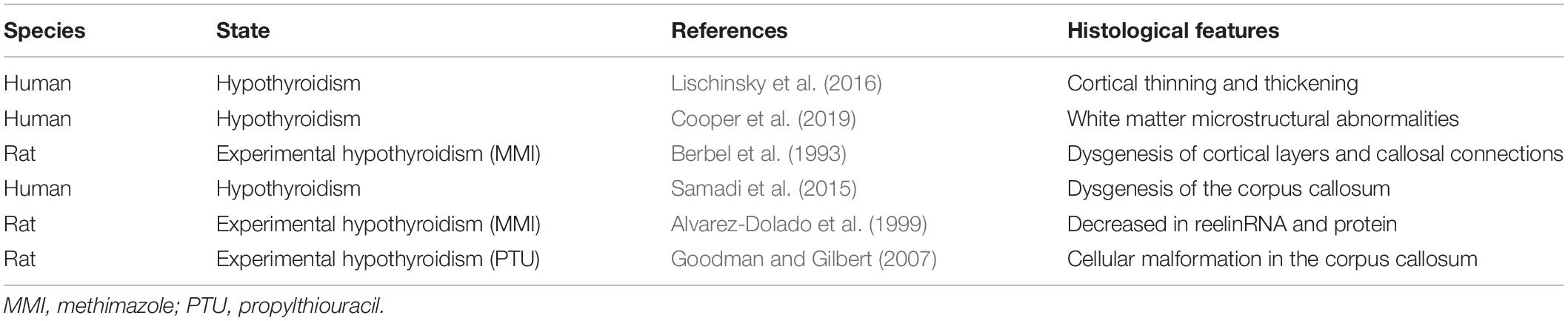
Table 1. Role of thyroid hormone on the brain architecture.
Involvement of Thyroid Hormone on Parvalbumin Expression
Parvalbumin is a calcium-binding protein and a type of albumin with a small molecular weight (12 kD) (Arif, 2009). Parvalbumin is expressed in GABAergic neurons in the central nervous system, and parvalbumin-expressing neurons are mainly observed in the cortex, hippocampus, cerebellum, and reticular hypothalamic nucleus (Celio, 1986, 1990; Kosaka et al., 1987). Parvalbumin-expressing GABAergic neurons are generated from the medial ganglionic eminence and migrate tangentially to their respective destination areas (Danglot et al., 2006). In the cortex and hippocampus, the developmental expression of parvalbumin mRNA is observed from approximately postnatal day 10, and gradually increases daily (de Lecea et al., 1995). Parvalbumin neurons differentiate into basket and chandelier cells and function as fast-spiking interneurons (Hu et al., 2014).
Berbel et al. (1996) first reported the microstructural differences in parvalbumin neurons in adult hypothyroid rats. This report indicated that hypothyroid rats showed dysgenesis of parvalbumin-positive terminal puncta in the neocortex, while there were no differences observed in the number of parvalbumin neurons. Guadano-Ferraz et al. (2003) also reported that thyroid hormone receptor alpha 1-deficient mice displayed a decrease in the density of parvalbumin-positive terminals in the hippocampus. These two reports mainly showed the effect of hypothyroidism on the nerve endings of parvalbumin neurons. In contrast, a decrease in the number of parvalbumin neurons in the cortex and other regions has been shown in monocarboxylate transporter 8 (MCT8) and deiodinase type 2 (Dio2)-double deficient mice (Barez-Lopez et al., 2019). MCT8 actively transports a variety of iodothyronines, including thyroid hormones (T3 and T4) (Friesema et al., 2003), and Dio2 activates thyroid hormones by converting the prohormone T4 to bioactive T3 (Croteau et al., 1996). In mice, single mutations of MCT8 or Dio2 do not cause a significant decrease in the number of parvalbumin neurons due to compensatory effects. Therefore, double deficiency of MCT8 and Dio2 as well as severe thyroid hormone dysfunction may lead to a decrease in the number of parvalbumin neurons in mice. Gilbert et al. (2007) reported in detail the effects of thyroid hormone insufficiency on parvalbumin expression. Interestingly, animal models of congenital hypothyroidism display a significant decrease in the number of parvalbumin neurons during the juvenile phase, and the levels of parvalbumin expression slightly catch up with those of normal animals, with recovery of serum thyroid hormone levels after the termination of treatment with antithyroid agents. However, the degree of the decrease in parvalbumin neuron number and its subsequent recovery is dependent on the concentration of antithyroid agents, and significant recovery of the number of parvalbumin-expressing neurons was observed in the low concentration exposure group, but not in the high concentration exposure group. Thus, even though the levels of serum thyroid hormone completely recover in adulthood, animals that experience thyroid hormone deficiency during the perinatal period still retain the signatures of temporary hormonal defects in parvalbumin neurons in the adult brain. As a result of this occurrence, dysfunction of neuron-specific K(+)/Cl(−) co-transporter (KCC2) and a delayed onset of synaptic inhibition have been observed accordingly (Friauf et al., 2008; Yi et al., 2014). In general, during the first 2 weeks after birth, synaptic transmission via the inhibitory transmitter changes from excitatory depolarizing to inhibitory hyperpolarizing effects in GABAergic neurons (Cherubini et al., 1991; Rivera et al., 1999). Therefore, delay in the functional conversion of inhibitory neurons may significantly perturb the integrative function of inhibitory neural circuits in the hypothyroid brain.
On the other hand, just as treatment with levothyroxine avoids developmental delay accompanying congenital hypothyroidism in humans, thyroid hormone replacement immediately after birth can prevent a decrease in the number of parvalbumin neurons in rodents (Gilbert et al., 2007; Uchida et al., 2014, 2021). Thyroid hormone replacement after postnatal day 14 has no effect on the number of parvalbumin neurons, suggesting that a critical period of thyroid hormone sensitivity exists before this day (Gilbert et al., 2007; Uchida et al., 2014, 2021). Interestingly, a transient increase in blood thyroid hormone levels was observed around postnatal day 14 (Fishman et al., 1982; Calikoglu et al., 1996; Hadj-Sahraoui et al., 2000). Hence, the hormonal surge and/or the abundance of hormones in the early postnatal period might be important for normal neurodevelopment, including maturation of parvalbumin neurons. As mentioned above, parvalbumin expression and its morphogenesis have been observed to correlate with thyroid hormone levels; however, the direct or indirect action of TH on the transcription of PV genes remains unclear.
Similar to observations in patients with MCT8 mutations, loss of parvalbumin expression has been observed in the brains of patients with schizophrenia and autism (Hashimoto et al., 2003; Lopez-Espindola et al., 2014; Filice et al., 2020). Since these psychiatric disorders are observed in perturbation of executive functions, parvalbumin expression in the human prefrontal cortex has been preferentially analyzed accordingly. Although there are differences in the results among studies, a decrease in parvalbumin expression in the prefrontal cortex has been confirmed in schizophrenia and autism (Beasley and Reynolds, 1997; Hashemi et al., 2017; Ariza et al., 2018; Kaar et al., 2019) (Table 2). Therefore, a parvalbumin hypothesis for developmental delay has been proposed (Filice et al., 2020).
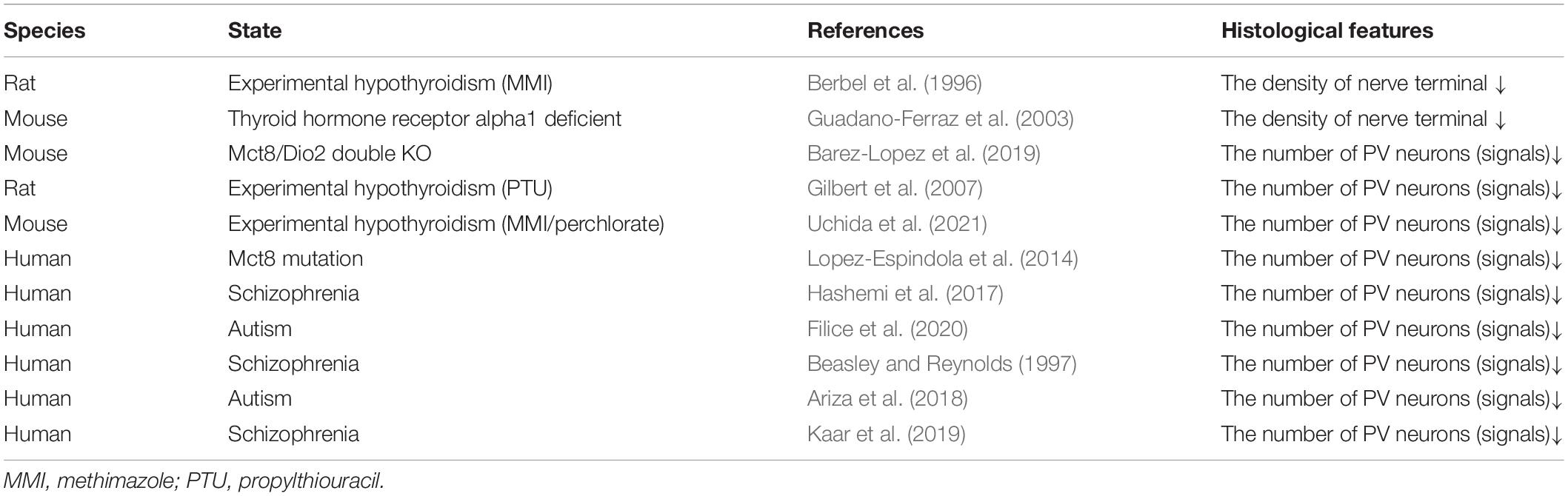
Table 2. Morphological abnormalities of parvalbumin neurons in the brain.
Relationship Between Thyroid Disease and Psychiatric Disorders (Schizophrenia and Autism)
An increased prevalence of thyroid disorders has been noted in families of individuals with schizophrenia (DeLisi et al., 2000; Palha and Goodman, 2006; Radhakrishnan et al., 2013; Gyllenberg et al., 2016; Sharif et al., 2018). Radhakrishnan et al. (2013) reported that in a retrospective hospital-based study, hypothyroidism was observed in 25% of patients with schizophrenia. Sharif et al. (2018) also reported that the population of patients with schizophrenia in hypothyroid patients was higher than that in controls. Generally, the prevalence of schizophrenia is approximately 1%, but there is a twofold increase in the incidence rate of schizophrenia in patients with hypothyroidism. These large-scale studies show a higher interaction between hypothyroidism and schizophrenia, although the functional relevance of these disorders remains unclear. Since antipsychotics affect thyroid hormone secretion and conversion of T4 toT3 (Terao et al., 1995; Langlois et al., 2001), it should also be considered whether medication affects thyroid status, so as to estimate the relationship between thyroid state and schizophrenia more accurately. According to Melamed et al. (2020), the increased rate of hypothyroidism in patients with schizophrenia after, but not before, the diagnosis of schizophrenia suggests that antipsychotic medications may affect thyroid hormone levels. However, the principal preoccupation is whether perinatal thyroid hormone deficiency is associated with the onset of schizophrenia. According to Gyllenberg et al. (2016), maternal hypothyroxinemia may be associated with an increased risk for the onset of schizophrenia, suggesting an association between low maternal thyroxine and increased odds of offspring schizophrenia. Therefore, congenital hypothyroidism (maternal hypothyroidism) is a potential risk factor for the onset of schizophrenia.
It has been reported that maternal hypothyroidism is associated with an increased risk of ASD (Roman et al., 2013; Chang and Shin, 2014; Getahun et al., 2018; Ge et al., 2020) and that hypothyroid animal models are useful for understanding the molecular mechanisms of ASD (Sadamatsu et al., 2006; Berbel et al., 2014). In a population-based study, there were no strong associations between neonatal thyroid hormones and ASD, but subgroups of newborns with the lowest T4 levels exhibited modestly increased ASD risk (Lyall et al., 2017). Although no significant differences were reported in the levels of serum T4, T3, and thyroid stimulating hormone (TSH) in patients with ASD compared to reference samples (Cohen et al., 1980), this study analyzed thyroid function in children aged 10–12 years with ASD in comparison to normal children. Since mild maternal thyroid hormone insufficiency during the perinatal period affects brain formation in fetuses and neonatal infants, measurement of postnatal hormone levels may provide a clue to its relevance to ASD. In fact, Lischinsky et al. (2016) reported that even mild variations in maternal thyroid hormones permanently affect the offspring cortex. Furthermore, although early treatment of congenital hypothyroidism prevents developmental delay, affected children still exhibit subtle persistent neurocognitive deficits, such as poor attention (Rovet and Hepworth, 2001). In an overall evaluation of offspring, there may be a rigid functional correlation between maternal hypothyroidism during the perinatal period and ASD in offspring (Table 3). In subsequent studies, single nucleotide polymorphisms in the ligand-binding domain of thyroid hormone receptors were found in patients with ASD (Kalikiri et al., 2017). Furthermore, alterations in thyroid hormone-dependent genes have been observed in the postmortem brains of humans with ASD (Khan et al., 2014). Hence, the risk of developing autism may be associated not only with an underactive the thyroid gland, but also with a defect in the process of hormone functioning or in the rate of transcribed products. More clinical studies and basic research using animal models are needed to better understand the detailed functional relationship between thyroid diseases and ASD.
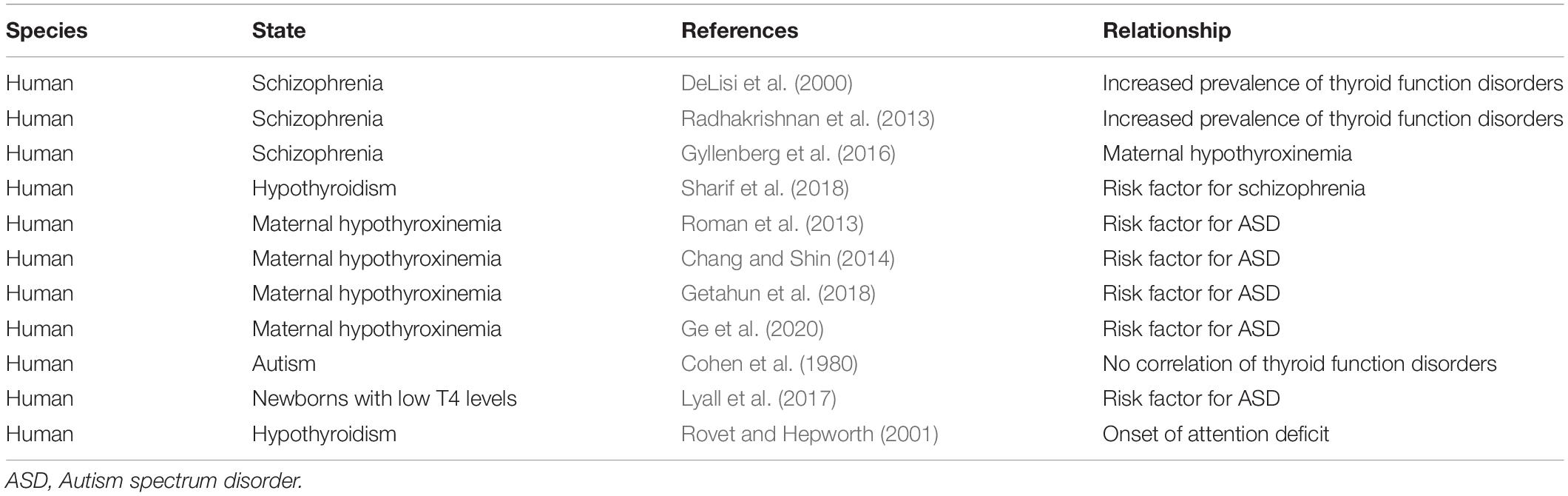
Table 3. Relationship between thyroid diseases and psychiatric disorders.
Thyroid Function Disorders and Rett Syndrome
The relationship between thyroid function disorders and RTT is unclear. RTT is a rare genetic disorder caused by mutations or deletions in a gene called MeCP2 on the X chromosome, resulting in severe mental and physical disabilities (Braddock et al., 1993; Gold et al., 2018). RTT is also rarely caused by abnormalities in the CDKL5 and FOXG1 genes (Guerrini and Parrini, 2012). Human autopsy brain tissue from with patients with RTT displays decreased in dendritic spines and neurotrophic factors (Belichenko and Dahlstrom, 1995; Lipani et al., 2000), and such histological impairment may be a leading cause of developmental delay. Partially common neuropathological findings are observed in both RTT and congenital hypothyroidism (Eayrs, 1960; Armstrong, 1997); this is of interest as a research subject for basic medical researchers. Currently, there are less than 10 publications on the relationship between RTT and thyroid state, and several papers have reported very interesting results. Cooke et al. (1995) were the first to report that patients with RTT have thyroid dysfunction, which displays a significant decrease in serum total T4 concentration compared to the reference range. Stagi et al. (2015) also reported abnormal thyroid function in RTT, showing that serum T4 levels are elevated in patients with RTT. These two studies have shown contradictory results regarding the serum T4 levels. We cannot describe whether the discrepancies depend on the blood sample or the technique, and these phenomena are of interest for the study of thyroid function on RTT. Future case reports are needed to better interpret the alterations in serum T4 levels with RTT. In contrast, the dysgenesis of neurite length in MeCP2-deficient cells was significantly restored by the administration of IGF-1, whereas IGF-1 concentration in culture media was enhanced by the administration of T3 (de Souza et al., 2017). Therefore, the neuropathological findings observed in RRTs may be due to a decrease in IGF-1 levels, and thyroid hormones may have an indirect effect. Furthermore, MeCP2-knockout cells show that thyroid hormone-related genes, such as hormone transporters and deiodinases, are altered in these cells as compared to normal cells (de Souza et al., 2019). Therefore, MeCP2 probably has a significant influence on the assembly of the thyroid system in the body. In contrast, experimental hypothyroidism leads to alterations in MeCP2 expression in the cortex and liver of rodents. Uchida et al. (2021) reported that hypothyroid pups indicated a decrease in MeCP2 staining signals in cortical layers II–IV; however, the expression of MeCP2 mRNA was not altered. In contrast, Bunker et al. (2017) showed that neonatal exposure to antithyroid agents led to a decrease in MeCP2 mRNA expression in the liver; however, translated products did not change. Although there is not a clear explanation regarding the behavior of MeCP2 transcripts and translated products differs between organs, these results suggest that thyroid hormones have a significant effect on the expression of MeCP2, at least, and the relationship between thyroid function and MeCP2 might be that of reciprocal interactions rather than one-way interactions (Table 4). Further studies are needed to clarify the molecular mechanisms by which thyroid hormones affect MeCP2 expression.

Table 4. Thyroid function disorders and RTT.
Fukuda et al. (2005) reported delayed corticogenesis in MeCP2-deficient mice. Interestingly, the cortex of MeCP2-deficient mice has no parvalbumin neurons at postnatal day 14, and its expression catches up at 6 weeks after birth (Fukuda et al., 2005). Parvalbumin-expressing neurons contribute to aspects of the RTT phenotype; genetically modified mice specifically defecting MeCP2 on parvalbumin neurons indicate distinct RTT-like phenotypes (Ito-Ishida et al., 2015). Hence, the functional defect of parvalbumin might have a profound causal relationship with the development of psychiatric disorders. Of course, this behavior of parvalbumin closely resembles that of histological alterations in thyroid hormone deficiency during the perinatal period. In addition, the article also reports immature cortical formation in the somatosensory cortex of MeCP2-deficient mice, a phenomenon also observed in hypothyroid mice. The histological abnormalities observed in MeCP2-deficient and hypothyroid mice share many similarities, suggesting that there are downstream overlapping molecular mechanisms. Further exploration of the common denominator with hypothyroidism may reveal the molecular mechanism of developmental delay in RTT.
Conclusion
In this review, we discussed the effects of thyroid hormones on the neural architecture of the brain, and then concisely mentioned their relationship with parvalbumin and/or MeCP2. The role of thyroid hormones in brain development has been studied since the early 20th century, and many studies have been published accordingly. Most of them are based on histomorphological evaluation by Nissl and Golgi staining, and the products of these histological studies have had a significant impact on the directionality of the current research. Furthermore, in recent years, researchers have been able to develop genetically modified mice with knockdown of target genes, and the importance of the thyroid system in brain development is becoming clearer. Nevertheless, just as the elementary processes of memory have been discovered, the mechanism of memory has not yet been elucidated, and the mechanism of developmental delay caused by congenital hypothyroidism has not been clarified. However, through animal experiments and clinical studies, a decrease in parvalbumin neurons has been reproducibly observed in hypothyroidism, and this phenomenon has also been observed in the postmortem brains of patients with schizophrenia and autism. Such comparative analysis with other psychiatric disorders may provide clues to the pathogenesis of developmental delay. In addition, since abnormal thyroid function is observed in patients with RTT, the onset of diseases associated with developmental disorders may have a common molecular mechanism. On the other hand, even though mild perinatal thyroid hormone deficiencies or even early treatment with levothyroxine, children who experience thyroid hormone deficiency during the perinatal period still retain the signatures of temporary hormonal defects in the adult brain. This case strongly indicates that thyroid hormones are essential for brain development. Hence, the study of congenital hypothyroidism and brain development remains a fascinating research topic.
Author Contributions
KU wrote the manuscript. KU and MS collected the relevant research manuscript for the review. Both authors contributed to the article and approved the submitted version.
Funding
This work was supported by the JSPS KAKENHI Grant Numbers JP19K08969 and JP16H06276 (AdAMS).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We would like to thank N. Katayama (Department of Humanities and Social Studies, Shokei Gakuin University) for advice on the manuscript. We would also like to thank Editage (http://www.editage.com) for English language editing.
References
Alvarez-Dolado, M., Ruiz, M., Del Rio, J. A., Alcantara, S., Burgaya, F., Sheldon, M., et al. (1999). Thyroid hormone regulates reelin and dab1 expression during brain development. J. Neurosci. 19, 6979–6993. doi: 10.1523/JNEUROSCI.19-16-06979.1999
Ameis, S. H., Lerch, J. P., Taylor, M. J., Lee, W., Viviano, J. D., Pipitone, J., et al. (2016). A Diffusion Tensor Imaging Study in Children With ADHD, Autism Spectrum Disorder, OCD, and Matched Controls: distinct and Non-Distinct White Matter Disruption and Dimensional Brain-Behavior Relationships. Am. J. Psychiatry 173, 1213–1222. doi: 10.1176/appi.ajp.2016.15111435
Arif, S. H. (2009). A Ca(2+)-binding protein with numerous roles and uses: parvalbumin in molecular biology and physiology. Bioessays 31, 410–421. doi: 10.1002/bies.200800170
Ariza, J., Rogers, H., Hashemi, E., Noctor, S. C., and Martinez-Cerdeno, V. (2018). The Number of Chandelier and Basket Cells Are Differentially Decreased in Prefrontal Cortex in Autism. Cereb. Cortex 28, 411–420. doi: 10.1093/cercor/bhw349
Barez-Lopez, S., Grijota-Martinez, C., Auso, E., Fernandez-De Frutos, M., Montero-Pedrazuela, A., and Guadano-Ferraz, A. (2019). Adult Mice Lacking Mct8 and Dio2 Proteins Present Alterations in Peripheral Thyroid Hormone Levels and Severe Brain and Motor Skill Impairments. Thyroid 29, 1669–1682. doi: 10.1089/thy.2019.0068
Beasley, C. L., and Reynolds, G. P. (1997). Parvalbumin-immunoreactive neurons are reduced in the prefrontal cortex of schizophrenics. Schizophr. Res. 24, 349–355. doi: 10.1016/s0920-9964(96)00122-3
Belichenko, P. V., and Dahlstrom, A. (1995). Confocal laser scanning microscopy and 3-D reconstructions of neuronal structures in human brain cortex. Neuroimage 2, 201–207. doi: 10.1006/nimg.1995.1026
Berbel, P., Guadano-Ferraz, A., Martinez, M., Quiles, J. A., Balboa, R., and Innocenti, G. M. (1993). Organization of auditory callosal connections in hypothyroid adult rats. Eur. J. Neurosci. 5, 1465–1478. doi: 10.1111/j.1460-9568.1993.tb00214.x
Berbel, P., Marco, P., Cerezo, J. R., and Defelipe, J. (1996). Distribution of parvalbumin immunoreactivity in the neocortex of hypothyroid adult rats. Neurosci. Lett. 204, 65–68. doi: 10.1016/0304-3940(96)12318-1
Berbel, P., Navarro, D., and Roman, G. C. (2014). An evo-devo approach to thyroid hormones in cerebral and cerebellar cortical development: etiological implications for autism. Front. Endocrinol. 5:146. doi: 10.3389/fendo.2014.00146
Braddock, S. R., Braddock, B. A., and Graham, J. M. Jr. (1993). Rett syndrome. An update and review for the primary pediatrician. Clin. Pediatr. 32, 613–626. doi: 10.1177/000992289303201011
Bunker, S. K., Dandapat, J., Chainy, G. B. N., Sahoo, S. K., and Nayak, P. K. (2017). Neonatal Exposure to 6-n-Propyl-Thiouracil, an Anti-Thyroid Drug, Alters Expression of Hepatic DNA Methyltransferases, Methyl CpG-Binding Proteins, Gadd45a, p53, and PCNA in Adult Male Rats. Eur. Thyroid J. 6, 281–291. doi: 10.1159/000479681
Calikoglu, A. S., Gutierrez-Ospina, G., and D’ercole, A. J. (1996). Congenital hypothyroidism delays the formation and retards the growth of the mouse primary somatic sensory cortex (S1). Neurosci. Lett. 213, 132–136. doi: 10.1016/0304-3940(96)12836-6
Casanova, M. F., El-Baz, A., Elnakib, A., Switala, A. E., Williams, E. L., Williams, D. L., et al. (2011). Quantitative analysis of the shape of the corpus callosum in patients with autism and comparison individuals. Autism 15, 223–238. doi: 10.1177/1362361310386506
Celio, M. R. (1986). Parvalbumin in most gamma-aminobutyric acid-containing neurons of the rat cerebral cortex. Science 231, 995–997. doi: 10.1126/science.3945815
Celio, M. R. (1990). Calbindin D-28k and parvalbumin in the rat nervous system. Neuroscience 35, 375–475. doi: 10.1016/0306-4522(90)90091-h
Chang, K., and Shin, J. I. (2014). Association of gestational maternal hypothyroxinemia and increased autism risk: the role of brain-derived neurotrophic factor. Ann. Neurol. 75:971. doi: 10.1002/ana.24143
Cherubini, E., Gaiarsa, J. L., and Ben-Ari, Y. (1991). GABA: an excitatory transmitter in early postnatal life. Trends Neurosci. 14, 515–519. doi: 10.1016/0166-2236(91)90003-d
Cohen, D. J., Young, J. G., Lowe, T. L., and Harcherik, D. (1980). Thyroid hormone in autistic children. J. Autism Dev. Disord. 10, 445–450. doi: 10.1007/bf02414820
Cooke, D. W., Naidu, S., Plotnick, L., and Berkovitz, G. D. (1995). Abnormalities of thyroid function and glucose control in subjects with Rett syndrome. Horm. Res. 43, 273–278. doi: 10.1159/000184309
Cooper, H. E., Kaden, E., Halliday, L. F., Bamiou, D. E., Mankad, K., Peters, C., et al. (2019). White matter microstructural abnormalities in children with severe congenital hypothyroidism. Neuroimage Clin. 24:101980. doi: 10.1016/j.nicl.2019.101980
Croteau, W., Davey, J. C., Galton, V. A., and St Germain, D. L. (1996). Cloning of the mammalian type II iodothyronine deiodinase. A selenoprotein differentially expressed and regulated in human and rat brain and other tissues. J. Clin. Invest. 98, 405–417. doi: 10.1172/JCI118806
Danglot, L., Triller, A., and Marty, S. (2006). The development of hippocampal interneurons in rodents. Hippocampus 16, 1032–1060. doi: 10.1002/hipo.20225
de Lecea, L., del Rio, J. A., and Soriano, E. (1995). Developmental expression of parvalbumin mRNA in the cerebral cortex and hippocampus of the rat. Brain Res. Mol. Brain Res. 32, 1–13. doi: 10.1016/0169-328x(95)00056-x
de Souza, J. S., Carromeu, C., Torres, L. B., Araujo, B. H., Cugola, F. R., Maciel, R. M., et al. (2017). IGF1 neuronal response in the absence of MECP2 is dependent on TRalpha 3. Hum. Mol. Genet. 26, 270–281. doi: 10.1093/hmg/ddw384
de Souza, J. S., Ferreira, D. R., Herai, R., Carromeu, C., Torres, L. B., Araujo, B. H. S., et al. (2019). Altered Gene Expression of Thyroid Hormone Transporters and Deiodinases in iPS MeCP2-Knockout Cells-Derived Neurons. Mol. Neurobiol. 56, 8277–8295. doi: 10.1007/s12035-019-01645-2
DeLisi, L. E., Smith, A. B., Razi, K., Stewart, J., Wang, Z., Sandhu, H. K., et al. (2000). Investigation of a candidate gene for schizophrenia on Xq13 previously associated with mental retardation and hypothyroidism. Am. J. Med. Genet. 96, 398–403.
Eayrs, J. T. (1960). Influence of the thyroid on the central nervous system. Br. Med. Bull. 16, 122–127. doi: 10.1093/oxfordjournals.bmb.a069810
Filice, F., Janickova, L., Henzi, T., Bilella, A., and Schwaller, B. (2020). The Parvalbumin Hypothesis of Autism Spectrum Disorder. Front. Cell. Neurosci. 14:577525. doi: 10.3389/fncel.2020.577525
Fishman, R. H., Gaathon, A., and Yanai, J. (1982). Early barbiturate treatment eliminates peak serum thyroxine levels in neonatal mice and produces ultrastructural damage in the brains of adults. Brain Res. 281, 202–205. doi: 10.1016/0165-3806(82)90158-4
Friauf, E., Wenz, M., Oberhofer, M., Nothwang, H. G., Balakrishnan, V., Knipper, M., et al. (2008). Hypothyroidism impairs chloride homeostasis and onset of inhibitory neurotransmission in developing auditory brainstem and hippocampal neurons. Eur. J. Neurosci. 28, 2371–2380. doi: 10.1111/j.1460-9568.2008.06528.x
Friesema, E. C., Ganguly, S., Abdalla, A., Manning Fox, J. E., Halestrap, A. P., and Visser, T. J. (2003). Identification of monocarboxylate transporter 8 as a specific thyroid hormone transporter. J. Biol. Chem. 278, 40128–40135. doi: 10.1074/jbc.m300909200
Fukuda, T., Itoh, M., Ichikawa, T., Washiyama, K., and Goto, Y. (2005). Delayed maturation of neuronal architecture and synaptogenesis in cerebral cortex of Mecp2-deficient mice. J. Neuropathol. Exp. Neurol. 64, 537–544. doi: 10.1093/jnen/64.6.537
Ge, G. M., Leung, M. T. Y., Man, K. K. C., Leung, W. C., Ip, P., Li, G. H. Y., et al. (2020). Maternal Thyroid Dysfunction During Pregnancy and the Risk of Adverse Outcomes in the Offspring: a Systematic Review and Meta-Analysis. J. Clin. Endocrinol. Metab. 105:dgaa555. doi: 10.1210/clinem/dgaa555
Getahun, D., Jacobsen, S. J., Fassett, M. J., Wing, D. A., Xiang, A. H., Chiu, V. Y., et al. (2018). Association between maternal hypothyroidism and autism spectrum disorders in children. Pediatr. Res. 83, 580–588.
Gilbert, M. E., Sui, L., Walker, M. J., Anderson, W., Thomas, S., Smoller, S. N., et al. (2007). Thyroid hormone insufficiency during brain development reduces parvalbumin immunoreactivity and inhibitory function in the hippocampus. Endocrinology 148, 92–102. doi: 10.1210/en.2006-0164
Gold, W. A., Krishnarajy, R., Ellaway, C., and Christodoulou, J. (2018). Rett Syndrome: a Genetic Update and Clinical Review Focusing on Comorbidities. ACS Chem. Neurosci. 9, 167–176. doi: 10.1021/acschemneuro.7b00346
Goodman, J. H., and Gilbert, M. E. (2007). Modest thyroid hormone insufficiency during development induces a cellular malformation in the corpus callosum: a model of cortical dysplasia. Endocrinology 148, 2593–2597. doi: 10.1210/en.2006-1276
Guadano-Ferraz, A., Benavides-Piccione, R., Venero, C., Lancha, C., Vennstrom, B., Sandi, C., et al. (2003). Lack of thyroid hormone receptor alpha1 is associated with selective alterations in behavior and hippocampal circuits. Mol. Psychiatry 8, 30–38. doi: 10.1038/sj.mp.4001196
Guerrini, R., and Parrini, E. (2012). Epilepsy in Rett syndrome, and CDKL5- and FOXG1-gene-related encephalopathies. Epilepsia 53, 2067–2078. doi: 10.1111/j.1528-1167.2012.03656.x
Gyllenberg, D., Sourander, A., Surcel, H. M., Hinkka-Yli-Salomaki, S., Mckeague, I. W., and Brown, A. S. (2016). Hypothyroxinemia During Gestation and Offspring Schizophrenia in a National Birth Cohort. Biol. Psychiatry 79, 962–970. doi: 10.1016/j.biopsych.2015.06.014
Hadj-Sahraoui, N., Seugnet, I., Ghorbel, M. T., and Demeneix, B. (2000). Hypothyroidism prolongs mitotic activity in the post-natal mouse brain. Neurosci. Lett. 280, 79–82. doi: 10.1016/s0304-3940(00)00768-0
Hashemi, E., Ariza, J., Rogers, H., Noctor, S. C., and Martinez-Cerdeno, V. (2017). The Number of Parvalbumin-Expressing Interneurons Is Decreased in the Prefrontal Cortex in Autism. Cereb. Cortex 27, 1931–1943.
Hashimoto, T., Volk, D. W., Eggan, S. M., Mirnics, K., Pierri, J. N., Sun, Z., et al. (2003). Gene expression deficits in a subclass of GABA neurons in the prefrontal cortex of subjects with schizophrenia. J. Neurosci. 23, 6315–6326. doi: 10.1523/JNEUROSCI.23-15-06315.2003
Hu, H., Gan, J., and Jonas, P. (2014). Interneurons. Fast-spiking, parvalbumin(+) GABAergic interneurons: from cellular design to microcircuit function. Science 345:1255263.
Ito-Ishida, A., Ure, K., Chen, H., Swann, J. W., and Zoghbi, H. Y. (2015). Loss of MeCP2 in Parvalbumin-and Somatostatin-Expressing Neurons in Mice Leads to Distinct Rett Syndrome-like Phenotypes. Neuron 88, 651–658. doi: 10.1016/j.neuron.2015.10.029
Kaar, S. J., Angelescu, I., Marques, T. R., and Howes, O. D. (2019). Pre-frontal parvalbumin interneurons in schizophrenia: a meta-analysis of post-mortem studies. J. Neural Transm. 126, 1637–1651. doi: 10.1007/s00702-019-02080-2
Kalikiri, M. K., Mamidala, M. P., Rao, A. N., and Rajesh, V. (2017). Analysis and functional characterization of sequence variations in ligand binding domain of thyroid hormone receptors in autism spectrum disorder (ASD) patients. Autism Res. 10, 1919–1928. doi: 10.1002/aur.1838
Khan, A., Harney, J. W., Zavacki, A. M., and Sajdel-Sulkowska, E. M. (2014). Disrupted brain thyroid hormone homeostasis and altered thyroid hormone-dependent brain gene expression in autism spectrum disorders. J. Physiol. Pharmacol. 65, 257–272.
Koibuchi, N., and Chin, W. W. (2000). Thyroid hormone action and brain development. Trends Endocrinol. Metab. 11, 123–128. doi: 10.1016/s1043-2760(00)00238-1
Kosaka, T., Katsumaru, H., Hama, K., Wu, J. Y., and Heizmann, C. W. (1987). GABAergic neurons containing the Ca2+-binding protein parvalbumin in the rat hippocampus and dentate gyrus. Brain Res. 419, 119–130. doi: 10.1016/0006-8993(87)90575-0
Langlois, M. C., Beaudry, G., Zekki, H., Rouillard, C., and Levesque, D. (2001). Impact of antipsychotic drug administration on the expression of nuclear receptors in the neocortex and striatum of the rat brain. Neuroscience 106, 117–128. doi: 10.1016/s0306-4522(01)00248-2
Lawrence, Y. A., Kemper, T. L., Bauman, M. L., and Blatt, G. J. (2010). Parvalbumin-, calbindin-, and calretinin-immunoreactive hippocampal interneuron density in autism. Acta Neurol. Scand. 121, 99–108. doi: 10.1111/j.1600-0404.2009.01234.x
Lipani, J. D., Bhattacharjee, M. B., Corey, D. M., and Lee, D. A. (2000). Reduced nerve growth factor in Rett syndrome postmortem brain tissue. J. Neuropathol. Exp. Neurol. 59, 889–895. doi: 10.1093/jnen/59.10.889
Lischinsky, J. E., Skocic, J., Clairman, H., and Rovet, J. (2016). Preliminary Findings Show Maternal Hypothyroidism May Contribute to Abnormal Cortical Morphology in Offspring. Front. Endocrinol. 7:16. doi: 10.3389/fendo.2016.00016
Lopez-Espindola, D., Morales-Bastos, C., Grijota-Martinez, C., Liao, X. H., Lev, D., Sugo, E., et al. (2014). Mutations of the thyroid hormone transporter MCT8 cause prenatal brain damage and persistent hypomyelination. J. Clin. Endocrinol. Metab. 99, E2799–E2804. doi: 10.1210/jc.2014-2162
Lyall, K., Anderson, M., Kharrazi, M., and Windham, G. C. (2017). Neonatal thyroid hormone levels in association with autism spectrum disorder. Autism Res. 10, 585–592.
Melamed, S. B., Farfel, A., Gur, S., Krivoy, A., Weizman, S., Matalon, A., et al. (2020). Thyroid function assessment before and after diagnosis of schizophrenia: a community-based study. Psychiatry Res. 293:113356. doi: 10.1016/j.psychres.2020.113356
Morreale de Escobar, G., Obregon, M. J., and Escobar del Rey, F. (1987). Fetal and maternal thyroid hormones. Horm. Res. 26, 12–27.
Nicholson, J. L., and Altman, J. (1972). Synaptogenesis in the rat cerebellum: effects of early hypo- and hyperthyroidism. Science 176, 530–532. doi: 10.1126/science.176.4034.530
Oppenheimer, J. H., and Schwartz, H. L. (1997). Molecular basis of thyroid hormone-dependent brain development. Endocr. Rev. 18, 462–475. doi: 10.1210/edrv.18.4.0309
Palha, J. A., and Goodman, A. B. (2006). Thyroid hormones and retinoids: a possible link between genes and environment in schizophrenia. Brain Res. Rev. 51, 61–71. doi: 10.1016/j.brainresrev.2005.10.001
Qiu, M. G., Ye, Z., Li, Q. Y., Liu, G. J., Xie, B., and Wang, J. (2011). Changes of brain structure and function in ADHD children. Brain Topogr. 24, 243–252.
Radhakrishnan, R., Calvin, S., Singh, J. K., Thomas, B., and Srinivasan, K. (2013). Thyroid dysfunction in major psychiatric disorders in a hospital based sample. Indian J. Med. Res. 138, 888–893.
Rivera, C., Voipio, J., Payne, J. A., Ruusuvuori, E., Lahtinen, H., Lamsa, K., et al. (1999). The K+/Cl- co-transporter KCC2 renders GABA hyperpolarizing during neuronal maturation. Nature 397, 251–255. doi: 10.1038/16697
Roman, G. C., Ghassabian, A., Bongers-Schokking, J. J., Jaddoe, V. W., Hofman, A., De Rijke, Y. B., et al. (2013). Association of gestational maternal hypothyroxinemia and increased autism risk. Ann. Neurol. 74, 733–742. doi: 10.1002/ana.23976
Rovet, J. F., and Hepworth, S. (2001). Attention problems in adolescents with congenital hypothyroidism: a multicomponential analysis. J. Int. Neuropsychol. Soc. 7, 734–744. doi: 10.1017/s135561770176609x
Sadamatsu, M., Kanai, H., Xu, X., Liu, Y., and Kato, N. (2006). Review of animal models for autism: implication of thyroid hormone. Congenit. Anom. 46, 1–9. doi: 10.1111/j.1741-4520.2006.00094.x
Samadi, A., Skocic, J., and Rovet, J. F. (2015). Children born to women treated for hypothyroidism during pregnancy show abnormal corpus callosum development. Thyroid 25, 494–502. doi: 10.1089/thy.2014.0548
Sharif, K., Tiosano, S., Watad, A., Comaneshter, D., Cohen, A. D., Shoenfeld, Y., et al. (2018). The link between schizophrenia and hypothyroidism: a population-based study. Immunol. Res. 66, 663–667. doi: 10.1007/s12026-018-9030-7
Soghomonian, J. J., Zhang, K., Reprakash, S., and Blatt, G. J. (2017). Decreased parvalbumin mRNA levels in cerebellar Purkinje cells in autism. Autism Res. 10, 1787–1796. doi: 10.1002/aur.1835
Stagi, S., Cavalli, L., Congiu, L., Scusa, M. F., Ferlini, A., Bigoni, S., et al. (2015). Thyroid function in Rett syndrome. Horm. Res. Paediatr. 83, 118–125. doi: 10.1159/000370066
Terao, T., Oga, T., Nozaki, S., Ohta, A., Otsubo, Y., Yamamoto, S., et al. (1995). Possible inhibitory effect of lithium on peripheral conversion of thyroxine to triiodothyronine: a prospective study. Int. Clin. Psychopharmacol. 10, 103–105. doi: 10.1097/00004850-199506000-00006
Uchida, K., Hasuoka, K., Fuse, T., Kobayashi, K., Moriya, T., Suzuki, M., et al. (2021). Thyroid hormone insufficiency alters the expression of psychiatric disorder-related molecules in the hypothyroid mouse brain during the early postnatal period. Sci. Rep. 11:6723. doi: 10.1038/s41598-021-86237-8
Uchida, K., Taguchi, Y., Sato, C., Miyazaki, H., Kobayashi, K., Kobayashi, T., et al. (2014). Amelioration of improper differentiation of somatostatin-positive interneurons by triiodothyronine in a growth-retarded hypothyroid mouse strain. Neurosci. Lett. 559, 111–116. doi: 10.1016/j.neulet.2013.11.052
Uchida, K., Yonezawa, M., Nakamura, S., Kobayashi, T., and Machida, T. (2005). Impaired neurogenesis in the growth-retarded mouse is reversed by T3 treatment. Neuroreport 16, 103–106. doi: 10.1097/00001756-200502080-00005
Wallis, K., Sjogren, M., Van Hogerlinden, M., Silberberg, G., Fisahn, A., Nordstrom, K., et al. (2008). Locomotor deficiencies and aberrant development of subtype-specific GABAergic interneurons caused by an unliganded thyroid hormone receptor alpha1. J. Neurosci. 28, 1904–1915. doi: 10.1523/JNEUROSCI.5163-07.2008
Keywords: thyroid hormone, hypothyroid, developmental disorder, parvalbumin, psychiatric disorder, MeCP2
Citation: Uchida K and Suzuki M (2021) Congenital Hypothyroidism and Brain Development: Association With Other Psychiatric Disorders. Front. Neurosci. 15:772382. doi: 10.3389/fnins.2021.772382
Received: 08 September 2021; Accepted: 17 November 2021;
Published: 09 December 2021.
Edited by:
Kazuhiko Sawada, Tsukuba International University, JapanReviewed by:
Farimah Beheshti, Torbat Heydarieh University of Medical Sciences, IranAaron Hanukoglu, Tel Aviv University, Israel
Copyright © 2021 Uchida and Suzuki. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Katsuya Uchida, dWNoaWRhQG0udG9ob2t1LmFjLmpw