 Di Hu
Di Hu Zunren Liu
Zunren Liu Xin Qi
Xin Qi- 1Department of Physiology and Biophysics, Case Western Reserve University School of Medicine, Cleveland, OH, United States
- 2Department of Biology, College of Arts and Sciences, Case Western Reserve University, Cleveland, OH, United States
- 3Center for Mitochondrial Disease, Case Western Reserve University School of Medicine, Cleveland, OH, United States
Many lines of evidence have indicated the therapeutic potential of rescuing mitochondrial integrity by targeting specific mitochondrial quality control pathways in neurodegenerative diseases, such as Parkinson’s disease, Huntington’s disease, and Alzheimer’s disease. In addition to ATP synthesis, mitochondria are critical regulators of ROS production, lipid metabolism, calcium buffering, and cell death. The mitochondrial unfolded protein response, mitochondrial dynamics, and mitophagy are the three main quality control mechanisms responsible for maintaining mitochondrial proteostasis and bioenergetics. The proper functioning of these complex processes is necessary to surveil and restore mitochondrial homeostasis and the healthy pool of mitochondria in cells. Mitochondrial dysfunction occurs early and causally in disease pathogenesis. A significant accumulation of mitochondrial damage resulting from compromised quality control pathways leads to the development of neuropathology. Moreover, genetic or pharmaceutical manipulation targeting the mitochondrial quality control mechanisms can sufficiently rescue mitochondrial integrity and ameliorate disease progression. Thus, therapies that can improve mitochondrial quality control have great promise for the treatment of neurodegenerative diseases. In this review, we summarize recent progress in the field that underscores the essential role of impaired mitochondrial quality control pathways in the pathogenesis of neurodegenerative diseases. We also discuss the translational approaches targeting mitochondrial function, with a focus on the restoration of mitochondrial integrity, including mitochondrial dynamics, mitophagy, and mitochondrial proteostasis.
Introduction
Neurodegenerative disorders (NDs) collectively affect more than 50 million worldwide (Gammon, 2014; GBD 2015 Neurological Disorders Collaborator Group, 2017). The most common NDs include Alzheimer’s disease (AD), Parkinson’s disease (PD), Huntington’s disease (HD), and amyotrophic lateral sclerosis (ALS). Although NDs have been studied for decades, the mechanisms underlying their pathogenesis are still elusive due to the complexity of the disease-causing factors (Bertram and Tanzi, 2005; Jellinger, 2010). Nevertheless, these diseases share some common pathological features: pathological protein aggregation (e.g., Amyloid beta [Aβ] in AD and Lewy bodies in PD) and mitochondrial damage in vulnerable brain regions (Ross and Poirier, 2004; Johri and Beal, 2012). Furthermore, ND-related proteins can directly impair mitochondrial function and trigger cell death. Thus, it is believed that mitochondrial dysfunction plays an important role in the neuronal loss observed with NDs.
Normal mitochondrial function is critical for energy production, calcium buffering, lipid metabolism, and redox regulation that govern cell growth, proliferation, and survival (Antico Arciuch et al., 2012; Giorgi et al., 2018). Sustained ATP production in neurons via the electron transport chain (ETC) on the mitochondrial inner membrane (IMM) secures the energy supply for their physiological functions (Mattson et al., 2008). Presynaptic mitochondria serve as a cytosolic calcium reservoir to mediate the release and recycling of neurotransmitters (Billups and Forsythe, 2002; Mattson et al., 2008). Moreover, mitochondria produce and eliminate reactive oxygen species (ROS) during oxidative phosphorylation (OXPHOS). The excessive production of mitochondrial ROS can impair protein function and induce inflammatory responses, leading to neuronal death (Johri and Beal, 2012). Therefore, under stress or disease conditions, quality control mechanisms are required to maintain mitochondrial function. To date, three major mitochondrial quality control (MQC) mechanisms that regulate mitochondrial integrity and maintain mitochondrial functions have been well-characterized: (1) activation of the mitochondrial unfolded protein response (UPRmt) can rescue protein homeostasis and bioenergetics (Nargund et al., 2015); (2) mitochondrial dynamics (fission and fusion) maintain mitochondrial morphology and bioenergetics (Chan, 2020; Giacomello et al., 2020); (3) mitophagy pathways eliminate damaged mitochondria via various adaptor proteins (e.g., Parkin/PTEN-induced kinase 1 [PINK1]), ensuring a pool of healthy mitochondria (Harper et al., 2018; Pickles et al., 2018).
In most cases of NDs, mitochondrial dysfunction is the result of abnormal MQC. For instance, mitochondrial fragmentation resulting from excessive mitochondrial fission causes bioenergetic deficits in the brain of patients with AD, PD, HD, and ALS (Bueler, 2009; Magrane et al., 2009; Manczak et al., 2011; Shirendeb et al., 2011; Wang W. et al., 2013). Conversely, pharmaceutical manipulations that normalize mitochondrial fission can efficiently rescue mitochondrial morphology and function and increase neuronal survival in various disease models (Guo X. et al., 2013; Filichia et al., 2016; Joshi et al., 2018a,b; Zhao Y. et al., 2019; Hu et al., 2021b). Recent studies have demonstrated that impaired mitophagy is one of the key aspects of the pathogenesis of AD, PD, HD, and ALS (Khalil et al., 2015; Grassi et al., 2018; Fang et al., 2019; Harding et al., 2021). Moreover, the activation of UPRmt has been observed in ND models, and genetic suppression of UPRmt exacerbated the development of neuropathology (Yi et al., 2018; Munoz-Carvajal and Sanhueza, 2020; Zhu et al., 2021). These findings collectively demonstrated the essential role of MQC in keeping neurons healthy and, more importantly, indicate potential therapeutic targets for the treatment of NDs.
In this review, we briefly introduce the current understanding of the molecular basis of MQC, including aspects of the mechanistic pathways, physiological functions, and pathological relevance. We then summarize the current findings of MQC impairment and potential MQC-targeted therapeutic strategies in several NDs. In Tables 1, 2, we summarize the MQC-related proteins and MQC-targeted therapeutic agents, respectively.
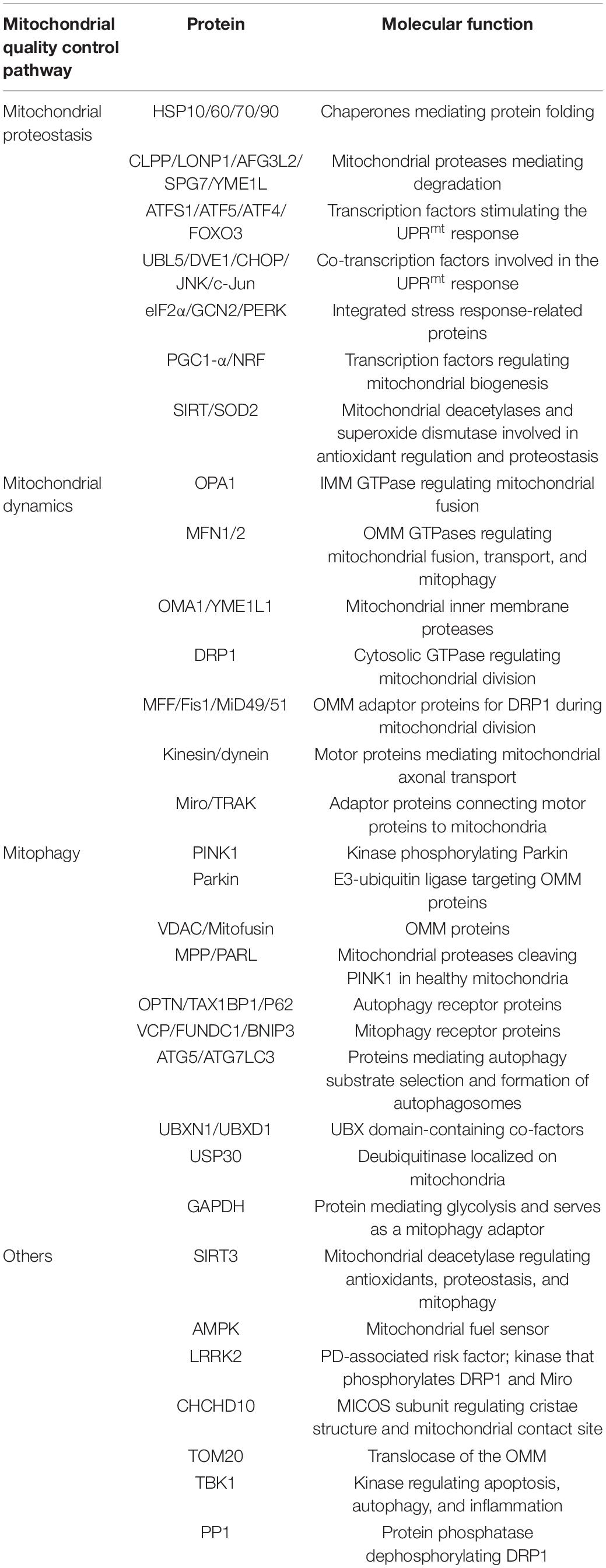
Table 1. Proteins associated with mitochondrial quality control.
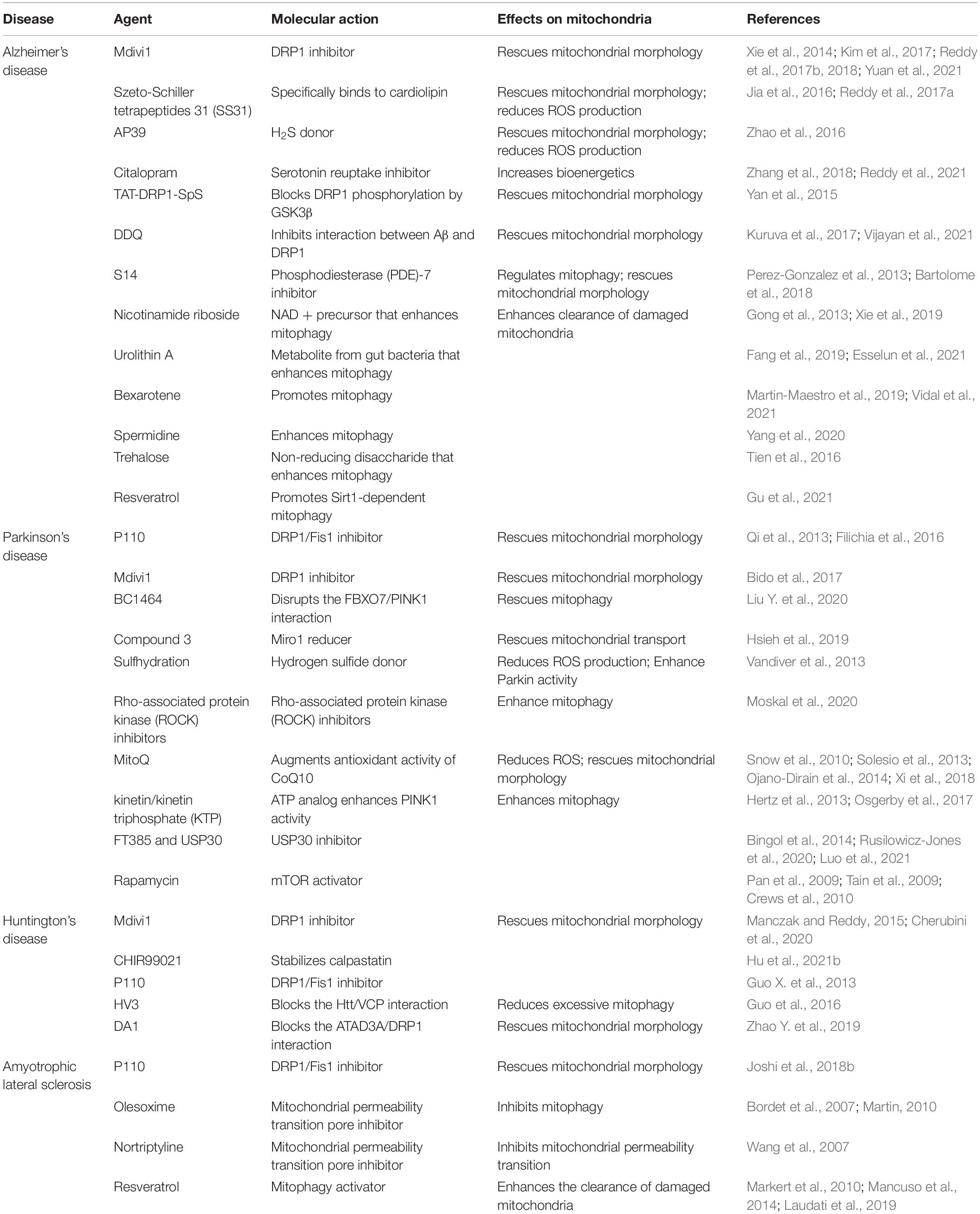
Table 2. Function of therapeutic agents in neurodegenerative diseases.
Mitochondrial Quality Control
Homeostasis of the Mitochondrial Proteome
Mitochondria contain 1100–1300 proteins encoded by either the mitochondrial genome or nuclear genes (Calvo and Mootha, 2010). Under the regulation of particular synchronization programs, mitochondrial- and nuclear-encoded proteins coordinate exquisitely to sustain mitochondrial functions (Couvillion et al., 2016; Soto et al., 2021). Disrupting mitochondrial import and translation of mitochondrial- and nuclear-encoded proteins could affect the precise stoichiometry of the OXPHOS complex subunits and lead to proteotoxicity (Gomes et al., 2013; Houtkooper et al., 2013; Tang et al., 2020). The accumulation of mitochondrial proteome damage (e.g., misfolded protein and protein carbonylation) directly impacts mitochondrial integrity and aging. Therefore, quality control mechanisms are required to maintain mitochondrial protein homeostasis.
Mitochondrial proteostasis is surveilled and regulated by chaperone proteins and proteases (Moehle et al., 2019). Mitochondrial heat shock protein 70 (mtHSP70), mtHSP90, and the large chaperonin complex HSP60/10 correct protein folding (Mayer and Bukau, 2005; Saibil, 2013). In the mitochondrial matrix, Lon peptidase 1 (LONP1) (mammalian) and endopeptidase Clp (ClpP) recognize and degrade misfolded and damaged proteins (Haynes et al., 2007; Shin et al., 2021). The IMM contains both m-AAA (AFG3-like matrix AAA peptidase subunit [AFG3L2] and hereditary spastic paraplegia type 7 [SPG7]) and i-AAA (YME1L1) protease complexes face the matrix and degrade the proteins in the intermembrane space (IMS) (Stiburek et al., 2012; Shanmughapriya et al., 2015; Pareek and Pallanck, 2020).
Unfolded protein response is one of the major mechanisms for maintaining mitochondrial proteostasis; its pathogenic relevance has recently been identified in NDs (Figure 1). UPRmt is the retrograde signaling between the mitochondria and nucleus that induces mitochondrial proteases and chaperones, which alleviate an overload of mitochondrial proteins (Zhao et al., 2002; Aldridge et al., 2007; Nargund et al., 2012). Conditions that increase mitochondrial proteotoxicity (e.g., mitochondrial DNA [mtDNA] depletion or OXPHOS perturbation) can provoke UPRmt (Lin et al., 2016; Naresh and Haynes, 2019). Though with some discrepancies, UPRmt has been demonstrated in both Caenorhabditis elegans (C. elegans) and mammalian systems. In C. elegans, UPRmt activation is mediated by activating transcription factor associated with stress-1 (ATFS-1) (Nargund et al., 2012). ATFS-1 has an amino-terminal mitochondrial-targeting sequence (MTS), which enables cells to evaluate mitochondrial protein import efficiency (Nargund et al., 2012). In cells with a healthy mitochondrial network, ATFS-1 is imported into mitochondria and degraded by the matrix-localized protease LON (Nargund et al., 2012). When mitochondria are impaired, the N-terminal nuclear localization signal (NLS) prevails and directs ATFS-1 to the nucleus to regulat transcription (Nargund et al., 2012). Importantly, MTS removal or inactivation results in the constitutive nuclear accumulation of ATFS-1 and UPRmt activation (Nargund et al., 2012). Other transcription co-factors also mediate UPRmt in C. elegans, including ubiquitin-like protein 5 (UBL5)/DVE-1 (Benedetti et al., 2006; Haynes et al., 2007). UPRmt activation stimulates the expression of mitochondrial protease CLPP-1 and chaperones (e.g., mtHSP70) that are transported into the mitochondria to relieve proteo-stress (Haynes et al., 2007). Interestingly, chromatin remodeling is required for UPRmt regulation in C. elegans (Merkwirth et al., 2016; Tian et al., 2016).
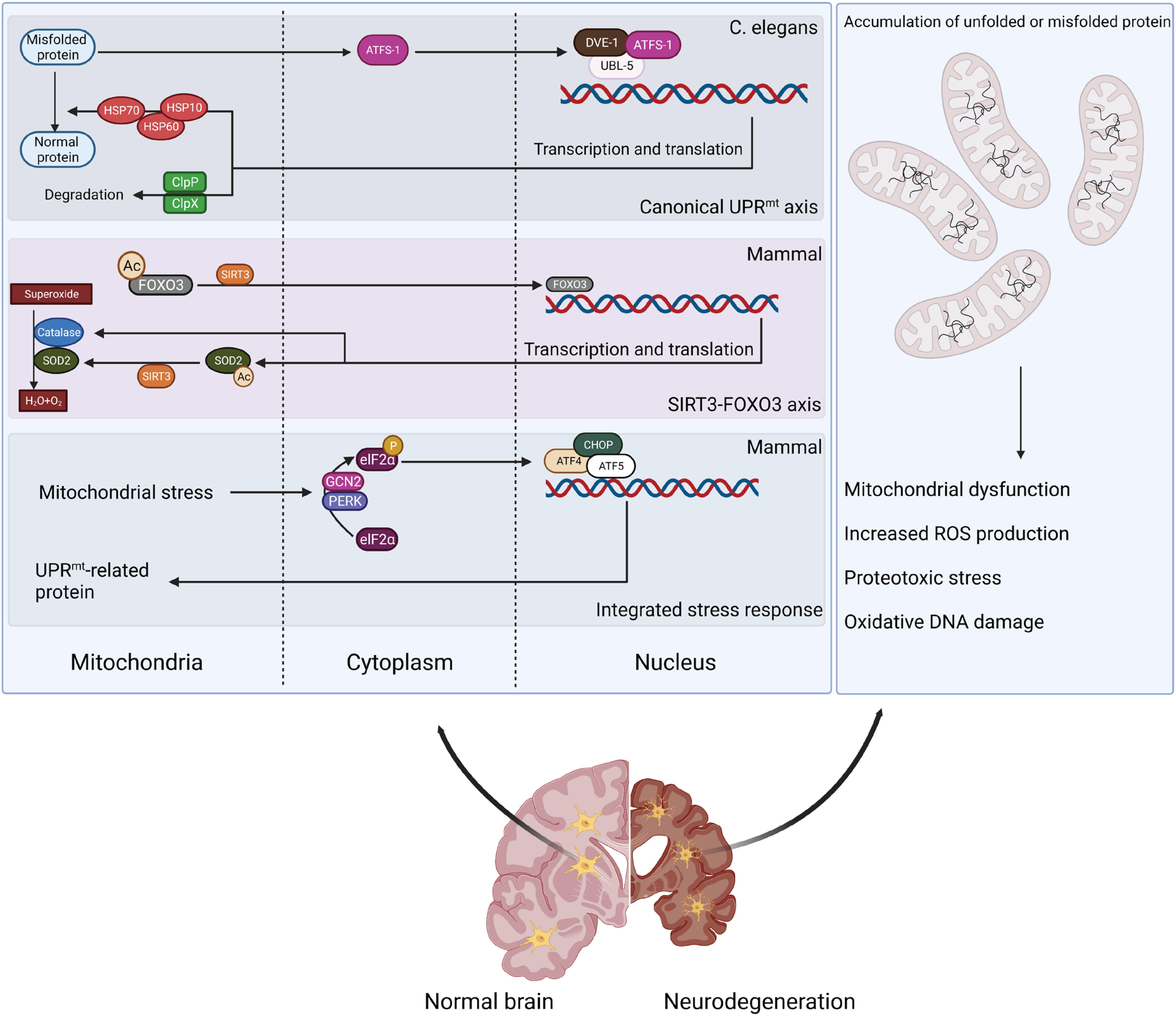
Figure 1. Retrograde signaling between mitochondria and nucleus to alleviate mitochondrial proteo-stress. Mitochondrial stress can stimulate defensive responses through different pathways. Canonical UPRmt activation is mediated by ATFS-1 and transcription co-factors DVE-1/UBL-5 to stimulate expression of mitochondrial chaperones (HSP10/60/70) and proteases (ClpP). Proteo-stress-induced production of mitochondrial ROS activates SIRT3 to deacetylate FOXO3, stimulating antioxidant responses. Mitochondrial stress also triggers ISR by stimulating eIF2α phosphorylation by GCN2 or PERK and activating ATF4/ATF5/CHOP-induced transcription of UPRmt-related genes. However, mitochondrial stress response is defective in neurodegenerative disease, leading to a disturbance in protein homeostasis and mitochondrial dysfunction.
The regulation of UPRmt is likely more complicated in mammalian cells. Recent studies suggest that three bZIP transcription factors (C/EBP homologous protein [CHOP] and activating transcription factors 4 and 5 [ATF4 and ATF5]) trigger UPRmt activation, which requires the integrated stress response (ISR)-associated phosphorylation of translation initiation factor 2A (eIF2α) by general control non-derepressible 2 (GCN2) and protein kinase R (PKR)-like endoplasmic reticulum kinase (PERK) (Aldridge et al., 2007; Fiorese et al., 2016; Fiorese and Haynes, 2017; Kaspar et al., 2021). A recent study indicated that ATF5 is the mammalian ortholog of ATFS-1 because its activity appears to be regulated by mitochondrial import efficiency (Fiorese et al., 2016). During mitochondrial stress, ATF5 is required to induce multiple mitochondrial proteases and chaperones, including HSP60, mtHSP70, and LONP1 (Fiorese et al., 2016). UPRmt activation can also be regulated by c-Jun N-terminal kinase (JNK) and the c-JUN pathway in mammals (Horibe and Hoogenraad, 2007; Nargund et al., 2012; Fiorese et al., 2016). The perturbation of protein homeostasis in the IMS can also stimulate the protective response mediated by mitochondrial sirtuin 3 (SIRT3) (Tseng et al., 2013; Qureshi et al., 2017). SIRT3 deacetylates and activates mitochondrial transcription factor forkhead box O3 (FOXO3), which then translocates to the nucleus to induce the expression of antioxidant enzymes (e.g., superoxide dismutase 2 [SOD2] and catalase) and biogenesis regulator peroxisome proliferator-activated receptor-γ coactivator-1α (PGC-1α) (Kenny et al., 2017; Weng et al., 2020). As a result, UPRmt activation stimulates mitochondrial biogenesis and promotes pathogen resistance and lifespan (Qureshi et al., 2017).
Mitochondrial proteostasis and biogenesis can also be indirectly regulated by the master transcription co-factor PGC-1α. PGC-1α modulates the nuclear respiratory factors (NRF1 and NRF2) that regulate the expression of the ETC subunits encoded by the nuclear genome and bind to the promoter of genes involved in mtDNA transcription (Scarpulla, 2008), including mitochondrial transcription factor A (TFAM) (Virbasius and Scarpulla, 1994). In addition, NRF2 can regulate the expression of other mitochondrial enzymes, such as translocase of the outer membrane (TOM20) that mediates mitochondrial protein import (Blesa and Hernandez-Yago, 2006). PGC-1α is considered a neuroprotective target because a pathogenic role for PGC-1α dysregulation has been ubiquitously found in NDs (i.e., AD, PD, HD, and ALS) (Qin et al., 2009; Zheng et al., 2010; Johri et al., 2012; Thau et al., 2012). In parallel with the UPRmt, other quality control strategies (e.g., mitochondrial precursor over-accumulation stress [mPOS], the unfolded protein response activated by the mistargeting of proteins [UPRam], mitochondrial ISR, and mitochondria-associated degradation [MAD]) have been implicated in regulating mitochondrial proteostasis and mitochondria-cytosol homeostasis (Silva et al., 2009; Wang and Chen, 2015; Wrobel et al., 2015; Liao et al., 2020). The pathological implications of these additional mechanisms in NDs are still under investigation. Thus, we mainly focus on UPRmt in NDs in this review.
Mitochondrial Fission and Fusion
Mitochondria are dynamic organelles that undergo continuous fission and fusion and are distributed in a tubular network in the cytoplasm (Giacomello et al., 2020). Mitochondrial fission segregates damaged parts from healthy mitochondria, whereas mitochondrial fusion allows the union of two mitochondria to enable genetic complementation, resulting in healthy and functional mitochondria (Ni et al., 2015; Giacomello et al., 2020). Thus, the balance between fission and fusion is critical for regulating mitochondrial size, number, and transport during cell proliferation and differentiation. It also ensures mitochondrial integrity coupled with appropriate bioenergetics to adapt to cellular stress.
Mitochondrial fission and fusion are regulated by the evolutionally conserved GTPase-dynamin superfamily distributed in the cytosol, mitochondrial outer membrane (OMM), and IMM (Figure 2). Mitochondrial fusion requires the cooperation of the mitofusins (MFN1 and MFN2) and optical atrophy 1 (OPA1) (Cipolat et al., 2004; Song et al., 2009; Gao and Hu, 2021). MFN1 and MFN2 are located on the OMM and coordinate the fusion of the outer membrane (Schrepfer and Scorrano, 2016). OPA1 mediates the inner membrane fusion (Mishra et al., 2014). As the central mediator of mitochondrial fusion, OPA1 exists as long and short forms mediated by alternative splicing or processed by the IMM metalloendopeptidases OMA1 and YME1L1 (Anand et al., 2014; Baker et al., 2014; Gilkerson et al., 2021). In addition, OPA1 plays an independent role in maintaining the cristae structure (Patten et al., 2014). Mitochondrial fission relies on GTPase dynamin-related protein 1 (DRP1). Upon stimulation, this cytosolic protein is recruited onto mitochondrial surface, where it self-assembles into a spiral structure to constrict mitochondrial tubules, mediating division (Taguchi et al., 2007; Kalia et al., 2018). The recruitment of DRP1 onto the mitochondrial surface requires adaptor proteins residing on the OMM. While mitochondrial fission factor (MFF) mainly governs the mid-zone fission for biogenesis of new mitochondria, mitochondrial fission 1 (Fis1) regulates the peripheral fission for lysosomal degradation of damaged mitochondria (Kleele et al., 2021). The adaptor proteins (e.g., MFF and mitochondrial elongation factors MiD49/51) facilitate the recruitment of DRP1 to the OMM. Depletion of any of these adaptors results in elongated mitochondrial morphology, which mimics the effects of DRP1 depletion (Loson et al., 2013). There are indications that MFF and MiD49/51 interact to form a complex that mediates DRP1-dependent fission (Palmer et al., 2011). Mitochondrial fission sites appear to be determined by the physical interaction between the endoplasmic reticulum (ER) and mitochondria at the contact site (Friedman et al., 2011; Korobova et al., 2013). Live-cell imaging has demonstrated that the ER tubules cross and wrap around the mitochondria at the fission site, where MFF, MiD49/51, and DRP1 are often colocalized (Friedman et al., 2011). The site for mitochondrial fission is also influenced by mtDNA replication (Kraus and Ryan, 2017). Evidence has shown that mitochondrial nucleoids active in gene replication are highly associated with the constriction, leading to mitochondrial fission (Murley et al., 2013; Lewis et al., 2016). In cells, the site of mitochondrial fission is close to the mitochondrial nucleoids (Lewis et al., 2016).
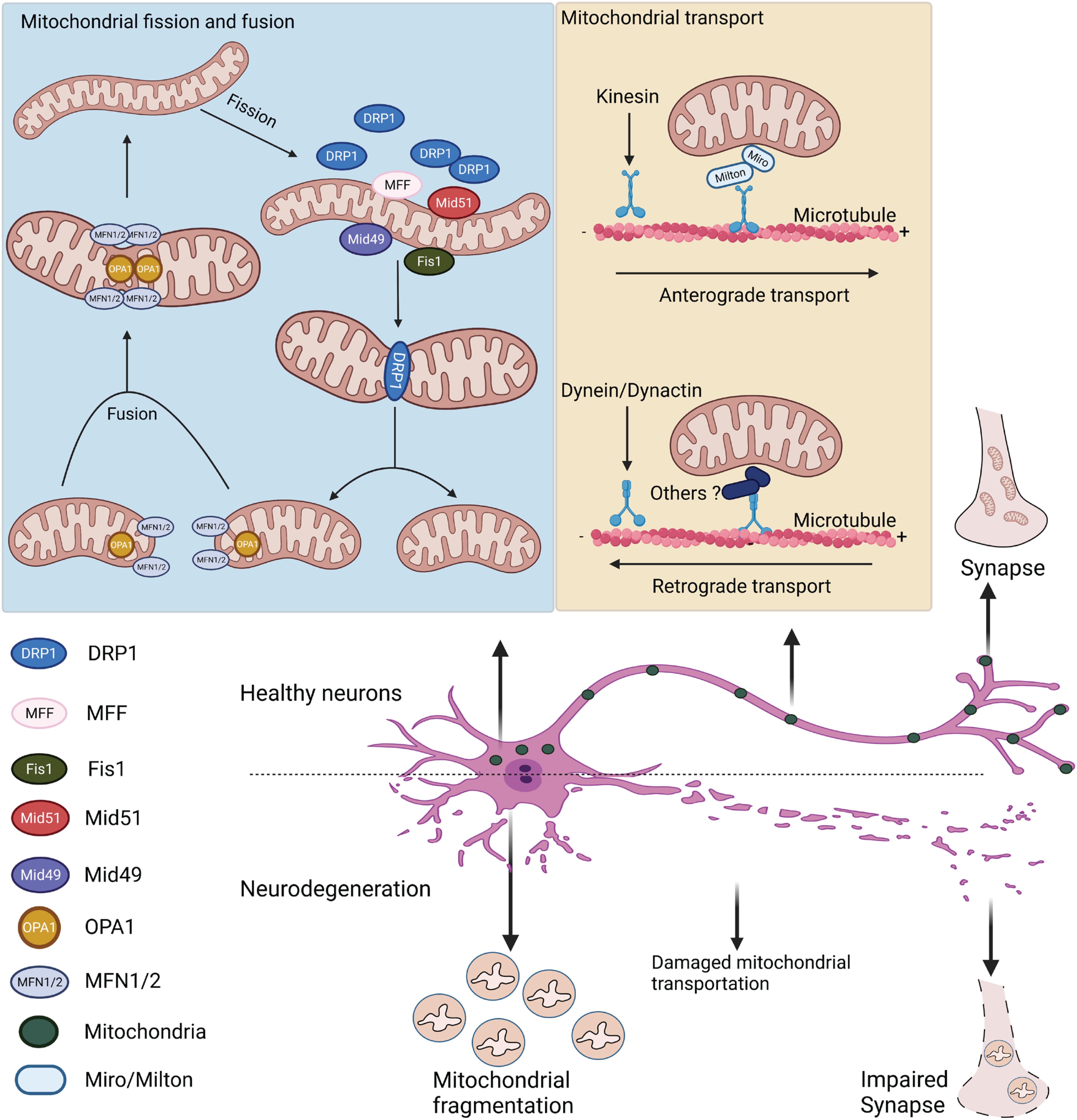
Figure 2. Mitochondrial fission, fusion, and transport in healthy and degenerative neurons. Mitochondrial fusion is mainly mediated by MFN1/2 and OPA1 on the outer and inner mitochondrial membranes, respectively. Mitochondrial fission is regulated by cytosolic DRP1 and its adaptor proteins (e.g., MFF, Fis1, and Mid49/51) on the OMM. After translocating onto mitochondria, DRP1 assembles to constrict the mitochondrial membrane. Mitochondrial axonal transport is mediated by the motor-adaptor protein complex. Anterograde transport is regulated by kinesin motor and Miro/Milton adaptors, whereas retrograde transport is regulated by the dynein/dynactin protein complex. In neurodegenerative diseases, dysregulated mitochondrial dynamics leads to mitochondrial fragmentation and impaired mitochondrial transport in neurons.
In addition to fission and fusion, intracellular mitochondrial dynamics usually involve the transport and re-distribution of mitochondrial units, which is critical in neuronal cells that are polarized with long axons and dendrites (Yu and Pekkurnaz, 2018). Neuronal mitochondria are commonly found at the synaptic terminals, where they provide sufficient ATP and regulate Ca2+ for neurotransmission (Sheng and Cai, 2012). The bidirectional transport of mitochondria is coordinated by microtubule-based machinery and the result of mitochondrial coupling to motor-adaptor-receptor protein complexes (Saxton and Hollenbeck, 2012; Figure 2). Typically, anterograde transport of mitochondria toward the (+) end of the microtubules is mainly facilitated by motor proteins of the kinesin-1 families (Wang and Schwarz, 2009; Mandal and Drerup, 2019). The attachment of motor proteins to mitochondria is mediated by the adaptor proteins Milton and Miro, a mitochondrial Rho-like GTPase (Tang, 2016; Eberhardt et al., 2020). Retrograde mitochondrial transport is regulated by dynein motor protein and dynactin adaptor protein TRAK (kinesin binding protein) (Schnapp and Reese, 1989; Loss and Stephenson, 2017). The detailed mechanism of how the dynein motor binds to mitochondria is still largely unknown. After transport, mitochondria are stabilized on the axons by anchor protein syntaphilin, whose downregulation results in an increased percentage of mobile mitochondria along the axon (Lin et al., 2017; Cardanho-Ramos et al., 2020). Notably, Miro participates in mitochondrial axonal transport and facilitates mitochondrial fission, fusion, and Ca2+ homeostasis (Eberhardt et al., 2020). Recent studies using super-resolution microscopy identified Miro localization at the mitochondria- and ER-associated membrane (MAM), associated with the mitochondrial contact site and cristae organizing system (MICOS) (Modi et al., 2019). Though the detailed mechanisms need further investigation, these data suggest that Miro regulates mitochondrial fission and couples MICOS to the TRAK motor protein adaptors to ensure the transport of mitochondria. Intriguingly, Miro interacts with MFN2 to regulate mitochondrial fusion and axonal transport (Misko et al., 2010). The close relationship between mitochondrial fission and fusion and mitochondrial transport is noteworthy. The mitochondrial fission-related protein DRP1 is also implicated in mitochondrial transport. DRP1 inhibition sufficiently disrupts mitochondrial transport in vitro and in vivo (Mandal and Drerup, 2019). DRP1 is also important for the distribution of mitochondria in nerve terminals of dopaminergic neurons (Berthet et al., 2014). In addition, one recent study showed that DRP1 interacts with the dynein-dynactin complex to modulate retrograde mitochondrial transport. Moreover, MFN2 can directly regulate mitochondrial axonal transport in neurons, as MFN2 downregulation causes a decreased rate of mitochondrial transport and increased pausing time (Misko et al., 2010). Previous studies suggested that MFN2 is the direct receptor of the TRAK1/2 adaptor protein mediating mitochondrial transport (Lee C.A. et al., 2018). Moreover, inhibition of MFN2 in neurons and in vivo significantly reduces both anterograde and retrograde mitochondrial transport (Misko et al., 2010; Mandal and Drerup, 2019).
Mitophagy
Mitophagy is the selective targeting and degradation of impaired mitochondria via lysosomes. It promotes the turnover of healthy mitochondria, regulates the number of mitochondria to meet metabolic demands, and eliminates dysfunctional mitochondria to prevent ROS production and the release of damage-associated patterns from mitochondria, which would further induce cellular stress and degeneration (Palikaras et al., 2018; Wang Y. et al., 2019). Mitophagy is regulated by several pathways in mammalian cells (Figure 3). PINK1-dependent Parkin activation is the best-characterized mitophagy pathway. In the healthy state, PINK1 is recruited and imported into mitochondria through the TOM-TIM (translocase of the inner membrane) complex, where it is cleaved by matrix processing peptidase (MPP) and presenilins-associated rhomboid-like protein (PARL) (Greene et al., 2012; Lazarou et al., 2012; Thomas et al., 2014; Kazlauskaite and Muqit, 2015; Pickrell and Youle, 2015). However, loss of mitochondrial membrane potential in dysfunctional mitochondria prevents the import of PINK1 and stabilizes it on the OMM, resulting in the phosphorylation of ubiquitin molecules (Greene et al., 2012; Pickles et al., 2018). Phosphorylated ubiquitin then recruits cytosolic Parkin to the mitochondria and activates its ubiquitin ligase activity (Kane et al., 2014; Pickles et al., 2018). Activated Parkin ubiquitinates OMM proteins, including voltage-dependent anion channel (VDAC) and mitofusins (Gegg et al., 2013; Yamano et al., 2016; McLelland et al., 2018; Ham et al., 2020), and recruits autophagy receptors (e.g., optineurin [OPTN], Tax1-binding protein [TAX1BP1], and sequestosome 1 [SQSTM1, or p62]), leading to the formation of microtubule-associated protein 1A/1B-light chain 3 (LC3)-positive phagophores (Geisler et al., 2010; Richter et al., 2016; Whang et al., 2017). Phagophores sequester damaged mitochondria and deliver them to the lysosomes for degradation. Recent findings suggested an emerging role for Miro in PINK1/Parkin-mediated mitophagy. It has been proposed that Miro is phosphorylated by leucine-rich repeat kinase-2 (LRRK2) in the presence of mitochondrial dysfunction, followed by the phosphorylation of both Miro and Parkin by PINK1 (Wang et al., 2011; Hsieh et al., 2016). Once activated, Parkin can ubiquitinate Miro, targeting it for proteasomal degradation and subsequent mitochondrial clearance by mitophagy (Panchal and Tiwari, 2021). In addition to PINK1 and Parkin, other receptor proteins can also mediate mitophagy. Notably, the AAA + ATPase valosin-containing protein (VCP)/p97 has emerged as a critical mitophagy receptor required for OMM-associated degradation and is involved in Parkin-dependent mitophagy (Sun and Qiu, 2020). Following mitochondrial depolarization, UBX domain-containing co-factor UBXN1/UBXD1 translocates alongside VCP to mitochondria in a Parkin-dependent manner and recruits LC3 to form phagophores (Bento et al., 2018; Mengus et al., 2021). VCP overexpression results in mitochondrial fragmentation and cell death (Fang et al., 2015). Furthermore, VCP mutations are associated with mitochondrial depolarization, oxidative stress in ALS and frontotemporal dementia (FTD) (Kakizuka, 2008; Meyer et al., 2012). FUN14 domain-containing-1 (FUNDC1) is an OMM protein that functions as a mitophagy receptor upon mitochondrial uncoupling and hypoxia (Zhang, 2021). BCL2-interacting protein-3 (BNIP3) is also involved in hypoxia-induced mitophagy (Ney, 2015).
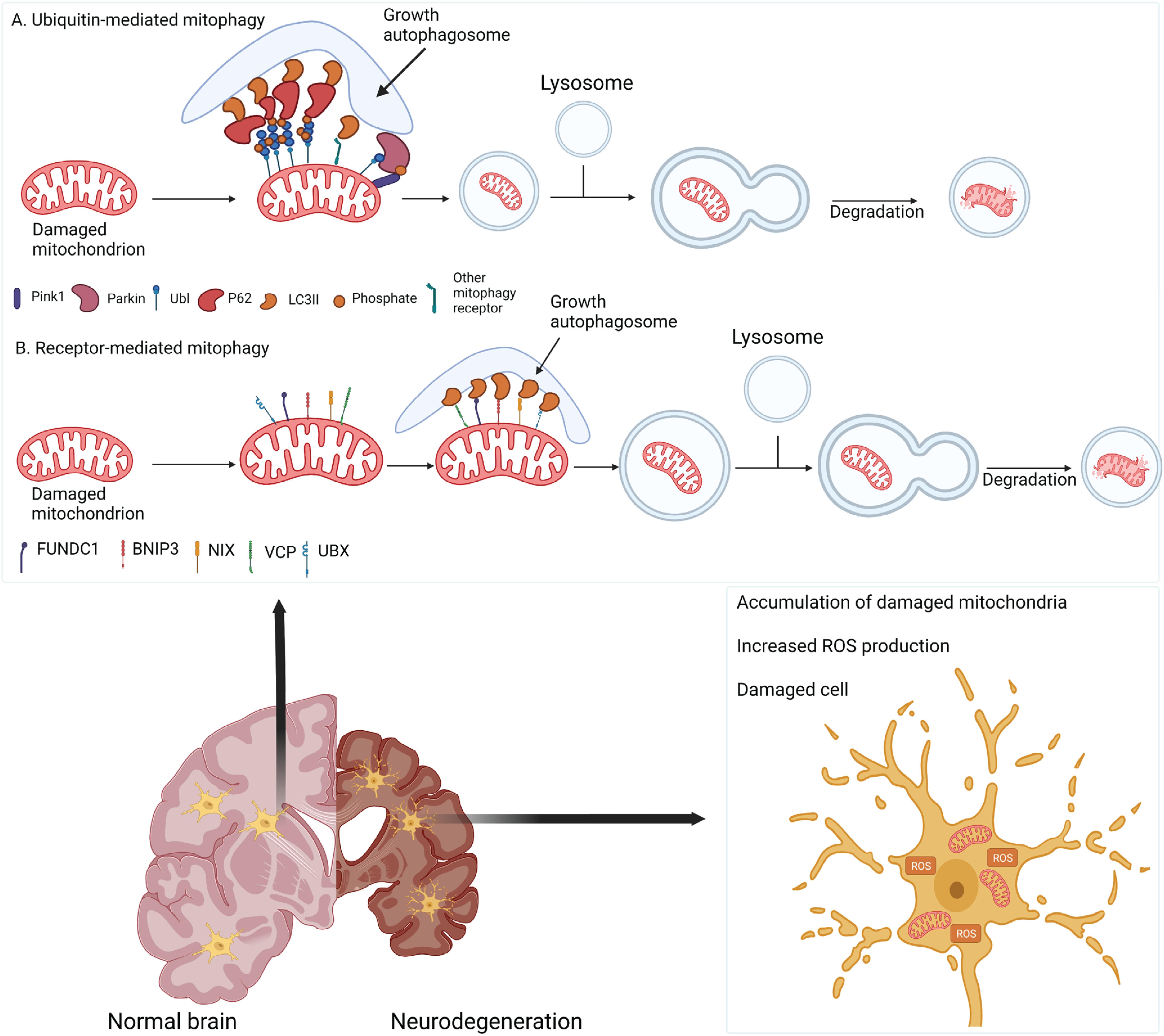
Figure 3. Damaged mitochondria are cleared by mitophagy. (A) Depolarized mitochondria retain PINK1 and Parkin to ubiquitinate OMM proteins, recruiting adaptor proteins (e.g., p62) to form autophagosomes that fuse with lysosomes for degradation. (B) In parallel, mitophagy can be mediated by receptor proteins (e.g., FUNDC1, BNIP3 and VCP) to recruit an autophagy adaptor. In neurodegenerative diseases, impaired mitophagy causes the accumulation of damaged mitochondria, increased ROS production, and neuronal death.
Alzheimer’s Disease
Alzheimer’s disease is the most common neurodegenerative disorder characterized by progressive memory loss and cognitive impairment, affecting 30 million people worldwide (Haque and Levey, 2019). The most prevalent pathological hallmarks of this disease include the accumulation of Aβ plaques and neurofibrillary tangles composed of misfolded microtubule-associated protein Tau. There is currently no cure for AD. Recent studies suggest that AD is not a linear downstream consequence of Aβ deposition but rather a multifactorial disease. In addition to Aβ toxicity, mitochondrial dysfunction has been suggested as a hallmark of AD because patients exhibit early metabolic changes (Moreira et al., 2010; Ortiz and Swerdlow, 2019). Furthermore, abnormal mitochondrial structure, accumulation of damaged mitochondria, and excessive ROS production are well-documented in various AD models (Ortiz and Swerdlow, 2019). Thus, impaired mitochondrial integrity might play an important role in AD pathogenesis. Understanding the mechanisms of mitochondrial dysregulation and impaired MQC may provide molecular targets for treating AD.
Mitochondrial Quality Control Impairment in Alzheimer’s Disease
Previous studies demonstrated an imbalance between mitochondrial fusion and fission in AD, contributing to disease pathogenesis. Multiple independent studies have found that in AD patient brains and mutant amyloid precursor protein (APP)-expressing AD animal models, the expression of the mitochondrial fission-related protein Fis1 and the GTPase activity of DRP1 are increased (Cho et al., 2009; Wang X.L. et al., 2009; Joshi et al., 2018a). Moreover, the protein levels of fusion-related proteins (e.g., MFN1, OPA1, and MFN2) are decreased (Wang X. et al., 2009). Increased expression of mitochondrial fission-related proteins is correlated with Tau protein accumulation in neurons derived from AD patient-induced pluripotent stem cells (iPSCs) (Wang X. et al., 2008; Lee J. et al., 2018). The mRNA expression levels for genes expressing mitochondrial fission-related proteins (e.g., Fis1) are also increased in the peripheral blood of AD patients brain (Pakpian et al., 2020). Moreover, treatment of hippocampal neurons with Aβ-oligomer can reduce mitochondrial fusion (Wang X.L. et al., 2009). Furthermore, overexpression of either WT APP or mutant APPswe (APP Swedish mutant) can alter mitochondrial morphology and induce mitochondrial fragmentation in AD cell culture models (Wang X.L. et al., 2008). In addition to neurons, astrocytes carrying APOE3/4 variants, which is strong risk factor of AD, have altered mitochondrial DRP1 (Schmukler et al., 2020).
Impaired axonal transport is a featured axon pathology in AD. Both Aβ and Tau can disrupt axonal mitochondrial transport (Zheng et al., 2019). For example, Aβ treatment reduces mitochondrial mobility in hippocampal neuronal cultures (Zhao C. et al., 2019), and Tau overexpression in Neuro-2a cells disrupts axonal mitochondrial trafficking (Ebneth et al., 1998). Intriguingly, anterograde mitochondrial transport is more vulnerable in AD, possibly because Tau inhibits kinesin-1 activity but has little effect on dynein (Ebneth et al., 1998). Moreover, the mitochondrial anchor protein syntaphilin is degraded in AD-related human APP-expressing neurons, triggering retrograde transport of mitochondria (Lin et al., 2017). These results suggest that mitochondrial dynamics and mitochondrial transport are impaired in AD, which may lead to synaptic dysfunction and neurodegeneration. Furthermore, the accumulated mitochondria in the soma of neurons cannot be cleared by autophagy due to the defective transport and exacerbate neuronal loss in AD. These findings indicate a contribution of dysregulated mitochondrial dynamics and axonal transport to AD pathogenesis.
Mitophagy impairment is associated with ROS production, synaptic damage, Aβ accumulation, and neuronal loss in AD (Tran and Reddy, 2020). Mitophagy defects have been widely observed in various animal and cellular mutant APP-associated AD models. In a clinical study, the expression of Parkin and autophagy-related 5 (ATG5) was slightly but significantly decreased in the sera and brains from 160 AD patients compared to 40 control subjects (Castellazzi et al., 2019). In Tau and APP-expressing neuroblastoma cells, overexpression of P301L mutant Tau or mutant APPswe impairs the translocation of mitochondrial Parkin, PINK1, LC3-II/I, and other mitophagy-related proteins (Ye et al., 2015; Cummins et al., 2019). Mutation in presenilin-1 (PS1) causes familial AD. Mitophagy is impaired in the presenilin-1 (PS1) missense mutation A246E AD model due to increased lysosomal pH (Coffey et al., 2014). In AD patient iPSC-derived neuronal cells, the expression levels of LC3 and transcription factor EB (TFEB) are decreased (Martin-Maestro et al., 2019). Thus, a reduction in mitophagy-related proteins or changes in the cellular environment can result in impaired mitophagy.
Disruptions in mitochondrial proteostasis have been repeatedly observed in AD. Aβ can be translocated into mitochondria, interrupting mitochondrial protein import and impairing preprotein maturation. These disturbances lead to mitochondrial dysfunction (Mossmann et al., 2014; Cenini et al., 2016). Devi et al. (2006) demonstrated that both full-length and C-terminal truncated APP could accumulate in the mitochondrial protein import channels (TOM40 and TIM23), causing mitochondrial protein import deficits and dysfunction exclusively in AD patient brains. In a yeast model, mitochondrial Aβ peptide inhibited the degradation of presequence peptides by presequence protease (PreP) homolog Cym1, leading to the accumulation of mitochondrial preprotein and processing intermediates and an imbalanced organellar proteome (Mossmann et al., 2014). The activation of UPRmt was recently reported in AD patients and other models, which may disrupt proteostasis through Aβ or APP. Accumulation of mitochondrial unfolded protein has been observed in AD patient brains and is a potential diagnostic biomarker for this disease (Beck et al., 2016). The mRNA levels of UPRmt-related components (e.g., DNAJA3, HSPD1, HSPE1, CLPP, and YME1L1) are all significantly increased in AD subjects (Beck et al., 2016). In addition, HSP60 protein levels are increased in AD patients, possibly resulting from the constitutive activation of the mitochondrial stress response (Walls et al., 2012). One recent study demonstrated that UPRmt responses are associated with Aβ toxicity in AD patients, mouse and C. elegans models of AD (Sorrentino et al., 2017). Notably, pharmaceutically or genetically improved mitochondrial proteostasis reduced amyloid aggregation in C. elegans AD model, and a transgenic AD mouse model, indicating a protective role for UPRmt in AD (Sorrentino et al., 2017). In Aβ peptide (Aβ25–35)-treated neuroblastoma cells and APPswe/PS1dE9 double transgenic mice, the HSP60, LONP1 and ClpP expression levels are elevated compared to the controls (Shen et al., 2019). The connection between UPRmt and Alzheimer’s disease was further demonstrated in PreP knock-out brain organoids, where a mitochondrial PreP deficiency induced UPRmt, increased Aβ accumulation, and triggered AD-like phenotypes (Perez et al., 2020). Further studies are needed to understand how APP or Aβ induces UPRmt activation and disrupts mitochondrial proteostasis in AD.
Targeting Mitochondrial Quality Control Prevents Alzheimer’s Disease-Associated Pathology
Mitochondrial quality control maintains mitochondrial function. In AD patients and model systems, expression levels of MQC components involved in mitochondrial dynamics, mitophagy, and UPRmt are significantly altered. Restoration or overexpression of these components may be possible therapeutic approaches for AD. Multiple evidences suggest the abnormal mitochondrial fission in AD patient and animal and cellular models of AD, inducing mitochondrial fragmentation (Calkins et al., 2011; Zhang L. et al., 2016). Thus, the inhibition of fission-related proteins or overexpression of fusion-related proteins may represent possible treatments (Xie et al., 2014). Several compounds can reduce the expression levels of mitochondrial fission-related proteins or induce fusion-related proteins. DRP1 inhibitor, mitochondrial division inhibitor-1 (Mdivi1), and the mitochondria-targeted antioxidant Szeto-Schiller tetrapeptide 31 (SS31) reduced the levels of hydrogen peroxide and GTPase DRP1 activity in mutant APPswe-overexpressing Neuro-2a cells (Reddy et al., 2017b, 2018). In senescence-accelerated mouse-prone 8 (SAMP8) mice that have an accelerated aging phenotype, SS31 can correct learning disabilities by decreasing the levels of the mitochondrial fission proteins DRP1 and Fis1 (Jia et al., 2016). Mdivi1 can attenuate mitochondrial fission, reverse the inhibition of mitochondrial complex I, and modulate reactive oxygen species levels by impairing DRP1 GTPase activity in Aβ-treated BV-2 microglia cells (Park et al., 2013). Treatment with the mitochondria-targeted hydrogen sulfide donor AP39 attenuates mitochondrial impairment by increasing OPA1 and MFN1 expression levels and decreasing Fis1 protein levels in APP/PS1 neurons and transgenic mice (Zhao et al., 2016). In addition, administration of citalopram, a selective serotonin reuptake inhibitor, can alleviate memory loss, cognitive decline, defective biogenesis, impaired dendritic spines and defective synaptic MQC in APP/PS1 double transgenic mice (Zhang et al., 2018). This compound also activates mitochondrial fusion and reduces mitochondrial fission (Zhang et al., 2017). Glycogen synthase kinase 3 (GSK3)-dependent phosphorylation of DRP1 at Ser40 and Ser44 can increase DRP1 GTPase activity, which induces mitochondrial fragmentation (Yan et al., 2015). The synthetic polypeptide TAT-DRP1-SpS can block this phosphorylation and reduce mitochondrial fragmentation in the APP/PS1 AD mouse model (Yan et al., 2015). DDQ and SS31 can enhance mitochondrial fusion by activating MFN1 and MFN2 and repress Bax activation, cytochrome c release, and the mitochondrial permeability transition pore in APPswe-expressing cells (Kuruva et al., 2017). DRP1 siRNA can also prevent mitochondrial fission, loss of mitochondrial membrane potential, and cell death (Grohm et al., 2012). Zhang et al. (2020) showed that conditional heterozygous depletion of DRP1 in oligodendrocytes could rescue mitochondrial morphology, abolish NLR family pyrin domain containing 3 (NLRP3)-mediated inflammatory injury, and restore axonal myelination in the 5xFAD AD mouse model. Rescue of mitochondrial transport may also have a beneficial effect in AD. Indeed, blockage of the mitochondrial permeability transition pore by genetic depletion of cyclophilin D or treatment with SS31 attenuates impaired mitochondrial trafficking, potentially due to reduced Ca2+ and ROS (Guo L. et al., 2013; Jia et al., 2016).
Enhanced mitophagy-related protein levels in mouse and cellular AD models can restore mitophagy and reduce neuronal stress caused by the accumulation of damaged mitochondria. For example, treatment of APP/PS1 mice with the phosphodiesterase (PDE)-7 inhibitor S14 has a neuroprotective effect by modulating Aβ-induced mitochondrial dysfunction through restored LC3-II expression levels (Bartolome et al., 2018). In addition, citalopram activates PINK1, ATG5, ATG7, p62, and LC3B-I/II, restoring mitophagy and abrogating synaptic toxicities in APP-Tg2576 transgenic mice (Reddy et al., 2021). Parkin overexpression in APP/PS1 mice and Aβ-treated cells can ameliorate mitochondrial dysfunction, restore PINK levels, increase ATP production, and decrease ubiquitinated Aβ levels (Hong et al., 2014; Martin-Maestro et al., 2016; Wang H.M. et al., 2020). Moreover, nicotinamide riboside (NR) increases mitophagy-related proteins (e.g., LC3) to enhance mitophagy and alleviate memory loss in APPswe/PS1dE9 AD mice (Aman et al., 2020). Furthermore, Fang et al. (2019) demonstrated that treatment with the mitophagy activator urolithin A restored mitophagy by increasing PINK1, Parkin, and Beclin-1 protein levels and attenuates the loss of cognitive ability and Aβ pathology in APP/PS1 mice. In the familial AD-related mutant PS1 iPSC-derived neural stem cells, bexarotene can restore mitophagy and rescue the damaged mitochondrial network morphology (Martin-Maestro et al., 2019). Treatment of C. elegans co-expressing Aβ and Tau with spermidine improved behavior, extend lifespan, and protect against memory loss via the PINK/Parkin pathway (Yang et al., 2020). Other enzymes (e.g., adenylate-activated protein kinase [AMPK] and SIRT) can also influence mitophagy activity. For instance, AMPK overexpression sufficiently reduces Tau phosphorylation, GSK3β activity, and brain impairment in streptozotocin (STZ) mice (Wang L. et al., 2020). In addition, SIRT activators (e.g., resveratrol) induce autophagy through the mTOR (Target of rapamycin)-ULK1 (Unc-51 Like Autophagy Activating Kinase 1) pathway (Park et al., 2016). Clinical trials have also shown that SIRT activators modulate Aβ levels and inflammatory markers in AD patients. Finally, compounds that can induce lysosomes and autophagosomes, such as trehalose, can also induce mitophagy and attenuate the accumulation of APP in AD (Tien et al., 2016).
Unfolded protein response maintains homeostasis and reduces the proteotoxicity caused by Aβ (Munoz-Carvajal and Sanhueza, 2020); its activation can remove unfolded proteins induced by APP mutants. Enhancement of UPRmt can restore memory and reduce Aβ toxicity in the APP/PS1 double transgenic mice (Shen et al., 2020). Recent studies have shown that the sirtuin family is essential for UPRmt, and a reduction in SIRT3 expression correlates with mitochondrial dysfunction in AD (Lee J. et al., 2018). NAD+ can act as a UPRmt inducer by activating SIRT3 (Papa and Germain, 2014). In C. elegans, an NAD+ booster could attenuate the proteotoxicity caused by Aβ (Mouchiroud et al., 2013). Induction of the expression of the chaperones mtHSP70 and mtHSP90 may inhibit Aβ aggregation and the formation of plaques in the AD brain (Roe et al., 2018; Evgen’ev et al., 2019).
Parkinson’s Disease
Parkinson’s disease is the second predominant neurodegenerative disorder that affects more than 10 million people worldwide (Ball et al., 2019). PD is characterized by a selective loss of dopaminergic neurons in the substantia nigra, resulting in rest tremor, bradykinesia, rigidity, and the accumulation of intracellular Lewy bodies composed of α-Synuclein protein (α-Syn). Although the pathogenic mechanisms of PD remain elusive, numerous studies suggest a dominant role for mitochondrial dysfunction in various PD models. Notably, most of the PD-related mutations were identified in proteins that regulate mitochondrial functions. Thus, improving mitochondrial function by targeting MQC might efficiently ameliorate PD pathogenesis.
Mitochondrial Quality Control Impairment in Parkinson’s Disease
Recent studies have demonstrated UPRmt impairment in multiple PD models. Mutant α-Syn (e.g., A30P/A53T) is prone to aggregate and associated with early onset of PD (Li et al., 2001). In transgenic mice, human dopaminergic SH-SY5Y cells, and iPSC-derived dopaminergic neurons, the α-Syn A53T mutant can preferentially accumulate in mitochondria and interact with matrix ClpP, suppressing its peptidase activity. These events result in mitochondrial dysfunction and neuronal damage (Hu et al., 2019). Mutation in PINK1/PRKN is associated with familial PD. In nematode PD models, missense mutations in pink-1/pdr-1 caused the accumulation of damaged mitochondria, which activated UPRmt to mitigate the detrimental effects of these mutations (Cooper et al., 2017). Treatment of SH-SY5Y cells with mitochondrial toxin 1-methyl-4-phenylpyridinium (MPP +) (metabolite of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine [MPTP]) that selectively impairs dopaminergic neuron by inhibiting ETC complex I impaired UPRmt and induced OXPHOS stress, which could be reversed by overexpression of PGC-1α or ATF5 (Cai et al., 2020; Hu et al., 2021a).
Abnormal mitochondrial dynamics have been reported in PD. α-Syn has been shown to directly regulate mitochondrial morphology. α-Syn overexpression causes mitochondrial fragmentation in multiple PD models, including WT α-Syn-expressing C. elegans, A53T α-Syn-expressing SH-SY5Y cells, and primary neurons from A53T α-Syn-expressing rat (Kamp et al., 2010; Li et al., 2013; Bido et al., 2017). Recent studies have illustrated the molecular mechanisms underlying α-Syn-induced abnormalities in mitochondrial morphology. Oligomeric α-Syn binds to the lipids in the OMM and distresses membrane curvature, reducing mitochondrial fusion rate (Ryan et al., 2018; Gilmozzi et al., 2020). Moreover, WT α-Syn can localize to the MAM to positively modulate mitochondrial morphology. This function is impaired by pathogenic mutations (e.g., A53T) in α-Syn (Guardia-Laguarta et al., 2014). Overexpression of A53T α-Syn activates mitogen-activated protein kinase (p38 MAPK), which phosphorylates DRP1 at serine 616 (S616) and triggers mitochondrial fission in vitro (Gui et al., 2020). In addition, MFN1 and MFN2 protein levels are decreased in α-Syn-expressing primary cortical neurons, which correlates with a decrease in mitochondrial fusion and smaller mitochondria (Faustini et al., 2019). In a Drosophila model of α-Syn-associated PD, mutant α-Syn proteins misallocate to mitochondria where they interact with spectrin and alter actin stabilization, resulting in DRP1 recruitment, mitochondrial fragmentation, and neuronal death (Ordonez et al., 2018).
Increased α-Syn levels have also been linked to the arrest of both anterograde and retrograde mitochondrial transport (Prots et al., 2018; Valdinocci et al., 2019). The anterograde trafficking of mitochondria is disrupted in early-stage sporadic PD followed by altered retrograde transport in late-stage disease (Zanellati et al., 2015). Prots et al. (2018) showed that the E57K α-Syn variant tends to form oligomers that directly bind to and disrupt the interactions between kinesin and the microtubules in human neuronal cells. The α-Syn A53T mutant can also directly bind to Miro, causing aberrant mitochondrial axonal transport in human neurons and Drosophila (Shaltouki et al., 2018). Mutations in other genes that are risk factors for familial PD can also cause dysfunctional mitochondrial dynamics. Vacuolar protein sorting-associated protein 35 (VPS35) is a component of the retromer complex, which is involved in retrograde transport from endosomes to the trans-Golgi network (Tang et al., 2015). Loss of function mutations in VPS35 impair the proteostasis including the enlargement of endolysosomes and is linked with autosomal dominant PD (Vilarino-Guell et al., 2011). The VPS35 R524W and D620N mutants increase the clearance of inactive DRP1, leading to mitochondrial fragmentation in human neurons and mouse brain (Wang et al., 2016). LRRK2 is a kinase localized to the cytosol and associated with the OMM (Biskup et al., 2006). Mutation in LRRK2 has been linked to the late-onset of PD (Clark et al., 2006). The LRRK2 G2019S mutant can directly phosphorylate DRP1 at threonine 595 (Thr595) to cause excessive mitochondrial fission in iPSC-derived neurons (Su and Qi, 2013). Moreover, this mutant appears to alter the polymerization/depolymerization cycles of microtubules, disrupting mitochondrial trafficking in PD (Kett et al., 2012; Godena et al., 2014; Singh et al., 2019). LRRK2 and PINK1 Drosophila mutants exhibit disturbed mitochondrial calcium homeostasis with functional involvement of Miro (Lee K.S. et al., 2018). Mutant LRRK2 can also induce PINK1 and Parkin-dependent Miro degradation and mitophagy, which inhibits mitochondrial axonal transport (Bonello et al., 2019; Liu et al., 2019). MPP + induces Parkin S-nitrosylation, triggering DRP1 phosphorylation at S616 and excessive mitochondrial fission (Zhang Z. et al., 2016). Moreover, MPP + can inhibit kinesin-1-mediated anterograde mitochondrial transport (Kim-Han et al., 2011).
Mitophagy is impaired in numerous ways in PD. PINK1 and Parkin mutations resulting in loss of function lead to autosomal recessive forms of PD (Lucking et al., 2000; Morais et al., 2009; Vives-Bauza et al., 2010). In addition to mitophagy, PINK1 and Parkin are also important for maintaining mitochondrial morphology (Poole et al., 2008). In A53T α-Syn transgenic mice, α-Syn accumulation on mitochondria causes increased mitophagy and neuronal death (Choubey et al., 2011). In A53T and E46K α-Syn transgenic mice, accumulation of α-Syn on the mitochondria promotes externalization of cardiolipin from the IMM to OMM to recruit LC3 to the mitochondria, inducing mitophagy (Ryan et al., 2018). In the neurons derived from PD patient iPSCs, α-Syn interacts with Miro via its N-terminus, leading to excessive Miro accumulation on the mitochondrial surface and delayed mitophagy (Shaltouki et al., 2018). These studies indicate the role of abnormal mitophagy in α-Syn-mediated toxicity. In addition to PINK1/Parkin, LRRK2 depletion or the LRRK2 G2019S mutant impair the autophagy/lysosomal pathway, leading to the accumulation of autophagosomes (Bonello et al., 2019; Obergasteiger et al., 2020; Wauters et al., 2020; Boecker et al., 2021). The levels of the autophagy markers p62 and LC3 are increased in induced pluripotent stem cell-derived dopaminergic neurons from PD patients with the LRRK2 G2019S mutation (Vermilyea et al., 2020). Several independent studies have shown PINK1/Parkin-dependent accumulation of Ras-related protein Rab-10 (RAB10), an LRRK2 substrate, on damaged mitochondria in PD patients with the LRRK2 G2019S mutation, suggesting that LRRK2 is involved in PINK1/Parkin-mediated mitophagy (Bonello et al., 2019; Wauters et al., 2020). Mutant DJ-1 causes a rare form of autosomal recessive PD. DJ-1 is regarded as a neuroprotective factor that translocates onto stressed mitochondria to regulate the clearance of ROS. DJ-1 loss-of-function increases the recruitment of Parkin to damaged mitochondria and mitophagy activity (Thomas et al., 2011).
Targeting Mitochondrial Quality Control Prevents Parkinson’s Disease-Associated Pathology
Given the importance of mitochondrial dysfunction in PD pathogenesis, therapeutics targeting mitochondria have been studied for the prevention and treatment of PD. Although the pathological phenotypes from mutation-associated and neurotoxin-induced PD models are different, the outcomes of mitochondrial dysfunction, including impaired mitochondrial dynamics, mitophagy, and UPRmt, are the same. Therefore, understanding the mechanisms of MQC impairment in PD could provide potential targets for developing novel PD treatments.
Several lines of evidence demonstrate the beneficial effects of UPRmt activation in both familial and idiopathic PD models. In SH-SY5Y cells, upregulation of UPRmt activity by overexpression of PGC-1α or ATF5 significantly improved mitochondrial function and cell survival after MPP + treatment (Cai et al., 2020; Hu et al., 2021a). UPRmt activation by ATFS-1 protected C. elegans against mutant PINK1/Parkin-induced mitochondrial fragmentation, oxidative stress, and cellular toxicity, promoting longevity and dopaminergic neuron survival (Cooper et al., 2017). Moreover, activation of UPRmt by ginseng total protein (GTP) from herbal extracts rescued PD-related pathologies in mutant PINK1B9-expressing Drosophila and prolonged their lifespan (Liu M. et al., 2020). Furthermore, the α-Syn A53T mutant preferentially accumulates in mitochondria and directly binds to UPRmt-related ClpP, suppressing its peptidase activity (Hu et al., 2019). Conversely, ClpP overexpression sufficiently decreases α-Syn A53T mutant-associated pathology (Hu et al., 2019).
Pharmaceutical and genetic approaches to modulate mitochondrial dynamics efficiently improve mitochondrial integrity and neuronal survival in PD. Filichia et al. (2016) showed that subcutaneous administration of rationally designed small peptide P110, a selective inhibitor of the DRP1/Fis1 interaction, to MPTP-treated mice blocked DRP1 mitochondrial translocation and protected dopaminergic neurons. Similarly, the small-molecule DRP1 inhibitor Mdivi-1 attenuated mitochondrial fragmentation and α-Syn aggregation and prevented motor deficits in the α-Syn A53T mutant-expressing rat model (Bido et al., 2017). Moreover, overexpression of MFN2 or a DRP1 K38A dominant-negative variant rescued mitochondrial deficits and neuropathology in the α-Syn A53T mutant rat model (Choubey et al., 2011; Rappold et al., 2014). 6-Hydroxydopamine (6-OHDA) is a neurotoxin that can selectively trigger dopaminergic neuronal loss (Simola et al., 2007). MitoQ is a mitochondria-targeted antioxidant that consists of a lipophilic triphenylphosphonium (TPP) cation linked to a ubiquinone antioxidant moiety of the endogenous antioxidant coenzyme Q10. In 6-OHDA-treated cells and a PD mouse model, MitoQ activated PGC-1α, enhancing MFN2-mediated mitochondrial fusion and the survival of dopaminergic neurons (Xi et al., 2018). Furthermore, LRRK2 inhibition can correct mitochondrial transport and morphology to preserve neuronal function in PD animal models (Xiong et al., 2017; Singh et al., 2019).
Because the pathogenic role of mitophagy defects in PD has been elucidated, improving the efficiency of mitochondrial clearance by mitophagy may represent a disease-modifying strategy for PD. Indeed, studies focusing on the development of mitophagy modulators have demonstrated their therapeutic potential in NDs. For instance, PINK1 can be targeted for cellular elimination through the ubiquitin E3 ligase subunit, F-box protein 7 (FBXO7) (Liu Y. et al., 2020). Indeed, Liu Y. et al. (2020) demonstrated that compound BC1464, which specifically disrupts the FBXO7/PINK1 interaction, could rescue mitophagy and confer neuroprotection in several PD culture models (e.g., primary cortical neurons, neuroblastoma cells, and patient-derived cells). Treatment with a Miro1 reducer (compound 3) decreased mitochondrial-localized Miro levels, rescuing mitochondrial transport and PD-related phenotypes in iPSC-derived neurons and Drosophila models (Hsieh et al., 2019). Parkin sulfhydration is markedly decreased in PD patients; however, the catalytic activity of Parkin can be improved through this modification, suggesting the therapeutic potential of hydrogen sulfide donors (Vandiver et al., 2013). A recent high-throughput screening for compounds that upregulate the mitochondrial recruitment of Parkin identified a series of neuroprotective Rho-associated protein kinase (ROCK) inhibitors (Moskal et al., 2020). In dopaminergic neurons and SH-SY5Y cells, activation of PINK1 with the ATP neo-substrate kinetin/kinetin triphosphate (KTP) significantly improves the activity of WT PINK1 and the PINK1 G309D mutant and enhances Parkin phosphorylation and its mitochondrial recruitment (Hertz et al., 2013). Ubiquitin-specific protease 30 (USP30) is a mitochondrial deubiquitinase that opposes Parkin-mediated mitophagy by removing the poly-ubiquitin chain from damaged mitochondria (Bingol et al., 2014). Knock-down of USP30 enhances mitophagy and rescues paraquat-induced dopamine neuronal loss in Drosophila (Bingol et al., 2014). In addition, two distinct USP30 inhibitors (FT385 and USP30i) can significantly improve mitophagy, indicating potential therapeutic approaches for PD (Bingol et al., 2014; Rusilowicz-Jones et al., 2020). Treatment of MPTP-treated mice with the mTOR activator rapamycin triggers mitophagy, preventing neuronal loss (Moors et al., 2017). Furthermore, genetic upregulation of PINK1 or Parkin can significantly alleviate MPTP-induced neurodegeneration and motor deficits in PD mice (Li and Chen, 2019).
Huntington’s Disease
Huntington’s disease is a devastating monogenic neurological disorder caused by the dominantly inherited CAG trinucleotide repeat expansion in the gene encoding the huntingtin (Htt) protein (Kerschbamer and Biagioli, 2016). Clinical symptoms of HD include hyperkinesia or chorea of the face, trunk, and legs, followed by cognitive and psychiatric disturbances (Roos, 2010). HD is characterized by the selective loss of GABAergic medium spiny neurons (MSN) and the presence of mutant huntingtin (mtHtt) aggregates in the striatum (Ehrlich, 2012). With polyglutamine expansion at the N-terminus, mtHtt protein gains a toxic function that disturbs multiple subcellular functions, leading to neuronal death (Schulte and Littleton, 2011). Mitochondrial dysfunction has been widely demonstrated to be correlated with the loss of MSN and HD pathogenesis. Thus, enhancing mitochondrial function is a potential therapeutic strategy for HD.
Mitochondrial Quality Control Impairment in Huntington’s Disease
Mutant huntingtin impairs mitochondrial function through multiple pathways. HD patients exhibit well-documented metabolic defects (Nambron et al., 2016). mtHtt represses the activity of PGC-1α, a transcriptional coactivator involved in mitochondrial biogenesis, glucose metabolism, β-oxidation of fatty acids, and adaptive thermogenesis (Cui et al., 2006). As a result, the activities of OXPHOS complexes I, II, III, and IV are affected in HD patient brains (Cui et al., 2006). Alternations in mitochondrial dynamics are well characterized in HD. Song et al. (2011) demonstrated that mtHtt could bind to and activate DRP1, leading to DRP1 mitochondrial translocation and mitochondrial fragmentation in HD patient brains and mice models. In addition to directly interacting with mtHtt, DRP1 can be activated by multiple kinases, including MAPK/ERK2 and cyclin-dependent kinase (CDK5) (Jahani-Asl et al., 2015; Roe and Qi, 2018). Moreover, mtHtt-induced DRP1 translocation can trigger the dimerization of ATPase Family AAA Domain Containing 3A (ATAD3A) in multiple neuronal and mouse models of HD, resulting in mitochondrial fragmentation and mtDNA damage (Zhao Y. et al., 2019). Interestingly, mtHtt can also affect mitochondrial movement when expressed in primary rat cortical neurons (Chang et al., 2006). Furthermore, the mtHtt binding partner huntingtin-associated protein (HAP1) can interact with kinesin and dynein to regulate mitochondrial transport (Rong et al., 2006).
It has recently been discovered that mtHtt can disrupt mitochondrial proteostasis in HD. mtHtt localizes to the IMS in mtHtt-expressing cells and HD patient brains where it binds with high affinity to the TIM23 complex, causing defective import of nuclear-encoded proteins and disrupting mitochondrial proteostasis (Yablonska et al., 2019). Moreover, mtHtt inhibits UPRmt in HD cells and HD R6/2 transgenic mice by impairing the mRNA stability of mitochondrial ATP Binding Cassette Subfamily B Member 10 (ABCB10), which suppresses UPRmt signaling (Fu et al., 2019). Several recent studies have suggested that the removal of defective mitochondria is compromised in HD. Indeed, mtHtt affects autophagosome formation, leading to the accumulation of damaged mitochondria (Lee et al., 2012; Franco-Iborra et al., 2021). While its physiological function is still unknown, glyceraldehyde-3-phosphate dehydrogenase (GAPDH)-mediated mitophagy is a novel micro-mitophagy pathway to eliminate damaged mitochondria via lysosomes following ischemia or reoxygenation-induced injury, which is independent of its glycolytic activity (Yogalingam et al., 2013). Interestingly, mtHtt can interact with mitochondrial GAPDH, which stalls GAPDH-mediated mitophagy and causes the accumulation of damaged mitochondria in HD cells (Hwang et al., 2015). One recent study also suggested a pathogenic role for excessive mitophagy in HD. In HD patients and transgenic mouse models, the interaction of VCP with mtHtt causes its translocation and accumulation in mitochondria, triggering excessive mitophagy via the recruitment of LC3 to the mitochondria (Guo et al., 2016). However, another study indicated that the interaction between mtHtt and autophagy receptor p62 disrupts the loading of damaged mitochondria into autophagosome and their transport to lysosomes (Ehrnhoefer et al., 2018).
Targeting Mitochondrial Quality Control Rescues Huntington’s Disease-Associated Pathology
Many lines of evidence have shown that improving mitochondrial function can efficiently rescue HD-related pathology, indicating the role of mitochondrial dysfunction in HD pathogenesis and the therapeutic potential of targeting MQC. SIRT3 activation can improve anterograde mitochondrial neurite transport and maintain the viability of primary striatal neurons from HD mice (Naia et al., 2021). Furthermore, the overexpression of the SIRT3 ortholog dSirt2 ameliorated neurodegeneration and extended lifespan of HD flies (Naia et al., 2021). Inhibition of DRP1 mitochondrial translocation can sufficiently rescue mitochondrial morphology, biogenesis, and neuronal viability in HD models. Recently, Hu et al. (2021b) identified CHIR99021 as a mitochondrial enhancer that can significantly improve mitochondrial function (e.g., mitochondrial membrane potential, respiration) by preventing DRP1 translocation via calpastatin stabilization in HD neurons, and rescue the neuropathology and motor dysfunctions in HD mouse models. Moreover, inhibition of DRP1 activity by small molecule inhibitor Mdivi-1 or overexpression of the dominant-negative DRP1 K38A mutant prevents mitochondrial fission and improves mitochondrial function (Song et al., 2011; Manczak and Reddy, 2015). In addition, Guo X. et al. (2013) demonstrated that long-term administration of peptide P110, which interferes with the interaction between DRP1 and Fis1, can rescue mitochondrial fragmentation, HD-related neuropathology, and motor deficits observed in HD mouse models. Treatment with peptide inhibitor DA1 that blocks Drp1/ATAD3A interaction can rescue mitochondrial fragmentation and mtDNA lesion, and HD-related pathologies in HD mouse models (Zhao Y. et al., 2019). Thus, antagonizing DRP1-mediated mitochondrial fission could represent an important therapeutic approach against HD.
Approaches mediating mitophagy can also protect against HD. Khalil et al. (2015) showed that PINK1 overexpression could ameliorate ATP levels and improve neuronal integrity and survival in an HD Drosophila model, counteracting mtHtt toxicity. Subcutaneous administration of the small peptide HV3 to HD mice abolished the mitochondrial translocation of VCP by blocking the interaction between mtHtt and VCP (Guo et al., 2016). This treatment also corrected excessive mitophagy and reduced cell death. In addition, GAPDH overexpression is sufficient to rescue defective mitophagy, enhance mitochondrial function, and promote cell survival (Hwang et al., 2015). Moreover, treatment with mitochondrial activators that induce PGC-1α expression promotes mitochondrial biogenesis and provides neuroprotection by activating autophagy and increasing the turnover of mtHtt aggregates (Tsunemi et al., 2012).
Amyotrophic Lateral Sclerosis
Amyotrophic lateral sclerosis is the most common subtype of motor neuron disease, with a worldwide prevalence of 4–6 in 100,000 people (Bucheli et al., 2013). It is an incurable, fatal neurodegenerative disorder with an average survival of 2-3 years from diagnosis (Bryukhovetskiy et al., 2020). ALS is characterized by rapid, progressive degeneration of upper and lower motor neurons, resulting in muscle atrophy, gradual paralysis, and death (Ravits et al., 2007). ALS is a multi-factorial disease. Proteins altered in ALS, such as superoxide dismutase 1 (SOD1), TAR DNA binding protein (TDP43), fused in sarcoma (FUS), and C9orf72, have been implicated in a wide range of cellular pathways (Bruijn et al., 1997; Neumann et al., 2006; Vance et al., 2009; Cooper-Knock et al., 2012). Notably, all these proteins can cause mitochondrial dysfunction. Thus, mitochondrial dysfunction is a crucial factor involved in ALS pathogenesis and rescuing mitochondrial integrity might protect motor neuron function.
Mitochondrial Quality Control Impairment in Amyotrophic Lateral Sclerosis
Though still under investigation, evidence has shown UPRmt activation in various ALS models. The SOD1 G93A mutant localizes to the IMS and activates two UPRmt axes in an ALS mouse model (Riar et al., 2017). TDP-43 dysregulation suppresses ETC complex I and activates UPRmt in cellular and mouse ALS models (Wang P. et al., 2019). Another study showed that TDP-43 is a potential substrate for UPRmt protease LONP1, and downregulation of LONP1 increases TDP-43 expression, resulting in mitochondrial dysfunction and neurodegeneration (Wang P. et al., 2019). More recently, mutations have been reported in the coiled-coil-helix-coiled-coil-helix domain containing 10 (CHCHD10) gene in ALS patients (Veldink and Consor, 2018; Harjuhaahto et al., 2020). CHCHD10 encodes a mitochondrial protein, which may maintain the MICOS (Genin et al., 2016). Mutant CHCHD10 is associated with mitochondrial dysfunction and the early death of motor neurons (Ryan et al., 2021). In mutant CHCHD10 ALS mice, aggregation of mutant CHCHD10 induces proteotoxic stress and the upregulation of the UPRmt transcriptional regulators ATF5 and CHOP (Anderson et al., 2019). A multi-OMICS study of CHCHD10 variants linked to ALS demonstrated metabolic disturbances and UPRmt activation (Straub et al., 2021). However, the connection between mutant SOD1, TDP-43, and CHCHD10 needs further investigation.
Studies of ALS patients and animal models indicate altered mitochondrial dynamics in ALS disease pathogenesis. Mitochondrial fragmentation has been observed in ALS models expressing mutant SOD1, potentially due to the downregulation of mitofusins and OPA1 and upregulation of DRP1 and Fis1 in the mouse spinal cord and skeletal muscles (Luo et al., 2013). Altered mitochondrial transport is also evident in ALS motor neurons, which precedes neuronal loss (Magrane et al., 2014). Abnormal mitochondrial transport has also been observed in mutant SOD1 transgenic mice; the mutant SOD1 reduced Miro levels and directly interacted with dynein-dynactin complexes (Shi et al., 2010; Smith et al., 2019). The use of in vitro and in vivo TDP-43 models has shown a tendency for mitochondria to fragment (Wang W.Z. et al., 2013). In addition, increased DRP1 phosphorylation and decreased OPA1 expression have been reported in mutant TDP-43 transgenic mice (Wang W.Z. et al., 2013). Moreover, several independent studies demonstrated that TDP-43 decreased the expression of mitofusins in patient muscles, transgenic mice, and Drosophila neurons by binding to MFN1 and MFN2 mRNA (Khalil et al., 2017). Mitochondrial fragmentation has also been reported in neurons and Drosophila models expressing WT or mutant FUS (Deng et al., 2015; Chen et al., 2016). Furthermore, shortened mitochondria have been observed in fibroblasts derived from ALS patients expressing mutant CHCHD10 or mutant C9orf72 (Genin et al., 2016; Onesto et al., 2016).
Mitophagy is the most affected MQC mechanism in ALS. Mutations in mitophagy regulators (e.g., VCP, TBK1, and OPTN) are directly linked to ALS (Maruyama et al., 2010; Johnson et al., 2011; Khalil and Lievens, 2017; Oakes et al., 2017). Mutant VCP cannot migrate to impaired mitochondria or recognize ubiquitinated proteins, resulting in the accumulation of damaged mitochondria (Yamano et al., 2016). OPTN and TBK1 mutations interfere with LC3 recruitment to depolarized mitochondria (Li et al., 2016; Ryan and Tumbarello, 2018; Harding et al., 2021). Autophagosomes accumulate in mouse motor neurons expressing mutant SOD1 and patient fibroblasts expressing mutant C9orf72 (Rudnick et al., 2017; Leskela et al., 2021). Mutant SOD1 also suppresses endogenous Miro levels through a Parkin-dependent pathway, resulting in impaired mitochondrial transport and mitophagy (Moller et al., 2017). In the G93A SOD1 mouse model of ALS, decreased MFN2 expression causes defective transport of mitochondria and the calpastatin protein (Wang et al., 2018). In addition, TDP-43 overexpression can cause abnormal aggregation of mitochondria in ALS mouse models (Xu et al., 2010). Furthermore, PINK1 and Parkin protein levels are increased in FUS-overexpressing HEK293 cells (Chen et al., 2016). Conversely, PINK1 or Parkin downregulation reduces abnormal phenotypes in FUS-expressing Drosophila (Chen et al., 2016).
Targeting Mitochondrial Quality Control Rescues Amyotrophic Lateral Sclerosis-Associated Pathology
Finding a cure for ALS has so far been unsuccessful. Previous studies have suggested that modulating MQC is a potential treatment option for ALS. However, treatments to improve mitochondrial function by reducing oxidative stress and apoptosis (e.g., CoQ10, olesoxime, and nortriptyline) have yielded disappointing results in clinical trials despite promising animal studies (Bordet et al., 2007; Wang et al., 2007; Kaufmann et al., 2009). Thus, it is important to determine which types of mitochondrial dysfunction are relevant to this disease and its progression in order to identify new molecular targets for the development of ALS therapies. Sustained treatment of G93A SOD1 transgenic mice with small peptide P110 rescued mitochondrial morphology and improved motor performance and survival (Joshi et al., 2018b). Protein phosphatase 1 (PP1) dephosphorylates DRP1; its suppression prevents mitochondrial fragmentation and ALS-related neuronal damage in primary mutant SOD1 neuronal cultures and iPSC-derived motor neurons (Choi et al., 2020). Inhibiting mitochondrial fission with a dominant-negative DRP1 K38A mutant construct precludes motor neuronal death in mutant SOD1-expressing ALS models (Song et al., 2013). Moreover, induction of mitochondrial fusion by MFN2 overexpression alleviates ALS-TDP-43-induced mitochondrial dysfunction and neuronal damage in spinal cord motor neurons (Wang W. et al., 2013). Miro overexpression also sufficiently rescues mitochondrial axonal transport defects in mutant SOD1 cortical and motor neurons (Moller et al., 2017). Promoting mitochondrial biogenesis with resveratrol or PGC-1α upregulation alleviates the ALS-related syndromes and extends the lifespan of SOD1 G93A and SOD1 G37R transgenic mice, respectively (Markert et al., 2010; Da Cruz et al., 2012). These findings collectively indicate the therapeutic potential of altering mitochondrial dynamics in ALS. Though some data have shown a connection between UPRmt alteration or mitophagy impairment and ALS pathogenesis, whether modulating these MQC pathways protects motor neurons remains to be elucidated.
Conclusion and Future Perspectives
Maintenance of neuronal homeostasis relies heavily on functional mitochondria, which is highlighted by the fact that mitochondrial dysfunction is often associated with NDs (e.g., AD, PD, HD, and ALS). In addition to producing ATP, mitochondria are home to multiple metabolic processes. Critical strategies exist to regulate mitochondrial integrity. Mitochondrial protein homeostasis is maintained by local chaperones and proteases. The GTPase superfamily proteins regulate mitochondrial morphology, and damaged mitochondria are removed by macroautophagy. These quality control mechanisms co-exist to detect and repair defects that affect mitochondrial function to maintain cellular physiology. Thus, MQC impairment leads to the accumulation of damaged mitochondria, excessive ROS production, energy deficits, and synaptic and neuronal degeneration. There are still many questions regarding the crosstalk between these different MQC mechanisms and their coordination in mitochondrial homeostasis and neurodegenerative disease. For example, the emerging role of UPRmt supports the concept of mitochondrial protein homeostasis, and more importantly, provides a potential mechanism to explain mitochondrial dysfunction observed in neurodegenerative diseases. However, although ATF5 appears to be a functional ortholog of ATFS-1 in C. elegans, there is much diversity in mammalian UPRmt. Two recent publications suggest that the cleavage of mitochondrial protein DELE1 (DAP3 binding cell death enhancer 1) by inner membrane protease OMA1 is a pathway to transduce the signal of a mitochondrial defect to the cytosol. After being released into the cytosol, the cleaved DELE1 binds to and activates the heme-regulated eIF2α kinase, which phosphorylates eIF2-α and induces the translation of transcription factors ATF4 and CHOP (Fessler et al., 2020; Guo et al., 2020). Therefore, additional transcription factors and proteases other than the canonical ATFS-1/ATF5 signaling pathway can respond to mitochondrial proteo-stress. These findings implicate the coordination of multiple molecular pathways in maintaining mitochondrial proteostasis in mammalian systems. Therefore, previous conclusions on the pathogenic relevance of UPRmt disturbances in neurodegenerative diseases in C. elegans should be further validated in mammalian systems. Establishing the mechanism by which UPRmt is impaired during neurodegeneration requires further investigation to identify potential molecular targets for treating NDs.
Despite substantial evidence of the therapeutic advances of targeting MQC in AD, PD, HD, and ALS, any critical discrepancies between experiment models and human subjects should be considered carefully. To date, there are no in vivo models recapitulating all the pathological features and disease progression observed in PD patients (Potashkin et al., 2010; Jagmag et al., 2015). Thus, approaches manipulating MQC in rodent or fly models of PD may not be efficacious in patients. In addition, although mitochondrial dysfunction is a common feature shared by different PD models, the underlying pathological mechanism leading to the impairment of MQC is somehow distinguishable among the different models. For example, while mitophagy is mainly affected in mutant PINK1/Parkin models, multiple MQC mechanisms are impaired in α-Syn A53T mutant-related PD models (Martin et al., 2006; Li et al., 2013; Pickrell and Youle, 2015). Therefore, mitophagy modulators identified using PINK1/Parkin models might only partially rescue mitochondrial function or maintain neuronal viability in other PD models. Thus, the therapeutic potential of novel small molecules or peptides targeting MQC should be evaluated in multiple familial and sporadic PD models. Similarly, treatments targeting MQC in AD, HD, and ALS should be validated in different models.
Data from several recent studies indicate that MQC mechanisms seem to regulate each other. Haeussler et al. (2020) found that UPRmt activation can stimulate mitochondrial fission, and reversely, whereas blocking mitochondrial fusion can induce the UPRmt response under physiological conditions in C. elegans. These data suggest that mitochondrial fission and UPRmt may be activated simultaneously, providing a cue for the concomitant activation of UPRmt and mitochondrial fragmentation in NDs. Nevertheless, because of such mutual regulation of UPRmt and mitochondrial dynamics, therapeutic approaches seeking to upregulate UPRmt in NDs should be considered carefully since excessive fission would impair mitochondrial function. Moreover, mitochondrial proteolytic stress can be rescued by Parkin and PINK1-mediated mitophagy (Jin and Youle, 2013; Burbulla et al., 2014), suggesting that mitophagy may alleviate mitochondrial proteotoxicity. It is clear that mitophagy functions to eliminate damaged mitochondria. Therefore the upregulation of mitophagy could enrich the pool of healthy mitochondria in NDs, such as PD, where there is an accumulation of stressed and damaged mitochondria. However, a recent study showed that the activation of mitophagy inhibited UPRmt activation in C. elegans (Haeussler et al., 2020). Unlike the mitophagy that targets mitochondria with irreversible damage, UPRmt activation affects the function of the overall mitochondrial pool in the cell as a consequence of transcriptional regulation. Given that UPRmt activation induces genes that promote mitochondrial biogenesis and functions, it is necessary to evaluate the side effects of therapeutic strategies aiming to upregulate mitophagy on the mitochondrial function within the healthy pool. Therefore, understanding the connection between these MQCs could provide cues for developing efficient and safe treatments for NDs.
Whether a combination treatment targeting multiple pathways may provide a better therapeutic effect against NDs needs further investigation. Although improving mitochondrial function by targeting MQC can prevent or slow disease progression in vivo, it remains unclear whether these modulations can reverse the volume of neurons in the CNS. Further investigation is also required to understand the side effects and administration methods of targeting MQC for neurodegenerative treatments. Our current knowledge of MQC continues to evolve, providing a novel research scheme for developing practical therapeutic approaches to combat NDs.
Author Contributions
DH made substantial contribution to the conception and design of the study. DH and ZL participated in drafting the manuscript. XQ revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by grants from the United States National Institutes of Health (R01AG065240, R01NS115903, and R21NS107897 to XQ), a Dr. Ralph and Marian Falk Medical Research Trust–Transformative Award (to XQ), and Harrington Rare Disease Scholar Award (to XQ).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Aldridge, J. E., Horibe, T., and Hoogenraad, N. J. (2007). Discovery of genes activated by the mitochondrial unfolded protein response (mtUPR) and cognate promoter elements. PLoS One 2:e874. doi: 10.1371/journal.pone.0000874
Aman, Y., Ryan, B., Torsetnes, S. B., Knapskog, A. B., Watne, L. O., Mcewan, W. A., et al. (2020). Enhancing mitophagy as a therapeutic approach for neurodegenerative diseases. Int. Rev. Neurobiol. 155, 169–202. doi: 10.1016/bs.irn.2020.02.008
Anand, R., Wai, T., Baker, M. J., Kladt, N., Schauss, A. C., Rugarli, E., et al. (2014). The i-AAA protease YME1L and OMA1 cleave OPA1 to balance mitochondrial fusion and fission. J. Cell Biol. 204, 919–929. doi: 10.1083/jcb.201308006
Anderson, C. J., Bredvik, K., Burstein, S. R., Davis, C., Meadows, S. M., Dash, J., et al. (2019). ALS/FTD mutant CHCHD10 mice reveal a tissue-specific toxic gain-of-function and mitochondrial stress response. Acta Neuropathol. 138, 103–121. doi: 10.1007/s00401-019-01989-y
Antico Arciuch, V. G., Elguero, M. E., Poderoso, J. J., and Carreras, M. C. (2012). Mitochondrial regulation of cell cycle and proliferation. Antioxid. Redox. Signal. 16, 1150–1180. doi: 10.1089/ars.2011.4085
Baker, M. J., Lampe, P. A., Stojanovski, D., Korwitz, A., Anand, R., Tatsuta, T., et al. (2014). Stress-induced OMA1 activation and autocatalytic turnover regulate OPA1-dependent mitochondrial dynamics. EMBO J. 33, 578–593. doi: 10.1002/embj.201386474
Ball, N., Teo, W. P., Chandra, S., and Chapman, J. (2019). Parkinson’s disease and the environment. Front. Neurol. 10:218. doi: 10.3389/fneur.2019.00218
Bartolome, F., De La Cueva, M., Pascual, C., Antequera, D., Fernandez, T., Gil, C., et al. (2018). Amyloid beta-induced impairments on mitochondrial dynamics, hippocampal neurogenesis, and memory are restored by phosphodiesterase 7 inhibition. Alzheimers Res. Ther. 10:24. doi: 10.1186/s13195-018-0352-4
Beck, J. S., Mufson, E. J., and Counts, S. E. (2016). Evidence for mitochondrial upr gene activation in familial and sporadic Alzheimer’s disease. Curr. Alzheimer Res. 13, 610–614. doi: 10.2174/1567205013666151221145445
Benedetti, C., Haynes, C. M., Yang, Y., Harding, H. P., and Ron, D. (2006). Ubiquitin-like protein 5 positively regulates chaperone gene expression in the mitochondrial unfolded protein response. Genetics 174, 229–239. doi: 10.1534/genetics.106.061580
Bento, A. C., Bippes, C. C., Kohler, C., Hemion, C., Frank, S., and Neutzner, A. (2018). UBXD1 is a mitochondrial recruitment factor for p97/VCP and promotes mitophagy. Sci. Rep. 8:12415. doi: 10.1038/s41598-018-30963-z
Berthet, A., Margolis, E. B., Zhang, J., Hsieh, I., Zhang, J. S., Hnasko, T. S., et al. (2014). Loss of mitochondrial fission depletes axonal mitochondria in midbrain dopamine neurons. J. Neurosci. 34, 14304–14317. doi: 10.1523/jneurosci.0930-14.2014
Bertram, L., and Tanzi, R. E. (2005). The genetic epidemiology of neurodegenerative disease. J. Clin. Invest. 115, 1449–1457. doi: 10.1172/jci24761
Bido, S., Soria, F. N., Fan, R. Z., Bezard, E., and Tieu, K. (2017). Mitochondrial division inhibitor-1 is neuroprotective in the A53T-alpha-synuclein rat model of Parkinson’s disease. Sci. Rep. 7:7495. doi: 10.1038/s41598-017-07181-0
Billups, B., and Forsythe, I. D. (2002). Presynaptic mitochondrial calcium sequestration influences transmission at mammalian central synapses. J. Neurosci. 22, 5840–5847. doi: 10.1523/JNEUROSCI.22-14-05840.2002
Bingol, B., Tea, J. S., Phu, L., Reichelt, M., Bakalarski, C. E., Song, Q. H., et al. (2014). The mitochondrial deubiquitinase USP30 opposes parkin-mediated mitophagy. Nature 510, 370–375.
Biskup, S., Moore, D. J., Celsi, F., Higashi, S., West, A. B., Andrabi, S. A., et al. (2006). Localization of LRRK2 to membranous and vesicular structures in mammalian brain. Ann. Neurol. 60, 557–569. doi: 10.1002/ana.21019
Blesa, J. R., and Hernandez-Yago, J. (2006). Distinct functional contributions of 2 GABP-NRF-2 recognition sites within the context of the human TOMM70 promoter. Biochem. Cell Biol. 84, 813–822. doi: 10.1139/o06-064
Boecker, C. A., Goldsmith, J., Dou, D., Cajka, G. G., and Holzbaur, E. L. F. (2021). Increased LRRK2 kinase activity alters neuronal autophagy by disrupting the axonal transport of autophagosomes. Curr. Biol. 31:e2146. doi: 10.1016/j.cub.2021.02.061
Bonello, F., Hassoun, S. M., Mouton-Liger, F., Shin, Y. S., Muscat, A., Tesson, C., et al. (2019). LRRK2 impairs PINK1/Parkin-dependent mitophagy via its kinase activity: pathologic insights into Parkinson’s disease. Hum. Mol. Genet. 28, 1645–1660. doi: 10.1093/hmg/ddz004
Bordet, T., Buisson, B., Michaud, M., Drouot, C., Galea, P., Delaage, P., et al. (2007). Identification and characterization of cholest-4-en-3-one, oxime (TRO19622), a novel drug candidate for amyotrophic lateral sclerosis. J. Pharmacol. Exp. Therapeut. 322, 709–720. doi: 10.1124/jpet.107.123000
Bruijn, L. I., Becher, M. W., Lee, M. K., Anderson, K. L., Jenkins, N. A., Copeland, N. G., et al. (1997). ALS-linked SOD1 mutant G85R mediates damage to astrocytes and promotes rapidly progressive disease with SOD1-containing inclusions. Neuron 18, 327–338. doi: 10.1016/s0896-6273(00)80272-x
Bryukhovetskiy, A. S., Grivtsova, L. Y., and Sharma, H. S. (2020). Is the ALS a motor neuron disease or a hematopoietic stem cell disease? Prog. Brain Res. 258, 381–396. doi: 10.1016/bs.pbr.2020.09.005
Bucheli, M. E., Calderon, A., Chicaiza, D., Franco, C., Lopez, R., Digga, E., et al. (2013). Feedback interaction of research, advocacy, and clinical care applied to ALS research in South America. Neurology 81, 1959–1961. doi: 10.1212/01.wnl.0000436610.28210.a5
Bueler, H. (2009). Impaired mitochondrial dynamics and function in the pathogenesis of Parkinson’s disease. Exp. Neurol. 218, 235–246. doi: 10.1016/j.expneurol.2009.03.006
Burbulla, L. F., Fitzgerald, J. C., Stegen, K., Westermeier, J., Thost, A. K., Kato, H., et al. (2014). Mitochondrial proteolytic stress induced by loss of mortalin function is rescued by Parkin and PINK1. Cell Death Dis. 5:e1180. doi: 10.1038/cddis.2014.103
Cai, Y., Shen, H., Weng, H., Wang, Y., Cai, G., Chen, X., et al. (2020). Overexpression of PGC-1alpha influences the mitochondrial unfolded protein response (mtUPR) induced by MPP(+) in human SH-SY5Y neuroblastoma cells. Sci. Rep. 10:10444. doi: 10.1038/s41598-020-67229-6
Calkins, M. J., Manczak, M., Mao, P., Shirendeb, U., and Reddy, P. H. (2011). Impaired mitochondrial biogenesis, defective axonal transport of mitochondria, abnormal mitochondrial dynamics and synaptic degeneration in a mouse model of Alzheimer’s disease. Hum. Mol. Genet. 20, 4515–4529. doi: 10.1093/hmg/ddr381
Calvo, S. E., and Mootha, V. K. (2010). The mitochondrial proteome and human disease. Ann. Rev. Genom. Hum. Genet. 11, 25–44.
Cardanho-Ramos, C., Faria-Pereira, A., and Morais, V. A. (2020). Orchestrating mitochondria in neurons: cytoskeleton as the conductor. Cytoskeleton 77, 65–75. doi: 10.1002/cm.21585
Castellazzi, M., Patergnani, S., Donadio, M., Giorgi, C., Bonora, M., Bosi, C., et al. (2019). Autophagy and mitophagy biomarkers are reduced in sera of patients with Alzheimer’s disease and mild cognitive impairment. Sci. Rep. 9:20009. doi: 10.1038/s41598-019-56614-5
Cenini, G., Rub, C., Bruderek, M., and Voos, W. (2016). Amyloid beta-peptides interfere with mitochondrial preprotein import competence by a coaggregation process. Mol. Biol. Cell 27, 3257–3272. doi: 10.1091/mbc.E16-05-0313
Chan, D. C. (2020). Mitochondrial dynamics and its involvement in disease. Annu. Rev. Pathol. 15, 235–259. doi: 10.1146/annurev-pathmechdis-012419-032711
Chang, D. T. W., Rintoula, G. L., Pandipati, S., and Reynolds, I. J. (2006). Mutant huntingtin aggregates impair mitochondrial movement and trafficking in cortical neurons. Neurobiol. Dis. 22, 388–400. doi: 10.1016/j.nbd.2005.12.007
Chen, Y. B., Deng, J. W., Wang, P., Yang, M. X., Chen, X. P., Zhu, L., et al. (2016). PINK1 and Parkin are genetic modifiers for FUS-induced neurodegeneration. Hum. Mol. Genet. 25, 5059–5068. doi: 10.1093/hmg/ddw310
Cherubini, M., Lopez-Molina, L., and Gines, S. (2020). Mitochondrial fission in Huntington’s disease mouse striatum disrupts ER-mitochondria contacts leading to disturbances in Ca(2+) efflux and reactive oxygen species (ROS) homeostasis. Neurobiol. Dis. 136:104741. doi: 10.1016/j.nbd.2020.104741
Cho, D. H., Nakamura, T., Fang, J. G., Cieplak, P., Godzik, A., Gu, Z., et al. (2009). S-Nitrosylation of Drp1 mediates beta-Amyloid-Related mitochondrial fission and neuronal injury. Science 324, 102–105. doi: 10.1126/science.1171091
Choi, S. Y., Lee, J. H., Chung, A. Y., Jo, Y., Shin, J. H., Park, H. C., et al. (2020). Prevention of mitochondrial impairment by inhibition of protein phosphatase 1 activity in amyotrophic lateral sclerosis. Cell Death Dis. 11:888. doi: 10.1038/s41419-020-03102-8
Choubey, V., Safiulina, D., Vaarmann, A., Cagalinec, M., Wareski, P., Kuum, M., et al. (2011). Mutant A53T alpha-synuclein induces neuronal death by increasing mitochondrial autophagy. J. Biol. Chem. 286, 10814–10824. doi: 10.1074/jbc.m110.132514
Cipolat, S., Martins De Brito, O., Dal Zilio, B., and Scorrano, L. (2004). OPA1 requires mitofusin 1 to promote mitochondrial fusion. Proc. Natl. Acad. Sci. U S A. 101, 15927–15932. doi: 10.1073/pnas.0407043101
Clark, L. N., Wang, Y., Karlins, E., Saito, L., Mejia-Santana, H., Harris, J., et al. (2006). Frequency of LRRK2 mutations in early- and late-onset Parkinson disease. Neurology 67, 1786–1791. doi: 10.1212/01.wnl.0000244345.49809.36
Coffey, E. E., Beckel, J. M., Laties, A. M., and Mitchell, C. H. (2014). Lysosomal alkalization and dysfunction in human fibroblasts with the Alzheimer’s disease-linked presenilin 1 A246e mutation can be reversed with camp. Neuroscience 263, 111–124. doi: 10.1016/j.neuroscience.2014.01.001
Cooper, J. F., Machiela, E., Dues, D. J., Spielbauer, K. K., Senchuk, M. M., and Van Raamsdonk, J. M. (2017). Activation of the mitochondrial unfolded protein response promotes longevity and dopamine neuron survival in Parkinson’s disease models. Sci. Rep. 7:16441. doi: 10.1038/s41598-017-16637-2
Cooper-Knock, J., Hewitt, C., Highley, J. R., Brockington, A., Milano, A., Man, S., et al. (2012). Clinico-pathological features in amyotrophic lateral sclerosis with expansions in C9ORF72. Brain 135, 751–764.
Couvillion, M. T., Soto, I. C., Shipkovenska, G., and Churchman, L. S. (2016). Synchronized mitochondrial and cytosolic translation programs. Nature 533, 499–503. doi: 10.1038/nature18015
Crews, L., Spencer, B., Desplats, P., Patrick, C., Paulino, A., Rockenstein, E., et al. (2010). Selective molecular alterations in the autophagy pathway in patients with Lewy body disease and in models of alpha-synucleinopathy. PLoS One 5:e9313. doi: 10.1371/journal.pone.0009313
Cui, L., Jeong, H., Borovecki, F., Parkhurst, C. N., Tanese, N., and Krainc, D. (2006). Transcriptional repression of PGC-1alpha by mutant huntingtin leads to mitochondrial dysfunction and neurodegeneration. Cell 127, 59–69. doi: 10.1016/j.cell.2006.09.015
Cummins, N., Tweedie, A., Zuryn, S., Bertran-Gonzalez, J., and Gotz, J. (2019). Disease-associated tau impairs mitophagy by inhibiting Parkin translocation to mitochondria. EMBO J. 38:e99360. doi: 10.15252/embj.201899360
Da Cruz, S., Parone, P. A., Lopes, V. S., Lillo, C., Mcalonis-Downes, M., Lee, S. K., et al. (2012). Elevated PGC-1 alpha activity sustains mitochondrial biogenesis and muscle function without extending survival in a mouse model of inherited ALS. Cell Metab. 15, 778–786. doi: 10.1016/j.cmet.2012.03.019
Deng, J. W., Yang, M. X., Chen, Y. B., Chen, X. P., Liu, J. H., Sun, S. F., et al. (2015). FUS interacts with HSP60 to promote mitochondrial damage. PLoS Genetics 11:e1005357. doi: 10.1371/journal.pgen.1005357
Devi, L., Prabhu, B. M., Galati, D. F., Avadhani, N. G., and Anandatheerthavarada, H. K. (2006). Accumulation of amyloid precursor protein in the mitochondrial import channels of human Alzheimer’s disease brain is associated with mitochondrial dysfunction. J. Neurosci. 26, 9057–9068. doi: 10.1523/JNEUROSCI.1469-06.2006
Eberhardt, E. L., Ludlam, A. V., Tan, Z. Y., and Cianfrocco, M. A. (2020). Miro: a molecular switch at the center of mitochondrial regulation. Protein Sci. 29, 1269–1284. doi: 10.1002/pro.3839
Ebneth, A., Godemann, R., Stamer, K., Illenberger, S., Trinczek, B., Mandelkow, E. M., et al. (1998). Overexpression of tau protein inhibits kinesin-dependent trafficking of vesicles, mitochondria, and endoplasmic reticulum: implications for Alzheimer’s disease. J. Cell Biol. 143, 777–794. doi: 10.1083/jcb.143.3.777
Ehrlich, M. E. (2012). Huntington’s disease and the striatal medium spiny neuron: cell-autonomous and non-cell-autonomous mechanisms of disease. Neurotherapeutics 9, 270–284. doi: 10.1007/s13311-012-0112-2
Ehrnhoefer, D. E., Martin, D. D. O., Schmidt, M. E., Qiu, X., Ladha, S., Caron, N. S., et al. (2018). Preventing mutant huntingtin proteolysis and intermittent fasting promote autophagy in models of Huntington disease. Acta Neuropathol. Commun. 6:16. doi: 10.1186/s40478-018-0518-0
Esselun, C., Theyssen, E., and Eckert, G. P. (2021). Effects of urolithin a on mitochondrial parameters in a cellular model of early Alzheimer disease. Int. J. Mol. Sci. 22:8333. doi: 10.3390/ijms22158333
Evgen’ev, M., Bobkova, N., Krasnov, G., Garbuz, D., Funikov, S., Kudryavtseva, A., et al. (2019). The effect of human HSP70 administration on a mouse model of Alzheimer’s disease strongly depends on transgenicity and age. J. Alzheimers. Dis. 67, 1391–1404. doi: 10.3233/JAD-180987
Fang, E. F., Hou, Y., Palikaras, K., Adriaanse, B. A., Kerr, J. S., Yang, B., et al. (2019). Mitophagy inhibits amyloid-beta and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nat. Neurosci. 22, 401–412. doi: 10.1038/s41593-018-0332-9
Fang, L., Hemion, C., Bento, A. C. P. F., Bippes, C. C., Flammer, J., and Neutzner, A. (2015). Mitochondrial function in neuronal cells depends on p97/VCP/Cdc48-mediated quality control. Front. Cell. Neurosci. 9:16. doi: 10.3389/fncel.2015.00016
Faustini, G., Marchesan, E., Zonta, L., Bono, F., Bottani, E., Longhena, F., et al. (2019). Alpha-Synuclein preserves mitochondrial fusion and function in neuronal cells. Oxid. Med. Cell Longev. 2019:4246350.
Fessler, E., Eckl, E. M., Schmitt, S., Mancilla, I. A., Meyer-Bender, M. F., Hanf, M., et al. (2020). A pathway coordinated by DELE1 relays mitochondrial stress to the cytosol. Nature 579, 433–434. doi: 10.1038/s41586-020-2076-4
Filichia, E., Hoffer, B., Qi, X., and Luo, Y. (2016). Inhibition of Drp1 mitochondrial translocation provides neural protection in dopaminergic system in a Parkinson’s disease model induced by MPTP. Sci. Rep. 6:32656. doi: 10.1038/srep32656
Fiorese, C. J., and Haynes, C. M. (2017). Integrating the UPRmt into the mitochondrial maintenance network. Crit. Rev. Biochem. Mol. Biol. 52, 304–313. doi: 10.1080/10409238.2017.1291577
Fiorese, C. J., Schulz, A. M., Lin, Y. F., Rosin, N., Pellegrino, M. W., and Haynes, C. M. (2016). The transcription factor ATF5 mediates a mammalian mitochondrial UPR. Curr. Biol. 26, 2037–2043. doi: 10.1016/j.cub.2016.06.002
Franco-Iborra, S., Plaza-Zabala, A., Montpeyo, M., Sebastian, D., Vila, M., and Martinez-Vicente, M. (2021). Mutant HTT (huntingtin) impairs mitophagy in a cellular model of Huntington disease. Autophagy 17, 672–689. doi: 10.1080/15548627.2020.1728096
Friedman, J. R., Lackner, L. L., West, M., Dibenedetto, J. R., Nunnari, J., and Voeltz, G. K. (2011). ER tubules mark sites of mitochondrial division. Science 334, 358–362. doi: 10.1126/science.1207385
Fu, Z., Liu, F., Liu, C., Jin, B., Jiang, Y., Tang, M., et al. (2019). Mutant huntingtin inhibits the mitochondrial unfolded protein response by impairing ABCB10 mRNA stability. Biochim Biophys. Acta Mol. Basis Dis. 1865, 1428–1435. doi: 10.1016/j.bbadis.2019.02.015
Gammon, K. (2014). Neurodegenerative disease: brain windfall. Nature 515, 299–300. doi: 10.1038/nj7526-299a
Gao, S., and Hu, J. (2021). Mitochondrial fusion: the machineries in and out. Trends Cell Biol. 31, 62–74. doi: 10.1016/j.tcb.2020.09.008
GBD 2015 Neurological Disorders Collaborator Group (2017). Global, regional, and national burden of neurological disorders during 1990-2015: a systematic analysis for the Global Burden of Disease Study 2015. Lancet Neurol. 16, 877–897.
Gegg, M. E., Cooper, J. M., Chau, K. Y., Rojo, M., Schapira, A. H. V., and Taanman, J. W. (2013). Mitofusin 1 and mitofusin 2 are ubiquitinated in a PINK1/parkin-dependent manner upon induction of mitophagy (vol 19, pg 4861, 2010). Hum. Mol. Genet. 22, 1697–1697. doi: 10.1093/hmg/ddq419
Geisler, S., Holmstrom, K. M., Skujat, D., Fiesel, F. C., Rothfuss, O. C., Kahle, P. J., et al. (2010). PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat. Cell Biol. 12, 119–U170. doi: 10.1038/ncb2012
Genin, E. C., Plutino, M., Bannwarth, S., Villa, E., Cisneros-Barroso, E., Roy, M., et al. (2016). CHCHD10 mutations promote loss of mitochondrial cristae junctions with impaired mitochondrial genome maintenance and inhibition of apoptosis. EMBO Mol. Med. 8, 58–72. doi: 10.15252/emmm.201505496
Giacomello, M., Pyakurel, A., Glytsou, C., and Scorrano, L. (2020). The cell biology of mitochondrial membrane dynamics. Nat. Rev. Mol. Cell Biol. 21, 204–224. doi: 10.1038/s41580-020-0210-7
Gilkerson, R., De La Torre, P., and Vallier, S. S. (2021). Mitochondrial OMA1 and OPA1 as gatekeepers of organellar structure/function and cellular stress response. Front. Cell Dev. Biol. 9:626117. doi: 10.3389/fcell.2021.626117
Gilmozzi, V., Gentile, G., Rueda, M. P. C., Hicks, A. A., Pramstaller, P. P., Zanon, A., et al. (2020). Interaction of alpha-synuclein with lipids: mitochondrial cardiolipin as a critical player in the pathogenesis of Parkinson’s disease. Front. Neurosci. 14:578993. doi: 10.3389/fnins.2020.578993
Giorgi, C., Marchi, S., and Pinton, P. (2018). The machineries, regulation and cellular functions of mitochondrial calcium. Nat. Rev. Mol. Cell Biol. 19, 713–730. doi: 10.1038/s41580-018-0052-8
Godena, V. K., Brookes-Hockookes-Hocking, N., Moller, A., Shaw, G., Oswald, M., Sancho, R. M., et al. (2014). Increasing microtubule acetylation rescues axonal transport and locomotor deficits caused by LRRK2 Roc-COR domain mutations. Nat. Commun. 5:5245. doi: 10.1038/ncomms6245
Gomes, A. P., Price, N. L., Ling, A. J., Moslehi, J. J., Montgomery, M. K., Rajman, L., et al. (2013). Declining NAD(+) induces a pseudohypoxic state disrupting nuclear-mitochondrial communication during aging. Cell 155, 1624–1638. doi: 10.1016/j.cell.2013.11.037
Gong, B., Pan, Y., Vempati, P., Zhao, W., Knable, L., Ho, L., et al. (2013). Nicotinamide riboside restores cognition through an upregulation of proliferator-activated receptor-gamma coactivator 1 alpha regulated beta-secretase 1 degradation and mitochondrial gene expression in Alzheimer’s mouse models. Neurobiol. Aging 34, 1581–1588. doi: 10.1016/j.neurobiolaging.2012.12.005
Grassi, D., Howard, S., Zhou, M., Diaz-Perez, N., Urban, N. T., Guerrero-Given, D., et al. (2018). Identification of a highly neurotoxic alpha-synuclein species inducing mitochondrial damage and mitophagy in Parkinson’s disease. Proc. Natl. Acad. Sci. U S A. 115, E2634–E2643. doi: 10.1073/pnas.1713849115
Greene, A. W., Grenier, K., Aguileta, M. A., Muise, S., Farazifard, R., Haque, M. E., et al. (2012). Mitochondrial processing peptidase regulates PINK1 processing, import and Parkin recruitment. EMBO Rep. 13, 378–385. doi: 10.1038/embor.2012.14
Grohm, J., Kim, S. W., Mamrak, U., Tobaben, S., Cassidy-Stone, A., Nunnari, J., et al. (2012). Inhibition of Drp1 provides neuroprotection in vitro and in vivo. Cell Death. Differ. 19, 1446–1458. doi: 10.1038/cdd.2012.18
Gu, J., Li, Z., Chen, H., Xu, X., Li, Y., and Gui, Y. (2021). Neuroprotective effect of trans-resveratrol in mild to moderate Alzheimer disease: a randomized, double-blind trial. Neurol. Ther. Online ahead of print. doi: 10.1007/s40120-021-00271-2
Guardia-Laguarta, C., Area-Gomez, E., Rub, C., Liu, Y. H., Magrane, J., Becker, D., et al. (2014). alpha-Synuclein is localized to mitochondria-associated ER membranes. J. Neurosci. 34, 249–259. doi: 10.1523/jneurosci.2507-13.2014
Gui, C., Ren, Y., Chen, J., Wu, X., Mao, K., Li, H., et al. (2020). p38 MAPK-DRP1 signaling is involved in mitochondrial dysfunction and cell death in mutant A53T alpha-synuclein model of Parkinson’s disease. Toxicol. Appl. Pharmacol. 388:114874. doi: 10.1016/j.taap.2019.114874
Guo, L., Du, H., Yan, S., Wu, X., Mckhann, G. M., Chen, J. X., et al. (2013). Cyclophilin D deficiency rescues axonal mitochondrial transport in Alzheimer’s neurons. PLoS One 8:e54914. doi: 10.1371/journal.pone.0054914
Guo, X., Disatnik, M. H., Monbureau, M., Shamloo, M., Mochly-Rosen, D., and Qi, X. (2013). Inhibition of mitochondrial fragmentation diminishes Huntington’s disease-associated neurodegeneration. J. Clin. Invest. 123, 5371–5388. doi: 10.1172/JCI70911
Guo, X., Sun, X., Hu, D., Wang, Y. J., Fujioka, H., Vyas, R., et al. (2016). VCP recruitment to mitochondria causes mitophagy impairment and neurodegeneration in models of Huntington’s disease. Nat. Commun. 7:12646. doi: 10.1038/ncomms12646
Guo, X. Y., Aviles, G., Liu, Y., Tian, R. L., Unger, B. A., Lin, Y. H. T., et al. (2020). Mitochondrial stress is relayed to the cytosol by an OMA1-DELE1-HRI pathway. Nature 579, 427–432. doi: 10.1038/s41586-020-2078-2
Haeussler, S., Kohler, F., Witting, M., Premm, M. F., Rolland, S. G., Fischer, C., et al. (2020). Autophagy compensates for defects in mitochondrial dynamics. PLoS Genet 16:e1008638. doi: 10.1371/journal.pgen.1008638
Ham, S. J., Lee, D., Yoo, H., Jun, K., Shin, H., and Chung, J. (2020). Decision between mitophagy and apoptosis by Parkin via VDAC1 ubiquitination. Proc. Natl. Acad. Sci. U S A. 117, 4281–4291. doi: 10.1073/pnas.1909814117
Haque, R. U., and Levey, A. I. (2019). Alzheimer’s disease: a clinical perspective and future nonhuman primate research opportunities. Proc. Natl. Acad. Sci. U S A. 116, 26224–26229. doi: 10.1073/pnas.1912954116
Harding, O., Evans, C. S., Ye, J. Q., Cheung, J., Maniatis, T., and Holzbaur, E. L. F. (2021). ALS- and FTD-associated missense mutations in TBK1 differentially disrupt mitophagy. Proc. Natl. Acad. Sci. U S A. 118:e2025053118. doi: 10.1073/pnas.2025053118
Harjuhaahto, S., Rasila, T. S., Molchanova, S. M., Woldegebriel, R., Kvist, J., Konovalova, S., et al. (2020). ALS and Parkinson’s disease genes CHCHD10 and CHCHD2 modify synaptic transcriptomes in human iPSC-derived motor neurons. Neurobiol. Dis. 141:104940. doi: 10.1016/j.nbd.2020.104940
Harper, J. W., Ordureau, A., and Heo, J. M. (2018). Building and decoding ubiquitin chains for mitophagy. Nat. Rev. Mol. Cell Biol. 19, 93–108. doi: 10.1038/nrm.2017.129
Haynes, C. M., Petrova, K., Benedetti, C., Yang, Y., and Ron, D. (2007). ClpP mediates activation of a mitochondrial unfolded protein response in C-elegans. Dev. Cell 13, 467–480. doi: 10.1016/j.devcel.2007.07.016
Hertz, N. T., Berthet, A., Sos, M. L., Thorn, K. S., Burlingame, A. L., Nakamura, K., et al. (2013). A Neo-substrate that amplifies catalytic activity of Parkinson’s-disease-related kinase PINK1. Cell 154, 737–747. doi: 10.1016/j.cell.2013.07.030
Hong, X., Liu, J., Zhu, G., Zhuang, Y., Suo, H., Wang, P., et al. (2014). Parkin overexpression ameliorates hippocampal long-term potentiation and beta-amyloid load in an Alzheimer’s disease mouse model. Hum. Mol. Genet. 23, 1056–1072. doi: 10.1093/hmg/ddt501
Horibe, T., and Hoogenraad, N. J. (2007). The chop gene contains an element for the positive regulation of the mitochondrial unfolded protein response. PLoS One 2:e835. doi: 10.1371/journal.pone.0000835
Houtkooper, R. H., Mouchiroud, L., Ryu, D., Moullan, N., Katsyuba, E., Knott, G., et al. (2013). Mitonuclear protein imbalance as a conserved longevity mechanism. Nature 497, 451–457. doi: 10.1038/nature12188
Hsieh, C. H., Li, L., Vanhauwaert, R., Nguyen, K. T., Davis, M. D., Bu, G., et al. (2019). Miro1 marks Parkinson’s disease subset and miro1 reducer rescues neuron loss in Parkinson’s models. Cell Metab. 30:e1137. doi: 10.1016/j.cmet.2019.08.023
Hsieh, C. H., Shaltouki, A., Gonzalez, A. E., Da Cruz, A. B., Burbulla, L. F., St Lawrence, E., et al. (2016). Functional impairment in miro degradation and mitophagy is a shared feature in familial and sporadic Parkinson’s disease. Cell Stem Cell 19, 709–724. doi: 10.1016/j.stem.2016.08.002
Hu, D., Sun, X., Magpusao, A., Fedorov, Y., Thompson, M., Wang, B., et al. (2021b). Small-molecule suppression of calpastatin degradation reduces neuropathology in models of Huntington’s disease. Nat. Commun. 12:5305. doi: 10.1038/s41467-021-25651-y
Hu, D., Liu, Z., and Qi, X. (2021a). UPR(mt) activation protects against MPP(+)-induced toxicity in a cell culture model of Parkinson’s disease. Biochem. Biophys. Res. Commun. 569, 17–22. doi: 10.1016/j.bbrc.2021.06.079
Hu, D., Sun, X., Liao, X., Zhang, X., Zarabi, S., Schimmer, A., et al. (2019). Alpha-synuclein suppresses mitochondrial protease ClpP to trigger mitochondrial oxidative damage and neurotoxicity. Acta Neuropathol. 137, 939–960. doi: 10.1007/s00401-019-01993-2
Hwang, S., Disatnik, M. H., and Mochly-Rosen, D. (2015). Impaired GAPDH-induced mitophagy contributes to the pathology of Huntington’s disease. EMBO Mol. Me 7, 1307–1326. doi: 10.15252/emmm.201505256
Jagmag, S. A., Tripathi, N., Shukla, S. D., Maiti, S., and Khurana, S. (2015). Evaluation of models of parkinson’s disease. Front. Neurosci. 9:503. doi: 10.3389/fnins.2015.00503
Jahani-Asl, A., Huang, E., Irrcher, I., Rashidian, J., Ishihara, N., Lagace, D. C., et al. (2015). CDK5 phosphorylates DRP1 and drives mitochondrial defects in NMDA-induced neuronal death. Hum. Mol. Genet. 24, 4573–4583. doi: 10.1093/hmg/ddv188
Jellinger, K. A. (2010). Basic mechanisms of neurodegeneration: a critical update. J. Cell Mol. Med. 14, 457–487.
Jia, Y. L., Sun, S. J., Chen, J. H., Jia, Q., Huo, T. T., Chu, L. F., et al. (2016). SS31, a small molecule antioxidant peptide, attenuates beta-amyloid elevation, mitochondrial/synaptic deterioration and cognitive deficit in SAMP8 mice. Curr. Alzheimer Res. 13, 297–306. doi: 10.2174/1567205013666151218150004
Jin, S. M., and Youle, R. J. (2013). The accumulation of misfolded proteins in the mitochondrial matrix is sensed by PINK1 to induce PARK2/Parkin-mediated mitophagy of polarized mitochondria. Autophagy 9, 1750–1757. doi: 10.4161/auto.26122
Johnson, J. O., Mandrioli, J., Benatar, M., Abramzon, Y., Van Deerlin, V. M., Trojanowski, J. Q., et al. (2011). Exome sequencing reveals VCP mutations as a cause of familial ALS. Neuron 69, 397–397.
Johri, A., and Beal, M. F. (2012). Mitochondrial dysfunction in neurodegenerative diseases. J. Pharmacol. Exp. Ther. 342, 619–630.
Johri, A., Calingasan, N. Y., Hennessey, T. M., Sharma, A., Yang, L., Wille, E., et al. (2012). Pharmacologic activation of mitochondrial biogenesis exerts widespread beneficial effects in a transgenic mouse model of Huntington’s disease. Hum. Mol. Genet. 21, 1124–1137. doi: 10.1093/hmg/ddr541
Joshi, A. U., Saw, N. L., Shamloo, M., and Mochly-Rosen, D. (2018a). Drp1/Fis1 interaction mediates mitochondrial dysfunction, bioenergetic failure and cognitive decline in Alzheimer’s disease. Oncotarget 9, 6128–6143. doi: 10.18632/oncotarget.23640
Joshi, A. U., Saw, N. L., Vogel, H., Cunnigham, A. D., Shamloo, M., and Mochly-Rosen, D. (2018b). Inhibition of Drp1/Fis1 interaction slows progression of amyotrophic lateral sclerosis. EMBO Mol. Med. 10:e8166. doi: 10.15252/emmm.201708166
Kakizuka, A. (2008). Roles of VCP in human neurodegenerative disorders. Biochem. Soc. Trans. 36, 105–108. doi: 10.1042/bst0360105
Kalia, R., Wang, R. Y., Yusuf, A., Thomas, P. V., Agard, D. A., Shaw, J. M., et al. (2018). Structural basis of mitochondrial receptor binding and constriction by DRP1. Nature 558, 401–405. doi: 10.1038/s41586-018-0211-2
Kamp, F., Exner, N., Lutz, A. K., Wender, N., Hegermann, J., Brunner, B., et al. (2010). Inhibition of mitochondrial fusion by alpha-synuclein is rescued by PINK1. Parkin and DJ-1. EMBO J. 29, 3571–3589. doi: 10.1038/emboj.2010.223
Kane, L. A., Lazarou, M., Fogel, A. I., Li, Y., Yamano, K., Sarraf, S. A., et al. (2014). PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J. Cell Biol. 205, 143–153. doi: 10.1083/jcb.201402104
Kaspar, S., Oertlin, C., Szczepanowska, K., Kukat, A., Senft, K., Lucas, C., et al. (2021). Adaptation to mitochondrial stress requires CHOP-directed tuning of ISR. Sci. Adv. 7:eabf0971. doi: 10.1126/sciadv.abf0971
Kaufmann, P., Thompson, J. L., Levy, G., Buchsbaum, R., Shefner, J., Krivickas, L. S., et al. (2009). Phase II trial of CoQ10 for ALS finds insufficient evidence to justify phase III. Ann. Neurol. 66, 235–244. doi: 10.1002/ana.21743
Kazlauskaite, A., and Muqit, M. M. K. (2015). PINK1 and Parkin - mitochondrial interplay between phosphorylation and ubiquitylation in Parkinson’s disease. FEBS J. 282, 215–223. doi: 10.1111/febs.13127
Kenny, T. C., Hart, P., Ragazzi, M., Sersinghe, M., Chipuk, J., Sagar, M. A. K., et al. (2017). Selected mitochondrial DNA landscapes activate the SIRT3 axis of the UPRmt to promote metastasis. Oncogene 36, 4393–4404. doi: 10.1038/onc.2017.52
Kerschbamer, E., and Biagioli, M. (2016). Huntington’s disease as neurodevelopmental disorder: altered chromatin regulation, coding, and non-coding RNA transcription. Front. Neurosci. 9:509. doi: 10.3389/fnins.2015.00509
Kett, L. R., Boassa, D., Ho, C. C., Rideout, H. J., Hu, J., Terada, M., et al. (2012). LRRK2 Parkinson disease mutations enhance its microtubule association. Hum. Mol. Genet. 21, 890–899. doi: 10.1093/hmg/ddr526
Khalil, B., Cabirol-Pol, M. J., Miguel, L., Whitworth, A. J., Lecourtois, M., and Lievens, J. C. (2017). Enhancing Mitofusin/Marf ameliorates neuromuscular dysfunction in Drosophila models of TDP-43 proteinopathies. Neurobiol. Aging 54, 71–83. doi: 10.1016/j.neurobiolaging.2017.02.016
Khalil, B., El Fissi, N., Aouane, A., Cabirol-Pol, M. J., Rival, T., and Lievens, J. C. (2015). PINK1-induced mitophagy promotes neuroprotection in Huntington’s disease. Cell Death Dis. 6:e1617. doi: 10.1038/cddis.2014.581
Khalil, B., and Lievens, J. C. (2017). Mitochondrial quality control in amyotrophic lateral sclerosis: towards a common pathway? Neural Regen. Res. 12, 1052–1061. doi: 10.4103/1673-5374.211179
Kim, H., Lee, J. Y., Park, K. J., Kim, W. H., and Roh, G. S. (2017). A mitochondrial division inhibitor, Mdivi-1, inhibits mitochondrial fragmentation and attenuates kainic acid-induced hippocampal cell death. BMC Neurosci. 17:33. doi: 10.1186/s12868-016-0270-y
Kim-Han, J. S., Antenor-Dorsey, J. A., and O’malley, K. L. (2011). The Parkinsonian mimetic, MPP+, specifically impairs mitochondrial transport in dopamine axons. J. Neurosci. 31, 7212–7221. doi: 10.1523/JNEUROSCI.0711-11.2011
Kleele, T., Rey, T., Winter, J., Zaganelli, S., Mahecic, D., Perreten Lambert, H., et al. (2021). Distinct fission signatures predict mitochondrial degradation or biogenesis. Nature 593, 435–439. doi: 10.1038/s41586-021-03510-6
Korobova, F., Ramabhadran, V., and Higgs, H. N. (2013). An actin-dependent step in mitochondrial fission mediated by the ER-associated formin INF2. Science 339, 464–467. doi: 10.1126/science.1228360
Kraus, F., and Ryan, M. T. (2017). The constriction and scission machineries involved in mitochondrial fission. J. Cell Sci. 130, 2953–2960. doi: 10.1242/jcs.199562
Kuruva, C. S., Manczak, M., Yin, X., Ogunmokun, G., Reddy, A. P., and Reddy, P. H. (2017). Aqua-soluble DDQ reduces the levels of Drp1 and Abeta and inhibits abnormal interactions between Abeta and Drp1 and protects Alzheimer’s disease neurons from Abeta- and Drp1-induced mitochondrial and synaptic toxicities. Hum. Mol. Genet. 26, 3375–3395. doi: 10.1093/hmg/ddx226
Laudati, G., Mascolo, L., Guida, N., Sirabella, R., Pizzorusso, V., Bruzzaniti, S., et al. (2019). Resveratrol treatment reduces the vulnerability of SH-SY5Y cells and cortical neurons overexpressing SOD1-G93A to Thimerosal toxicity through SIRT1/DREAM/PDYN pathway. Neurotoxicology 71, 6–15. doi: 10.1016/j.neuro.2018.11.009
Lazarou, M., Jin, S. M., Kane, L. A., and Youle, R. J. (2012). Role of PINK1 binding to the TOM complex and alternate intracellular membranes in recruitment and activation of the E3 ligase parkin. Dev. Cell 22, 320–333. doi: 10.1016/j.devcel.2011.12.014
Lee, C. A., Chin, L. S., and Li, L. (2018). Hypertonia-linked protein Trak1 functions with mitofusins to promote mitochondrial tethering and fusion. Protein Cell 9, 693–716. doi: 10.1007/s13238-017-0469-4
Lee, J., Kim, Y., Liu, T., Hwang, Y. J., Hyeon, S. J., Im, H., et al. (2018). SIRT3 deregulation is linked to mitochondrial dysfunction in Alzheimer’s disease. Aging Cell 17, e12679. doi: 10.1111/acel.12679
Lee, K. S., Huh, S., Lee, S., Wu, Z., Kim, A. K., Kang, H. Y., et al. (2018). Altered ER-mitochondria contact impacts mitochondria calcium homeostasis and contributes to neurodegeneration in vivo in disease models. Proc. Natl. Acad. Sci. U S A. 115, E8844–E8853.
Lee, H., Noh, J. Y., Oh, Y., Kim, Y., Chang, J. W., Chung, C. W., et al. (2012). IRE1 plays an essential role in ER stress-mediated aggregation of mutant huntingtin via the inhibition of autophagy flux. Hum. Mol. Genet. 21, 101–114. doi: 10.1093/hmg/ddr445
Leskela, S., Hoffmann, D., Rostalski, H., Huber, N., Wittrahm, R., Hartikainen, P., et al. (2021). FTLD patient-derived fibroblasts show defective mitochondrial function and accumulation of p62. Mol. Neurobiol. Online ahead of print. doi: 10.1007/s12035-021-02475-x
Lewis, S. C., Uchiyama, L. F., and Nunnari, J. (2016). ER-mitochondria contacts couple mtDNA synthesis with mitochondrial division in human cells. Science 353:aaf5549. doi: 10.1126/science.aaf5549
Li, F. X., Xie, X. Q., Wang, Y. L., Liu, J. P., Cheng, X. F., Guo, Y. J., et al. (2016). Structural insights into the interaction and disease mechanism of neurodegenerative disease-associated optineurin and TBK1 proteins. Nat. Commun. 7:12708.
Li, J., Uversky, V. N., and Fink, A. L. (2001). Effect of familial Parkinson’s disease point mutations A30P and A53T on the structural properties, aggregation, and fibrillation of human alpha-synuclein. Biochemistry 40, 11604–11613. doi: 10.1021/bi010616g
Li, L., Nadanaciva, S., Berger, Z., Shen, W., Paumier, K., Schwartz, J., et al. (2013). Human A53T alpha-synuclein causes reversible deficits in mitochondrial function and dynamics in primary mouse cortical neurons. PLoS One 8:e85815. doi: 10.1371/journal.pone.0085815
Li, R. R., and Chen, J. Z. (2019). Salidroside protects dopaminergic neurons by enhancing PINK1/Parkin-Mediated mitophagy. Oxid. Med. Cell. Longev. 2019:9341018. doi: 10.1155/2019/9341018
Liao, P. C., Wolken, D. M. A., Serrano, E., Srivastava, P., and Pon, L. A. (2020). Mitochondria-Associated Degradation Pathway (MAD) function beyond the outer membrane. Cell Rep. 32:107902. doi: 10.1016/j.celrep.2020.107902
Lin, M. Y., Cheng, X. T., Tammineni, P., Xie, Y., Zhou, B., Cai, Q., et al. (2017). Releasing syntaphilin removes stressed mitochondria from axons independent of mitophagy under pathophysiological conditions. Neuron 94:e596.
Lin, Y. F., Schulz, A. M., Pellegrino, M. W., Lu, Y., Shaham, S., and Haynes, C. M. (2016). Maintenance and propagation of a deleterious mitochondrial genome by the mitochondrial unfolded protein response. Nature 533, 416–419. doi: 10.1038/nature17989
Liu, J., Liu, W. J., Li, R. L., and Yang, H. (2019). Mitophagy in Parkinson’s disease: from pathogenesis to treatment. Cells 8:712.
Liu, M., Yu, S., Wang, J., Qiao, J., Liu, Y., Wang, S., et al. (2020). Ginseng protein protects against mitochondrial dysfunction and neurodegeneration by inducing mitochondrial unfolded protein response in Drosophila melanogaster PINK1 model of Parkinson’s disease. J. Ethnopharmacol. 247:112213. doi: 10.1016/j.jep.2019.112213
Liu, Y., Lear, T. B., Verma, M., Wang, K. Z., Otero, P. A., Mckelvey, A. C., et al. (2020). Chemical inhibition of FBXO7 reduces inflammation and confers neuroprotection by stabilizing the mitochondrial kinase PINK1. JCI Insight 5:e131834. doi: 10.1172/jci.insight.131834
Loson, O. C., Song, Z. Y., Chen, H. C., and Chan, D. C. (2013). Fis1, Mff, MiD49, and MiD51 mediate Drp1 recruitment in mitochondrial fission. Mol. Biol. Cell 24, 659–667. doi: 10.1091/mbc.e12-10-0721
Loss, O., and Stephenson, F. A. (2017). Developmental changes in trak-mediated mitochondrial transport in neurons. Mol. Cell. Neurosci. 80, 134–147. doi: 10.1016/j.mcn.2017.03.006
Lucking, C. B., Durr, A., Bonifati, V., Vaughan, J., De Michele, G., Gasser, T., et al. (2000). Association between early-onset Parkinson’s disease and mutations in the parkin gene. N. Engl. J. Med. 342, 1560–1567. doi: 10.1056/NEJM200005253422103
Luo, G., Yi, J. X., Ma, C. L., Xiao, Y. J., Yi, F., Yu, T., et al. (2013). Defective mitochondrial dynamics is an early event in skeletal muscle of an amyotrophic lateral sclerosis mouse model. PLoS One 8:e82112. doi: 10.1371/journal.pone.0082112
Luo, H. K., Krigman, J., Zhang, R. H., Yang, M. C., and Sun, N. (2021). Pharmacological inhibition of USP30 activates tissue-specific mitophagy. Acta Physiol. 232:e13666. doi: 10.1111/apha.13666
Magrane, J., Cortez, C., Gan, W. B., and Manfredi, G. (2014). Abnormal mitochondrial transport and morphology are common pathological denominators in SOD1 and TDP43 ALS mouse models. Hum. Mol. Genet. 23, 1413–1424. doi: 10.1093/hmg/ddt528
Magrane, J., Hervias, I., Henning, M. S., Damiano, M., Kawamata, H., and Manfredi, G. (2009). Mutant SOD1 in neuronal mitochondria causes toxicity and mitochondrial dynamics abnormalities. Hum. Mol. Genet. 18, 4552–4564.
Mancuso, R., Del Valle, J., Modol, L., Martinez, A., Granado-Serrano, A. B., Ramirez-Nunez, O., et al. (2014). Resveratrol improves motoneuron function and extends survival in SOD1(G93A) ALS mice. Neurotherapeutics 11, 419–432. doi: 10.1007/s13311-013-0253-y
Manczak, M., Calkins, M. J., and Reddy, P. H. (2011). Impaired mitochondrial dynamics and abnormal interaction of amyloid beta with mitochondrial protein Drp1 in neurons from patients with Alzheimer’s disease: implications for neuronal damage. Hum. Mol. Genet. 20, 2495–2509. doi: 10.1093/hmg/ddr139
Manczak, M., and Reddy, P. H. (2015). Mitochondrial division inhibitor 1 protects against mutant huntingtin-induced abnormal mitochondrial dynamics and neuronal damage in Huntington’s disease. Hum. Mol. Genet. 24, 7308–7325. doi: 10.1093/hmg/ddv429
Mandal, A., and Drerup, C. M. (2019). Axonal transport and mitochondrial function in neurons. Front. Cell Neurosci. 13:373. doi: 10.3389/fncel.2019.00373
Markert, C. D., Kim, E., Gifondorwa, D. J., Childers, M. K., and Milligan, C. E. (2010). A single-dose resveratrol treatment in a mouse model of amyotrophic lateral sclerosis. J. Med. Food 13, 1081–1085. doi: 10.1089/jmf.2009.0243
Martin, L. J. (2010). Olesoxime, a cholesterol-like neuroprotectant for the potential treatment of amyotrophic lateral sclerosis. Idrugs 13, 568–580.
Martin, L. J., Pan, Y., Price, A. C., Sterling, W., Copeland, N. G., Jenkins, N. A., et al. (2006). Parkinson’s disease alpha-synuclein transgenic mice develop neuronal mitochondrial degeneration and cell death. J. Neurosci. 26, 41–50. doi: 10.1523/jneurosci.4308-05.2006
Martin-Maestro, P., Gargini, R., Perry, G., Avila, J., and Garcia-Escudero, V. (2016). PARK2 enhancement is able to compensate mitophagy alterations found in sporadic Alzheimer’s disease. Hum. Mol. Genet. 25, 792–806. doi: 10.1093/hmg/ddv616
Martin-Maestro, P., Sproul, A., Martinez, H., Paquet, D., Gerges, M., Noggle, S., et al. (2019). Autophagy induction by bexarotene promotes mitophagy in presenilin 1 familial Alzheimer’s disease iPSC-Derived neural stem cells. Mol. Neurobiol. 56, 8220–8236. doi: 10.1007/s12035-019-01665-y
Maruyama, H., Morino, H., Ito, H., Izumi, Y., Kato, H., Watanabe, Y., et al. (2010). Mutations of optineurin in amyotrophic lateral sclerosis. Nature 465, 223–226. doi: 10.3109/17482968.2010.545952
Mattson, M. P., Gleichmann, M., and Cheng, A. (2008). Mitochondria in neuroplasticity and neurological disorders. Neuron 60, 748–766. doi: 10.1016/j.neuron.2008.10.010
Mayer, M. P., and Bukau, B. (2005). Hsp70 chaperones: cellular functions and molecular mechanism. Cell. Mol. Life Sci. 62, 670–684. doi: 10.1007/s00018-004-4464-6
McLelland, G. L., Goiran, T., Yi, W., Dorval, G., Chen, C. X., Lauinger, N. D., et al. (2018). Mfn2 ubiquitination by PINK1/parkin gates the p97-dependent release of ER from mitochondria to drive mitophagy. eLife 7:e32866. doi: 10.7554/eLife.32866
Mengus, C., Neutzner, M., Bento, A. C. P. F., Bippes, C. C., Kohler, C., Decembrini, S., et al. (2021). VCP/p97 cofactor UBXN1/SAKS1 regulates mitophagy by modulating MFN2 removal from mitochondria. Autophagy. Online ahead of print. doi: 10.1080/15548627.2021.1922982
Merkwirth, C., Jovaisaite, V., Durieux, J., Matilainen, O., Jordan, S. D., Quiros, P. M., et al. (2016). Two conserved histone demethylases regulate mitochondrial stress-induced longevity. Cell 165, 1209–1223. doi: 10.1016/j.cell.2016.04.012
Meyer, H., Bug, M., and Bremer, S. (2012). Emerging functions of the VCP/p97 AAA-ATPase in the ubiquitin system. Nat. Cell Biol. 14, 117–123. doi: 10.1038/ncb2407
Mishra, P., Carelli, V., Manfredi, G., and Chan, D. C. (2014). Proteolytic cleavage of Opa1 stimulates mitochondrial inner membrane fusion and couples fusion to oxidative phosphorylation. Cell Metab. 19, 630–641. doi: 10.1016/j.cmet.2014.03.011
Misko, A., Jiang, S. R., Wegorzewska, I., Milbrandt, J., and Baloh, R. H. (2010). Mitofusin 2 is necessary for transport of axonal mitochondria and interacts with the Miro/Milton complex. J. Neurosci. 30, 4232–4240. doi: 10.1523/JNEUROSCI.6248-09.2010
Modi, S., Lopez-Domenech, G., Halff, E. F., Covill-Cooke, C., Ivankovic, D., Melandri, D., et al. (2019). Miro clusters regulate ER-mitochondria contact sites and link cristae organization to the mitochondrial transport machinery. Nat. Commun. 10:4399. doi: 10.1038/s41467-019-12382-4
Moehle, E. A., Shen, K., and Dillin, A. (2019). Mitochondrial proteostasis in the context of cellular and organismal health and aging. J. Biol. Chem. 294, 5396–5407. doi: 10.1074/jbc.TM117.000893
Moller, A., Bauer, C. S., Cohen, R. N., Webster, C. P., and De Vos, K. J. (2017). Amyotrophic lateral sclerosis-associated mutant SOD1 inhibits anterograde axonal transport of mitochondria by reducing Miro1 levels. Hum. Mol. Genet. 26, 4668–4679. doi: 10.1093/hmg/ddx348
Moors, T. E., Hoozemans, J. J. M., Ingrassia, A., Beccari, T., Pametti, L., Chartier-Harlin, M. C., et al. (2017). Therapeutic potential of autophagy-enhancing agents in Parkinson’s disease. Mol. Neurodegenerat. 12:11.
Morais, V. A., Verstreken, P., Roethig, A., Smet, J., Snellinx, A., Vanbrabant, M., et al. (2009). Parkinson’s disease mutations in PINK1 result in decreased complex I activity and deficient synaptic function. EMBO Mol. Med. 1, 99–111. doi: 10.1002/emmm.200900006
Moreira, P. I., Carvalho, C., Zhu, X. W., Smith, M. A., and Perry, G. (2010). Mitochondrial dysfunction is a trigger of Alzheimer’s disease pathophysiology. Biochim. Biophys. Acta-Mol. Basis Dis. 1802, 2–10. doi: 10.1016/j.bbadis.2009.10.006
Moskal, N., Riccio, V., Bashkurov, M., Taddese, R., Datti, A., Lewis, P. N., et al. (2020). ROCK inhibitors upregulate the neuroprotective parkin-mediated mitophagy pathway. Nat. Commun. 11:88. doi: 10.1038/s41467-019-13781-3
Mossmann, D., Vogtle, F. N., Taskin, A. A., Teixeira, P. F., Ring, J., Burkhart, J. M., et al. (2014). Amyloid-beta peptide induces mitochondrial dysfunction by inhibition of preprotein maturation. Cell Metab. 20, 662–669. doi: 10.1016/j.cmet.2014.07.024
Mouchiroud, L., Houtkooper, R. H., Moullan, N., Katsyuba, E., Ryu, D., Canto, C., et al. (2013). The NAD(+)/Sirtuin pathway modulates longevity through activation of mitochondrial UPR and FOXO signaling. Cell 154, 430–441. doi: 10.1016/j.cell.2013.06.016
Munoz-Carvajal, F., and Sanhueza, M. (2020). The mitochondrial unfolded protein response: a hinge between healthy and pathological aging. Front. Aging Neurosci. 12:581849. doi: 10.3389/fnagi.2020.581849
Murley, A., Lackner, L. L., Osman, C., West, M., Voeltz, G. K., Walter, P., et al. (2013). ER-associated mitochondrial division links the distribution of mitochondria and mitochondrial DNA in yeast. eLife 2:e00422. doi: 10.7554/eLife.00422
Naia, L., Carmo, C., Campesan, S., Fao, L. G., Cotton, V. E., Valero, J., et al. (2021). Mitochondrial SIRT3 confers neuroprotection in Huntington’s disease by regulation of oxidative challenges and mitochondrial dynamics. Free Radic. Biol. Med. 163, 163–179. doi: 10.1016/j.freeradbiomed.2020.11.031
Nambron, R., Silajdzic, E., Kalliolia, E., Ottolenghi, C., Hindmarsh, P., Hill, N. R., et al. (2016). A metabolic study of Huntington’s disease. PLoS One 11:e0146480. doi: 10.1371/journal.pone.0146480
Naresh, N. U., and Haynes, C. M. (2019). Signaling and regulation of the mitochondrial unfolded protein response. Cold Spring Harb. Perspect. Biol. 11:a033944. doi: 10.1101/cshperspect.a033944
Nargund, A. M., Fiorese, C. J., Pellegrino, M. W., Deng, P., and Haynes, C. M. (2015). Mitochondrial and nuclear accumulation of the transcription factor ATFS-1 promotes OXPHOS recovery during the UPR(mt). Mol. Cell 58, 123–133. doi: 10.1016/j.molcel.2015.02.008
Nargund, A. M., Pellegrino, M. W., Fiorese, C. J., Baker, B. M., and Haynes, C. M. (2012). Mitochondrial import efficiency of ATFS-1 regulates mitochondrial UPR activation. Science 337, 587–590. doi: 10.1126/science.1223560
Neumann, M., Sampathu, D. M., Kwong, L. K., Truax, A. C., Micsenyi, M. C., Chou, T. T., et al. (2006). Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314, 130–133.
Ney, P. A. (2015). Mitochondrial autophagy: origins, significance, and role of BNIP3 and NIX. Biochim. Biophys. Acta 1853, 2775–2783. doi: 10.1016/j.bbamcr.2015.02.022
Ni, H. M., Williams, J. A., and Ding, W. X. (2015). Mitochondrial dynamics and mitochondrial quality control. Redox Biol. 4, 6–13. doi: 10.1016/j.redox.2014.11.006
Oakes, J. A., Davies, M. C., and Collins, M. O. (2017). TBK1: a new player in ALS linking autophagy and neuroinflammation. Mol. Brain 10:5. doi: 10.1186/s13041-017-0287-x
Obergasteiger, J., Frapporti, G., Lamonaca, G., Pizzi, S., Picard, A., Lavdas, A. A., et al. (2020). Kinase inhibition of G2019S-LRRK2 enhances autolysosome formation and function to reduce endogenous alpha-synuclein intracellular inclusions. Cell Death Discov. 6:45. doi: 10.1038/s41420-020-0279-y
Ojano-Dirain, C. P., Antonelli, P. J., and Le Prell, C. G. (2014). Mitochondria-Targeted antioxidant MitoQ reduces gentamicin-induced ototoxicity. Otol. Neurotol. 35, 533–539. doi: 10.1097/MAO.0000000000000192
Onesto, E., Colombrita, C., Gumina, V., Borghi, M. O., Dusi, S., Doretti, A., et al. (2016). Gene-specific mitochondria dysfunctions in human TARDBP and C9ORF72 fibroblasts. Acta Neuropathol. Commun. 4:47. doi: 10.1186/s40478-016-0316-5
Ordonez, D. G., Lee, M. K., and Feany, M. B. (2018). alpha-synuclein induces mitochondrial dysfunction through spectrin and the actin cytoskeleton. Neuron 97, 108–121.e6. doi: 10.1016/j.neuron.2017.11.036
Ortiz, J. M. P., and Swerdlow, R. H. (2019). Mitochondrial dysfunction in Alzheimer’s disease: role in pathogenesis and novel therapeutic opportunities. Br. J. Pharmacol. 176, 3489–3507. doi: 10.1111/bph.14585
Osgerby, L., Lai, Y. C., Thornton, P. J., Amalfitano, J., Le Duff, C. S., Jabeen, I., et al. (2017). Kinetin riboside and its protides activate the Parkinson’s disease associated PTEN-induced putative kinase 1 (PINK1) independent of mitochondrial depolarization. J. Med. Chem. 60, 3518–3524. doi: 10.1021/acs.jmedchem.6b01897
Pakpian, N., Phopin, K., Kitidee, K., Govitrapong, P., and Wongchitrat, P. (2020). Alterations in mitochondrial dynamic-related genes in the peripheral blood of Alzheimer’s disease patients. Curr. Alzheimer Res. 17, 616–625. doi: 10.2174/1567205017666201006162538
Palikaras, K., Lionaki, E., and Tavernarakis, N. (2018). Mechanisms of mitophagy in cellular homeostasis, physiology and pathology. Nat. Cell Biol. 20, 1013–1022. doi: 10.1038/s41556-018-0176-2
Palmer, C. S., Osellame, L. D., Laine, D., Koutsopoulos, O. S., Frazier, A. E., and Ryan, M. T. (2011). MiD49 and MiD51, new components of the mitochondrial fission machinery. EMBO Rep. 12, 565–573. doi: 10.1038/embor.2011.54
Pan, T., Rawal, P., Wu, Y., Xie, W., Jankovic, J., and Le, W. (2009). Rapamycin protects against rotenone-induced apoptosis through autophagy induction. Neuroscience 164, 541–551. doi: 10.1016/j.neuroscience.2009.08.014
Panchal, K., and Tiwari, A. K. (2021). Miro (Mitochondrial Rho GTPase), a key player of mitochondrial axonal transport and mitochondrial dynamics in neurodegenerative diseases. Mitochondrion 56, 118–135. doi: 10.1016/j.mito.2020.10.005
Papa, L., and Germain, D. (2014). SirT3 regulates the mitochondrial unfolded protein response. Mol. Cell. Biol. 34, 1378–1378. doi: 10.1128/mcb.00190-14
Pareek, G., and Pallanck, L. J. (2020). Inactivation of the mitochondrial protease Afg3l2 results in severely diminished respiratory chain activity and widespread defects in mitochondrial gene expression. PLoS Genetics 16:e1009118. doi: 10.1371/journal.pgen.1009118
Park, D., Jeong, H., Lee, M. N., Koh, A., Kwon, O., Yang, Y. R., et al. (2016). Resveratrol induces autophagy by directly inhibiting mTOR through ATP competition. Sci. Rep. 6:21772. doi: 10.1038/srep21772
Park, J., Choi, H., Min, J. S., Park, S. J., Kim, J. H., Park, H. J., et al. (2013). Mitochondrial dynamics modulate the expression of pro-inflammatory mediators in microglial cells. J. Neurochem. 127, 221–232. doi: 10.1111/jnc.12361
Patten, D. A., Wong, J., Khacho, M., Soubannier, V., Mailloux, R. J., Pilon-Larose, K., et al. (2014). OPA1-dependent cristae modulation is essential for cellular adaptation to metabolic demand. EMBO J. 33, 2676–2691. doi: 10.15252/embj.201488349
Perez, M. J., Ivanyuk, D., Panagiotakopoulou, V., Di Napoli, G., Kalb, S., Brunetti, D., et al. (2020). Loss of function of the mitochondrial peptidase PITRM1 induces proteotoxic stress and Alzheimer’s disease-like pathology in human cerebral organoids. Mol. Psychiatry. doi: 10.1038/s41380-020-0807-4
Perez-Gonzalez, R., Pascual, C., Antequera, D., Bolos, M., Redondo, M., Perez, D. I., et al. (2013). Phosphodiesterase 7 inhibitor reduced cognitive impairment and pathological hallmarks in a mouse model of Alzheimer’s disease. Neurobiol. Aging 34, 2133–2145. doi: 10.1016/j.neurobiolaging.2013.03.011
Pickles, S., Vigie, P., and Youle, R. J. (2018). Mitophagy and quality control mechanisms in mitochondrial maintenance. Curr. Biol. 28, R170–R185.
Pickrell, A. M., and Youle, R. J. (2015). The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron 85, 257–273. doi: 10.1016/j.neuron.2014.12.007
Poole, A. C., Thomas, R. E., Andrews, L. A., Mcbride, H. M., Whitworth, A. J., and Pallanck, L. J. (2008). The PINK1/Parkin pathway regulates mitochondrial morphology. Proc. Natl. Acad. Sci. U S A. 105, 1638–1643. doi: 10.1073/pnas.0709336105
Potashkin, J. A., Blume, S. R., and Runkle, N. K. (2010). Limitations of animal models of Parkinson’s disease. Parkinsons Dis. 2011:658083.
Prots, I., Grosch, J., Brazdis, R. M., Simmnacher, K., Veber, V., Havlicek, S., et al. (2018). alpha-Synuclein oligomers induce early axonal dysfunction in human iPSC-based models of synucleinopathies. Proc. Natl. Acad. Sci. U S A. 115, 7813–7818. doi: 10.1073/pnas.1713129115
Qi, X., Qvit, N., Su, Y. C., and Mochly-Rosen, D. (2013). A novel Drp1 inhibitor diminishes aberrant mitochondrial fission and neurotoxicity. J. Cell Sci. 126, 789–802. doi: 10.1242/jcs.114439
Qin, W., Haroutunian, V., Katsel, P., Cardozo, C. P., Ho, L., Buxbaum, J. D., et al. (2009). PGC-1alpha expression decreases in the Alzheimer disease brain as a function of dementia. Arch. Neurol. 66, 352–361.
Qureshi, M. A., Haynes, C. M., and Pellegrino, M. W. (2017). The mitochondrial unfolded protein response: signaling from the powerhouse. J. Biol. Chem. 292, 13500–13506. doi: 10.1074/jbc.r117.791061
Rappold, P. M., Cui, M., Grima, J. C., Fan, R. Z., De Mesy-Bentley, K. L., Chen, L. A., et al. (2014). Drp1 inhibition attenuates neurotoxicity and dopamine release deficits in vivo. Nat. Commun. 5:5244. doi: 10.1038/ncomms6244
Ravits, J., Paul, P., and Jorg, C. (2007). Focality of upper and lower motor neuron degeneration at the clinical onset of ALS. Neurology 68, 1571–1575. doi: 10.1212/01.wnl.0000260965.20021.47
Reddy, A. P., Sawant, N., Morton, H., Kshirsagar, S., Bunquin, L. E., Yin, X., et al. (2021). Selective serotonin reuptake inhibitor citalopram ameliorates cognitive decline and protects against amyloid beta-induced mitochondrial dynamics, biogenesis, autophagy, mitophagy and synaptic toxicities in a mouse model of Alzheimer’s disease. Hum. Mol. Genet. 30, 789–810. doi: 10.1093/hmg/ddab091
Reddy, P. H., Manczak, M., and Kandimalla, R. (2017a). Mitochondria-targeted small molecule SS31: a potential candidate for the treatment of Alzheimer’s disease. Hum. Mol. Genet. 26:1597. doi: 10.1093/hmg/ddx129
Reddy, P. H., Manczak, M., and Yin, X. (2017b). Mitochondria-Division inhibitor 1 protects against amyloid-beta induced mitochondrial fragmentation and synaptic damage in Alzheimer’s disease. J. Alzheimers. Dis. 58, 147–162. doi: 10.3233/JAD-170051
Reddy, P. H., Manczak, M., Yin, X., and Reddy, A. P. (2018). Synergistic protective effects of mitochondrial division inhibitor 1 and mitochondria-targeted small peptide SS31 in Alzheimer’s disease. J. Alzheimers. Dis. 62, 1549–1565. doi: 10.3233/JAD-170988
Riar, A. K., Burstein, S. R., Palomo, G. M., Arreguin, A., Manfredi, G., and Germain, D. (2017). Sex specific activation of the ERalpha axis of the mitochondrial UPR (UPRmt) in the G93A-SOD1 mouse model of familial ALS. Hum. Mol. Genet. 26, 1318–1327. doi: 10.1093/hmg/ddx049
Richter, B., Sliter, D. A., Herhaus, L., Stolz, A., Wang, C. X., Beli, P., et al. (2016). Phosphorylation of OPTN by TBK1 enhances its binding to Ub chains and promotes selective autophagy of damaged mitochondria. Proc. Natl. Acad. Sci. U S A. 113, 4039–4044. doi: 10.1073/pnas.1523926113
Roe, A. J., and Qi, X. (2018). Drp1 phosphorylation by MAPK1 causes mitochondrial dysfunction in cell culture model of Huntington’s disease. Biochem. Biophys. Res. Commun. 496, 706–711. doi: 10.1016/j.bbrc.2018.01.114
Roe, M. S., Wahab, B., Torok, Z., Horvath, I., Vigh, L., and Prodromou, C. (2018). Dihydropyridines allosterically modulate Hsp90 providing a novel mechanism for heat shock protein co-induction and neuroprotection. Front. Mol. Biosci. 5:51. doi: 10.3389/fmolb.2018.00051
Rong, J., Mcguire, J. R., Fang, Z. H., Sheng, G. Q., Shin, J. Y., Li, S. H., et al. (2006). Regulation of intracellular trafficking of huntingtin-associated protein-1 is critical for TrkA protein levels and neurite outgrowth. J. Neurosci. 26, 6019–6030. doi: 10.1523/JNEUROSCI.1251-06.2006
Ross, C. A., and Poirier, M. A. (2004). Protein aggregation and neurodegenerative disease. Nat. Med. 10(Suppl.), S10–S17.
Rudnick, N. D., Griffey, C. J., Guarnieri, P., Gerbino, V., Wang, X. Y., Piersaint, J. A., et al. (2017). Distinct roles for motor neuron autophagy early and late in the SOD1(G93A) mouse model of ALS. Proc. Natl. Acad. Sci. U S A. 114, E8294–E8303. doi: 10.1073/pnas.1704294114
Rusilowicz-Jones, E. V., Jardine, J., Kallinos, A., Pinto-Fernandez, A., Guenther, F., Giurrandino, M., et al. (2020). USP30 sets a trigger threshold for PINK1-PARKIN amplification of mitochondrial ubiquitylation. Life Sci. Alliance 3:e202000768. doi: 10.26508/lsa.202000768
Ryan, E. B., Yan, J., Miller, N., Dayanidhi, S., Ma, Y. C., Deng, H. X., et al. (2021). Early death of ALS-linked CHCHD10-R15L transgenic mice with central nervous system, skeletal muscle, and cardiac pathology. iScience 24:102061. doi: 10.1016/j.isci.2021.102061
Ryan, T., Bamm, V. V., Stykel, M. G., Coackley, C. L., Humphries, K. M., Jamieson-Williams, R., et al. (2018). Cardiolipin exposure on the outer mitochondrial membrane modulates alpha-synuclein. Nat. Commun. 9:817. doi: 10.1038/s41467-018-03241-9
Ryan, T. A., and Tumbarello, D. A. (2018). Optineurin: a coordinator of membrane-associated cargo trafficking and autophagy. Front. Immunol. 9:1024. doi: 10.3389/fimmu.2018.01024
Saibil, H. (2013). Chaperone machines for protein folding, unfolding and disaggregation. Nat. Rev. Mol. Cell Biol. 14, 630–642. doi: 10.1038/nrm3658
Saxton, W. M., and Hollenbeck, P. J. (2012). The axonal transport of mitochondria. J. Cell Sci. 125, 2095–2104.
Scarpulla, R. C. (2008). Transcriptional paradigms in mammalian mitochondrial biogenesis and function. Physiol. Rev. 88, 611–638. doi: 10.1152/physrev.00025.2007
Schmukler, E., Solomon, S., Simonovitch, S., Goldshmit, Y., Wolfson, E., Michaelson, D. M., et al. (2020). Altered mitochondrial dynamics and function in APOE4-expressing astrocytes. Cell Death Dis. 11:578. doi: 10.1038/s41419-020-02776-4
Schnapp, B. J., and Reese, T. S. (1989). Dynein is the motor for retrograde axonal-transport of organelles. Proc. Natl. Acad. Sci. U S A. 86, 1548–1552. doi: 10.1073/pnas.86.5.1548
Schrepfer, E., and Scorrano, L. (2016). Mitofusins, from mitochondria to metabolism. Mol. Cell. 61, 683–694. doi: 10.1016/j.molcel.2016.02.022
Schulte, J., and Littleton, J. T. (2011). The biological function of the Huntingtin protein and its relevance to Huntington’s disease pathology. Curr. Trends Neurol. 5, 65–78.
Shaltouki, A., Hsieh, C. H., Kim, M. J., and Wang, X. (2018). Alpha-synuclein delays mitophagy and targeting Miro rescues neuron loss in Parkinson’s models. Acta Neuropathol. 136, 607–620. doi: 10.1007/s00401-018-1873-4
Shanmughapriya, S., Rajan, S., Hoffman, N. E., Higgins, A. M., Tomar, D., Nemani, N., et al. (2015). SPG7 is an essential and conserved component of the mitochondrial permeability transition pore. Mol. Cell. 60, 47–62. doi: 10.1016/j.molcel.2015.08.009
Shen, Y., Ding, M., Xie, Z., Liu, X., Yang, H., Jin, S., et al. (2019). Activation of mitochondrial unfolded protein response in SHSY5Y expressing APP cells and APP/PS1 mice. Front. Cell Neurosci. 13:568. doi: 10.3389/fncel.2019.00568
Shen, Y., Ding, M., Xie, Z. H., Liu, X. T., Yang, H., Jin, S. Q., et al. (2020). Activation of mitochondrial unfolded protein response in SHSY5Y expressing APP cells and APP/PS1 mice. Front. Cell. Neurosci. 13:568.
Sheng, Z. H., and Cai, Q. (2012). Mitochondrial transport in neurons: impact on synaptic homeostasis and neurodegeneration. Nat. Rev. Neurosci. 13, 77–93. doi: 10.1038/nrn3156
Shi, P., Strom, A. L., Gal, J., and Zhu, H. (2010). Effects of ALS-related SOD1 mutants on dynein- and KIF5-mediated retrograde and anterograde axonal transport. Biochim. Biophys. Acta 1802, 707–716. doi: 10.1016/j.bbadis.2010.05.008
Shin, C. S., Meng, S., Garbis, S. D., Moradian, A., Taylor, R. W., Sweredoski, M. J., et al. (2021). LONP1 and mtHSP70 cooperate to promote mitochondrial protein folding. Nat. Commun. 12:265. doi: 10.1038/s41467-020-20597-z
Shirendeb, U., Reddy, A. P., Manczak, M., Calkins, M. J., Mao, P., Tagle, D. A., et al. (2011). Abnormal mitochondrial dynamics, mitochondrial loss and mutant huntingtin oligomers in Huntington’s disease: implications for selective neuronal damage. Hum. Mol. Genet. 20, 1438–1455. doi: 10.1093/hmg/ddr024
Silva, J. M., Wong, A., Carelli, V., and Cortopassi, G. A. (2009). Inhibition of mitochondrial function induces an integrated stress response in oligodendroglia. Neurobiol. Dis. 34, 357–365. doi: 10.1016/j.nbd.2009.02.005
Simola, N., Morelli, M., and Carta, A. R. (2007). The 6-hydroxydopamine model of Parkinson’s disease. Neurotox. Res. 11, 151–167.
Singh, A., Zhi, L., and Zhang, H. (2019). LRRK2 and mitochondria: recent advances and current views. Brain Res. 1702, 96–104. doi: 10.1016/j.brainres.2018.06.010
Smith, E. F., Shaw, P. J., and De Vos, K. J. (2019). The role of mitochondria in amyotrophic lateral sclerosis. Neurosci. Lett. 710:132933.
Snow, B. J., Rolfe, F. L., Lockhart, M. M., Frampton, C. M., O’sullivan, J. D., Fung, V., et al. (2010). A double-blind, placebo-controlled study to assess the mitochondria-targeted antioxidant MitoQ as a disease-modifying therapy in Parkinson’s disease. Mov. Disord. 25, 1670–1674. doi: 10.1002/mds.23148
Solesio, M. E., Prime, T. A., Logan, A., Murphy, M. P., Arroyo-Jimenez, M. D., Jordan, J., et al. (2013). The mitochondria-targeted anti-oxidant MitoQ reduces aspects of mitochondrial fission in the 6-OHDA cell model of Parkinson’s disease. Biochimica Biophys. Acta-Mol. Basis Dis. 1832, 174–182. doi: 10.1016/j.bbadis.2012.07.009
Song, W., Chen, J., Petrilli, A., Liot, G., Klinglmayr, E., Zhou, Y., et al. (2011). Mutant huntingtin binds the mitochondrial fission GTPase dynamin-related protein-1 and increases its enzymatic activity. Nat. Med. 17, 377–382. doi: 10.1038/nm.2313
Song, W., Song, Y., Kincaid, B., Bossy, B., and Bossy-Wetzel, E. (2013). Mutant SOD1G93A triggers mitochondrial fragmentation in spinal cord motor neurons: neuroprotection by SIRT3 and PGC-1alpha. Neurobiol. Dis. 51, 72–81. doi: 10.1016/j.nbd.2012.07.004
Song, Z., Ghochani, M., Mccaffery, J. M., Frey, T. G., and Chan, D. C. (2009). Mitofusins and OPA1 mediate sequential steps in mitochondrial membrane fusion. Mol. Biol. Cell 20, 3525–3532. doi: 10.1091/mbc.e09-03-0252
Sorrentino, V., Romani, M., Mouchiroud, L., Beck, J. S., Zhang, H., D’amico, D., et al. (2017). Enhancing mitochondrial proteostasis reduces amyloid-beta proteotoxicity. Nature 552, 187–193. doi: 10.1038/nature25143
Soto, I., Couvillion, M., Mcshane, E., Hansen, K. G., Moran, J. C., Barrientos, A., et al. (2021). Balanced mitochondrial and cytosolic translatomes underlie the biogenesis of human respiratory complexes. bioRxiv [preprint]. doi: 10.1101/2021.05.31.446345
Stiburek, L., Cesnekova, J., Kostkova, O., Fornuskova, D., Vinsova, K., Wenchich, L., et al. (2012). YME1L controls the accumulation of respiratory chain subunits and is required for apoptotic resistance, cristae morphogenesis, and cell proliferation. Mol. Biol. Cell 23, 1010–1023. doi: 10.1091/mbc.E11-08-0674
Straub, I. R., Weraarpachai, W., and Shoubridge, E. A. (2021). Multi-OMICS study of a CHCHD10 variant causing ALS demonstrates metabolic rewiring and activation of endoplasmic reticulum and mitochondrial unfolded protein responses. Hum. Mol. Genet. 30, 687–705. doi: 10.1093/hmg/ddab078
Su, Y. C., and Qi, X. (2013). Inhibition of excessive mitochondrial fission reduced aberrant autophagy and neuronal damage caused by LRRK2 G2019S mutation. Hum. Mol. Genet. 22, 4545–4561. doi: 10.1093/hmg/ddt301
Sun, X. N., and Qiu, H. Y. (2020). Valosin-Containing protein, a calcium-associated ATPase protein, in endoplasmic reticulum and mitochondrial function and its implications for diseases. Int. J. Mol. Sci. 21:3842. doi: 10.3390/ijms21113842
Taguchi, N., Ishihara, N., Jofuku, A., Oka, T., and Mihara, K. (2007). Mitotic phosphorylation of dynamin-related GTPase Drp1 participates in mitochondrial fission. J. Biol. Chem. 282, 11521–11529. doi: 10.1074/jbc.M607279200
Tain, L. S., Mortiboys, H., Tao, R. N., Ziviani, E., Bandmann, O., and Whitworth, A. J. (2009). Rapamycin activation of 4E-BP prevents parkinsonian dopaminergic neuron loss. Nat. Neurosci. 12, 1129–U1113. doi: 10.1038/nn.2372
Tang, B. L. (2016). MIRO GTPases in mitochondrial transport. Homeostasis Pathol. Cells 5:1. doi: 10.3390/cells5010001
Tang, F. L., Erion, J. R., Tian, Y., Liu, W., Yin, D. M., Ye, J., et al. (2015). VPS35 in dopamine neurons is required for endosome-to-golgi retrieval of Lamp2a, a receptor of chaperone-mediated autophagy that is critical for alpha-synuclein degradation and prevention of pathogenesis of Parkinson’s disease. J. Neurosci. 35, 10613–10628. doi: 10.1523/JNEUROSCI.0042-15.2015
Tang, J. X., Thompson, K., Taylor, R. W., and Olahova, M. (2020). Mitochondrial OXPHOS biogenesis: co-regulation of protein synthesis. import, and assembly pathways. Int. J. Mol. Sci. 21:3820. doi: 10.3390/ijms21113820
Thau, N., Knippenberg, S., Korner, S., Rath, K. J., Dengler, R., and Petri, S. (2012). Decreased mRNA expression of PGC-1alpha and PGC-1alpha-regulated factors in the SOD1G93A ALS mouse model and in human sporadic ALS. J. Neuropathol. Exp. Neurol. 71, 1064–1074. doi: 10.1097/NEN.0b013e318275df4b
Thomas, K. J., Mccoy, M. K., Blackinton, J., Beilina, A., Van Der Brug, M., Sandebring, A., et al. (2011). DJ-1 acts in parallel to the PINK1/parkin pathway to control mitochondrial function and autophagy. Hum. Mol. Genet. 20, 40–50. doi: 10.1093/hmg/ddq430
Thomas, R. E., Andrews, L. A., Burman, J. L., Lin, W. Y., and Pallanck, L. J. (2014). PINK1-Parkin pathway activity is regulated by degradation of PINK1 in the mitochondrial matrix. PLoS Genetics 10:e1004279. doi: 10.1371/journal.pgen.1004279
Tian, Y., Garcia, G., Bian, Q., Steffen, K. K., Joe, L., Wolff, S., et al. (2016). Mitochondrial stress induces chromatin reorganization to promote longevity and UPRmt. Cell 165, 1197–1208. doi: 10.1016/j.cell.2016.04.011
Tien, N. T., Karaca, I., Tamboli, I. Y., and Walter, J. (2016). Trehalose alters subcellular trafficking and the metabolism of the alzheimer-associated amyloid precursor protein. J. Biol. Chem. 291, 10528–10540. doi: 10.1074/jbc.M116.719286
Tran, M., and Reddy, P. H. (2020). Defective autophagy and mitophagy in aging and Alzheimer’s disease. Front. Neurosci. 14:612757. doi: 10.3389/fnins.2020.612757
Tseng, A. H. H., Shieh, S. S., and Wang, D. L. (2013). SIRT3 deacetylates FOXO3 to protect mitochondria against oxidative damage. Free Radic. Biol. Med. 63, 222–234. doi: 10.1016/j.freeradbiomed.2013.05.002
Tsunemi, T., Ashe, T. D., Morrison, B. E., Soriano, K. R., Au, J., Roque, R. A., et al. (2012). PGC-1alpha rescues Huntington’s disease proteotoxicity by preventing oxidative stress and promoting TFEB function. Sci. Transl. Med. 4:142ra197. doi: 10.1126/scitranslmed.3003799
Valdinocci, D., Simoes, R. F., Kovarova, J., Cunha-Oliveira, T., Neuzil, J., and Pountney, D. L. (2019). Intracellular and intercellular mitochondrial dynamics in Parkinson’s disease. Front. Neurosci. 13:930. doi: 10.3389/fnins.2019.00930
Vance, C., Rogelj, B., Hortobagyi, T., De Vos, K. J., Nishimura, A. L., Sreedharan, J., et al. (2009). Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 323, 1208–1211. doi: 10.1126/science.1165942
Vandiver, M. S., Paul, B. D., Xu, R., Karuppagounder, S., Rao, F., Snowman, A. M., et al. (2013). Sulfhydration mediates neuroprotective actions of parkin. Nat. Commun. 4:1626. doi: 10.1038/ncomms2623
Veldink, J. H., and Consor, P. M. A. S. (2018). CHCHD10 variants in amyotrophic lateral sclerosis: where is the evidence? Ann. Neurol. 84, 110–116. doi: 10.1002/ana.25273
Vermilyea, S. C., Babinski, A., Tran, N., To, S., Guthrie, S., Kluss, J. H., et al. (2020). In vitro CRISPR/Cas9-Directed gene editing to model LRRK2 G2019S Parkinson’s disease in common marmosets. Sci. Rep. 10:3447. doi: 10.1038/s41598-020-60273-2
Vidal, V., Puente, A., Garcia-Cerro, S., Garcia Unzueta, M. T., Rueda, N., Riancho, J., et al. (2021). Bexarotene impairs cognition and produces hypothyroidism in a mouse model of down syndrome and Alzheimer’s disease. Front. Pharmacol. 12:613211. doi: 10.3389/fphar.2021.613211
Vijayan, M., Bose, C., and Reddy, P. H. (2021). Protective effects of a small molecule inhibitor, DDQ against amyloid beta in Alzheimer’s disease. Mitochondrion 59, 17–29. doi: 10.1016/j.mito.2021.04.005
Vilarino-Guell, C., Wider, C., Ross, O. A., Dachsel, J. C., Kachergus, J. M., Lincoln, S. J., et al. (2011). VPS35 mutations in Parkinson disease. Am. J. Hum. Genet. 89, 162–167.
Virbasius, J. V., and Scarpulla, R. C. (1994). Activation of the human mitochondrial transcription factor a gene by nuclear respiratory factors - a potential regulatory link between nuclear and mitochondrial gene-expression in organelle biogenesis. Proc. Natl. Acad. Sci. U S A. 91, 1309–1313. doi: 10.1073/pnas.91.4.1309
Vives-Bauza, C., Zhou, C., Huang, Y., Cui, M., De Vries, R. L., Kim, J., et al. (2010). PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc. Natl. Acad. Sci. U.S.A. 107, 378–383.
Walls, K. C., Coskun, P., Gallegos-Perez, J. L., Zadourian, N., Freude, K., Rasool, S., et al. (2012). Swedish Alzheimer mutation induces mitochondrial dysfunction mediated by HSP60 mislocalization of amyloid precursor protein (APP) and beta-amyloid. J. Biol. Chem. 287, 30317–30327. doi: 10.1074/jbc.M112.365890
Wang, H., Guan, Y., Wang, X., Smith, K., Cormier, K., Zhu, S., et al. (2007). Nortriptyline delays disease onset in models of chronic neurodegeneration. Eur. J. Neurosci. 26, 633–641. doi: 10.1111/j.1460-9568.2007.05663.x
Wang, L. W., Gao, J., Liu, J. Y., Siedlak, S. L., Torres, S., Fujioka, H., et al. (2018). Mitofusin 2 regulates axonal transport of calpastatin to prevent neuromuscular synaptic elimination in skeletal muscles. Cell Metab. 28, 400–418.e8. doi: 10.1016/j.cmet.2018.06.011
Wang, P., Deng, J., Dong, J., Liu, J., Bigio, E. H., Mesulam, M., et al. (2019). TDP-43 induces mitochondrial damage and activates the mitochondrial unfolded protein response. PLoS Genet 15:e1007947. doi: 10.1371/journal.pgen.1007947
Wang, Y., Liu, N., and Lu, B. (2019). Mechanisms and roles of mitophagy in neurodegenerative diseases. CNS Neurosci. Ther. 25, 859–875.
Wang, W., Li, L., Lin, W. L., Dickson, D. W., Petrucelli, L., Zhang, T., et al. (2013). The ALS disease-associated mutant TDP-43 impairs mitochondrial dynamics and function in motor neurons. Hum. Mol. Genet. 22, 4706–4719. doi: 10.1093/hmg/ddt319
Wang, W. Z., Li, L., Lin, W. L., Dickson, D. W., Petrucelli, L., Zhang, T., et al. (2013). The ALS disease-associated mutant TDP-43 impairs mitochondrial dynamics and function in motor neurons. Hum. Mol. Genet. 22, 4706–4719.
Wang, W., Wang, X., Fujioka, H., Hoppel, C., Whone, A. L., Caldwell, M. A., et al. (2016). Parkinson’s disease-associated mutant VPS35 causes mitochondrial dysfunction by recycling DLP1 complexes. Nat. Med. 22, 54–63. doi: 10.1038/nm.3983
Wang, X., and Chen, X. J. (2015). A cytosolic network suppressing mitochondria-mediated proteostatic stress and cell death. Nature 524, 481–484. doi: 10.1038/nature14859
Wang, X., and Schwarz, T. L. (2009). The mechanism of Ca2+ -dependent regulation of kinesin-mediated mitochondrial motility. Cell 136, 163–174. doi: 10.1016/j.cell.2008.11.046
Wang, X., Su, B., Lee, H. G., Li, X., Perry, G., Smith, M. A., et al. (2009). Impaired balance of mitochondrial fission and fusion in Alzheimer’s disease. J. Neurosci. 29, 9090–9103.
Wang, X. L., Su, B., Lee, H. G., Li, X. Y., Perry, G., Smith, M. A., et al. (2009). Impaired balance of mitochondrial fission and fusion in Alzheimer’s disease. J. Neurosci. 29, 9090–9103. doi: 10.1523/jneurosci.1357-09.2009
Wang, X., Su, B., Siedlak, S. L., Moreira, P. I., Fujioka, H., Wang, Y., et al. (2008). Amyloid-beta overproduction causes abnormal mitochondrial dynamics via differential modulation of mitochondrial fission/fusion proteins. Proc. Natl. Acad. Sci. U.S.A. 105, 19318–19323. doi: 10.1073/pnas.0804871105
Wang, X. L., Su, B., Siedlak, S. L., Moreira, P. I., Fujioka, H., Wang, Y., et al. (2008). Amyloid-beta overproduction causes abnormal mitochondrial dynamics via differential modulation of mitochondrial fission/fusion proteins. Proc. Natl. Acad. Sci. U S A. 105, 19318–19323.
Wang, X., Winter, D., Ashrafi, G., Schlehe, J., Wong, Y. L., Selkoe, D., et al. (2011). PINK1 and Parkin target Miro for phosphorylation and degradation to arrest mitochondrial motility. Cell 147, 893–906. doi: 10.1016/j.cell.2011.10.018
Wang, H. M., Zhang, T., Ge, X. H., Chen, J. J., Zhao, Y. W., and Fu, J. L. (2020). Parkin overexpression attenuates a beta-induced mitochondrial dysfunction in HEK293 cells by restoring impaired mitophagy. Life Sci. 244:117322. doi: 10.1016/j.lfs.2020.117322
Wang, L., Li, N., Shi, F. X., Xu, W. Q., Cao, Y., Lei, Y., et al. (2020). Upregulation of AMPK ameliorates Alzheimer’s disease-like tau pathology and memory impairment. Mol. Neurobiol. 57, 3349–3361. doi: 10.1007/s12035-020-01955-w
Wauters, F., Cornelissen, T., Imberechts, D., Martin, S., Koentjoro, B., Sue, C., et al. (2020). LRRK2 mutations impair depolarization-induced mitophagy through inhibition of mitochondrial accumulation of RAB10. Autophagy 16, 203–222. doi: 10.1080/15548627.2019.1603548
Weng, H. D., Ma, Y. H., Chen, L. N., Cai, G. E., Chen, Z. T., Zhang, S. C., et al. (2020). A new vision of mitochondrial unfolded protein response to the sirtuin family. Curr. Neuropharmacol. 18, 613–623. doi: 10.2174/1570159X18666200123165002
Whang, M. I., Tavares, R. M., Benjamin, D. I., Kattah, M. G., Advincula, R., Nomura, D. K., et al. (2017). The ubiquitin binding protein TAX1BP1 mediates autophagasome induction and the metabolic transition of activated T cells. Immunity 46, 405–420. doi: 10.1016/j.immuni.2017.02.018
Wrobel, L., Topf, U., Bragoszewski, P., Wiese, S., Sztolsztener, M. E., Oeljeklaus, S., et al. (2015). Mistargeted mitochondrial proteins activate a proteostatic response in the cytosol. Nature 524, 485–488. doi: 10.1038/nature14951
Xi, Y., Feng, D., Tao, K., Wang, R., Shi, Y., Qin, H., et al. (2018). MitoQ protects dopaminergic neurons in a 6-OHDA induced PD model by enhancing Mfn2-dependent mitochondrial fusion via activation of PGC-1alpha. Biochim Biophys. Acta Mol. Basis Dis. 1864, 2859–2870. doi: 10.1016/j.bbadis.2018.05.018
Xie, N., Wang, C., Lian, Y., Wu, C., Zhang, H., and Zhang, Q. (2014). Inhibition of mitochondrial fission attenuates a beta-induced microglia apoptosis. Neuroscience 256, 36–42. doi: 10.1016/j.neuroscience.2013.10.011
Xie, X., Gao, Y., Zeng, M., Wang, Y., Wei, T. F., Lu, Y. B., et al. (2019). Nicotinamide ribose ameliorates cognitive impairment of aged and Alzheimer’s disease model mice. Metab. Brain Dis. 34, 353–366. doi: 10.1007/s11011-018-0346-8
Xiong, Y., Dawson, T. M., and Dawson, V. L. (2017). Models of LRRK2-Associated Parkinson’s disease. Adv. Neurobiol. 14, 163–191. doi: 10.1007/978-3-319-49969-7_9
Xu, Y. F., Gendron, T. F., Zhang, Y. J., Lin, W. L., D’alton, S., Sheng, H., et al. (2010). Wild-Type human TDP-43 expression causes TDP-43 phosphorylation, mitochondrial aggregation, motor deficits, and early mortality in transgenic mice. J. Neurosci. 30, 10851–10859. doi: 10.1523/JNEUROSCI.1630-10.2010
Yablonska, S., Ganesan, V., Ferrando, L. M., Kim, J., Pyzel, A., Baranova, O. V., et al. (2019). Mutant huntingtin disrupts mitochondrial proteostasis by interacting with TIM23. Proc. Natl. Acad. Sci. U S A. 116, 16593–16602. doi: 10.1073/pnas.1904101116
Yamano, K., Matsuda, N., and Tanaka, K. (2016). The ubiquitin signal and autophagy: an orchestrated dance leading to mitochondrial degradation. EMBO Rep. 17, 300–316. doi: 10.15252/embr.201541486
Yan, J., Liu, X. H., Han, M. Z., Wang, Y. M., Sun, X. L., Yu, N., et al. (2015). Blockage of GSK3 beta-mediated Drp1 phosphorylation provides neuroprotection in neuronal and mouse models of Alzheimer’s disease. Neurobiol. Aging 36, 211–227. doi: 10.1016/j.neurobiolaging.2014.08.005
Yang, X., Zhang, M., Dai, Y., Sun, Y., Aman, Y., Xu, Y., et al. (2020). Spermidine inhibits neurodegeneration and delays aging via the PINK1-PDR1-dependent mitophagy pathway in C. elegans. Aging (Albany NY) 12, 16852–16866. doi: 10.18632/aging.103578
Ye, X., Sun, X., Starovoytov, V., and Cai, Q. (2015). Parkin-mediated mitophagy in mutant hAPP neurons and Alzheimer’s disease patient brains. Hum. Mol. Genet. 24, 2938–2951. doi: 10.1093/hmg/ddv056
Yi, H. S., Chang, J. Y., and Shong, M. (2018). The mitochondrial unfolded protein response and mitohormesis: a perspective on metabolic diseases. J. Mol. Endocrinol. 61, R91–R105.
Yogalingam, G., Hwang, S., Ferreira, J. C., and Mochly-Rosen, D. (2013). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) phosphorylation by protein kinase Cdelta (PKCdelta) inhibits mitochondria elimination by lysosomal-like structures following ischemia and reoxygenation-induced injury. J. Biol. Chem. 288, 18947–18960. doi: 10.1074/jbc.M113.466870
Yu, S. B., and Pekkurnaz, G. (2018). Mechanisms orchestrating mitochondrial dynamics for energy homeostasis. J. Mol. Biol. 430, 3922–3941.
Yuan, Y., Chen, J., Ge, X., Deng, J., Xu, X., Zhao, Y., et al. (2021). Activation of ERK-Drp1 signaling promotes hypoxia-induced Abeta accumulation by upregulating mitochondrial fission and BACE1 activity. FEBS Open Bio 11, 2740–2755. doi: 10.1002/2211-5463.13273
Zanellati, M. C., Monti, V., Barzaghi, C., Reale, C., Nardocci, N., Albanese, A., et al. (2015). Mitochondrial dysfunction in Parkinson disease: evidence in mutant PARK2 fibroblasts. Front. Genet. 6:78. doi: 10.3389/fgene.2015.00078
Zhang, L., Trushin, S., Christensen, T. A., Bachmeier, B. V., Gateno, B., Schroeder, A., et al. (2016). Altered brain energetics induces mitochondrial fission arrest in Alzheimer’s disease. Sci. Rep. 6:18725. doi: 10.1038/srep18725
Zhang, Z., Liu, L., Jiang, X., Zhai, S., and Xing, D. (2016). The essential role of drp1 and its regulation by S-Nitrosylation of parkin in dopaminergic neurodegeneration: implications for Parkinson’s disease. Antioxid. Redox. Signal. 25, 609–622. doi: 10.1089/ars.2016.6634
Zhang, Q., Yang, C., Liu, T. Y., Liu, L., Li, F., Cai, Y. L., et al. (2018). Citalopram restores short-term memory deficit and non-cognitive behaviors in APP/PS1 mice while halting the advance of Alzheimer’s disease-like pathology. Neuropharmacology 131, 475–486. doi: 10.1016/j.neuropharm.2017.12.021
Zhang, W. (2021). The mitophagy receptor FUN14 domain-containing 1 (FUNDC1): a promising biomarker and potential therapeutic target of human diseases. Genes Dis. 8, 640–654. doi: 10.1016/j.gendis.2020.08.011
Zhang, W., Guo, J. H., Niu, X. Y., Wang, J., Wang, Z. J., Wu, M. N., et al. (2017). Citalopram ameliorates impairments in spatial memory and synaptic plasticity in female 3xTgAD mice. Biomed. Res. Int. 2017:1238687. doi: 10.1155/2017/1238687
Zhang, X., Wang, R., Hu, D., Sun, X., Fujioka, H., Lundberg, K., et al. (2020). Oligodendroglial glycolytic stress triggers inflammasome activation and neuropathology in Alzheimer’s disease. Sci. Adv. 6:eabb8680. doi: 10.1126/sciadv.abb8680
Zhao, C., Su, P., Lv, C., Guo, L., Cao, G., Qin, C., et al. (2019). Berberine alleviates amyloid beta-induced mitochondrial dysfunction and synaptic loss. Oxid. Med. Cell Longev. 2019:7593608. doi: 10.1155/2019/7593608
Zhao, Y., Sun, X., Hu, D., Prosdocimo, D. A., Hoppel, C., Jain, M. K., et al. (2019). ATAD3A oligomerization causes neurodegeneration by coupling mitochondrial fragmentation and bioenergetics defects. Nat. Commun. 10:1371. doi: 10.1038/s41467-019-09291-x
Zhao, F. L., Fang, F., Qiao, P. F., Yan, N., Gao, D., and Yan, Y. (2016). AP39, a mitochondria-targeted hydrogen sulfide donor, supports cellular bioenergetics and protects against Alzheimer’s disease by preserving mitochondrial function in APP/PS1 mice and neurons. Oxid. Med. Cell. Longev. 2016:8360738. doi: 10.1155/2016/8360738
Zhao, Q., Wang, J. H., Levichkin, I. V., Stasinopoulos, S., Ryan, M. T., and Hoogenraad, N. J. (2002). A mitochondrial specific stress response in mammalian cells. EMBO J. 21, 4411–4419.
Zheng, B., Liao, Z. X., Locascio, J. J., Lesniak, K. A., Roderick, S. S., Watt, M. L., et al. (2010). PGC-1 alpha, a potential therapeutic target for early intervention in Parkinson’s disease. Sci. Trans. Med. 2:52ra73. doi: 10.1126/scitranslmed.3001059
Zheng, Y. R., Zhang, X. N., and Chen, Z. (2019). Mitochondrial transport serves as a mitochondrial quality control strategy in axons: implications for central nervous system disorders. CNS Neurosci. Therapeut. 25, 876–886. doi: 10.1111/cns.13122
Keywords: neurodegenerative diseases, mitochondrial quality control, mitochondrial proteostasis, mitochondrial dynamics, mitophagy
Citation: Hu D, Liu Z and Qi X (2021) Mitochondrial Quality Control Strategies: Potential Therapeutic Targets for Neurodegenerative Diseases? Front. Neurosci. 15:746873. doi: 10.3389/fnins.2021.746873
Received: 25 July 2021; Accepted: 19 October 2021;
Published: 12 November 2021.
Edited by:
Shabab B. Hannan, University of Tübingen, GermanyReviewed by:
Angelika Harbauer, Max Planck Institute of Neurobiology, GermanyMaria Jose Perez, German Center for Neurodegeneratives, Helmholtz Association of German Research Centers (HZ), Germany
Copyright © 2021 Hu, Liu and Qi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xin Qi, eHhxMzhAY2FzZS5lZHU=