 Basharat Hussain
Basharat Hussain Cheng Fang
Cheng Fang Junlei Chang
Junlei Chang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Neurosci., 19 August 2021
Sec. Neurodegeneration
Volume 15 - 2021 | https://doi.org/10.3389/fnins.2021.688090
This article is part of the Research TopicDeciphering the biomarkers of Alzheimer’s diseaseView all 20 articles
The blood–brain barrier (BBB) plays a vital role in maintaining the specialized microenvironment of the neural tissue. It separates the peripheral circulatory system from the brain parenchyma while facilitating communication. Alterations in the distinct physiological properties of the BBB lead to BBB breakdown associated with normal aging and various neurodegenerative diseases. In this review, we first briefly discuss the aging process, then review the phenotypes and mechanisms of BBB breakdown associated with normal aging that further cause neurodegeneration and cognitive impairments. We also summarize dementia such as Alzheimer's disease (AD) and vascular dementia (VaD) and subsequently discuss the phenotypes and mechanisms of BBB disruption in dementia correlated with cognition decline. Overlaps between AD and VaD are also discussed. Techniques that could identify biomarkers associated with BBB breakdown are briefly summarized. Finally, we concluded that BBB breakdown could be used as an emerging biomarker to assist to diagnose cognitive impairment associated with normal aging and dementia.
The central nervous system (CNS) comprises the brain and spinal cord that control all the essential functions of the body. The distinctive physiological and anatomical structure of the brain and spinal cord makes the CNS a largely immune-privileged organ (Engelhardt and Coisne, 2011; Ransohoff and Engelhardt, 2012). Blood vessels are essential to transport oxygen and nutrients, remove CO2 and other waste products, and, thus, maintain homeostasis in the body. Blood vessels that vascularize the CNS acquire specific anatomical and functional characteristics that collectively form the blood–brain barrier (BBB) (Obermeier et al., 2013; Zhao et al., 2015).
At the cellular level, the BBB is developed by continuous non-fenestrated endothelial cells (ECs) encompassed by pericytes, smooth muscle cells, astrocytes, microglia, oligodendroglia, and neurons that are altogether called the neurovascular unit (NVU) (Zlokovic, 2011; Blanchette and Daneman, 2015; Chow and Gu, 2015). At the molecular level, the BBB ECs are compacted by claudins, occludins, and ZO-1 [tight junction (TJ) proteins] and junction adhesion molecules (JAM) proteins to restrict the paracellular and transcellular diffusion of molecules in the CNS. In addition, the BBB ECs mediate influx transporters to select metabolite uptake from the blood and efflux transporters to remove toxins and waste products from the brain into the blood. In BBB ECs, leukocyte adhesion molecules (LAMs) express very low to suppress immune surveillance in the brain (Quaegebeur et al., 2011; Engelhardt and Ransohoff, 2012; Chow and Gu, 2015; Xiao et al., 2020). Thus, the BBB confines the access of neurotoxic compounds, blood cells, and pathogens to the brain (Winkler et al., 2011). In addition, the BBB sustains the homeostasis of the brain through tight regulation of the transport of molecules between the brain parenchyma and peripheral circulation (Abbott, 2013). Figure 1A shows the normal BBB.
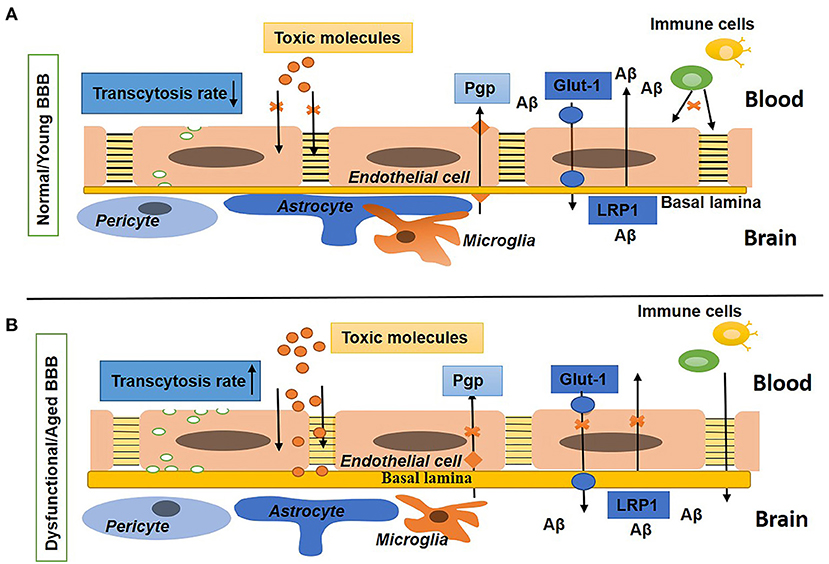
Figure 1. Schematic diagram shows the normal/young blood–brain barrier (BBB) and dysfunctional/aged BBB. (A) Shows BBB in a young or normal state with tight and adherens junctions, a low rate of transcytosis, no diffusion of toxins, the presence of influx (Glut-1) and efflux (P-gp) transporters, and a low expression of leukocytes adhesion molecules (LAMs). The basal lamina is thin and surrounded by pericytes, astrocyte endfeet, and microglia. (B) Shows BBB in an aged or disease state with a high rate of transcytosis and diffusion of toxins, repression in influx and efflux transporters, upregulated expression of LAMs, and increased density of the extracellular matrix (ECM). Pericytes, astrocytes, and microglia are not associated with the basal lamina.
Hence, the BBB is a fundamental and crucial element of normal and healthy brain function. Any impairment in the cellular or molecular components causes BBB breakdown that results in BBB dysfunction. Aging is one of several factors involved in the breaking of the BBB and was first observed in aged patients reported in the 1970s (Tibbling et al., 1977). In dysfunctional BBBs, the possibility of permeability increases; thus, toxic and blood-borne inflammatory substances that infiltrate the brain could change the biochemical microenvironment of the neurons, thus leading to neurodegenerative diseases and dementia (Abbott et al., 2010; Zeevi et al., 2010; Zlokovic, 2011; Rosenberg, 2014; Sweeney et al., 2018b). It has been reported that BBB disruption in aged people is strongly related to Alzheimer's disease (AD) and cognitive impairment (Farrall and Wardlaw, 2009; Van De Haar et al., 2016; Skillbäck et al., 2017; Zenaro et al., 2017; Sweeney et al., 2018b; Nation et al., 2019). Figure 1B shows the impaired BBB.
In this review, we first briefly discuss the aging process, and then review the phenotypes and mechanisms of BBB breakdown associated with normal aging that further cause neurodegeneration and cognitive impairments. We also summarize dementia such as AD and vascular dementia (VaD); then, we discuss the phenotypes and mechanisms of BBB disruption in dementia correlated with cognition decline. Subsequently, we also discuss the overlap between AD and VaD. Furthermore, we mention biomarkers associated with BBB breakdown during aging and dementia; additionally, we also briefly discuss various techniques to identify BBB biomarkers. Finally, we conclude that BBB breakdown could be used as a novel biomarker to diagnose cognitive impairment associated with normal aging and dementia.
The universal process in an organism leads to the cumulation of biological variations responsible for progressively diminishing bodily functions over time, which is known as aging (Kritsilis et al., 2018). Because of the advancement in medicine and the living standard of humans, life expectancy has doubled worldwide (Aw et al., 2007). Aged people are estimated to make up approximately 20% of the world population in the next 50 years (Ellison et al., 2015). In terms of the brain and the BBB, normal aging can be defined as a retrogression in the activities of the body with no cognitive ailment and dementia. Although ailments do not occur in this case, the frequency of age-related diseases increases with the aging process. Alzheimer's, cardiovascular, Parkinson's disease, stroke, and various other neurological diseases commonly occur in aged people (Erdo et al., 2017). A recent study demonstrated that BBB breakdown could be considered a biomarker for the normal aging process (Verheggen et al., 2020). Furthermore, BBB breakdown also impairs the influx of nutrients (glucose) and oxygen and efflux of waste products, which may cause hypoxia-associated inflammation (Elahy et al., 2015; Raja et al., 2018). Subsequently, age-related BBB pathology makes the brain more susceptible to neuronal impairment and even causes neurodegeneration (Levit et al., 2020; Banks et al., 2021). It has been reported that aged people with prior cognitive impairment were more vulnerable to BBB disruption than people with no cognitive dysfunction of the same age; hence, BBB disruption can be considered an early biomarker related to declines in human cognition (Nation et al., 2019). All these studies show that the way alterations in BBB components progress with time might be an interesting research topic to explore in association with the normal aging brain.
During aging, various changes occur in the structure and function of brain vasculature. In the aged brain, the BBB becomes broken; hence, the permeability of the BBB elevates (Villeda et al., 2011; Hyman et al., 2012) and declines in the cerebral blood flow (CBF) occur (Tarumi and Zhang, 2018). The potency of neovascularization diminishes (Rivard et al., 2000; Gao et al., 2009) and the density of capillary of brain vasculature reduces with age (Reeson et al., 2018). It has been observed that, during aging, BBB breakdown is the first incident that starts in the hippocampus, which may lead to declines in cognition (Montagne et al., 2015). In normal aging, the main changes that are strongly correlated to BBB breakdown are presented in Table 1.
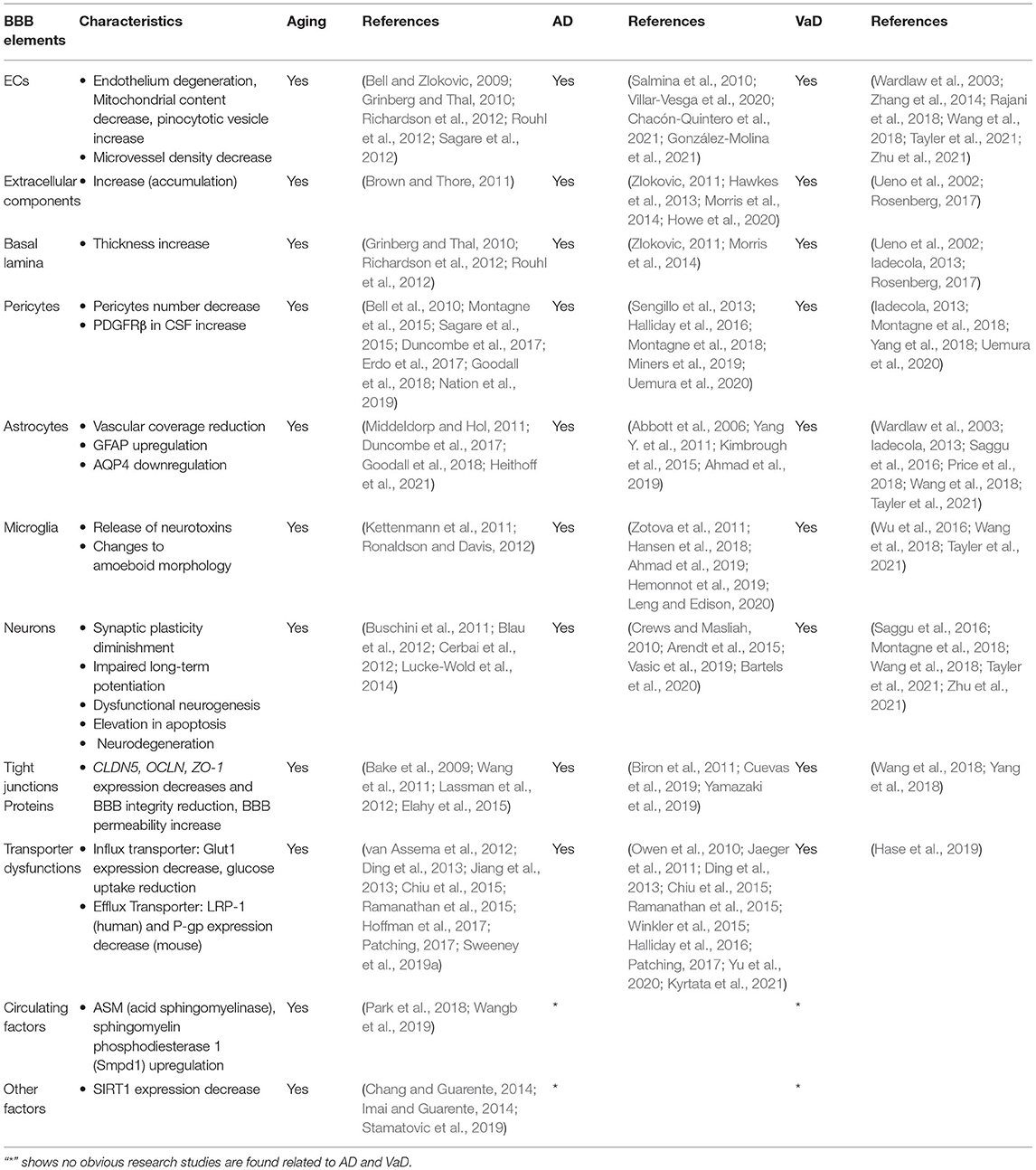
Table 1. Changes associated with blood–brain barrier (BBB) breakdown in normal aging, Alzheimer's disease (AD), and vascular dementia (VaD).
It has been reported that, in aging, the brain endothelium becomes progressively dysfunctional, which is correlated with aberrant changes in the BBB (Cai W. et al., 2017; Edwards et al., 2019). The extracellular matrix (ECM) of the basal membrane or basal lamina covers the brain endothelium and is considered uniform and thin. In normal aging, the thickness of the ECM increases with the increase in collagen IV and argin but decreases in laminin concentrations (Candiello et al., 2010). Although the ECM has a role in maintaining BBB integrity by inducing TJ (occludin) protein expression, changes in the ECM cause BBB disruption, and thus result in increased BBB permeability (Hawkins and Davis, 2005; Candiello et al., 2010; Sanchez-Covarrubias et al., 2014).
In the CNS BBB, ECs associated with pericytes, astrocytes, neurons, and glial cells that develop and maintain their specific phenotype led to BBB integrity (Erickson and Banks, 2018). However, with aging, this association caused BBB breakdown. During aging, physiological ultrastructure changes have been reported in pericytes, such as an increase in mitochondria size (Hicks et al., 1983), vesicular and lipofuscin-like inclusions (Rascher and Wolburg, 2002), and foamy transformations (Sturrock, 1980). In addition, the protrusions on the basal lamina or the ECM membrane of the microvessels have been observed to result in the degeneration of pericytes (Ueno et al., 1998). A loss of pericytes has also been reported in aging mice, rats, and the human brain (Stewart et al., 1987; De Jong et al., 1990; Bell et al., 2010; Duncombe et al., 2017; Goodall et al., 2018) but some studies observed that the number of pericytes increases in aged rat brains (Heinsen and Heinsen, 1983; Peinado et al., 1998). However, no change was observed in the number of pericytes in aged monkey brains (Peters et al., 1991).
Platelet-derived growth factor receptor beta (PDGFRβ) maintains the phenotype of pericytes in the brains of aged mice with PDGFRβ+/−, which shows that the loss of pericytes leads to BBB breakdown and increased BBB permeability (Bell et al., 2010). It has been reported, in the aged human brain, the level of soluble PDGFRβ in cerebrospinal fluid (CSF) increases, showing damage to the pericyte associated with BBB disruption (Montagne et al., 2015; Sagare et al., 2015; Nation et al., 2019). In addition, it has been reported that, in APOE4 carriers, the elevated PDGFRβ in the CSF may be used as a biomarker of cognitive impairment (Montagne et al., 2020).
The endfeet of astrocytes that ensheath the pericytes have a contribution to BBB development and maintenance. With the age, vascular coverage and aquaporin-4 (AQP4) expression of astrocyte endfeet are reduced whereas glial fibrillary acidic protein (GFAP) expression and endfeet sizes are increased (Middeldorp and Hol, 2011; Duncombe et al., 2017; Goodall et al., 2018), leading to increase in reactive astrogliosis.
Microglia are distributed ubiquitously in the CNS and activated during aging and pathology (Kettenmann et al., 2011; Kofler and Wiley, 2011; Harry, 2013; Sanchez-Covarrubias et al., 2014). Microglia have a ramified structure in the resting state, but when activated, this structure changes into an amoeboid morphology during aging or a pathophysiological state (Kettenmann et al., 2011). During aging or stress, the activated microglia produce tumor necrosis factor-α (TNF-α), proteases, nitric oxide (NO), and peroxide (Ronaldson and Davis, 2012), which are associated with an alteration in the TJ protein. This alteration induces BBB leakage (Huber et al., 2006), which, in turn, leads to cell injury and neurodegeneration (Ronaldson and Davis, 2012).
Studies have shown that neurons directly connect with brain ECs and astrocytes (Ben-Menachem et al., 1982; Cohen et al., 1996, 1997; Tong and Hamel, 1999; Vaucher et al., 2000; Sanchez-Covarrubias et al., 2014). Impairment in this association results in BBB breakdown and leads to an increase in BBB permeability to albumin (Berezowski et al., 2004). Figure 1 shows the difference between young or normal BBB and aged or dysfunctional BBB.
During aging, various mechanisms cause BBB breakdown and increase BBB permeability. For example, in aging, oxidative stress induces ECs to produce TNF-α that cause the degradation of the basement membrane, and TJs (Occludin, Zonula occludins-1), which, in turn, results in BBB disruption and an increase in BBB permeability (Donato et al., 2007; Bake et al., 2009; Lee et al., 2012; Elahy et al., 2015; Cai W. et al., 2017). In addition, the activity of caspase 3/7 in the aged brain increases, which causes the suppression of cell viability and the upregulation of apoptosis in pericytes (Schultz et al., 2018), resulting in the reduction of the number of pericytes in the BBB (Bell et al., 2010). The senile pericytes produce NO and react with O2, causing increased oxidative stress and compromised BBB integrity (Hughes et al., 2006; Sweeney et al., 2016; Cai W. et al., 2017). Similarly, in aging, oxidative stress enhances astrocytes to upregulate the expression of cytokines and chemokines, such as matrix metalloproteinase 3 (MMP3) and p16INK4A [senescence-associated secretory phenotype (SASP)], that induce BBB disruption, neuroinflammation, and cognitive impairments (Simpson et al., 2010; Salminen et al., 2011; Cai Z. et al., 2017; Bussian et al., 2018). In aging, oxidative stress also activates the microglia to release cytokines, chemokines IL-6, IL1ß, and TNF-α, which results in the elevation of reactive oxygen and nitrogen species; this ultimately causes the breakdown of the BBB (Gredilla et al., 2010; Choi et al., 2014; Fivenson et al., 2017). With age, oxidative stress causes the production of reactive oxygen species (ROS) to elevate in the CNS, but the capability of neurons to clear ROS decreases, resulting in neurodegeneration (Nicholls and Budd, 2000; Mattson and Magnus, 2006). Furthermore, with age, calcium dysregulation in neurons occurs, which represses calcium-binding proteins correlated with the elevation of ROS. This results in BBB degradation and neuronal loss (He et al., 1997) as shown (Figure 2).
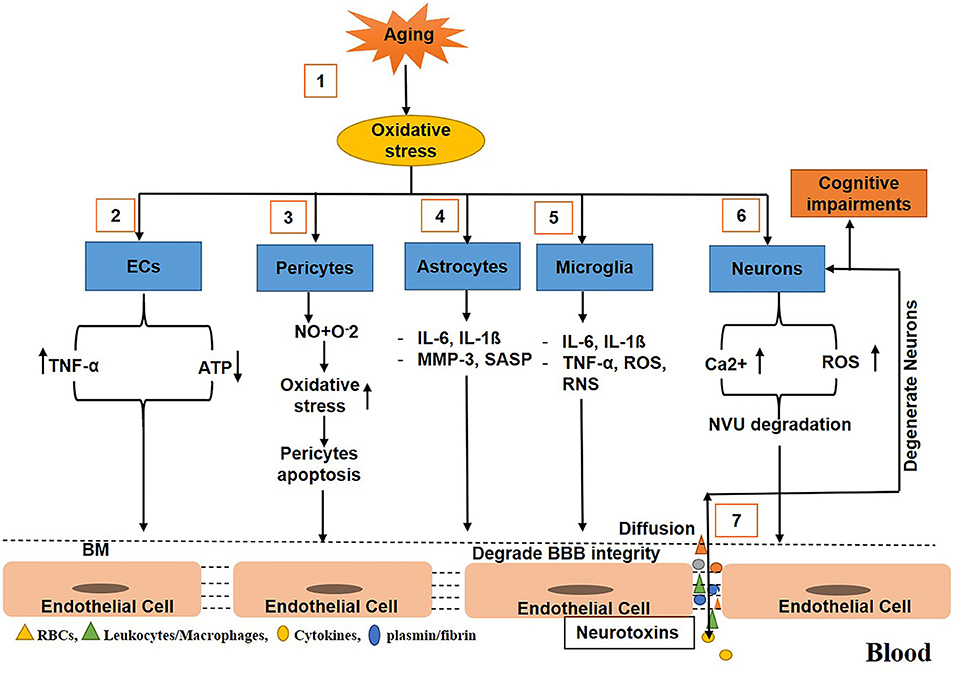
Figure 2. Schematic diagram shows the mechanisms of blood–brain barrier (BBB) breakdown in normal aging. (1) Oxidative stress increases with age. (2) Oxidative stress triggers endothelial cells ECs to release tumor necrosis factor-α (TNF-α) and consume more ATP. (3) Oxidative stress induces pericytes to release nitric oxides that react with reactive oxygen to further upregulate the oxidative stress causing pericytes apoptosis associated with loss in BBB integrity. (4) During aging, oxidative stress stimulates and activates astrocytes to release cytokines and chemokines that degrade the basement membrane and tight junction leading to BBB impairment. (5) Oxidative stress also activates the microglia to secrete cytokines, chemokines, reactive oxygen, and nitrogen species, causing degradation in BBB integrity. (6) Oxidative stress also induces neurons to release reactive oxygen species (ROS) and calcium ions accumulation that cause to degrade the neurovascular unit (NVU). (7) Toxins freely diffuse to and from the brain, causing neurodegeneration and decline in cognition.
Once the BBB integrity becomes compromised, blood-derived proteins such as fibrinogen and plasminogen cross the BBB, and the pro-inflammatory fibrin aggregates in the brain (Cortes-Canteli et al., 2015). A study using a mouse model showed that accumulated fibrin bind with CD11b/CD18 and activate microglia, which then triggers a decline in cognition (Merlini et al., 2019). Accumulated fibrin in brain also induce increased ROS level and activates nicotinamide adenine dinucleotide phosphate (NADPH oxidase), which upregulates pro-inflammatory gene expression and causes damage to neuronal axons (Ryu et al., 2018; Merlini et al., 2019). In addition, fibrinogen phosphorylates Smad 1/5/8 represses oligodendrocyte progenitor cells (OPCs) (Ryu et al., 2015). Furthermore, the complex of Aβ-fibrinogen activates microglia via CD11b/CD18, which inhibits the breakdown of fibrinogen and promotes neuronal degeneration (Cortes-Canteli et al., 2010; Zhao et al., 2017) as shown (Figure 3A).
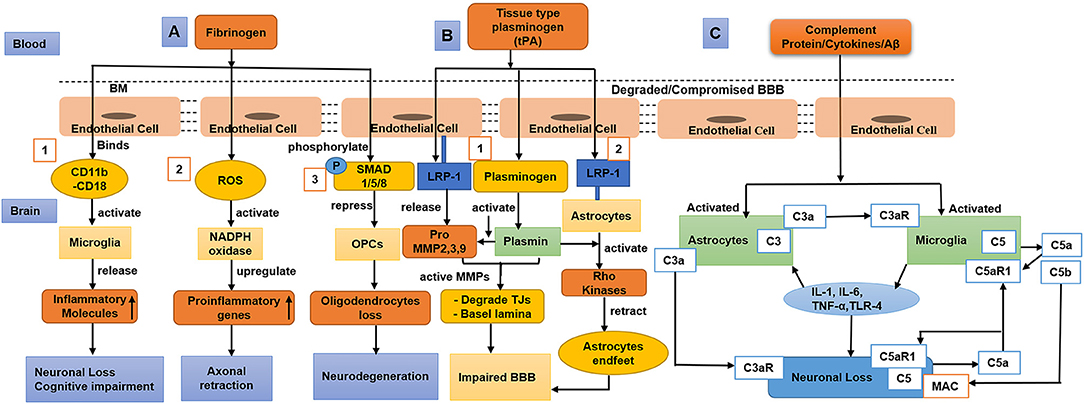
Figure 3. Diagram that shows fibrinogen, tissue-type plasminogen activator (tPA), plasmin, and complement proteins/cytokines/Aβ cross compromised the blood–brain barrier (BBB), causing neuronal loss and decline in cognition. (A) (1) Fibrinogen activates the microglia via CD11b/CD18, which releases inflammatory/toxic molecules causing neurodegeneration and cognitive impairment. (2) Fibrinogen induces reactive oxygen species (ROS), activates nicotinamide adenine dinucleotide phosphate (NADPH oxidase), and upregulates proinflammatory genes causing axonal retraction and cognitive impairment. (3) Fibrinogen phosphorylates SMAD 1/5/8 and represses oligodendrocyte progenitor cells (OPCs) leading to oligodendrocyte loss. (B) (1) tPA binds with low-density lipoprotein receptor-related protein-1 (LRP-1) of endothelial cells (ECs), which secretes pro-matrix metalloproteinases (MMPs). tPA activates plasminogen into plasmin, which further activates pro-MMPs that degrade tight junctions (TJs) and basal lamina. (2) tPA also binds with the LRP-1 of astrocytes, induces plasmin mediated activation of Rho kinase, and results in astrocyte retraction leading to BBB impairment. (C) During aging and dementia such as Alzheimer's disease (AD), complement proteins, oxidative stress, and the aggregated amyloid-beta (Aβ) activate the astrocytes and microglia, leading to neuroinflammation. The C3aR and C5aR1 signalings on activated microglia cause the release of cytokines [IL-1, IL-6, tumor necrosis factor-α (TNF-α), and toll-like receptor 4 (TLR-4)] and result in neurodegeneration. To aggravate neuronal apoptosis, C5a as a neuronal-derived signal that interacts with C5aR1 on neurons in an autocrine way. Furthermore, C3a as an astrocytic-derived signal binds to C3aR on neurons to exacerbate neuronal morphology. During neuroinflammation, microglia-derived C5b binds with the membrane attack complex (MAC) that enhances neuronal loss.
The tissue-type plasminogen activator (tPA) binds and activates low-density lipoprotein receptor-related protein-1 (LRP-1) on ECs. In run, these ECs produce pro-MMPs (MMP-2, MMP-3, and MMP-9) (Wang et al., 2003; Cheng et al., 2006; Suzuki et al., 2009). Subsequently, tPA converts the surface-bound inactive plasminogen (Plg) into active plasmin (Plm) (Doeuvre et al., 2010; Yepes et al., 2021). Plasmin, in turn, activates the MMPs, leading to the degradation of TJs and basal lamina (Mazzieri et al., 1997; Ramos-DeSimone et al., 1999; Monea et al., 2002; Rosenberg and Yang, 2007; Yang Y. et al., 2011). Furthermore, tPA also binds with LRP-1 on astrocytes, which induces plasmin-mediated activation of Rho kinases and retracts the endfeet of the astrocytes from the blood vessel wall, thus resulting in BBB dysfunction (Niego et al., 2012). In addition, a study suggested that plasminogen might regulate brain inflammation during AD (Baker et al., 2018) as shown (Figure 3B).
Human brain ECs continuously produce complement regulatory proteins and components (Wu et al., 2016), which are elevated by CNS injury or infiltration into the brain when the BBB is dysfunctional. However, at a young age or in a normal state, complement proteins mostly do not cross the BBB (Hoarau et al., 2011; Veerhuis et al., 2011). Once the complement proteins cross the compromised BBB, they could alter the functions of the microglia, oligodendrocytes, and neurons (Orsini et al., 2014). Complement activation produces C3a and C5a that interact with C3aR and C5aR1, respectively, which play a significant role in the infiltration of inflammatory cells into the brain and the induction of cytokine cascades (IL-1, TNF-α, IL-6, IL-8, IL-17) subsequently leading to neurodegeneration (Jacob and Alexander, 2014; Alexander, 2018). In AD, amyloid-beta (Aβ) activates the complement signaling by binding to C1q. Inhibition of the C5/C5aR1 pathway was also reported to be a protective therapeutic target in AD (Fonseca et al., 2009) as shown (Figure 3C).
Dementia is a group of conditions or disorders that affect the functions of the brain. It is a progressive neurological disease associated with impairments in cognition and deterioration of the everyday life activities of an affected individual (Mills et al., 2007; Kirshner, 2009). In dementia, BBB breakdown and cerebral hypoperfusion cause brain damage and a decline in cognition (Nation et al., 2019; Tayler et al., 2021). Dementia is a considerable health complication affecting millions of people worldwide. In developed countries, AD and VaD are two significant types of dementia with a prevalence of about 4.4 and 1–2%, respectively (Ray et al., 2013), with AD being the most common type of dementia in aged people (Ballaed et al., 2011; Hyman et al., 2012). The World Alzheimer Report 2018 estimated that approximately 50 million people of the global population suffer from dementia, which can increased to 82 million in 2030 and triple to 152 million by 2050 (Patterson, 2018). Alzheimer's is considered to account for 60–70% of all dementia cases worldwide (Leng and Edison, 2020). As the BBB has vital contributions to maintaining the microenvironment of the CNS, any impairment in the cellular or molecular components of the BBB can cause various neurodegenerative diseases, including AD (Zlokovic, 2005; Erickson and Banks, 2013; Zenaro et al., 2017). After AD, VaD is the second most common type of dementia, accounting for 15% of all dementia cases worldwide (O'Brien and Thomas, 2015). Vascular dementia is a type of neurological disease with a defect in cognition caused by impairment in the vascular system, such as a reduction in CBF (Sabayan et al., 2012). Various vascular pathologies are associated with VaD, such as infarcts and white matter (WM) alterations (O'Brien and Thomas, 2015). In addition, brain hemorrhage, ischemia, and hypoxia may be the causing factors of VaD (Kirshner, 2009; Grinberg and Heinsen, 2010).
Aging is responsible for pathophysiological changes that aggravate neurological diseases. It causes the thickening of the wall of the blood vessel and increases blood vessel tortuosity, which may lead to BBB disruption (Rosenberg, 2012). The BBB breakdown in AD results in the accumulation of insoluble extracellular plaques of β-amyloid (Aβ) along the walls of blood vessels and causes inflammation in the NVU (Kinnecom et al., 2007; Kang et al., 2017). In neuronal cytoplasms, the accumulation of neurofibrillary tangles (NFT) of P-tau is also associated with AD (Kang et al., 2017). It has been observed that, in AD, the reduction of Aβ clearance is correlated with declines in CBF and cognitive impairment (Sagare et al., 2012). These pathological markers are associated with BBB impairment, which causes microglial activation, neuroinflammation, degeneration of neurons, and cognitive impairment (Bhaskar et al., 2010; Iadecola, 2013). As pericytes have a crucial role in the development and maintenance of BBB, their number and density decreased in the cortex and hippocampus of AD patients (Sengillo et al., 2013), subsequently leading to the upregulation of the expression of Aβ and p-tau protein (Sagare et al., 2013).
Vascular (stroke, hypertension, diabetes, etc.) and genetic factors (APOE4) are two pathways that cause BBB impairment and oligemia (reduced CBF) that result in dementia. In the Aβ-independent pathway (blue), the BBB breakdown causes a release of neurotoxins from one side and leads to CBF reduction on another side. In the Aβ-dependent pathway (green), the BBB breakdown impairs the clearance of Aβ and APP (amyloid precursor protein), leading to the aggregation of Aβ in the brain. The accumulated Aβ and vascular hypoperfusion phosphorylate tau, leading to the formation of NFTs. In addition, the deposited Aβ also cause inflammation in the brain. In conclusion, both factors and pathways cause neurodegeneration leading to dementia (AD) (Iadecola and Davisson, 2008; Jack, 2010; Winkler et al., 2011; Sagare et al., 2013; Edwards et al., 2019) as shown (Figure 4).
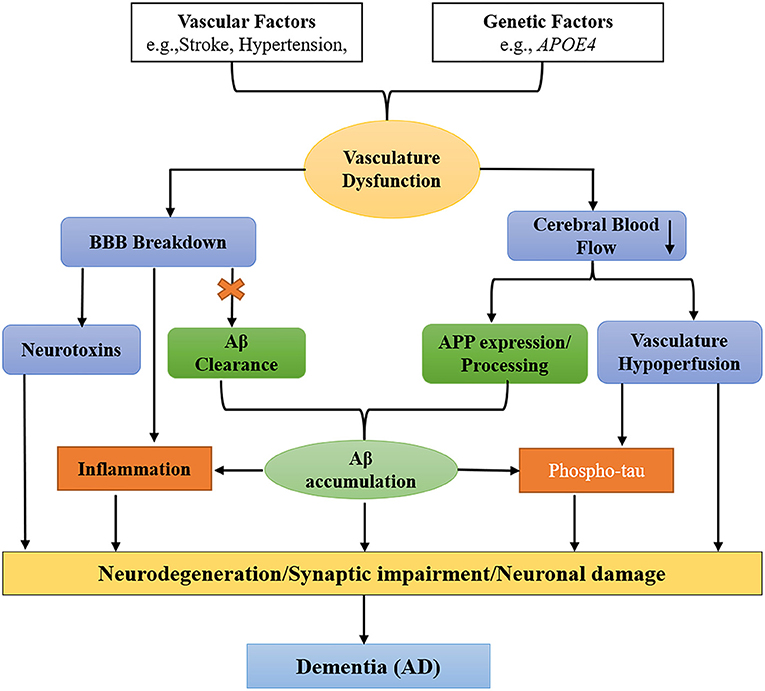
Figure 4. Schematic diagram shows two pathways that cause dementia [specifically Alzheimer's disease (AD)]. Vascular factors (stroke, hypertension, diabetes, etc.) and genetic factors (such as APOE4) cause defects in the vascular system leading to blood–brain barrier (BBB) impairment and oligemia (reduced cerebral blood flow), which finally correlate with dementia and neuronal degeneration. In the amyloid-beta (Aβ)-independent pathway (blue), vascular and genetic factors cause BBB breakdown and the secretion of neurotoxins from one side and oligemia from the other side. While in the Aβ-dependent pathway (green), the first BBB breakage impairs Aβ clearance and the amyloid precursor protein (APP), resulting in the accumulation of Aβ in the brain. Vascular hypoperfusion and Aβ phosphorylate tau (p-tau) that forms neurofibrillary tangles (NFTs). Finally, both pathways cause neurodegeneration/synaptic impairment/neuronal damage leading to dementia (specifically AD).
In AD patients, the BBB is shown as leakages in brain vasculature, the perivascular aggregation of fibrinogen, albumin, thrombin, and immunoglobulin (IgG), the loss of TJs, and the degeneration of ECs and pericytes (Nelson et al., 2016). Furthermore, identical phenotypes were also observed in Apoe−/− mice due to BBB impairment (Nishitsuji et al., 2011; Bell et al., 2012; Hammer et al., 2014; Soto et al., 2015; Castillo-Gomez et al., 2016; Di Cataldo et al., 2016), indicating that ApoE is vital for maintaining BBB integrity.
As pericytes are crucial for maintaining the BBB, any dysfunction in the signaling pathways of pericytes results in the breakdown of BBB, which causes dementia and other neurodegenerative diseases (Sagare et al., 2013; Nikolakopoulou et al., 2019). Brain microvascular endothelial cells (BMEC) secrete platelet-derived growth factor BB (PDGF-BB) and activate PDGFRβ signaling, which is essential for the proliferation, migration, and survival of pericytes (Stratman et al., 2010). An impairment in PDGFRβ signaling leads to pericyte degeneration (Stratman et al., 2010; Nation et al., 2019). According to a previous study, PDGFRβ signaling was decreased in adult Foxf2 deficient mice, thus resulting in high BBB permeability (Reyahi et al., 2015). Impairment in the BBB was also reported in Pdgfrβ+/− pericyte-deficient mice, which subsequently caused neuronal degeneration (Bell et al., 2010). In the AD murine model (APPsw/0), the deterioration of pericytes results in the dysfunction of the BBB, leading to amyloid β accumulation and tau protein (p-tau) phosphorylation (Sagare et al., 2013). The breakdown of the BBB was also reported in AD patients associated with the reduction in pericytes (Sengillo et al., 2013). Studies also showed that the leakage of the BBB in AD patients starts at the hippocampus, resulting in an increase of soluble PDGFRβ (sPDGFRβ) in the CSF (Montagne et al., 2015; Miners et al., 2019). Additionally, the level of sPDGFRβ in the CSF can be used as a biomarker to predict dementia and other neurodegenerative diseases such as AD (Nation et al., 2019).
Astrocytes are one of the main components of the NVU and are essential for the integrity of the BBB. In an in vitro study, it was observed that Sonic hedgehog (Shh) signaling released from astrocytes plays a vital role in the maintenance of BBB integrity by upregulation of CLDN5 and OCLN (Alvarez et al., 2011; Wang et al., 2014). Recently, it was also reported that, in the stroke mouse model, ischemia-induced astrogliosis led to the downregulation of the expression of TJ protein claudin-5 and occludin (Matthes et al., 2021), suggesting that astrocytes have a role in the regulation of TJ proteins. Another recent study reported that, in the tamoxifen-induced astrocyte ablation adult mouse model, the expression of TJ protein ZO-1 was downregulated in vessel regions where astrocyte loss occurred, which may show the role of astrocytes in maintaining the integrity of the BBB in adult brains (Heithoff et al., 2021). A conditional knockout mouse study also showed that the deletion of laminins in astrocytes caused a decline in astrocytic AQP4 (Aquaporin4) expression, thus leading to a loss of TJ in ECs (Yao et al., 2014). In the AD brain, various changes in the morphology of astrocytes have been reported to cause BBB breakdown (Cai Z. et al., 2017). The depolarization of astrocyte endfeet may diminish the integrity of BBB, which was reported in the tg-ArcSwe mouse model of AD(Yang J. et al., 2011). In AD models, researchers also identified several changes in the morphology of astrocytes endfeet near aggregated vascular Aβ (Kimbrough et al., 2015).
A mouse study showed that microglia stimulate TJ protein claudin-5 expression and maintain BBB integrity (Haruwaka et al., 2019). However, the BBB integrity becomes compromised with prolonged inflammation through the changing of the morphology of microglia (Lassman et al., 2012; Haruwaka et al., 2019). In the AD brain, due to the accumulation of Aβ, microglia activate and secret inflammatory cytokines, such as interleukins (IL-1 and IL-6) and tumor necrosis factor (TNF-α, and TNF-β) (Zhou et al., 2012) that cause BBB impairment (Wang et al., 2014). As a result, the trafficking of neutrophils through the BBB becomes elevated due to BBB breakdown (Allen et al., 2012; Wang et al., 2014; Zenaro et al., 2015). Furthermore, it has been observed that, in the tamoxifen-induced astrocyte knockout adult mouse model, the loss of astrocytes causes the activation of microglia (Heithoff et al., 2021); in turn, the activated microglia produce reactive oxygen and reactive nitrogen species (RNS), leading to BBB dysfunction and neurodegeneration (Block, 2008; Sumi et al., 2010).
In the physiological state, perivascular macrophages (PVMs) have a significant role in the maintenance of TJs between ECs. They also decrease vessel leakage, degrade pathogens, and limit inflammation (Lapenna et al., 2018) while contributing to BBB breakdown in the disease state (Boyle et al., 2018). These PVMs are enriched with scavenger receptors, might be involved in the clearance of toxin products from the brain parenchyma (Faraco et al., 2017), and have a diverse role in disease states such as AD (Lapenna et al., 2018). Perivascular macrophages have been shown to phagocytose and alleviate Aβ plaques, and the PVM-deficient mouse model showed an increased aggregation of Aβ42 and cerebral amyloid angiopathy (CAA) related with AD (Yang et al., 2019). Another study showed that PVMs that are deficient in CD36 and Nox2 abrogated the production of ROS and Aβ cerebrovascular impairment compared with wild-type mice (Park et al., 2017).
In addition, perivascular fibroblasts (FBs) express the ECM genes col1a2 and col5a1 and are considered to mediate blood vessel integrity. Zebrafish deficient with col5a1 showed spontaneous hemorrhage in the presence of the additional genetic ablation of the col1a2 gene, suggesting the role of perivascular FBs in stabilizing vascular integrity (Rajan et al., 2020). Perivascular FBs also express Lama2, Lamb1, and Lamc1, which encode laminin 211 that interacts with astrocytic dystrophin, resulting in the regulation of AQP4 in astrocytic endfeet. This study suggests that any impairment in perivascular FBs result in the dysregulation of AQP4, which may cause Aβ aggregation and AD as reviewed by Lendahl et al. (2019). Furthermore, it has been reported that alteration in the activity of perivascular FBs also leads to other neurological disorders (Månberg et al., 2021).
Various pathological and aberrant events such as oxidative stress, inflammation, and the ApoE4 genotype cause BBB breakdown associated with AD. Research has shown that, in AD, the activation of the inflammatory and oxidative stress signaling pathways is the primary event that causes BBB disruption (Perry et al., 2002; Candore et al., 2010; Eikelenboom et al., 2012). Cytokines (Pan et al., 2011), Aβ (Gonzalez-Velasquez et al., 2008; Deli et al., 2010; Carrano et al., 2011), LPS (Bannerman and Goldblum, 1999; Verma et al., 2006), and p-tau proteins (Kovac et al., 2009) are the stimulus of inflammation for the activation of inflammatory pathways in BBB ECs. Hence, increases of the pro-inflammatory mediators and ROS/RNS in BBB ECs, astrocytes (Tada et al., 1994), and pericytes (Kovac et al., 2011; Takata et al., 2011) ultimately cause BBB breakdown. Recently we reviewed the role of peripheral inflammation in BBB breakdown (Huang et al., 2021). Glucose transporter protein (GLUT1) is repressed in the endothelium of AD, which causes a decline in the glucose level of the CNS (Mooradian et al., 1997; Winkler et al., 2015). In human AD, LRP1, which is a primary receptor for the clearance of amyloid-β, is downregulated with an increase in oxidative stress (Deane et al., 2004; Donahue et al., 2006; Sagare et al., 2007; Miller et al., 2008; Owen et al., 2010; Halliday et al., 2016); as a result, the transport of Aβ from the brain becomes reduced and leads to amyloid-β accumulation in the brain (Deane et al., 2004, 2008; Storck et al., 2016). In mice, systemic inflammation with LPS has been observed to downregulate both LRP-1 and P-gp efflux transporters and block the Aβ clearance from the brain (Jaeger et al., 2011; Erickson et al., 2012). Furthermore, it has been reported that the expression levels of the receptor for advanced glycosylation end products (RAGE) in both mural cells and brain endothelium were elevated (Deane et al., 2003; Donahue et al., 2006; Miller et al., 2008). The function of the RAGE is to transfer Aβ from blood to the brain (opposite to LRP1), which enhances neuronal inflammation. In AD patients, the RAGE is observed as a significant therapeutic target (Bell et al., 2012). A transgenic mouse with overexpressed APP (amyloid precursor protein) has been reported to show vascular impairment due to the elevation of Aβ40 (Niwa et al., 2000).
Aquaporin-4 (AQP4) is the prime water channel expressed in the CNS and is primarily expressed in astrocytes, thus playing a vital role in normal brain homeostasis and various neurological diseases (Lan et al., 2016b). Specifically, AQP4 facilitates the clearance of Aβ, and alteration in AQP4 expression leads to the accumulation of amyloid-β in the brain (Hoshi et al., 2012; Yang et al., 2012). Furthermore astrocytic AQP4-deficient animals cannot efficiently remove Aβ from the brain (Iliff et al., 2012). A study showed that AQP4 is crucial to regulate fluid flow in the brain interstitial required to maintain the microenvironment for neurons to function properly. The perturbed AQP4 expression has been observed to cause Aβ deposition and inflammation in the human brain, which leads to AD (Rasmussen et al., 2018). In AD patients and animal models, the expression and distribution of AQP4 were altered, leading to amyloid-β accumulation, which plays a vital role in the pathogenesis of AD as reviewed by Yang et al. (2016). Furthermore, it has been observed that, in AD patients, the localization of AQP4 in the perivascular space was reduced and is associated with an increase in neurofibrillary and amyloid-β pathology (Zeppenfeld et al., 2017). In addition, AQP4 facilitates the transport of potassium and calcium ions, which plays an essential role in the pathogenesis of AD as reviewed by Lan et al. (2016a). In AD, the chronic activation of microglia leads to the release of abundant pro-inflammatory cytokines and abolishes phagocytosis, thus causing the deposition of Aβ and neuroinflammation (Krabbe et al., 2013; Heneka et al., 2015) and subsequently producing ROS that causes BBB dysfunction and neurotoxicity (Block, 2008; Sumi et al., 2010). The activated microglia also release IL-1β (a pro-inflammatory cytokine) that amplify the BBB leakage and diminish the ability of the astrocytes to maintain the BBB (Wang et al., 2014). Therefore, AQP4 can be a fascinating therapeutic target for AD and other CNS diseases.
Apolipoprotein E (ApoE) is a protein encoded by the APOE gene, located on chromosome 9 and associated with lipid transport. APOE consists of three alleles, namely, ε2, ε3, and ε4, translated to ApoE2, ApoE3, and ApoE4 isoforms. The APOE isoform distributed as APOE3 is the most abundant in humans at approximately 77.9%, while APOE4 and APOE2 distributions are 13.7 and 8.4%, respectively (Farrer et al., 1997). In the CNS, astrocytes produce ApoE, whereas, in peripheral tissue, ApoE production occurs in the liver (Liu et al., 2013).
Studies reported that ApoE plays an essential role in maintaining BBB integrity (Nishitsuji et al., 2011). An in vivo study showed that ApoE2/3 induces BBB integrity by interacting with LRP-1 on pericytes to block the cyclophilin-A nuclear factor kβ matrix metalloproteinase 9 (CypA-NF-kβ-MMP-9) pathway, thus resulting in the inhibition of MMPs (Bell et al., 2012). Researchers also observed that the APOE4 isoform is a major risk factor for AD, and that the binding of Aβ with apoE4 shifts fast clearance of soluble Aβ40/42 from LRP1 to VLDLR; hence, Aβ-apoE4 complexes at the BBB are cleared with a slower rate than LRP1 (Deane et al., 2008; Tachibana et al., 2019). The expression of APOE4 causes a reduction in BBB integrity by promoting pericyte degeneration in AD (Bell et al., 2012), which is correlated with high BBB permeability to IgG and fibrin (Halliday et al., 2016). In a transgenic mouse study, the mice that had Apoe replaced with human APOE (TR-APOE) showed astrocytes that secreted ApoE4 blocks pericytic LRP-1, resulting in the activation of the proinflammatory CypA-NF-kB MMP9 pathway, BBB disruption, and brain hemorrhage through the enzymatic breakdown of the TJ and basement membrane (Nishitsuji et al., 2011; Bell et al., 2012). A study showed that an LRP1 endothelial knockout caused the activation of the CypA–MMP9 pathway in the endothelium, which led to damage to TJs and BBB breakdown (Nikolakopoulou et al., 2021). In TR-APOE4 mice, the repression of Glut1 and upregulation of RAGE expression were also observed compared with TR-APOE3 or TR-APOE2 (Alata et al., 2015). It has been reported that humans carrying APOE4 are more prone to breakdown in the BBB and loss of pericytes than non-APOE4 carriers (Hultman et al., 2013; Zonneveld et al., 2014; Halliday et al., 2016). Furthermore, CypA and MMP-9 levels increase in APOE4 carriers, leading to the elevation of IgG and fibrinogen leakages (Halliday et al., 2016). Overall, these results suggest that ApoE2/3 represses inflammation by interacting with pericyte LRP-1, subsequently inducing BBB integrity. In contrast, the ApoE4 might have BBB impairment properties or cause a higher risk of BBB breakdown. The repression of ApoE4 or inhibition of the CypA–MMP9 pathway in humans with AD might be an exciting topic in the future for the reduction the neurodegenerative process (Figure 5).
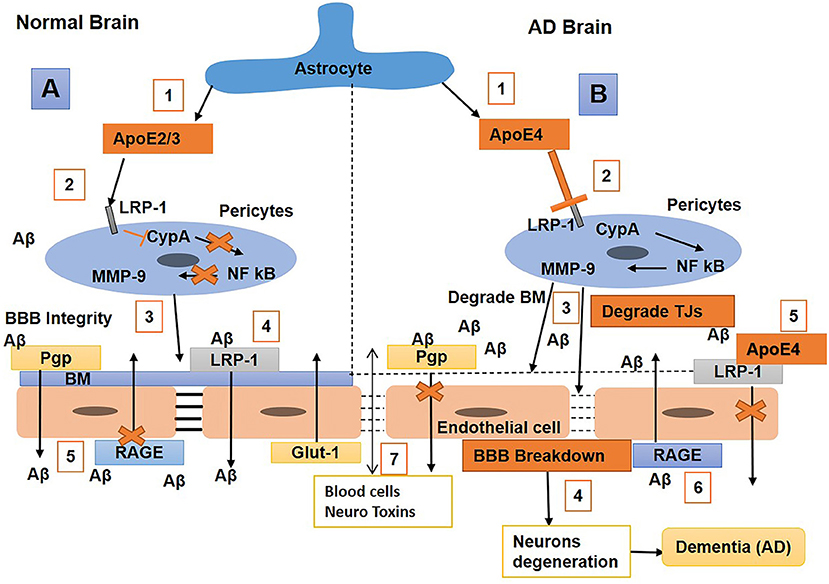
Figure 5. Schematic diagram shows the mechanisms of blood–brain barrier (BBB) breakdown in a normal brain and one with Alzheimer's disease (AD). (A) Normal brain (1) Astrocytes release ApoE2/3, (2) bind with the low-density lipoprotein receptor-related protein-1 (LRP-1) on pericytes and repress CypA-NFkB, which, in turn, stops matrix metalloproteinase 9 (MMP9) secretion in pericytes; (3) hence, maintaining BM and BBB integrity, with (4) LRP-1 and P-gp on endothelial cells (ECs) also helping in amyloid-beta (Aβ) clearance. (5) Receptor for advanced glycosylation end products (RAGE) expression is repressed to stop the transport of Aβ into the brain. (B) AD brain (1) Astrocytes secret ApoE4, (2) weakly interact with LRP-1 on pericytes which activates the cyclophilin-A nuclear factor kβ matrix metalloproteinase 9 (CypA-NFkB-MMP9 pathways), (3) and result in BM and tight junctions (TJs) degradation leading to BBB breakdown, (4) associated with neurodegeneration and dementia. (5) ApoE4 also weakly interacts with LRP-1 on ECs that cannot significantly clear Aβ from the brain; hence, Aβ accumulates in the brain, causing neuronal damage. (6) Also, RAGE expression is upregulated, which promotes the transport of Aβ from blood to brain. (7) Blood cells and neurotoxins diffuse into the brain and cause neuronal loss and dementia.
Vascular dementia is a neurodegenerative disease caused by reduced CBF to the brain resulting in cognitive dysfunction. After AD, VaD is considered the second most common dementia, accounting for ~15–30% of all dementia (Sloane et al., 2002; Abou-Saleh et al., 2011; Gorelick et al., 2011; Goodman et al., 2017).
Chronic hypoperfusion and thrombosis are the main factors in VaD that cause reduced CBF and promote oxidative stress, hypoxia, and inflammatory molecule expression (cytokines/chemokines). These chronic events cause damage to the periventricular WM, basal ganglia, and hippocampus. Cerebrovascular pathology has a significant contribution to the pathogenesis of VaD by damaging the brain. Vascular impairments include large vessel atherosclerosis (AS), small vessel AS, and CAA. These cerebrovascular pathologies cause microinfarcts in gray matter, WM lesions, and microbleeds (Thal et al., 2012). These vascular abnormalities can occur throughout the brain, resulting in VaD (Grinberg and Heinsen, 2010).
Hypertension is one of the factors that cause BBB breakdown in VaD with the accumulation of perivascular collagen in the hippocampus and WM lesions (Verhaaren et al., 2013). Toxic molecules or high blood pressure cause damage to the BBB endothelium. Hypertension also causes a reduction in the integrity of ECs and pericytes, astrocytes endfeet swelling, and retraction from the vessel wall, which results in BBB breakdown and subsequently leading to a reduction in CBF (Wardlaw et al., 2003). Studies reported that acute ischemia induces BBB permeability by the secretion of ROS (Abboud et al., 2007; Simpkins et al., 2016). A study also showed that, during vascular pathology, chronic hypoperfusion causes BBB disruption in WM lesions (Tomimoto et al., 1996). Another study showed that BBB disruption due to the degeneration of pericytes results in the disruption of WM circulation, deposition of fibrinogen, and reduction of CBF that further induces damage to the myelin, axons, and oligodendrocytes (Montagne et al., 2018) (Figure 6). Furthermore, animal experiments showed that chronic cerebral hypoperfusion (CCH) increases BBB leakage to intravenously injected horseradish peroxidase (HRP) in the corpus callosum. In animals, perivascular collagen was also accumulated in the corpus callosum associated with WM lesion formation and elevated BBB permeability (Ueno et al., 2002). In VaD, PVMs have been reported to induce oxidative stress leading to hypertension (Yang et al., 2019).
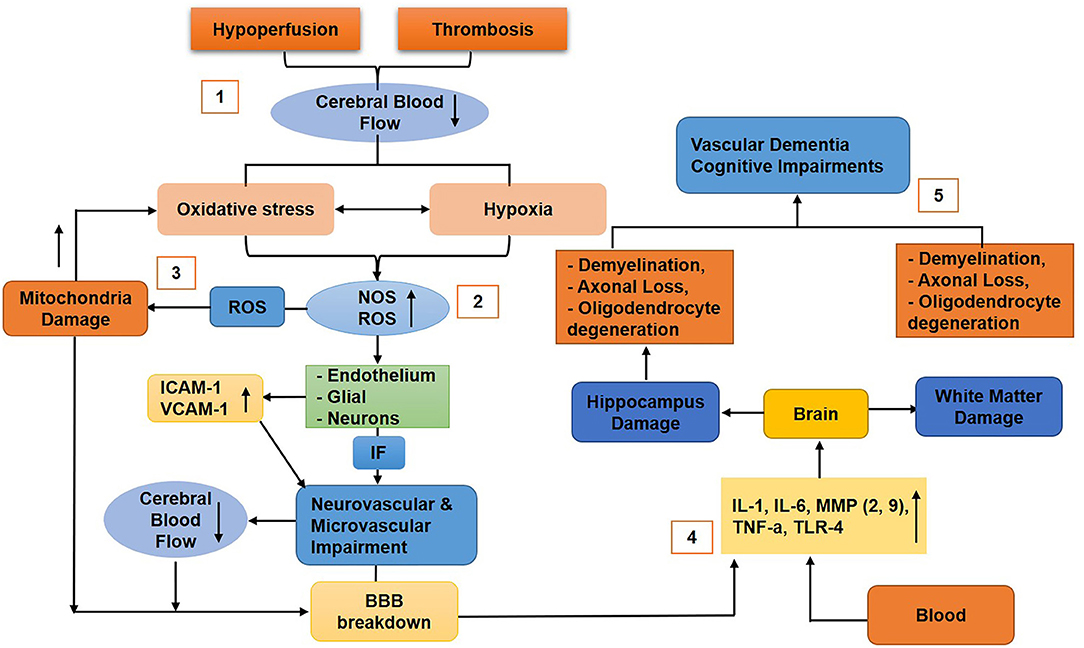
Figure 6. Diagram that shows the molecular mechanism of blood–brain barrier (BBB) breakdown in vascular dementia. (1) Hypoperfusion and thrombosis cause reduced cerebral blood flow (CBF), thus generating oxidative stress and hypoxia (2), which upregulate nitric oxide and reactive oxygen species (ROS) that stimulate the endothelial (ICAM-1, VCAM-1 upregulated), glial, and neuronal cells to release inflammatory factors that cause neurovascular unit (NVU) impairment and reduce CBF and BBB disruption. (3) ROS damage mitochondria of the BBB cells that will further upregulate oxidative stress, resulting in BBB impairments. (4) Cytokines, chemokines, toxins, and other inflammatory molecules infiltrate the brain and cause damage to the hippocampus and white matter (5) associated with neuronal loss, vascular dementia, and cognitive impairments.
Hypoxia upregulates oxidative stress, which produces NO, ROS, and free radicals (Li et al., 2013; Ma et al., 2013; Zhang et al., 2014). In addition, oxidative stress disrupts the ratio of antioxidants, NO, and ROS and causes damage to the endothelial, glial, and neuronal cells, resulting in the impairment of the NVU, BBB disruption, and mediation of a reduction in CBF (Liu and Zhang, 2012). In particular, ROS can further lead to mitochondrial dysfunction resulting in cerebral hypoxia that induces oxidative stress (Zhang et al., 2014). Cerebral vascular hypoxia produces inflammatory molecules that cause apoptosis and impairments in the function of microvessels. The cytokines/chemokines cause damage to the endothelium, glial, and neurons cells and, hence, enhance BBB permeability (Gill et al., 2010). The inflammatory molecules such as IL-1, IL-6, MMPs (MMP-2, MMP-9), TNFα, and TLR4 (toll-like receptor 4) infiltrate the brain (Li and Lai, 2007; Gill et al., 2010; Candelario-Jalil et al., 2011; Reuter et al., 2015), cause demyelination, and damage the axons and oligodendrocytes associated with the hippocampus and WM lesions (Chen et al., 2011).
Damage to oligodendrocytes represses remyelination (Ihara et al., 2010), and demyelination retains the transmission of neural signals, thus resulting in cognitive impairment. Overall, hypoxia, oxidative stress, and inflammation cause defects in neurogenesis, impairment in the proliferation of neuronal progenitor cell, synaptic plasticity, and reduced spine density in the hippocampus, thus resulting in cognitive impairment (Stranahan et al., 2008; Park et al., 2010) (Figure 6). Furthermore, several studies reported that CCH causes AD and VaD (Du et al., 2017). It has also been reported that intercellular adhesion molecule 1 (ICAM-1) and vascular adhesion molecule 1 (VCAM-1) were significantly upregulated in the vascular ECs of the CCH animal model associated with cognitive impairment (Won et al., 2013; Khan et al., 2015).
As discussed above and in other studies, significant clinical heterogeneity has been shown between AD and VaD (Sachdev et al., 2014; Chui and Ramirez-Gomez, 2015); however, recent studies reported that these two diseases co-occur in what is called mixed dementia (Emrani et al., 2020). In mixed dementia, vascular pathology not only mediates AD progression, but the pathology of AD also potentiates vascular impairments, suggesting that pure AD or VaD rarely occur (Emrani et al., 2020). In addition, another study reported that, in aged individuals, there is genetic overlap between vascular dysfunction and AD that is primarily associated with apolipoprotein E (Lin et al., 2019).
Community and epidemiological studies reported the mixed neuropathology that is quite common in both AD and VaD (Schneider et al., 2009; Wharton et al., 2011). The clinical study observed that only 9% of 1,000 patients with cognitive impairments have pure AD pathology; however, AD pathology is mainly associated with vascular dysfunction or other neurodegenerative diseases (Boyle et al., 2018). Another clinical study examined 63 patients with mild cognitive impairment (MCI), in which only 28% were reported as pure AD and approximately 24% were diagnosed with mixed dementia (AD and VaD) (Silbert et al., 2012). Researchers observed that frontal lobe lesions and vascular pathology, e.g., white matter hyperintensities (WMH), are associated with neuropsychiatric symptoms and are common in both AD and VaD (Anor et al., 2017). Alzheimer's disease and VaD share many similar clinical pathologies that lead to cognitive impairment and neuropsychiatric symptoms associated with behavioral alterations (Kalaria, 2002) as shown in Table 2. Hence, these studies suggest that there might be considerable overlaps between AD and VaD, and comprehensive studies should be considered to understand the pathophysiology of dementia instead of segregating AD from VaD.
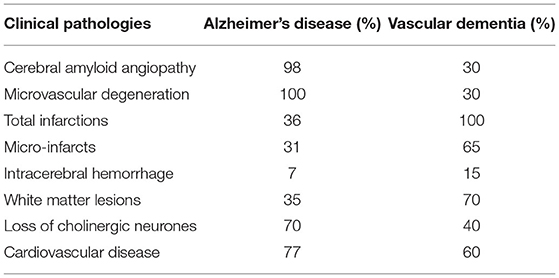
Table 2. Pathologies associated to both Alzheimer's disease and vascular dementia.
Various imaging techniques and other methods are currently being used to identify biomarkers associated with BBB breakdown in different neurological disorders, which are helpful in healthcare decisions. However, during the acute phase of BBB disruption, some of the clinical care places may lack the facilities to perform MRIs; hence, the detection of peripheral blood biomarkers is the best approach to identifying the status of BBB.
Studies showed that, while the blood/CSF albumin ratio can be used as a biomarker to detect BBB permeability, it cannot distinguish BBB and blood-CSF permeability nor locate leakage as reviewed by Farrall and Wardlaw (2009). Hence, nowadays, the dynamic contrast-enhanced MRI (DCE-MRI) technique is used to directly identify and localize these elusive permeability values (Raja et al., 2018). A study in healthy, aged individuals using DCE-MRI with a gadolinium-based contrast agent injected intravenously identified that BBB leakage was high and localized in the brain regions most vulnerable to damage from aging (Verheggen et al., 2020). It has been observed that, by using DCE-MRI, the BBB permeability index Ktrans was increased in the hippocampus and some of its sub-regions, CA1 and dentate gyrus (DG), but not in CA3. This study showed that, in the hippocampus, the BBB integrity was lost progressively with age. Still, no significant BBB leakage was observed in the cortical and sub-cortical regions (Montagne et al., 2015), suggesting that, in terms of aging, the BBB breakdown starts in the hippocampus. A study using CSF biomarkers and the DCE-MRI technique reported that aged people with prior cognitive impairment had higher BBB permeability than healthy individuals (Nation et al., 2019). These studies suggest that it is possible to detect and localize BBB leakage by using DCE-MRI.
It has been observed that, in epileptic patients, the levels of serum Visinin-like protein 1 (sVILIP-1) and serum caveolin 1 (sCAV-1) are higher, which may be used as biomarkers for the diagnosis of BBB breakdown (Tan et al., 2020). Another protein biomarker is s100β, which is produced by astrocyte endfeet; when the BBB becomes compromised, s100β is immediately released into the peripheral blood (Kadry et al., 2020). Furthermore, a study identified that the expression levels of A-kinase anchoring protein 7 (AKAP7) were high in the peripheral blood (lymphocyte), and thus might be considered to identify BBB breakdown during ischemic stroke or post-stroke (O'Connell et al., 2017). Neuron-specific enolase (NSE) and GFAP are also promising biomarkers that can be detected in the CSF to identify BBB breakdown (Kadry et al., 2020). A study reported that the elevated level of sPDGFRβ is associated with damage to the pericytes and BBB disruption leading to a decline in cognition (Sweeney et al., 2020). Soluble PDGFRβ as a biomarker was also observed in VaD (Iadecola, 2017; Sweeney et al., 2019a) and various other neurological diseases (Sweeney et al., 2018a, 2019b). Soluble cell adhesion molecules (CAMS), zonulin, and soluble 4-1BBL (transmembrane protein receptor) have also been identified to be associated with BBB damage. PECAM-1, P-selectin, and E-selectin are soluble adhesion molecules reported to be upregulated in individuals with compromised BBB and can be used as biomarkers for BBB breakdown (D'Ambrosio et al., 2015). Increased leakage of gadolinium (DCE-MRI; Ktrans), microbleeds (T2*-weighted and SWI-MRI), reduced glucose transport (FDG-PET), diminished P-glycoprotein 1 function (verapamil-PET), and CNS leukocyte infiltration (MMP inhibitor-PET) are some of the techniques that can be used to identify biomarkers associated with BBB damages in various CNS diseases (Sweeney et al., 2018b).
The BBB consists of a set of physiological properties that tightly regulate the normal microenvironment essential for proper neuronal activities. Any impairment in these properties either at the cellular or molecular level causes BBB breakdown. Aging is one of the factors that contribute to BBB disruption. During aging, the various physiological properties of the BBB are impaired, leading to BBB dysfunction. The neurotoxins infiltrating the brain can also cause cognitive impairments and neurodegeneration. Furthermore, BBB breakdown also contributes to dementia that includes ADs and VaD. In AD with disruption of BBB, Aβ and NFT of p-tau accumulate in the blood vessel, causing further inflammation in the NVU that, in turn, induces the release inflammatory factors to degenerate neurons associated with a decline in cognition. Another factor that degrades the integrity of BBB associated with AD is APOE4. In dementia, VaD accounts for the most cases next to AD caused by BBB breakdown. In VaD, the CBF is reduced and inflammatory molecules infiltrate the brain due to BBB impairment, subsequently causing neuronal loss and, thus, cognitive impairment. Hence, BBB breakdown can be used as a novel biomarker to study various neurological impairments such as AD, VaD, and other associated declines in cognition.
Recently, RepSox was identified to inhibit TGF-B, VEGFA, and inflammatory gene networks (Roudnicky et al., 2020). Furthermore, RepSox significantly elevated BBB resistance, induced TJs and transporters, reduced paracellular permeability by activating Notch and Wnt pathways, and, thus, might be used as an emerging BBB therapeutics to treat neurological diseases such as AD (Roudnicky et al., 2020). In addition, secreted protein acidic and rich in cysteine (SPARC) was identified to decrease transendothelial electrical resistance (TEER) and TJ proteins (ZO-1, OCLN) and increase paracellular permeability by regulating the tyrosine kinase pathway (Alkabie et al., 2016). Hence, the SPARC–collagen binding domain might be a potential therapeutic target to treat AD (Pilozzi et al., 2020). Furthermore, SPARC/Hevin normalization may also be considered as a novel therapeutic target for the modulation of AD progression (Strunz et al., 2019).
Although researchers have reported the contributions of BBB disruption to the pathogenesis of cognitive impairment associated with normal aging and dementia, more research is needed to elucidate the precisely causing factors and the cellular and molecular mechanisms of BBB maintenance, breakdown, and repair correlated with neurodegeneration and cognition decline. In the future, how aging and dementia affect BBB function in health and disease state, thus leading to neurodegeneration and cognitive impairment, should be explored in living organisms. Clinical research pertaining to this will boost our knowledge and help us better understand the association between BBB breakdown and cognitive decline. Such studies pave the way for the use of the BBB as a novel biomarker and therapeutic target to treat dementia and other neurological diseases associated with cognitive impairment. Furthermore, these studies suggest that amelioration in the cerebrovascular pathways (particularly BBB breakdown) can alleviate neurodegeneration in dementia (particularly AD) associated with cognitive impairment. Hence, the characterization of the cellular and molecular constituents of the cerebrovascular systems that contribute to the pathophysiology of dementia will provide a systematic methodology of dementia diagnosis. More profound knowledge of the vascular system will also help design emerging efficient strategies that can be used for the therapeutic interventions of cognitive impairment and dementia.
BH drafted the manuscript and made the figures and table. CF and JC discussed and revised the manuscript. All authors contributed to the article and approved the final manuscript.
This study was supported by the National Natural Science Foundation of China (81771293), the Science Technology and Innovation Commission of Shenzhen Municipality (ZDSYS20190902093409851 and SGLH20180625142404672), the international collaboration project of the Chinese Academy of Sciences (172644KYSB20200045), the CAS-Croucher Funding Scheme for Joint Laboratories, and the Guangdong Innovation Platform of Translational Research for Cerebrovascular Diseases. BH is supported by the Chinese Government Scholarship (CSC No. 2018SLJ023241) for International Students.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbott, N. J. (2013). Blood-brain barrier structure and function and the challenges for CNS drug delivery. J. Inherit. Metab. Dis. 36, 437–449. doi: 10.1007/s10545-013-9608-0
Abbott, N. J., Patabendige, A. A., Dolman, D. E., Yusof, S. R., and Begley, D. J. (2010). Structure and function of the blood-brain barrier. Neurobiol. Dis. 37, 13–25. doi: 10.1016/j.nbd.2009.07.030
Abbott, N. J., Rönnbäck, L., and Hansson, E. (2006). Astrocyte-endothelial interactions at the blood-brain barrier. Nat. Rev. Neurosci. 7, 41–53. doi: 10.1038/nrn1824
Abboud, H., Labreuche, J., Meseguer, E., Lavallee, P. C., Simon, O., Olivot, J.-M., et al. (2007). Ischemia-modified albumin in acute stroke. Cerebrovasc. Dis. 23, 216–220. doi: 10.1159/000097644
Abou-Saleh, M. T., Katona, C. L., and Kumar, A. (2011). Principles and Practice of Geriatric Psychiatry. Hoboken, NJ: Wiley–Blackwell.
Ahmad, M. H., Fatima, M., and Mondal, A. C. (2019). Influence of microglia and astrocyte activation in the neuroinflammatory pathogenesis of Alzheimer's disease: rational insights for the therapeutic approaches. J. Clin. Neurosci. 59, 6–11. doi: 10.1016/j.jocn.2018.10.034
Alata, W., Ye, Y., St-Amour, I., Vandal, M., and Calon, F. (2015). Human apolipoprotein e ε4 expression impairs cerebral vascularization and blood-brain barrier function in mice. J. Cereb. Blood Flow Metab. 35, 86–94. doi: 10.1038/jcbfm.2014.172
Alexander, J. J. (2018). Blood-brain barrier (BBB) and the complement landscape. Mol. Immunol. 102, 26–31. doi: 10.1016/j.molimm.2018.06.267
Alkabie, S., Basivireddy, J., Zhou, L., Roskams, J., Rieckmann, P., and Quandt, J. A. (2016). SPARC expression by cerebral microvascular endothelial cells in vitro and its influence on blood-brain barrier properties. J. Neuroinflammation 13, 1–17. doi: 10.1186/s12974-016-0657-9
Allen, C., Thornton, P., Denes, A., McColl, B. W., Pierozynski, A., Monestier, M., et al. (2012). Neutrophil cerebrovascular transmigration triggers rapid neurotoxicity through release of proteases associated with decondensed DNA. J. Immunol. 189, 381–392. doi: 10.4049/jimmunol.1200409
Alvarez, J. I., Dodelet-Devillers, A., Kebir, H., Ifergan, I., Fabre, P. J., Terouz, S., et al. (2011). The Hedgehog pathway promotes blood-brain barrier integrity and CNS immune quiescence. Science 334, 1727–1731. doi: 10.1126/science.1206936
Anor, C. J., O'Connor, S., Saund, A., Tang-Wai, D. F., Keren, R., and Tartaglia, M. C. (2017). Neuropsychiatric symptoms in Alzheimer disease, vascular dementia, and mixed dementia. Neurodegener. Dis. 17, 127–134. doi: 10.1159/000455127
Arendt, T., Brückner, M. K., Morawski, M., Jäger, C., and Gertz, H.-J. (2015). Early neurone loss in Alzheimer's disease: cortical or subcortical? Acta Neuropathol. Commun. 3, 1–11. doi: 10.1186/s40478-015-0187-1
Aw, D., Silva, A. B., and Palmer, D. B. (2007). Immunosenescence: emerging challenges for an ageing population. Immunology 120, 435–446. doi: 10.1111/j.1365-2567.2007.02555.x
Bake, S., Friedman, J. A., and Sohrabji, F. (2009). Reproductive age-related changes in the blood brain barrier: expression of IgG and tight junction proteins. Microvasc. Res. 78, 413–424. doi: 10.1016/j.mvr.2009.06.009
Baker, S. K., Chen, Z.-L., Norris, E. H., Revenko, A. S., MacLeod, A. R., and Strickland, S. (2018). Blood-derived plasminogen drives brain inflammation and plaque deposition in a mouse model of Alzheimer's disease. Proc. Natl. Acad. Sci. U.S.A. 115, E9687–E9696. doi: 10.1073/pnas.1811172115
Ballaed, C., Gauthier, S., Corbett, A., Brayne, C., and Aarsland, D. (2011). Jones e. Alzheimer's disease. Lancet 377, 1019–1031. doi: 10.1016/S0140-6736(10)61349-9
Banks, W. A., Reed, M. J., Logsdon, A. F., Rhea, E. M., and Erickson, M. A. (2021). Healthy aging and the blood-brain barrier. Nature Aging 1, 243–254. doi: 10.1038/s43587-021-00043-5
Bannerman, D. D., and Goldblum, S. E. (1999). Direct effects of endotoxin on the endothelium: barrier function and injury. Lab. Invest. 79, 1181–1199.
Bartels, T., De Schepper, S., and Hong, S. (2020). Microglia modulate neurodegeneration in Alzheimer's and Parkinson's diseases. Science 370, 66–69. doi: 10.1126/science.abb8587
Bell, R. D., Winkler, E. A., Sagare, A. P., Singh, I., LaRue, B., Deane, R., et al. (2010). Pericytes control key neurovascular functions and neuronal phenotype in the adult brain and during brain aging. Neuron 68, 409–427. doi: 10.1016/j.neuron.2010.09.043
Bell, R. D., Winkler, E. A., Singh, I., Sagare, A. P., Deane, R., Wu, Z., et al. (2012). Apolipoprotein E controls cerebrovascular integrity via cyclophilin A. Nature 485, 512–516. doi: 10.1038/nature11087
Bell, R. D., and Zlokovic, B. V. (2009). Neurovascular mechanisms and blood-brain barrier disorder in Alzheimer's disease. Acta Neuropathol. 118, 103–113. doi: 10.1007/s00401-009-0522-3
Ben-Menachem, E., Johansson, B. B., and Svensson, T. (1982). Increased vulnerability of the blood-brain barrier to acute hypertension following depletion of brain noradrenaline. J. Neural Transm. 53, 159–167. doi: 10.1007/BF01243407
Berezowski, V., Landry, C., Dehouck, M.-P., Cecchelli, R., and Fenart, L. (2004). Contribution of glial cells and pericytes to the mRNA profiles of P-glycoprotein and multidrug resistance-associated proteins in an in vitro model of the blood-brain barrier. Brain Res. 1018, 1–9. doi: 10.1016/j.brainres.2004.05.092
Bhaskar, K., Konerth, M., Kokiko-Cochran, O. N., Cardona, A., Ransohoff, R. M., and Lamb, B. T. (2010). Regulation of tau pathology by the microglial fractalkine receptor. Neuron 68, 19–31. doi: 10.1016/j.neuron.2010.08.023
Biron, K. E., Dickstein, D. L., Gopaul, R., and Jefferies, W. A. (2011). Amyloid triggers extensive cerebral angiogenesis causing blood brain barrier permeability and hypervascularity in Alzheimer's disease. PLoS ONE 6:e23789. doi: 10.1371/journal.pone.0023789
Blanchette, M., and Daneman, R. (2015). Formation and maintenance of the BBB. Mech. Dev. 138, 8–16. doi: 10.1016/j.mod.2015.07.007
Blau, C. W., Cowley, T. R., O'Sullivan, J., Grehan, B., Browne, T. C., Kelly, L., et al. (2012). The age-related deficit in LTP is associated with changes in perfusion and blood-brain barrier permeability. Neurobiol. Aging 33, 1005.e1023–1005.e1035. doi: 10.1016/j.neurobiolaging.2011.09.035
Block, M. L. (2008). NADPH oxidase as a therapeutic target in Alzheimer's disease. BMC Neurosci. 9:S8. doi: 10.1186/1471-2202-9-S2-S8
Boyle, P. A., Yu, L., Wilson, R. S., Leurgans, S. E., Schneider, J. A., and Bennett, D. A. (2018). Person-specific contribution of neuropathologies to cognitive loss in old age. Ann. Neurol. 83, 74–83. doi: 10.1002/ana.25123
Brown, W. R., and Thore, C. R. (2011). Cerebral microvascular pathology in ageing and neurodegeneration. Neuropathol. Appl. Neurobiol. 37, 56–74. doi: 10.1111/j.1365-2990.2010.01139.x
Buschini, E., Piras, A., Nuzzi, R., and Vercelli, A. (2011). Age related macular degeneration and drusen: neuroinflammation in the retina. Prog. Neurobiol. 95, 14–25. doi: 10.1016/j.pneurobio.2011.05.011
Bussian, T. J., Aziz, A., Meyer, C. F., Swenson, B. L., van Deursen, J. M., and Baker, D. J. (2018). Clearance of senescent glial cells prevents tau-dependent pathology and cognitive decline. Nature 562, 578–582. doi: 10.1038/s41586-018-0543-y
Cai, W., Zhang, K., Li, P., Zhu, L., Xu, J., Yang, B., et al. (2017). Dysfunction of the neurovascular unit in ischemic stroke and neurodegenerative diseases: an aging effect. Ageing Res. Rev. 34, 77–87. doi: 10.1016/j.arr.2016.09.006
Cai, Z., Wan, C.-Q., and Liu, Z. (2017). Astrocyte and Alzheimer's disease. J. Neurol. 264, 2068–2074. doi: 10.1007/s00415-017-8593-x
Candelario-Jalil, E., Thompson, J., Taheri, S., Grossetete, M., Adair, J. C., Edmonds, E., et al. (2011). Matrix metalloproteinases are associated with increased blood-brain barrier opening in vascular cognitive impairment. Stroke 42, 1345–1350. doi: 10.1161/STROKEAHA.110.600825
Candiello, J., Cole, G. J., and Halfter, W. (2010). Age-dependent changes in the structure, composition and biophysical properties of a human basement membrane. Matrix Biol. 29, 402–410. doi: 10.1016/j.matbio.2010.03.004
Candore, G., Bulati, M., Caruso, C., Castiglia, L., Colonna-Romano, G., Di Bona, D., et al. (2010). Inflammation, cytokines, immune response, apolipoprotein E, cholesterol, and oxidative stress in Alzheimer disease: therapeutic implications. Rejuvenation Res. 13, 301–313. doi: 10.1089/rej.2009.0993
Carrano, A., Hoozemans, J. J., van der Vies, S. M., Rozemuller, A. J., van Horssen, J., and de Vries, H. E. (2011). Amyloid beta induces oxidative stress-mediated blood-brain barrier changes in capillary amyloid angiopathy. Antioxid. Redox Signal. 15, 1167–1178. doi: 10.1089/ars.2011.3895
Castillo-Gomez, E., Kästner, A., Steiner, J., Schneider, A., Hettling, B., Poggi, G., et al. (2016). The brain as immunoprecipitator of serum autoantibodies against N-Methyl-D-aspartate receptor subunit NR1. Ann. Neurol. 79, 144–151. doi: 10.1002/ana.24545
Cerbai, F., Lana, D., Nosi, D., Petkova-Kirova, P., Zecchi, S., Brothers, H. M., et al. (2012). The neuron-astrocyte-microglia triad in normal brain ageing and in a model of neuroinflammation in the rat hippocampus. PLoS ONE 7:e45250. doi: 10.1371/journal.pone.0045250
Chacón-Quintero, M. V., Pineda-López, L. G., Villegas-Lanau, C. A., Posada-Duque, R., and Cardona-Gómez, G. P. (2021). Beta-secretase 1 underlies reactive astrocytes and endothelial disruption in neurodegeneration. Front. Cell. Neurosci. 15:656832. doi: 10.3389/fncel.2021.656832
Chang, H.-C., and Guarente, L. (2014). SIRT1 and other sirtuins in metabolism. Trends Endocrinol. Metab. 25, 138–145. doi: 10.1016/j.tem.2013.12.001
Chen, J., Cui, X., Zacharek, A., Cui, Y., Roberts, C., and Chopp, M. (2011). White matter damage and the effect of matrix metalloproteinases in type 2 diabetic mice after stroke. Stroke 42, 445–452. doi: 10.1161/STROKEAHA.110.596486
Cheng, T., Petraglia, A. L., Li, Z., Thiyagarajan, M., Zhong, Z., Wu, Z., et al. (2006). Activated protein C inhibits tissue plasminogen activator-induced brain hemorrhage. Nat. Med. 12, 1278–1285. doi: 10.1038/nm1498
Chiu, C., Miller, M. C., Monahan, R., Osgood, D. P., Stopa, E. G., and Silverberg, G. D. (2015). P-glycoprotein expression and amyloid accumulation in human aging and Alzheimer's disease: preliminary observations. Neurobiol. Aging 36, 2475–2482. doi: 10.1016/j.neurobiolaging.2015.05.020
Choi, D.-H., Kim, J.-H., Seo, J.-H., Lee, J., Choi, W. S., and Kim, Y.-S. (2014). Matrix metalloproteinase-3 causes dopaminergic neuronal death through Nox1-regenerated oxidative stress. PLoS ONE 9:e115954. doi: 10.1371/journal.pone.0115954
Chow, B. W., and Gu, C. (2015). The molecular constituents of the blood-brain barrier. Trends Neurosci. 38, 598–608. doi: 10.1016/j.tins.2015.08.003
Chui, H. C., and Ramirez-Gomez, L. (2015). Clinical and imaging features of mixed Alzheimer and vascular pathologies. Alzheimers Res. Ther. 7:21. doi: 10.1186/s13195-015-0104-7
Cohen Z. Bonvento G. Lacombe P. and Hamel, E. (1996). Serotonin in the regulation of brain microcirculation. Prog. Neurobiol. 50, 335–362. doi: 10.1016/S0301-0082(96)00033-0
Cohen, Z., Molinatti, G., and Hamel, E. (1997). Astroglial and vascular interactions of noradrenaline terminals in the rat cerebral cortex. J. Cereb. Blood Flow Metab. 17, 894–904. doi: 10.1097/00004647-199708000-00008
Cortes-Canteli, M., Mattei, L., Richards, A. T., Norris, E. H., and Strickland, S. (2015). Fibrin deposited in the Alzheimer's disease brain promotes neuronal degeneration. Neurobiol. Aging 36, 608–617. doi: 10.1016/j.neurobiolaging.2014.10.030
Cortes-Canteli, M., Paul, J., Norris, E. H., Bronstein, R., Ahn, H. J., Zamolodchikov, D., et al. (2010). Fibrinogen and β-amyloid association alters thrombosis and fibrinolysis: a possible contributing factor to Alzheimer's disease. Neuron 66, 695–709. doi: 10.1016/j.neuron.2010.05.014
Crews, L., and Masliah, E. (2010). Molecular mechanisms of neurodegeneration in Alzheimer's disease. Hum. Mol. Genet. 19, R12–R20. doi: 10.1093/hmg/ddq160
Cuevas, E., Rosas-Hernandez, H., Burks, S. M., Ramirez-Lee, M. A., Guzman, A., Imam, S. Z., et al. (2019). Amyloid Beta 25-35 induces blood-brain barrier disruption in vitro. Metab. Brain Dis. 34, 1365–1374. doi: 10.1007/s11011-019-00447-8
D'Ambrosio, A., Pontecorvo, S., Colasanti, T., Zamboni, S., Francia, A., and Margutti, P. (2015). Peripheral blood biomarkers in multiple sclerosis. Autoimmun. Rev. 14, 1097–1110. doi: 10.1016/j.autrev.2015.07.014
De Jong, G., Horvath, E., and Luiten, P. (1990). Effects of early onset of nimodipine treatment on microvascular integrity in the aging rat brain. Stroke 21(12 Suppl), IV113–IV116.
Deane, R., Du Yan, S., Submamaryan, R. K., LaRue, B., Jovanovic, S., Hogg, E., et al. (2003). RAGE mediates amyloid-β peptide transport across the blood-brain barrier and accumulation in brain. Nat. Med. 9, 907–913. doi: 10.1038/nm890
Deane, R., Sagare, A., Hamm, K., Parisi, M., Lane, S., Finn, M. B., et al. (2008). apoE isoform-specific disruption of amyloid β peptide clearance from mouse brain. J. Clin. Invest. 118, 4002–4013. doi: 10.1172/JCI36663
Deane, R., Wu, Z., Sagare, A., Davis, J., Du Yan, S., Hamm, K., et al. (2004). LRP/amyloid β-peptide interaction mediates differential brain efflux of Aβ isoforms. Neuron 43, 333–344. doi: 10.1016/j.neuron.2004.07.017
Deli, M. A., Veszelka, S., Csiszár, B., Tóth, A., Kittel, A., Csete, M., et al. (2010). Protection of the blood-brain barrier by pentosan against amyloid-β-induced toxicity. J. Alzheimers Dis. 22, 777–794. doi: 10.3233/JAD-2010-100759
Di Cataldo, V., Géloën, A., Langlois, J.-B., Chauveau, F., Thézé, B., Hubert, V., et al. (2016). Exercise does not protect against peripheral and central effects of a high cholesterol diet given ad libitum in old ApoE–/– mice. Front. Physiol. 7:453. doi: 10.3389/fphys.2016.00453
Ding, F., Yao, J., Rettberg, J. R., Chen, S., and Brinton, R. D. (2013). Early decline in glucose transport and metabolism precedes shift to ketogenic system in female aging and Alzheimer's mouse brain: implication for bioenergetic intervention. PLoS ONE 8:e79977. doi: 10.1371/journal.pone.0079977
Doeuvre, L., Plawinski, L., Goux, D., Vivien, D., and Anglés-Cano, E. (2010). Plasmin on adherent cells: from microvesiculation to apoptosis. Biochem. J. 432, 365–373. doi: 10.1042/BJ20100561
Donahue, J. E., Flaherty, S. L., Johanson, C. E., Duncan, J. A., Silverberg, G. D., Miller, M. C., et al. (2006). RAGE, LRP-1, and amyloid-beta protein in Alzheimer's disease. Acta Neuropathol. 112, 405–415. doi: 10.1007/s00401-006-0115-3
Donato, A. J., Eskurza, I., Silver, A. E., Levy, A. S., Pierce, G. L., Gates, P. E., et al. (2007). Direct evidence of endothelial oxidative stress with aging in humans: relation to impaired endothelium-dependent dilation and upregulation of nuclear factor-κB. Circ. Res. 100, 1659–1666. doi: 10.1161/01.RES.0000269183.13937.e8
Du, S.-Q., Wang, X.-R., Xiao, L.-Y., Tu, J.-F., Zhu, W., He, T., et al. (2017). Molecular mechanisms of vascular dementia: what can be learned from animal models of chronic cerebral hypoperfusion? Mol. Neurobiol. 54, 3670–3682. doi: 10.1007/s12035-016-9915-1
Duncombe, J., Lennen, R. J., Jansen, M. A., Marshall, I., Wardlaw, J. M., and Horsburgh, K. (2017). Ageing causes prominent neurovascular dysfunction associated with loss of astrocytic contacts and gliosis. Neuropathol. Appl. Neurobiol. 43, 477–491. doi: 10.1111/nan.12375
Edwards, G. A. III., Gamez, N., Escobedo Jr, G., Calderon, O., and Moreno-Gonzalez, I. (2019). Modifiable risk factors for Alzheimer's disease. Front. Aging Neurosci. 11:146. doi: 10.3389/fnagi.2019.00146
Eikelenboom, P., Van Exel, E., Veerhuis, R., Rozemuller, A. J., Van Gool, W. A., and Hoozemans, J. J. (2012). Innate immunity and the etiology of late-onset Alzheimer's disease. Neurodegener. Dis. 10, 271–273. doi: 10.1159/000334287
Elahy, M., Jackaman, C., Mamo, J. C., Lam, V., Dhaliwal, S. S., Giles, C., et al. (2015). Blood-brain barrier dysfunction developed during normal aging is associated with inflammation and loss of tight junctions but not with leukocyte recruitment. Immunity Ageing 12, 1–9. doi: 10.1186/s12979-015-0029-9
Ellison, D., White, D., and Farrar, F. C. (2015). Aging population. Nurs. Clin. 50, 185–213. doi: 10.1016/j.cnur.2014.10.014
Emrani, S., Lamar, M., Price, C. C., Wasserman, V., Matusz, E., Au, R., et al. (2020). Alzheimer's/vascular spectrum dementia: classification in addition to diagnosis. J. Alzheimers Dis. 73, 63–71. doi: 10.3233/JAD-190654
Engelhardt, B., and Coisne, C. (2011). Fluids and barriers of the CNS establish immune privilege by confining immune surveillance to a two-walled castle moat surrounding the CNS castle. Fluids Barriers CNS 8, 1–9. doi: 10.1186/2045-8118-8-4
Engelhardt, B., and Ransohoff, R. M. (2012). Capture, crawl, cross: the T cell code to breach the blood-brain barriers. Trends Immunol. 33, 579–589. doi: 10.1016/j.it.2012.07.004
Erdo, F., Denes, L., and de Lange, E. (2017). Age-associated physiological and pathological changes at the blood-brain barrier: a review. J. Cereb. Blood Flow Metab. 37, 4–24. doi: 10.1177/0271678X16679420
Erickson, M. A., and Banks, W. A. (2013). Blood-brain barrier dysfunction as a cause and consequence of Alzheimer's disease. J. Cereb. Blood Flow Metab. 33, 1500–1513. doi: 10.1038/jcbfm.2013.135
Erickson, M. A., and Banks, W. A. (2018). Neuroimmune axes of the blood-brain barriers and blood-brain interfaces: bases for physiological regulation, disease states, and pharmacological interventions. Pharmacol. Rev. 70, 278–314. doi: 10.1124/pr.117.014647
Erickson, M. A., Hansen, K., and Banks, W. A. (2012). Inflammation-induced dysfunction of the low-density lipoprotein receptor-related protein-1 at the blood-brain barrier: protection by the antioxidant N-acetylcysteine. Brain Behav. Immun. 26, 1085–1094. doi: 10.1016/j.bbi.2012.07.003
Faraco, G., Park, L., Anrather, J., and Iadecola, C. (2017). Brain perivascular macrophages: characterization and functional roles in health and disease. J. Mol. Med. 95, 1143–1152. doi: 10.1007/s00109-017-1573-x
Farrall, A. J., and Wardlaw, J. M. (2009). Blood-brain barrier: ageing and microvascular disease-systematic review and meta-analysis. Neurobiol. Aging 30, 337–352. doi: 10.1016/j.neurobiolaging.2007.07.015
Farrer, L. A., Cupples, L. A., Haines, J. L., Hyman, B., Kukull, W. A., Mayeux, R., et al. (1997). Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease: a meta-analysis. JAMA 278, 1349–1356. doi: 10.1001/jama.1997.03550160069041
Fivenson, E. M., Lautrup, S., Sun, N., Scheibye-Knudsen, M., Stevnsner, T., Nilsen, H., et al. (2017). Mitophagy in neurodegeneration and aging. Neurochem. Int. 109, 202–209. doi: 10.1016/j.neuint.2017.02.007
Fonseca, M. I., Ager, R. R., Chu, S.-H., Yazan, O., Sanderson, S. D., LaFerla, F. M., et al. (2009). Treatment with a C5aR antagonist decreases pathology and enhances behavioral performance in murine models of Alzheimer's disease. J. Immunol. 183, 1375–1383. doi: 10.4049/jimmunol.0901005
Gao, T., Lin, Z., and Jin, X. (2009). Hydrocortisone suppression of the expression of VEGF may relate to toll-like receptor (TLR) 2 and 4. Curr. Eye Res. 34, 777–784. doi: 10.1080/02713680903067919
Gill, R., Tsung, A., and Billiar, T. (2010). Linking oxidative stress to inflammation: Toll-like receptors. Free Radic. Biol. Med. 48, 1121–1132. doi: 10.1016/j.freeradbiomed.2010.01.006
González-Molina, L. A., Villar-Vesga, J., Henao-Restrepo, J., Villegas, A., Lopera, F., Cardona-Gómez, G. P., et al. (2021). Extracellular vesicles from 3xTg-AD mouse and Alzheimer's disease patient astrocytes impair neuroglial and vascular components. Front. Aging Neurosci. 13:593927. doi: 10.3389/fnagi.2021.593927
Gonzalez-Velasquez, F. J., Kotarek, J. A., and Moss, M. A. (2008). Soluble aggregates of the amyloid-β protein selectively stimulate permeability in human brain microvascular endothelial monolayers. J. Neurochem. 107, 466–477. doi: 10.1111/j.1471-4159.2008.05618.x
Goodall, E. F., Wang, C., Simpson, J. E., Baker, D. J., Drew, D. R., Heath, P. R., et al. (2018). Age-associated changes in the blood-brain barrier: comparative studies in human and mouse. Neuropathol. Appl. Neurobiol. 44, 328–340. doi: 10.1111/nan.12408
Goodman, R. A., Lochner, K. A., Thambisetty, M., Wingo, T. S., Posner, S. F., and Ling, S. M. (2017). Prevalence of dementia subtypes in United States Medicare fee-for-service beneficiaries, 2011-2013. Alzheimers Dement. 13, 28–37. doi: 10.1016/j.jalz.2016.04.002
Gorelick, P. B., Scuteri, A., Black, S. E., DeCarli, C., Greenberg, S. M., Iadecola, C., et al. (2011). Vascular contributions to cognitive impairment and dementia: a statement for healthcare professionals from the American Heart Association/American Stroke Association. Stroke 42, 2672–2713. doi: 10.1161/STR.0b013e3182299496
Gredilla, R., Bohr, V. A., and Stevnsner, T. (2010). Mitochondrial DNA repair and association with aging-an update. Exp. Gerontol. 45, 478–488. doi: 10.1016/j.exger.2010.01.017
Grinberg, L. T., and Heinsen, H. (2010). Toward a pathological definition of vascular dementia. J. Neurol. Sci. 299, 136–138. doi: 10.1016/j.jns.2010.08.055
Grinberg, L. T., and Thal, D. R. (2010). Vascular pathology in the aged human brain. Acta Neuropathol. 119, 277–290. doi: 10.1007/s00401-010-0652-7
Halliday, M. R., Rege, S. V., Ma, Q., Zhao, Z., Miller, C. A., Winkler, E. A., et al. (2016). Accelerated pericyte degeneration and blood-brain barrier breakdown in apolipoprotein E4 carriers with Alzheimer's disease. J. Cereb. Blood Flow Metab. 36, 216–227. doi: 10.1038/jcbfm.2015.44
Hammer, C., Stepniak, B., Schneider, A., Papiol, S., Tantra, M., Begemann, M., et al. (2014). Neuropsychiatric disease relevance of circulating anti-NMDA receptor autoantibodies depends on blood-brain barrier integrity. Mol. Psychiatry 19, 1143–1149. doi: 10.1038/mp.2013.110
Hansen, D. V., Hanson, J. E., and Sheng, M. (2018). Microglia in Alzheimer's disease. J. Cell Biol. 217, 459–472. doi: 10.1083/jcb.201709069
Harry, G. J. (2013). Microglia during development and aging. Pharmacol. Therapeut. 139, 313–326. doi: 10.1016/j.pharmthera.2013.04.013
Haruwaka, K., Ikegami, A., Tachibana, Y., Ohno, N., Konishi, H., Hashimoto, A., et al. (2019). Dual microglia effects on blood brain barrier permeability induced by systemic inflammation. Nat. Commun. 10, 1–17. doi: 10.1038/s41467-019-13812-z
Hase, Y., Ding, R., Harrison, G., Hawthorne, E., King, A., Gettings, S., et al. (2019). White matter capillaries in vascular and neurodegenerative dementias. Acta Neuropathol. Commun. 7, 1–12. doi: 10.1186/s40478-019-0666-x
Hawkes, C. A., Gatherer, M., Sharp, M. M., Dorr, A., Yuen, H. M., Kalaria, R., et al. (2013). Regional differences in the morphological and functional effects of aging on cerebral basement membranes and perivascular drainage of amyloid-β from the mouse brain. Aging Cell 12, 224–236. doi: 10.1111/acel.12045
Hawkins, B. T., and Davis, T. P. (2005). The blood-brain barrier/neurovascular unit in health and disease. Pharmacol. Rev. 57, 173–185. doi: 10.1124/pr.57.2.4
He, H., Lam, M., McCormick, T. S., and Distelhorst, C. W. (1997). Maintenance of calcium homeostasis in the endoplasmic reticulum by Bcl-2. J. Cell Biol. 138, 1219–1228. doi: 10.1083/jcb.138.6.1219
Heinsen, H., and Heinsen, Y. (1983). Cerebellar capillaries. Anat. Embryol. 168, 101–116. doi: 10.1007/BF00305402
Heithoff, B. P., George, K. K., Phares, A. N., Zuidhoek, I. A., Munoz-Ballester, C., and Robel, S. (2021). Astrocytes are necessary for blood-brain barrier maintenance in the adult mouse brain. Glia 69, 436–472. doi: 10.1002/glia.23908
Hemonnot, A.-L., Hua, J., Ulmann, L., and Hirbec, H. (2019). Microglia in Alzheimer disease: well-known targets and new opportunities. Front. Aging Neurosci. 11:233. doi: 10.3389/fnagi.2019.00233
Heneka, M. T., Carson, M. J., El Khoury, J., Landreth, G. E., Brosseron, F., Feinstein, D. L., et al. (2015). Neuroinflammation in Alzheimer's disease. Lancet Neurol. 14, 388–405. doi: 10.1016/S1474-4422(15)70016-5
Hicks, P., Rolsten, C., Brizzee, D., and Samorajski, T. (1983). Age-related changes in rat brain capillaries. Neurobiol. Aging 4, 69–75. doi: 10.1016/0197-4580(83)90057-X
Hoarau, J.-J., Krejbich-Trotot, P., Jaffar-Bandjee, M.-C., Das, T., Thon-Hon, G.-V., Kumar, S., et al. (2011). Activation and control of CNS innate immune responses in health and diseases: a balancing act finely tuned by neuroimmune regulators (NIReg). CNS Neurol. Disord. Drug Targets 10, 25–43. doi: 10.2174/187152711794488601
Hoffman, J. D., Parikh, I., Green, S. J., Chlipala, G., Mohney, R. P., Keaton, M., et al. (2017). Age drives distortion of brain metabolic, vascular and cognitive functions, and the gut microbiome. Front. Aging Neurosci. 9:298. doi: 10.3389/fnagi.2017.00298
Hoshi, A., Yamamoto, T., Shimizu, K., Ugawa, Y., Nishizawa, M., Takahashi, H., et al. (2012). Characteristics of aquaporin expression surrounding senile plaques and cerebral amyloid angiopathy in Alzheimer disease. J. Neuropathol. Exp. Neurol. 71, 750–759. doi: 10.1097/NEN.0b013e3182632566
Howe, M. D., McCullough, L. D., and Urayama, A. (2020). The role of basement membranes in cerebral amyloid angiopathy. Front. Physiol. 11:601320. doi: 10.3389/fphys.2020.601320
Huang, X., Hussain, B., and Chang, J. (2021). Peripheral inflammation and blood-brain barrier disruption: effects and mechanisms. CNS Neurosci. Therapeut. 27, 36–47. doi: 10.1111/cns.13569
Huber, J. D., Campos, C. R., Mark, K. S., and Davis, T. P. (2006). Alterations in blood-brain barrier ICAM-1 expression and brain microglial activation after λ-carrageenan-induced inflammatory pain. Am. J. Physiol. Heart Circ. Physiol. 290, H732–H740. doi: 10.1152/ajpheart.00747.2005
Hughes, S., Gardiner, T., Hu, P., Baxter, L., Rosinova, E., and Chan-Ling, T. (2006). Altered pericyte-endothelial relations in the rat retina during aging: implications for vessel stability. Neurobiol. Aging 27, 1838–1847. doi: 10.1016/j.neurobiolaging.2005.10.021
Hultman, K., Strickland, S., and Norris, E. H. (2013). The APOE ε4/ε4 genotype potentiates vascular fibrin (ogen) deposition in amyloid-laden vessels in the brains of Alzheimer's disease patients. J. Cereb. Blood Flow Metab. 33, 1251–1258. doi: 10.1038/jcbfm.2013.76
Hyman, B. T., Phelps, C. H., Beach, T. G., Bigio, E. H., Cairns, N. J., Carrillo, M. C., et al. (2012). National Institute on Aging-Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease. Alzheimers Dement. 8, 1–13. doi: 10.1016/j.jalz.2011.10.007
Iadecola, C. (2013). The pathobiology of vascular dementia. Neuron 80, 844–866. doi: 10.1016/j.neuron.2013.10.008
Iadecola, C. (2017). The neurovascular unit coming of age: a journey through neurovascular coupling in health and disease. Neuron 96, 17–42. doi: 10.1016/j.neuron.2017.07.030
Iadecola, C., and Davisson, R. L. (2008). Hypertension and cerebrovascular dysfunction. Cell Metab. 7, 476–484. doi: 10.1016/j.cmet.2008.03.010
Ihara, M., Polvikoski, T. M., Hall, R., Slade, J. Y., Perry, R. H., Oakley, A. E., et al. (2010). Quantification of myelin loss in frontal lobe white matter in vascular dementia, Alzheimer's disease, and dementia with Lewy bodies. Acta Neuropathol. 119, 579–589. doi: 10.1007/s00401-009-0635-8
Iliff, J. J., Wang, M., Liao, Y., Plogg, B. A., Peng, W., Gundersen, G. A., et al. (2012). A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid β. Sci. Transl. Med. 4:147ra111. doi: 10.1126/scitranslmed.3003748
Imai, S.-I., and Guarente, L. (2014). NAD+ and sirtuins in aging and disease. Trends Cell Biol. 24, 464–471. doi: 10.1016/j.tcb.2014.04.002
Jack, C. (2010). Vascular risk factor detection and control may prevent Alzheimer's disease. Ageing Res. Rev. 9, 218–225. doi: 10.1016/j.arr.2010.04.002
Jacob, A., and Alexander, J. J. (2014). Complement and blood-brain barrier integrity. Mol. Immunol. 61, 149–152. doi: 10.1016/j.molimm.2014.06.039
Jaeger, L., Dohgu, S., Sultana, R., Lynch, J., Owen, J., Erickson, M., et al. (2011). Lipopolysaccharide alters the blood-brain barrier transport of amyloid β protein: a mechanism for inflammation in the progression of Alzheimer's disease. Brain Behav. Immun. 23, 507–517. doi: 10.1016/j.bbi.2010.11.019
Jiang, T., Yin, F., Yao, J., Brinton, R. D., and Cadenas, E. (2013). Lipoic acid restores age-associated impairment of brain energy metabolism through the modulation of A kt/JNK signaling and PGC 1α transcriptional pathway. Aging Cell 12, 1021–1031. doi: 10.1111/acel.12127
Kadry, H., Noorani, B., and Cucullo, L. (2020). A blood-brain barrier overview on structure, function, impairment, and biomarkers of integrity. Fluids Barriers CNS 17, 1–24. doi: 10.1186/s12987-020-00230-3
Kalaria, R. (2002). Similarities between Alzheimer's disease and vascular dementia. J. Neurol. Sci. 203, 29–34. doi: 10.1016/S0022-510X(02)00256-3
Kang, S., Lee, Y.-H., and Lee, J. E. (2017). Metabolism-centric overview of the pathogenesis of Alzheimer's disease. Yonsei Med. J. 58, 479–488. doi: 10.3349/ymj.2017.58.3.479
Kettenmann, H., Hanisch, U.-K., Noda, M., and Verkhratsky, A. (2011). Physiology of microglia. Physiol. Rev. 91, 461–553. doi: 10.1152/physrev.00011.2010
Khan, M. B., Hoda, M. N., Vaibhav, K., Giri, S., Wang, P., Waller, J. L., et al. (2015). Remote ischemic postconditioning: harnessing endogenous protection in a murine model of vascular cognitive impairment. Transl. Stroke Res. 6, 69–77. doi: 10.1007/s12975-014-0374-6
Kimbrough, I. F., Robel, S., Roberson, E. D., and Sontheimer, H. (2015). Vascular amyloidosis impairs the gliovascular unit in a mouse model of Alzheimer's disease. Brain 138, 3716–3733. doi: 10.1093/brain/awv327
Kinnecom, C., Lev, M., Wendell, L., Smith, E., Rosand, J., Frosch, M., et al. (2007). Course of cerebral amyloid angiopathy-related inflammation. Neurology 68, 1411–1416. doi: 10.1212/01.wnl.0000260066.98681.2e
Kirshner, H. S. (2009). Vascular dementia: a review of recent evidence for prevention and treatment. Curr. Neurol. Neurosci. Rep. 9, 437–442. doi: 10.1007/s11910-009-0065-y
Kofler, J., and Wiley, C. A. (2011). Microglia: key innate immune cells of the brain. Toxicol. Pathol. 39, 103–114. doi: 10.1177/0192623310387619
Kovac, A., Erickson, M. A., and Banks, W. A. (2011). Brain microvascular pericytes are immunoactive in culture: cytokine, chemokine, nitric oxide, and LRP-1 expression in response to lipopolysaccharide. J. Neuroinflammation 8, 1–9. doi: 10.1186/1742-2094-8-139
Kovac, A., Zilkova, M., Deli, M. A., Zilka, N., and Novak, M. (2009). Human truncated tau is using a different mechanism from amyloid-β to damage the blood-brain barrier. J. Alzheimers Dis. 18, 897–906. doi: 10.3233/JAD-2009-1197
Krabbe, G., Halle, A., Matyash, V., Rinnenthal, J. L., Eom, G. D., Bernhardt, U., et al. (2013). Functional impairment of microglia coincides with Beta-amyloid deposition in mice with Alzheimer-like pathology. PLoS ONE 8:e60921. doi: 10.1371/journal.pone.0060921
Kritsilis, M. V, Rizou, S., Koutsoudaki, P. N., Evangelou, K., Gorgoulis, V. G., et al. (2018). Ageing, cellular senescence and neurodegenerative disease. Int. J. Mol. Sci. 19:2937. doi: 10.3390/ijms19102937
Kyrtata, N., Emsley, H. C., Sparasci, O., Parkes, L. M., and Dickie, B. R. (2021). A Systematic review of glucose transport alterations in Alzheimer's disease. Front. Neurosci. 15:626636. doi: 10.3389/fnins.2021.626636
Lan, Y.-L., Zhao, J., Ma, T., and Li, S. (2016a). The potential roles of aquaporin 4 in Alzheimer's disease. Mol. Neurobiol. 53, 5300–5309. doi: 10.1007/s12035-015-9446-1
Lan, Y.-L., Zou, S., Chen, J.-J., Zhao, J., and Li, S. (2016b). The neuroprotective effect of the association of aquaporin-4/glutamate transporter-1 against Alzheimer's disease. Neural Plast. 2016:4626593. doi: 10.1155/2016/4626593
Lapenna, A., De Palma, M., and Lewis, C. E. (2018). Perivascular macrophages in health and disease. Nat. Rev. Immunol. 18, 689–702. doi: 10.1038/s41577-018-0056-9
Lassman, M. E., McLaughlin, T. M., Somers, E. P., Stefanni, A. C., Chen, Z., Murphy, B. A., et al. (2012). A rapid method for cross-species quantitation of apolipoproteins A1, B48 and B100 in plasma by ultra-performance liquid chromatography/tandem mass spectrometry. Rapid Commun. Mass Spectrom. 26, 101–108. doi: 10.1002/rcm.5296
Lee, P., Kim, J., Williams, R., Sandhir, R., Gregory, E., Brooks, W. M., et al. (2012). Effects of aging on blood brain barrier and matrix metalloproteases following controlled cortical impact in mice. Exp. Neurol. 234, 50–61. doi: 10.1016/j.expneurol.2011.12.016
Lendahl, U., Nilsson, P., and Betsholtz, C. (2019). Emerging links between cerebrovascular and neurodegenerative diseases-a special role for pericytes. EMBO Rep. 20:e48070. doi: 10.15252/embr.201948070
Leng, F., and Edison, P. (2020). Neuroinflammation and microglial activation in Alzheimer disease: where do we go from here? Nat. Rev. Neurol. 17, 157–172. doi: 10.1038/s41582-020-00435-y
Levit, A., Hachinski, V., and Whitehead, S. N. (2020). Neurovascular unit dysregulation, white matter disease, and executive dysfunction: the shared triad of vascular cognitive impairment and Alzheimer disease. GeroScience 42, 445–465. doi: 10.1007/s11357-020-00164-6
Li, W., and Lai, X.-S. (2007). Changes of interleukin-1beta and TNF-alpha contents in the hippocampus and the interventional effect of electroacupuncture in vascular dementia rats. Zhen Ci Yan Jiu 32, 34–37.
Li, W. Z., Wu, W. Y., Huang, H., Wu, Y. Y., and Yin, Y. Y. (2013). Protective effect of bilobalide on learning and memory impairment in rats with vascular dementia. Mol. Med. Rep. 8, 935–941. doi: 10.3892/mmr.2013.1573
Lin, Y.-F., Smith, A. V., Aspelund, T., Betensky, R. A., Smoller, J. W., Gudnason, V., et al. (2019). Genetic overlap between vascular pathologies and Alzheimer's dementia and potential causal mechanisms. Alzheimers Dement. 15, 65–75. doi: 10.1016/j.jalz.2018.08.002
Liu, C.-C., Kanekiyo, T., Xu, H., and Bu, G. (2013). Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy. Nat. Rev. Neurol. 9, 106–118. doi: 10.1038/nrneurol.2012.263
Liu, H., and Zhang, J. (2012). Cerebral hypoperfusion and cognitive impairment: the pathogenic role of vascular oxidative stress. Int. J. Neurosci. 122, 494–499. doi: 10.3109/00207454.2012.686543
Lucke-Wold, B. P., Logsdon, A. F., Turner, R. C., Rosen, C. L., and Huber, J. D. (2014). Aging, the metabolic syndrome, and ischemic stroke: redefining the approach for studying the blood-brain barrier in a complex neurological disease. Adv. Pharmacol. 71, 411–449. doi: 10.1016/bs.apha.2014.07.001
Månberg, A., Skene, N., Sanders, F., Trusohamn, M., Remnestål, J., Szczepińska, A., et al. (2021). Altered perivascular fibroblast activity precedes ALS disease onset. Nat. Med. 27, 640–646. doi: 10.1038/s41591-021-01295-9
Ma, X., Sun, Z., Liu, Y., Jia, Y., Zhang, B., and Zhang, J. (2013). Resveratrol improves cognition and reduces oxidative stress in rats with vascular dementia. Neural Regen. Res. 8, 2050–2059. doi: 10.3969/j.issn.1673-5374.2013.22.004
Matthes, F., Matuskova, H., Arkelius, K., Ansar, S., Lundgaard, I., and Meissner, A. (2021). An improved method for physical separation of cerebral vasculature and parenchyma enables detection of blood-brain-barrier dysfunction. NeuroSci 2, 59–74. doi: 10.3390/neurosci2010004
Mattson, M. P., and Magnus, T. (2006). Ageing and neuronal vulnerability. Nat. Rev. Neurosci. 7, 278–294. doi: 10.1038/nrn1886
Mazzieri, R., Masiero, L., Zanetta, L., Monea, S., Onisto, M., Garbisa, S., et al. (1997). Control of type IV collagenase activity by components of the urokinase-plasmin system: a regulatory mechanism with cell-bound reactants. EMBO J. 16, 2319–2332. doi: 10.1093/emboj/16.9.2319
Merlini, M., Rafalski, V. A., Coronado, P. E. R., Gill, T. M., Ellisman, M., Muthukumar, G., et al. (2019). Fibrinogen induces microglia-mediated spine elimination and cognitive impairment in an Alzheimer's disease model. Neuron 101, 1099.e6–1108.e6. doi: 10.1016/j.neuron.2019.01.014
Middeldorp, J., and Hol, E. (2011). GFAP in health and disease. Prog. Neurobiol. 93, 421–443. doi: 10.1016/j.pneurobio.2011.01.005
Miller, M. C., Tavares, R., Johanson, C. E., Hovanesian, V., Donahue, J. E., Gonzalez, L., et al. (2008). Hippocampal RAGE immunoreactivity in early and advanced Alzheimer's disease. Brain Res. 1230, 273–280. doi: 10.1016/j.brainres.2008.06.124
Mills, S., Cain, J., Purandare, N., and Jackson, A. (2007). Biomarkers of cerebrovascular disease in dementia. Brit. J. Radiol. 80, S128–S145. doi: 10.1259/bjr/79217686
Miners, J., Kehoe, P., Love, S., Zetterberg, H., and Blennow, K. (2019). CSF evidence of pericyte damage in Alzheimer's disease is associated with markers of blood-brain barrier dysfunction and disease pathology. Alzheimers Res. Ther. 11, 1–6. doi: 10.1186/s13195-019-0534-8
Monea, S., Lehti, K., Keski-Oja, J., and Mignatti, P. (2002). Plasmin activates pro-matrix metalloproteinase-2 with a membrane-type 1 matrix metalloproteinase-dependent mechanism. J. Cell. Physiol. 192, 160–170. doi: 10.1002/jcp.10126
Montagne, A., Barnes, S. R., Sweeney, M. D., Halliday, M. R., Sagare, A. P., Zhao, Z., et al. (2015). Blood-brain barrier breakdown in the aging human hippocampus. Neuron 85, 296–302. doi: 10.1016/j.neuron.2014.12.032
Montagne, A., Nation, D. A., Sagare, A. P., Barisano, G., Sweeney, M. D., Chakhoyan, A., et al. (2020). APOE4 leads to blood-brain barrier dysfunction predicting cognitive decline. Nature 581, 71–76. doi: 10.1038/s41586-020-2247-3
Montagne, A., Nikolakopoulou, A. M., Zhao, Z., Sagare, A. P., Si, G., Lazic, D., et al. (2018). Pericyte degeneration causes white matter dysfunction in the mouse central nervous system. Nat. Med. 24, 326–337. doi: 10.1038/nm.4482
Mooradian, A., Chung, H., and Shah, G. (1997). GLUT-1 expression in the cerebra of patients with Alzheimer's disease. Neurobiol. Aging 18, 469–474. doi: 10.1016/S0197-4580(97)00111-5
Morris, A. W., Carare, R. O., Schreiber, S., and Hawkes, C. A. (2014). The cerebrovascular basement membrane: role in the clearance of β-amyloid and cerebral amyloid angiopathy. Front. Aging Neurosci. 6:251. doi: 10.3389/fnagi.2014.00251
Nation, D. A., Sweeney, M. D., Montagne, A., Sagare, A. P., D'Orazio, L. M., Pachicano, M., et al. (2019). Blood-brain barrier breakdown is an early biomarker of human cognitive dysfunction. Nat. Med. 25, 270–276. doi: 10.1038/s41591-018-0297-y
Nelson, A. R., Sweeney, M. D., Sagare, A. P., and Zlokovic, B. V. (2016). Neurovascular dysfunction and neurodegeneration in dementia and Alzheimer's disease. Biochim. Biophys. Acta 1862, 887–900. doi: 10.1016/j.bbadis.2015.12.016
Nicholls, D. G., and Budd, S. L. (2000). Mitochondria and neuronal survival. Physiol. Rev. 80, 315–360. doi: 10.1152/physrev.2000.80.1.315
Niego, B. E., Freeman, R., Puschmann, T. B., Turnley, A. M., and Medcalf, R. L. (2012). t-PA-specific modulation of a human blood-brain barrier model involves plasmin-mediated activation of the Rho kinase pathway in astrocytes. Blood 119, 4752–4761. doi: 10.1182/blood-2011-07-369512
Nikolakopoulou, A. M., Montagne, A., Kisler, K., Dai, Z., Wang, Y., Huuskonen, M. T., et al. (2019). Pericyte loss leads to circulatory failure and pleiotrophin depletion causing neuron loss. Nat. Neurosci. 22, 1089–1098. doi: 10.1038/s41593-019-0434-z
Nikolakopoulou, A. M., Wang, Y., Ma, Q., Sagare, A. P., Montagne, A., Huuskonen, M. T., et al. (2021). Endothelial LRP1 protects against neurodegeneration by blocking cyclophilin A. J. Exp. Med. 218:e20202207. doi: 10.1084/jem.20202207
Nishitsuji, K., Hosono, T., Nakamura, T., Bu, G., and Michikawa, M. (2011). Apolipoprotein E regulates the integrity of tight junctions in an isoform-dependent manner in an in vitro blood-brain barrier model. J. Biol. Chem. 286, 17536–17542. doi: 10.1074/jbc.M111.225532
Niwa, K., Younkin, L., Ebeling, C., Turner, S. K., Westaway, D., Younkin, S., et al. (2000). Aβ1-40-related reduction in functional hyperemia in mouse neocortex during somatosensory activation. Proc. Nat. Acad. Sci.U.S.A. 97, 9735–9740. doi: 10.1073/pnas.97.17.9735
Obermeier, B., Daneman, R., and Ransohoff, R. M. (2013). Development, maintenance and disruption of the blood-brain barrier. Nat. Med. 19, 1584–1596. doi: 10.1038/nm.3407
O'Brien, T., and Thomas, J. A. (2015). Vascular dementia. Lancet 386, 1698–1706. doi: 10.1016/S.0140-6736(15)00463-8
O'Connell, G. C., Treadway, M. B., Petrone, A. B., Tennant, C. S., Lucke-Wold, N., Chantler, P. D., et al. (2017). Peripheral blood AKAP7 expression as an early marker for lymphocyte-mediated post-stroke blood brain barrier disruption. Sci. Rep. 7, 1–13. doi: 10.1038/s41598-017-01178-5
Orsini, F., De Blasio, D., Zangari, R., Zanier, E. R., and De Simoni, M.-G. (2014). Versatility of the complement system in neuroinflammation, neurodegeneration and brain homeostasis. Front. Cell. Neurosci. 8:380. doi: 10.3389/fncel.2014.00380
Owen, J. B., Sultana, R., Aluise, C. D., Erickson, M. A., Price, T. O., Bu, G., et al. (2010). Oxidative modification to LDL receptor-related protein 1 in hippocampus from subjects with Alzheimer disease: implications for Aβ accumulation in AD brain. Free Radic. Biol. Med. 49, 1798–1803. doi: 10.1016/j.freeradbiomed.2010.09.013
Pan, W. P, Stone, K., Hsuchou, H. K, Manda, V., et al. (2011). Cytokine signaling modulates blood-brain barrier function. Curr. Pharm. Des. 17, 3729–3740. doi: 10.2174/138161211798220918
Park, H. R., Park, M., Choi, J., Park, K.-Y., Chung, H. Y., and Lee, J. (2010). A high-fat diet impairs neurogenesis: involvement of lipid peroxidation and brain-derived neurotrophic factor. Neurosci. Lett. 482, 235–239. doi: 10.1016/j.neulet.2010.07.046
Park, L., Uekawa, K., Garcia-Bonilla, L., Koizumi, K., Murphy, M., Pistik, R., et al. (2017). Brain perivascular macrophages initiate the neurovascular dysfunction of Alzheimer Aβ peptides. Circ. Res. 121, 258–269. doi: 10.1161/CIRCRESAHA.117.311054
Park, M. H., Lee, J. Y., Park, K. H., Jung, I. K., Kim, K.-T., Lee, Y.-S., et al. (2018). Vascular and neurogenic rejuvenation in aging mice by modulation of ASM. Neuron 100, 167.e9–182.e9. doi: 10.1016/j.neuron.2018.09.010
Patching, S. G. (2017). Glucose transporters at the blood-brain barrier: function, regulation and gateways for drug delivery. Mol. Neurobiol. 54, 1046–1077. doi: 10.1007/s12035-015-9672-6
Peinado, M. A., Quesada, A., Pedrosa, J. A., Torres, M. I., Martinez, M., Esteban, F. J., et al. (1998). Quantitative and ultrastructural changes in glia and pericytes in the parietal cortex of the aging rat. Microsc. Res. Tech. 43, 34–42. doi: 10.1002/(SICI)1097-0029(19981001)43:1<34::AID-JEMT6>3.0.CO;2-G
Perry, G., Cash, A. D., and Smith, M. A. (2002). Alzheimer disease and oxidative stress. J. Biomed. Biotechnol. 2, 120–123. doi: 10.1155/S1110724302203010
Peters, A., Josephson, K., and Vincent, S. L. (1991). Effects of aging on the neuroglial cells and pericytes within area 17 of the rhesus monkey cerebral cortex. Anat. Rec. 229, 384–398. doi: 10.1002/ar.1092290311
Pilozzi, A., Carro, C., Whalen, M., and Huang, X. (2020). “Blood-brain barrier degradation and the implication of SPARC protein as a potential therapeutic target for Alzheimer's disease,” in Alzheimer's Disease: Drug Discovery, ed X. Huang (Brisbane, QLD: Exon Publications), 135–149. doi: 10.36255/exonpublications.alzheimersdisease.2020.ch8
Price, B. R., Norris, C. M., Sompol, P., and Wilcock, D. M. (2018). An emerging role of astrocytes in vascular contributions to cognitive impairment and dementia. J. Neurochem. 144, 644–650. doi: 10.1111/jnc.14273
Quaegebeur, A., Lange, C., and Carmeliet, P. (2011). The neurovascular link in health and disease: molecular mechanisms and therapeutic implications. Neuron 71, 406–424. doi: 10.1016/j.neuron.2011.07.013
Raja, R., Rosenberg, G. A., and Caprihan, A. (2018). MRI measurements of blood-brain barrier function in dementia: a review of recent studies. Neuropharmacology 134, 259–271. doi: 10.1016/j.neuropharm.2017.10.034
Rajan, A. M., Ma, R. C., Kocha, K. M., Zhang, D. J., and Huang, P. (2020). Dual function of perivascular fibroblasts in vascular stabilization in zebrafish. PLoS Genet. 16:e1008800. doi: 10.1371/journal.pgen.1008800
Rajani, R. M., Quick, S., Ruigrok, S. R., Graham, D., Harris, S. E., Verhaaren, B. F., et al. (2018). Reversal of endothelial dysfunction reduces white matter vulnerability in cerebral small vessel disease in rats. Sci. Transl. Med. 10:eaam9507. doi: 10.1126/scitranslmed.aam9507
Ramanathan, A., Nelson, A. R., Sagare, A. P., and Zlokovic, B. V. (2015). Impaired vascular-mediated clearance of brain amyloid beta in Alzheimer's disease: the role, regulation and restoration of LRP1. Front. Aging Neurosci. 7:136. doi: 10.3389/fnagi.2015.00136
Ramos-DeSimone, N., Hahn-Dantona, E., Sipley, J., Nagase, H., French, D. L., and Quigley, J. P. (1999). Activation of matrix metalloproteinase-9 (MMP-9) via a converging plasmin/stromelysin-1 cascade enhances tumor cell invasion. J. Biol. Chem. 274, 13066–13076. doi: 10.1074/jbc.274.19.13066
Ransohoff, R. M., and Engelhardt, B. (2012). The anatomical and cellular basis of immune surveillance in the central nervous system. Nat. Rev. Immunol. 12, 623–635. doi: 10.1038/nri3265
Rascher, G., and Wolburg, H. (2002). “The blood-brain barrier in the aging brain.” in Neuroglia in the Aging Brain, ed J. S. de Vellis (Totowa, NJ: Humana Press), 305–320. doi: 10.1385/1-59259-105-1:305
Rasmussen, M. K., Mestre, H., and Nedergaard, M. (2018). The glymphatic pathway in neurological disorders. Lancet Neurol. 17, 1016–1024. doi: 10.1016/S1474-4422(18)30318-1
Ray, L., Khemka, V. K., Behera, P., Bandyopadhyay, K., Pal, S., Pal, K., et al. (2013). Serum homocysteine, dehydroepiandrosterone sulphate and lipoprotein (a) in Alzheimer's disease and vascular dementia. Aging Dis. 4, 57–64.
Reeson, P., Choi, K., and Brown, C. E. (2018). VEGF signaling regulates the fate of obstructed capillaries in mouse cortex. Elife 7:e33670. doi: 10.7554/eLife.33670.029
Reuter, B., Rodemer, C., Grudzenski, S., Meairs, S., Bugert, P., Hennerici, M. G., et al. (2015). Effect of simvastatin on MMPs and TIMPs in human brain endothelial cells and experimental stroke. Transl. Stroke Res. 6, 156–159. doi: 10.1007/s12975-014-0381-7
Reyahi, A., Nik, A. M., Ghiami, M., Gritli-Linde, A., Pontén, F., Johansson, B. R., et al. (2015). Foxf2 is required for brain pericyte differentiation and development and maintenance of the blood-brain barrier. Dev. Cell 34, 19–32. doi: 10.1016/j.devcel.2015.05.008
Richardson, K. C. M, Stephan, B. G, Ince, P., Brayne, C., et al. (2012). The neuropathology of vascular disease in the medical research council cognitive function and ageing study (MRC CFAS). Curr. Alzheimer Res. 9, 687–696. doi: 10.2174/156720512801322654
Rivard, A., Berthou-Soulie, L., Principe, N., Kearney, M., Curry, C., Branellec, D., et al. (2000). Age-dependent defect in vascular endothelial growth factor expression is associated with reduced hypoxia-inducible factor 1 activity. J. Biol. Chem. 275, 29643–29647. doi: 10.1074/jbc.M001029200
Ronaldson, T., and Davis, P. (2012). Blood-brain barrier integrity and glial support: mechanisms that can be targeted for novel therapeutic approaches in stroke. Curr. Pharm. Des. 18, 3624–3644. doi: 10.2174/138161212802002625
Rosenberg, G. (2014). Blood-brain barrier permeability in aging and Alzheimer's disease. J. Prevent. Alzheimers Dis. 1, 138–139. doi: 10.14283/2Fjpad.2014.25
Rosenberg, G. A. (2012). Neurological diseases in relation to the blood-brain barrier. J. Cereb. Blood Flow Metab. 32, 1139–1151. doi: 10.1038/jcbfm.2011.197
Rosenberg, G. A. (2017). Extracellular matrix inflammation in vascular cognitive impairment and dementia. Clin. Sci. 131, 425–437. doi: 10.1042/CS20160604
Rosenberg, G. A., and Yang, Y. (2007). Vasogenic edema due to tight junction disruption by matrix metalloproteinases in cerebral ischemia. Neurosurg. Focus 22, 1–9. doi: 10.3171/foc.2007.22.5.5
Roudnicky, F., Zhang, J. D., Kim, B. K., Pandya, N. J., Lan, Y., Sach-Peltason, L., et al. (2020). Inducers of the endothelial cell barrier identified through chemogenomic screening in genome-edited hPSC-endothelial cells. Proc. Nat. Acad. Sci. U.S.A. 117, 19854–19865. doi: 10.1073/pnas.1911532117
Rouhl, R. P., Damoiseaux, J. G., Lodder, J., Theunissen, R. O., Knottnerus, I. L., Staals, J., et al. (2012). Vascular inflammation in cerebral small vessel disease. Neurobiol. Aging 33, 1800–1806. doi: 10.1016/j.neurobiolaging.2011.04.008
Ryu, J. K., Petersen, M. A., Murray, S. G., Baeten, K. M., Meyer-Franke, A., Chan, J. P., et al. (2015). Blood coagulation protein fibrinogen promotes autoimmunity and demyelination via chemokine release and antigen presentation. Nat. Commun. 6, 1–15. doi: 10.1038/ncomms9164
Ryu, J. K., Rafalski, V. A., Meyer-Franke, A., Adams, R. A., Poda, S. B., Coronado, P. E. R., et al. (2018). Fibrin-targeting immunotherapy protects against neuroinflammation and neurodegeneration. Nat. Immunol. 19, 1212–1223. doi: 10.1038/s41590-018-0232-x
Sabayan, B., Jansen, S., Oleksik, A. M., van Osch, M. J., van Buchem, M. A., van Vliet, P., et al. (2012). Cerebrovascular hemodynamics in Alzheimer's disease and vascular dementia: a meta-analysis of transcranial Doppler studies. Ageing Res. Rev. 11, 271–277. doi: 10.1016/j.arr.2011.12.009
Sachdev, P., Kalaria, R., O'Brien, J., Skoog, I., Alladi, S., Black, S. E., et al. (2014). Diagnostic criteria for vascular cognitive disorders: a VASCOG statement. Alzheimer Dis. Assoc. Disord. 28, 206–218. doi: 10.1097/WAD.0000000000000034
Sagare, A., Deane, R., Bell, R. D., Johnson, B., Hamm, K., Pendu, R., et al. (2007). Clearance of amyloid-β by circulating lipoprotein receptors. Nat. Med. 13, 1029–1031. doi: 10.1038/nm1635
Sagare, A. P., Bell, R. D., Zhao, Z., Ma, Q., Winkler, E. A., Ramanathan, A., et al. (2013). Pericyte loss influences Alzheimer-like neurodegeneration in mice. Nat. Commun. 4, 1–14. doi: 10.1038/ncomms3932
Sagare, A. P., Bell, R. D., and Zlokovic, B. V. (2012). Neurovascular dysfunction and faulty amyloid β-peptide clearance in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2:a011452. doi: 10.1101/cshperspect.a011452
Sagare, A. P., Sweeney, M. D., Makshanoff, J., and Zlokovic, B. V. (2015). Shedding of soluble platelet-derived growth factor receptor-β from human brain pericytes. Neurosci. Lett. 607, 97–101. doi: 10.1016/j.neulet.2015.09.025
Saggu, R., Schumacher, T., Gerich, F., Rakers, C., Tai, K., Delekate, A., et al. (2016). Astroglial NF-kB contributes to white matter damage and cognitive impairment in a mouse model of vascular dementia. Acta Neuropathol. Commun. 4, 1–10. doi: 10.1186/s40478-016-0350-3
Salmina, A. B., Inzhutova, A. I., Malinovskaya, N. A., and Petrova, M. M. (2010). Endothelial dysfunction and repair in Alzheimer-type neurodegeneration: neuronal and glial control. J. Alzheimers Dis. 22, 17–36. doi: 10.3233/JAD-2010-091690
Salminen, A., Ojala, J., Kaarniranta, K., Haapasalo, A., Hiltunen, M., and Soininen, H. (2011). Astrocytes in the aging brain express characteristics of senescence-associated secretory phenotype. Eur. J. Neurosci. 34, 3–11. doi: 10.1111/j.1460-9568.2011.07738.x
Sanchez-Covarrubias, L., Slosky, L. M., Thompson, B. J., Davis, T. P., and Ronaldson, P. T. (2014). Transporters at CNS barrier sites: obstacles or opportunities for drug delivery? Curr. Pharm. Des. 20, 1422–1449. doi: 10.2174/13816128113199990463
Schneider, J. A., Arvanitakis, Z., Leurgans, S. E., and Bennett, D. A. (2009). The neuropathology of probable Alzheimer disease and mild cognitive impairment. Ann. Neurol. 66, 200–208. doi: 10.1002/ana.21706
Schultz, N., Brännström, K., Byman, E., Moussaud, S., Nielsen, H. M., Bank, N. B., et al. (2018). Amyloid-beta 1-40 is associated with alterations in NG2+ pericyte population ex vivo and in vitro. Aging Cell 17:e12728. doi: 10.1111/acel.12728
Sengillo, J. D., Winkler, E. A., Walker, C. T., Sullivan, J. S., Johnson, M., and Zlokovic, B. V. (2013). Deficiency in mural vascular cells coincides with blood-brain barrier disruption in Alzheimer's disease. Brain Pathol. 23, 303–310. doi: 10.1111/bpa.12004
Silbert, L. C., Dodge, H. H., Perkins, L. G., Sherbakov, L., Lahna, D., Erten-Lyons, D., et al. (2012). Trajectory of white matter hyperintensity burden preceding mild cognitive impairment. Neurology 79, 741–747. doi: 10.1212/WNL.0b013e3182661f2b
Simpkins, A. N., Dias, C., Leigh, R., Benson, R. T., Hsia, A. W., Latour, L. L., et al. (2016). Identification of reversible disruption of the human blood-brain barrier following acute ischemia. Stroke 47, 2405–2408. doi: 10.1161/STROKEAHA.116.013805
Simpson, J., Ince, P., Haynes, L., Theaker, R., Gelsthorpe, C., Baxter, L., et al. (2010). Population variation in oxidative stress and astrocyte DNA damage in relation to Alzheimer-type pathology in the ageing brain. Neuropathol. Appl. Neurobiol. 36, 25–40. doi: 10.1111/j.1365-2990.2009.01030.x
Skillbäck, T., Delsing, L., Synnergren, J., Mattsson, N., Janelidze, S., Nägga, K., et al. (2017). CSF/serum albumin ratio in dementias: a cross-sectional study on 1861 patients. Neurobiol. Aging 59, 1–9. doi: 10.1016/j.neurobiolaging.2017.06.028
Sloane, P. D., Zimmerman, S., Suchindran, C., Reed, P., Wang, L., Boustani, M., et al. (2002). The public health impact of Alzheimer's disease, 2000-2050: potential implication of treatment advances. Annu. Rev. Public Health 23, 213–231. doi: 10.1146/annurev.publhealth.23.100901.140525
Soto, I., Graham, L. C., Richter, H. J., Simeone, S. N., Radell, J. E., Grabowska, W., et al. (2015). APOE stabilization by exercise prevents aging neurovascular dysfunction and complement induction. PLoS Biol. 13:e1002279. doi: 10.1371/journal.pbio.1002279
Stamatovic, S. M., Martinez-Revollar, G., Hu, A., Choi, J., Keep, R. F., and Andjelkovic, A. V. (2019). Decline in Sirtuin-1 expression and activity plays a critical role in blood-brain barrier permeability in aging. Neurobiol. Dis. 126, 105–116. doi: 10.1016/j.nbd.2018.09.006
Stewart, P., Magliocco, M., Hayakawa, K., Farrell, C., Del Maestro, R., Girvin, J., et al. (1987). A quantitative analysis of blood-brain barrier ultrastructure in the aging human. Microvasc. Res. 33, 270–282. doi: 10.1016/0026-2862(87)90022-7
Storck, S. E., Meister, S., Nahrath, J., Meißner, J. N., Schubert, N., Di Spiezio, A., et al. (2016). Endothelial LRP1 transports amyloid-β 1-42 across the blood-brain barrier. J. Clin. Invest. 126, 123–136. doi: 10.1172/JCI81108
Stranahan, A. M., Norman, E. D., Lee, K., Cutler, R. G., Telljohann, R. S., Egan, J. M., et al. (2008). Diet-induced insulin resistance impairs hippocampal synaptic plasticity and cognition in middle-aged rats. Hippocampus 18, 1085–1088. doi: 10.1002/hipo.20470
Stratman, A. N., Schwindt, A. E., Malotte, K. M., and Davis, G. E. (2010). Endothelial-derived PDGF-BB and HB-EGF coordinately regulate pericyte recruitment during vasculogenic tube assembly and stabilization. Blood 116, 4720–4730. doi: 10.1182/blood-2010-05-286872
Strunz, M., Jarrell, J. T., Cohen, D. S., Rosin, E. R., Vanderburg, C. R., and Huang, X. (2019). Modulation of SPARC/Hevin proteins in Alzheimer's disease brain injury. J. Alzheimers Dis. 68, 695–710. doi: 10.3233/JAD-181032
Sturrock, R. (1980). A comparative quantitative and morphological study of ageing in the mouse neostriatum, indusium griseum and anterior commissure. Neuropathol. Appl. Neurobiol. 6, 51–68. doi: 10.1111/j.1365-2990.1980.tb00204.x
Sumi, N., Nishioku, T., Takata, F., Matsumoto, J., Watanabe, T., Shuto, H., et al. (2010). Lipopolysaccharide-activated microglia induce dysfunction of the blood-brain barrier in rat microvascular endothelial cells co-cultured with microglia. Cell. Mol. Neurobiol. 30, 247–253. doi: 10.1007/s10571-009-9446-7
Suzuki, Y., Nagai, N., Yamakawa, K., Kawakami, J., Lijnen, H. R., and Umemura, K. (2009). Tissue-type plasminogen activator (t-PA) induces stromelysin-1 (MMP-3) in endothelial cells through activation of lipoprotein receptor-related protein. Blood 114, 3352–3358. doi: 10.1182/blood-2009-02-203919
Sweeney, M. D., Ayyadurai, S., and Zlokovic, B. V. (2016). Pericytes of the neurovascular unit: key functions and signaling pathways. Nat. Neurosci. 19, 771–783. doi: 10.1038/nn.4288
Sweeney, M. D., Kisler, K., Montagne, A., Toga, A. W., and Zlokovic, B. V. (2018a). The role of brain vasculature in neurodegenerative disorders. Nat. Neurosci. 21, 1318–1331. doi: 10.1038/s41593-018-0234-x
Sweeney, M. D., Montagne, A., Sagare, A. P., Nation, D. A., Schneider, L. S., Chui, H. C., et al. (2019a). Vascular dysfunction-the disregarded partner of Alzheimer's disease. Alzheimers Dement. 15, 158–167. doi: 10.1016/j.jalz.2018.07.222
Sweeney, M. D., Sagare, A. P., Pachicano, M., Harrington, M. G., Joe, E., Chui, H. C., et al. (2020). A novel sensitive assay for detection of a biomarker of pericyte injury in cerebrospinal fluid. Alzheimers Dement. 16, 821–830. doi: 10.1002/alz.12061
Sweeney, M. D., Sagare, A. P., and Zlokovic, B. V. (2018b). Blood-brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat. Rev. Neurol. 14, 133–150. doi: 10.1038/nrneurol.2017.188
Sweeney, M. D., Zhao, Z., Montagne, A., Nelson, A. R., and Zlokovic, B. V. (2019b). Blood-brain barrier: from physiology to disease and back. Physiol. Rev. 99, 21–78. doi: 10.1152/physrev.00050.2017
Tachibana, M., Holm, M.-L., Liu, C.-C., Shinohara, M., Aikawa, T., Oue, H., et al. (2019). APOE4-mediated amyloid-β pathology depends on its neuronal receptor LRP1. J. Clin. Invest. 129, 1272–1277. doi: 10.1172/JCI124853
Tada, M., Diserens, A.-C., Desbaillets, I., and de Tribolet, N. (1994). Analysis of cytokine receptor messenger RNA expression in human glioblastoma cells and normal astrocytes by reverse-transcription polymerase chain reaction. J. Neurosurg. 80, 1063–1073. doi: 10.3171/jns.1994.80.6.1063
Takata, F., Dohgu, S., Matsumoto, J., Takahashi, H., Machida, T., Wakigawa, T., et al. (2011). Brain pericytes among cells constituting the blood-brain barrier are highly sensitive to tumor necrosis factor-α, releasing matrix metalloproteinase-9 and migrating in vitro. J. Neuroinflammation 8, 1–12. doi: 10.1186/1742-2094-8-106
Tan, Z., Jiang, J., Tian, F., Peng, J., Yang, Z., Li, S., et al. (2020). Serum visinin-like protein 1 is a better biomarker than neuron-specific enolase for seizure-induced neuronal injury: a prospective and observational study. Front. Neurol. 11:567587. doi: 10.3389/fneur.2020.567587
Tarumi, T., and Zhang, R. (2018). Cerebral blood flow in normal aging adults: cardiovascular determinants, clinical implications, and aerobic fitness. J. Neurochem. 144, 595–608. doi: 10.1111/jnc.14234
Tayler, H., Miners, J. S., Güzel, Ö., MacLachlan, R., and Love, S. (2021). Mediators of cerebral hypoperfusion and blood-brain barrier leakiness in Alzheimer's disease, vascular dementia and mixed dementia. Brain Pathol. 31:e12935. doi: 10.1111/bpa.12935
Thal, D. R., Grinberg, L. T., and Attems, J. (2012). Vascular dementia: different forms of vessel disorders contribute to the development of dementia in the elderly brain. Exp. Gerontol. 47, 816–824. doi: 10.1016/j.exger.2012.05.023
Tibbling, G., Link, H., and Öhman, S. (1977). Principles of albumin and IgG analyses in neurological disorders. I. Establishment of reference values. Scand. J. Clin. Lab. Invest. 37, 385–390. doi: 10.3109/00365517709091496
Tomimoto, H., Akiguchi, I., Suenaga, T., Nishimura, M., Wakita, H., Nakamura, S., et al. (1996). Alterations of the blood-brain barrier and glial cells in white-matter lesions in cerebrovascular and Alzheimer's disease patients. Stroke 27, 2069–2074. doi: 10.1161/01.STR.27.11.2069
Tong, X., and Hamel, E. (1999). Regional cholinergic denervation of cortical microvessels and nitric oxide synthase-containing neurons in Alzheimer's disease. Neuroscience 92, 163–175. doi: 10.1016/S0306-4522(98)00750-7
Uemura, M. T., Maki, T., Ihara, M., Lee, V. M., and Trojanowski, J. Q. (2020). Brain microvascular pericytes in vascular cognitive impairment and dementia. Front. Aging Neurosci. 12:80. doi: 10.3389/fnagi.2020.00080
Ueno, M., Akiguchi, I., Hosokawa, M., Shinnou, M., Sakamoto, H., Takemura, M., et al. (1998). Ultrastructural and permeability features of microvessels in the olfactory bulbs of SAM mice. Acta Neuropathol. 96, 261–270. doi: 10.1007/s004010050893
Ueno, M., Tomimoto, H., Akiguchi, I., Wakita, H., and Sakamoto, H. (2002). Blood-brain barrier disruption in white matter lesions in a rat model of chronic cerebral hypoperfusion. J. Cereb. Blood Flow Metab. 22, 97–104. doi: 10.1097/00004647-200201000-00012
van Assema, D. M., Lubberink, M., Boellaard, R., Schuit, R. C., Windhorst, A. D., Scheltens, P., et al. (2012). P-glycoprotein function at the blood-brain barrier: effects of age and gender. Mol. Imaging Biol. 14, 771–776. doi: 10.1007/s11307-012-0556-0
Van De Haar, H. J., Burgmans, S., Jansen, J. F., Van Osch, M. J., Van Buchem, M. A., Muller, M., et al. (2016). Blood-brain barrier leakage in patients with early Alzheimer disease. Radiology 281, 527–535. doi: 10.1148/radiol.2016152244
Vasic, V., Barth, K., and Schmidt, M. H. (2019). Neurodegeneration and neuro-regeneration-Alzheimer's disease and stem cell therapy. Int. J. Mol. Sci. 20, 4272. doi: 10.3390/ijms20174272
Vaucher, E., Tong, X. K., Cholet, N., Lantin, S., and Hamel, E. (2000). GABA neurons provide a rich input to microvessels but not nitric oxide neurons in the rat cerebral cortex: a means for direct regulation of local cerebral blood flow. J. Compar. Neurol. 421, 161–171. doi: 10.1002/(SICI)1096-9861(20000529)421:2<161::AID-CNE3>3.0.CO;2-F
Veerhuis, R., Nielsen, H. M., and Tenner, A. J. (2011). Complement in the brain. Mol. Immunol. 48, 1592–1603. doi: 10.1016/j.molimm.2011.04.003
Verhaaren, B. F., Vernooij, M. W., de Boer, R., Hofman, A., Niessen, W. J., van der Lugt, A., et al. (2013). High blood pressure and cerebral white matter lesion progression in the general population. Hypertension 61, 1354–1359. doi: 10.1161/HYPERTENSIONAHA.111.00430
Verheggen, I. C., de Jong, J. J., van Boxtel, M. P., Gronenschild, E. H., Palm, W. M., Postma, A. A., et al. (2020). Increase in blood-brain barrier leakage in healthy, older adults. GeroScience 42, 1183–1193. doi: 10.1007/s11357-020-00211-2
Verma, S., Nakaoke, R., Dohgu, S., and Banks, W. A. (2006). Release of cytokines by brain endothelial cells: a polarized response to lipopolysaccharide. Brain Behav. Immun. 20, 449–455. doi: 10.1016/j.bbi.2005.10.005
Villar-Vesga, J., Henao-Restrepo, J., Voshart, D. C., Aguillon, D., Villegas, A., Castaño, D., et al. (2020). Differential profile of systemic extracellular vesicles from sporadic and familial Alzheimer's disease leads to neuroglial and endothelial cell degeneration. Front. Aging Neurosci. 12:587989. doi: 10.3389/fnagi.2020.587989
Villeda, S. A., Luo, J., Mosher, K. I., Zou, B., Britschgi, M., Bieri, G., et al. (2011). The ageing systemic milieu negatively regulates neurogenesis and cognitive function. Nature 477, 90–94. doi: 10.1038/nature10357
Wang, F., Cao, Y., Ma, L., Pei, H., Rausch, W. D., and Li, H. (2018). Dysfunction of cerebrovascular endothelial cells: prelude to vascular dementia. Front. Aging Neurosci. 10:376. doi: 10.3389/fnagi.2018.00376
Wang, W., Lv, S., Zhou, Y., Fu, J., Li, C., and Liu, P. (2011). Tumor necrosis factor-α affects blood-brain barrier permeability in acetaminophen-induced acute liver failure. Eur. J. Gastroenterol. Hepatol. 23, 552–558. doi: 10.1097/MEG.0b013e3283470212
Wang, X., Lee, S.-R., Arai, K., Lee, S.-R., Tsuji, K., Rebeck, G. W., et al. (2003). Lipoprotein receptor-mediated induction of matrix metalloproteinase by tissue plasminogen activator. Nat. Med. 9, 1313–1317. doi: 10.1038/nm926
Wang, Y., Jin, S., Sonobe, Y., Cheng, Y., Horiuchi, H., Parajuli, B., et al. (2014). Interleukin-1β induces blood-brain barrier disruption by downregulating sonic hedgehog in astrocytes. PLoS ONE 9:e110024. doi: 10.1371/journal.pone.0110024
Wangb, C. L. A., Wuc, Y., Gulbinsc, E., Grassméc, H., and Zhaoa, Z. (2019). Acid sphingomyelinase-ceramide system in bacterial infections. Cell. Physiol. Biochem. 52, 280–301. doi: 10.33594/000000021
Wardlaw, J., Sandercock, P., Dennis, M., and Starr, J. (2003). Is breakdown of the blood-brain barrier responsible for lacunar stroke, leukoaraiosis, and dementia? Stroke 34, 806–812. doi: 10.1161/01.STR.0000058480.77236.B3
Wharton, S. B., Brayne, C., Savva, G. M., Matthews, F. E., Forster, G., Simpson, J., et al. (2011). Epidemiological neuropathology: the MRC cognitive function and aging study experience. J. Alzheimers Dis. 25, 359–372. doi: 10.3233/JAD-2011-091402
Winkler, E. A., Bell, R. D., and Zlokovic, B. V. (2011). Central nervous system pericytes in health and disease. Nat. Neurosci. 14, 1398–1405. doi: 10.1038/nn.2946
Winkler, E. A., Nishida, Y., Sagare, A. P., Rege, S. V., Bell, R. D., Perlmutter, D., et al. (2015). GLUT1 reductions exacerbate Alzheimer's disease vasculo-neuronal dysfunction and degeneration. Nat. Neurosci. 18, 521–530. doi: 10.1038/nn.3966
Won, J.-S., Kim, J., Annamalai, B., Shunmugavel, A., Singh, I., and Singh, A. K. (2013). Protective role of S-nitrosoglutathione (GSNO) against cognitive impairment in rat model of chronic cerebral hypoperfusion. J. Alzheimers Dis. 34, 621–635. doi: 10.3233/JAD-121786
Wu, F., Zou, Q., Ding, X., Shi, D., Zhu, X., Hu, W., et al. (2016). Complement component C3a plays a critical role in endothelial activation and leukocyte recruitment into the brain. J. Neuroinflammation 13, 1–14. doi: 10.1186/s12974-016-0485-y
Xiao, M., Xiao, Z. J., Yang, B., Lan, Z., and Fang, F. (2020). Blood-brain barrier: more contributor to disruption of central nervous system homeostasis than victim in neurological disorders. Front. Neurosci. 14:764. doi: 10.3389/fnins.2020.00764
Yamazaki, Y., Shinohara, M., Shinohara, M., Yamazaki, A., Murray, M. E., Liesinger, A. M., et al. (2019). Selective loss of cortical endothelial tight junction proteins during Alzheimer's disease progression. Brain 142, 1077–1092. doi: 10.1093/brain/awz011
Yang, C., Huang, X., Huang, X., Mai, H., Li, J., Jiang, T., et al. (2016). Aquaporin-4 and Alzheimer's disease. J. Alzheimers Dis. 52, 391–402. doi: 10.3233/JAD-150949
Yang, J., Lunde, L. K., Nuntagij, P., Oguchi, T., Camassa, L., Nilsson, L. N., et al. (2011). Loss of astrocyte polarization in the tg-ArcSwe mouse model of Alzheimer's disease. J. Alzheimers Dis. 27, 711–722. doi: 10.3233/JAD-2011-110725
Yang, T., Guo, R., and Zhang, F. (2019). Brain perivascular macrophages: recent advances and implications in health and diseases. CNS Neurosci. Therapeut. 25, 1318–1328. doi: 10.1111/cns.13263
Yang, W., Wu, Q., Yuan, C., Gao, J., Xiao, M., Gu, M., et al. (2012). Aquaporin-4 mediates astrocyte response to β-amyloid. Mol. Cell. Neurosci. 49, 406–414. doi: 10.1016/j.mcn.2012.02.002
Yang, Y., Hill, J. W., and Rosenberg, G. A. (2011). Multiple roles of metalloproteinases in neurological disorders. Prog. Mol. Biol. Transl. Sci. 99, 241–263. doi: 10.1016/B978-0-12-385504-6.00006-3
Yang, Y., Kimura-Ohba, S., Thompson, J. F., Salayandia, V. M., Cosse, M., Raz, L., et al. (2018). Vascular tight junction disruption and angiogenesis in spontaneously hypertensive rat with neuroinflammatory white matter injury. Neurobiol. Dis. 114, 95–110. doi: 10.1016/j.nbd.2018.02.012
Yao, Y., Chen, Z.-L., Norris, E. H., and Strickland, S. (2014). Astrocytic laminin regulates pericyte differentiation and maintains blood brain barrier integrity. Nat. Commun. 5, 1–12. doi: 10.1038/ncomms4413
Yepes, M., Woo, Y., and Martin-Jimenez, C. (2021). Plasminogen activators in neurovascular and neurodegenerative disorders. Int. J. Mol. Sci. 22:4380. doi: 10.3390/ijms22094380
Yu, X., Ji, C., and Shao, A. (2020). Neurovascular unit dysfunction and neurodegenerative disorders. Front. Neurosci. 14:334. doi: 10.3389/fnins.2020.00334
Zeevi, N., Pachter, J., McCullough, L. D., Wolfson, L., and Kuchel, G. A. (2010). The blood-brain barrier: geriatric relevance of a critical brain-body interface. J. Am. Geriatr. Soc. 58, 1749–1757. doi: 10.1111/j.1532-5415.2010.03011.x
Zenaro, E., Piacentino, G., and Constantin, G. (2017). The blood-brain barrier in Alzheimer's disease. Neurobiol. Dis. 107, 41–56. doi: 10.1016/j.nbd.2016.07.007
Zenaro, E., Pietronigro, E., Della Bianca, V., Piacentino, G., Marongiu, L., Budui, S., et al. (2015). Neutrophils promote Alzheimer's disease-like pathology and cognitive decline via LFA-1 integrin. Nat. Med. 21, 880–886. doi: 10.1038/nm.3913
Zeppenfeld, D. M., Simon, M., Haswell, J. D., D'Abreo, D., Murchison, C., Quinn, J. F., et al. (2017). Association of perivascular localization of aquaporin-4 with cognition and Alzheimer disease in aging brains. JAMA Neurol. 74, 91–99. doi: 10.1001/jamaneurol.2016.4370
Zhang, X., Wu, B., Nie, K., Jia, Y., and Yu, J. (2014). Effects of acupuncture on declined cerebral blood flow, impaired mitochondrial respiratory function and oxidative stress in multi-infarct dementia rats. Neurochem. Int. 65, 23–29. doi: 10.1016/j.neuint.2013.12.004
Zhao, Z., Nelson, A. R., Betsholtz, C., and Zlokovic, B. V. (2015). Establishment and dysfunction of the blood-brain barrier. Cell 163, 1064–1078. doi: 10.1016/j.cell.2015.10.067
Zhao, Z., Ong, L. K., Johnson, S., Nilsson, M., and Walker, F. R. (2017). Chronic stress induced disruption of the peri-infarct neurovascular unit following experimentally induced photothrombotic stroke. J. Cereb. Blood Flow Metab. 37, 3709–3724. doi: 10.1177/0271678X17696100
Zhou, X., Spittau, B., and Krieglstein, K. (2012). TGFβ signalling plays an important role in IL4-induced alternative activation of microglia. J. Neuroinflammation 9, 1–14. doi: 10.1186/1742-2094-9-210
Zhu, H.-Y., Hong, F.-F., and Yang, S.-L. (2021). The roles of nitric oxide synthase/nitric oxide pathway in the pathology of vascular dementia and related therapeutic approaches. Int. J. Mol. Sci. 22:4540. doi: 10.3390/ijms22094540
Zlokovic, B. V. (2005). Neurovascular mechanisms of Alzheimer's neurodegeneration. Trends Neurosci. 28, 202–208. doi: 10.1016/j.tins.2005.02.001
Zlokovic, B. V. (2011). Neurovascular pathways to neurodegeneration in Alzheimer's disease and other disorders. Nat. Rev. Neurosci. 12, 723–738. doi: 10.1038/nrn3114
Zonneveld, H. I., Goos, J. D., Wattjes, M. P., Prins, N. D., Scheltens, P., van der Flier, W. M., et al. (2014). Prevalence of cortical superficial siderosis in a memory clinic population. Neurology 82, 698–704. doi: 10.1212/WNL.0000000000000150
Keywords: blood-brain barrier, biomarkers, cognitive impairment, aging, dementia
Citation: Hussain B, Fang C and Chang J (2021) Blood–Brain Barrier Breakdown: An Emerging Biomarker of Cognitive Impairment in Normal Aging and Dementia. Front. Neurosci. 15:688090. doi: 10.3389/fnins.2021.688090
Received: 30 March 2021; Accepted: 14 July 2021;
Published: 19 August 2021.
Edited by:
Kin Ying Mok, University College London, United KingdomReviewed by:
Berislav Zlokovic, University of Southern California, United StatesCopyright © 2021 Hussain, Fang and Chang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Junlei Chang, amwuY2hhbmdAc2lhdC5hYy5jbg==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.