 Marie Sprengell
Marie Sprengell Britta Kubera
Britta Kubera Achim Peters
Achim Peters
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
SYSTEMATIC REVIEW article
Front. Neurosci., 24 June 2021
Sec. Neuroendocrine Science
Volume 15 - 2021 | https://doi.org/10.3389/fnins.2021.685031
This article is part of the Research TopicCentral Regulation of MetabolismView all 15 articles
This work joins a series that methodically tests the predictions of the Selfish-Brain theory. The theory postulates a vital ability of the mammalian brain, namely to give priority to its own energy metabolism. The brain behaves “selfishly” in this respect. For the cerebral artery occlusion studied here, the theory predicts an increase in blood glucose concentration, what becomes the hypothesis to be tested. We conducted a systematic review of cerebral-artery-occlusion papers to test whether or not the included studies could confirm this hypothesis. We identified 239 records, screened 231 works by title or abstract, and analyzed 89 by full text. According to strict selection criteria (set out in our PROSPERO preregistration, complying with PRISMA guidelines), 7 papers provided enough information to decide on the hypothesis. Our hypothesis could be fully confirmed for the 3 to 24 h after the onset of a transient 2 h or permanent occlusion. As for the mechanism, the theory predicts that the energy-deprived brain suppresses insulin secretion via the sympathoadrenal system, thereby preventing insulin-mediated glucose uptake into muscle and fat and, as a result, enhancing insulin-independent glucose uptake via the blood-brain barrier. Evidence from our included studies actually demonstrated cerebral insulin suppression. In all, the current work confirms the second major prediction of the Selfish-Brain theory that relates to a proximal bottleneck of the cerebral supply chain, cerebral artery occlusion. Its first major prediction relates to a distal supply bottleneck, caloric restriction, and is fulfilled as shown by our previous work, whereas the prediction of the long held gluco-lipostatic theory, which sees the brain as only passively supplied, is violated (Sprengell et al., 2021). The crucial point was that caloric restriction elicits smaller changes in mass (energy) in the brain than in the body. Taken together, the evidence from the current and previous work clearly shows that the most accurate predictions are possible with a theory that views the brain as an independently self-regulating energy compartment occupying a primary position in energy metabolism.
This work is based on the concept of the “cerebral supply chain” where the brain is the end consumer (Peters and Langemann, 2009) (Figure 1). The key principle of supply chains is on-demand procurement, which makes the delivery process more economical and less susceptible to disturbances (Slack et al., 2004). We performed a systematic review on the most critical disturbance of the cerebral supply chain: a proximal supply bottleneck caused by cerebral arterial occlusion. It is known that disruption of cerebral fuel supply results in an intraneuronal adenosine triphosphate (ATP) depletion that ultimately leads to ischemic stroke (Wagner et al., 1992). It is also known that patients with acute ischemic stroke often present with hyperglycemia (Bravata et al., 2003), but it remains unclear to what extent this is due to the stroke or undiagnosed diabetes mellitus. What exactly a proximal bottleneck does to the more distal parts of the cerebral supply chain has not yet been studied systematically.
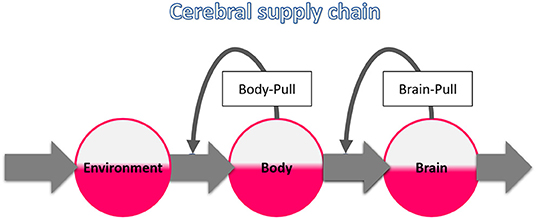
Figure 1. The cerebral supply chain. Energy is transferred from the environment through the body to the brain, the end consumer of the supply chain. The characteristic of supply chains is the procurement on demand, also called pull principle. When the brain needs energy, the brain-pull demands it from the body. When the body needs energy, the body-pull demands it from the environment.
This is our second systematic review on the cerebral supply chain; our first examined the effect of a more distal bottleneck in the supply chain, i.e. caloric restriction (Figure 2A). We found that with caloric restriction, the mass (energy) changes in the brain were particularly small (Sprengell et al., 2021). Thus, peripheral energetic intervention has little effect on the central nervous system. In the current systematic review, we examine the reverse case, namely, what effect central energetic interventions have on peripheral energy metabolism. More precisely, we examined what effect the interruption of the brain's energy supply has on the systemic blood glucose concentration. We hypothesize that disruption of brain arterial supply causes a marked rise in blood glucose concentration.
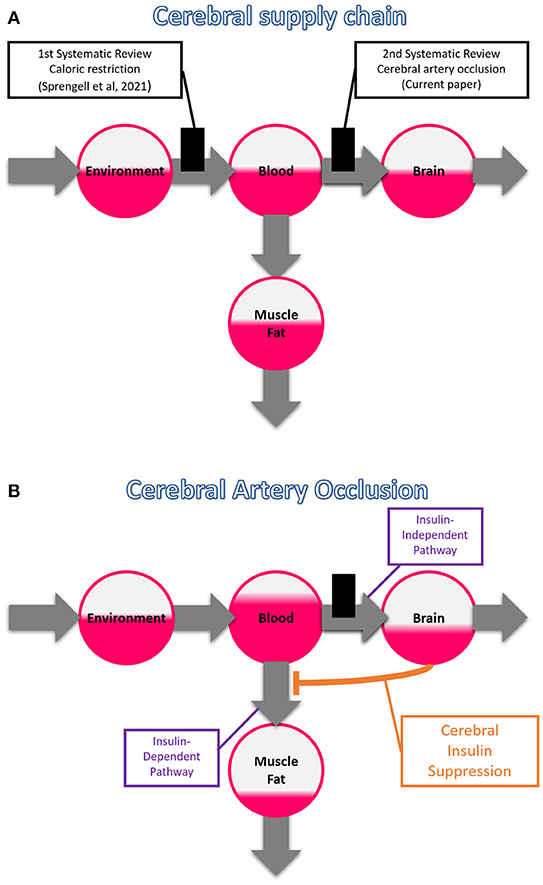
Figure 2. The branched cerebral supply chain. The branching originates from the blood, transmitting energy either to the brain or to muscle or fat tissue. (A) Systematic reviews on two different bottlenecks in the cerebral supply chain. The first one deals with a distal bottleneck, i.e. caloric restriction, and is already published (Sprengell et al., 2021); the second one, the current work, deals with a proximal bottleneck, i.e. cerebral artery occlusion. (B) Cerebral insulin suppression induced by cerebral artery occlusion. Intraneuronal ATP depletion activates the sympathetic nervous system and the hypothalamic-pituitary-adrenal axis, which both rigorously suppress pancreatic ß-cell secretion, and in so doing block insulin-dependent glucose uptake in muscle and fat tissue. The portion of glucose that is prevented from entering muscle and fat tissue accumulates in the blood, facilitating transfer across the blood-brain barrier via the insulin-independent pathway, allowing partial compensation when brain supply is compromised.
Our hypothesis is a prediction of the Selfish-Brain theory (Peters et al., 2004). This theory deals with the energy allocation within the mammalian organism. It postulates that in case of energy shortage there is a vital ability of the brain, namely to give priority to its own energy metabolism. Rival positions include the long-held gluco-lipostatic theory (Kennedy, 1953; Mayer, 1953) and its modern variants (Chaput and Tremblay, 2009; Schwartz et al., 2017), which all view the brain as only passively supplied. Our first systematic review showed that caloric restriction causes only minor mass (energy) changes in the brain [of the order of −4%] as opposed to major changes in the body [of about −30%] (Sprengell et al., 2021). This finding completely fulfilled the predictions of the Selfish-Brain theory, while those of the gluco-lipostatic theory and its variants were violated. The most accurate predictions are therefore possible with a theory that views the brain as an independently self-regulating energy compartment that occupies a primary position in a hierarchically organized metabolism. Our second systematic review, the current one, aims to examine evidence of this vital ability of the brain. That is, if the hypothesis formulated here can be confirmed by our systematic literature search, an important prediction of the Selfish-Brain theory is fulfilled.
To illustrate how our hypothesis can be deduced from theory, we refer to the cerebral supply chain. It is a mathematical representation of the Selfish-Brain theory (Peters and Langemann, 2009). Logistic supply chains have played an important role in the economy for 80 years, e.g. in automobile production. The mathematical principles of these supply chains could be directly applied to energy metabolism. A general rule for all supply chains is that the flow of goods or energy is composed of two components, one determined by the supplier (push component) and the other by the subsequent recipient (pull component) (Slack et al., 2004). In the cerebral supply chain, energy is transferred from the environment, through the blood, to the end consumer – the brain. As this supply chain is branched, a portion of the energy is transferred from the blood to the muscle and fat tissue. In addition to the antegrade flow of energy toward the brain as the end consumer, there is also a retrograde flow of information consisting of the pull commands.
Brain-pull refers to the brain's ability to procure itself with energy on demand (Figure 1). The energy content of the brain (intraneuronal ATP) determines how much energy is demanded from the body. In contrast, the body-pull demands energy from the environment for the body. Both the energy content of the blood (glucose) and that of the body stores (triglycerides) determine how much energy is pulled from the environment. To date, several redundant brain-pull mechanisms have been identified (Peters and McEwen, 2015). These are neuroendocrine in nature and provide the brain with additional energy sources as needed.
Among brain-pull mechanisms, “cerebral insulin suppression” (CIS) is one of the most important (Woods and Porte, 1974; Hitze et al., 2010). Energy sensors in brain regions such as the amygdala (Zhou et al., 2010) and VMH (Spanswick et al., 1997; Routh et al., 2014; Toda et al., 2016) detect even the slightest drop in neuronal ATP and set brain-pull mechanisms in action. VMH activation lowers insulin concentrations and increases blood glucose concentrations (Meek et al., 2016; Stanley et al., 2016). In detail, the VMH activates the sympathoadrenal system, which strongly suppresses insulin secretion from pancreatic ß-cells (Ahren, 2000). Insulin-dependent glucose uptake (GLUT4) in muscle and fat tissue is suppressed, while the remaining circulating glucose is almost completely available to insulin-independent glucose transport (GLUT1) across the blood-brain barrier. For the brain needs virtually no insulin to take up glucose (Hom et al., 1984; Hasselbalch et al., 1999; Seaquist et al., 2001). In summary, GLUT1 glucose uptake safeguards basal energy supply of vital organs, like brain and immune cells (Deng et al., 2014), while GLUT4 allows the storage of surplus energy in muscle and fat cells (Shepherd and Kahn, 1999). Thus, CIS allocates more energy to the brain when needed.
In the case of cerebral artery occlusion, our theory-based prediction is as follows (Figure 2B). Occlusion of a cerebral artery restricts the energy flow to the brain, which in the supply chain model corresponds to a reduced blood-push component. Reduced supply (i.e. decreased blood-push) leads to cerebral ATP depletion, resulting in enhanced CIS (i.e. increased brain-pull), which prevents glucose uptake in muscle and fat tissue, and eventually causes glucose accumulation in the blood. While a portion of the circulating glucose cannot enter muscle or fat tissue, this is instead made available to the energy-depleted brain. In this way, the cerebral supply chain model generates a prediction that becomes the hypothesis to be tested here: Cerebral artery occlusion increases glucose concentration in the blood.
To this end, we performed a systematic review to examine whether or not the experimental studies found on cerebral artery occlusion can actually confirm this hypothesis.
The protocol for this systematic review was pre-registered on PROSPERO on 30th of January 2020, and updated versions were published on 28th of September 2020 and 14th of December 2020 (International prospective register of systematic reviews; CRD42020156816). We complied with the PRISMA (Preferred Reporting Items for Systematic Reviews and Meta-analyses) guidelines for systematic reviews of interventions (Moher et al., 2009). Furthermore, the Cochrane Handbook for systematic reviews of interventions was used (Higgins and Thomas, 2019).
We conducted a systematic search of the literature to identify studies in mammals that focused on how the experimental cerebral artery occlusion affects blood glucose concentration. One reviewer developed the search strategies, which were then discussed with two other reviewers. The databases of MEDLINE and BIOSIS Previews were searched from their inception to 20th December 2020, using a combination of key words and in case of the first database MeSH terms. The full MEDLINE and BIOSIS search strategies are provided in the Supplementary Material. Briefly, the search strategies included terms relating to the intervention (cerebral ischemia), to the outcome (blood glucose concentration) and to the methodical approach (experimental study), combined by the Boolean operator AND. Synonyms for terms were combined with the operator OR.
We used the following criteria to include or exclude articles for our systematic review. Only original full research papers published in English or German were included. We included studies that examined mammals of any species or sex. We included only interventional studies that were standardized laboratory experiments or clinical trials and that examined two groups, an interventional group undergoing cerebral artery occlusion and a non-exposed, sham-operated control group. Since we had included clinical trials in our first systematic review (Sprengell et al., 2021), we did the same here for the sake of consistency, but of course did not expect to identify clinical trials, since cerebral artery occlusion in humans is not ethically defensible. We only included studies that measured blood glucose concentration. We did not include studies that measured outcomes in the intervention and control groups at different time points. We did not include studies, that occluded the vertebral arteries, as cerebral control centers of energy metabolism could have been affected. We did not include studies, in which clots were injected in the aorta, because of unpredictable consequences for other parts of the body. We did not include studies with combined interventions such as cerebral artery occlusion and systemic hypotension. We did not include studies in which the individuals had diseases or were on medication that had been shown to affect energy metabolism; for more details see Sprengell et al. (2021). We did not include trials in pregnant individuals or fetuses, nor in ovariectomized or genetically modified mammals with altered energy metabolism.
The selection of the articles was performed in two steps. We have indicated the reasons for exclusion at each step (Figure 3). First, one reviewer screened the article titles or abstracts against the inclusion and exclusion criteria. This first step of article selection was checked by another reviewer. When a discrepancy occurred regarding the inclusion or exclusion of an article, the two reviewers discussed it until agreement was reached. Otherwise, disagreements were resolved by consulting the third reviewer. Second, two reviewers independently selected the remaining articles by analyzing the full text. Again, disagreements regarding the inclusion or exclusion of an article were resolved by discussion among each other or, if necessary, by consultation with the third reviewer.
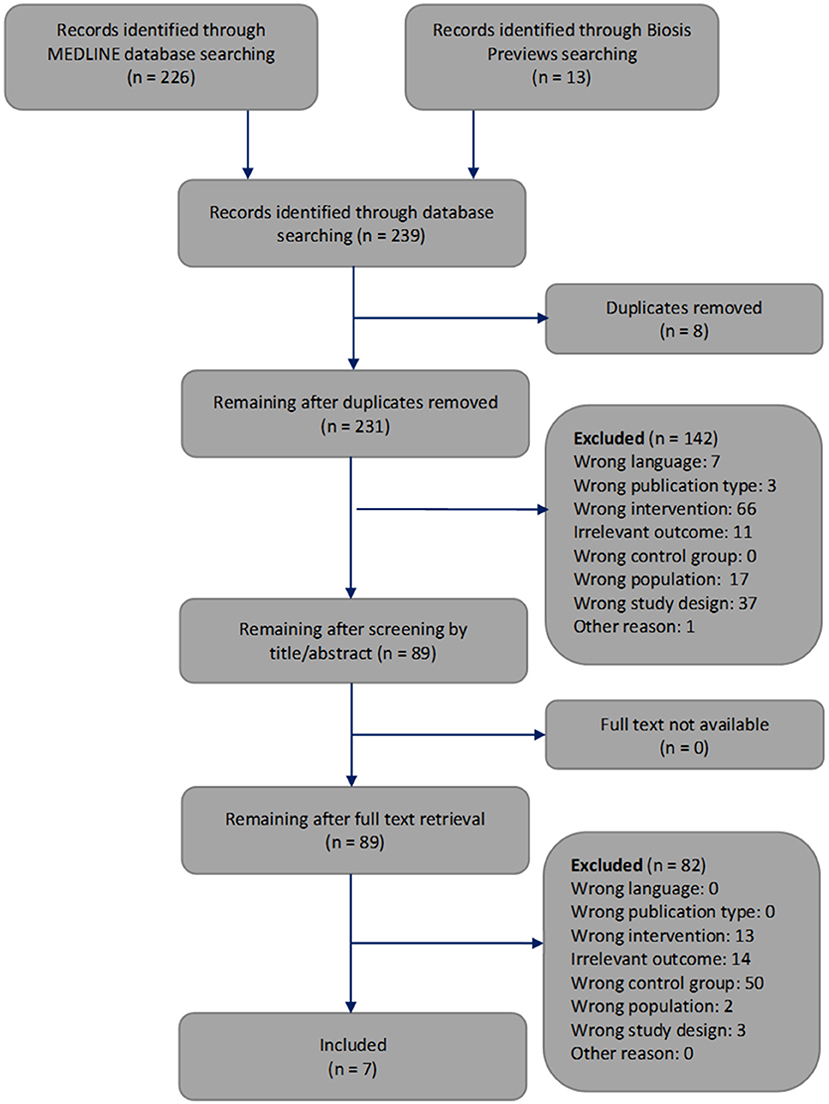
Figure 3. Flowchart through different phases of the systematic review modified according to Moher's publication (Moher et al., 2009).
Data from all of the 7 included studies were extracted by one reviewer, independently checked by two other reviewers, and tabulated alphabetically. We recorded the population, sample size, kind of intervention and duration, study design, statistical test applied, baseline blood glucose concentrations as well as blood glucose concentrations after onset of cerebral artery occlusion and secondary outcomes.
To assess the risk of bias of non-human studies the SYRCLE‘s tool was used (Hooijmans et al., 2014). One reviewer assessed the risk of bias of the 7 included studies. The results were independently checked by two other reviewers. All differences were clarified by discussion.
Based on the respective statistical test, we decided whether the hypothesis could be confirmed (p < 0.05) or not (p ≥ 0.05).
The systematic search of the literature generated 239 articles, which were processed as summarized in Figure 3. Two hundred and thirty one works were screened by title or abstract, and 89 articles were analyzed by full text. We identified seven studies that met all inclusion criteria and focused on how cerebral artery occlusion affects blood glucose concentrations (Harada et al., 2009; Yamazaki et al., 2012, 2014; Wang et al., 2013, 2014; Li et al., 2016; Boujon et al., 2019).
Table 1 provides details of the 7 included studies. Four studies investigated mice (Harada et al., 2009; Yamazaki et al., 2012, 2014; Boujon et al., 2019) and 3 studies investigated rats (Wang et al., 2013, 2014; Li et al., 2016). The sample sizes varied between 9 and 53.
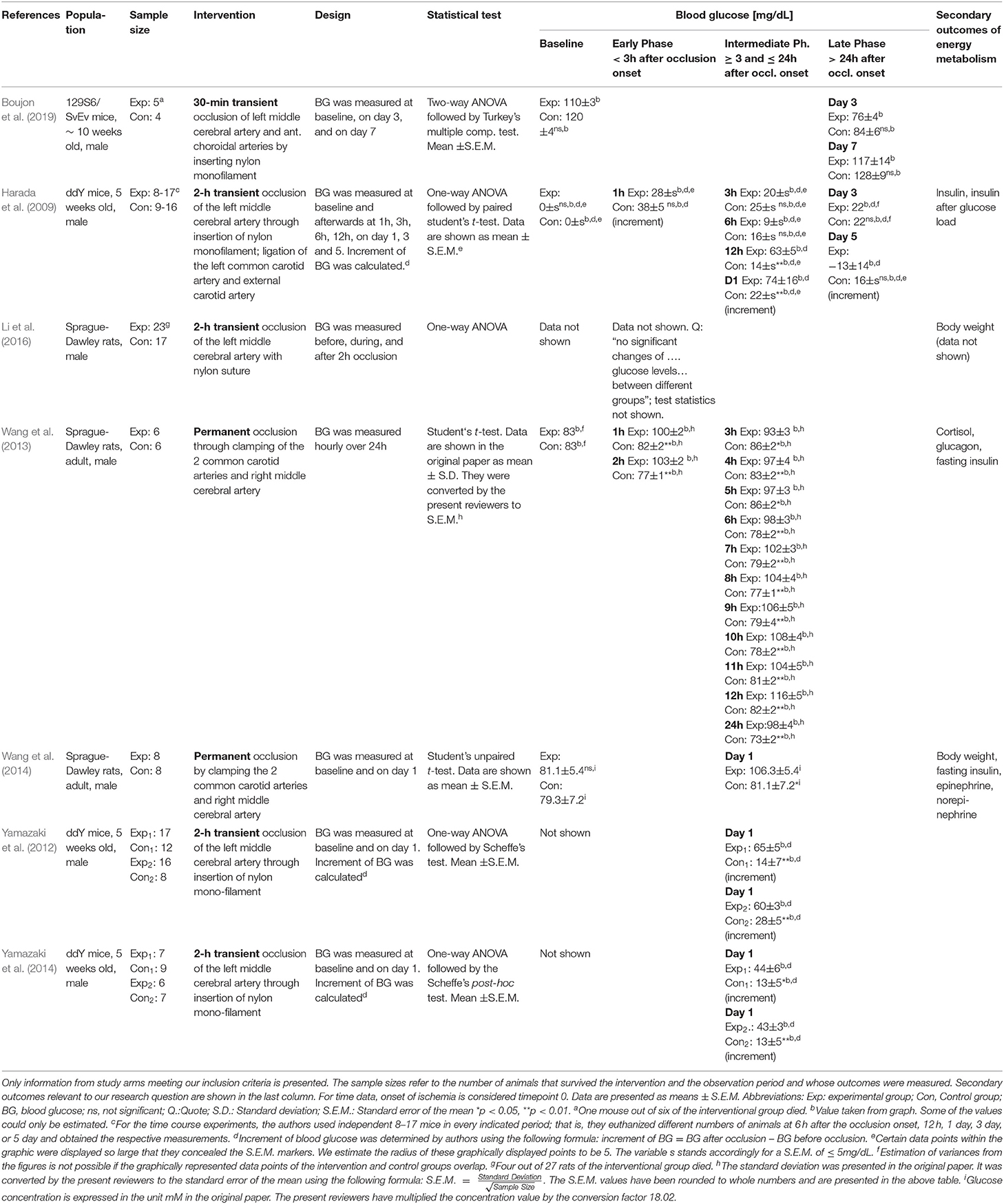
Table 1. Characteristics and results of included studies.
All included studies provided details on how cerebral artery occlusion was implemented. In 2 studies, the left middle cerebral artery was occluded by insertion of a nylon monofilament (Yamazaki et al., 2012, 2014). Harada and colleagues also occluded the left middle cerebral artery and additionally ligated the left common carotid artery and external carotid artery (Harada et al., 2009). Two other studies performed occlusion by clamping the two common carotid arteries and right middle cerebral artery (Wang et al., 2013, 2014). In one study, the left middle cerebral artery and anterior choroidal arteries were occluded by insertion of a nylon monofilament (Boujon et al., 2019). In a further study, the middle cerebral artery was occluded with nylon suture (Li et al., 2016). The duration of cerebral artery occlusion ranged from 30 min (Boujon et al., 2019) and 2 h (Harada et al., 2009; Yamazaki et al., 2012, 2014; Li et al., 2016) to permanent (Wang et al., 2013, 2014).
Blood glucose concentrations were measured during three phases, where we distinguished the early phase lasting less than 3 h after occlusion onset, the intermediate phase 3 to 24 h after occlusion onset, and the late phase lasting more than 7 days after reperfusion. Work examining the intermediate phase included, firstly, a paper by Wang and coworkers, which reported hourly measurements over 24 h of permanent ischemia, finding that as early as 1 h after the onset of occlusion, the blood glucose profile was elevated and remained elevated above that of controls (all p < 0.05) (Wang et al., 2013); secondly, another paper by Wang and coworkers, which reported measurements of blood glucose concentrations at baseline and after 24 h of permanent ischemia, finding that 24 h after the onset of occlusion, blood glucose was higher than in controls (p < 0.05) (Wang et al., 2014); thirdly, 2 papers by Yamazaki and coworkers, each reporting in 2 experiments the change in blood glucose concentration 24 h after the onset of a transient 2 h cerebral artery occlusion, finding that blood glucose increased more than in controls (all p < 0.05) (Yamazaki et al., 2012, 2014); and finally the work by Harada and coworkers that examined not only the intermediate phase, but also early and late phase, reporting changes in blood glucose concentration at baseline and 1, 3, 6, 12, and 24 h, 3 and 5 days after the onset of transient 2 h cerebral artery occlusion, finding that the blood glucose increase was more pronounced than in controls at 12 and 24 h (p < 0.01), while no increase could be detected within the first 6 h or on day 3 or 5 (Harada et al., 2009).
Work examining only the late phase was that of Boujon and co-workers, which reported measurements of blood glucose concentration at baseline and on days 3 and 7 after the onset of transient 30 min cerebral artery occlusion, and found that no increase could be detected on either day 3 or 7 (Boujon et al., 2019). Work examining only the early phase was that of Li and coworkers, who could not demonstrate a difference in blood glucose levels between the intervention and sham-operated groups either during the 2 h occlusion or right afterward (Li et al., 2016).
Secondary outcomes relevant to our research question were provided by (Harada et al., 2009) (insulin concentrations and insulin after glucose load), (Wang et al., 2013) (cortisol, glucagon, fasting insulin concentrations) and (Wang et al., 2014) (fasting insulin, blood norepinephrine, blood epinephrine concentrations, and body weight).
Table 2 provides the risk of bias assessments for all 7 included studies.
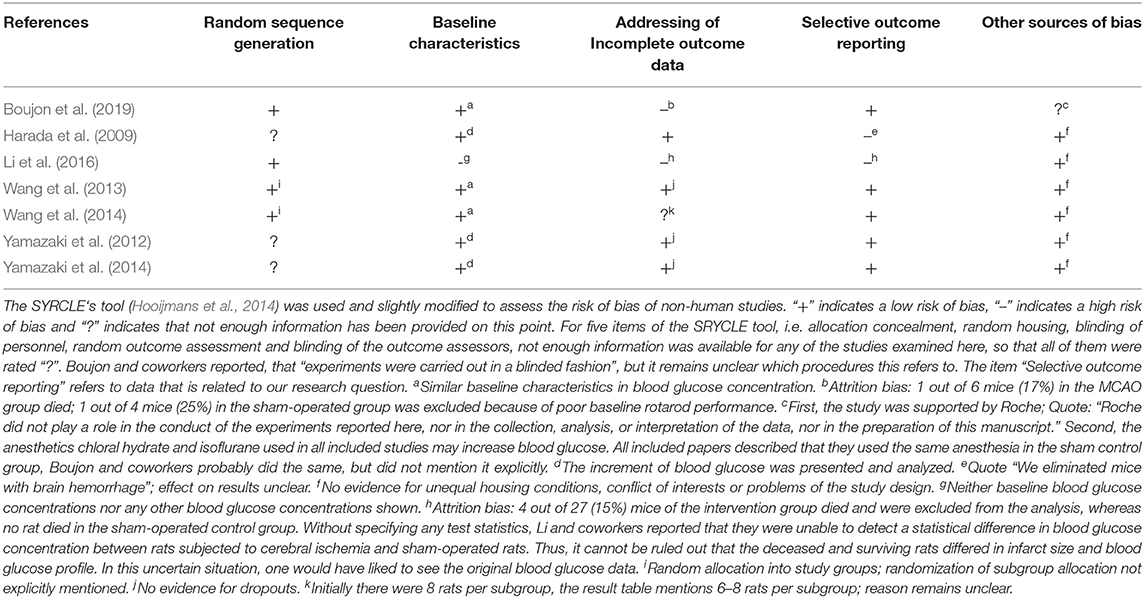
Table 2. Risk of bias assessment.
All papers which examined the intermediate phase 3 to 24 h after occlusion onset could confirm our hypothesis (Harada et al., 2009; Yamazaki et al., 2012, 2014; Wang et al., 2013, 2014) (Figure 4). All of these intermediate phase studies showed that blood glucose concentrations were significantly higher or had changed more in the intervention group than in the sham-operated group (Table 3). One work could confirm the hypothesis already for the early phase 1 h after occlusion onset (Wang et al., 2013).
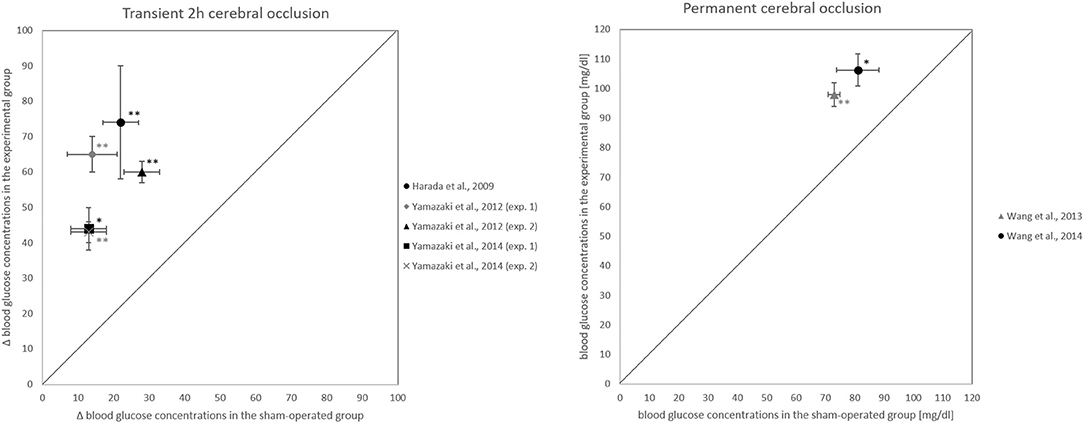
Figure 4. Blood glucose changes 24 h after occlusion onset. (Left) Comparison of how experimental and sham-operated groups differ in their blood glucose changes from before to 24 h after the onset of transient 2 h cerebral artery occlusion. (Right) Comparison of how experimental and sham-operated groups differ in blood glucose concentrations at 24 h after onset of permanent occlusion. *P < 0.05, **P < 0.01, significant difference in blood glucose concentrations or their changes between experimental and sham-operated groups.
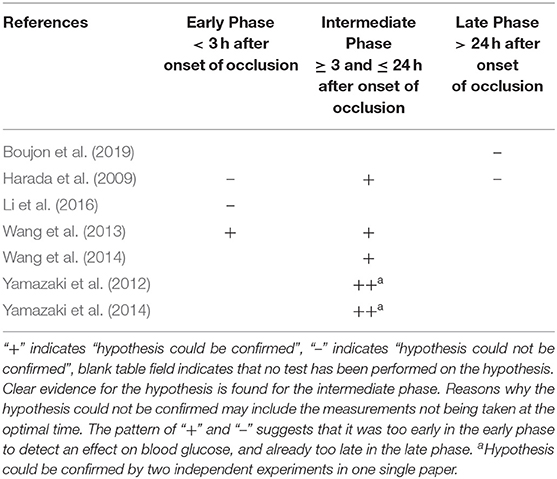
Table 3. Hypothesis decision in the different ischemia phases.
However, the papers that examined the late phase 3 to 7 days after reperfusion onset failed to confirm the hypothesis for the phase (Harada et al., 2009; Boujon et al., 2019). Two papers that examined the early phase could not confirm the hypothesis for this phase (Harada et al., 2009; Li et al., 2016), in contrast to Wang and coworkers who, as noted, could confirm it (Wang et al., 2013).
A total of 231 works was screened by title and abstract, and 89 were analyzed in full text. According to strict selection criteria defined in our PROSPERO pre-announcement and complying with PRISMA guidelines, 7 studies met all inclusion criteria. Of the 7 papers, all 5 that examined the intermediate phase 3 to 24 h after occlusion onset could confirm the hypothesis (Harada et al., 2009; Yamazaki et al., 2012, 2014; Wang et al., 2013, 2014) (Figure 4). For the late phase 3 to 7 days after reperfusion onset, the hypothesis could not be confirmed (Harada et al., 2009; Boujon et al., 2019). This evidence suggests that blood glucose elevation during and after cerebral artery occlusion is a temporary phenomenon. In all, our hypothesis could be fully confirmed for the period 3 to 24 h after the onset of a transient 2 h or permanent occlusion, so it holds that cerebral artery occlusion increases blood glucose concentration.
The 7 included studies represent a spectrum of diverse experiments which can be summarized as follows. All studies were published between 2009 and 2019, were performed on mice or rats, and the cerebral artery occlusion was either transient (ranging from 30 min to 2 h) or permanent (Harada et al., 2009; Yamazaki et al., 2012, 2014; Wang et al., 2013, 2014; Li et al., 2016; Boujon et al., 2019). Blood glucose was monitored with varying frequency over a period of about 2 h to 7 days. The papers that examined the intermediate phase reported that blood glucose concentrations 24 h after occlusion onset were 25 to 52 mg/dl higher than those of the control group (all p < 0.05). The papers that examined the late phase could no longer demonstrate the blood glucose increase seen in the intermediate phase (Harada et al., 2009; Boujon et al., 2019).
The mechanisms underlying stroke hyperglycemia are unknown, as some authors point out (Arnberg et al., 2015), while others hold overly complex beliefs (Dungan et al., 2009). Given that the Selfish-Brain theory makes accurate predictions in cases where the predictions of conventional theories failed (Sprengell et al., 2021), there is reason to believe that the neuroendocrine mechanisms as assigned by the Selfish-Brain theory provide a reliable explanation for the development of hyperglycemia in stroke. Typically, the Selfish-Brain theory covers topics such as psychosocial stress, depression, anorexia nervosa, obesity, and the development of atherosclerosis, myocardial infarction and stroke (Peters and McEwen, 2015). What is new here is that the theory addresses the consequences of stroke.
In terms of mechanisms, the Selfish-Brain theory refers to the principle of supply chains stating that when “push” fails, “pull” takes over. For the cerebral supply chain, this means that a reduction in the blood-push component (e.g. cerebral artery occlusion) is at least partially compensated by an enhanced brain-pull component. Brain-pull function is exerted by the sympathetic nervous system (SNS) and the hypothalamic-pituitary-adrenal (HPA) axis, which provide additional energy substrates for brain supply when needed. Among the studies found in our current systematic review, 2 studies measured blood concentrations of epinephrine, norepinephrine, and cortisol and showed that the concentrations of these stress hormones 24 h after occlusion onset were two times higher than those of the control group (Wang et al., 2013, 2014).
As mentioned above, several redundant brain-pull mechanisms are at work when the brain needs energy. Here we name three of them: The first brain-pull mechanism is that the SNS and HPA axis cause insulin suppression, which results in more glucose being delivered to the brain (Woods and Porte, 1974; Ahren, 2000). In fact, among the included studies was one that focused on insulin changes, showing that on day 1 after cerebral artery occlusion, an oral glucose load failed to increase plasma insulin concentrations, whereas in the sham-operated control group, the same load caused a 6-fold increase in plasma insulin concentrations (Harada et al., 2009). Thus, Harada and coworkers were able to confirm cerebral artery occlusion to induce CIS. CIS can be diagnosed on the basis of an inappropriately low insulin concentration at a given blood glucose concentration (Hitze et al., 2010). Importantly, CIS also occurs in many other critical situations, where brain energy homeostasis is challenged, including myocardial infarction (Taylor et al., 1969), psychosocial stress (Hitze et al., 2010), mental stress during anticipation of electric shock (Mason et al., 1968), acute hypoxia (Baum and Porte, 1969), deep hypothermia (Baum and Porte, 1971), caloric restriction (Peters et al., 2011) and sleep debt (Spiegel et al., 1999).
The second brain-pull mechanism is that SNS and HPA increase muscular proteolysis and hepatic gluconeogenesis, procuring the brain with even more glucose (Zhang et al., 2018). The third brain-pull mechanism is that SNS and HPA axis increase visceral lipolysis and hepatic ketogenesis, providing ketones as alternative brain substrate (Kubera et al., 2014). Indeed, it has been shown that brain ischemia via SNS activation induces the formation of ß-hydroxybutyrate (ketogenesis) in the liver and the consumption of ß-hydroxybutyrate in the brain (Koch et al., 2017). Ketones were also shown to exert beneficial effects on pathological and functional outcomes after experimental stroke (Gibson et al., 2012). Overall, brain-pull mechanisms cause the body stores to take in less energy and deliver more.
Replenishing the brain at the expense of body stores is what the supply chain model predicts will lead to weight loss (Figure 2B). One of the studies we included showed that compared to sham-operated controls, rats that underwent cerebral ischemia lost 9% of their body weight within 24 h (Wang et al., 2014). This is only a single experiment, and moreover, body weight was only a secondary endpoint. Nevertheless, these results match the prediction that weight loss in cerebral ischemia is caused by energy transfer from the periphery to the brain. Taken together, occlusion of the central cerebral arteries profoundly affects peripheral energy metabolism, with the strongest evidence for an increase in blood glucose concentration.
Hyperglycemia persists even after reperfusion, for which we here provide evidence of both cause and mechanism. In 3 of our included studies, hyperglycemia is still detectable 22 h after reperfusion (Harada et al., 2009; Yamazaki et al., 2012, 2014). While it is clear that impaired cerebral supply during occlusion leads to cerebral ATP deficiency (Wagner et al., 1992), the persistence of this deficiency state after reperfusion seems surprising at first glance. Regarding the cause of such a prolonged hyperglycemia there is evidence demonstrating that cerebral energy consumption increases from the onset of ischemia, particularly in the penumbra, but also in the core ischemic brain regions that undergo infarction (Arnberg et al., 2015). Cerebral energy consumption does not decrease until most ischemic brain regions have succumbed to infarction. As long as the penumbra exhibits increased energy consumption, cerebral ATP concentration as monitored in the VMH can recover only slowly. Thus, even after reperfusion, a prolonged hyperglycemic course seems plausible.
Regarding the mechanism accounting for increased energy consumption in undersupplied brain regions, the Selfish-Brain theory made the following predictions (Peters et al., 2004). ATP binds to low- and high-affinity ATP-sensitive-potassium channels localized on presynaptic GABAergic and postsynaptic glutamatergic neurons, respectively. This type of multi-site neuronal ensemble leads to biphasic responses when neuronal ATP concentrations fall. Mild ATP deficiency hyperpolarizes GABAergic neurons, disinhibits postsynaptic glutamatergic neurons, and thus increases glutamatergic activity and energy expenditure, whereas severe ATP deficiency hyperpolarizes glutamatergic neurons and thus suspends glutamatergic activity. Such a theory-predicted biphasic response to falling ATP levels could indeed be confirmed experimentally (Steinkamp et al., 2007). The clinical correlate of this biphasic course becomes evident when seizure susceptibility increases in moderate hypoglycemia due to facilitated glutamatergic activity, and coma develops in profound hypoglycemia due to ubiquitous silencing of glutamatergic activity (Arieff et al., 1974; Mobbs et al., 2001).
Post-stroke hyperglycemia has long been controversial, with either its deleterious-toxic, its beneficial neuroenergetic, or its Janus-faced dual aspect being held. The deleterious-toxic aspect was supported by the observation that higher blood glucose concentrations were associated with more severe ischemic strokes (Capes et al., 2001). On this basis, it was hypothesized that using insulin to normalize post-stroke hyperglycemia would improve infarct outcome. Several randomized controlled trials were conducted on this issue. Just as diabetologists and intensivists were disappointed with intensified insulin therapy because it caused more deaths in their type 2 diabetic and critically ill patients (Gerstein et al., 2008; The-NICE-SUGAR-Study-Investigators, 2009), so too were neurologists disappointed with intensified insulin treatment of post-stroke hyperglycemia. Specifically, a Cochrane review and the more recent SHINE trial showed that in ischemic stroke, intensified insulin therapy did not improve the outcomes of death, neurological deficit or dependency, but increased risk of symptomatic hypoglycemia (Bellolio et al., 2014; Johnston et al., 2019). While these findings from human studies challenged the deleterious-toxic aspect of post-stroke hyperglycemia, animal studies could indeed demonstrate glucotoxic mechanisms (Kruyt et al., 2010; Khan et al., 2016). This contradiction can be resolved with the Janus-faced dual aspect of post-stroke hyperglycemia, which allows for both deleterious-toxic and beneficial effects (Endres et al., 2008).
The beneficial neuroenergetic aspect of post-stoke hyperglycemia is also advocated (Arnberg et al., 2015). This position was supported by one of the larger randomized controlled trials on intensified insulin therapy, which showed that normalization of post-stroke hyperglycemia led to a 2.5-fold increase in infarct growth (Rosso et al., 2012). In line with this finding, Ginsberg and coworkers could show that rats rendered hyperglycemic by dextrose injection prior to infarct induction developed higher brain glucose concentrations and also smaller infarct volumes than normoglycemic controls (Ginsberg et al., 1987). At this point, a look at the evidence from diabetology and intensive medicine may provide further insight. Hypoglycemia and hypovolemia are among the situations critical for brain supply, in which CIS is essential for increasing blood glucose concentrations (Jarhult and Holst, 1978; Corrall and Frier, 1981). Presumably, neuroenergetic mechanisms such as CIS evolved under evolutionary pressure to survive periods of starvation or injury with blood loss, rather than to survive stroke. Nevertheless, hypoglycemia, hypovolemia, and ischemic stroke display the same neuroenergetic pattern: CIS with increase in blood glucose. This commonality supports the notion that infarct-related systemic hyperglycemia, despite its toxic aspect, is manifestation of an adaptive process that replenishes the energy-depleted brain.
Our systematic review has weaknesses and strengths. The weakness is that our search does not completely represent all relevant studies. However, no systematic review can claim to be complete unless the authors have viewed every paper in full text from all databases – which is not feasible. Authors of systematic reviews must therefore accept a certain degree of incompleteness due to the constraints imposed by their search algorithm. They need to find an optimal trade-off between sensitivity and specificity for their search. It became clear that our systematic review was not complete either when we had finished the data extraction of all papers. To our surprise, the work of Chen et al. (2016) surfaced. This work would have met all of our inclusion criteria, yet our search algorithm did not detect it. The reason for this was that the search term “ligation” did not appear in this paper, as Chen et al. referred to their previous work regarding their methods, nor was the term indexed for this paper in PubMed. We did not expect such a case when designing our search algorithm. We nevertheless adhered to the protocol and refrained from revising our search algorithm and performing a second search run, as such an approach risks biasing and undermining hypothesis testing.
One of the strengths of our systematic review is the neutrality of its search. We designed our search algorithm to the best of our knowledge and after that was set, had no influence on which studies were found. Even in the case of an incomplete search, this approach would provide us with an unbiased, well-defined set of experimental data against which we could perform hypothesis tests. Basically, compared to hand searching, systematic database searching is much less susceptible to researcher bias in paper selection. Now that we knew the results of Chen's study (Chen et al., 2016), a freedom of choice to either rerun a modified search or to stick with the existing search results would have provided us with an opportunity to influence hypothesis testing. To avoid such bias, we strictly followed our pre-registered PROSPERO protocol and stuck to our original search results. Nevertheless, and for the sake of completeness, we believe it is important to show whether Chen's results, had they been taken into account, would have affected the statement of our work. This was not the case (see Supplementary Material C). These considerations point out that when testing a scientific hypothesis in a systematic review it is the degree of completeness that matters, but even more so the neutrality of the search.
In conclusion, our systematic review confirms a major prediction of the Selfish-Brain theory, namely that cerebral artery occlusion elevates blood glucose concentrations. For the causes and effects involved in this blood glucose elevation, the theory makes further predictions such as increase in stress hormones epinephrine, norepinephrine and cortisol, suppression of insulin secretion, and acute body weight loss (Peters and McEwen, 2015), all of which could be fulfilled by findings from our included papers. This is the second major prediction of the Selfish-Brain theory that has been confirmed and that relates to a proximal bottleneck of the cerebral supply chain, the occlusion of the cerebral arteries. The first major prediction, which had also been confirmed, related to a distal supply bottleneck of the cerebral supply chain, caloric restriction (Sprengell et al., 2021). We did not only systematically search the literature databases, but also followed a systematic plan to capture the potential bottlenecks of the cerebral supply chain and predict their impact on cerebral and peripheral energy states. By passing this second test, centered on cerebral ischemia, the Selfish-Brain theory continues to demonstrate the accuracy of its predictions.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
MS developed the search strategies that BK and AP approved. MS screened the article titles or abstracts against the inclusion and exclusion criteria. BK checked this step, and disagreements were resolved where necessary by consulting the third reviewer AP. MS and BK independently analyzed the full text, and disagreements were resolved where necessary by consultation with the third reviewer AP. MS extracted the data, which BK and AP independently checked. MS assessed the risk of bias, which BK and AP independently checked and approved. BK and AP wrote the manuscript. All authors have read and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Our special thanks go to Professor Markus Schwaninger, Chair of the Institute of Experimental and Clinical Pharmacology and Toxicology, University of Luebeck, for his criticism and invaluable comments on this work. We also thank Sabine Wittnebel for expert assistance in the retrieval and acquisition of full text articles.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnins.2021.685031/full#supplementary-material
Ahren, B. (2000). Autonomic regulation of islet hormone secretion-implications for health and disease. Diabetologia 43, 393–410. doi: 10.1007/s001250051322
Arieff, A. I., Doerner, T., Zelig, H., and Massry, S. G. (1974). Mechanisms of seizures and coma in hypoglycemia. Evidence for a direct effect of insulin on electrolyte transport in brain. J. Clin. Invest. 54, 654–663. doi: 10.1172/JCI107803
Arnberg, F., Grafstrom, J., Lundberg, J., Nikkhou-Aski, S., Little, P., Damberg, P., et al. (2015). Imaging of a clinically relevant stroke model: glucose hypermetabolism revisited. Stroke 46, 835–842. doi: 10.1161/STROKEAHA.114.008407
Baum, D., and Porte, D. (1969). Adrenergic regulation of hyperglycemia in acute hypoxia. Diabetes 18:346.
Baum, D., and Porte, D. Jr. (1971). Alpha-adrenergic inhibition of immunoreactive insulin release during deep hypothermia. Am. J. Physiol. 221, 303–311. doi: 10.1152/ajplegacy.1971.221.1.303
Bellolio, M. F., Gilmore, R. M., and Ganti, L. (2014). Insulin for glycaemic control in acute ischaemic stroke. Cochrane Database Syst. Rev. 23:CD005346. doi: 10.1002/14651858.CD005346.pub4
Boujon, V., Uhlemann, R., Wegner, S., Wright, M. B., Laufs, U., Endres, M., et al. (2019). Dual PPARalpha/gamma agonist aleglitazar confers stroke protection in a model of mild focal brain ischemia in mice. J. Mol. Med. 97, 1127–1138. doi: 10.1007/s00109-019-01801-0
Bravata, D. M., Kim, N., Concato, J., and Brass, L. M. (2003). Hyperglycaemia in patients with acute ischaemic stroke: how often do we screen for undiagnosed diabetes? QJM 96, 491–497. doi: 10.1093/qjmed/hcg087
Capes, S. E., Hunt, D., Malmberg, K., Pathak, P., and Gerstein, H. C. (2001). Stress hyperglycemia and prognosis of stroke in non-diabetic and diabetic patients: a systematic overview. Stroke 32, 2426–2432. doi: 10.1161/hs1001.096194
Chaput, J. P., and Tremblay, A. (2009). The glucostatic theory of appetite control and the risk of obesity and diabetes. Int. J. Obes. 33, 46–53. doi: 10.1038/ijo.2008.221
Chen, W. Y., Mao, F. C., Liu, C. H., Kuan, Y. H., Lai, N. W., Wu, C. C., et al. (2016). Chromium supplementation improved post-stroke brain infarction and hyperglycemia. Metab. Brain Dis. 31, 289–297. doi: 10.1007/s11011-015-9749-y
Corrall, R. J., and Frier, B. M. (1981). Acute hypoglycemia in man: neural control of pancreatic islet cell function. Metabolism 30, 160–164. doi: 10.1016/0026-0495(81)90166-9
Deng, D., Xu, C., Sun, P., Wu, J., Yan, C., Hu, M., et al. (2014). Crystal structure of the human glucose transporter GLUT1. Nature 510, 121–125. doi: 10.1038/nature13306
Dungan, K. M., Braithwaite, S. S., and Preiser, J. C. (2009). Stress hyperglycaemia. Lancet 373, 1798–1807. doi: 10.1016/S0140-6736(09)60553-5
Endres, M., Engelhardt, B., Koistinaho, J., Lindvall, O., Meairs, S., Mohr, J. P., et al. (2008). Improving outcome after stroke: overcoming the translational roadblock. Cerebrovasc. Dis. 25, 268–278. doi: 10.1159/000118039
Gerstein, H. C., Miller, M. E., Byington, R. P., Goff, D. C. Jr., Bigger, J. T., Buse, J. B., et al. (2008). Effects of intensive glucose lowering in type 2 diabetes. N. Engl. J. Med. 358, 2545–2559. doi: 10.1056/NEJMoa0802743
Gibson, C. L., Murphy, A. N., and Murphy, S. P. (2012). Stroke outcome in the ketogenic state–a systematic review of the animal data. J. Neurochem. 123, 52–57. doi: 10.1111/j.1471-4159.2012.07943.x
Ginsberg, M. D., Prado, R., Dietrich, W. D., Busto, R., and Watson, B. D. (1987). Hyperglycemia reduces the extent of cerebral infarction in rats. Stroke 18, 570–574. doi: 10.1161/01.STR.18.3.570
Harada, S., Fujita, W. H., Shichi, K., and Tokuyama, S. (2009). The development of glucose intolerance after focal cerebral ischemia participates in subsequent neuronal damage. Brain Res. 1279, 174–181. doi: 10.1016/j.brainres.2009.05.014
Hasselbalch, S. G., Knudsen, G. M., Videbaek, C., Pinborg, L. H., Schmidt, J. F., Holm, S., et al. (1999). No effect of insulin on glucose blood-brain barrier transport and cerebral metabolism in humans. Diabetes 48, 1915–1921. doi: 10.2337/diabetes.48.10.1915
Higgins, J. P. T., and Thomas, J. M. (2019). Cochrane Handbook for Systematic Reviews of Interventions, 2nd Edn. Chichester, UK: John Wiley & Sons Ltd. doi: 10.1002/9781119536604
Hitze, B., Hubold, C., van Dyken, R., Schlichting, K., Lehnert, H., Entringer, S., et al. (2010). How the selfish brain organizes its supply and demand. Front. Neuroenergetics 2. doi: 10.3389/fnene.2010.00007
Hom, F. G., Goodner, C. J., and Berrie, M. A. (1984). A [3H]2-deoxyglucose method for comparing rates of glucose metabolism and insulin responses among rat tissues in vivo. Validation of the model and the absence of an insulin effect on brain. Diabetes 33, 141–152. doi: 10.2337/diab.33.2.141
Hooijmans, C. R., Rovers, M. M., de Vries, R. B., Leenaars, M., Ritskes-Hoitinga, M., and Langendam, M. W. (2014). SYRCLE's risk of bias tool for animal studies. BMC Med. Res. Methodol. 14, 43. doi: 10.1186/1471-2288-14-43
Jarhult, J., and Holst, J. J. (1978). Reflex adrenergic control of endocrine pancreas evoked by unloading of carotid baroreceptors in cats. Acta Physiol. Scand. 104, 188–202. doi: 10.1111/j.1748-1716.1978.tb06266.x
Johnston, K. C., Bruno, A., Pauls, Q., Hall, C. E., Barrett, K. M., Barsan, W., Fansler, A., Van de Bruinhorst, K., Janis, S., Durkalski-Mauldin, V. L., and Neurological Emergencies Treatment Trials N, the STI (2019). Intensive vs standard treatment of hyperglycemia and functional outcome in patients with acute ischemic stroke: the SHINE randomized clinical trial. JAMA 322, 326–335. doi: 10.1001/jama.2019.9346
Kennedy, G. C. (1953). The role of depot fat in the hypothalamic control of food intake in the rat. Proc. R. Soc. London Ser. 140, 578–592. doi: 10.1098/rspb.1953.0009
Khan, M. A., Schultz, S., Othman, A., Fleming, T., Lebron-Galan, R., Rades, D., et al. (2016). hyperglycemia in stroke impairs polarization of monocytes/macrophages to a protective non-inflammatory cell type. J. Neurosci. 36, 9313–9325. doi: 10.1523/JNEUROSCI.0473-16.2016
Koch, K., Berressem, D., Konietzka, J., Thinnes, A., Eckert, G. P., and Klein, J. (2017). Hepatic ketogenesis induced by middle cerebral artery occlusion in mice. J. Am. Heart Assoc. 6. doi: 10.1161/JAHA.117.005556
Kruyt, N. D., Biessels, G. J., Devries, J. H., and Roos, Y. B. (2010). Hyperglycemia in acute ischemic stroke: pathophysiology and clinical management. Nat. Rev. Neurol. 6, 145–155. doi: 10.1038/nrneurol.2009.231
Kubera, B., Hubold, C., Wischnath, H., Zug, S., and Peters, A. (2014). Rise of ketone bodies with psychosocial stress in normal weight men. Psychoneuroendocrinology 45, 43–48. doi: 10.1016/j.psyneuen.2014.03.008
Li, L., Tian, J., Long, M. K., Chen, Y., Lu, J., Zhou, C., et al. (2016). Protection against experimental stroke by ganglioside GM1 is associated with the inhibition of autophagy. PLoS One 11:e0144219. doi: 10.1371/journal.pone.0144219
Mason, J. W., Wherry, F. E., Brady, J. V., Beer, B., Pennington, L. L., and Goodman, A. C. (1968). Plasma insulin response to 72-hr. Avoidance sessions in the monkey. Psychosom. Med. 30, 746–759. doi: 10.1097/00006842-196809000-00030
Mayer, J. (1953). Glucostatic mechanism of regulation of food intake. N. Engl. J. Med. 249, 13–16. doi: 10.1056/NEJM195307022490104
Meek, T. H., Nelson, J. T., Matsen, M. E., Dorfman, M. D., Guyenet, S. J., Damian, V., et al. (2016). Functional identification of a neurocircuit regulating blood glucose. Proc. Natl. Acad. Sci. U. S. A. 113, E2073–E2082. doi: 10.1073/pnas.1521160113
Mobbs, C. V., Kow, L. M., and Yang, X. J. (2001). Brain glucose-sensing mechanisms: ubiquitous silencing by aglycemia vs. hypothalamic neuroendocrine responses. Am. J. Physiol. Endocrinol. Metab. 281, E649–E654. doi: 10.1152/ajpendo.2001.281.4.E649
Moher, D., Liberati, A., Tetzlaff, J., Altman, D. G., and PRISMA-P Group (2009). Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. PLoS Med 6:e1000097. doi: 10.1371/journal.pmed.1000097
Peters, A., Bosy-Westphal, A., Kubera, B., Langemann, D., Goele, K., Later, W., et al. (2011). Why doesn't the brain lose weight, when obese people diet? Obesity Facts. 4:151–7.
Peters, A., and Langemann, D. (2009). Build-ups in the supply chain of the brain: on the neuroenergetic cause of obesity and type 2 diabetes mellitus. Front. Neuroenergetics 1:2. doi: 10.3389/neuro.14.002.2009
Peters, A., and McEwen, B. S. (2015). Stress habituation, body shape and cardiovascular mortality. Neurosci. Biobehav. Rev. 56, 139–150. doi: 10.1016/j.neubiorev.2015.07.001
Peters, A., Schweiger, U., Pellerin, L., Hubold, C., Oltmanns, K. M., Conrad, M., et al. (2004). The selfish brain: competition for energy resources. Neurosci. Biobehav. Rev. 28, 143–180. doi: 10.1016/j.neubiorev.2004.03.002
Rosso, C., Corvol, J. C., Pires, C., Crozier, S., Attal, Y., Jacqueminet, S., et al. (2012). Intensive versus subcutaneous insulin in patients with hyperacute stroke: results from the randomized INSULINFARCT trial. Stroke 43, 2343–2349. doi: 10.1161/STROKEAHA.112.657122
Routh, V. H., Hao, L., Santiago, A. M., Sheng, Z., and Zhou, C. (2014). Hypothalamic glucose sensing: making ends meet. Front. Syst. Neurosci. 8, 236. doi: 10.3389/fnsys.2014.00236
Schwartz, M. W., Seeley, R. J., Zeltser, L. M., Drewnowski, A., Ravussin, E., Redman, L. M., et al. (2017). Obesity pathogenesis: an endocrine society scientific statement. Endocr. Rev. 38, 267–296. doi: 10.1210/er.2017-00111
Seaquist, E. R., Damberg, G. S., Tkac, I., and Gruetter, R. (2001). The effect of insulin on in vivo cerebral glucose concentrations and rates of glucose transport/metabolism in humans. Diabetes 50, 2203–2209. doi: 10.2337/diabetes.50.10.2203
Shepherd, P. R., and Kahn, B. B. (1999). Glucose transporters and insulin action-implications for insulin resistance and diabetes mellitus. N. Engl. J. Med. 341, 248–257. doi: 10.1056/NEJM199907223410406
Slack, N., Chambers, S., and Johnston, R. (2004). Operations Management, 4 Edn. Harlow: FT Prentice Hall.
Spanswick, D., Smith, M. A., Groppi, V. E., Logan, S. D., and Ashford, M. L. (1997). Leptin inhibits hypothalamic neurons by activation of ATP-sensitive potassium channels. Nature 390, 521–525. doi: 10.1038/37379
Spiegel, K., Leproult, R., and Van Cauter, E. (1999). Impact of sleep debt on metabolic and endocrine function. Lancet 354, 1435–1439. doi: 10.1016/S0140-6736(99)01376-8
Sprengell, M., Kubera, B., and Peters, A. (2021). Brain more resistant to energy restriction than body: a systematic review. Front. Neurosci. 15:639617. doi: 10.3389/fnins.2021.639617
Stanley, S. A., Kelly, L., Latcha, K. N., Schmidt, S. F., Yu, X., Nectow, A. R., et al. (2016). Bidirectional electromagnetic control of the hypothalamus regulates feeding and metabolism. Nature 531, 647–650. doi: 10.1038/nature17183
Steinkamp, M., Li, T., Fuellgraf, H., and Moser, A. (2007). K(ATP)-dependent neurotransmitter release in the neuronal network of the rat caudate nucleus. Neurochem. Int. 50, 159–163. doi: 10.1016/j.neuint.2006.07.011
Taylor, S. H., Saxton, C., Majid, P. A., Dykes, J. R., Ghosh, P., and Stoker, J. B. (1969). Insulin secretion following myocardial infarction with particular respect to the pathogenesis of cardiogenic shock. Lancet 2, 1373–1378. doi: 10.1016/S0140-6736(69)90928-3
The-NICE-SUGAR-Study-Investigators (2009). Intensive versus conventional glucose control in critically ill patients. N. Engl. J. Med. 360,1283–1297. doi: 10.1056/NEJMoa0810625
Toda, C., Kim, J. D., Impellizzeri, D., Cuzzocrea, S., Liu, Z. W., and Diano, S. (2016). UCP2 regulates mitochondrial fission and ventromedial nucleus control of glucose responsiveness. Cell 164, 872–883. doi: 10.1016/j.cell.2016.02.010
Wagner, K. R., Kleinholz, M., de Courten-Myers, G. M., and Myers, R. E. (1992). Hyperglycemic versus normoglycemic stroke: topography of brain metabolites, intracellular pH, and infarct size. J. Cereb. Blood Flow Metab. 12, 213–222. doi: 10.1038/jcbfm.1992.31
Wang, Y. Y., Chen, C. J., Lin, S. Y., Chuang, Y. H., Sheu, W. H., and Tung, K. C. (2013). Hyperglycemia is associated with enhanced gluconeogenesis in a rat model of permanent cerebral ischemia. Mol. Cell Endocrinol. 367, 50–56. doi: 10.1016/j.mce.2012.12.016
Wang, Y. Y., Lin, S. Y., Chuang, Y. H., Sheu, W. H., Tung, K. C., and Chen, C. J. (2014). Activation of hepatic inflammatory pathways by catecholamines is associated with hepatic insulin resistance in male ischemic stroke rats. Endocrinology 155, 1235–1246. doi: 10.1210/en.2013-1593
Woods, S. C., and Porte, D. Jr. (1974). Neural control of the endocrine pancreas. Physiol. Rev. 54, 596–619. doi: 10.1152/physrev.1974.54.3.596
Yamazaki, Y., Harada, S., and Tokuyama, S. (2012). Post-ischemic hyperglycemia exacerbates the development of cerebral ischemic neuronal damage through the cerebral sodium-glucose transporter. Brain Res. 1489, 113–120. doi: 10.1016/j.brainres.2012.10.020
Yamazaki, Y., Harada, S., and Tokuyama, S. (2014). Sodium-glucose transporter type 3-mediated neuroprotective effect of acetylcholine suppresses the development of cerebral ischemic neuronal damage. Neuroscience 269, 134–142. doi: 10.1016/j.neuroscience.2014.03.046
Zhang, X., Yang, S., Chen, J., and Su, Z. (2018). Unraveling the regulation of hepatic gluconeogenesis. Front. Endocrinol. 9:802. doi: 10.3389/fendo.2018.00802
Keywords: brain energy metabolism, cerebral artery occlusion, blood glucose, cerebral insulin suppression, selfish-brain theory, systematic review
Citation: Sprengell M, Kubera B and Peters A (2021) Proximal Disruption of Brain Energy Supply Raises Systemic Blood Glucose: A Systematic Review. Front. Neurosci. 15:685031. doi: 10.3389/fnins.2021.685031
Received: 24 March 2021; Accepted: 28 May 2021;
Published: 24 June 2021.
Edited by:
Srinivas Sriramula, The Brody School of Medicine at East Carolina University, United StatesReviewed by:
Arturo Ortega, Centro de Investigación y de Estudios Avanzados del Instituto Politécnico Nacional, MexicoCopyright © 2021 Sprengell, Kubera and Peters. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Achim Peters, YWNoaW0ucGV0ZXJzQHVrc2guZGU=
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.