
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Neurosci., 23 April 2021
Sec. Neuropharmacology
Volume 15 - 2021 | https://doi.org/10.3389/fnins.2021.653651
This article is part of the Research TopicTargeting Neuroinflammation in Central Nervous System Disorders: Uncovering Mechanisms, Pharmacological Targets, and Neuropharmaceutical DevelopmentsView all 26 articles
 Angeles Vinuesa1,2
Angeles Vinuesa1,2 Carlos Pomilio1,2
Carlos Pomilio1,2 Amal Gregosa1,2
Amal Gregosa1,2 Melisa Bentivegna1,2
Melisa Bentivegna1,2 Jessica Presa1,2Melina Bellotto1,2
Jessica Presa1,2Melina Bellotto1,2 Flavia Saravia1,2
Flavia Saravia1,2 Juan Beauquis1,2*
Juan Beauquis1,2*Overnutrition and modern diets containing high proportions of saturated fat are among the major factors contributing to a low-grade state of inflammation, hyperglycemia and dyslipidemia. In the last decades, the global rise of type 2 diabetes and obesity prevalence has elicited a great interest in understanding how changes in metabolic function lead to an increased risk for premature brain aging and the development of neurodegenerative disorders such as Alzheimer’s disease (AD). Cognitive impairment and decreased neurogenic capacity could be a consequence of metabolic disturbances. In these scenarios, the interplay between inflammation and insulin resistance could represent a potential therapeutic target to prevent or ameliorate neurodegeneration and cognitive impairment. The present review aims to provide an update on the impact of metabolic stress pathways on AD with a focus on inflammation and insulin resistance as risk factors and therapeutic targets.
Modern lifestyle is associated with detrimental behavioral and dietary habits such as sedentarism and high dietary intake of saturated fats and refined sugars. These, among other habits, are determinants for the development of obesity, type 2 diabetes (T2D) and associated conditions that have a straightforward impact on several systems including the central nervous system (CNS), thereby heightening the risk for cognitive impairment and neuropsychiatric diseases.
Obesity is a multifactorial disease mainly defined as an excessive accumulation and abnormal distribution of adipose tissue in the body. In terms of prevalence, overweight and obesity rates have increased alarmingly in the last decades currently reaching global epidemic levels (Who, 2018). Almost 40% of the world’s adult population was overweight in 2016 and 13% was obese, with the aggravating fact of this trend being featured also at juvenile stages. Moreover, overweight and obesity constitute the major risk factors for the development of T2D and metabolic syndrome (MetS). As for diabetes, the main hallmark is the presence of increased blood glucose levels and its prevalence was estimated to be close to 9% of the world’s population (Idf, 2017). The most common form is the T2D, representing 85–95% of the cases, closely associated with metabolic factors such as obesity. Metabolic syndrome encompasses a variety of conditions associated with cardiometabolic risk, a cluster of alterations including high blood pressure, hyperglycemia, hyperinsulinemia, dyslipidemia and increased abdominal fat or obesity. However, these conditions may not manifest in the same way in all patients. Its prevalence has dramatically increased in a similar fashion as obesity in all age groups (Bussler et al., 2017). Although each of the pathologies stated above has its own features, they are intimately related, sharing components, common risk factors and also being part of the etiology of one another. Therefore, one could attempt to focus on their shared pathways in order to discuss potential mechanisms involved in the development of associated pathologies.
Inflammation and insulin resistance are among the most common phenomena underlying the pathophysiology of obesity- and T2D- related diseases and, interestingly, also consistently found in several neuropsychiatric conditions (Parimisetty et al., 2016; O’Brien et al., 2017; Ying et al., 2020). As it will be further discussed ahead, in neurodegenerative conditions associated with brain aging, the hippocampus is one of the most vulnerable and primarily affected structures, showing atrophy and alterations in neuroplasticity mechanisms related to synaptic connectivity and adult neurogenesis leading to cognitive dysfunction (Mu and Gage, 2011). The fact that chronic metabolic disturbances can also negatively affect such plasticity mechanisms in addition to their association with cognitive deficits and mood alterations (Biro and Wien, 2010; Puder and Munsch, 2010), suggests there are converging pathways of metabolic stress and aging.
In the last decades, it has been reported that T2D, obesity, hypertension and sedentarism are among the main modifiable risk factors for neurodegenerative diseases (Bruce-Keller et al., 2009). In particular, in this review we will focus on Alzheimer’s disease (AD) as it is the most prevalent neurodegenerative disease, the major cause of dementia and has a strong association with metabolic disorders and related cardiovascular risk factors.
From several points of view, AD and obesity constitute very relevant public health concerns due to the economic and social burden of the diseases. The economic burden of AD, that was estimated to be more than $300 billion in 2020 only in the United States, is not only related to direct costs such as diagnosis, drug therapy, hospitalization and specialized nursing or home care but also with the quality of life of both patients and caregivers in such a progressive and long- term disease (Alzheimer’s-Association., 2020). Global obesity economic burden has been estimated to be two trillion USD in 2014, accounting for a 20% of the total health care and is also related to decreased productivity due to associated disease disability and promotion of premature aging (Tremmel et al., 2017). These diseases’ impact over public health could be even much greater if we considered at least four important factors: increasing obesity rates in younger generations, diagnosis underestimation, obesity comorbidities and the lack of effective therapies to mitigate AD-like neurodegeneration.
Taking the above into account, the relevance and interrelation of both metabolic and neurodegenerative health domains leads to the development of a research field devoted to the study of shared pathways in order to better comprehend their dynamics and identify potential solutions (Figure 1). Obesity-induced brain inflammation, as a common phenomenon involved in the increased risk for brain dysfunction, could act as a trigger and also as an exacerbating factor in the development of neurodegenerative disorders. Therefore, in this review we will discuss the particular effect of the mentioned metabolic disorders on brain plasticity and the potentiality for inflammatory and associated insulin-resistance mediators as therapeutic targets in AD. Analyzing existing interventions that address either insulin signaling or inflammation might contribute to the understanding of brain dysfunction mechanisms and neurodegenerative diseases as potential metabolic pathologies.
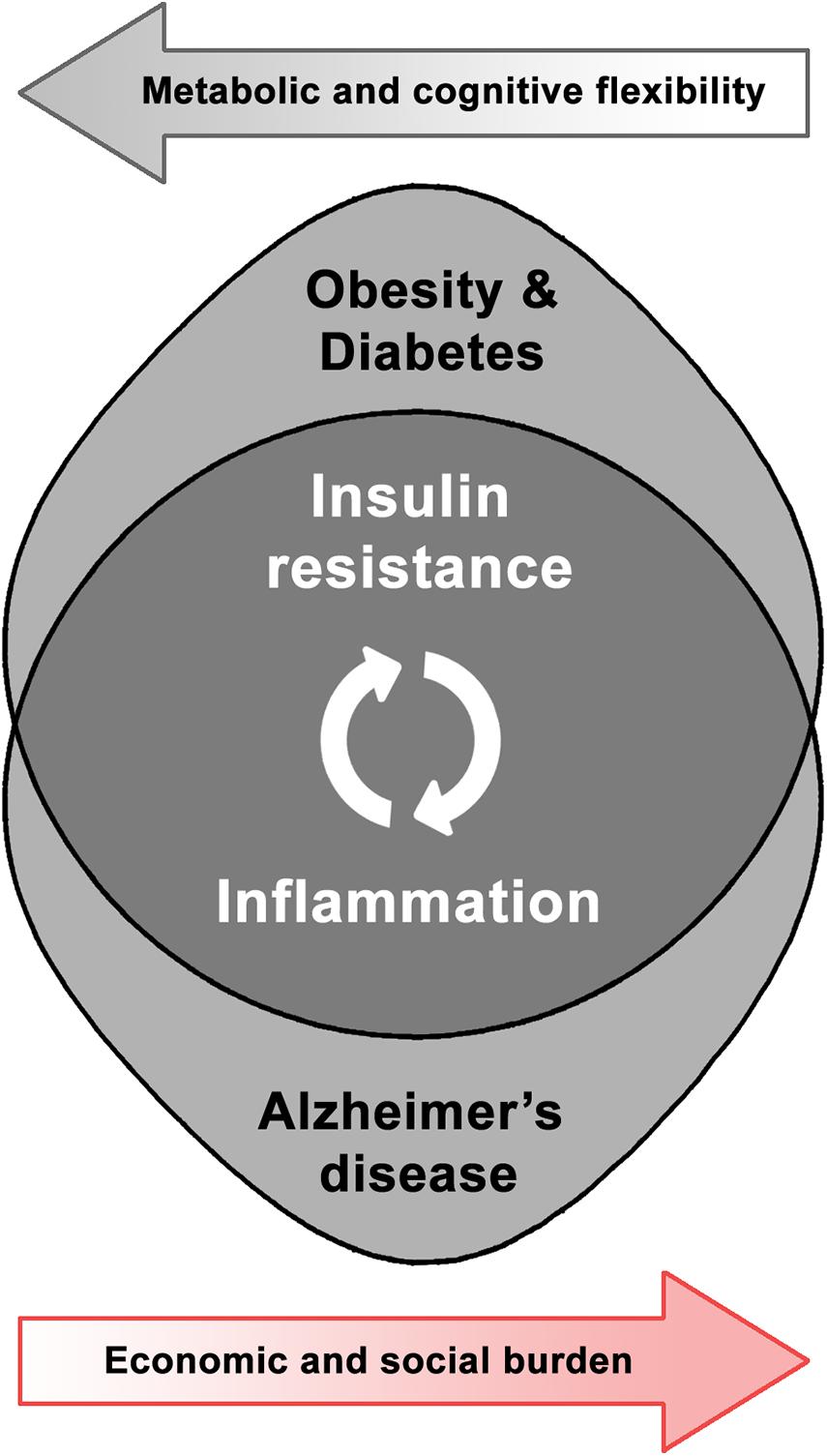
Figure 1. Shared pathophysiological pathways and synergic burden of obesity-related disorders and Alzheimer’s disease.
Together with obesity, both mood and neurocognitive disorders are highly prevalent public health concerns and a bidirectional relationship among them has been suggested. Several studies have recently shown a strong association between metabolic disturbances and the development of anxiety-related disorders and poor cognitive outcomes (Avinun and Hariri, 2019; Ronan et al., 2019; van Duinkerken and Ryan, 2019), with higher rates of anxiety and depression found in overweight and obese people (Luppino et al., 2010; Gomes et al., 2019; James et al., 2020). Interestingly, both anxiety and depression are associated with an increased likelihood of developing AD (Becker et al., 2018; Canton-Habas et al., 2020).
As life expectancy has constantly grown, brain aging-related diseases have increased in a similar fashion. Among neurodegenerative disorders, AD is the most common form of age-related dementia. There are approximately 50 million people worldwide suffering from dementia and 60–70% of the cases may be attributed to AD, according to the World Health Organization (Who, 2020). While age is the main risk factor associated to the development of sporadic AD, increasing the chances by 50% every 5 years after 65 years old and reaching a 30–50% prevalence after 85 years of age (Alzheimer’s-Association., 2020), it has been reported that T2D and obesity, independently, are associated with an approximate two-to-threefold increased chance of developing AD (Bruce-Keller et al., 2009). Other risk factors for sporadic AD, which constitute more than 95% of AD cases, are the presence of the ε4 allele for apolipoprotein E (APOE) (Morris et al., 2017), hypertension, atherosclerosis, hypercholesterolemia, traumatic brain injury, among others (Edwards et al., 2019). Familial AD, which accounts for 5% of total AD cases, is associated with inherited mutations in the amyloid precursor protein (APP) gene or in the genes for the enzymes that process APP to produce amyloid β (Aβ). Clinically, AD is characterized by the progressive loss of memory and cognitive abilities that ultimately lead to severe impairments in daily life activities. Cerebral extracellular accumulation of Aβ peptide plaques and intraneuronal neurofibrillary tangles of hyperphosphorylated Tau are the main histopathological hallmarks. Among the shared brain alterations between metabolic pathologies and neurodegeneration, insulin resistance constitutes one of the main factors. A considerable body of evidence suggests a relevant overlap across T2D and AD including risk, comorbidity and pathophysiological pathways. As a matter of fact, though controversial, some authors propose that AD might represent the so-called “Type 3 diabetes,” given that initial stages of the disease related to cognitive dysfunction go along with multiple alterations of the insulin pathway in the hippocampus (de la Monte and Tong, 2014; Arnold et al., 2018).
The impact of midlife obesity in cognitive decline has been extensively studied and even considered to be an early predictor of AD (Dahl et al., 2010; Chuang et al., 2016; Gottesman et al., 2017). High levels of saturated fatty acid in the cerebrospinal fluid (CSF) of overweight patients with related metabolic comorbidities correlate with amnestic mild cognitive impairment (MCI) (Melo et al., 2020). However, the relevance of obesity may be somewhat controversial across different age groups, particularly depending on the phenotypic parameter assessed. For instance, while a high body mass index (BMI > 25 kg/m2) in early and midlife obese patients has been associated with the development of cognitive impairment, it is more controversial in the elderly since some reports support the same relationship between both phenomena (Biro and Wien, 2010; Benito-Leon et al., 2013; Melo et al., 2020) while others have not found a significant association (Dahl and Hassing, 2013; Wang et al., 2016; Deckers et al., 2017). In that sense, some studies claim that indirect parameters associated with obesity, such as body fat distribution, hyperglycemia or high-fat diet (HFD) consumption, exhibit higher correlation with cognitive dysfunction in aged cohorts (Assuncao et al., 2018). A large longitudinal analysis by Mac Giollabhui et al. (2020) has shown that higher BMI in a cohort of adolescents predicts a worse performance of executive function and greater depression risk, in a context of peripheral inflammation.
Another obesity-related factor that has been associated with an increased risk of AD is dyslipidemia. Studies in animal models have shown that high consumption of cholesterol and hypercholesterolemia are associated with increased Aβ aggregation and neuroinflammation (Sparks et al., 1994; Zatta et al., 2002; Xue et al., 2007). Correspondingly, high cholesterolemia in middle-aged humans is linked with an increased risk for AD in later life compared with a control population (Solomon et al., 2009) and a lipid profile showing high levels of low-density lipoprotein (LDL) cholesterol and low levels of high-density lipoprotein (HDL) cholesterol is associated with increased Aβ levels in the brain (Reed et al., 2014). However, a recent meta-analysis has found that AD risk is increased in individuals with high LDL cholesterol blood levels but not in those with high levels of HDL cholesterol, total cholesterol or triglycerides (Saiz-Vazquez et al., 2020). Even though cholesterol does not cross the blood–brain barrier (BBB) in physiological conditions, cholesterol oxidized products like oxysterols can cross it and may play a role in memory consolidation and AD pathophysiology (Lutjohann et al., 1996; Kotti et al., 2006).
Regarding experimental data, our group and others have shown neurological consequences associated with cognitive dysfunction and emotional processing alterations in obesogenic contexts. Exposure to a HFD promotes an altered behavioral profile with diminished spatial memory, impaired daily life activities, as well as anxiety and depressive-like behaviors in young adult mice (Vinuesa et al., 2016) and cognitive decline in aged mice (Wei et al., 2018). Likewise, HFD has shown to exacerbate brain pathology and related behavioral impairment in APP/PS1 and 3xTg-AD mouse models of AD (Theriault et al., 2016; Sah et al., 2017; Bracko et al., 2020), reinforcing the relevant crosstalk between metabolic and age-associated brain dysfunction.
In the following sections of this review, we will provide an overview of the phenomena in common between the aforementioned metabolic and neurodegenerative domains and associated therapeutic approaches, in order to ascertain the relevance of metabolic derangements as drivers of brain dysfunction in AD.
Although many cellular and molecular pathways are affected in the context of diabetes, obesity and metabolic syndrome, one of the primarily occurring phenomena is inflammation. High peripheral levels of inflammatory biomarkers such as C-reactive protein (CRP), IL-6 and TNF-α have been described in obesogenic contexts (Ellulu et al., 2017). Low-grade systemic inflammation is one of the earliest and main pathological events that might lead to the development of insulin resistance in most insulin-sensitive tissues (Kloting and Bluher, 2014; Parimisetty et al., 2016). The activation of the immune system and chronic inflammation are not only featured by increased circulating pro-inflammatory molecules but also by infiltration of macrophages and other immune cells into the affected tissues.
In the context of obesity, the hypertrophied adipose tissue experiments a functional switch, changing the secretory profile of endocrine factors from homeostatic to pathologic or pro-inflammatory, thereby leading to a decreased metabolic flexibility (Ronan et al., 2019), with lower levels of the pro-homeostatic adiponectin and increased levels of pro-inflammatory adipokines, generating a context of chronic low grade inflammation affecting not only adipose tissue but also the liver, insulin-producing pancreatic β-cells, skeletal muscle, the heart and the brain (Ridker et al., 2000; Aleffi et al., 2005; Le et al., 2011). Regarding systemic inflammatory markers, a positive association has been found not only with high BMI and obesity-related parameters but also with CNS impairment. In fact, Mac Giollabhui et al. (2020) reported increased levels of the proinflammatory cytokine IL-6 as a relevant predictor factor of both depression and executive dysfunction in individuals with elevated BMI. In fact, chronic sterile, low-grade inflammation is associated with aging and age-related diseases, leading to the emergent concept of “inflamm-aging” in order to describe a dysregulation in the homeostasis of cytokines and oxidative stress (Franceschi et al., 2018). In the case of AD for instance, there have been described certain genetic variants leading to increased levels of IL-1β, IL-6, and TNF-α that are associated with a greater risk of developing the disease (Wang, 2015; Miwa et al., 2016; Mun et al., 2016). Circulating levels of these mediators, together with CRP, constitute relevant damage biomarkers connecting chronic metabolic diseases such as obesity and T2D with incident dementia or late-onset AD (Franceschi and Campisi, 2014). Moreover, in preclinical AD models, peripheral immune challenges with lipopolysaccharides (LPS) or polyI:C have shown to accelerate or increase Aβ deposition in 3xTg-AD mice and APPswe Tg2576 (Sheng et al., 2003; Kitazawa et al., 2005; Krstic et al., 2012), supporting a role for peripheral inflammatory mediators in brain pathology.
Additionally, in reference to the metabolic-induced inflammatory response, alterations in gut microbiota have also emerged critical for pathogenesis. In the last decade, the gut microbiome has gained increasing interest due to its role in the physiology of humans and several animal models including rodents (Bruce-Keller et al., 2009). The composition and diversity of gut microbiota can be modulated by a wide range of factors including colonization at birth and lifestyle events across the lifespan such as feeding behavior, toxins and antibiotic exposure in addition to aging, experiencing a shift to a decreased diversity in the elderly and thereby contributing to systemic inflammation and detrimental signaling to the brain (Claesson et al., 2012; Bokulich et al., 2016; Sochocka et al., 2019). For instance, HFD exposure has shown to reduce microbiome diversity and transplantation of obese-derived microbiota into control germ-free mice was able to induce metabolic alterations associated with obesity (Turnbaugh et al., 2006; Cani et al., 2008). An unhealthy microbiome could promote a “leaky gut” and thus enable the increase of immunogenic components such as LPS in plasma, a phenomenon known as endotoxemia, contributing to the metabolic inflammation (Carvalho et al., 2012). In a similar fashion, as the gastrointestinal barrier disruption could lead to the increase of potentially toxic effectors in circulation, the BBB integrity has also shown to be affected in the context of metabolic disorders, aging and neurodegeneration (Erickson and Banks, 2013; Salameh et al., 2019). Therefore, the gut-brain axis disruption might as well contribute to the negative impact of peripheral insults on the CNS.
Neuroinflammation is a shared hallmark among diverse pathologies and should not be considered an isolated or independent phenomenon from systemic inflammation since different experimental approaches have shown that peripheral inflammatory challenges as well as chronic low grade inflammation trigger inflammation in the CNS (Heneka et al., 2015). In relation to obesity for instance, circulating pro-inflammatory cytokines, free-fatty acids and ceramides have shown to impact also on limbic structures. This effect could be worsened by BBB increased permeability, as it has been described in HFD exposed rodents, leading to the alteration of hippocampal function (Hargrave et al., 2016). Hypothalamic inflammation has been found to be involved in the development as well as in the maintenance of an obese phenotype, promoting and perpetuating unhealthy feeding behavior, altering the reward circuitry (Guillemot-Legris and Muccioli, 2017) and has actually been acutely detected after high-fat feeding, even before the establishment of obesity (Maric et al., 2014). In this context, reactive gliosis can be found in the arcuate nucleus, being microglial cells the main cell type proposed to sense nutrient overload and trigger the inflammatory response since long-chain fatty acids bind to toll-like receptors (TLR4) and downstream cytokine synthesis and endoplasmic reticulum stress (ERS) pathway (Milanski et al., 2009). On the other hand, several reports have shown that metabolic-induced inflammation is also present in extra-hypothalamic structures such as hippocampus, cortex and amygdala, constituting a relevant link between the neuropsychiatric alterations and obesity or insulin resistance-associated disorders (Parimisetty et al., 2016; Vinuesa et al., 2016; Solas et al., 2017).
As regards age-related cognitive decline, neuroinflammation and oxidative stress are known to be present in the hippocampus and potentially related to the reduced plasticity and associated memory impairment (Frank et al., 2006; Patterson, 2015; Stebbings et al., 2016). Nevertheless, in the particular case of AD-like aging, it is not yet elucidated if inflammation should be considered a trigger, consequence or aggravating factor. Neuroinflammation (mostly given by hyperreactivity of glial cells secreting pro-inflammatory molecules) is consistently found in AD patients and animal models (Heneka et al., 2015). In fact, in the case of animal models, neuroinflammation has been detected even prior to amyloid deposition (Heneka et al., 2005; Beauquis et al., 2013). Reactive glial cells and increased pro-inflammatory mediators have shown to affect both Aβ production and clearance, two processes directly associated with the main histopathological hallmark of AD. For instance, acute neuroinflammation after traumatic brain injury or hypoxia have shown to increase levels of relevant actors for APP amyloidogenic enzymatic cleavage such as the beta-site amyloid precursor protein cleaving enzyme 1 (BACE1), as well as presenilin 1 (PS1) or nicastrin, components of the γ-secretase complex in rodents and pigs (Blasko et al., 2004; Chen et al., 2004; Nadler et al., 2008). Moreover, the deletion of TNF-α in transgenic 5XFAD mice promoted a decreased deposition of Aβ in association with a decreased expression of PS1 and β-secretase (Paouri et al., 2017). Interestingly, Aβ per se acts as an important TLR4 ligand, constituting a positive feed-forward loop with neuroinflammation thus contributing to Aβ plaque formation (Stewart et al., 2010). In reference to Aβ clearance, there are several mechanisms affected in the context of chronic or unsolved inflammatory response. Microglial cells, as the first-line defense brain cells, are mostly in charge of removing Aβ deposits by phagocytosis and intracellular proteolysis mechanisms such as proteasome-degradation and autophagy.
During the last decades, several authors emphasized the relevance of the autophagy-lysosomal system and its regulation in age-associated diseases like AD, proposing the modulation of cellular metabolism and the activation of autophagy as a valid therapeutic approach. Autophagy involves the semi-specific isolation of intracellular content -mainly misfolded proteins and dysfunctional organelles- into a double-membrane vesicle called autophagosome. Later, the autophagosome is fused to a lysosome, forming the autolysosome, where the content of the vesicle is degraded and further recycled into the cytosol (Liu and Li, 2019). Autophagy is negatively affected by age, and particularly impaired in AD. The impairment of autophagy involves the accumulation of autophagosomes that do not evolve into autolysosomes, as it was evidenced in neuronal and glial cells, both in patients and experimental models of AD (Gregosa et al., 2019; Uddin et al., 2019; Pomilio et al., 2020; Stavoe and Holzbaur, 2020). While there is no concluding information regarding if autophagy impairment is a cause or a consequence for AD, its dysfunction is related to a loss of amyloid degradation capacity and an accumulation of toxic waste that could lead to reduced neuronal viability and neuroinflammation, reinforcing the degenerative character of this pathology. Chronic activation of autophagy by rapamycin administration is associated with cognitive improvement, decreased amyloid pathology and enhanced autophagy in transgenic mouse models of AD (Spilman et al., 2010; Majumder et al., 2011). Neuroinflammation is also associated with this scenario. Our group has recently shown that microglial autophagy is impaired in AD patients and experimental models (Pomilio et al., 2020), and that this condition is associated with the assembly of inflammasomes and the production of the proinflammatory cytokine IL-1β (Cho et al., 2014). Interestingly, a recent article by Liu et al. (2020) showed that promoting microglial autophagy in APP/PS1 mice using nanoparticles not only enhances amyloid degradation but also reduces neuroinflammation by causing a decrease in brain levels of proinflammatory cytokines and an increase in anti inflammatory cytokines, finally conducing to a better performance in cognitive tests.
Autophagy in the brain is not independent of peripheral stimuli. In the last years, several studies showed that gut microbiota is altered in transgenic mouse models of AD, and that restoring microbiota enhances the autophagic flux in the brain, amyloid degradation and cognitive performance (Bonfili et al., 2017, 2018; Chen et al., 2017). Particularly, Bonfili et al. (2017) showed that microbiota restoration causes a decrease in plasma levels of proinflammatory cytokines, suggesting that peripheral inflammation, brain autophagy and neurodegeneration are closely related.
Overall, the canonical function of insulin is to maintain glucose homeostasis and to regulate cellular division and growth through the PI3K/Akt pathway and mitogenic activity through the RAS/MAPK signal transduction pathway. The concept of insulin resistance emerges when insulin levels, whether elevated or within normal range, are associated with a diminished metabolic response or sensitivity (Wilcox, 2005).
Several reports indicate that inflammation affects insulin signaling pathways in insulin-sensitive tissues (Kloting and Bluher, 2014; Parimisetty et al., 2016). Hence, it seems evident that obesity-induced inflammation is closely related to the development of T2D, mainly featured by an inefficient action of insulin or insulin resistance, thus placing insulin in the center of the scene. Among the candidate molecules involved in this inflammatory response are reactive oxygen species (ROS), pro-inflammatory cytokines such as TNF-α, IL-1β, IL-6 and stress kinases as Jun N-terminal kinase (JNK), inhibitor of kappaB kinase (IKK) and protein kinase C (PKC) (Boucher et al., 2014). Regarding IL-1β, there are relevant factors to be considered particularly associated with acute and chronic effects in relation to glycemic control. It has been shown that postprandial-induced hyperglycemia triggers inflammasome-mediated maturation and secretion of IL-1β in adipose macrophages as an homeostatic mechanism to favor insulin-dependent glucose uptake and protect the organism against potential food microbes (Dror et al., 2017). Nonetheless, when the pro-inflammatory cytokine levels together with hyperglycemia-induced ROS are sustained chronically, deleterious effects arise and contribute to the development of insulin resistance (Maedler et al., 2002). The latter has been proposed not only for IL-1β but other inflammatory cytokines such as TNF-α and IL-6. Hotamisligil et al. (1993), one of the first groups in assigning a relevant role for inflammatory cytokines such as TNF-α in the context of experimental obesity or diabetes, also proposed that the endoplasmic reticulum (ER) should be considered as an essential component in the coordination of metabolic responses (Hotamisligil, 2010; Kloting and Bluher, 2014) and could also be involved in the disruption of insulin signaling. Under physiological conditions, proteins are synthesized at the ER, folded, modified and ultimately targeted to the appropriate cellular compartment. However, when there is an uncontrolled or excessive increase in protein synthesis and presence of incorrectly folded proteins, the ERS response or unfolded protein response (UPR) is triggered to activate JNK/AP1, IKK, NFκB and oxidative stress pathways. In the context of metabolic stress, nutrient overload or elevated free fatty acids constitute potential inductors of this response (Velloso et al., 2015).
Another relevant phenomenon underlying inflammation-induced insulin resistance is the activation of TLRs, members of the pattern recognition receptor family responsible for the innate immune response. Interestingly, different rodent models of TLR4 ablation, either genetically modified by knock-out approaches (Poggi et al., 2007; Jia et al., 2014; Li et al., 2014) or by pharmacological inhibition (Zhang et al., 2015), have shown to prevent HFD-induced insulin resistance. As for TLR4 ligands in obesogenic contexts, it has been reported that long chain fatty acids increased in obesity-related contexts are able to stimulate TLR4 signaling leading to the development of insulin resistance (Velloso et al., 2015). Regarding lipotoxic stimuli as insulin-resistance inducers, ceramides have also been found to be very relevant. Ceramides are important bioactive lipidic messengers regulating cellular functions such as stress response, inflammation and survival. Levels are increased in obesogenic and diabetogenic contexts and were found to induce insulin resistance both in vitro and in vivo (Holland et al., 2011; Chavez et al., 2014). Even though ERS and TLRs activation have been originally studied as independent mechanisms in the pathophysiology of metabolic disorders, there is a considerable body of evidence suggesting a constant crosstalk between them in the modulation of metabolic dysfunction and inflammation associated to a decreased insulin sensitivity and downstream actions (Guillemot-Legris and Muccioli, 2017).
During insulin resistance, the activity of Akt -a main effector of the insulin receptor pathway- remains low. Akt is a multi-substrate kinase that regulates the activity of the AMP-dependent kinase (AMPK). Although AMPK is activated by several stimuli, it acts as a master regulator for cellular metabolism and its activation is necessary as a mediator for insulin effects on skeletal muscle and adipose tissue, as it was recently reviewed by Chen et al. (2020). Particularly, AMPK activation directly inhibits de novo lipid synthesis and enhances fatty acid oxidation, linking its activity with insulin resistance and macrophage activation in the context of obesity and T2D (Herzig and Shaw, 2018). In fact, activation of AMPK is proposed as the mechanism responsible for the therapeutic effects of metformin and other antidiabetic treatments, as it is mentioned ahead in Section “Therapeutic Approaches.” Akt and AMPK altogether regulate the activity of the mammalian target of rapamycin (mTOR) complex. The activation of Akt leads to the activation of mTOR, while AMPK directly inhibits its function. When activated, mTOR upregulates major anabolic pathways like protein synthesis while inhibiting protein degradation through the downregulation of the autophagic-lysosomal system (Herzig and Shaw, 2018).
Insulin sensitivity, among other factors, is regulated by a number of small insulin-sensitive adipocytes (Leonardini et al., 2009), via PPARγ, a member of the peroxisome proliferator-activated receptors (PPAR) family that regulates the expression of several genes, mainly related to metabolism and inflammation. It is highly expressed in adipose tissue, and also in the liver and skeletal muscle (Vidal-Puig et al., 1996). The endogenous ligands of PPARs are free fatty acids and eicosanoids, favoring lipid storage in adipose tissue while reducing lipotoxicity, promoting an anti-inflammatory response and enhancing insulin sensitivity (Varga et al., 2011). It has been hypothesized that PPARγ function is also neuroprotective. It was found to be expressed in the hippocampus, cortex and hypothalamus and is associated with decreased levels of pro-inflammatory mediators, ROS and Aβ in AD models (Inestrosa et al., 2005; Warden et al., 2016).
Mainly known for its role in peripheral glucose homeostasis, insulin also has a significant impact within the brain, functioning as a key neuromodulator in behavioral, cellular, biochemical and molecular studies. Emerging evidence from human and animal studies indicate that insulin influences cerebral bioenergetics and neural functioning (Kellar and Craft, 2020). The brain is now regarded as an insulin-sensitive organ with widespread, yet selective, expression of the insulin receptor in the olfactory bulb, hypothalamus, hippocampus, cerebellum, amygdala and cerebral cortex. In the hypothalamus, insulin signaling modulates metabolism, food intake and energy balance (Lee and Mattson, 2014), while in extra-hypothalamic structures such as the hippocampus, insulin actions are associated with neurotrophic and neuromodulatory functions. Insulin receptor signaling in the brain is important for neuronal development, glucose regulation, feeding behavior, body weight, and cognitive processes such as executive functioning, learning and memory (Kellar and Craft, 2020). Insulin receptors are abundantly expressed in neurons and glial cells (Frolich et al., 1998) and, together with insulin-like growth factors (IGFs) and receptors, regulate functions like neuron differentiation, dendritic growth, neuronal survival, circuit development, synaptic plasticity and postsynaptic neurotransmitter receptor trafficking (Kleinridders et al., 2014; Gralle, 2017), playing a key role in memory and learning (Blazquez et al., 2014; Ferrario and Reagan, 2017). Consistently, individuals with diabetes or insulin resistance show hippocampal atrophy, alterations in brain connectivity and variable degrees of cognitive impairment, shared phenomena between metabolic and neurodegenerative disorders (Beauquis et al., 2008, 2010; Dey et al., 2017).
Insulin signaling has been shown to be impaired in experimental models of AD (Chua et al., 2012; Ferreira et al., 2018) and in patients (Baker et al., 2011). Brain insulin resistance is a cause for dysregulation of brain bioenergetics, and also leads to enhanced Aβ production, impaired axonal transport, apoptosis and neuroinflammation, as it was recently reviewed by Nguyen et al. (2020). Particularly, impaired hippocampal insulin signaling is associated with impairments in memory and other executive functions, and it is proposed as a mediator between peripheral insulin resistance in T2D and brain dysfunction in AD. In line with this, Li et al. (2018) demonstrated that an animal model presenting both T2D and AD exhibits accelerated neurodegeneration and cognitive impairment, in association with dysregulation of the insulin degrading enzyme (IDE) expression. IDE is an extracellular protease that degrades not only insulin, but also Aβ, in a competitive way. Several authors have proposed that Aβ clearance by IDE could be impaired as IDE is saturated by the high insulin levels in the extracellular space due to insulin resistance, finally leading to Aβ accumulation and aggregation (Qiu and Folstein, 2006; Mayeux and Stern, 2012; Li et al., 2018).
Insulin resistance has also been associated with the modulation of amyloid production, tau phosphorylation and neuroinflammation through the regulation of glycogen synthase kinase (GSK)−3 activity (Jope et al., 2007). This kinase is one of the main targets of the PI3K/Akt pathway and is inactive upon phosphorylation by Akt. Hence, it has been hypothesized that a defect in insulin signaling would lead to GSK3 activation through diminished PI3K/Akt signaling. Subsequently, activation of GSK3 would lead to a number of effects among which tau hyperphosphorylation (Ishiguro et al., 1993) and increased Aβ production through BACE1 activation (Ly et al., 2013) or APP phosphorylation (Rockenstein et al., 2007) could play a significant role in AD pathogenesis. However, there are conflicting results in studies addressing this issue. While some support this hypothesis (Steen et al., 2005) others have found that inactive forms of GSK3 are present in AD brains (Griffin et al., 2005) and this discrepancy would depend on the progression (early vs. late stages) of the disease (Jimenez et al., 2011).
It has been shown that insulin resistance, induced by chronic exposure to a HFD promotes short-term memory impairments and decreases brain-derived neurotrophic factor (BDNF) and phospho-Akt levels in the prefrontal cortex and hippocampus. BDNF, a neurotrophin associated with synaptic and neural plasticity, shares components of the PI3K/Akt insulin pathway through the interaction with tropomyosin receptor kinase B (TrkB) (Kowianski et al., 2018). This fact stresses the relevance of these signaling cascades’ crosstalk and the consequent impact of the potential impairment induced by metabolic alterations.
Even though there is a solid body of literature supporting a mainly pro-homeostatic role of insulin on brain function, contradicting evidence in relation to aging and neurodegeneration should not be ignored. Particularly, and in conflict with the previously mentioned deleterious effects of insulin resistance, there is evidence from animal models suggesting that the downregulation of insulin signaling may have protective effects in aging and AD. In Tg2576 mice, a commonly used model of AD, neuronal deletion of insulin receptor substrate 2 (IRS2) is associated with decreased Aβ deposition in the brain, improved cognitive performance and increased survival of adult animals (Freude et al., 2009; Stohr et al., 2013). However, it should be noted that the expression of Akt and other downstream components of the insulin signaling pathway are unaffected by the deletion of IRS2 in this model. Similarly, using the nematode Caenorhabditis elegans expressing a minigene for human Aβ1–42, Cohen et al. (2006) have shown that amyloid aggregation is decreased and aging is slowed when insulin signaling is reduced after a treatment with a daf-2 RNAi. Regarding human studies, Harries et al. (2012) have shown that the expression of genes participating in insulin signaling as PI3K, PTEN, FOXO and PDK1 is decreased in aged human populations in a similar fashion to that seen under the pro-longevity mTOR inhibition therapy. Thus, these results may suggest that decreased insulin signaling may have a role extending lifespan and modulating amyloid aggregation, in potential conflict with therapeutic strategies aimed at promoting insulin signaling. However it is not clear yet if the impairment of insulin signaling is a consequence of neurodegeneration or a protective response, stressing the need for more mechanistic and functional studies to clarify this central point.
The brain is exposed to a highly dynamic environment and requires concerted mechanisms to exert appropriate responses. In that sense, brain plasticity represents the CNS intrinsic ability to act upon constantly changing conditions by means of structural and functional processes. Among the phenomena involved in such flexibility, synapse formation, elimination or strength modulation, as well as the promotion of hippocampal neurogenesis are essential mechanisms (Kelly et al., 2006; Selemon, 2013; Lucassen et al., 2015). In line with this, alterations of such mechanisms of structural plasticity are associated with cognitive decline and memory dysfunction in aging and neurodegenerative disorders.
Hippocampal neurogenesis is a key neuroplastic process that involves the proliferation of neural precursor cells (NPCs) in the subgranular zone (SGZ), differentiation, survival and proper integration into the hippocampal circuit. It is a thoroughly relevant process underlying emotional behavior and cognitive function mainly associated with memory and learning (Clelland et al., 2009; Toda and Gage, 2017). Neurogenesis occurs during development and continues throughout life, persisting even in aged adults and AD patients. Nevertheless, it declines with age and is strongly affected by the environment (Klempin and Kempermann, 2007). Tobin et al. (2019) recently showed that adult neurogenesis exhibits a positive correlation with cognitive status in MCI and AD versus aged patients and could render an early sign of the disease. Animal models of AD show decreased rates of hippocampal neurogenesis (Mu and Gage, 2011; Myhre et al., 2019). Interestingly, overactivation of GSK3β by genetic blockade of the kinase inhibition in NPCs has been associated with diminished neurogenesis together with impaired hippocampus-dependent behavior in mice (Eom and Jope, 2009; Pardo et al., 2016), reinforcing the relevance of neurotrophin-induced signaling in hippocampal plasticity affected in age-related neurodegeneration.
Taking into account metabolic disturbances as negative modulating factors, the aforementioned conditions associated with obesity and T2D, insulin resistance and sustained inflammation have consistently shown to promote neural connectivity dysfunction. Several studies from our group and others have reported alterations in hippocampal neurogenesis as a consequence of HFD exposure, occurring in association with impaired cognitive and emotional processing (Boitard et al., 2012; Hao et al., 2016; Vinuesa et al., 2016, 2019). In particular, the early-onset of HFD exposure in rodents has shown to be especially detrimental; both C57BL6/J mice after 6 (Vinuesa et al., 2019) or 11 (Boitard et al., 2012) weeks of HFD exposure and 12-week-old rats exposed to a cafeteria diet (Ferreira et al., 2018), showed a drastic reduction of the total number of doublecortin (DCX+) cells in the SGZ of the DG. Besides, hippocampal neurogenesis appears to be affected also in genetic models of obesity such as the leptin-deficient ob/ob mice, with alteration of proliferation and differentiation processes in the SGZ (Bracke et al., 2019). Leptin receptor-deficient db/db mice, that develop diabetes in addition to obesity, also show a reduction of neurogenic capability in association to a cognitively impaired phenotype (Stranahan et al., 2008).
As regards structural changes in dendritic spines and synaptic plasticity upon metabolic disturbances, HFD exposure for instance has been associated to cause alterations either in the density or the morphology patterns of dendritic spines in the hippocampus. Hao et al. (2016) showed that animals exposed to HFD since 6 weeks of age suffered a decrease in the density of dendritic spines in dentate granule neurons in adulthood which was reversible by switching to a low fat diet. Additionally, work from our group has shown that a moderate HFD exposure since weaning, was associated with a predominance of immature dendritic spines in CA1 pyramidal neurons in young adult C57BL/6 mice, even though the total density of dendritic spines remained unchanged (Vinuesa et al., 2019). Moreover, dendritic spine reduction was also detected in pyramidal neurons of the parietal cortex in the context of streptozotocin-induced diabetes in hyperglycemic rats, together with an impaired memory performance assessed by the Morris Water Maze (MWM) (Malone et al., 2008).
Synaptic failure and loss are key factors underlying cognitive impairment in AD patients (Scheff et al., 2006, 2015). Progressive dendritic spine alterations were found to correlate with intracellular Tau deposits in postmortem analysis (Merino-Serrais et al., 2013), thereby associating functional and histological hallmarks of the disease. Several studies have also shown dendritic spine loss and synaptic dysfunction in animal models of AD, where Aβ toxicity appears to be the main contributor (Tu et al., 2014; Subramanian et al., 2020). Apart from Aβ-induced toxicity, dendritic spine and associated synaptic alterations could be associated with reduced levels of BDNF in MCI and AD patients, reinforced by the neuroprotective effects of the neurotrophin reported in experimental AD models (Peng et al., 2005; O’Bryant et al., 2009; Caccamo et al., 2010; Jiao et al., 2016). BDNF signaling through TRKB receptor and PI3K-Akt-GSK3β pathway are central regulators mediating synaptic plasticity-required dendritic spine turnover and preserving long-term potentiation (LTP) (Peineau et al., 2007; Kowianski et al., 2018). The fact that neurotrophin-induced regulation shares the main components of insulin pathway and is counteracted by proinflammatory mediators such as IL-1β (Tong et al., 2012), stresses the relevance of insulin resistance in AD pathophysiology.
The fact that the mentioned alterations of structural plasticity present in AD are also induced by metabolic pathologies would support a further exacerbation of neuroplasticity decline by interaction of age-related neurodegeneration with obesity and associated disturbances.
Given the relevance of insulin resistance and its close association with inflammation as common pathophysiological mechanisms present in metabolic and neurological disorders, in the following sections we will address the aforementioned phenomena as potential therapeutic targets for CNS dysfunction in AD-related pathology. The crosstalk between these pathways on the brain is schematized in Figure 2, showing the main actors involved and identifying with letters A-I the potential targets that will be discussed below, classified as insulin signaling-, inflammation- or lifestyle-based approaches.
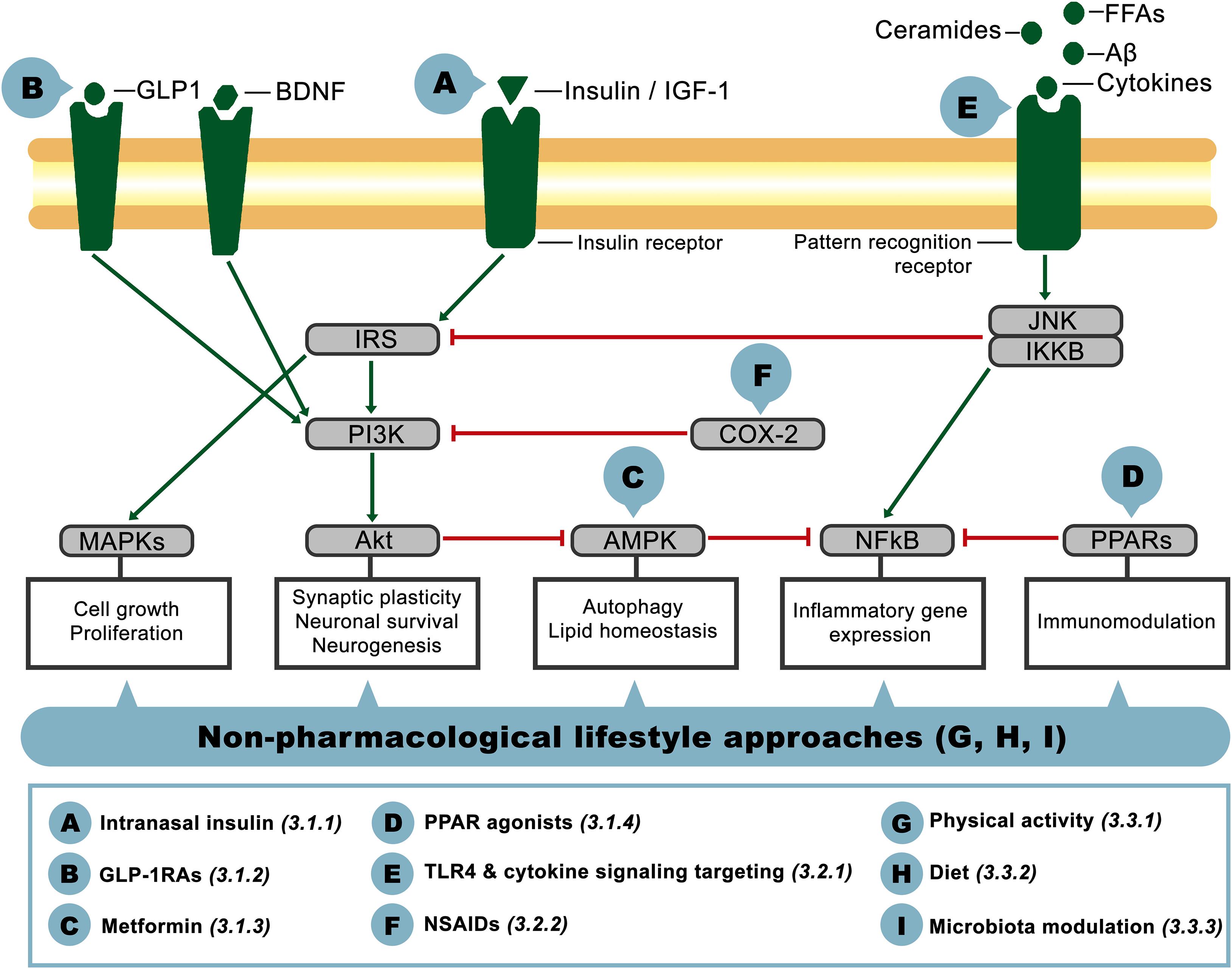
Figure 2. Schematic representation of interacting components of the insulin signaling and inflammatory pathways on the brain. Pointed-head arrows depict activation and blunt-end arrows inhibition, while letter circles A-I point the level at which the potential therapeutic approaches discussed in this article would act. Treatment definition and corresponding reference section are defined in the inferior square.
Among the existing treatments that address insulin resistance mainly used in diabetic patients, several clinical and pre-clinical studies have assessed the effect of insulin signaling-based approaches as potential mitigators of brain dysfunction and cognitive impairment.
As it was previously mentioned, the bioavailability and downstream actions of insulin in the brain can be diminished in the context of MCI, dementia and particularly in AD, supporting the concept of the disease as “Type 3 Diabetes.” The administration of insulin in the context of diabetes is a well-known treatment. Nevertheless, regarding potentially positive neurological outcomes, the systemic administration of insulin results in low transport levels to the brain and, on the other hand, may account for undesirable side effects such as hypoglycemia (Schmid et al., 2018). In that sense, intranasal (IN) insulin delivery represents a safe and non-invasive method that has gained increased interest in neurodegeneration research, both in patients and animal models. Intranasal administration of insulin has shown a rapid and widespread distribution through perineural spaces of the trigeminal nerve and the ability to activate the insulin receptor (Lochhead et al., 2019). The common doses of IN insulin range from 20 to 160 IU according to different studies, with different outcomes even though there is a growing consensus that IN administration is able to promote changes on CNS as well as peripheral pathways. There are many positive metabolic effects of IN insulin such as body weight management, food intake and fat composition, with variations according to sex, age and protocol of administration (Derkach et al., 2017). However, in this review we will focus on cognitive-related outcomes. Several clinical studies, some of which are explicated in Table 1, have shown that either IN insulin or its long-lasting analog Detemir were associated with memory improvement both in cognitively intact (Benedict et al., 2004) and impaired individuals. Memory amelioration in AD subjects was observed for different doses of IN insulin and treatment duration with APOE-ε4 allele being a strong though controversial moderating factor. Different trials assessing acute (Reger et al., 2008) or 4-month chronic IN insulin treatment (Craft et al., 2017) showed improved memory outcomes for APOE-ε4 non-carriers, albeit the latter report also exhibited minor but significant changes in APOE-ε4 carriers. Contrary to these studies, Claxton et al. (2015) only detected IN insulin positive modulation of verbal and visuospatial working memory in AD or MCI adults carrying APOE-ε4 genetic variant. Additionally, in this cohort of patients a greater degree of insulin resistance at baseline was associated with greater cognitive improvement after the treatment (Claxton et al., 2015).
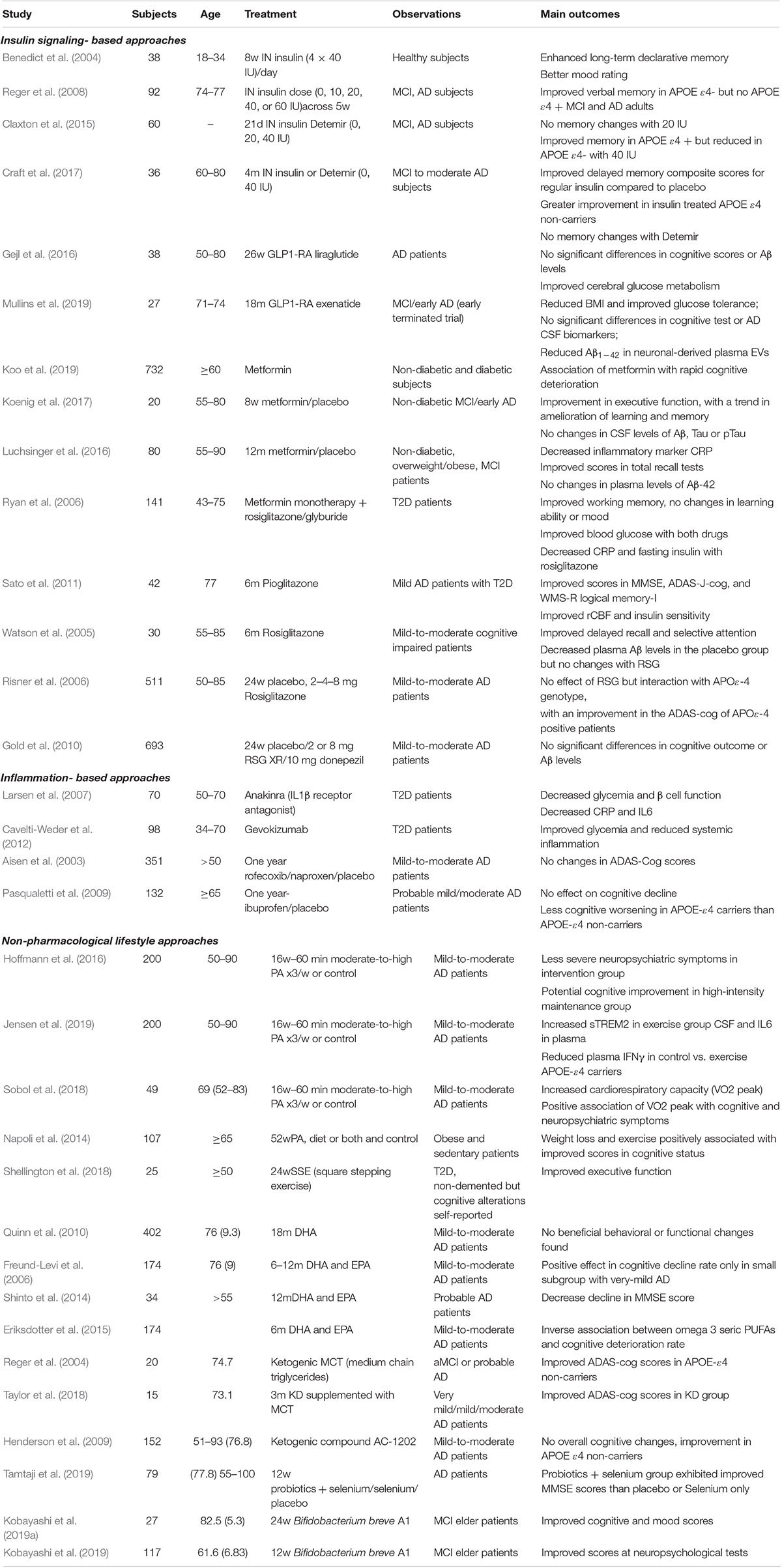
Table 1. Clinical trials targeting insulin resistance and/or inflammation for the treatment of cognitive impairment and AD.
As regards animal models, IN insulin treatment has been associated with positive results in the behavioral profile, brain metabolic pathways, neuroinflammation and plasticity of several rodent models of AD-like aging. For instance, 4- to 5-month-old APP/PS1 transgenic mice treated with IN insulin for 6 weeks presented enhanced memory performance and decreased anxiety-like behavior, along with improved histopathological signs such as decreased levels of soluble Aβ and Aβ plaques in the hippocampus and cortex, insulin-signaling rescue and enhanced hippocampal neurogenesis in relation to vehicle-treated mice (Mao et al., 2016). Moreover, IN insulin treatment has shown beneficial effects in the non-transgenic model SAMP8 AD-like mice. Twelve-month-old AD-like mice exhibited cognitive improvement assessed by spatial memory and object recognition paradigms (Salameh et al., 2015). The same insulin dose rendered hippocampal gene expression changes associated with the inflammatory response in young and aged SAMP8 mice (Rhea et al., 2019). Consistent with this, IN insulin treatment exhibited a positive cognitive, metabolic and histopathological outcome in 14-month-old HFD-induced 3xTG-AD mice (Sanguinetti et al., 2019). Furthermore, in the intracerebroventricular (i.c.v) streptozotocin-induced model, claimed by several authors to promote an AD-like phenotype, IN insulin has shown to prevent memory impairment, neuroinflammation and insulin resistance in the hippocampus and cortex (Rajasekar et al., 2017a), as well as promoting restored cerebral blood flow (CBF), decreased ROS and increased BDNF expression (Rajasekar et al., 2017b).
Glucagon-like peptide 1 is a peptide hormone mainly synthesized in intestinal L-cells and is one of the incretin hormones promoting glucose-induced insulin secretion by the pancreas after food intake, reducing the release of glucagon and also protecting the pancreas by preventing apoptosis of β-cells (Farilla et al., 2003; de Heer et al., 2008). Apart from these effects, it has been shown that this hormone plays an important role in different organs. For instance, GLP-1 receptors are also present in the liver, heart and brain. GLP-1 function has been associated to the prevention of cardiovascular diseases (Bethel et al., 2018) and constitutes a key component of the gut-brain axis, behaving as a relevant neuropeptide, not only for the regulation of food intake and glucose homeostasis but as a neuroprotective factor in relation to synaptic function and plasticity (Gault and Holscher, 2008). As regards T2D and obesity treatment, there are currently approved GLP-1 RAs such as exenatide, liraglutide and lixisenatide which are resistant to the enzymatic cleavage of dipeptidyl peptidase-4 (DPP-4), thus accounting for an increased half-life in comparison to endogenous GLP-1, that have shown positive management of the disease (Aroda, 2018). Given the pleiotropic character of GLP-1 R and in particular its role in neuromodulation and associated cognitive-related outcome, the use of GLP-1 RAs has been proposed as a potential therapy for neurodegenerative diseases (Gejl et al., 2016). Interestingly, Parkinson’s disease patients that participated in a randomized controlled trial (RCT) for exenatide exhibited improved motor and cognitive scores 12 months after the trial endpoint (Aviles-Olmos et al., 2014).
Although there is an important research gap regarding clinical evidence in AD given the limitation of sample size or unfinished trials (Mullins et al., 2019), as it is shown in Table 1 for the GLP-1RAs exenatide and liraglutide, there is very promising data drawn mainly from preclinical animal models of AD.
In that sense, Cai et al. (2018) showed that the GLP-1 RA lixisenatide treatment for 60 days in 12-month-old APP/PS1/Tau AD female mice promoted a decreased Aβ plaque load and p-Tau levels in the hippocampus, along with a decreased neuroinflammatory status. In a previous work, the same group also showed the ability of lixisenatide to ameliorate memory performance assessed by the MWM in intrahippocampal Aβ- injected rats (Cai et al., 2014). Regarding exenatide and cognitive outcome in AD rodent models, its use has been associated to memory improvement in i.c.v streptozotocin (STZ)-induced rats when treated during 2 weeks (Solmaz et al., 2015) and Bomba et al. (2013) showed that 3-month-old PS1-KI mice treated for 9 months with the GLP-1RA had improved performance in MWM while the 3xTg-AD assessed within the same protocol, did not show cognitive amelioration. Despite the lack of cognitive improvement in 3xTg-AD mice, the authors later showed that exenatide promoted an enhanced BDNF signaling and reduced inflammation in HFD-induced 3xTg-AD mice (Bomba et al., 2019). Moreover, Batista et al. (2018) have recently reported neuroprotective features of liraglutide not only in mice i.c.v-injected with Aβ oligomers (AβO) but also in non-human primates (NHP). Interestingly, the GLP-1 RA was able to revert AβO-induced cognitive impairment as well as to prevent insulin receptor loss in mice hippocampus, the latter also being found in NHP, along with synapse loss prevention and decreased p-Tau levels (Batista et al., 2018). Finally, a dual therapy targeting both GLP-1 and GIP incretin receptors, proposed as improved candidates for T2D treatment, has shown to ameliorate memory impairment, Aβ deposition, glial reactivity and expression of synaptic marker PSD95 in 10-month-old APP/PS1 mice, potentially due to the rescue of proteostatic functions associated to the enhancement of autophagy and decreased ERS response (Panagaki et al., 2018).
Metformin constitutes one of the most common treatments for T2D. It is an oral biguanide and anti-hyperglycemic drug that acts as an insulin sensitizer, lowering blood glucose by increasing its uptake and reducing hepatic gluconeogenesis (Zhou et al., 2001). Although metformin’s mechanism of action is not thoroughly understood, it has been associated with the activation of Ampk. The use of metformin in diabetic patients has been associated not only with blood glucose control but also with an improved lipidic profile and decreased inflammatory markers (Lin et al., 2018). Concerning the impact of metformin in cognitive decline, either induced in the context of diabetes or among age-related AD patients, there is promising evidence albeit contradictory data should be also taken into account. In Table 1, we can find a representative selection of clinical studies addressing brain-related outcomes of metformin treatment in aged patients with/without diabetic medical history. For instance, in a pilot study, Koenig et al. (2017) showed that metformin exposure in non-diabetic patients with MCI or early signs of AD was associated with an ameliorated cognitive function, even though no changes in AD CSF biomarkers such as Aβ, Tau or p-Tau levels were observed. Moreover, in non-diabetic patients with amnestic MCI co-occurring with overweight or obesity, metformin appeared to ameliorate systemic inflammation lowering CRP levels and a partial improvement in recall memory (Luchsinger et al., 2016). However, in a prospective cohort report with a 1–7.6 years range follow-up, comparing older non-diabetic patients and diabetic patients treated or not with metformin, the exposure to the biguanide drug was not associated with changes in Ad assessment as shown by the Korean version of the Consortium to Establish a Registry for Alzheimer’s Disease Assessment Packet (Cerad-K), the short geriatric depression scale (SGDS) or activities of daily living score (Barthel ADL index). In fact, when analyzing annual “rapid deterioration” among diabetic patients, the authors found metformin to be associated with a more marked deterioration (Koo et al., 2019). Nonetheless, more longitudinal and larger cohort studies should be assessed since there are confounding factors such as other antidiabetic treatments and lifestyle factors (diet and exercise), that may account for the variability and conclusions of such complex studies.
Concerning experimental assessment of metformin in animal models, the use of the anti-hyperglycemic drug has shown to rescue hippocampal plasticity alterations and associated cognitive outcomes in both metabolic dysfunction-induced and AD-like paradigms. As an example, oral administration of metformin in middle-aged C57BL/6J mice was able to rescue spatial memory impairment, adult neurogenesis in the DG, presumably by the enhanced signaling of IR/IRS1 and AMPK/PKC detected in the hippocampus of metformin chronic exposure (Tanokashira et al., 2018). In the same line, db/db mice exposed to metformin exhibited an improved spatial memory assessed by the MWM and a lesser degree of hippocampal atrophy (Chen et al., 2019). In the context of experimental AD, 26-week-old APP/PS1 female mice were treated during 14 days with i.p metformin and several pathophysiological parameters were changed. Along with hippocampus-dependent memory improvement, metformin exposure in APP/PS1 mice was associated with an increased neurogenic ability, reduced Aβ soluble levels and deposits as well as an amelioration of the inflammatory status in hippocampus and cortex, in relation to vehicle-treated APP/PS1 mice. As regards the potential mechanisms involved, the authors showed the relevance of the reported induction of AMPK/mTOR/PKC pathway since at least the inflammatory response and soluble Aβ levels decreased by metformin in the hippocampus and cortex of APP/PS1 mice was impaired in the presence of AMPK inhibitor (Ou et al., 2018). Interestingly, in a sporadic AD model, 12-month-old SAMP8 mice treated with metformin for 8 weeks, also exhibited an ameliorated behavioral profile with improved learning and memory parameters together with decreased levels of Aβ and p-Tau, even though pGSK3β was found to be slightly increased (Farr et al., 2019).
Among insulin sensitizing drugs, another relevant category is represented by thiazolidinediones such as rosiglitazone (RSG) and pioglitazone (PIO). Such compounds, commonly used in the management of T2D, are agonists/ligands of PPARγ receptor, a master regulator of adipose tissue differentiation. As regards clinical evidence of pharmacological ligands of PPARγ, several reports addressed the impact of glitazones on the cognitive status of patients with diabetes and/or MCI-AD (Table 1). Ryan et al. (2006) analyzed diabetic patients with metformin monotherapy that were exposed during 24 weeks to RSG or glyburide treatment. In the metabolic sphere, both drugs were able to improve blood glucose and fasting insulin levels and C-peptide were only reduced in the rosiglitazone group. Both drugs were associated with improved working memory, even though no differences were registered in learning ability or mood assessment (Ryan et al., 2006). Concerning T2D patients co-occurring with mild AD, antidiabetic add-on therapy with PIO for 6 months enhanced insulin sensitivity, improved patients performance in the following cognitive tests: Mini- Mental State Examination (MMSE), Alzheimer’s Disease Assessment Scale- Cognitive Subscale Japanese version (ADAS-J-Cog), and Wechsler Memory Scale-revised (WMS-R) logical memory-I and regional CBF in the parietal lobe (Sato et al., 2011). The effect of RSG in AD patients is rather controversial yet. Although certain preliminary or explorative analyses showed an association with cognitive improvement (Watson et al., 2005) or an interaction of the treatment with APOE-ε4 genotype (Risner et al., 2006), this was not further supported by Gold et al. (2010) in their large clinical study and future clinical evidence is needed.
In relation to experimental data from animal models and potential mechanisms involved, PPAR agonists have shown positive effects in AD pathophysiology and cognitive status. For instance, the treatment of 3xTG-AD female mice with PIO was associated with several beneficial effects such as learning improvement, decreased Aβ and Tau deposits in the hippocampus and enhanced hippocampal plasticity shown by increased LTP, along with decreased plasma cholesterol levels (Searcy et al., 2012). Neuroprotective outcomes in AD models were also found by combining different PPAR agonists. For example, the use of PPAR-δ and PPAR-γ agonists L165,041 and F-L-Leu, respectively, has shown positive changes in brain slices from i.c.v STZ injected rats: decreased inflammation, improved mitochondrial function and reduced Aβ neurotoxicity, although lipid peroxidation and cholinergic function were not rescued (Reich et al., 2018), suggesting that PPAR ligand therapies should be thought as complementary treatments in AD-like pathology. In the same line, a recent report showed that targeting PPAR-α with fenofibrate and PPAR-γ with PIO during 21 days ameliorated memory impairment induced by Aβ1–40 i.c.v injection. Additionally, authors found the Wnt/β catenin to be enhanced with the treatment, proposing this signaling pathway relevant for neuron plasticity and synaptic remodeling altered in AD and mood disorders to be involved (Assaf et al., 2020).
As mentioned in previous sections, inflammation is a crucial phenomenon in the development of insulin resistance-related pathologies and targeting different components of the inflammatory response has been a promising yet challenging approach in chronic metabolic pathologies such as obesity and diabetes. Furthermore, taking into consideration that inflammation is a common factor occurring also in a wide variety of neuropsychiatric pathologies, and with microglia being a fundamental element in such response, many authors have undertaken experimental animal approaches to inhibit inflammation and ultimately microglial cells with the aim to reverse cognitive decline. In the present section, we will analyze available inflammation-based therapies used in diabetes or obesity and the impact as potential treatment for AD.
As it was previously described, inflammatory mediators are intimately related to the promotion and propagation of metabolic-induced damage in insulin-resistant contexts both in the periphery and the CNS. Even though anti-inflammatory therapies are not primary treatments for diabetes, obesity or AD, there is interesting clinical and preclinical evidence that may as well contribute to strengthen the role of inflammation and enable the design of future strategies.
As regards TLR4 direct targeting, neurological available data is scarce and comes mostly from animal models possibly due to its relevance in innate immunity and potential secondary effects. Moser et al. (2018) showed that in pathologic contexts such as diet-induced obesity, pharmacological microglia TLR4 inhibition with TAK-242 prevented adverse neurological outcomes by decreasing microgliosis, enhancing neurogenesis and potentially modifying amyloidogenic pathways. Consistent with this, the neutralization of HMGB1, a TLR4 ligand has shown to reverse cognitive decline and brain pathology in the 5xFAD mouse model. The subcutaneous administration of HMGB1 monoclonal antibody was able to recover dendritic spine density and decrease DNA damage in 6-month-old 5xFAD cortex (Fujita et al., 2016). In the same line, inhibiting obesity-induced microglia activation has shown to ameliorate cognitive decline in two different approaches: pharmacologically with the use of minocycline and genetically by the downregulation of microglial fractalkine receptor (Cope et al., 2018). Accordingly, in a very recent report, Melo et al. (2020) showed that targeting microglia inflammation induced by the exposure to the saturated fatty acid palmitate prevented both the defective insulin signaling in hippocampal cultures and also in rodents, in addition to amelioration of the cognitively impaired phenotype. This was achieved by the use of two different approaches: minocycline or TNFα- neutralizing antibody therapy with infliximab. The latter also prevented memory impairment in mice exposed to chronic HFD feeding (Melo et al., 2020), reinforcing the role of pro-inflammatory cytokines as mediators of metabolic-induced impairment of hippocampal flexibility, as it will be further discussed.
In relation to TLR4 downstream signaling, there is an increasing body of literature addressing NFκB induced-cytokines targeting. One of the most studied is IL-1β due to its close association with metabolic inflammation and insulin resistance. IL-1β receptor antagonists (IL-1β RA) have been used in clinical trials focused both on diabetic and AD-like scenarios as it can be seen in Table 1. For instance, T2D adult patients treated with the IL-1β RA anakinra presented ameliorated glucose levels and pancreatic function with decreased systemic inflammation markers such as CRP and IL6 (Larsen et al., 2007) and similar results were observed with the use of IL-1β neutralizing antibody Gevokizumab (Cavelti-Weder et al., 2012). As regards the impact of cytokine inhibition in cognitive decline and AD pathology, evidence comes mostly from animal models. APP/PS1 mice lacking either NLRP3 or caspase 1, which are key components for the maturation and secretion of IL-1β, showed improved spatial memory, synaptic plasticity and decreased Aβ levels (Heneka et al., 2013), suggesting a relevant role of the pro-inflammatory cytokine in the progression of AD pathology. In the same line, LTP analysis in a rat model of amyloidosis (McGill-RThy1- APP TG rat) showed improved synaptic plasticity when animals were treated with NLRP3 inhibitor Mcc950, IL-1β RA anakinra or anti- TNF-α (etanercept) (Qi et al., 2018). TNF-α is also one of the most relevant pro-inflammatory cytokines and its neutralization has been associated with improved cognition and synaptic flexibility in different AD models. XPro 1595 anti- TNF-α antibody rescued LTP, Aβ deposition and inflammatory response in 5xFAD (MacPherson et al., 2017) and the neutralization TNFSF10 pro-apoptotic cytokine belonging to TNF family was also able to improve spatial memory, decrease Aβ levels and inflammation in the hippocampus of 3xTG-AD mice (Cantarella et al., 2015). Interestingly, XPro 1595 anti- TNF-α antibody also exhibited interesting results in a diet-induced obesity paradigm by exposing C57BL/6 mice to a high-fat high-carbohydrate diet. Authors have recently found not only an improvement of metabolic parameters associated to dyslipidemia and insulin resistance in the periphery but also central insulin signaling was improved in the hypothalamus and prefrontal cortex, together with behavioral changes in anxiety-like behavior (De Sousa Rodrigues et al., 2019), reinforcing the potential of anti-inflammatory strategies in metabolic- and brain-dysfunction contexts.
Non-steroidal anti-inflammatory drugs (NSAIDs) constitute a well-known strategy for targeting both acute and chronic inflammation in diverse contexts. There are different types of compounds in this category with primarily anti-inflammatory and analgesic consequences, associated with the inhibition of cyclooxygenase (COX) activity and downstream prostaglandin synthesis (Rao and Knaus, 2008). Given the relevance of the inflammatory response in the pathophysiology and progression of chronic metabolic diseases such as obesity and T2D as well as in the context of AD-like dementia, there has been a genuine interest in preclinical and clinical exploration of NSAIDs as potential therapies. However, there is controversial or insufficient data as regards their efficacy. In relation to AD risk and progression, there is observational data suggesting a potential benefit of NSAIDs usage in the prevention of the disease (Etminan et al., 2003) and small clinical trials providing evidence in the same line (Rogers et al., 1993), albeit further RCTs should be conducted. For instance, in a quite large multicenter RCT, Aisen et al. (2003) showed that the selective Cox-2 inhibitor rofecoxib or traditional NSAID naproxen failed to ameliorate cognitive decline after 12-month exposure to the drugs in mild-to-moderate AD patients. Additionally, drug-exposed groups reported adverse effects such as dizziness, hypertension and fatigue more frequently than placebo (Aisen et al., 2003). Consistent with this work, 12-month exposure to ibuprofen was also unable to slow time-dependent cognitive decline, even though an interestingly favorable different outcome was seen for APOE-ε4 carriers in comparison with non-carriers (Pasqualetti et al., 2009). In the same line, the large longitudinal trial assessing naproxen or celecoxib NSAIDs contributed to the “non-beneficial” body of evidence of these drugs for improvement of AD cognitive impairment (Group et al., 2008).
Despite the predominance of disappointing clinic results, animal models of AD have shown positive effects of NSAIDs in the progression of the disease. For example, ibuprofen treatment was associated to decreased Aβ deposition, microglial activation and inflammatory cytokine levels in the APP Tg2576 AD mice model, together with improved signs of degeneration shown by decreased dystrophic neurites (Lim et al., 2000; Yan et al., 2003). Likewise, decreased microglial activation and Aβ deposition was observed in 12-month-old APP/PS1 transgenic mice treated with the NCX-2216 NSAID agent (Jantzen et al., 2002). Moreover, and in relation to diabetes and cognitive impairment, Wang et al. (2011) showed that aspirin treatment during 4 and 8 weeks promoted decreased levels of pro-inflammatory cytokines in the hippocampus and improved spatial memory in STZ-induced diabetic mice.
As mentioned, cholesterol and its oxidized forms called oxysterols are associated with increased risk for AD, promotion of amyloid pathology and induced inflammation. Oxysterols are ligands for liver X receptors (LXRs), nuclear receptors that function as lipid-activated transcription factors and regulate cholesterol homeostasis (Wang and Tontonoz, 2018). Besides their role on lipid metabolism, LXRs can regulate inflammation and neuroinflammation. Evidence from in vitro models showed that LXR agonists inhibit the production of pro-inflammatory mediators from microglia and astroglia at least in part through interfering with the capacity of NFκB to bind DNA and induce transcription (Zhang-Gandhi and Drew, 2007). Also LXR agonists can interfere with Aβ production and decrease soluble amyloid peptides in the brain of the APP23 mouse model of AD (Koldamova et al., 2005). Some drug candidates have been proposed for AD treatment but due to the mixed nature of LXRs effects and adverse effects such as dyslipidemia and hepatic steatosis (Grefhorst et al., 2002; Tai et al., 2014), clinical translation of these strategies has been unsuccessful. Novel agonists overcoming these effects can have potential as AD therapies.
A more direct approach has been proposed using statins and cholesterol lowering therapies. First known for their protective effects on cardiovascular disease, statins were then proposed as risk-reducing therapies for AD, with many studies describing protection against the risk of dementia and AD but failing when tested in controlled randomized trials, probably due to the complexity of AD, to the variety of lipids involved (HDL cholesterol, LDL-cholesterol, triglycerides) and the separate cholesterol pools (circulation vs. brain) that coexist (reviewed in Reitz, 2013; Samant and Gupta, 2021). Another strategy aims at enhancing CYP46A1 activity, the enzyme that converts cholesterol to 24S-hydroxycholesterol (24S-OHC), the major oxysterol in the brain. This oxysterol, besides its effects on cholesterol metabolism and function as LXR agonist, is an α-secretase activator that hence decreases the rate of Aβ production and plaque formation (Bjorkhem et al., 2009). Efavirenz, L-glutamine and dapagliflozin are some of the CYP46A1 activators that showed promising results in experimental models and are being tested in clinical trials (Mast et al., 2017; van der Kant et al., 2019; Samant and Gupta, 2021).
As it can be appreciated in the previous sections, on a general basis insulin function-based therapies appear to downregulate inflammation and inflammation-based therapies seem to improve insulin sensitivity, supporting the notion of inflammation and insulin resistance as bidirectionally affecting phenomena. In that line, in the present section we will discuss non-pharmacological lifestyle approaches mainly associated to diet and physical activity interventions in aged adults either with obesity comorbidity or age-related cognitive impairment. Interestingly, such approaches can be potentially proposed not only as treatments but also as relevant preventive strategies in order to promote a healthier aging and improved quality of life.
Among lifestyle modifiable risk factors associated with AD, great attention has been devoted to physical activity since sedentarism is a crucial risk factor for obesity, cardiovascular disease and is also associated with impaired cognitive function. Sustained physical activity has shown to ameliorate overall health and particularly maintain cognitive function in disease-free elders even when taken up at late stages (Elosua et al., 2005; Hamer et al., 2014). Moreover, non-demented patients with physical and cognitive frailty exhibited positive results regarding both physical parameters such as muscle strength and speed and also in cognitive flexibility when exposed to a 4 months high-speed resistance exercise training (Yoon et al., 2018). Given the lack of successful therapies for age-related dementia, and in particular due to AD, several clinical trials have addressed exercise-based interventions as a potential treatment of AD symptoms, some of which are cited in Table 1. For instance, a RCT assessing the impact of high-to-moderate exercise in patients diagnosed with probable mild or moderate AD has shown to improve neuropsychiatric symptoms and cognitive status after 16 weeks of continuous attendance to the program (Hoffmann et al., 2016). From the same trial, Jensen et al. (2019) recently reported the effect of the intervention on inflammatory biomarkers. Although they did not find many significant changes, the exercise group presented increased CSF levels of sTREM2 and IL-6 in plasma and, interestingly, APOE-ε4 non-carriers had reduced levels of IFNγ than APOE-ε4 carriers after exercise intervention (Jensen et al., 2019). A subgroup of the patients enrolled in the trial were subjected to cardiorespiratory fitness assessment and an improvement was found in the intervention group, together with a positive association with mood and cognitive outcomes (Sobol et al., 2018). In close association with the physical activity approach, dance therapy constitutes a non-pharmacological alternative that has exhibited positive outcomes in the amelioration of several symptoms related to AD pathology. Exposure to different types of dance sessions has shown to promote improvement in diverse domains including physical well-being, mood and cognitive status, irrespective of the practice duration (Lazarou et al., 2017; Marquez et al., 2017). However, it should be noted that this kind of intervention not only entails moderate exercise but also social interaction. Therefore, the improvement assessed would be the result of mixed stimuli resembling an enriched sensory-motor environment, as proposed by Kattenstroth et al. (2010).
In reference to the interaction with obesity or insulin resistance in the older population, lifestyle interventions also showed to ameliorate cognitive function. Even though the impact of weight loss in the elderly is controversial, in the case of obese older patients, weight loss associated to diet and exercise has shown positive outcomes in several measures assessing global cognition. In fact, the combination of both interventions appeared to be better, although for several parameters the combination of diet and exercise was no different than exercise alone (Napoli et al., 2014). Additionally, albeit being a rather small sample-sized study, a light exercise intervention promoted mild improvement in executive functioning in non-demented T2D patients with self-reported cognitive impairment (Shellington et al., 2018).
Regarding experimental models, there is an extensive body of literature addressing the impact of exercise paradigms on brain function (Ryan and Kelly, 2016). Exercise has elicited positive effects on cognition, brain metabolism and plasticity. For instance, 6-month-old APP/PS1 mice subjected to regular exercise during 4 weeks exhibited improved learning and spatial memory together with decreased levels of Aβ and p-Tau in the cortex and hippocampus. Additionally, it promoted enhanced mitochondrial function and increased synaptic density (Pang et al., 2019). Likewise, it was recently reported that hippocampus-dependent memory and synaptic puncta were positively modulated in 12-month-old mice of the same strain subjected to running during 4 months (Zhang et al., 2020). Furthermore, running in the 5xFAD mice promoted beneficial effects in spatial memory and Aβ burden along with increased BDNF levels and enhanced adult neurogenesis. Interestingly enough, these effects were partially mimicked by the combination of adult neurogenesis enhancement via NPCs’ pharmacologically induced survival and BDNF overexpression in the hippocampus. These effects were not detected when neurogenesis alone was stimulated, suggesting a crucial role for BDNF in the exercise-induced response (Choi et al., 2018). Moreover, exercise has also shown to ameliorate hippocampus-dependent behavior, together with inflammation and oxidative stress in the “sporadic” rat model of AD induced with SZT (Lu et al., 2017). Finally, exercise has also promoted pro-cognitive, pro-neuroplastic and anti-inflammatory effects in metabolically challenged contexts such as diabetes or diet-induced obesity (Ruegsegger et al., 2019; Wang et al., 2019), becoming a quite strongly recommendable lifestyle approach not only to prevent the development of the mentioned chronic disorders but also being potential therapies in the amelioration of life quality by targeting inflammation and insulin resistance derangements.
In the same sense that highly processed fat and sugar-enriched western diets negatively affect metabolism, inflammatory status and cognitive function, modulating dietary composition can be a powerful strategy to positively modify the aforementioned health domains. For instance, Mediterranean diet (MD) has been associated with general wellbeing and preserved cognitive function in the elderly (McGrattan et al., 2019). MD is predominantly constituted by fruits, vegetables, wholegrains and legumes, fish and poultry to a lesser extent and a very low intake of red meats and processed food where olive oil is the main source of fat, thereby associated with antioxidant and anti-inflammatory components. Although MD has exhibited an important adherence among commonly proposed diets, the use of certain supplements addressing some of its components has also been related to beneficial effects. As an example, lycopene is a potent antioxidant carotenoid present in fruits and vegetables that has shown protective effects in several chronic diseases such as cancer, cardiovascular disease and neurodegeneration (Saini et al., 2020). As regards dietary interventions, CNS impact of lipid metabolism-based supplements was given attention. Not only approaches to decrease cholesterol levels were studied (Chu et al., 2018) but also administration of fish oil or similar supplements containing omega3 polyunsaturated fatty acids (PUFAs) for their potentiality to maintain cognitive function. The most commonly used PUFAs are docosahexaenoic acid (DHA), eicosapentaenoic acid (EPA), and alpha-linolenic acid (ALA), which are reported as relevant precursors of neuronal membrane components, fluidity and signaling (Chappus-McCendie et al., 2019). As it can be appreciated in Table 1, several clinical trials analyzed the impact of DHA and EPA in patients with mild-to-moderate AD and mixed results were obtained. Quinn et al. (2010) were not able to find significant effects of DHA in a quite large RCT. Nevertheless, when administration of DHA was combined with EPA, other clinical studies found either a subtle cognitive improvement in a subgroup of very-mild AD (Freund-Levi et al., 2006) or a decrease of cognitive decline rates in mild-to-moderate AD patients (Shinto et al., 2014; Eriksdotter et al., 2015).
Among dietary interventions, fasting, calorie restriction and ketogenic diets (KD) have also been widely proposed to reverse obesity and T2D, being able not only to promote weight loss but also to enhance insulin sensitivity and reduce inflammation (Harvie et al., 2011; Myette-Cote et al., 2018). In the present section, we will discuss the impact of these particular dietary approaches on brain plasticity and pathological hallmarks as potential therapies for AD-like neurodegeneration. Fasting can be defined as a period of food deprivation. It has been associated with different socio-cultural factors from religion to periods of food scarcity and it was later related to increased lifespan in several species (Brandhorst et al., 2015). There are different fasting protocols described in the literature, mainly due to length and frequency variations. Essentially, intermittent or periodic fasting and dietary restriction are very similar approaches since the main phenomenon is the time restriction of food intake. At the level of the metabolic response and in a similar fashion as ketogenic diets, these dietary interventions lead to an increase of ketone bodies, producing relevant changes in glucose metabolism (Wilhelmi de Toledo et al., 2019). Ketogenic diet is mainly featured by a very high-fat and low-carbohydrate intake and can be difficult to sustain, therefore the increase of ketone bodies can also be achieved by administering medium chain triglycerides (MCT) without changing the diet (Reger et al., 2004; Henderson et al., 2009; Taylor et al., 2018). In Table 1, we can find a selection of relevant clinical studies addressing their impact on cognitive function.
As regards experimental studies, KD, intermittent fasting (IF) or dietary restriction (DR) have shown to promote improved glucose metabolism together with anti-inflammatory and cognitive amelioration in animal models of AD. For instance, KD exposed 3xTg-AD mice exhibited enhanced memory and learning ability in addition to decreased hippocampal Aβ deposition (Kashiwaya et al., 2013), while APP/V7/171 transgenic female mice also showed decreased levels of Aβ along with positive metabolic changes in response to KD, though the behavioral alterations were not significantly improved (Van der Auwera et al., 2005). As for intermittent fasting, it was recently shown that App NL-G-F mice maintained in an IF protocol alternating ad libitum and fasting days for a month, exhibited an adaptive response to acute fasting with improved memory and anxiety-related outcomes. The behavioral profile amelioration occurred concomitantly with synaptic remodeling dependent of SIRT3 mitochondrial deacetylase (Liu et al., 2019), suggesting that IF can modulate oxidative stress, thereby constituting a potential approach to mitigate AD-related cognitive impairment and synaptic dysfunction. In the same line, our group and others have shown that DR represents a moderate and feasible schedule capable of improving cognitive function in aged wild type (Ma et al., 2018) and PDAPP-J20 transgenic mice (Gregosa et al., 2019). In the latter, we found that three cycles of DR alternated with ad libitum feeding between 6.5 and 8 months of age not only was able to improve spatial memory but was also associated with enhanced neurogenesis and decreased Aβ load in the hippocampus. Moreover, periodic DR prevented microglial activation and promoted a decrease in the levels of hippocampal IL-1β, thus counteracting the neuroinflammatory status.
The inflammatory status in the mentioned structures of the CNS present in the context of metabolic diseases has been associated with a low-grade chronic inflammation response both detected and potentially triggered in the periphery. One of the possible mechanisms corresponds to alterations of the gut microbiota affecting the microbiome-gut-brain axis. Obesity-related disorders are often reported to co-occur with dysbiosis-associated inflammation and interventions with probiotics and related bioactive compounds have been able to reverse inflammatory parameters and ameliorate cognitive decline and hippocampal plasticity alterations, such as LTP and synaptic activity (Solas et al., 2017). Gut microbiome diversity alterations have been associated with the AD-like pathological context, not only in relation to the gut but also BBB dysfunction (Vogt et al., 2017). Therefore, probiotic supplementation has been proposed as a potentially beneficial strategy to improve AD symptoms by modulating microbiota and ultimately the gut-brain axis. Probiotics are living microorganisms that have shown to elicit beneficial effects in the host, preventing infections, obesity-related alterations as well as promoting CNS positive modulation (Hutchinson et al., 2020). As it can be appreciated in the studies cited in Table 1, different probiotic formulations have shown to ameliorate cognitive function in patients with MCI or AD. For instance, Tamtaji et al. (2019) showed that the administration of a probiotic therapy containing L. acidophilus, B. bifidum, and Bifidobacterium longum co-supplemented with selenium, present in several beneficial probiotic formulations, was able to promote improved cognitive performance as well as an ameliorated metabolic profile. Peripheral inflammation decreased together with enhanced insulin sensitivity and an ameliorated lipidemic profile (Tamtaji et al., 2019). Consistently with this data, there is also experimental evidence of positive outcomes in 3x-Tg mice that were given selenium-enriched yeast for 3 months. Interestingly, spatial memory was improved concomitantly with reduced neuroinflammation, GSK3β-dependent Tau phosphorylation and increased expression of synaptic proteins in the cortex and hippocampus (Zhang et al., 2017). In the same line, in a pilot study and a larger RCT (Kobayashi et al., 2019a,b), analyzed the impact of a 6- or 3-month probiotic supplementation with Bifidobacterium breve A1 in aged patients with MCI, respectively. In both cases, administration of the probiotics resulted in improved scores at neuropsychological tests assessing cognitive and mood status. Likewise, Bifidobacterium breve A1 administration to mice that were i.c.v- injected with Aβ elicited an anti-inflammatory response, along with an improvement of cognitive decline (Kobayashi et al., 2017). Taken together, this experimental and clinical data suggests that probiotic supplementation may constitute a beneficial lifestyle approach to enrich and reshape gut microbiome, thereby exerting an anti-inflammatory and anti-neurodegenerative response to AD pathological context.
The alarming prevalence of both AD and obesity- related metabolic disorders and the consequent implications in public health strongly require the rise of efficient therapeutic solutions. The fact that metabolic alterations are risk factors for the development of age-related dementia not only reinforces the relevance of the problem but also provides useful insight as regards potential research fields to further explore. Therefore, this review intended to highlight the shared pathways at the interface of these pathologies as potential targets to override AD burden.
Insulin resistance and chronic inflammation are two synergic phenomena underlying the pathogenesis of metabolic diseases as well as the development of the wide variety of associated disorders, including AD-like neurodegeneration. Therapeutic approaches targeting these interacting factors could be proposed as potential AD treatments even though data is variable and there is an important gap between experimental evidence from animal models and replication in clinical trials. Several factors should be taken into account. Firstly, the specific origin or etiology of the diseased context might differ considerably, with a variable substrate to address mainly according to age but also as regards pre-existing or co-occurring morbidities, namely developmental traits, obesity, diabetes and genetic predisposition. Secondly, and in association with the lack of etiology certainty in AD patients, the proposed therapies may be targeting either causes or mere symptoms, with an additional factor: the targeted pathways redundancy and compensatory mechanisms. In that sense, lifestyle approaches may tackle different pathological components in a more extensive/holistic manner, serving also as powerful preventive strategies.
AV, CP, AG, MnB, JP, MsB, FS, and JB discussed, wrote and reviewed this manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by Williams, René Barón and Alberto J. Roemmers Foundations, ANPCyT PICT Grants 2013-2645, 2014-1168, 2016-1046, and 2016-1572, CONICET PIP Grant 2013-2015, and UBACyT 2018.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
AV and CP are postdoctoral CONICET fellows, MsB is Ph.D. CONICET fellow, AG and JP are Ph.D. ANPCyT fellows, FS and JB are CONICET researchers.
Aisen, P. S., Schafer, K. A., Grundman, M., Pfeiffer, E., Sano, M., Davis, K. L., et al. (2003). Effects of rofecoxib or naproxen vs placebo on Alzheimer disease progression: a randomized controlled trial. JAMA 289, 2819–2826. doi: 10.1001/jama.289.21.2819
Aleffi, S., Petrai, I., Bertolani, C., Parola, M., Colombatto, S., Novo, E., et al. (2005). Upregulation of proinflammatory and proangiogenic cytokines by leptin in human hepatic stellate cells. Hepatology 42, 1339–1348. doi: 10.1002/hep.20965
Alzheimer’s-Association. (2020). 2020 Alzheimer’s disease facts and figures. Alzheimer’s and Dementia. The Journal of the Alzheimer’s Association.
Arnold, S. E., Arvanitakis, Z., Macauley-Rambach, S. L., Koenig, A. M., Wang, H. Y., Ahima, R. S., et al. (2018). Brain insulin resistance in type 2 diabetes and Alzheimer disease: concepts and conundrums. Nat Rev Neurol 14, 168–181. doi: 10.1038/nrneurol.2017.185
Aroda, V. R. (2018). A review of GLP-1 receptor agonists: Evolution and advancement, through the lens of randomised controlled trials. Diabetes. Obes. Metab 20(Suppl. 1), 22–33. doi: 10.1111/dom.13162
Assaf, N., El-Shamarka, M. E., Salem, N. A., Khadrawy, Y. A., and El Sayed, N. S. (2020). Neuroprotective effect of PPAR alpha and gamma agonists in a mouse model of amyloidogenesis through modulation of the Wnt/beta catenin pathway via targeting alpha- and beta-secretases. Prog Neuropsychopharmacol Biol Psychiatry 97, 109793. doi: 10.1016/j.pnpbp.2019.109793
Assuncao, N., Sudo, F. K., Drummond, C., De Felice, F. G., and Mattos, P. (2018). Metabolic Syndrome and cognitive decline in the elderly: A systematic review. PLoS ONE 13, e0194990. doi: 10.1371/journal.pone.0194990
Aviles-Olmos, I., Dickson, J., Kefalopoulou, Z., Djamshidian, A., Kahan, J., Ell, P., et al. (2014). Motor and cognitive advantages persist 12 months after exenatide exposure in Parkinson’s disease. J Parkinsons Dis 4, 337–344. doi: 10.3233/jpd-140364
Avinun, R., and Hariri, A. R. (2019). A polygenic score for body mass index is associated with depressive symptoms via early life stress: Evidence for gene-environment correlation. J. Psychiatr. Res. 118, 9–13. doi: 10.1016/j.jpsychires.2019.08.008
Baker, L. D., Cross, D. J., Minoshima, S., Belongia, D., Watson, G. S., and Craft, S. (2011). Insulin resistance and Alzheimer-like reductions in regional cerebral glucose metabolism for cognitively normal adults with prediabetes or early type 2 diabetes. Arch. Neurol. 68, 51–57.
Batista, A. F., Forny-Germano, L., Clarke, J. R., Lyra, E. S. N. M., Brito-Moreira, J., Boehnke, S. E., et al. (2018). The diabetes drug liraglutide reverses cognitive impairment in mice and attenuates insulin receptor and synaptic pathology in a non-human primate model of Alzheimer’s disease. J. Pathol. 245, 85–100. doi: 10.1002/path.5056
Beauquis, J., Homo-Delarche, F., Giroix, M. H., Ehses, J., Coulaud, J., Roig, P., et al. (2010). Hippocampal neurovascular and hypothalamic-pituitary-adrenal axis alterations in spontaneously type 2 diabetic GK rats. Exp.Neurol. 222, 125–134. doi: 10.1016/j.expneurol.2009.12.022
Beauquis, J., Pomilio, C., Vinuesa, A., Pavía, P., Galvan, V., and Saravia, F. (2013). “Early astroglial and neuronal changes in the hippocampus of PDAPP-J20 transgenic mice, model of Alzheimer’s disease,” in Comunicación oral en la Reunión Anual de la Society for Neuroscience, noviembre 2013, (San Diego).
Beauquis, J., Saravia, F., Coulaud, J., Roig, P., Dardenne, M., Homo-Delarche, F., et al. (2008). Prominently decreased hippocampal neurogenesis in a spontaneous model of type 1 diabetes, the nonobese diabetic mouse. Exp.Neurol. 210, 359–367. doi: 10.1016/j.expneurol.2007.11.009
Becker, E., Orellana Rios, C. L., Lahmann, C., Rucker, G., Bauer, J., and Boeker, M. (2018). Anxiety as a risk factor of Alzheimer’s disease and vascular dementia. Br. J. Psychiatry 213, 654–660.
Benedict, C., Hallschmid, M., Hatke, A., Schultes, B., Fehm, H. L., Born, J., et al. (2004). Intranasal insulin improves memory in humans. Psychoneuroendocrinology 29, 1326–1334. doi: 10.1016/j.psyneuen.2004.04.003
Benito-Leon, J., Mitchell, A. J., Hernandez-Gallego, J., and Bermejo-Pareja, F. (2013). Obesity and impaired cognitive functioning in the elderly: a population-based cross-sectional study (NEDICES). Eur. J. Neurol. 20.
Bethel, M. A., Patel, R. A., Merrill, P., Lokhnygina, Y., Buse, J. B., Mentz, R. J., et al. (2018). Cardiovascular outcomes with glucagon-like peptide-1 receptor agonists in patients with type 2 diabetes: a meta-analysis. Lancet Diabetes Endocrinol 6, 105–113.
Biro, F. M., and Wien, M. (2010). Childhood obesity and adult morbidities. Am.J.Clin.Nutr. 91, 1499S–1505S.
Bjorkhem, I., Cedazo-Minguez, A., Leoni, V., and Meaney, S. (2009). Oxysterols and neurodegenerative diseases. Mol. Aspects Med. 30, 171–179. doi: 10.1016/j.mam.2009.02.001
Blasko, I., Beer, R., Bigl, M., Apelt, J., Franz, G., Rudzki, D., et al. (2004). Experimental traumatic brain injury in rats stimulates the expression, production and activity of Alzheimer’s disease beta-secretase (BACE-1). J Neural Transm (Vienna) 111, 523–536. doi: 10.1007/s00702-003-0095-6
Blazquez, E., Velazquez, E., Hurtado-Carneiro, V., and Ruiz-Albusac, J. M. (2014). Insulin in the brain: its pathophysiological implications for States related with central insulin resistance, type 2 diabetes and Alzheimer’s disease. Front Endocrinol (Lausanne) 5, 161.
Boitard, C., Etchamendy, N., Sauvant, J., Aubert, A., Tronel, S., Marighetto, A., et al. (2012). Juvenile, but not adult exposure to high-fat diet impairs relational memory and hippocampal neurogenesis in mice. Hippocampus 22, 2095–2100. doi: 10.1002/hipo.22032
Bokulich, N. A., Chung, J., Battaglia, T., Henderson, N., Jay, M., Li, H., et al. (2016). Antibiotics, birth mode, and diet shape microbiome maturation during early life. Sci Transl Med 8, 343ra382.
Bomba, M., Ciavardelli, D., Silvestri, E., Canzoniero, L. M., Lattanzio, R., Chiappini, P., et al. (2013). Exenatide promotes cognitive enhancement and positive brain metabolic changes in PS1-KI mice but has no effects in 3xTg-AD animals. Cell Death Dis 4, e612. doi: 10.1038/cddis.2013.139
Bomba, M., Granzotto, A., Castelli, V., Onofrj, M., Lattanzio, R., Cimini, A., et al. (2019). Exenatide Reverts the High-Fat-Diet-Induced Impairment of BDNF Signaling and Inflammatory Response in an Animal Model of Alzheimer’s Disease. J. Alzheimers. Dis. 70, 793–810. doi: 10.3233/jad-190237
Bonfili, L., Cecarini, V., Berardi, S., Scarpona, S., Suchodolski, J. S., Nasuti, C., et al. (2017). Microbiota modulation counteracts Alzheimer’s disease progression influencing neuronal proteolysis and gut hormones plasma levels. Sci Rep 7, 2426.
Bonfili, L., Cecarini, V., Cuccioloni, M., Angeletti, M., Berardi, S., Scarpona, S., et al. (2018). SLAB51 Probiotic Formulation Activates SIRT1 Pathway Promoting Antioxidant and Neuroprotective Effects in an AD Mouse Model. Mol. Neurobiol. 55, 7987–8000. doi: 10.1007/s12035-018-0973-4
Boucher, J., Kleinridders, A., and Kahn, C. R. (2014). Insulin receptor signaling in normal and insulin-resistant states. Cold Spring Harb. Perspect. Biol 6.
Bracke, A., Domanska, G., Bracke, K., Harzsch, S., Van Den Brandt, J., Broker, B., et al. (2019). Obesity Impairs Mobility and Adult Hippocampal Neurogenesis. J Exp Neurosci 13, 1179069519883580.
Bracko, O., Vinarcsik, L. K., Cruz Hernandez, J. C., Ruiz-Uribe, N. E., Haft-Javaherian, M., Falkenhain, K., et al. (2020). High fat diet worsens Alzheimer’s disease-related behavioral abnormalities and neuropathology in APP/PS1 mice, but not by synergistically decreasing cerebral blood flow. Sci Rep 10, 9884.
Brandhorst, S., Choi, I. Y., Wei, M., Cheng, C. W., Sedrakyan, S., Navarrete, G., et al. (2015). A Periodic Diet that Mimics Fasting Promotes Multi-System Regeneration, Enhanced Cognitive Performance, and Healthspan. Cell Metab 22, 86–99. doi: 10.1016/j.cmet.2015.05.012
Bruce-Keller, A. J., Keller, J. N., and Morrison, C. D. (2009). Obesity and vulnerability of the CNS. Biochim. Biophys. Acta 1792, 395–400.
Bussler, S., Penke, M., Flemming, G., Elhassan, Y. S., Kratzsch, J., Sergeyev, E., et al. (2017). Novel Insights in the Metabolic Syndrome in Childhood and Adolescence. Horm Res Paediatr 88, 181–193.
Caccamo, A., Maldonado, M. A., Bokov, A. F., Majumder, S., and Oddo, S. (2010). CBP gene transfer increases BDNF levels and ameliorates learning and memory deficits in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. U.S.A. 107, 22687–22692. doi: 10.1073/pnas.1012851108
Cai, H. Y., Holscher, C., Yue, X. H., Zhang, S. X., Wang, X. H., Qiao, F., et al. (2014). Lixisenatide rescues spatial memory and synaptic plasticity from amyloid beta protein-induced impairments in rats. Neuroscience 277, 6–13. doi: 10.1016/j.neuroscience.2014.02.022
Cai, H. Y., Yang, J. T., Wang, Z. J., Zhang, J., Yang, W., Wu, M. N., et al. (2018). Lixisenatide reduces amyloid plaques, neurofibrillary tangles and neuroinflammation in an APP/PS1/tau mouse model of Alzheimer’s disease. Biochem. Biophys. Res. Commun. 495, 1034–1040. doi: 10.1016/j.bbrc.2017.11.114
Cani, P. D., Bibiloni, R., Knauf, C., Waget, A., Neyrinck, A. M., Delzenne, N. M., et al. (2008). Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes Metab. Res. Rev 57, 1470–1481. doi: 10.2337/db07-1403
Cantarella, G., Di Benedetto, G., Puzzo, D., Privitera, L., Loreto, C., Saccone, S., et al. (2015). Neutralization of TNFSF10 ameliorates functional outcome in a murine model of Alzheimer’s disease. Brain 138, 203–216. doi: 10.1093/brain/awu318
Canton-Habas, V., Rich-Ruiz, M., Romero-Saldana, M., and Carrera-Gonzalez, M. D. P. (2020). Depression as a Risk Factor for Dementia and Alzheimer’s Disease. Biomedicines 8.
Carvalho, B. M., Guadagnini, D., Tsukumo, D. M. L., Schenka, A. A., Latuf-Filho, P., Vassallo, J., et al. (2012). Modulation of gut microbiota by antibiotics improves insulin signalling in high-fat fed mice. Diabetologia 55, 2823–2834. doi: 10.1007/s00125-012-2648-4
Cavelti-Weder, C., Babians-Brunner, A., Keller, C., Stahel, M. A., Kurz-Levin, M., Zayed, H., et al. (2012). Effects of gevokizumab on glycemia and inflammatory markers in type 2 diabetes. Diabetes Care 35, 1654–1662. doi: 10.2337/dc11-2219
Chappus-McCendie, H., Chevalier, L., Roberge, C., and Plourde, M. (2019). Omega-3 PUFA metabolism and brain modifications during aging. Prog Neuropsychopharmacol Biol Psychiatry 94, 109662. doi: 10.1016/j.pnpbp.2019.109662
Chavez, J. A., Siddique, M. M., Wang, S. T., Ching, J., Shayman, J. A., and Summers, S. A. (2014). Ceramides and glucosylceramides are independent antagonists of insulin signaling. J. Biol. Chem. 289, 723–734. doi: 10.1074/jbc.m113.522847
Chen, D., Yang, X., Yang, J., Lai, G., Yong, T., Tang, X., et al. (2017). Prebiotic Effect of Fructooligosaccharides from Morinda officinalis on Alzheimer’s Disease in Rodent Models by Targeting the Microbiota-Gut-Brain Axis. Front Aging Neurosci 9, 403.
Chen, J. L., Luo, C., Pu, D., Zhang, G. Q., Zhao, Y. X., Sun, Y., et al. (2019). Metformin attenuates diabetes-induced tau hyperphosphorylation in vitro and in vivo by enhancing autophagic clearance. Exp. Neurol. 311, 44–56. doi: 10.1016/j.expneurol.2018.09.008
Chen, M., Huang, N., Liu, J., Huang, J., Shi, J., and Jin, F. (2020). AMPK: A bridge between diabetes mellitus and Alzheimer’s disease. Behav. Brain Res. 400, 113043. doi: 10.1016/j.bbr.2020.113043
Chen, X. H., Siman, R., Iwata, A., Meaney, D. F., Trojanowski, J. Q., and Smith, D. H. (2004). Long-term accumulation of amyloid-beta, beta-secretase, presenilin-1, and caspase-3 in damaged axons following brain trauma. Am. J. Pathol. 165, 357–371. doi: 10.1016/s0002-9440(10)63303-2
Cho, M. H., Cho, K., Kang, H. J., Jeon, E. Y., Kim, H. S., Kwon, H. J., et al. (2014). Autophagy in microglia degrades extracellular beta-amyloid fibrils and regulates the NLRP3 inflammasome. Autophagy. 10, 1761–1775. doi: 10.4161/auto.29647
Choi, S. H., Bylykbashi, E., Chatila, Z. K., Lee, S. W., Pulli, B., Clemenson, G. D., et al. (2018). Combined adult neurogenesis and BDNF mimic exercise effects on cognition in an Alzheimer’s mouse model. Science 361.
Chu, C. S., Tseng, P. T., Stubbs, B., Chen, T. Y., Tang, C. H., Li, D. J., et al. (2018). Use of statins and the risk of dementia and mild cognitive impairment: A systematic review and meta-analysis. Sci Rep 8, 5804.
Chua, L. M., Lim, M. L., Chong, P. R., Hu, Z. P., Cheung, N. S., and Wong, B. S. (2012). Impaired neuronal insulin signaling precedes Abeta42 accumulation in female AbetaPPsw/PS1DeltaE9 mice. J. Alzheimers. Dis. 29, 783–791. doi: 10.3233/jad-2012-111880
Chuang, Y. F., An, Y., Bilgel, M., Wong, D. F., Troncoso, J. C., O’brien, R. J., et al. (2016). Midlife adiposity predicts earlier onset of Alzheimer’s dementia, neuropathology and presymptomatic cerebral amyloid accumulation. Mol. Psychiatry 21, 910–915. doi: 10.1038/mp.2015.129
Claesson, M. J., Jeffery, I. B., Conde, S., Power, S. E., O’connor, E. M., Cusack, S., et al. (2012). Gut microbiota composition correlates with diet and health in the elderly. Nature 488, 178–184.
Claxton, A., Baker, L. D., Hanson, A., Trittschuh, E. H., Cholerton, B., Morgan, A., et al. (2015). Long Acting Intranasal Insulin Detemir Improves Cognition for Adults with Mild Cognitive Impairment or Early-Stage Alzheimer’s Disease Dementia. J. Alzheimers. Dis. 45, 1269–1270. doi: 10.3233/jad-159002
Clelland, C. D., Choi, M., Romberg, C., Clemenson, G. D. Jr., Fragniere, A., et al. (2009). A functional role for adult hippocampal neurogenesis in spatial pattern separation. Science 325, 210–213. doi: 10.1126/science.1173215
Cohen, E., Bieschke, J., Perciavalle, R. M., Kelly, J. W., and Dillin, A. (2006). Opposing activities protect against age-onset proteotoxicity. Science 313, 1604–1610. doi: 10.1126/science.1124646
Cope, E. C., Lamarca, E. A., Monari, P. K., Olson, L. B., Martinez, S., Zych, A. D., et al. (2018). Microglia Play an Active Role in Obesity-Associated Cognitive Decline. J. Neurosci. 38, 8889–8904. doi: 10.1523/jneurosci.0789-18.2018
Craft, S., Claxton, A., Baker, L. D., Hanson, A. J., Cholerton, B., Trittschuh, E. H., et al. (2017). Effects of Regular and Long-Acting Insulin on Cognition and Alzheimer’s Disease Biomarkers: A Pilot Clinical Trial. J. Alzheimers. Dis. 57, 1325–1334. doi: 10.3233/jad-161256
Dahl, A., Hassing, L. B., Fransson, E., Berg, S., Gatz, M., Reynolds, C. A., et al. (2010). Being overweight in midlife is associated with lower cognitive ability and steeper cognitive decline in late life. J. Gerontol. A Biol. Sci. Med. Sci. 65, 57–62. doi: 10.1093/gerona/glp035
Dahl, A. K., and Hassing, L. B. (2013). Obesity and cognitive aging. Epidemiol. Rev. 35, 22–32. doi: 10.1093/epirev/mxs002
de Heer, J., Rasmussen, C., Coy, D. H., and Holst, J. J. (2008). Glucagon-like peptide-1, but not glucose-dependent insulinotropic peptide, inhibits glucagon secretion via somatostatin (receptor subtype 2) in the perfused rat pancreas. Diabetologia 51, 2263–2270. doi: 10.1007/s00125-008-1149-y
de la Monte, S. M., and Tong, M. (2014). Brain metabolic dysfunction at the core of Alzheimer’s disease. Biochem. Pharmacol 88, 548–559. doi: 10.1016/j.bcp.2013.12.012
De Sousa Rodrigues, M. E., Houser, M. C., Walker, D. I., Jones, D. P., Chang, J., Barnum, C. J., et al. (2019). Targeting soluble tumor necrosis factor as a potential intervention to lower risk for late-onset Alzheimer’s disease associated with obesity, metabolic syndrome, and type 2 diabetes. Alzheimers Res Ther 12, 1. doi: 10.1007/s11010-011-0917-z
Deckers, K., Van Boxtel, M. P. J., Verhey, F. R. J., and Kohler, S. (2017). Obesity and Cognitive Decline in Adults: Effect of Methodological Choices and Confounding by Age in a Longitudinal Study. J. Nutr. Health Aging 21, 546–553. doi: 10.1007/s12603-016-0757-3
Derkach, K. V., Ivantsov, A. O., Chistyakova, O. V., Sukhov, I. B., Buzanakov, D. M., Kulikova, A. A., et al. (2017). Intranasal Insulin Restores Metabolic Parameters and Insulin Sensitivity in Rats with Metabolic Syndrome. Bull. Exp. Biol. Med. 163, 184–189. doi: 10.1007/s10517-017-3762-6
Dey, A., Hao, S., Wosiski-Kuhn, M., and Stranahan, A. M. (2017). Glucocorticoid-mediated activation of GSK3beta promotes tau phosphorylation and impairs memory in type 2 diabetes. Neurobiol. Aging 57, 75–83. doi: 10.1016/j.neurobiolaging.2017.05.010
Dror, E., Dalmas, E., Meier, D. T., Wueest, S., Thevenet, J., Thienel, C., et al. (2017). Postprandial macrophage-derived IL-1beta stimulates insulin, and both synergistically promote glucose disposal and inflammation. Nat. Immunol. 18, 283–292. doi: 10.1038/ni.3659
Edwards, P. K., Mears, S. C., Stambough, J. B., Foster, S. E., and Barnes, C. L. (2019). Response to Letter to the Editor on “Choices, Compromises, and Controversies in Total Knee and Total Hip Arthroplasty Modifiable Risk Factors: What You Need to Know”.. J. Arthroplasty 34, 1039. doi: 10.1016/j.arth.2019.02.015
Ellulu, M. S., Patimah, I., Khaza’ai, H., Rahmat, A., and Abed, Y. (2017). Obesity and inflammation: the linking mechanism and the complications. Arch Med Sci 13, 851–863. doi: 10.5114/aoms.2016.58928
Elosua, R., Bartali, B., Ordovas, J. M., Corsi, A. M., Lauretani, F., Ferrucci, L., et al. (2005). Association between physical activity, physical performance, and inflammatory biomarkers in an elderly population: the InCHIANTI study. J Gerontol A Biol Sci Med Sci 60, 760–767. doi: 10.1093/gerona/60.6.760
Eom, T. Y., and Jope, R. S. (2009). Blocked inhibitory serine-phosphorylation of glycogen synthase kinase-3alpha/beta impairs in vivo neural precursor cell proliferation. Biol. Psychiatry 66, 494–502. doi: 10.1016/j.biopsych.2009.04.015
Erickson, M. A., and Banks, W. A. (2013). Blood-brain barrier dysfunction as a cause and consequence of Alzheimer’s disease. J. Cereb. Blood Flow Metab. 33, 1500–1513. doi: 10.1038/jcbfm.2013.135
Eriksdotter, M., Vedin, I., Falahati, F., Freund-Levi, Y., Hjorth, E., Faxen-Irving, G., et al. (2015). Plasma Fatty Acid Profiles in Relation to Cognition and Gender in Alzheimer’s Disease Patients During Oral Omega-3 Fatty Acid Supplementation: The OmegAD Study. J. Alzheimers. Dis. 48, 805–812. doi: 10.3233/jad-150102
Etminan, M., Gill, S., and Samii, A. (2003). Effect of non-steroidal anti-inflammatory drugs on risk of Alzheimer’s disease: systematic review and meta-analysis of observational studies. BMJ 327, 128. doi: 10.1136/bmj.327.7407.128
Farilla, L., Bulotta, A., Hirshberg, B., Li Calzi, S., Khoury, N., Noushmehr, H., et al. (2003). Glucagon-like peptide 1 inhibits cell apoptosis and improves glucose responsiveness of freshly isolated human islets. Endocrinology 144, 5149–5158. doi: 10.1210/en.2003-0323
Farr, S. A., Roesler, E., Niehoff, M. L., Roby, D. A., Mckee, A., and Morley, J. E. (2019). Metformin Improves Learning and Memory in the SAMP8 Mouse Model of Alzheimer’s Disease. J. Alzheimers. Dis. 68, 1699–1710. doi: 10.3233/jad-181240
Ferrario, C. R., and Reagan, L. P. (2017). Insulin-mediated synaptic plasticity in the CNS: Anatomical, functional and temporal contexts. Neuropharmacology.
Ferreira, L. S. S., Fernandes, C. S., Vieira, M. N. N., and De Felice, F. G. (2018). Insulin Resistance in Alzheimer’s Disease. Front Neurosci 12, 830.
Franceschi, C., and Campisi, J. (2014). Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. J. Gerontol. A Biol. Sci. Med. Sci. 69(Suppl. 1), S4–S9.
Franceschi, C., Garagnani, P., Parini, P., Giuliani, C., and Santoro, A. (2018). Inflammaging: a new immune-metabolic viewpoint for age-related diseases. Nat Rev Endocrinol 14, 576–590. doi: 10.1038/s41574-018-0059-4
Frank, M. G., Barrientos, R. M., Biedenkapp, J. C., Rudy, J. W., Watkins, L. R., and Maier, S. F. (2006). mRNA up-regulation of MHC II and pivotal pro-inflammatory genes in normal brain aging. Neurobiol. Aging 27, 717–722. doi: 10.1016/j.neurobiolaging.2005.03.013
Freude, S., Hettich, M. M., Schumann, C., Stohr, O., Koch, L., Kohler, C., et al. (2009). Neuronal IGF-1 resistance reduces Abeta accumulation and protects against premature death in a model of Alzheimer’s disease. FASEB J. 23, 3315–3324. doi: 10.1096/fj.09-132043
Freund-Levi, Y., Eriksdotter-Jonhagen, M., Cederholm, T., Basun, H., Faxen-Irving, G., Garlind, A., et al. (2006). Omega-3 fatty acid treatment in 174 patients with mild to moderate Alzheimer disease: OmegAD study: a randomized double-blind trial. Arch. Neurol. 63, 1402–1408. doi: 10.1001/archneur.63.10.1402
Frolich, L., Blum-Degen, D., Bernstein, H. G., Engelsberger, S., Humrich, J., Laufer, S., et al. (1998). Brain insulin and insulin receptors in aging and sporadic Alzheimer’s disease. J Neural Transm (Vienna) 105, 423–438. doi: 10.1007/s007020050068
Fujita, K., Motoki, K., Tagawa, K., Chen, X., Hama, H., Nakajima, K., et al. (2016). HMGB1, a pathogenic molecule that induces neurite degeneration via TLR4-MARCKS, is a potential therapeutic target for Alzheimer’s disease. Sci Rep 6, 31895.
Gault, V. A., and Holscher, C. (2008). GLP-1 agonists facilitate hippocampal LTP and reverse the impairment of LTP induced by beta-amyloid. Eur. J. Pharmacol 587, 112–117. doi: 10.1016/j.ejphar.2008.03.025
Gejl, M., Gjedde, A., Egefjord, L., Moller, A., Hansen, S. B., Vang, K., et al. (2016). In Alzheimer’s Disease, 6-Month Treatment with GLP-1 Analog Prevents Decline of Brain Glucose Metabolism: Randomized, Placebo-Controlled, Double-Blind Clinical Trial. Front Aging Neurosci 8, 108.
Gold, M., Alderton, C., Zvartau-Hind, M., Egginton, S., Saunders, A. M., Irizarry, M., et al. (2010). Rosiglitazone monotherapy in mild-to-moderate Alzheimer’s disease: results from a randomized, double-blind, placebo-controlled phase III study. Dement. Geriatr. Cogn. Disord 30, 131–146. doi: 10.1159/000318845
Gomes, A. P., Soares, A. L. G., Menezes, A. M. B., Assuncao, M. C., Wehrmeister, F. C., Howe, L. D., et al. (2019). Adiposity, depression and anxiety: interrelationship and possible mediators. Rev Saude Publica 53, 103. doi: 10.11606/s1518-8787.2019053001119
Gottesman, R. F., Albert, M. S., Alonso, A., Coker, L. H., Coresh, J., Davis, S. M., et al. (2017). Associations Between Midlife Vascular Risk Factors and 25-Year Incident Dementia in the Atherosclerosis Risk in Communities (ARIC) Cohort. JAMA Neurol 74, 1246–1254. doi: 10.1001/jamaneurol.2017.1658
Gralle, M. (2017). The neuronal insulin receptor in its environment. J. Neurochem. 140, 359–367. doi: 10.1111/jnc.13909
Grefhorst, A., Elzinga, B. M., Voshol, P. J., Plosch, T., Kok, T., Bloks, V. W., et al. (2002). Stimulation of lipogenesis by pharmacological activation of the liver X receptor leads to production of large, triglyceride-rich very low density lipoprotein particles. J. Biol. Chem. 277, 34182–34190. doi: 10.1074/jbc.m204887200
Gregosa, A., Vinuesa, A., Todero, M. F., Pomilio, C., Rossi, S. P., Bentivegna, M., et al. (2019). Periodic dietary restriction ameliorates amyloid pathology and cognitive impairment in PDAPP-J20 mice: Potential implication of glial autophagy. Neurobiol. Dis 104542. doi: 10.1016/j.nbd.2019.104542
Griffin, R. J., Moloney, A., Kelliher, M., Johnston, J. A., Ravid, R., Dockery, P., et al. (2005). Activation of Akt/PKB, increased phosphorylation of Akt substrates and loss and altered distribution of Akt and PTEN are features of Alzheimer’s disease pathology. J. Neurochem. 93, 105–117. doi: 10.1111/j.1471-4159.2004.02949.x
Group, A. R., Martin, B. K., Szekely, C., Brandt, J., Piantadosi, S., Breitner, J. C., et al. (2008). Cognitive function over time in the Alzheimer’s Disease Anti-inflammatory Prevention Trial (ADAPT): results of a randomized, controlled trial of naproxen and celecoxib. Arch. Neurol. 65, 896–905. doi: 10.1001/archneur.2008.65.7.nct70006
Guillemot-Legris, O., and Muccioli, G. G. (2017). Obesity-Induced Neuroinflammation: Beyond the Hypothalamus. Trends Neurosci 40, 237–253. doi: 10.1016/j.tins.2017.02.005
Hamer, M., Lavoie, K. L., and Bacon, S. L. (2014). Taking up physical activity in later life and healthy ageing: the English longitudinal study of ageing. Br. J. Sports Med. 48, 239–243. doi: 10.1136/bjsports-2013-092993
Hao, S., Dey, A., Yu, X., and Stranahan, A. M. (2016). Dietary obesity reversibly induces synaptic stripping by microglia and impairs hippocampal plasticity. Brain Behav. Immun. 51, 230–239. doi: 10.1016/j.bbi.2015.08.023
Hargrave, S. L., Davidson, T. L., Zheng, W., and Kinzig, K. P. (2016). Western diets induce blood-brain barrier leakage and alter spatial strategies in rats. Behav. Neurosci. 130, 123–135. doi: 10.1037/bne0000110
Harries, L. W., Fellows, A. D., Pilling, L. C., Hernandez, D., Singleton, A., Bandinelli, S., et al. (2012). Advancing age is associated with gene expression changes resembling mTOR inhibition: evidence from two human populations. Mech. Ageing Dev. 133, 556–562. doi: 10.1016/j.mad.2012.07.003
Harvie, M. N., Pegington, M., Mattson, M. P., Frystyk, J., Dillon, B., Evans, G., et al. (2011). The effects of intermittent or continuous energy restriction on weight loss and metabolic disease risk markers: a randomized trial in young overweight women. Int J Obes (Lond) 35, 714–727. doi: 10.1038/ijo.2010.171
Henderson, S. T., Vogel, J. L., Barr, L. J., Garvin, F., Jones, J. J., and Costantini, L. C. (2009). Study of the ketogenic agent AC-1202 in mild to moderate Alzheimer’s disease: a randomized, double-blind, placebo-controlled, multicenter trial. Nutr Metab (Lond) 6, 31. doi: 10.1186/1743-7075-6-31
Heneka, M. T., Carson, M. J., El Khoury, J., Landreth, G. E., Brosseron, F., Feinstein, D. L., et al. (2015). Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 14, 388–405.
Heneka, M. T., Kummer, M. P., Stutz, A., Delekate, A., Schwartz, S., Vieira-Saecker, A., et al. (2013). NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 493, 674–678. doi: 10.1038/nature11729
Heneka, M. T., Sastre, M., Dumitrescu-Ozimek, L., Dewachter, I., Walter, J., Klockgether, T., et al. (2005). Focal glial activation coincides with increased BACE1 activation and precedes amyloid plaque deposition in APP[V717I] transgenic mice. J Neuroinflammation 2, 22.
Herzig, S., and Shaw, R. J. (2018). AMPK: guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell Biol. 19, 121–135. doi: 10.1038/nrm.2017.95
Hoffmann, K., Sobol, N. A., Frederiksen, K. S., Beyer, N., Vogel, A., Vestergaard, K., et al. (2016). Moderate-to-High Intensity Physical Exercise in Patients with Alzheimer’s Disease: A Randomized Controlled Trial. J. Alzheimers. Dis. 50, 443–453.
Holland, W. L., Bikman, B. T., Wang, L. P., Yuguang, G., Sargent, K. M., Bulchand, S., et al. (2011). Lipid-induced insulin resistance mediated by the proinflammatory receptor TLR4 requires saturated fatty acid-induced ceramide biosynthesis in mice. J. Clin. Invest. 121, 1858–1870. doi: 10.1172/jci43378
Hotamisligil, G. S. (2010). Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell 140, 900–917. doi: 10.1016/j.cell.2010.02.034
Hotamisligil, G. S., Shargill, N. S., and Spiegelman, B. M. (1993). Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science 259, 87–91. doi: 10.1126/science.7678183
Hutchinson, A. N., Tingo, L., and Brummer, R. J. (2020). The Potential Effects of Probiotics and omega-3 Fatty Acids on Chronic Low-Grade Inflammation. Nutrients 12.
Idf. (2017). IDF DIABETES ATLAS [Online]. Available: https://www.diabetesatlas.org/en/[Accessed] (accessed December 20, 2020).
Inestrosa, N. C., Godoy, J. A., Quintanilla, R. A., Koenig, C. S., and Bronfman, M. (2005). Peroxisome proliferator-activated receptor gamma is expressed in hippocampal neurons and its activation prevents beta-amyloid neurodegeneration: role of Wnt signaling. Exp. Cell Res. 304, 91–104. doi: 10.1016/j.yexcr.2004.09.032
Ishiguro, K., Shiratsuchi, A., Sato, S., Omori, A., Arioka, M., Kobayashi, S., et al. (1993). Glycogen synthase kinase 3 beta is identical to tau protein kinase I generating several epitopes of paired helical filaments. FEBS Lett. 325, 167–172. doi: 10.1016/0014-5793(93)81066-9
James, S. L., Lucchesi, L. R., Bisignano, C., Castle, C. D., Dingels, Z. V., Fox, J. T., et al. (2020). Morbidity and mortality from road injuries: results from the Global Burden of Disease Study 2017. Inj. Prev. 26, i46–i56.
Jantzen, P. T., Connor, K. E., Dicarlo, G., Wenk, G. L., Wallace, J. L., Rojiani, A. M., et al. (2002). Microglial activation and beta -amyloid deposit reduction caused by a nitric oxide-releasing nonsteroidal anti-inflammatory drug in amyloid precursor protein plus presenilin-1 transgenic mice. J. Neurosci. 22, 2246–2254. doi: 10.1523/jneurosci.22-06-02246.2002
Jensen, C. S., Bahl, J. M., Ostergaard, L. B., Hogh, P., Wermuth, L., Heslegrave, A., et al. (2019). Exercise as a potential modulator of inflammation in patients with Alzheimer’s disease measured in cerebrospinal fluid and plasma. Exp. Gerontol. 121, 91–98. doi: 10.1016/j.exger.2019.04.003
Jia, L., Vianna, C. R., Fukuda, M., Berglund, E. D., Liu, C., Tao, C., et al. (2014). Hepatocyte Toll-like receptor 4 regulates obesity-induced inflammation and insulin resistance. Nat Commun 5, 3878.
Jiao, S. S., Shen, L. L., Zhu, C., Bu, X. L., Liu, Y. H., Liu, C. H., et al. (2016). Brain-derived neurotrophic factor protects against tau-related neurodegeneration of Alzheimer’s disease. Transl Psychiatry 6, e907. doi: 10.1038/tp.2016.186
Jimenez, S., Torres, M., Vizuete, M., Sanchez-Varo, R., Sanchez-Mejias, E., Trujillo-Estrada, L., et al. (2011). Age-dependent accumulation of soluble amyloid beta (Abeta) oligomers reverses the neuroprotective effect of soluble amyloid precursor protein-alpha (sAPP(alpha)) by modulating phosphatidylinositol 3-kinase (PI3K)/Akt-GSK-3beta pathway in Alzheimer mouse model. J. Biol. Chem. 286, 18414–18425. doi: 10.1074/jbc.m110.209718
Jope, R. S., Yuskaitis, C. J., and Beurel, E. (2007). Glycogen synthase kinase-3 (GSK3): inflammation, diseases, and therapeutics. Neurochem. Res. 32, 577–595. doi: 10.1007/s11064-006-9128-5
Kashiwaya, Y., Bergman, C., Lee, J. H., Wan, R., King, M. T., Mughal, M. R., et al. (2013). A ketone ester diet exhibits anxiolytic and cognition-sparing properties, and lessens amyloid and tau pathologies in a mouse model of Alzheimer’s disease. Neurobiol. Aging 34, 1530–1539. doi: 10.1016/j.neurobiolaging.2012.11.023
Kattenstroth, J. C., Kolankowska, I., Kalisch, T., and Dinse, H. R. (2010). Superior sensory, motor, and cognitive performance in elderly individuals with multi-year dancing activities. Front Aging Neurosci 2.
Kellar, D., and Craft, S. (2020). Brain insulin resistance in Alzheimer’s disease and related disorders: mechanisms and therapeutic approaches. Lancet Neurol. 19, 758–766. doi: 10.1016/s1474-4422(20)30231-3
Kelly, K. M., Nadon, N. L., Morrison, J. H., Thibault, O., Barnes, C. A., and Blalock, E. M. (2006). The neurobiology of aging. Epilepsy Res. 68(Suppl. 1), S5–S20.
Kitazawa, M., Oddo, S., Yamasaki, T. R., Green, K. N., and Laferla, F. M. (2005). Lipopolysaccharide-induced inflammation exacerbates tau pathology by a cyclin-dependent kinase 5-mediated pathway in a transgenic model of Alzheimer’s disease. J. Neurosci. 25, 8843–8853. doi: 10.1523/jneurosci.2868-05.2005
Kleinridders, A., Ferris, H. A., Cai, W., and Kahn, C. R. (2014). Insulin action in brain regulates systemic metabolism and brain function. Diabetes Metab. Res. Rev 63, 2232–2243. doi: 10.2337/db14-0568
Klempin, F., and Kempermann, G. (2007). Adult hippocampal neurogenesis and aging. Eur. Arch. Psychiatry Clin. Neurosci. 257, 271–280.
Kloting, N., and Bluher, M. (2014). Adipocyte dysfunction, inflammation and metabolic syndrome. Rev Endocr Metab Disord 15, 277–287. doi: 10.1007/s11154-014-9301-0
Kobayashi, Y., Kinoshita, T., Matsumoto, A., Yoshino, K., Saito, I., and Xiao, J. Z. (2019a). Bifidobacterium Breve A1 Supplementation Improved Cognitive Decline in Older Adults with Mild Cognitive Impairment: An Open-Label, Single-Arm Study. J Prev Alzheimers Dis 6, 70–75.
Kobayashi, Y., Kuhara, T., Oki, M., and Xiao, J. Z. (2019b). Effects of Bifidobacterium breve A1 on the cognitive function of older adults with memory complaints: a randomised, double-blind, placebo-controlled trial. Benef Microbes 10, 511–520. doi: 10.3920/bm2018.0170
Kobayashi, Y., Sugahara, H., Shimada, K., Mitsuyama, E., Kuhara, T., Yasuoka, A., et al. (2017). Therapeutic potential of Bifidobacterium breve strain A1 for preventing cognitive impairment in Alzheimer’s disease. Sci Rep 7, 13510.
Koenig, A. M., Mechanic-Hamilton, D., Xie, S. X., Combs, M. F., Cappola, A. R., Xie, L., et al. (2017). Effects of the Insulin Sensitizer Metformin in Alzheimer Disease: Pilot Data From a Randomized Placebo-controlled Crossover Study. Alzheimer Dis. Assoc. Disord. 31, 107–113. doi: 10.1097/wad.0000000000000202
Koldamova, R. P., Lefterov, I. M., Staufenbiel, M., Wolfe, D., Huang, S., Glorioso, J. C., et al. (2005). The liver X receptor ligand T0901317 decreases amyloid beta production in vitro and in a mouse model of Alzheimer’s disease. J. Biol. Chem. 280, 4079–4088. doi: 10.1074/jbc.m411420200
Koo, B. K., Kim, L. K., Lee, J. Y., and Moon, M. K. (2019). Taking metformin and cognitive function change in older patients with diabetes. Geriatr Gerontol Int 19, 755–761. doi: 10.1111/ggi.13692
Kotti, T. J., Ramirez, D. M., Pfeiffer, B. E., Huber, K. M., and Russell, D. W. (2006). Brain cholesterol turnover required for geranylgeraniol production and learning in mice. Proc. Natl. Acad. Sci. U.S.A. 103, 3869–3874. doi: 10.1073/pnas.0600316103
Kowianski, P., Lietzau, G., Czuba, E., Waskow, M., Steliga, A., and Morys, J. (2018). BDNF: A Key Factor with Multipotent Impact on Brain Signaling and Synaptic Plasticity. Cell Mol. Neurobiol 38, 579–593. doi: 10.1007/s10571-017-0510-4
Krstic, D., Madhusudan, A., Doehner, J., Vogel, P., Notter, T., Imhof, C., et al. (2012). Systemic immune challenges trigger and drive Alzheimer-like neuropathology in mice. J Neuroinflammation 9, 151.
Larsen, C. M., Faulenbach, M., Vaag, A., Volund, A., Ehses, J. A., Seifert, B., et al. (2007). Interleukin-1-receptor antagonist in type 2 diabetes mellitus. N. Engl. J. Med. 356, 1517–1526.
Lazarou, I., Parastatidis, T., Tsolaki, A., Gkioka, M., Karakostas, A., Douka, S., et al. (2017). International Ballroom Dancing Against Neurodegeneration: A Randomized Controlled Trial in Greek Community-Dwelling Elders With Mild Cognitive impairment. Am. J. Alzheimers Dis. Other Demen. 32, 489–499. doi: 10.1177/1533317517725813
Le, K. A., Ventura, E. E., Fisher, J. Q., Davis, J. N., Weigensberg, M. J., Punyanitya, M., et al. (2011). Ethnic differences in pancreatic fat accumulation and its relationship with other fat depots and inflammatory markers. Diabetes Care 34, 485–490. doi: 10.2337/dc10-0760
Lee, E. B., and Mattson, M. P. (2014). The neuropathology of obesity: insights from human disease. Acta Neuropathol. 127, 3–28. doi: 10.1007/s00401-013-1190-x
Leonardini, A., Laviola, L., Perrini, S., Natalicchio, A., and Giorgino, F. (2009). Cross-Talk between PPARgamma and Insulin Signaling and Modulation of Insulin Sensitivity. PPAR Res 2009, 818945.
Li, H., Yang, S., Wu, J., Ji, L., Zhu, L., Cao, L., et al. (2018). cAMP/PKA signaling pathway contributes to neuronal apoptosis via regulating IDE expression in a mixed model of type 2 diabetes and Alzheimer’s disease. J. Cell. Biochem. 119, 1616–1626. doi: 10.1002/jcb.26321
Li, J., Chen, S., Qiang, J., Wang, X., Chen, L., and Zou, D. (2014). Diet-induced obesity mediates a proinflammatory response in pancreatic beta cell via toll-like receptor 4. Cent Eur J Immunol 39, 306–315. doi: 10.5114/ceji.2014.45940
Lim, G. P., Yang, F., Chu, T., Chen, P., Beech, W., Teter, B., et al. (2000). Ibuprofen suppresses plaque pathology and inflammation in a mouse model for Alzheimer’s disease. J. Neurosci. 20, 5709–5714. doi: 10.1523/jneurosci.20-15-05709.2000
Lin, S. H., Cheng, P. C., Tu, S. T., Hsu, S. R., Cheng, Y. C., and Liu, Y. H. (2018). Effect of metformin monotherapy on serum lipid profile in statin-naive individuals with newly diagnosed type 2 diabetes mellitus: a cohort study. PeerJ 6, e4578. doi: 10.7717/peerj.4578
Liu, J., and Li, L. (2019). Targeting Autophagy for the Treatment of Alzheimer’s Disease: Challenges and Opportunities. Front Mol Neurosci 12, 203.
Liu, R., Yang, J., Liu, L., Lu, Z., Shi, Z., Ji, W., et al. (2020). An “Amyloid-beta Cleaner” for the Treatment of Alzheimer’s Disease by Normalizing Microglial Dysfunction. Adv Sci (Weinh) 7, 1901555. doi: 10.1002/advs.201901555
Liu, Y., Cheng, A., Li, Y. J., Yang, Y., Kishimoto, Y., Zhang, S., et al. (2019). SIRT3 mediates hippocampal synaptic adaptations to intermittent fasting and ameliorates deficits in APP mutant mice. Nat Commun 10, 1886.
Lochhead, J. J., Kellohen, K. L., Ronaldson, P. T., and Davis, T. P. (2019). Distribution of insulin in trigeminal nerve and brain after intranasal administration. Sci Rep 9, 2621.
Lu, Y., Dong, Y., Tucker, D., Wang, R., Ahmed, M. E., Brann, D., et al. (2017). Treadmill Exercise Exerts Neuroprotection and Regulates Microglial Polarization and Oxidative Stress in a Streptozotocin-Induced Rat Model of Sporadic Alzheimer’s Disease. J. Alzheimers. Dis. 56, 1469–1484. doi: 10.3233/jad-160869
Lucassen, P. J., Oomen, C. A., Naninck, E. F., Fitzsimons, C. P., Van Dam, A. M., Czeh, B., et al. (2015). Regulation of Adult Neurogenesis and Plasticity by (Early) Stress, Glucocorticoids, and Inflammation. Cold Spring Harb. Perspect. Biol. 7, a021303. doi: 10.1101/cshperspect.a021303
Luchsinger, J. A., Perez, T., Chang, H., Mehta, P., Steffener, J., Pradabhan, G., et al. (2016). Metformin in Amnestic Mild Cognitive Impairment: Results of a Pilot Randomized Placebo Controlled Clinical Trial. J. Alzheimers. Dis. 51, 501–514. doi: 10.3233/jad-150493
Luppino, F. S., De Wit, L. M., Bouvy, P. F., Stijnen, T., Cuijpers, P., Penninx, B. W., et al. (2010). Overweight, obesity, and depression: a systematic review and meta-analysis of longitudinal studies. Arch. Gen. Psychiatry 67, 220–229. doi: 10.1001/archgenpsychiatry.2010.2
Lutjohann, D., Breuer, O., Ahlborg, G., Nennesmo, I., Siden, A., Diczfalusy, U., et al. (1996). Cholesterol homeostasis in human brain: evidence for an age-dependent flux of 24S-hydroxycholesterol from the brain into the circulation. Proc. Natl. Acad. Sci. U.S.A. 93, 9799–9804. doi: 10.1073/pnas.93.18.9799
Ly, P. T., Wu, Y., Zou, H., Wang, R., Zhou, W., Kinoshita, A., et al. (2013). Inhibition of GSK3beta-mediated BACE1 expression reduces Alzheimer-associated phenotypes. J. Clin. Invest. 123, 224–235. doi: 10.1172/jci64516
Ma, L., Wang, R., Dong, W., and Zhao, Z. (2018). Caloric restriction can improve learning and memory in C57/BL mice probably via regulation of the AMPK signaling pathway. Exp. Gerontol. 102, 28–35. doi: 10.1016/j.exger.2017.11.013
Mac Giollabhui, N., Swistun, D., Murray, S., Moriarity, D. P., Kautz, M. M., Ellman, L. M., et al. (2020). Executive dysfunction in depression in adolescence: the role of inflammation and higher body mass. Psychol Med 50, 683–691. doi: 10.1017/s0033291719000564
MacPherson, K. P., Sompol, P., Kannarkat, G. T., Chang, J., Sniffen, L., Wildner, M. E., et al. (2017). Peripheral administration of the soluble TNF inhibitor XPro1595 modifies brain immune cell profiles, decreases beta-amyloid plaque load, and rescues impaired long-term potentiation in 5xFAD mice. Neurobiol. Dis. 102, 81–95. doi: 10.1016/j.nbd.2017.02.010
Maedler, K., Sergeev, P., Ris, F., Oberholzer, J., Joller-Jemelka, H. I., Spinas, G. A., et al. (2002). Glucose-induced beta cell production of IL-1beta contributes to glucotoxicity in human pancreatic islets. J. Clin. Invest. 110, 851–860. doi: 10.1172/jci200215318
Majumder, S., Richardson, A., Strong, R., and Oddo, S. (2011). Inducing autophagy by rapamycin before, but not after, the formation of plaques and tangles ameliorates cognitive deficits. PLoS ONE 6, e25416. doi: 10.1371/journal.pone.0025416
Malone, J. I., Hanna, S., Saporta, S., Mervis, R. F., Park, C. R., Chong, L., et al. (2008). Hyperglycemia not hypoglycemia alters neuronal dendrites and impairs spatial memory. Pediatr. Diabetes 9, 531–539. doi: 10.1111/j.1399-5448.2008.00431.x
Mao, Y. F., Guo, Z., Zheng, T., Jiang, Y., Yan, Y., Yin, X., et al. (2016). Intranasal insulin alleviates cognitive deficits and amyloid pathology in young adult APPswe/PS1dE9 mice. Aging Cell 15, 893–902. doi: 10.1111/acel.12498
Maric, T., Woodside, B., and Luheshi, G. N. (2014). The effects of dietary saturated fat on basal hypothalamic neuroinflammation in rats. Brain Behav. Immun. 36, 35–45. doi: 10.1016/j.bbi.2013.09.011
Marquez, D. X., Wilson, R., Aguinaga, S., Vasquez, P., Fogg, L., Yang, Z., et al. (2017). Regular Latin Dancing and Health Education May Improve Cognition of Late Middle-Aged and Older Latinos. J Aging Phys Act 25, 482–489. doi: 10.1123/japa.2016-0049
Mast, N., Saadane, A., Valencia-Olvera, A., Constans, J., Maxfield, E., Arakawa, H., et al. (2017). Cholesterol-metabolizing enzyme cytochrome P450 46A1 as a pharmacologic target for Alzheimer’s disease. Neuropharmacology 123, 465–476. doi: 10.1016/j.neuropharm.2017.06.026
Mayeux, R., and Stern, Y. (2012). Epidemiology of Alzheimer disease. Cold Spring Harb Perspect Med 2.
McGrattan, A. M., Mcguinness, B., Mckinley, M. C., Kee, F., Passmore, P., Woodside, J. V., et al. (2019). Diet and Inflammation in Cognitive Ageing and Alzheimer’s Disease. Curr Nutr Rep 8, 53–65.
Melo, H. M., Seixas, Da Silva, G. D. S., Sant’ana, M. R., Teixeira, C. V. L., Clarke, J. R., et al. (2020). Palmitate Is Increased in the Cerebrospinal Fluid of Humans with Obesity and Induces Memory Impairment in Mice via Pro-inflammatory TNF-alpha. Cell Rep 30.
Merino-Serrais, P., Benavides-Piccione, R., Blazquez-Llorca, L., Kastanauskaite, A., Rabano, A., Avila, J., et al. (2013). The influence of phospho-tau on dendritic spines of cortical pyramidal neurons in patients with Alzheimer’s disease. Brain 136, 1913–1928. doi: 10.1093/brain/awt088
Milanski, M., Degasperi, G., Coope, A., Morari, J., Denis, R., Cintra, D. E., et al. (2009). Saturated fatty acids produce an inflammatory response predominantly through the activation of TLR4 signaling in hypothalamus: implications for the pathogenesis of obesity. J. Neurosci. 29, 359–370. doi: 10.1523/jneurosci.2760-08.2009
Miwa, K., Okazaki, S., Sakaguchi, M., Mochizuki, H., and Kitagawa, K. (2016). Interleukin-6, interleukin-6 receptor gene variant, small-vessel disease and incident dementia. Eur. J. Neurol. 23, 656–663. doi: 10.1111/ene.12921
Morris, J. K., Uy, R. A. Z., Vidoni, E. D., Wilkins, H. M., Archer, A. E., Thyfault, J. P., et al. (2017). Effect of APOE epsilon4 Genotype on Metabolic Biomarkers in Aging and Alzheimer’s Disease. J. Alzheimers. Dis 58, 1129–1135. doi: 10.3233/jad-170148
Moser, V. A., Uchoa, M. F., and Pike, C. J. (2018). TLR4 inhibitor TAK-242 attenuates the adverse neural effects of diet-induced obesity. J Neuroinflammation 15, 306.
Mu, Y., and Gage, F. H. (2011). Adult hippocampal neurogenesis and its role in Alzheimer’s disease. Mol.Neurodegener. 6, 85. doi: 10.1186/1750-1326-6-85
Mullins, R. J., Mustapic, M., Chia, C. W., Carlson, O., Gulyani, S., Tran, J., et al. (2019). A Pilot Study of Exenatide Actions in Alzheimer’s Disease. Curr Alzheimer Res 16, 741–752. doi: 10.2174/1567205016666190913155950
Mun, M. J., Kim, J. H., Choi, J. Y., and Jang, W. C. (2016). Genetic polymorphisms of interleukin genes and the risk of Alzheimer’s disease: An update meta-analysis. Meta Gene 8, 1–10. doi: 10.1016/j.mgene.2016.01.001
Myette-Cote, E., Durrer, C., Neudorf, H., Bammert, T. D., Botezelli, J. D., Johnson, J. D., et al. (2018). The effect of a short-term low-carbohydrate, high-fat diet with or without postmeal walks on glycemic control and inflammation in type 2 diabetes: a randomized trial. Am. J. Physiol. Regul. Integr. Comp. Physiol. 315, R1210–R1219.
Myhre, C. L., Thygesen, C., Villadsen, B., Vollerup, J., Ilkjaer, L., Krohn, K. T., et al. (2019). Microglia Express Insulin-Like Growth Factor-1 in the Hippocampus of Aged APPswe/PS1DeltaE9 Transgenic Mice. Front Cell Neurosci 13, 308.
Nadler, Y., Alexandrovich, A., Grigoriadis, N., Hartmann, T., Rao, K. S., Shohami, E., et al. (2008). Increased expression of the gamma-secretase components presenilin-1 and nicastrin in activated astrocytes and microglia following traumatic brain injury. Glia 56, 552–567. doi: 10.1002/glia.20638
Napoli, N., Shah, K., Waters, D. L., Sinacore, D. R., Qualls, C., and Villareal, D. T. (2014). Effect of weight loss, exercise, or both on cognition and quality of life in obese older adults. Am. J. Clin. Nutr. 100, 189–198. doi: 10.3945/ajcn.113.082883
Nguyen, T. T., Ta, Q. T. H., Nguyen, T. T. D., Le, T. T., and Vo, V. G. (2020). Role of Insulin Resistance in the Alzheimer’s Disease Progression. Neurochem. Res. 45, 1481–1491.
O’Brien, P. D., Hinder, L. M., Callaghan, B. C., and Feldman, E. L. (2017). Neurological consequences of obesity. Lancet Neurol. 16, 465–477.
O’Bryant, S. E., Hobson, V., Hall, J. R., Waring, S. C., Chan, W., Massman, P., et al. (2009). Brain-derived neurotrophic factor levels in Alzheimer’s disease. J. Alzheimers. Dis. 17, 337–341.
Ou, Z., Kong, X., Sun, X., He, X., Zhang, L., Gong, Z., et al. (2018). Metformin treatment prevents amyloid plaque deposition and memory impairment in APP/PS1 mice. Brain Behav. Immun. 69, 351–363. doi: 10.1016/j.bbi.2017.12.009
Panagaki, T., Gengler, S., and Holscher, C. (2018). The Novel DA-CH3 Dual Incretin Restores Endoplasmic Reticulum Stress and Autophagy Impairments to Attenuate Alzheimer-Like Pathology and Cognitive Decrements in the APPSWE/PS1DeltaE9 Mouse Model. J. Alzheimers. Dis. 66, 195–218. doi: 10.3233/jad-180584
Pang, R., Wang, X., Pei, F., Zhang, W., Shen, J., Gao, X., et al. (2019). Regular Exercise Enhances Cognitive Function and Intracephalic GLUT Expression in Alzheimer’s Disease Model Mice. J. Alzheimers. Dis. 72, 83–96. doi: 10.3233/jad-190328
Paouri, E., Tzara, O., Zenelak, S., and Georgopoulos, S. (2017). Genetic Deletion of Tumor Necrosis Factor-alpha Attenuates Amyloid-beta Production and Decreases Amyloid Plaque Formation and Glial Response in the 5XFAD Model of Alzheimer’s Disease. J. Alzheimers. Dis. 60, 165–181. doi: 10.3233/jad-170065
Pardo, M., Abrial, E., Jope, R. S., and Beurel, E. (2016). GSK3beta isoform-selective regulation of depression, memory and hippocampal cell proliferation. Genes Brain Behav. 15, 348–355. doi: 10.1111/gbb.12283
Parimisetty, A., Dorsemans, A. C., Awada, R., Ravanan, P., Diotel, N., and Lefebvre D’hellencourt, C. (2016). Secret talk between adipose tissue and central nervous system via secreted factors-an emerging frontier in the neurodegenerative research. J Neuroinflammation 13, 67.
Pasqualetti, P., Bonomini, C., Dal Forno, G., Paulon, L., Sinforiani, E., Marra, C., et al. (2009). A randomized controlled study on effects of ibuprofen on cognitive progression of Alzheimer’s disease. Aging Clin. Exp. Res. 21, 102–110. doi: 10.1007/bf03325217
Patterson, S. L. (2015). Immune dysregulation and cognitive vulnerability in the aging brain: Interactions of microglia, IL-1beta, BDNF and synaptic plasticity. Neuropharmacology 96, 11–18. doi: 10.1016/j.neuropharm.2014.12.020
Peineau, S., Taghibiglou, C., Bradley, C., Wong, T. P., Liu, L., Lu, J., et al. (2007). LTP inhibits LTD in the hippocampus via regulation of GSK3beta. Neuron 53, 703–717. doi: 10.1016/j.neuron.2007.01.029
Peng, S., Wuu, J., Mufson, E. J., and Fahnestock, M. (2005). Precursor form of brain-derived neurotrophic factor and mature brain-derived neurotrophic factor are decreased in the pre-clinical stages of Alzheimer’s disease. J. Neurochem. 93, 1412–1421. doi: 10.1111/j.1471-4159.2005.03135.x
Poggi, M., Bastelica, D., Gual, P., Iglesias, M. A., Gremeaux, T., Knauf, C., et al. (2007). C3H/HeJ mice carrying a toll-like receptor 4 mutation are protected against the development of insulin resistance in white adipose tissue in response to a high-fat diet. Diabetologia 50, 1267–1276. doi: 10.1007/s00125-007-0654-8
Pomilio, C., Gorojod, R. M., Riudavets, M., Vinuesa, A., Presa, J., Gregosa, A., et al. (2020). Microglial autophagy is impaired by prolonged exposure to beta-amyloid peptides: evidence from experimental models and Alzheimer’s disease patients. Geroscience 42, 613–632. doi: 10.1007/s11357-020-00161-9
Puder, J. J., and Munsch, S. (2010). Psychological correlates of childhood obesity. Int.J.Obes.(Lond) 34(Suppl. 2), S37–S43.
Qi, Y., Klyubin, I., Cuello, A. C., and Rowan, M. J. (2018). NLRP3-dependent synaptic plasticity deficit in an Alzheimer’s disease amyloidosis model in vivo. Neurobiol. Dis. 114, 24–30. doi: 10.1016/j.nbd.2018.02.016
Qiu, W. Q., and Folstein, M. F. (2006). Insulin, insulin-degrading enzyme and amyloid-beta peptide in Alzheimer’s disease: review and hypothesis. Neurobiol. Aging 27, 190–198. doi: 10.1016/j.neurobiolaging.2005.01.004
Quinn, J. F., Raman, R., Thomas, R. G., Yurko-Mauro, K., Nelson, E. B., Van Dyck, C., et al. (2010). Docosahexaenoic acid supplementation and cognitive decline in Alzheimer disease: a randomized trial. JAMA 304, 1903–1911. doi: 10.1001/jama.2010.1510
Rajasekar, N., Nath, C., Hanif, K., and Shukla, R. (2017a). Intranasal Insulin Administration Ameliorates Streptozotocin (ICV)-Induced Insulin Receptor Dysfunction, Neuroinflammation, Amyloidogenesis, and Memory Impairment in Rats. Mol. Neurobiol. 54, 6507–6522. doi: 10.1007/s12035-016-0169-8
Rajasekar, N., Nath, C., Hanif, K., and Shukla, R. (2017b). Intranasal insulin improves cerebral blood flow, Nrf-2 expression and BDNF in STZ (ICV)-induced memory impaired rats. Life Sci. 173, 1–10. doi: 10.1016/j.lfs.2016.09.020
Rao, P., and Knaus, E. E. (2008). Evolution of nonsteroidal anti-inflammatory drugs (NSAIDs): cyclooxygenase (COX) inhibition and beyond. J. Pharm. Pharm. Sci 11, 81s–110s.
Reed, B., Villeneuve, S., Mack, W., Decarli, C., Chui, H. C., and Jagust, W. (2014). Associations between serum cholesterol levels and cerebral amyloidosis. JAMA Neurol 71, 195–200. doi: 10.1001/jamaneurol.2013.5390
Reger, M. A., Henderson, S. T., Hale, C., Cholerton, B., Baker, L. D., Watson, G. S., et al. (2004). Effects of beta-hydroxybutyrate on cognition in memory-impaired adults. Neurobiol. Aging 25, 311–314. doi: 10.1016/s0197-4580(03)00087-3
Reger, M. A., Watson, G. S., Green, P. S., Baker, L. D., Cholerton, B., Fishel, M. A., et al. (2008). Intranasal insulin administration dose-dependently modulates verbal memory and plasma amyloid-beta in memory-impaired older adults. J. Alzheimers. Dis. 13, 323–331. doi: 10.3233/jad-2008-13309
Reich, D., Gallucci, G., Tong, M., and De La Monte, S. M. (2018). Therapeutic Advantages of Dual Targeting of PPAR-delta and PPAR-gamma in an Experimental Model of Sporadic Alzheimer’s Disease. J Parkinsons Dis Alzheimers Dis 5.
Rhea, E. M., Nirkhe, S., Nguyen, S., Pemberton, S., Bammler, T. K., Beyer, R., et al. (2019). Molecular Mechanisms of Intranasal Insulin in SAMP8 Mice. J. Alzheimers. Dis. 71, 1361–1373. doi: 10.3233/jad-190707
Ridker, P. M., Rifai, N., Stampfer, M. J., and Hennekens, C. H. (2000). Plasma concentration of interleukin-6 and the risk of future myocardial infarction among apparently healthy men. Circulation 101, 1767–1772. doi: 10.1161/01.cir.101.15.1767
Risner, M. E., Saunders, A. M., Altman, J. F., Ormandy, G. C., Craft, S., Foley, I. M., et al. (2006). Efficacy of rosiglitazone in a genetically defined population with mild-to-moderate Alzheimer’s disease. Pharmacogenomics J. 6, 246–254. doi: 10.1038/sj.tpj.6500369
Rockenstein, E., Torrance, M., Adame, A., Mante, M., Bar-On, P., Rose, J. B., et al. (2007). Neuroprotective effects of regulators of the glycogen synthase kinase-3beta signaling pathway in a transgenic model of Alzheimer’s disease are associated with reduced amyloid precursor protein phosphorylation. J. Neurosci. 27, 1981–1991. doi: 10.1523/jneurosci.4321-06.2007
Rogers, J., Kirby, L. C., Hempelman, S. R., Berry, D. L., Mcgeer, P. L., Kaszniak, A. W., et al. (1993). Clinical trial of indomethacin in Alzheimer’s disease. Neurology 43, 1609–1611.
Ronan, L., Alexander-Bloch, A., and Fletcher, P. C. (2019). Childhood Obesity, Cortical Structure, and Executive Function in Healthy Children. Cereb Cortex.
Ruegsegger, G. N., Vanderboom, P. M., Dasari, S., Klaus, K. A., Kabiraj, P., Mccarthy, C. B., et al. (2019). Exercise and metformin counteract altered mitochondrial function in the insulin-resistant brain. JCI Insight 4.
Ryan, C. M., Freed, M. I., Rood, J. A., Cobitz, A. R., Waterhouse, B. R., and Strachan, M. W. (2006). Improving metabolic control leads to better working memory in adults with type 2 diabetes. Diabetes Care 29, 345–351. doi: 10.2337/diacare.29.02.06.dc05-1626
Ryan, S. M., and Kelly, A. M. (2016). Exercise as a pro-cognitive, pro-neurogenic and anti-inflammatory intervention in transgenic mouse models of Alzheimer’s disease. Ageing Res. Rev. 27, 77–92. doi: 10.1016/j.arr.2016.03.007
Sah, S. K., Lee, C., Jang, J. H., and Park, G. H. (2017). Effect of high-fat diet on cognitive impairment in triple-transgenic mice model of Alzheimer’s disease. Biochem. Biophys. Res. Commun. 493, 731–736. doi: 10.1016/j.bbrc.2017.08.122
Saini, R. K., Rengasamy, K. R. R., Mahomoodally, F. M., and Keum, Y. S. (2020). Protective effects of lycopene in cancer, cardiovascular, and neurodegenerative diseases: An update on epidemiological and mechanistic perspectives. Pharmacol. Res. 155, 104730. doi: 10.1016/j.phrs.2020.104730
Saiz-Vazquez, O., Puente-Martinez, A., Ubillos-Landa, S., Pacheco-Bonrostro, J., and Santabarbara, J. (2020). Cholesterol and Alzheimer’s Disease Risk: A Meta-Meta-Analysis. Brain Sci 10.
Salameh, T. S., Bullock, K. M., Hujoel, I. A., Niehoff, M. L., Wolden-Hanson, T., Kim, J., et al. (2015). Central Nervous System Delivery of Intranasal Insulin: Mechanisms of Uptake and Effects on Cognition. J. Alzheimers. Dis. 47, 715–728. doi: 10.3233/jad-150307
Salameh, T. S., Mortell, W. G., Logsdon, A. F., Butterfield, D. A., and Banks, W. A. (2019). Disruption of the hippocampal and hypothalamic blood-brain barrier in a diet-induced obese model of type II diabetes: prevention and treatment by the mitochondrial carbonic anhydrase inhibitor, topiramate. Fluids Barriers CNS 16, 1.
Samant, N. P., and Gupta, G. L. (2021). Novel therapeutic strategies for Alzheimer’s disease targeting brain cholesterol homeostasis. Eur. J. Neurosci. 53, 673–686. doi: 10.1111/ejn.14949
Sanguinetti, E., Guzzardi, M. A., Panetta, D., Tripodi, M., De Sena, V., Quaglierini, M., et al. (2019). Combined Effect of Fatty Diet and Cognitive Decline on Brain Metabolism, Food Intake, Body Weight, and Counteraction by Intranasal Insulin Therapy in 3xTg Mice. Front Cell Neurosci 13, 188.
Sato, T., Hanyu, H., Hirao, K., Kanetaka, H., Sakurai, H., and Iwamoto, T. (2011). Efficacy of PPAR-gamma agonist pioglitazone in mild Alzheimer disease. Neurobiol. Aging 32, 1626–1633. doi: 10.1016/j.neurobiolaging.2009.10.009
Scheff, S. W., Price, D. A., Ansari, M. A., Roberts, K. N., Schmitt, F. A., Ikonomovic, M. D., et al. (2015). Synaptic change in the posterior cingulate gyrus in the progression of Alzheimer’s disease. J. Alzheimers. Dis. 43, 1073–1090. doi: 10.3233/jad-141518
Scheff, S. W., Price, D. A., Schmitt, F. A., and Mufson, E. J. (2006). Hippocampal synaptic loss in early Alzheimer’s disease and mild cognitive impairment. Neurobiol. Aging 27, 1372–1384. doi: 10.1016/j.neurobiolaging.2005.09.012
Schmid, V., Kullmann, S., Gfrorer, W., Hund, V., Hallschmid, M., Lipp, H. P., et al. (2018). Safety of intranasal human insulin: A review. Diabetes. Obes. Metab 20, 1563–1577. doi: 10.1111/dom.13279
Searcy, J. L., Phelps, J. T., Pancani, T., Kadish, I., Popovic, J., Anderson, K. L., et al. (2012). Long-term pioglitazone treatment improves learning and attenuates pathological markers in a mouse model of Alzheimer’s disease. J. Alzheimers. Dis. 30, 943–961. doi: 10.3233/jad-2012-111661
Selemon, L. D. (2013). A role for synaptic plasticity in the adolescent development of executive function. Transl Psychiatry 3, e238. doi: 10.1038/tp.2013.7
Shellington, E. M., Reichert, S. M., Heath, M., Gill, D. P., Shigematsu, R., and Petrella, R. J. (2018). Results From a Feasibility Study of Square-Stepping Exercise in Older Adults With Type 2 Diabetes and Self-Reported Cognitive Complaints to Improve Global Cognitive Functioning. Can J Diabetes 42.
Sheng, J. G., Bora, S. H., Xu, G., Borchelt, D. R., Price, D. L., and Koliatsos, V. E. (2003). Lipopolysaccharide-induced-neuroinflammation increases intracellular accumulation of amyloid precursor protein and amyloid beta peptide in APPswe transgenic mice. Neurobiol. Dis. 14, 133–145. doi: 10.1016/s0969-9961(03)00069-x
Shinto, L., Quinn, J., Montine, T., Dodge, H. H., Woodward, W., Baldauf-Wagner, S., et al. (2014). A randomized placebo-controlled pilot trial of omega-3 fatty acids and alpha lipoic acid in Alzheimer’s disease. J. Alzheimers. Dis. 38, 111–120. doi: 10.3233/jad-130722
Sobol, N. A., Dall, C. H., Hogh, P., Hoffmann, K., Frederiksen, K. S., Vogel, A., et al. (2018). Change in Fitness and the Relation to Change in Cognition and Neuropsychiatric Symptoms After Aerobic Exercise in Patients with Mild Alzheimer’s Disease. J. Alzheimers. Dis. 65, 137–145. doi: 10.3233/jad-180253
Sochocka, M., Donskow-Lysoniewska, K., Diniz, B. S., Kurpas, D., Brzozowska, E., and Leszek, J. (2019). The Gut Microbiome Alterations and Inflammation-Driven Pathogenesis of Alzheimer’s Disease-a Critical Review. Mol. Neurobiol. 56, 1841–1851. doi: 10.1007/s12035-018-1188-4
Solas, M., Milagro, F. I., Ramirez, M. J., and Martinez, J. A. (2017). Inflammation and gut-brain axis link obesity to cognitive dysfunction: plausible pharmacological interventions. Curr. Opin. Pharmacol 37, 87–92. doi: 10.1016/j.coph.2017.10.005
Solmaz, V., Cinar, B. P., Yigitturk, G., Cavusoglu, T., Taskiran, D., and Erbas, O. (2015). Exenatide reduces TNF-alpha expression and improves hippocampal neuron numbers and memory in streptozotocin treated rats. Eur. J. Pharmacol 765, 482–487. doi: 10.1016/j.ejphar.2015.09.024
Solomon, A., Kivipelto, M., Wolozin, B., Zhou, J., and Whitmer, R. A. (2009). Midlife serum cholesterol and increased risk of Alzheimer’s and vascular dementia three decades later. Dement. Geriatr. Cogn. Disord 28, 75–80. doi: 10.1159/000231980
Sparks, D. L., Scheff, S. W., Hunsaker, J. C. III, Liu, H., Landers, T., and Gross, D. R. (1994). Induction of Alzheimer-like beta-amyloid immunoreactivity in the brains of rabbits with dietary cholesterol. Exp. Neurol. 126, 88–94. doi: 10.1006/exnr.1994.1044
Spilman, P., Podlutskaya, N., Hart, M. J., Debnath, J., Gorostiza, O., Bredesen, D., et al. (2010). Inhibition of mTOR by rapamycin abolishes cognitive deficits and reduces amyloid-beta levels in a mouse model of Alzheimer’s disease. PLoS.One. 5, e9979. doi: 10.1371/journal.pone.0009979
Stavoe, A. K. H., and Holzbaur, E. L. F. (2020). Neuronal autophagy declines substantially with age and is rescued by overexpression of WIPI2. Autophagy 16, 371–372. doi: 10.1080/15548627.2019.1695401
Stebbings, K. A., Choi, H. W., Ravindra, A., and Llano, D. A. (2016). The impact of aging, hearing loss, and body weight on mouse hippocampal redox state, measured in brain slices using fluorescence imaging. Neurobiol. Aging 42, 101–109. doi: 10.1016/j.neurobiolaging.2016.03.006
Steen, E., Terry, B. M., Rivera, E. J., Cannon, J. L., Neely, T. R., Tavares, R., et al. (2005). Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer’s disease–is this type 3 diabetes? J Alzheimers Dis 7, 63–80. doi: 10.3233/jad-2005-7107
Stewart, C. R., Stuart, L. M., Wilkinson, K., Van Gils, J. M., Deng, J., Halle, A., et al. (2010). CD36 ligands promote sterile inflammation through assembly of a Toll-like receptor 4 and 6 heterodimer. Nat. Immunol. 11, 155–161. doi: 10.1038/ni.1836
Stohr, O., Schilbach, K., Moll, L., Hettich, M. M., Freude, S., Wunderlich, F. T., et al. (2013). Insulin receptor signaling mediates APP processing and beta-amyloid accumulation without altering survival in a transgenic mouse model of Alzheimer’s disease. Age (Dordr) 35, 83–101. doi: 10.1007/s11357-011-9333-2
Stranahan, A. M., Arumugam, T. V., Cutler, R. G., Lee, K., Egan, J. M., and Mattson, M. P. (2008). Diabetes impairs hippocampal function through glucocorticoid-mediated effects on new and mature neurons. Nat.Neurosci. 11, 309–317. doi: 10.1038/nn2055
Subramanian, J., Savage, J. C., and Tremblay, M. E. (2020). Synaptic Loss in Alzheimer’s Disease: Mechanistic Insights Provided by Two-Photon in vivo Imaging of Transgenic Mouse Models. Front Cell Neurosci 14, 592607.
Tai, L. M., Koster, K. P., Luo, J., Lee, S. H., Wang, Y. T., Collins, N. C., et al. (2014). Amyloid-beta pathology and APOE genotype modulate retinoid X receptor agonist activity in vivo. J. Biol. Chem. 289, 30538–30555. doi: 10.1074/jbc.m114.600833
Tamtaji, O. R., Heidari-Soureshjani, R., Mirhosseini, N., Kouchaki, E., Bahmani, F., Aghadavod, E., et al. (2019). Probiotic and selenium co-supplementation, and the effects on clinical, metabolic and genetic status in Alzheimer’s disease: A randomized, double-blind, controlled trial. Clin. Nutr. 38, 2569–2575. doi: 10.1016/j.clnu.2018.11.034
Tanokashira, D., Kurata, E., Fukuokaya, W., Kawabe, K., Kashiwada, M., Takeuchi, H., et al. (2018). Metformin treatment ameliorates diabetes-associated decline in hippocampal neurogenesis and memory via phosphorylation of insulin receptor substrate 1. FEBS Open Bio 8, 1104–1118. doi: 10.1002/2211-5463.12436
Taylor, M. K., Sullivan, D. K., Mahnken, J. D., Burns, J. M., and Swerdlow, R. H. (2018). Feasibility and efficacy data from a ketogenic diet intervention in Alzheimer’s disease. Alzheimers Dement (N Y) 4, 28–36. doi: 10.1016/j.trci.2017.11.002
Theriault, P., Elali, A., and Rivest, S. (2016). High fat diet exacerbates Alzheimer’s disease-related pathology in APPswe/PS1 mice. Oncotarget 7, 67808–67827.
Tobin, M. K., Musaraca, K., Disouky, A., Shetti, A., Bheri, A., Honer, W. G., et al. (2019). Human Hippocampal Neurogenesis Persists in Aged Adults and Alzheimer’s Disease Patients. Cell Stem Cell.
Toda, T., and Gage, F. H. (2017). Review: adult neurogenesis contributes to hippocampal plasticity. Cell Tissue Res.
Tong, L., Prieto, G. A., Kramar, E. A., Smith, E. D., Cribbs, D. H., Lynch, G., et al. (2012). Brain-derived neurotrophic factor-dependent synaptic plasticity is suppressed by interleukin-1beta via p38 mitogen-activated protein kinase. J. Neurosci. 32, 17714–17724.
Tremmel, M., Gerdtham, U. G., Nilsson, P. M., and Saha, S. (2017). Economic Burden of Obesity: A Systematic Literature Review. Int J Environ Res Public Health 14.
Tu, S., Okamoto, S., Lipton, S. A., and Xu, H. (2014). Oligomeric Abeta-induced synaptic dysfunction in Alzheimer’s disease. Mol Neurodegener 9, 48.
Turnbaugh, P. J., Ley, R. E., Mahowald, M. A., Magrini, V., Mardis, E. R., and Gordon, J. I. (2006). An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 444, 1027–1031.
Uddin, M. S., Mamun, A. A., Labu, Z. K., Hidalgo-Lanussa, O., Barreto, G. E., and Ashraf, G. M. (2019). Autophagic dysfunction in Alzheimer’s disease: Cellular and molecular mechanistic approaches to halt Alzheimer’s pathogenesis. J. Cell. Physiol. 234, 8094–8112.
Van der Auwera, I., Wera, S., Van Leuven, F., and Henderson, S. T. (2005). A ketogenic diet reduces amyloid beta 40 and 42 in a mouse model of Alzheimer’s disease. Nutr Metab (Lond) 2, 28.
van der Kant, R., Langness, V. F., Herrera, C. M., Williams, D. A., Fong, L. K., Leestemaker, Y., et al. (2019). Cholesterol Metabolism Is a Druggable Axis that Independently Regulates Tau and Amyloid-beta in iPSC-Derived Alzheimer’s Disease Neurons. Cell Stem Cell 24.
van Duinkerken, E., and Ryan, C. M. (2019). Diabetes mellitus in the young and the old: Effects on cognitive functioning across the life span. Neurobiol. Dis. 134, 104608.
Varga, T., Czimmerer, Z., and Nagy, L. (2011). PPARs are a unique set of fatty acid regulated transcription factors controlling both lipid metabolism and inflammation. Biochim. Biophys. Acta 1812, 1007–1022.
Velloso, L. A., Folli, F., and Saad, M. J. (2015). TLR4 at the Crossroads of Nutrients, Gut Microbiota, and Metabolic Inflammation. Endocr.Rev. 36, 245–271.
Vidal-Puig, A., Jimenez-Linan, M., Lowell, B. B., Hamann, A., Hu, E., Spiegelman, B., et al. (1996). Regulation of PPAR gamma gene expression by nutrition and obesity in rodents. J. Clin. Invest. 97, 2553–2561.
Vinuesa, A., Bentivegna, M., Calfa, G., Filipello, F., Pomilio, C., Bonaventura, M. M., et al. (2019). Early Exposure to a High-Fat Diet Impacts on Hippocampal Plasticity: Implication of Microglia-Derived Exosome-like Extracellular Vesicles. Mol. Neurobiol. 56, 5075–5094.
Vinuesa, A., Pomilio, C., Menafra, M., Bonaventura, M. M., Garay, L., Mercogliano, M. F., et al. (2016). Juvenile exposure to a high fat diet promotes behavioral and limbic alterations in the absence of obesity. Psychoneuroendocrinology 72, 22–33.
Vogt, N. M., Kerby, R. L., Dill-Mcfarland, K. A., Harding, S. J., Merluzzi, A. P., Johnson, S. C., et al. (2017). Gut microbiome alterations in Alzheimer’s disease. Sci Rep 7, 13537.
Wang, B., and Tontonoz, P. (2018). Liver X receptors in lipid signalling and membrane homeostasis. Nat Rev Endocrinol 14, 452–463.
Wang, Q., Hu, J., Liu, Y., Li, J., Liu, B., Li, M., et al. (2019). Aerobic Exercise Improves Synaptic-Related Proteins of Diabetic Rats by Inhibiting FOXO1/NF-kappaB/NLRP3 Inflammatory Signaling Pathway and Ameliorating PI3K/Akt Insulin Signaling Pathway. J. Mol. Neurosci. 69, 28–38.
Wang, T. (2015). TNF-alpha G308A polymorphism and the susceptibility to Alzheimer’s disease: an updated meta-analysis. Arch. Med. Res. 46:24.
Wang, T., Fu, F. H., Han, B., Zhang, L. M., and Zhang, X. M. (2011). Long-term but not short-term aspirin treatment attenuates diabetes-associated learning and memory decline in mice. Exp. Clin. Endocrinol. Diabetes 119, 36–40.
Wang, Z., Dong, B., Hu, J., Adegbija, O., and Arnold, L. W. (2016). Exploring the non-linear association between BMI and mortality in adults with and without diabetes: the US National Health Interview Survey. Diabet. Med. 33, 1691–1699.
Warden, A., Truitt, J., Merriman, M., Ponomareva, O., Jameson, K., Ferguson, L. B., et al. (2016). Localization of PPAR isotypes in the adult mouse and human brain. Sci Rep 6, 27618.
Watson, G. S., Cholerton, B. A., Reger, M. A., Baker, L. D., Plymate, S. R., Asthana, S., et al. (2005). Preserved cognition in patients with early Alzheimer disease and amnestic mild cognitive impairment during treatment with rosiglitazone: a preliminary study. Am. J. Geriatr. Psychiatry 13, 950–958.
Wei, L., Yao, M., Zhao, Z., Jiang, H., and Ge, S. (2018). High-fat diet aggravates postoperative cognitive dysfunction in aged mice. BMC Anesthesiol 18, 20.
Who. (2018). Obesity and overweight [Online]. Available: http://www.who.int/mediacentre/factsheets/fs311/en/[Accessed] (accessed January 10, 2021).
Who. (2020). https://www.who.int/news-room/fact-sheets/detail/dementia. WHO. (accessed January 10, 2021).
Wilhelmi de Toledo, F., Grundler, F., Bergouignan, A., Drinda, S., and Michalsen, A. (2019). Safety, health improvement and well-being during a 4 to 21-day fasting period in an observational study including 1422 subjects. PLoS ONE 14, e0209353.
Xue, Q. S., Sparks, D. L., and Streit, W. J. (2007). Microglial activation in the hippocampus of hypercholesterolemic rabbits occurs independent of increased amyloid production. J Neuroinflammation 4, 20.
Yan, Q., Zhang, J., Liu, H., Babu-Khan, S., Vassar, R., Biere, A. L., et al. (2003). Anti-inflammatory drug therapy alters beta-amyloid processing and deposition in an animal model of Alzheimer’s disease. J. Neurosci. 23, 7504–7509.
Ying, W., Fu, W., Lee, Y. S., and Olefsky, J. M. (2020). The role of macrophages in obesity-associated islet inflammation and beta-cell abnormalities. Nat Rev Endocrinol 16, 81–90.
Yoon, D. H., Lee, J. Y., and Song, W. (2018). Effects of Resistance Exercise Training on Cognitive Function and Physical Performance in Cognitive Frailty: A Randomized Controlled Trial. J. Nutr. Health Aging 22, 944–951.
Zatta, P., Zambenedetti, P., Stella, M. P., and Licastro, F. (2002). Astrocytosis, microgliosis, metallothionein-I-II and amyloid expression in high cholesterol-fed rabbits. J. Alzheimers. Dis. 4, 1–9.
Zhang-Gandhi, C. X., and Drew, P. D. (2007). Liver X receptor and retinoid X receptor agonists inhibit inflammatory responses of microglia and astrocytes. J. Neuroimmunol. 183, 50–59.
Zhang, L., Tang, W., Chao, F. L., Zhou, C. N., Jiang, L., Zhang, Y., et al. (2020). Four-month treadmill exercise prevents the decline in spatial learning and memory abilities and the loss of spinophilin-immunoreactive puncta in the hippocampus of APP/PS1 transgenic mice. Neurobiol. Dis. 136, 104723.
Zhang, N., Liang, H., Farese, R. V., Li, J., Musi, N., and Hussey, S. E. (2015). Pharmacological TLR4 Inhibition Protects against Acute and Chronic Fat-Induced Insulin Resistance in Rats. PLoS ONE 10, e0132575.
Zhang, Z. H., Wen, L., Wu, Q. Y., Chen, C., Zheng, R., Liu, Q., et al. (2017). Long-Term Dietary Supplementation with Selenium-Enriched Yeast Improves Cognitive Impairment, Reverses Synaptic Deficits, and Mitigates Tau Pathology in a Triple Transgenic Mouse Model of Alzheimer’s Disease. J. Agric. Food Chem. 65, 4970–4979.
Keywords: Alzheimer’s disease, metabolic disorders, cognitive impairment, insulin resistance, inflammation, therapies
Citation: Vinuesa A, Pomilio C, Gregosa A, Bentivegna M, Presa J, Bellotto M, Saravia F and Beauquis J (2021) Inflammation and Insulin Resistance as Risk Factors and Potential Therapeutic Targets for Alzheimer’s Disease. Front. Neurosci. 15:653651. doi: 10.3389/fnins.2021.653651
Received: 14 January 2021; Accepted: 31 March 2021;
Published: 23 April 2021.
Edited by:
Con Stough, Swinburne University of Technology, AustraliaReviewed by:
Maite Solas, University of Navarra, SpainCopyright © 2021 Vinuesa, Pomilio, Gregosa, Bentivegna, Presa, Bellotto, Saravia and Beauquis. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Juan Beauquis, anVhbmJlYXVxdWlzQGdtYWlsLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.