 Samir Abu-Rumeileh
Samir Abu-Rumeileh Piero Parchi
Piero Parchi
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Neurosci., 12 March 2021
Sec. Neurodegeneration
Volume 15 - 2021 | https://doi.org/10.3389/fnins.2021.648743
This article is part of the Research TopicThe Impact of Neurofilament Light Chain (NFL) Quantification in Serum and Cerebrospinal Fluid in Neurodegenerative DiseasesView all 17 articles
Rapidly progressive dementia (RPD) is an umbrella term referring to several conditions causing a rapid neurological deterioration associated with cognitive decline and short disease duration. They comprise Creutzfeldt–Jakob disease (CJD), the archetypal RPD, rapidly progressive variants of the most common neurodegenerative dementias (NDs), and potentially treatable conditions such as infectious or autoimmune encephalitis and cerebrovascular disease. Given the significant clinical and, sometimes, neuroradiological overlap between these different disorders, biofluid markers also contribute significantly to the differential diagnosis. Among them, the neurofilament light chain protein (NfL) has attracted growing attention in recent years as a biofluid marker of neurodegeneration due to its sensitivity to axonal damage and the reliability of its measurement in both cerebrospinal fluid (CSF) and blood. Here, we summarize current knowledge regarding biological and clinical implications of NfL evaluation in biofluids across RPDs, emphasizing CJD, and other prion diseases. In the latter, NfL demonstrated a good diagnostic and prognostic accuracy and a potential value as a marker of proximity to clinical onset in pre-symptomatic PRNP mutation carriers. Similarly, in Alzheimer’s disease and other NDs, higher NfL concentrations seem to predict a faster disease progression. While increasing evidence indicates a potential clinical value of NfL in monitoring cerebrovascular disease, the association between NfL and prediction of outcome and/or disease activity in autoimmune encephalitis and infectious diseases has only been investigated in few cohorts and deserves confirmatory studies. In the era of precision medicine and evolving therapeutic options, CSF and blood NfL might aid the diagnostic and prognostic assessment of RPDs and the stratification and management of patients according to disease progression in clinical trials.
Rapidly progressive dementia (RPD) is an umbrella term that comprehends several heterogenous diseases characterized by a rapidly progressive cognitive decline and a short disease duration, typically 1–2 years between onset and occurrence of dementia and/or of 2–3 years from onset to death (Cohen et al., 2015; Grau-Rivera et al., 2015; Geschwind and Murray, 2018; Rudge et al., 2018; Zerr and Hermann, 2018). Evidence from epidemiological studies indicates that RPD is not a rare condition. However, data concerning the actual prevalence are lacking. The most frequent causes of RPDs comprise prion disease and other neurodegenerative dementias (NDs), autoimmune and infectious encephalitis, vascular and metabolic encephalopathies, and malignancies (Geschwind and Murray, 2018; Zerr and Hermann, 2018).
Sporadic Creutzfeldt–Jakob’s disease (CJD) represents the prototype of RPDs and is often referred to as the “great mimicker” given its wide phenotypic heterogeneity (Baiardi et al., 2018; Geschwind and Murray, 2018). The extensive clinical overlaps between RPDs, the urgency and severity of the clinical scenario, and the existence of treatable forms make the availability of reliable diagnostic and prognostic biomarkers of primary importance. Brain magnetic resonance imaging (MRI), cerebrospinal fluid (CSF) surrogate markers such as proteins total (t)-tau and 14-3-3, and electroencephalographic (EEG) examination, support the clinical diagnosis of probable CJD according to current criteria (Zerr and Parchi, 2018; Rhoads et al., 2020). However, the overall sensitivity and specificity of these diagnostic investigations are not optimal. Indeed, a significant proportion of non-prion RPDs may show positivity in one or more of these tests, whereas, on the other hand, atypical disease subtypes with slower progression may escape recognition (Hamlin et al., 2012; Lattanzio et al., 2017; Rudge et al., 2018). In this regard, the prion real-time quacking-induced conversion (RT-QuIC) assay with its virtually full specificity for prion disease significantly contributed to the improvement of the diagnostic assessment of RPDs (Candelise et al., 2020). However, the assay is still not fully available and represents in many laboratories a second-step diagnostic assay to be applied when screening tests are suggestive for prion disease (Abu-Rumeileh et al., 2019a). Thus, the availability of sensitive and readily accessible analytes for large and rapid screening remains of critical importance. Besides, there is also urgent need for markers to use as surrogate end points in ongoing clinical trials showing positive associations with prognostic variables, such as overall survival, disease severity, and progression rate.
The assessment of neurofilament light chain protein (NfL) has attracted growing attention in recent years due to its sensitivity to neuronal damage and reliability of its measurement in both CSF and blood (Gaetani et al., 2019; Barro et al., 2020). NfL is a subunit of neurofilaments, which are cytoskeletal proteins mainly located in large myelinated axons where they play an important role in maintaining neuronal structure. After neuronal damage, NfL reaches the interstitial fluid, which communicates freely with the CSF and the blood (Gaetani et al., 2019). Several studies have explored the added values of this biomarker in the diagnostic and prognostic assessment of suspected CJD cases, with promising results. Moreover, given the high awareness of clinical neurologists for the treatable forms of RPD, particularly for autoimmune encephalitis (AE), attempts to evaluate the diagnostic and prognostic role of NfL have also been recently extended to this field.
In the present review, we summarize the current knowledge regarding the biological and clinical implications of CSF and blood NfL across the most prevalent etiologies of RPD.
Prion diseases or transmissible spongiform encephalopathies are rare, phenotypically heterogeneous, rapidly progressive neurodegenerative diseases (Parchi and Saverioni, 2012). The pathogenesis of prion diseases relies on the templated seeded conversion of the cellular prion protein (PrPC), encoded by the prion protein (PRNP) gene, into a pathological isoform with abnormal conformation (PrPSc), which shows a tendency to aggregate and forms amyloid fibrils (Parchi and Saverioni, 2012; Puoti et al., 2012; Baiardi et al., 2019).
Sporadic CJD (sCJD) represents the most common form of human prion disease and accounts for about 85–90% of cases. The second most common variant (10–15% of cases) is genetic prion disease, which is related to pathogenic mutations in PRNP and encompasses three distinct clinical–pathological phenotypes, the genetic form of CJD (gCJD), fatal familial insomnia (FFI), and Gerstmann–Sträussler–Scheinker syndrome (GSS). Finally, a small proportion of cases (1–2%) is acquired (Baiardi et al., 2019). In turn, sCJD includes six major clinicopathological subtypes that are mainly determined by the genotype at the methionine (M)/valine (V) polymorphic codon 129 of the PRNP gene and the type (1 or 2) of PrPSc accumulating in the brain. The subtypes are named and classified according to these two main molecular variables into MM(V)1, MM2 cortical (MM2C), MM2 thalamic (MM2T), MV2 kuru (MV2K), VV1, and VV2 subtypes (Parchi et al., 1999, 2012; Baiardi et al., 2019).
Several studies (Table 1) evaluated CSF NfL in the prion disease spectrum. They showed significantly increased mean levels in most disease subtypes compared to non-neurodegenerative controls and other NDs (Steinacker et al., 2016; Kovacs et al., 2017; Abu-Rumeileh et al., 2018b, 2019a, 2020a; Zerr et al., 2018; Kanata et al., 2019; Vallabh et al., 2020). Interestingly, CSF NfL concentration varied significantly among sCJD subtypes and only partially correlated with t-tau levels (Abu-Rumeileh et al., 2018b; Zerr et al., 2018), suggesting that the two markers reflect distinct pathophysiological mechanisms. Both t-tau and NfL levels seem to be influenced by the neuronal degeneration occurring in a given period (i.e., speed of disease progression). Still, NfL raises more significantly than t-tau in the diseases with more widespread subcortical pathology (i.e., in deep nuclei, brainstem, cerebellum, and spinal cord), likely because of a more significant involvement of myelinated axons in white matter tracts in these regions compared to the cerebral cortex (Abu-Rumeileh et al., 2018b). Indeed, sCJD subtypes VV2 and MV2K showed significantly higher CSF NfL levels in comparison to the MM(V)1 group (Abu-Rumeileh et al., 2018b, 2020a; Zerr et al., 2018), which correlates with the more widespread and severe subcortical pathology and the higher amount of PrPSc accumulation and microglial activation in subcortical white matter in the former subtypes (Parchi et al., 2012; Baiardi et al., 2017; Franceschini et al., 2018). The divergent behavior of NfL and t-tau is especially evident in the MV2K subtype, which shows significantly lower concentrations of 14-3-3 and t-tau in CSF (Lattanzio et al., 2017) and a slower disease progression than the VV2. This might also explain the very high sensitivity of NfL for other slowly progressive prion disease subtypes showing low CSF values of t-tau and protein 14-3-3 [e.g., sCJD MM2C, gCJD E200K, GSS, FFI, and variable protease-sensitive prionopathy (VPSPr)] (Abu-Rumeileh et al., 2018b, 2019a; Zerr et al., 2018).
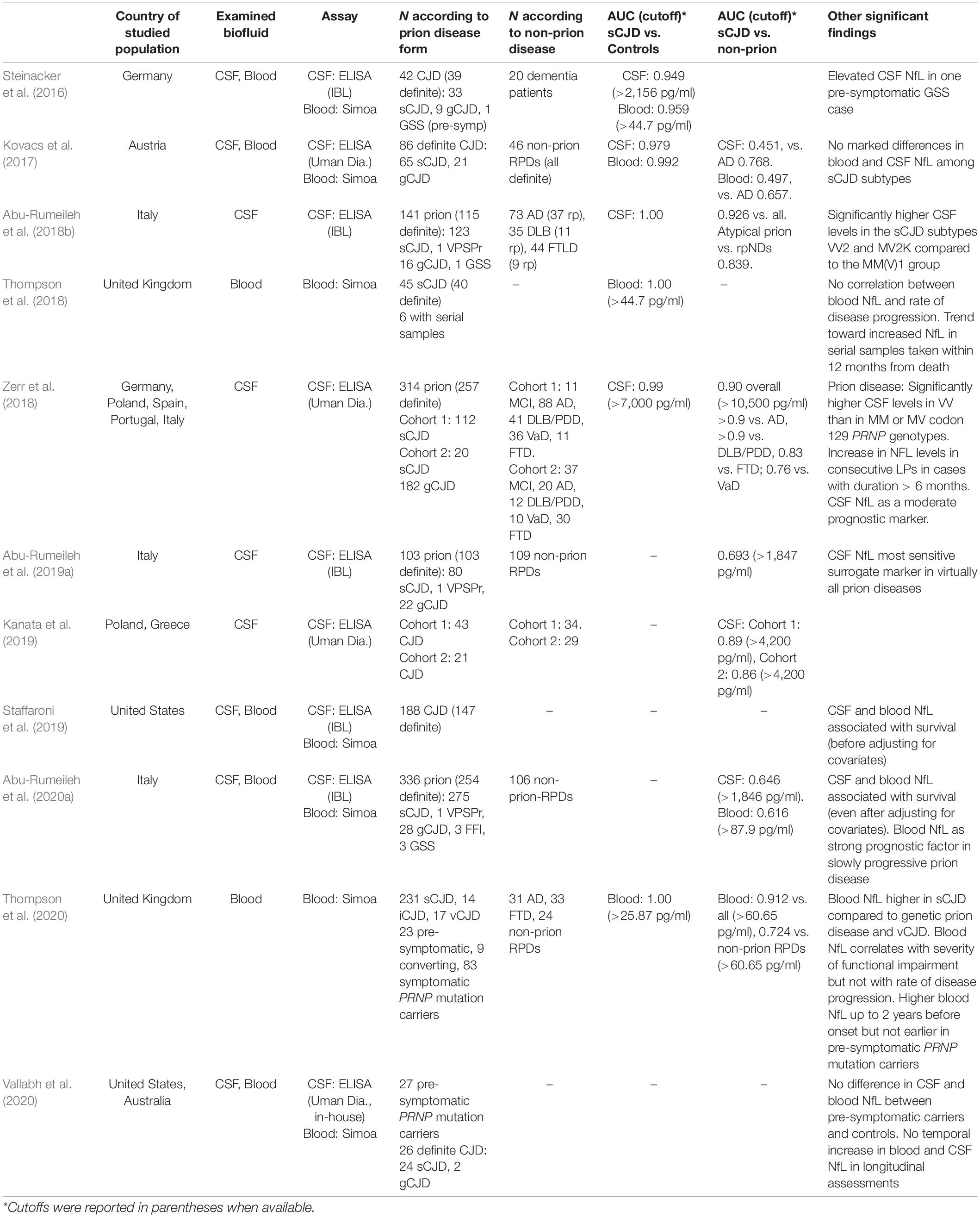
Table 1. Main findings of studies evaluating CSF and blood NfL in prion disease.
CSF NfL showed good diagnostic value across different studies on prion disease and full discrimination between CJD patients and controls (AUC 0.949–1.000) (Steinacker et al., 2016; Kovacs et al., 2017; Abu-Rumeileh et al., 2018b; Zerr et al., 2018). Interestingly, CSF NfL, either alone or in combination with other biomarkers, yielded a performance similar to t-tau in the distinction of prion disease from other NDs (AUC 0.926 vs. 0.939) and showed even a higher diagnostic value than t-tau in the specific comparisons between atypical prion disease and other rpNDs (AUC 0.839 vs. 0.722) (Abu-Rumeileh et al., 2018b). However, the biomarker accuracy for an early clinical diagnosis should be ideally assessed in a clinically or neuropathological based cohort of patients with heterogeneous RPD etiologies raising the clinical suspicion of prion disease, but only a few studies considered this approach (Kovacs et al., 2017; Abu-Rumeileh et al., 2019a, 2020a; Kanata et al., 2019). In this type of studies, CSF NfL diagnostic accuracy (AUC 0.451–0.890) was significantly lower than that of CSF t-tau (AUC 0.849–0.918) and/or protein 14-3-3 (AUC 0.711–0.908) (Kovacs et al., 2017; Abu-Rumeileh et al., 2018b, 2020a; Kanata et al., 2019). Indeed, increased levels of NfL are detected in most vascular, and neuroinflammatory diseases, and even in neurodegenerative disorders commonly associated with a slower progressive course than RPDs. However, NfL levels overlapping significantly with those seen in CJD mainly characterize frontotemporal dementia, amyotrophic lateral sclerosis, and atypical parkinsonism (Gaetani et al., 2019). Therefore, given its low specificity, CSF NfL has a limited role as an isolated test in the differential diagnosis of RPDs (Abu-Rumeileh et al., 2019a, 2020a; Kanata et al., 2019). Nevertheless, when specific tests such as RT-QuIC are also adopted in series, CSF NfL might be useful as a first step screening assay for suspected CJD cases as an alternative to t-tau or 14-3-3.
More recently, blood NfL levels were also found significantly higher in patients with prion disease belonging to all CJD forms and subtypes than both controls and other NDs (Table 1; Steinacker et al., 2016; Kovacs et al., 2017; Thompson et al., 2018, 2020; Abu-Rumeileh et al., 2020a). However, in contrast to the CSF analyte, blood NfL levels did not significantly differ among the most prevalent sCJD subtypes (Kovacs et al., 2017; Thompson et al., 2018; Abu-Rumeileh et al., 2020a). Interestingly, sCJD VV2, typically characterized by the highest CSF NfL and t-tau values among sCJD subtypes (Lattanzio et al., 2017; Abu-Rumeileh et al., 2018b, 2020a; Zerr et al., 2018), showed similar blood NfL and reduced blood tau levels compared with the MM(V)1 type (Kovacs et al., 2017; Abu-Rumeileh et al., 2020a). These data suggest that the different regional lesion profiles between the two CJD subtypes (Parchi et al., 1999; Baiardi et al., 2017), particularly the early cortical neuronal damage featuring MM(V)1 but not VV2, might be responsible for a higher spill over of these molecules in the blood compared to other brain regions (Abu-Rumeileh et al., 2020a). An overview of the distribution of CSF and blood NfL and a comparison with other classic and new biofluid markers across distinct prion disease forms and subtypes are provided in Table 2.
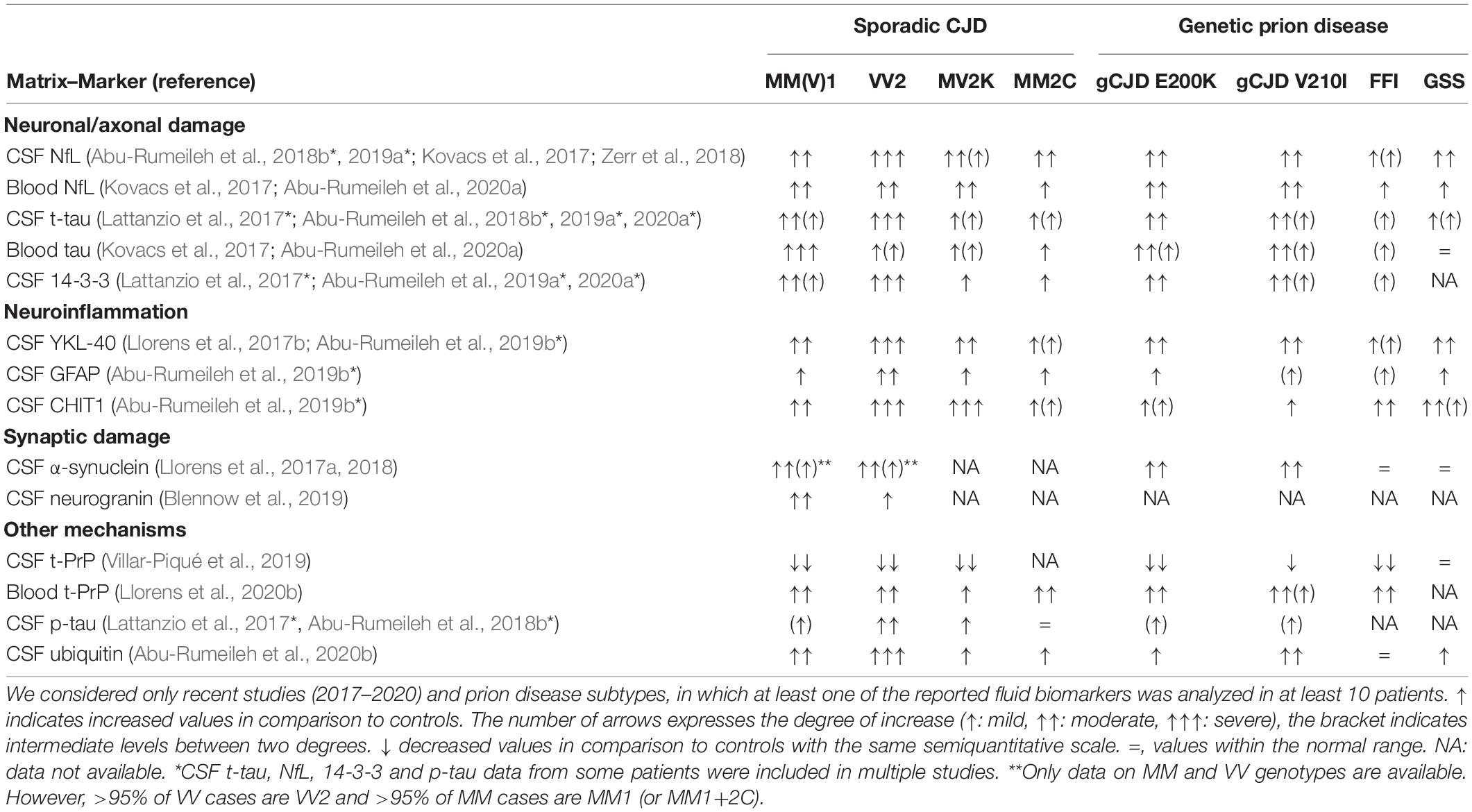
Table 2. Distribution of CSF and blood markers across prion disease subtypes.
Regarding the diagnostic value of blood NfL, only three studies to date evaluated the marker in clinically and/or neuropathological heterogeneous cohorts of RPDs (Kovacs et al., 2017; Abu-Rumeileh et al., 2020a; Thompson et al., 2020). They obtained similar results, namely, the lower value of blood NfL compared to blood tau in the discrimination between prion (or sCJD) and non-prion RPDs (NfL: AUC 0.497–0.724 vs. tau: AUC 0.722–0.837) (Kovacs et al., 2017; Abu-Rumeileh et al., 2020a; Thompson et al., 2020). Notably, the performance of classic CSF markers such as CSF t-tau and 14-3-3 appeared significantly superior to both plasma tau and NfL, raising the question of the real utility of blood surrogate markers in the diagnostic assessment of RPDs (Abu-Rumeileh et al., 2020a).
In conclusion, the very high negative predictive value of NfL for prion disease, due to its high sensitivity for neural damage, may justify its use as an early screening marker. Indeed, low or normal CSF and/or blood levels of NfL in a patient with RPD might exclude with very high certainty the diagnosis of prion disease and induce clinicians to consider other etiologies and to introduce an ex adiuvantibus therapy. In contrast, a significant increase of its levels in at least one biofluid should prompt a second more specific test such as the RT-QuIC.
Different methods have been adopted to assess the rate of clinical progression in prion disease. The MRC Prion Disease Rating Scale, a validated scale of functional impairment, has been used to model linear slopes of functional decline as a measure of the rate of clinical progression (Thompson et al., 2013). Despite the lack of a significant association between NfL values and the rate of disease progression (MRC slope), Thompson et al. (2018, 2020) found a slight correlation between blood NfL values and the degree of functional impairment, along with a tendency toward blood NfL levels raising over time in serial samples of symptomatic patients with increasingly functional impairment. Moreover, they found elevated blood NfL concentrations in all disease stages, with no overlapping values with controls in every sample tested longitudinally, suggesting a potential role of the marker as an outcome measure in clinical trials (Thompson et al., 2020). Similarly, in another study, CSF and blood NfL levels significantly correlated with another scale (i.e., Barthel Index) of functional impairment (Staffaroni et al., 2019).
To date, only three studies explored the association between NfL in CSF and/or blood and survival in prion disease (Zerr et al., 2018; Staffaroni et al., 2019; Abu-Rumeileh et al., 2020a). Zerr and colleagues originally reported that CSF NfL performs only moderately as a prognostic marker (Zerr et al., 2018). However, two studies later showed that both CSF and blood NfL were significantly associated with survival in prion disease/CJD (Staffaroni et al., 2019; Abu-Rumeileh et al., 2020a). However, only Abu-rumeileh et al. stratified the analysis according to the disease subtype, a strong independent factor in prion disease. The collected data showed that while blood and/or CSF t-tau best predicted survival in sCJD MM(V)1 and VV2 subgroups, blood NfL (but not CSF NfL) was significantly associated with survival in the slowly progressive prion diseases. Although a test for in vivo PrPSc typing is currently unavailable, codon 129 genotyping allows partial patient stratification since early disease onset. In this regard, multivariate analyses adjusted for age and codon 129 genotype showed that CSF and blood NfL, CSF tau, and CSF 14-3-3 were still predictive of survival (Staffaroni et al., 2019; Abu-Rumeileh et al., 2020a; Llorens et al., 2020a). Therefore, current evidence strongly recommends a stratification according to disease subtype or at least codon 129 genotype (in the clinical setting) to select outcomes and endpoints in future clinical trials (Abu-Rumeileh et al., 2020a).
Any future treatment aimed to prevent prion disease cannot prescind from the identification and longitudinal evaluation of pre-symptomatic PRNP mutation carriers. The significant heterogeneity of the age at onset in the prion disease spectrum combined with the rarity of the disease strongly limits the use of clinical onset as an outcome in preventive clinical trials. In this regard, identifying a biomarker of proximity to disease onset might be critical to overcome the issue (Minikel et al., 2019, 2020; Vallabh et al., 2020).
To date, only two studies explored neurofilament dynamics in cross-sectional and/or longitudinal samples from pre-symptomatic and symptomatic PRNP mutation carriers. One reported no difference in CSF and/or blood NfL levels between pre-symptomatic PRNP mutation carriers and healthy controls and no temporal increase in longitudinal blood and CSF samples in pre-symptomatic cases (Vallabh et al., 2020). Similarly, in the study of Thompson et al. (2020), NfL concentrations did not differ between pre-symptomatic PRNP mutation carriers with samples collected more than 2 years before symptom onset and those of controls. However, the marker increased progressively in the longitudinal evaluation from 2 years from onset to the symptomatic phase. In summary, the available data on this aspect are, to some extent, controversial. They potentially reflect a common selection bias, namely, the inclusion of subjects with several different PRNP mutations and heterogeneous clinical progression rates.
Nevertheless, recent evidence indicates that blood NfL may perform well as a surrogate endpoint of response to antisense oligonucleotides (ASOs) therapy in a prion disease mouse model (Minikel et al., 2020), where a significant reduction of the biomarker level was documented after drug administration. These findings resembled those obtained in the II phase trial for ASOs in SOD1 ALS (Miller et al., 2020) and open new possible horizons for implementing blood NfL as a reliable and accessible marker of disease monitoring in clinical trials for neurodegenerative diseases.
NDs represent the most common etiology in non-prion RPD patients’ cohorts and a frequent source of CJD misdiagnosis (Geschwind and Murray, 2018; Zerr and Hermann, 2018). In this regard, rapidly progressive variants of AD (rpAD), dementia with Lewy bodies (DLB), and frontotemporal dementia (FTD) may represent distinct subtypes of these disorders characterized by rapid cognitive decline, short disease duration (1–3 years), and subtle distinctive neuropathological and/or biochemical features (Cohen et al., 2015; Drummond et al., 2017; Lee et al., 2017; Abu-Rumeileh et al., 2018a; Geut et al., 2019; Zerr and Hermann, 2018).
The differential diagnosis between CJD and rp-NDs is still challenging, given the partial overlap in clinical features and CSF levels of the surrogate protein markers 14-3-3 and total-tau (Abu-Rumeileh et al., 2018b; Geschwind and Murray, 2018). For this purpose, other CSF biomarkers, including NfL (Abu-Rumeileh et al., 2018b), were tested, especially in the comparison between prion disease and rpAD. However, the evaluation of NfL levels in slowly and rp-ND forms failed to reveal a significant difference between the two groups, apart from FTD (Abu-Rumeileh et al., 2018b).
Another crucial open issue concerns the a priori identification of subjects with faster cognitive decline in AD and FTD cohorts to improve patient stratification for future clinical trials. In this regard, few studies showed an association between high CSF NfL levels and rapid cognitive decline in AD’s mild cognitive impairment stage (Zetterberg et al., 2016; Pillai et al., 2020). Similarly, in the FTD spectrum, higher basal CSF and/or blood NfL values have been related to faster disease progression (in some clinical syndromes) (Ljubenkov et al., 2018; Rojas et al., 2018) and shorter survival (Meeter et al., 2018; van der Ende et al., 2019; Benussi et al., 2020; Cajanus et al., 2020), suggesting the utility of an early NfL assessment in the prediction of future disease aggressiveness.
AE represents an increasingly recognized common cause of RPD (Geschwind and Murray, 2018).
Apart from the relevant diagnostic issue of the exclusion of prion disease (Abu-Rumeileh et al., 2019a), CSF and blood NfL have been explored in AE as easily accessible tools for disease monitoring, prognosis, and response to therapy. Although data are scanty and inconsistent, some findings deserve to be mentioned. In analogy with other RPDs, patients with AE typically showed higher CSF and/or blood NfL values than controls or reference intervals (Constantinescu et al., 2016, 2017; Li et al., 2019; Mariotto et al., 2019). However, concentrations in the normal range have also been reported (Körtvelyessy et al., 2018).
Interestingly, patients on active disease/progression, those on recovery and/or after disease treatment, and those with a stable clinical picture seem to show increased, decreased, or steady levels, respectively (Constantinescu et al., 2016; Li et al., 2019; Mariotto et al., 2019). Given the good correlation between CSF NfL and the disability grade (Constantinescu et al., 2016; Li et al., 2019), these data suggest a role for NfL as a marker of disease activity in AE.
CSF NfL concentration also predicted long-term outcomes in patients with AE (Constantinescu et al., 2016, 2017; Macher et al., 2020). However, most studies showed no associations between NfL values, CSF parameters, and MRI variables (Constantinescu et al., 2016; Mariotto et al., 2019; Macher et al., 2020), reflecting a possible discrepancy between clinical severity and radiological or biochemical features in these conditions (Mariotto et al., 2019).
On a significant issue, all the studies mentioned above merged patients with several subtypes of AE, raising concerns about a possible selection bias that may have influenced the results discussed above.
In RPDs secondary to infectious etiologies, CSF NfL has been reported to predict long-term neurocognitive status in herpes simplex encephalitis (Westman et al., 2020). In contrast, the measurement of NfL concentrations in patients with varicella-zoster encephalitis showed discordant results between studies (Eckerström et al., 2020; Tyrberg et al., 2020). Moreover, despite an inconsistent correlation between NfL levels and degree of cognitive impairment, NfL concentrations have been reported to increase 2 years before the appearance of cognitive decline in patients with HIV–dementia complex, suggesting a possible prognostic role for the marker (Gisslén et al., 2007; McLaurin et al., 2019).
Interestingly, NfL data have also been recently obtained in cerebrovascular disease forms that are occasionally associated with a rapid course, such as small vessel disease. Here, blood NfL concentrations have been associated with disability and neurological symptoms, including future cognitive impairment (Gattringer et al., 2017; Duering et al., 2018; Peters et al., 2020). Moreover, the marker correlated with disease severity, disease progression, and survival in CADASIL patients (Gravesteijn et al., 2018).
Here, we have provided an overview of currently available CSF and blood NfL data on RPDs. Evidence suggests the potential value of NfL as a first-step diagnostic test in the assessment of RPD patients to quickly screen for the presence of ongoing neuronal damage before performing more specific and/or expensive investigations such as RT-QuIC and/or MRI. Moreover, NfL measurement in biofluids might be useful to track the disease trajectory in prion disease and other neurodegenerative dementias. In this regard, higher levels of CSF and/or blood protein seemed to predict a faster disease progression, higher grade of functional impairment, and/or shorter survival, which are all relevant variables for the stratification and management of patients in ongoing clinical trials. Furthermore, preliminary evidence suggests that blood NfL may have a role as a marker of proximity to clinical onset and, possibly, as the surrogate endpoint of response to ASOs in pre-symptomatic PRNP mutation carriers, which are currently the best candidates for new therapeutic options. While in cerebrovascular disease, the evidence of an association between NfL, disease severity, and/or outcome is growing, the role of the marker in AE and infectious encephalitis is not well-clarified, given that current data sets include small cohorts of heterogeneous etiologies and/or insufficient data. In the era of precision medicine, the imminent application of biofluid markers in the routine flow chart may significantly improve the diagnostic and prognostic assessment of patients with RPD.
SA-R and PP assembled all the data and wrote the manuscript. Both authors contributed to the article and approved the submitted version.
The reviewer PH declared a past co-authorship with one of the authors PP to the handling editor.
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abu-Rumeileh, S., Baiardi, S., Ladogana, A., Zenesini, C., Bartoletti-Stella, A., Poleggi, A., et al. (2020a). Comparison between plasma and cerebrospinal fluid biomarkers for the early diagnosis and association with survival in prion disease. J. Neurol. Neurosurg. Psychiatry 91, 1181–1188. doi: 10.1136/jnnp-2020-323826
Abu-Rumeileh, S., Baiardi, S., Polischi, B., Mammana, A., Franceschini, A., Green, A., et al. (2019a). Diagnostic value of surrogate CSF biomarkers for Creutzfeldt-Jakob disease in the era of RT-QuIC. J. Neurol. 266, 3136–3143. doi: 10.1007/s00415-019-09537-0
Abu-Rumeileh, S., Capellari, S., and Parchi, P. (2018a). Rapidly Progressive Alzheimer’s Disease: Contributions to Clinical-Pathological Definition and Diagnosis. J. Alzheimers Dis. 63, 887–897. doi: 10.3233/JAD-171181
Abu-Rumeileh, S., Capellari, S., Stanzani-Maserati, M., Polischi, B., Martinelli, P., Caroppo, P., et al. (2018b). The CSF neurofilament light signature in rapidly progressive neurodegenerative dementias. Alzheimers Res. Ther. 10:3. doi: 10.1186/s13195-017-0331-1
Abu-Rumeileh, S., Oeckl, P., Baiardi, S., Halbgebauer, S., Steinacker, P., Capellari, S., et al. (2020b). CSF Ubiquitin Levels Are Higher in Alzheimer’s Disease than in Frontotemporal Dementia and Reflect the Molecular Subtype in Prion Disease. Biomolecules 10:497. doi: 10.3390/biom10040497
Abu-Rumeileh, S., Steinacker, P., Polischi, B., Mammana, A., Bartoletti-Stella, A., Oeckl, P., et al. (2019b). CSF biomarkers of neuroinflammation in distinct forms and subtypes of neurodegenerative dementia. Alzheimers Res. Ther. 12:2. doi: 10.1186/s13195-019-0562-4
Baiardi, S., Capellari, S., Bartoletti Stella, A., and Parchi, P. (2018). Unusual Clinical Presentations Challenging the Early Clinical Diagnosis of Creutzfeldt-Jakob Disease. J. Alzheimers Dis. 64, 1051–1065. doi: 10.3233/JAD-180123
Baiardi, S., Magherini, A., Capellari, S., Redaelli, V., Ladogana, A., Rossi, M., et al. (2017). Towards an early clinical diagnosis of sporadic CJD VV2 (ataxic type). J. Neurol. Neurosurg. Psychiatry 88, 764–772. doi: 10.1136/jnnp-2017-315942
Baiardi, S., Rossi, M., Capellari, S., and Parchi, P. (2019). Recent advances in the histo-molecular pathology of human prion disease. Brain Pathol. 29, 278–300. doi: 10.1111/bpa.12695
Barro, C., Chitnis, T., and Weiner, H. L. (2020). Blood neurofilament light: a critical review of its application to neurologic disease. Ann. Clin. Transl. Neurol. 7, 2508–2523. doi: 10.1002/acn3.51234
Benussi, A., Karikari, T. K., Ashton, N., Gazzina, S., Premi, E., Benussi, L., et al. (2020). Diagnostic and prognostic value of serum NfL and p-Tau181 in frontotemporal lobar degeneration. J. Neurol. Neurosurg. Psychiatry 91, 960–967. doi: 10.1136/jnnp-2020-323487
Blennow, K., Diaz-Lucena, D., Zetterberg, H., Villar-Pique, A., Karch, A., Vidal, E., et al. (2019). CSF neurogranin as a neuronal damage marker in CJD: a comparative study with AD. J. Neurol. Neurosurg. Psychiatry 90, 846–853. doi: 10.1136/jnnp-2018-320155
Cajanus, A., Katisko, K., Kontkanen, A., Jääskeläinen, O., Hartikainen, P., Haapasalo, A., et al. (2020). Serum neurofilament light chain in FTLD: association with C9orf72, clinical phenotype, and prognosis. Ann. Clin. Transl. Neurol. 7, 903–910. doi: 10.1002/acn3.51041
Candelise, N., Baiardi, S., Franceschini, A., Rossi, M., and Parchi, P. (2020). Towards an improved early diagnosis of neurodegenerative diseases: the emerging role of in vitro conversion assays for protein amyloids. Acta Neuropathol. Commun. 8:117. doi: 10.1186/s40478-020-00990-x
Cohen, M. L., Kim, C., Haldiman, T., ElHag, M., Mehndiratta, P., Pichet, T., et al. (2015). Rapidly progressive Alzheimer’s disease features distinct structures of amyloid-. Brain 138, 1009–1022.
Constantinescu, R., Krýsl, D., Andrén, K., Asztély, F., Bergquist, F., Zetterberg, H., et al. (2017). Cerebrospinal fluid markers of neuronal and glial cell damage in patients with autoimmune neurologic syndromes with and without underlying malignancies. J. Neuroimmunol. 306, 25–30. doi: 10.1016/j.jneuroim.2017.02.018
Constantinescu, R., Krýsl, D., Bergquist, F., Andrén, K., Malmeström, C., Asztély, F., et al. (2016). Cerebrospinal fluid markers of neuronal and glial cell damage to monitor disease activity and predict long-term outcome in patients with autoimmune encephalitis. Eur. J. Neurol. 23, 796–806. doi: 10.1111/ene.12942
Drummond, E., Nayak, S., Faustin, A., Pires, G., Hickman, R. A., Askenazi, M., et al. (2017). Proteomic differences in amyloid plaques in rapidly progressive and sporadic Alzheimer’s disease. Acta Neuropathol. 133, 933–954. doi: 10.1007/s00401-017-1691-0
Duering, M., Konieczny, M. J., Tiedt, S., Baykara, E., Tuladhar, A. M., Leijsen, E. V., et al. (2018). Serum Neurofilament Light Chain Levels Are Related to Small Vessel Disease Burden. J. Stroke 20, 228–238. doi: 10.5853/jos.2017.02565
Eckerström, M., Nilsson, S., Zetterberg, H., Blennow, K., and Grahn, A. (2020). Cognitive impairment without altered levels of cerebrospinal fluid biomarkers in patients with encephalitis caused by varicella-zoster virus: a pilot study. Sci. Rep. 10:22400. doi: 10.1038/s41598-020-79800-2
Franceschini, A., Strammiello, R., Capellari, S., Giese, A., and Parchi, P. (2018). Regional pattern of microgliosis in sporadic Creutzfeldt-Jakob disease in relation to phenotypic variants and disease progression. Neuropathol. Appl. Neurobiol. 44, 574–589. doi: 10.1111/nan.12461
Gaetani, L., Blennow, K., Calabresi, P., Di Filippo, M., Parnetti, L., and Zetterberg, H. (2019). Neurofilament light chain as a biomarker in neurological disorders. J. Neurol. Neurosurg. Psychiatry 90, 870–881. doi: 10.1136/jnnp-2018-320106
Gattringer, T., Pinter, D., Enzinger, C., Seifert-Held, T., Kneihsl, M., Fandler, S., et al. (2017). Serum neurofilament light is sensitive to active cerebral small vessel disease. Neurology 89, 2108–2114. doi: 10.1212/WNL.0000000000004645
Geschwind, M. D., and Murray, K. (2018). Differential diagnosis with other rapid progressive dementias in human prion diseases. Handb. Clin. Neurol. 153, 371–397. doi: 10.1016/B978-0-444-63945-5.00020-9
Geut, H., Vergouw, L. J. M., Galis, Y., Ingrassia, A., de Jong, F. J., Quadri, M., et al. (2019). Neuropathological and genetic characteristics of a post-mortem series of cases with dementia with Lewy bodies clinically suspected of Creutzfeldt-Jakob’s disease. Parkinsonism Relat. Disord. 63, 162–168. doi: 10.1016/j.parkreldis.2019.02.011
Gisslén, M., Hagberg, L., Brew, B. J., Cinque, P., Price, R. W., and Rosengren, L. (2007). Elevated cerebrospinal fluid neurofilament light protein concentrations predict the development of AIDS dementia complex. J. Infect. Dis. 195, 1774–1778. doi: 10.1086/518043
Grau-Rivera, O., Gelpi, E., Nos, C., Gaig, C., Ferrer, I., Saiz, A., et al. (2015). Clinicopathological Correlations and Concomitant Pathologies in Rapidly Progressive Dementia: A Brain Bank Series. Neurodegener. Dis. 15, 350–360. doi: 10.1159/000439251
Gravesteijn, G., Rutten, J. W., Verberk, I. M. W., Böhringer, S., Liem, M. K., van der Grond, J., et al. (2018). Serum Neurofilament light correlates with CADASIL disease severity and survival. Ann. Clin. Transl. Neurol. 6, 46–56. doi: 10.1002/acn3.678
Hamlin, C., Puoti, G., Berri, S., Sting, E., Harris, C., Cohen, M., et al. (2012). A comparison of tau and 14-3-3 protein in the diagnosis of Creutzfeldt-Jakob disease. Neurology 79, 547–552. doi: 10.1212/WNL.0b013e318263565f
Kanata, E., Golanska, E., Villar-Piqué, A., Karsanidou, A., Dafou, D., Xanthopoulos, K., et al. (2019). Cerebrospinal fluid neurofilament light in suspected sporadic Creutzfeldt-Jakob disease. J. Clin. Neurosci. 60, 124–127. doi: 10.1016/j.jocn.2018.09.031
Körtvelyessy, P., Prüss, H., Thurner, L., Maetzler, W., Vittore-Welliong, D., Schultze-Amberger, J., et al. (2018). Biomarkers of Neurodegeneration in Autoimmune-Mediated Encephalitis. Front. Neurol. 9:668. doi: 10.3389/fneur.2018.00668
Kovacs, G. G., Andreasson, U., Liman, V., Regelsberger, G., Lutz, M. I., Danics, K., et al. (2017). Plasma and cerebrospinal fluid tau and neurofilament concentrations in rapidly progressive neurological syndromes: a neuropathology-based cohort. Eur. J. Neurol. 24, 1326–e77. doi: 10.1111/ene.13389
Lattanzio, F., Abu-Rumeileh, S., Franceschini, A., Kai, H., Amore, G., Poggiolini, I., et al. (2017). Prion-specific and surrogate CSF biomarkers in Creutzfeldt-Jakob disease: diagnostic accuracy in relation to molecular subtypes and analysis of neuropathological correlates of p-tau and Aβ42 levels. Acta Neuropathol. 133, 559–578. doi: 10.1007/s00401-017-1683-0
Lee, E. B., Porta, S., Michael Baer, G., Xu, Y., Suh, E., Kwong, L. K., et al. (2017). Expansion of the classification of FTLD-TDP: distinct pathology associated with rapidly progressive frontotemporal degeneration. Acta Neuropathol. 134, 65–78. doi: 10.1007/s00401-017-1679-9
Li, J., Gu, Y., An, H., Zhou, Z., Zheng, D., Wang, Z., et al. (2019). Cerebrospinal fluid light and heavy neurofilament level increased in anti-N-methyl-d-aspartate receptor encephalitis. Brain Behav. 9:e01354. doi: 10.1002/brb3.1354
Ljubenkov, P. A., Staffaroni, A. M., Rojas, J. C., Allen, I. E., Wang, P., Heuer, H., et al. (2018). Cerebrospinal fluid biomarkers predict frontotemporal dementia trajectory. Ann. Clin. Transl. Neurol. 5, 1250–1263. doi: 10.1002/acn3.643
Llorens, F., Kruse, N., Karch, A., Schmitz, M., Zafar, S., Gotzmann, N., et al. (2018). Validation of α-Synuclein as a CSF Biomarker for Sporadic Creutzfeldt-Jakob Disease. Mol. Neurobiol. 55, 2249–2257. doi: 10.1007/s12035-017-0479-5
Llorens, F., Kruse, N., Schmitz, M., Gotzmann, N., Golanska, E., Thüne, K., et al. (2017a). Evaluation of α-synuclein as a novel cerebrospinal fluid biomarker in different forms of prion diseases. Alzheimers Dement. 13, 710–719. doi: 10.1016/j.jalz.2016.09.013
Llorens, F., Rübsamen, N., Hermann, P., Schmitz, M., Villar-Piqué, A., Goebel, S., et al. (2020a). A prognostic model for overall survival in sporadic Creutzfeldt-Jakob disease. Alzheimers Dement. 16, 1438–1447. doi: 10.1002/alz.12133
Llorens, F., Thüne, K., Tahir, W., Kanata, E., Diaz-Lucena, D., Xanthopoulos, K., et al. (2017b). YKL-40 in the brain and cerebrospinal fluid of neurodegenerative dementias. Mol. Neurodegener. 12:83. doi: 10.1186/s13024-017-0226-4
Llorens, F., Villar-Piqué, A., Schmitz, M., Diaz-Lucena, D., Wohlhage, M., Hermann, P., et al. (2020b). Plasma total prion protein as a potential biomarker for neurodegenerative dementia: diagnostic accuracy in the spectrum of prion diseases. Neuropathol. Appl. Neurobiol. 46, 240–254. doi: 10.1111/nan.12573
Macher, S., Zrzavy, T., Höftberger, R., Altmann, P., Pataraia, E., Zimprich, F., et al. (2020). Longitudinal measurement of CSF neurofilament light in anti-NMDAR encephalitis. Eur. J. Neurol. 2020:14631. doi: 10.1111/ene.14631
Mariotto, S., Gajofatto, A., Zuliani, L., Zoccarato, M., Gastaldi, M., Franciotta, D., et al. (2019). Serum and CSF neurofilament light chain levels in antibody-mediated encephalitis. J. Neurol. 266, 1643–1648. doi: 10.1007/s00415-019-09306-z
McLaurin, K. A., Booze, R. M., and Mactutus, C. F. (2019). Diagnostic and prognostic biomarkers for HAND. J. Neurovirol. 25, 686–701. doi: 10.1007/s13365-018-0705-6
Meeter, L. H. H., Vijverberg, E. G., Del Campo, M., Rozemuller, A. J. M., Donker Kaat, L., de Jong, F. J., et al. (2018). Clinical value of neurofilament and phospho-tau/tau ratio in the frontotemporal dementia spectrum. Neurology 90, 1231–1239e. doi: 10.1212/WNL.0000000000005261
Miller, T., Cudkowicz, M., Shaw, P. J., Andersen, P. M., Atassi, N., Bucelli, R. C., et al. (2020). Phase 1-2 Trial of Antisense Oligonucleotide Tofersen for SOD1 ALS. N. Engl. J. Med. 383, 109–119. doi: 10.1056/NEJMoa2003715
Minikel, E. V., Vallabh, S. M., Orseth, M. C., Brandel, J. P., Haïk, S., Laplanche, J. L., et al. (2019). Age at onset in genetic prion disease and the design of preventive clinical trials. Neurology 93, 125–134e. doi: 10.1212/WNL.0000000000007745
Minikel, E. V., Zhao, H. T., Le, J., O’Moore, J., Pitstick, R., Graffam, S., et al. (2020). Prion protein lowering is a disease-modifying therapy across prion disease stages, strains and endpoints. Nucleic Acids Res. 48:gkaa616. doi: 10.1093/nar/gkaa616
Parchi, P., and Saverioni, D. (2012). Molecular pathology, classification, and diagnosis of sporadic human prion disease variants. Folia Neuropathol. 50, 20–45.
Parchi, P., de Boni, L., Saverioni, D., Cohen, M. L., Ferrer, I., Gambetti, P., et al. (2012). Consensus classification of human prion disease histotypes allows reliable identification of molecular subtypes: an inter-rater study among surveillance centres in Europe and USA. Acta Neuropathol. 124, 517–529. doi: 10.1007/s00401-012-1002-8
Parchi, P., Giese, A., Capellari, S., Brown, P., Schulz-Schaeffer, W., Windl, O., et al. (1999). Classification of sporadic Creutzfeldt-Jakob disease based on molecular and phenotypic analysis of 300 subjects. Ann. Neurol. 46, 224–233.
Peters, N., van Leijsen, E., Tuladhar, A. M., Barro, C., Konieczny, M. J., Ewers, M., et al. (2020). Serum Neurofilament Light Chain Is Associated with Incident Lacunes in Progressive Cerebral Small Vessel Disease. J. Stroke 22, 369–376. doi: 10.5853/jos.2019.02845
Pillai, J. A., Bena, J., Bebek, G., Bekris, L. M., Bonner-Jackson, A., Kou, L., et al. (2020). Inflammatory pathway analytes predicting rapid cognitive decline in MCI stage of Alzheimer’s disease. Ann. Clin. Transl. Neurol. 7, 1225–1239. doi: 10.1002/acn3.51109
Puoti, G., Bizzi, A., Forloni, G., Safar, J. G., Tagliavini, F., and Gambetti, P. (2012). Sporadic human prion diseases: molecular insights and diagnosis. Lancet Neurol. 11, 618–628. doi: 10.1016/S1474-4422(12)70063-7
Rhoads, D. D., Wrona, A., Foutz, A., Blevins, J., Glisic, K., Person, M., et al. (2020). Diagnosis of prion diseases by RT-QuIC results in improved surveillance. Neurology 95, 1017–1026e. doi: 10.1212/WNL.0000000000010086
Rojas, J. C., Bang, J., Lobach, I. V., Tsai, R. M., Rabinovici, G. D., Miller, B. L., et al. (2018). CSF neurofilament light chain and phosphorylated tau 181 predict disease progression in PSP. Neurology 90, 273–281e. doi: 10.1212/WNL.0000000000004859
Rudge, P., Hyare, H., Green, A., Collinge, J., and Mead, S. (2018). Imaging and CSF analyses effectively distinguish CJD from its mimics. J. Neurol. Neurosurg. Psychiatry 89, 461–466. doi: 10.1136/jnnp-2017-316853
Staffaroni, A. M., Kramer, A. O., Casey, M., Kang, H., Rojas, J. C., Orrú, C. D., et al. (2019). Association of Blood and Cerebrospinal Fluid Tau Level and Other Biomarkers With Survival Time in Sporadic Creutzfeldt-Jakob Disease. JAMA Neurol. 76, 969–977. doi: 10.1001/jamaneurol.2019.1071
Steinacker, P., Blennow, K., Halbgebauer, S., Shi, S., Ruf, V., Oeckl, P., et al. (2016). Neurofilaments in blood and CSF for diagnosis and prediction of onset in Creutzfeldt-Jakob disease. Sci. Rep. 6:38737. doi: 10.1038/srep38737
Thompson, A. G. B., Luk, C., Heslegrave, A. J., Zetterberg, H., Mead, S. H., Collinge, J., et al. (2018). Neurofilament light chain and tau concentrations are markedly increased in the serum of patients with sporadic Creutzfeldt-Jakob disease, and tau correlates with rate of disease progression. J. Neurol. Neurosurg. Psychiatry 89, 955–961. doi: 10.1136/jnnp-2017-317793
Thompson, A. G., Anastasiadis, P., Druyeh, R., Whitworth, I., Nayak, A., Nihat, A., et al. (2020). Evaluation of plasma tau and neurofilament light chain biomarkers in a 12-year clinical cohort of human prion diseases. medRxiv 2020:20157594.
Thompson, A. G., Lowe, J., Fox, Z., Lukic, A., Porter, M. C., Ford, L., et al. (2013). The Medical Research Council prion disease rating scale: a new outcome measure for prion disease therapeutic trials developed and validated using systematic observational studies. Brain 136, 1116–1127. doi: 10.1093/brain/awt048
Tyrberg, T., Nilsson, S., Blennow, K., Zetterberg, H., and Grahn, A. (2020). Serum and cerebrospinal fluid neurofilament light chain in patients with central nervous system infections caused by varicella-zoster virus. J. Neurovirol. 26, 719–726. doi: 10.1007/s13365-020-00889-2
Vallabh, S. M., Minikel, E. V., Williams, V. J., Carlyle, B. C., McManus, A. J., Wennick, C. D., et al. (2020). Cerebrospinal fluid and plasma biomarkers in individuals at risk for genetic prion disease. BMC Med. 18:140. doi: 10.1186/s12916-020-01608-8
van der Ende, E. L., Meeter, L. H., Poos, J. M., Panman, J. L., Jiskoot, L. C., Dopper, E. G. P., et al. (2019). Serum neurofilament light chain in genetic frontotemporal dementia: a longitudinal, multicentre cohort study. Lancet Neurol. 18, 1103–1111. doi: 10.1016/S1474-4422(19)30354-0
Villar-Piqué, A., Schmitz, M., Lachmann, I., Karch, A., Calero, O., Stehmann, C., et al. (2019). Cerebrospinal Fluid Total Prion Protein in the Spectrum of Prion Diseases. Mol. Neurobiol. 56, 2811–2821. doi: 10.1007/s12035-018-1251-1
Westman, G., Aurelius, E., Ahlm, C., Blennow, K., Eriksson, K., Lind, L., et al. (2020). Cerebrospinal fluid biomarkers of brain injury, inflammation and synaptic autoimmunity predict long-term neurocognitive outcome in herpes simplex encephalitis. Clin. Microbiol. Infect. 2020:31. doi: 10.1016/j.cmi.2020.09.031
Zerr, I., and Hermann, P. (2018). Diagnostic challenges in rapidly progressive dementia. Expert Rev. Neurother. 18, 761–772. doi: 10.1080/14737175.2018.1519397
Zerr, I., and Parchi, P. (2018). Sporadic Creutzfeldt-Jakob disease. Handb. Clin. Neurol. 153, 155–174. doi: 10.1016/B978-0-444-63945-5.00009-X
Zerr, I., Schmitz, M., Karch, A., Villar-Piqué, A., Kanata, E., Golanska, E., et al. (2018). Cerebrospinal fluid neurofilament light levels in neurodegenerative dementia: Evaluation of diagnostic accuracy in the differential diagnosis of prion diseases. Alzheimers Dement. 14, 751–763. doi: 10.1016/j.jalz.2017.12.008
Keywords: Creutzfeldt-Jakob disease, prion disease, neurofilament light chain, Alzheimer’s disease, frontotemporal dementia, encephalitis, autoimmune, biofluid biomarkers
Citation: Abu-Rumeileh S and Parchi P (2021) Cerebrospinal Fluid and Blood Neurofilament Light Chain Protein in Prion Disease and Other Rapidly Progressive Dementias: Current State of the Art. Front. Neurosci. 15:648743. doi: 10.3389/fnins.2021.648743
Received: 01 January 2021; Accepted: 02 February 2021;
Published: 12 March 2021.
Edited by:
Helene Blasco, Université de Tours, FranceReviewed by:
Matthias Schmitz, University Medical Center Göttingen, GermanyCopyright © 2021 Abu-Rumeileh and Parchi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Piero Parchi, cGllcm8ucGFyY2hpQHVuaWJvLml0
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.